

Abstract
Objective:
To improve phenotype definition in genetic studies of epilepsy, we assessed the familial aggregation of focal seizure types and of specific seizure symptoms within the focal epilepsies in families from the Epilepsy Phenome/Genome Project.
Methods:
We studied 302 individuals with nonacquired focal epilepsy from 149 families. Familial aggregation was assessed by logistic regression analysis of relatives’ traits (dependent variable) by probands’ traits (independent variable), estimating the odds ratio for each symptom in a relative given presence vs absence of the symptom in the proband.
Results:
In families containing multiple individuals with nonacquired focal epilepsy, we found significant evidence for familial aggregation of ictal motor, autonomic, psychic, and aphasic symptoms. Within these categories, ictal whole body posturing, diaphoresis, dyspnea, fear/anxiety, and déjà vu/jamais vu showed significant familial aggregation. Focal seizure type aggregated as well, including complex partial, simple partial, and secondarily generalized tonic-clonic seizures.
Conclusion:
Our results provide insight into genotype–phenotype correlation in the nonacquired focal epilepsies and a framework for identifying subgroups of patients likely to share susceptibility genes.
The search for epilepsy genes is complicated by clinical and genetic heterogeneity. Although research has identified genes that cause rare Mendelian epilepsies, progress has been slower in common, genetically complex epilepsies.1–3 A major impediment is the difficulty in defining genetically homogenous subgroups, leading to reduced power for gene identification.4–6 Phenotype definition, the systematic identification of features likely to result from distinct genetic influences, is essential for addressing this problem.3,7
Identification of symptom profiles that exhibit familial aggregation can provide a scientific basis to define informative phenotypes for molecular genetic studies. Prior studies have provided evidence for distinct genetic influences on localization-related vs generalized epilepsy (GE), on myoclonic vs absence seizures, and on generalized tonic-clonic seizures within the idiopathic GEs (IGEs).8–13 Although distinctive phenotypes have helped direct the search for some focal epilepsy genes,14–18 no systematic study has examined familial aggregation of focal seizure types or specific semiology of focal seizures. We examined familial aggregation of focal seizure types and seizure symptoms among relatives with nonacquired focal epilepsy. We performed this analysis in families from the Epilepsy Phenome/Genome Project (EPGP), a large-scale collaboration to advance understanding of epilepsy genetics. Our results offer insight into genotype–phenotype correlation in genetically complex epilepsies and identify symptoms that may stratify the epilepsies into groups more likely to share susceptibility genes.
METHODS
The Epilepsy Phenome/Genome Project.
The EPGP recruited, phenotyped, and collected genetic samples from over 4,000 participants with epilepsy. The study focused on several family types: sibling and parent–child pairs with GE and nonacquired focal epilepsy (NAFE) and triads of an affected child and 2 unaffected parents with symptomatic epilepsies including infantile spasms, Lennox-Gastaut syndrome, and malformations of cortical development (periventricular nodular heterotopias and polymicrogyria). This analysis is restricted to pairs with NAFE.
EPGP consortium.
EPGP is an international collaboration with 27 sites in the United States, Canada, Argentina, Australia, and New Zealand. Participants were ascertained from local sites, referrals, and a national recruitment campaign between 2009 and 2013. We collected data on demographics, epilepsy phenotype, family history, EEG, neuroimaging, and medication response. To ensure consistency across sites, 6 cores (phenotyping, pharmacogenomics, imaging, electrophysiology, data review, data analysis) monitored data collection. Phenotypic data reside in a centralized repository19 and DNA in the National Institute of Neurological Disorders and Stroke Human Genetics DNA and Cell Line Repository at the Coriell Institute for Medical Research.
Participants.
Eligibility requirements for NAFE participants included enrollment age 4 weeks–60 years, age ≤40 years at first unprovoked seizure, and clear diagnosis of epilepsy, defined as a lifetime history of at least 2 unprovoked seizures or 1 seizure with epileptiform activity on EEG. Individuals were excluded if they had only febrile seizures or acute symptomatic seizures or an acquired CNS injury before epilepsy onset. Also excluded were individuals diagnosed with autistic disorder, pervasive developmental disorder, or severe developmental delay before seizure onset. Severe delay was characterized by at least 50% delay in motor, social, language, or cognitive areas or activities of daily living, assessed by clinical judgment of treating physicians.
Potential participants with a known epilepsy-associated pathogenic mutation were ineligible, unless they had genomic variants of unknown significance or benign polymorphisms. Genetic testing was not required. Participants were required to have at least 2 years of high-quality clinical data, or a complete history if onset was <2 years prior to enrollment. Phenome Core (PC) adjudication was required if records were insufficient.
NAFE classification required neuroimaging to be normal or to demonstrate mesial temporal sclerosis (MTS) or focal cortical dysplasia (FCD). MTS and FCD were not exclusion criteria because these lesions may have genetic associations rather than resulting from exogenous injury. Patients with other structural lesions, such as tumor or stroke, were excluded. In rare cases where MRI was unavailable, CT scans were accepted, with adjudication by the PC. Participants with benign rolandic epilepsy or other clinically defined self-limited epilepsy syndromes did not require neuroimaging. NAFE classification required focal EEG abnormalities (focal slowing or sharp waves on interictal EEG or focal epileptiform ictal EEG abnormality). If a focal EEG abnormality was not present, unambiguous focal seizure semiology was required. PC consensus established epilepsy diagnoses based on clinical history and available records.
The first participant recruited in a family was classified as the proband; if we identified multiple participants simultaneously, the participant with the most complete records became the proband. Probands and relatives had identical inclusion and exclusion criteria. This analysis excluded individuals with both generalized and focal epilepsy, if data were missing for broad seizure symptom categories, or if medical record quality was deemed inadequate or barely adequate.
Data collection.
We collected phenotypic data using in-person or telephone interviews and medical records. We ascertained age at seizure onset, types and frequencies of seizures, semiology and epilepsy syndrome, history of status epilepticus, and history of antiepileptic drug use and response using a semi-structured diagnostic interview adapted from a validated instrument.20,21 We abstracted clinical and laboratory data from medical records and recorded on standardized forms. Blood samples were sent to Coriell for DNA extraction. Genomic analysis is underway via the Epi4K Center without Walls for Collaborative Research in the Epilepsies.22 We uploaded representative EEG tracings, MRI scans, and relevant medical records to the EPGP Data Repository23 for review by the respective cores.
For each participant, the local site principal investigator (PI) completed a final diagnosis form. To improve consistency across sites in classification of focal seizure symptoms, the final diagnosis form defined each symptom.24 Two members of the Data Review Core independently reviewed all data for participants with NAFE, and discussed edits to the final diagnosis form with data reviewers and site PIs.
We characterized ictal NAFE symptoms into broad groups for analysis—motor, sensory, autonomic, psychic, and aphasia—based on the classification scheme reported previously.24 We also examined familial aggregation of specific ictal symptoms. NAFE seizure types were defined as simple partial seizures (aura), complex partial (dyscognitive) seizures, and secondarily generalized tonic-clonic seizures (SGTC).
Standard protocol approvals, registrations, and patient consents.
The institutional review board of each clinical center approved all research. All participants (or their legal representative) provided written informed consent. EPGP is registered with clinicaltrials.gov (NCT00552045).
Statistical analysis.
We restricted analyses to families containing 2 or more individuals with NAFE. We assessed associations of phenotypes within families by logistic regression analysis of relatives’ traits (dependent variable) by probands’ traits (independent variable) in pairs in which both participants had NAFE. Odds ratios (ORs) were estimated as the odds of a relative having the symptom given that the proband had the symptom, divided by the odds of a relative having the symptom given that the proband did not have the symptom. We computed both unadjusted ORs and ORs adjusted for relative’s age, sex, and relationship to the proband by including these variables as additional covariates in the logistic regression models. Relationship to the proband was included as a covariate to control for possible confounding resulting from differences in symptom prevalence according to relationship to the proband.
Analysis of families with more than 2 affected individuals was performed in a pairwise fashion; we assessed the presence of the symptom in relative 1 vs proband, then relative 2 vs proband. To account for multiple testing (we tested for familial aggregation of 26 symptoms and seizure types), we report false discovery rates (FDRs) using the Benjamini and Hochberg linear step-up method.25 A FDR <0.05 was considered significant and <0.10 to be suggestive.
As an internal control, bootstrap resampling was used to test for symptom aggregation between pairs of randomly selected individuals (regardless of family membership). Individuals were selected randomly from half of the total sample and paired with randomly selected individuals from the other half without regard to proband/relative status. Association of focal seizure symptoms in these pairs was assessed using the unadjusted regression method as above. Using the boot package in R, this process was repeated with 1,000 replications with replacement to calculate bootstrap ORs and 95% confidence intervals (CIs) using the bias-corrected and accelerated method.
To examine potential bias from testing the same proband twice in families of 3 individuals with NAFE, analyses were repeated after excluding the second proband–relative pair in each such family. To evaluate potential bias from individuals who might be too young to describe subjective symptoms, analyses were repeated excluding individuals under age 4.
To evaluate the effect of possible within-site correlations in phenotyping methods, we repeated analyses using regression models with generalized estimating equations (GEEs) with clinical site as the clustering variable. GEE analysis was performed using the geepack package in R. All analyses were performed using R (version 3.1.1).
RESULTS
The analysis included 302 participants with NAFE (table 1). These participants belonged to 149 families containing 153 proband–relative pairs concordant for NAFE. Most families (n = 142) contained a single proband–relative pair. Seven families contained 3 affected individuals each. Three of those families had 2 individuals with focal epilepsy and a third individual with generalized epilepsy; the other 4 families contained 3 individuals with focal epilepsy. The distribution of phenotypes in the families containing 3 affected individuals was not consistent with any known genetic epilepsy syndrome.
Table 1.
Demographics and clinical characteristics
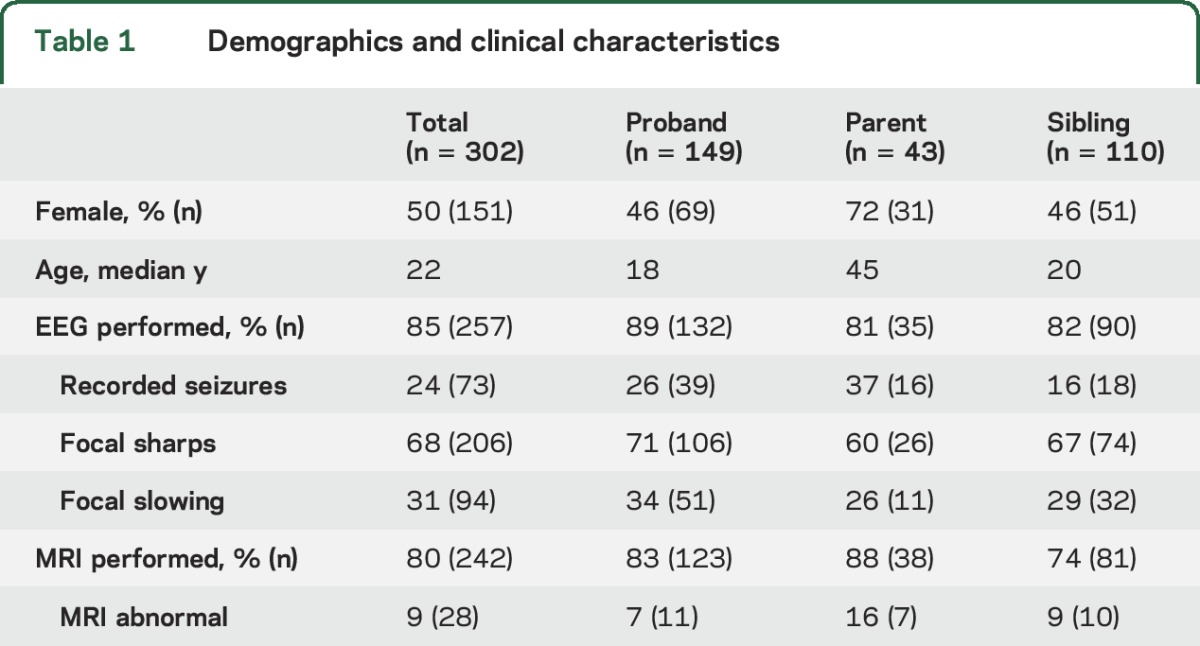
MRI abnormalities were present in fewer than 4% of participants in EPGP overall; approximately one-third of these were MTS. The frequency of abnormal MRI among individuals with NAFE in this analysis was 9% (table 1). Three cases had focal cortical dysplasia, 9 hippocampal sclerosis, 6 cortical atrophy/hypoplasia, 3 a migration abnormality, 2 cortical thinning, and 4 superficial focal cortical T2 hyperintensity. Sample sizes of specific MRI abnormalities were too small for analysis; there were no obvious associations between relative pairs.
Focal seizure symptoms.
Analyses of concordance in proband–relative pairs provided evidence for familial aggregation of motor, autonomic, psychic, and aphasic symptoms. Evidence for familial aggregation of sensory symptoms was not significant (table 2). To confirm that aggregation of symptoms in relative pairs was greater than expected by chance, we performed a bootstrap resampling analysis to test the co-occurrence of symptoms between randomly selected individuals. This revealed no aggregation of motor symptoms (OR 1.2, 95% CI 0.25–4.93), sensory symptoms (OR 1.1, 95% CI 0.50–3.76), autonomic symptoms (OR 0.4, 95% CI 0.12–0.50), psychic symptoms (OR 0.7, 95% CI 0.14–1.35), or aphasia (OR 0.7, 95% CI 0.06–1.59), confirming that these symptoms were aggregating based on family membership.
Table 2.
Familial aggregation of focal seizure characteristics
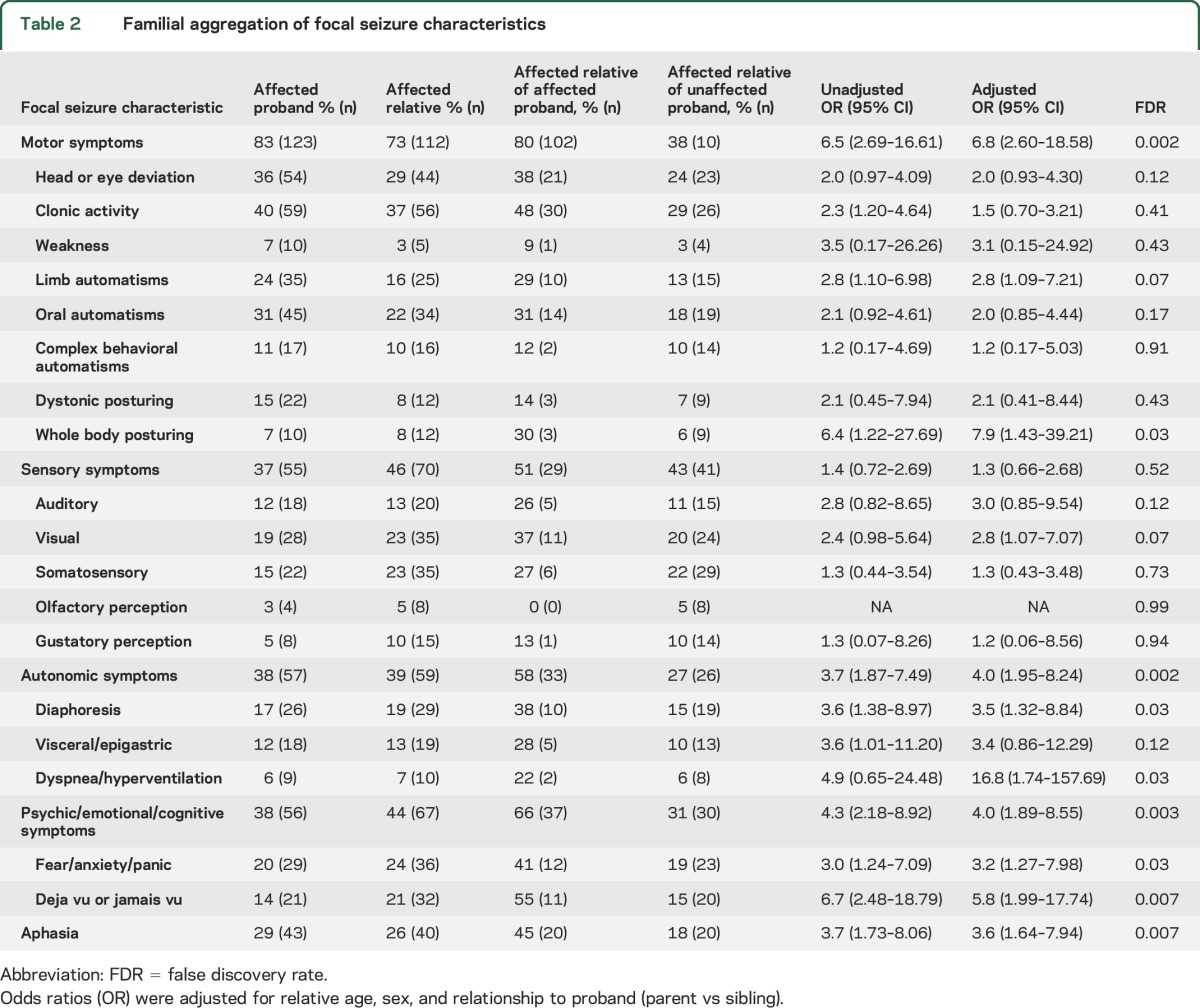
In unadjusted analyses of specific subcategories, we identified evidence for familial aggregation of focal clonus, limb automatisms, and whole body posturing (motor), ictal visual symptoms (sensory), ictal diaphoresis/flushing/pallor and visceral symptoms (autonomic), and déjà vu/jamais vu and fear/panic (psychic). After adjustment for relative age, sex, and relationship to proband, focal clonus and visceral symptoms became nonsignificant. After correction of adjusted models for multiple testing using FDRs, associations remained significant (FDR < 0.05) for motor symptoms (FDR = 0.002), whole body posturing (FDR = 0.03), autonomic symptoms (FDR = 0.002), diaphoresis (FDR = 0.03), dyspnea (FDR = 0.03), psychic symptoms (FDR = 0.003), fear/panic (FDR = 0.03), déjà vu/jamais vu (FDR = 0.007), and aphasia (FDR = 0.007).
Across probands and relatives, the median number of specific seizure symptoms reported was 3. Since combinations of focal symptoms help determine focal epilepsy syndromes, we evaluated familial aggregation of temporal lobe epilepsy (TLE), the most common syndrome (24%) in this cohort. TLE aggregated in families (adjusted OR 5.0, 95% CI 2.02–12.96). Sample sizes of individuals with frontal, parietal, and occipital lobe epilepsy were too small for interpretation.
Since subjective symptoms are difficult to detect in young children, we performed a sensitivity analysis removing pairs with children under age 4, with similar results (table e-1 at Neurology.org). To exclude potential bias from testing the same proband twice in families of 3, we performed a sensitivity analysis removing the second proband–relative pair from those families, again with similar results (table e-2). Finally, to examine possible differences in phenotyping methods across clinical sites, we tested regressions in GEE models using site as the clustering variable. We found no differences in familial aggregation compared to the primary analysis, suggesting results were not explained by within-site correlations in phenotyping (table e-3).
Focal seizure types.
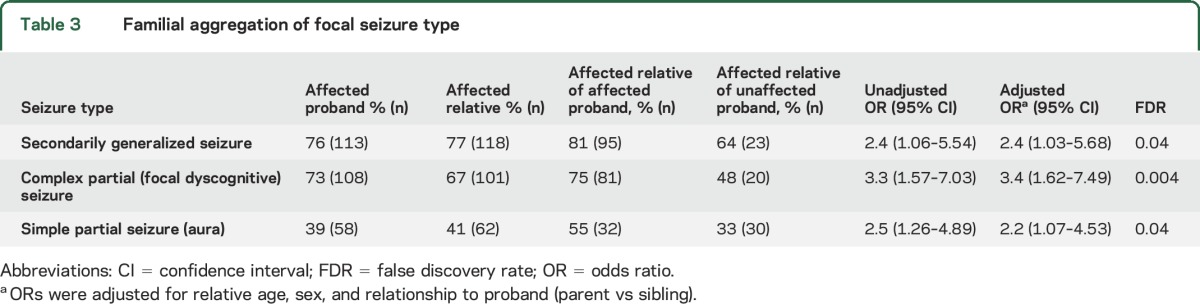
Complex partial (dyscognitive) seizures, simple partial seizures (aura), and SGTCs showed significant familial aggregation in unadjusted and adjusted analyses (table 3). Correcting for multiple comparisons, these associations remained significant for all focal seizure types.
Table 3.
Familial aggregation of focal seizure type
DISCUSSION
We report familial aggregation of specific seizure semiology and seizure types within nonacquired focal epilepsies, expanding on prior findings of familial aggregation of seizure types within the IGEs.11 We found familial aggregation of motor, autonomic, psychic, and aphasic seizure semiology/auras. In analysis of subsymptoms within these broad categories, we also found strong evidence (FDR < 0.05) for familial aggregation of whole body posturing, diaphoresis, dyspnea, fear/panic, and déjà vu/jamais vu, and suggestive evidence (FDR < 0.10) for limb automatisms and visual symptoms. These findings apply specifically to families containing multiple individuals with NAFE. The EPGP dataset’s ability to detect genetic contributions to specific phenotypic characteristics, particularly symptoms or seizure types, underscores the importance of large-scale, detailed phenotyping.
Although we cannot completely exclude study bias, we have minimized it. We considered 3 specific potential sources of bias. First, artefactual familial aggregation might occur if families were more likely to be enrolled if they were concordant for epilepsy features. This is unlikely to have influenced the results because instructions to site personnel explained that eligibility did not depend on concordance of epilepsy type or features within families. Moreover, site personnel were unlikely to restrict enrollment to concordant families because identification of eligible pairs was challenging.
A second potential source of bias was misclassification of focal seizure symptoms based on an assumption that relatives had the same type and symptoms of epilepsy. We attempted to minimize this bias in our data collection by including standardized questionnaires to ascertain symptoms in addition to routine clinical assessment, and by instructing site personnel to classify each participant without taking into account the features observed in relatives. The data review process also addressed this issue; reviewers noted evidence of bias in classification based on a relative’s diagnosis and requested reclassification in such cases. Because diagnoses were not made blind to family relationship, it is impossible to fully eliminate this source of bias. However, although site personnel might have assumed that affected members of the same family both had GE vs NAFE in ambiguous cases, they are less likely to have ascribed specific seizure symptoms to patients, such as déjà vu, based on symptoms in their relatives.
A third source of potential bias is selective recall (or reporting) of symptoms by members of the same family. This is a difficult source of bias to eliminate as communication among family members cannot be avoided. However, our diagnostic interview asks about each symptom systematically, rather than relying on open-ended descriptions, which may lead to reporting bias more frequently. Participants may be more likely to report a specific symptom that is also reported by a relative, but this should be mitigated by asking each participant about the occurrence of each symptom. The extent to which this type of bias may have influenced our results is difficult to estimate. To rule out this source of bias, special study designs might be needed; e.g., using adopted siblings or twins (with different degrees of genetic relatedness but the same symptom familiarity).
Because all participants here had NAFE defined by an electroclinical syndrome, ictal symptoms reported should align with their focal epilepsy syndrome, and familial aggregation of focal symptoms may reflect aggregation of specific syndromes not yet recognized. However, defining epilepsies by syndrome may not be the only way to identify genes, and we have previously demonstrated that a symptom or seizure type–based approach may be valuable in the IGEs.
While we provide evidence for symptom and seizure type–specific effects, some epilepsy genes clearly cause multiple different phenotypes, both within and across families, such as mutations in SCN1A, which are responsible for a wide variety of epilepsy phenotypes in genetic epilepsy with febrile seizures plus,26,27 or mutations in SLC2A1, which give rise to both varied epilepsy and other neurologic phenotypes in GLUT1 deficiency syndrome.28–30 Both types of effect—specific to a narrow phenotype and across phenotypes—have also been demonstrated to occur in the IGEs, in which it appears that some genetic factors raise risk for IGE overall, while others are specific for seizure type.9–12
The familial aggregation of focal ictal features (such as clonic motor manifestations likely to originate from the frontal lobe/motor strip, or auditory symptoms likely to originate in the lateral temporal lobe) suggests that some epilepsy genes play a role in region-specific neuroanatomic development or function. The role of genetic factors in involvement of specific neuroanatomic networks has been established by Mendelian syndromes such as autosomal dominant epilepsy with auditory features (ADEAF)31 and autosomal dominant nocturnal frontal lobe epilepsy (renamed autosomal dominant sleep-related hypermotor epilepsy).16,32 Some of the symptoms that did show familiality (déjà vu and autonomic symptoms) are common in mesial TLE (mTLE). These findings may facilitate gene discovery in mTLE, which is of considerable research interest. Focusing on those specific symptoms of mTLE that appear to aggregate in families may help identify mTLE cases with a shared genetic etiology, and therefore contribute to identification of causative genes, as occur in ADEAF, in which auditory symptoms are a defining feature. In other cases, genetic variants may lead to downstream effects involving intermediate anatomic lesions (e.g., FCD). The small number of lesional epilepsies in this analysis precluded evaluation of such etiologies.
Phenotype definition can also transform understanding of molecular mechanisms that may provide therapeutic targets. Certain genes influence specific clinical characteristics of epilepsy, and evidence to determine which features these phenotype-specific genes dictate is growing.7–11,33 Using a large collection of carefully phenotyped individuals with epilepsy, we have identified attributes of epilepsy that are familial and likely genetic. Systematic efforts to define clinical features that reflect shared genetic susceptibility can help classify the epilepsies in future genetic studies and reduce the heterogeneity that impedes progress in epilepsy genetics research. These results provide strong support for in-depth phenotyping in genetic studies of epilepsy,33 and can help direct the search for epilepsy genes.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the individuals with epilepsy and their families who contributed to this research effort; the EPGP clinical site coordinators, principal investigators, neurologists, and support staff who contributed substantial effort into recruitment, data management, and phenotyping; and the EPGP Community Referral Network (CRN), which consists of health care professionals who refer eligible families to EPGP and are not paid by the EPGP grant. A list of individual contributors can be found at www.epgp.org.
GLOSSARY
- ADEAF
autosomal dominant epilepsy with auditory features
- CI
confidence interval
- EPGP
Epilepsy Phenome/Genome Project
- FCD
focal cortical dysplasia
- FDR
false discovery rate
- GE
generalized epilepsy
- GEE
generalized estimating equation
- IGE
idiopathic generalized epilepsy
- mTLE
mesial temporal lobe epilepsy
- MTS
mesial temporal sclerosis
- NAFE
nonacquired focal epilepsy
- OR
odds ratio
- PC
Phenome Core
- PI
principal investigator
- SGTC
secondarily generalized tonic-clonic seizures
- TLE
temporal lobe epilepsy
Footnotes
Supplemental data at Neurology.org
Editorial, page 14
AUTHOR CONTRIBUTIONS
Melodie Winawer was a coinvestigator of EPGP and served on the EPGP Data Review Core. She conceived of the concordance study purpose and design, supervised analysis and interpretation of the data, and wrote the manuscript. Steven Tobochnik performed the new statistical analyses for the revision and edited the manuscript. Robyn Fahlstrom was the data manager for EPGP. She created datasets, worked collaboratively on analysis of data, and edited the manuscript. Catherine Shain collaborated with Robyn Fahlstrom on data analysis and edited the manuscript. Ruth Ottman supervised and carried out statistical analyses and edited the manuscript, in addition to her role as site principal investigator, as noted in appendix 1. Individual contributions of the other EPGP investigators are listed in appendix 1.
STUDY FUNDING
Supported by The National Institute for Neurologic Diseases and Stroke (grant U01 NS053998) and Finding a Cure for Epilepsy and Seizures (FACES).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ottman R. Analysis of genetically complex epilepsies. Epilepsia 2005;46(suppl 10):7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson MR, Shorvon SD. Heredity in epilepsy: neurodevelopment, comorbidity, and the neurological trait. Epilepsy Behav 2011;22:421–427. [DOI] [PubMed] [Google Scholar]
- 4.Sisodiya SM, Mefford HC. Genetic contribution to common epilepsies. Curr Opin Neurol 2011;24:140–145. [DOI] [PubMed] [Google Scholar]
- 5.Mullen SA, Crompton DE, Carney PW, Helbig I, Berkovic SF. A neurologist's guide to genome-wide association studies. Neurology 2009;72:558–565. [DOI] [PubMed] [Google Scholar]
- 6.Kasperaviciute D, Catarino CB, Heinzen EL, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain 2010;133:2136–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winawer MR. Phenotype definition in epilepsy. Epilepsy Behav 2006;8:462–476. [DOI] [PubMed] [Google Scholar]
- 8.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol 1998;43:435–445. [DOI] [PubMed] [Google Scholar]
- 9.Winawer MR, Marini C, Grinton BE, et al. Familial clustering of seizure types within the idiopathic generalized epilepsies. Neurology 2005;65:523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winawer MR, Rabinowitz D, Barker-Cummings C, et al. Evidence for distinct genetic influences on generalized and localization-related epilepsy. Epilepsia 2003;44:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winawer MR, Rabinowitz D, Pedley TA, Hauser WA, Ottman R. Genetic influences on myoclonic and absence seizures. Neurology 2003;61:1576–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durner M, Keddache MA, Tomasini L, et al. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol 2001;49:328–335. [PubMed] [Google Scholar]
- 13.Kinirons P, Rabinowitz D, Gravel M, et al. Phenotypic concordance in 70 families with IGE-implications for genetic studies of epilepsy. Epilepsy Res 2008;82:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology 2004;62:1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winawer MR, Ottman R, Hauser WA, Pedley TA. Autosomal dominant partial epilepsy with auditory features: defining the phenotype. Neurology 2000;54:2173–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 1995;11:201–203. [DOI] [PubMed] [Google Scholar]
- 17.Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Sutherland GR, Mulley JC. Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2. Nat Genet 1995;10:117–118. [DOI] [PubMed] [Google Scholar]
- 18.Scheffer IE, Bhatia KP, Lopes-Cendes I, et al. Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain 1995;118:61–73. [DOI] [PubMed] [Google Scholar]
- 19.Nesbitt G, McKenna K, Mays V, Carpenter A, Miller K, Williams M. The Epilepsy Phenome/Genome Project (EPGP) informatics platform. Int J med Inform 2013;82:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottman R, Hauser WA, Stallone L. Semistructured interview for seizure classification: agreement with physicians' diagnoses. Epilepsia 1990;31:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ottman R, Lee JH, Hauser WA, et al. Reliability of seizure classification using a semistructured interview. Neurology 1993;43:2526–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epi4K Consortium. Epi4K: gene discovery in 4,000 genomes. Epilepsia 2012;53:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesbitt G, McKenna K, Mays V, et al. The Epilepsy Phenome/Genome Project (EPGP) informatics platform. Int J Med Inform 2013;82:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi H, Winawer MR, Kalachikov S, Pedley TA, Hauser WA, Ottman R. Classification of partial seizure symptoms in genetic studies of the epilepsies. Neurology 2006;66:1648–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 1995;B:289–300. [Google Scholar]
- 26.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490. [DOI] [PubMed] [Google Scholar]
- 27.Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev 2009;31:394–400. [DOI] [PubMed] [Google Scholar]
- 28.Mullen SA, Marini C, Suls A, et al. Glucose transporter 1 deficiency as a treatable cause of myoclonic astatic epilepsy. Arch Neurol 2011;68:1152–1155. [DOI] [PubMed] [Google Scholar]
- 29.Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev 2009;31:545–552. [DOI] [PubMed] [Google Scholar]
- 30.Arsov T, Mullen SA, Damiano JA, et al. Early onset absence epilepsy: 1 in 10 cases is caused by GLUT1 deficiency. Epilepsia 2012;53:e204–e207. [DOI] [PubMed] [Google Scholar]
- 31.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinuper P, Bisulli F, Cross JH, et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 2016;86:1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg DA, Subaran R. Blinders, phenotype, and fashionable genetic analysis: a critical examination of the current state of epilepsy genetic studies. Epilepsia 2011;52:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.