

Abstract
The deep sea is one of the most extensive ecosystems on earth. Organisms living there survive in an extremely harsh environment, and their mitochondrial energy metabolism might be a result of evolution. As one of the most important organelles, mitochondria generate energy through energy metabolism and play an important role in almost all biological activities. In this study, the mitogenome of a deep‐sea sea anemone (Bolocera sp.) was sequenced and characterized. Like other metazoans, it contained 13 energy pathway protein‐coding genes and two ribosomal RNAs. However, it also exhibited some unique features: just two transfer RNA genes, two group I introns, two transposon‐like noncanonical open reading frames (ORFs), and a control region‐like (CR‐like) element. All of the mitochondrial genes were coded by the same strand (the H‐strand). The genetic order and orientation were identical to those of most sequenced actiniarians. Phylogenetic analyses showed that this species was closely related to Bolocera tuediae. Positive selection analysis showed that three residues (31 L and 42 N in ATP6, 570 S in ND5) of Bolocera sp. were positively selected sites. By comparing these features with those of shallow sea anemone species, we deduced that these novel gene features may influence the activity of mitochondrial genes. This study may provide some clues regarding the adaptation of Bolocera sp. to the deep‐sea environment.
Keywords: adaptation, Bolocera sp, deep‐sea sea anemone, mitochondrial genome
1. INTRODUCTION
The deep sea is the part of the ocean below the continental shelves, and it is the most extensive ecosystem on earth (Rex, 1981). The organisms living there survive in an extremely harsh environment, tolerating hundreds of bars of pressure, small amounts of oxygen, very little food, constant darkness, and low temperature (Sanders & Hessler, 1969). Because of the sparseness of animal life and the technical difficulties in sampling the deep‐sea benthos, our knowledge of deep‐sea organisms is based almost entirely on morphological distinctions at the species level (Etter, Rex, Chase, & Quattro, 1999). There is little information about the adaptive molecular mechanisms of the organisms living in the deep‐sea environment (Sanders & Hessler, 1969).
As the powerhouse of the cell, mitochondria generate energy by oxidative phosphorylation (OXPHOS) (Luo et al., 2008) and play important roles in energy metabolism and various biosynthetic pathways (Green & Reed, 1998; Newmeyer & Ferguson‐Miller, 2003). Mitochondria, with their own genetic material, are present in nearly all eukaryotic cells (Bernt, Braband, Schierwater, & Stadler, 2013), and they are autonomously replicated and transcribed (Boore, 1999). Mitogenomes have been widely used for studies of population genetics, phylogeography, phylogeny, and species identification (Brown, Brooke, Fordyce, & McCracken, 2011; Feng, Li, Kong, & Zheng, 2011; Gvoždík, Moravec, Klütsch, & Kotlik, 2010; Keskin & Can, 2009; Lei et al., 2010; Ma et al., 2013). The mitogenome, especially the 13 energy pathway protein‐coding genes, represents a particularly useful genetic marker for investigating the molecular basis of organismal adaptation to an extreme environment (Yu, Wang, Ting, & Zhang, 2011). In recent years, several mitochondrial genes have been shown to contain signatures of adaptive evolution, including the cytochrome b gene of alpacas (da Fonseca, Johnson, O'Brien, Ramos, & Antunes, 2008), the cytochrome c oxidase gene of plateau pikas (Luo et al., 2008), the NADH dehydrogenase six gene of domestic horses (Ning, Xiao, Li, Hua, & Zhang, 2010), and the ATP synthase genes of Caprinae (Hassanin, Ropiquet, Couloux, & Cruaud, 2009).
Most metazoan mitogenomes are circle molecules, between 14 and 18 kb in length that encode 37 genes (13 protein‐coding genes, 22 transfer RNA genes, and two ribosomal RNA genes) as well as a putative control region (Boore, 1999; Wolstenholme, 1992). As sea anemones are primitive animals (Putnam et al., 2007), their mitogenomes exhibit some differences. Similar to most metazoan mitogenomes, the typical sea anemone mitogenome is a circular molecule, 16–20 kb in length and encodes 13 energy pathway protein‐coding genes and two ribosomal RNA genes (Beagley, Okimoto, & Wolstenholme, 1998; Emblem et al., 2014; Osigus, Eitel, Bernt, Donath, & Schierwater, 2013). However, some distinctive features have been identified in these mitogenomes. Only 2 of the 22 essential transfer RNAs (tRNAs) are present, one or two genes are interrupted by a group I intron, the open reading frames (ORFs) encode unknown proteins, and there is a very slow nucleotide substitution rate (Beagley, Okada, & Wolstenholme, 1996; Beagley & Wolstenholme, 2013; Emblem et al., 2011; Johansen et al., 2010; Nielsen & Johansen, 2009; Osigus et al., 2013).
Although several complete or partial mitogenomes of sea anemones have been sequenced in recent years, the members of these genera are highly diverse (Emblem et al., 2014), and the information regarding these mitogenomes remains incomplete. For deep‐water species from extreme environments in particular, little mitochondrial data have been reported. To evaluate the variation in deep‐sea sea anemone gene structures and their adaptations to the deep‐sea environment, this study determined the complete mitogenome of a deep‐sea sea anemone (Bolocera sp.), identified its mitochondrial gene structures and arrangements, and elucidated their evolutionary characteristics.
2. MATERIALS AND METHODS
2.1. Specimens and DNA extraction
The specimen was collected on December 15, 2014, from a seamount on the Pacific Ocean (137.44˚E/8.52˚N) at a depth of 1106 m, fixed in 99% ethanol, and stored at 4°C. DNA was extracted using a Genomic DNA Kit (Tiangen Co. Beijing, China) according to the manufacturer's instructions. The specimen was not an endangered or protected species, and no specific permits were required for our collection process.
2.2. PCR and sequencing
To identify the subspecies of the specimen, two conserved genes (COI and cytb) were sequenced. The complete mitogenomes of closely related species were downloaded from the NCBI Entrez Database and amplified to search for conserved regions where primers for the complete mitogenome clone were designed. The complete mitogenome was amplified by overlapping PCR. All PCRs were performed in a 50 μl volume, which included 1 μl template DNA (approximately 100 ng), each primer at a concentration of 0.3 μmol/L, 5 μl of 10 × LA Taq buffer (Mg2+ plus), 5 μl of dNTP Mix (2.5 mmol/L), and 1 U of LA Taq (TaKaRa, Japan). The PCR amplifications used the following procedure: one cycle of denaturation for 5 min at 94°C; 35 cycles of 40 s at 94°C, 40 s at the primer‐specific annealing temperature, and 5 min at 72°C; and finally a 10‐min extension at 72°C. After purification, the PCR products were directly sequenced in both directions three times with the PCR primers. Sequencing was performed by ThermoFisher Scientific (Guangzhou, China).
2.3. Complete mitogenome analysis
The sequence alignment was conducted using Clustal X. The protein‐coding genes and rRNA genes were determined by BLAST and the NCBI Entrez Database and by comparison with the mitogenome sequences of homologous species. The tRNA genes and their secondary structures were predicted by the Web‐based tRNAscan‐SE 1.21 (Lowe & Eddy, 1997). The skew in nucleotide composition was calculated by AT skew and GC skew and measured according to the following formulae: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C) (Perna & Kocher, 1995), where A, T, C, and G are the occurrences of the corresponding bases. Codon usage was calculated by the Codon Usage Database (http://www.kazusa.or.jp/codon/). The gene map of the complete mitogenome was depicted by OGDRAW (http://ogdraw.mpimp-golm.mpg.de/).
2.4. Phylogenetic analysis
To illustrate the phylogenetic relationships among sea anemone, the complete mitogenomes of 23 Anthozoa species were downloaded from the GenBank database, including Aiptasia pulchella (HG423148), Alicia sansibarensis (KR051001), Antholoba achates (KR051002), Bolocera tuediae (HG423145), Halcampoides purpurea (KR051003), Hormathia digitata (HG423146), Isosicyonis striata (KR051006), Metridium senile (HG423143), Nematostella sp. (DQ643835), Sagartia ornata (KR051008), Urticina eques (HG423144), Chrysopathes formosa (NC_008411), Myriopathes japonica (NC_027667), Stichopathes lutkeni (NC_018377), Savalia savaglia (NC_008827), Acropora aculeus (KT001202), Acropora tenuis (NC003522), Pocillopora damicornis (EF526302), Siderastrea radians (DQ643838), Heliopora coerulea (NC_020375), Dendronephthya suensoni (NC_022809), Renilla muelleri (NC_018378), and Stylatula elongata (JX023275). Geodia neptuni (AY320032) (Demospongiae) was selected as the out‐group. The concatenated nucleotide sequences of 13 energy pathway protein‐coding genes were aligned using Clustal X with the default settings. The maximum likelihood (ML) method was employed to analyze the phylogenetic tree. The GTR + I + G model was selected as the best nucleotide substitution model by ModelTest 3.7 (Posada & Crandall, 1998). The ML analysis was performed by MEGA 5.1 with 1,000 bootstrap replicates.
2.5. Positive selection analysis
The selective pressure imposed on the mitogenomes of sea anemones was evaluated using CODEML from the PAML package. Two different tree‐building methods were used because the CODEML likelihood analysis is sensitive to tree topology. The two‐ratio and free‐ratio model (M1 model) was used in the mitogenome analysis. The branch‐site model was used to determine whether these genes have undergone positive selection in the foreground lineage. Bayes Empirical Bayes (BEB) analysis was used to calculate the Bayesian posterior probability of the positively selected sites.
3. RESULTS AND DISCUSSION
3.1. Genome organization
Similar to most metazoan mitogenomes, the mitogenome of Bolocera sp. is a circular molecule. The complete mitochondrial DNA of Bolocera sp. contained 19,463 bp. It shared the highest overall similarity (96.43%) with the B. tuediae mitochondrial DNA sequence (Emblem et al., 2014). In addition to the common individual base composition differences, the alignment of the two complete mitogenomes also showed several insertions or deletions of genetic fragments. Therefore, we speculated that Bolocera sp. was a new subspecies of Bolocera. Similar to those of other anthozoan species, the mitogenome of Bolocera sp. showed a different evolution pattern than most metazoans (Shearer, Van Oppen, Romano, & Wörheide, 2002). It encoded 16 protein‐coding genes (13 energy pathway protein‐coding genes, a heg gene, and two unknown ORFs), two tRNA genes, and two rRNA genes (Figure 1, Table 1). All of the genes were coded by the same strand (the H‐strand) and transcribed in the same direction. The gene order and orientation were identical to other sequenced actiniarians. In all the mitogenomes studied, no gene overlaps were observed, and the intergenic spacers varied. Several typical anthozoan mitogenome features (Beagley & Wolstenholme, 2013; Beagley et al., 1998; Emblem et al., 2014) were also observed in Bolocera sp., including the presence of two group I introns, two tRNA genes, and large intergenic spacers. In addition, two noncanonical protein‐coding genes (ORFC and ORFD) were observed. A genetic fragment similar to the control region (CR) of metazoans was also observed, which was first reported in sea anemones. The complete mitochondrial DNA sequence was deposited in the GenBank database under the accession number KU507297.
Figure 1.
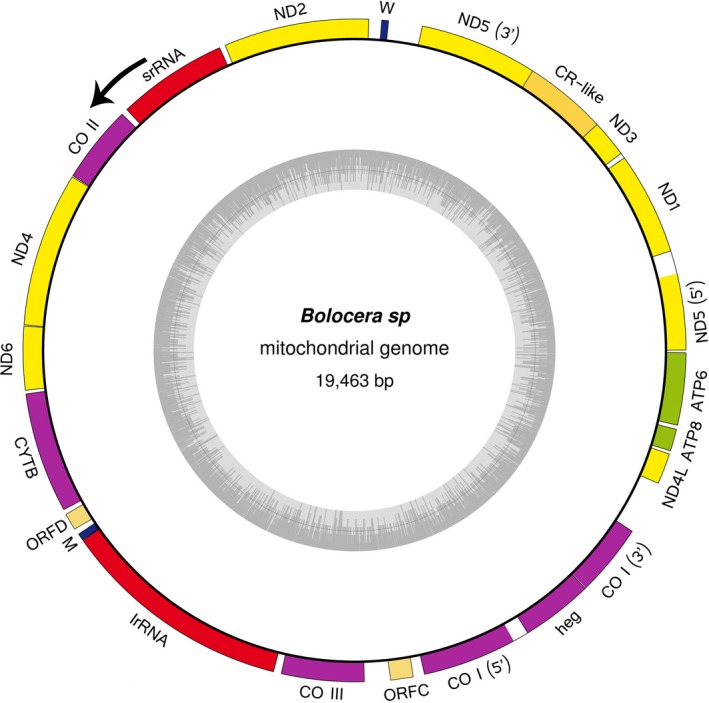
Graphical map of complete mitogenome of Bolocera sp. Different genes are represented by different boxes in different colors. tRNAs are displayed according to the one‐letter code. Genes encoded by the heavy strand are shown outside the circle, and those encoded by the light strand are shown inside. The direction of the arrows shows the direction of transcription. The inner ring shows the GC content of the mitogenome
Table 1.
Gene structure of the mitogenome of Bolocera sp
| Feature | Position numbers | Size | Codon | Intergenic nucleotides | Strand | ||
|---|---|---|---|---|---|---|---|
| Nucleotides | Amino acid | Start | Stop | ||||
| ND5 |
1–717, 3,115–4,230 |
1,833 | 610 | ATG | TAG | 18 | H |
| ND1 | 941–1,924 | 984 | 327 | ATG | TAA | 223 | H |
| ND3 | 1,969–2,325 | 357 | 118 | ATG | TAA | 44 | H |
| tRNA Trp | 4,544–4,613 | 70 | 313 | H | |||
| ND2 | 4,729–6,111 | 1,383 | 460 | ATG | TAG | 115 | H |
| srRNA | 6,161–7,215 | 1,055 | 49 | H | |||
| COII | 7,269–8,015 | 747 | 248 | ATG | TAA | 53 | H |
| ND4 | 8,021–9,496 | 1,476 | 491 | ATG | TAA | 5 | H |
| ND6 | 9,501–10,109 | 609 | 202 | ATG | TAA | 4 | H |
| cytb | 10,137–11,288 | 1,152 | 383 | ATG | TAG | 27 | H |
| ORFD | 11,371–11,505 | 135 | 44 | ATG | TAG | 82 | H |
| tRNA Met | 11,549–11,619 | 71 | 43 | H | |||
| lrRNA | 11,620–13,835 | 2,216 | 0 | H | |||
| COIII | 13,904–14,692 | 789 | 262 | ATG | TAA | 68 | H |
| ORFC | 14,946–15,146 | 201 | 66 | ATG | TAG | 253 | H |
| COI |
15,253–16,142, 16,996–17,677 |
1,572 | 523 | ATG | TAA | 106 | H |
| heg | 16,297–16,986 | 690 | 229 | GTG | TAA | 154 | H |
| ND4L | 18,182–18,481 | 300 | 99 | ATG | TAA | 504 | H |
| ATP8 | 18,506–18,721 | 216 | 71 | ATG | TAA | 24 | H |
| ATP6 | 18,755–19,444 | 690 | 229 | ATG | TAA | 33 | H |
3.2. Group I introns and unknown ORFs
In addition to the standard set of 13 energy pathway protein‐coding genes, an additional heg gene and two group I introns as well as two unknown ORFs were identified in the Bolocera sp. mitogenome (Figures 1 and 2, Table 1). Group I introns are genetic insertion elements that are extremely rare in metazoans and have only been identified within the mitogenomes of hexacorals and some sponges (Boore, 1999). In Lophelia pertusa (a scleractinian coral), a complex group I intron is inserted in the ND5 gene to host seven essential mitochondrial protein genes and a rRNA gene (Emblem et al., 2011). In this study, two group I introns were detected (ND5 intron and CO I intron). Both of the group I introns contained the same gene components as those in other hexacorallian species except for scleractinians (Figure 2). A gene structure analysis of hexacorallians showed that the ND5 intron absorbed more genes but that the CO I intron lost the heg gene. The reason for this difference might be that the scleractinian ancestor had a different evolutionary pathway than other hexacorallian suborder species before significant mitogenome rearrangement, which is supported by the opinion of Mónica (Medina, Collins, Takaoka, Kuehl, & Boore, 2006).
Figure 2.
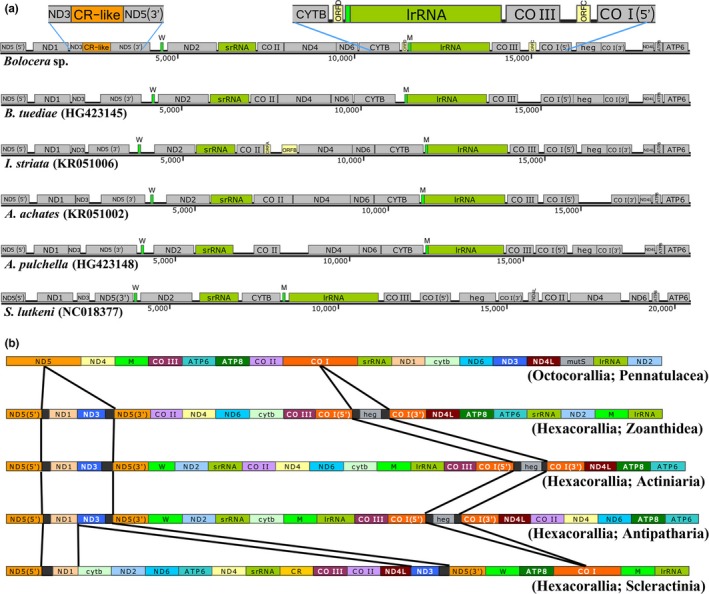
Linearized schemes of mitochondrial gene arrangements in anthozoans. (a) Linearized mitochondrial gene arrangements in actiniarians, Stichopathes lutkeni (Antipatharia) as outgroups. (b) Linearized mitochondrial gene arrangements in different suborders of anthozoa. Lengths of the genes correspond to relative lengths of the genomes in a. tRNAs are displayed according to the one‐letter code. Species names and NCBI accession numbers are given under each of the linearized schemes
Emblem et al. (2014) observed two ORFs (orfA‐51 and orfB‐26) of unknown function in the mitogenome of the cold‐water sea anemone U. eques, and a truncated version of orfA was also found in tropical reef A. pulchella and cold‐water H. digitate mitogenome. Foox, Brugler, Siddall, & Rodríguez (2016) also observed homologous ORFs in other actinioidean species, but no obvious similarity was noted within the ORFs. Instead of the ORFs described above, two new truncated ORFs (ORFC and ORFD) were observed in the Bolocera sp. mitogenome. Consistent with previous studies, no obvious similarity was detected. Flot & Tillier (2007) strongly suggested that the ORFs are expressed proteins, and Emblem even identified the ORFs to be functional elements (Emblem et al., 2014). The ORFs appear to be evolving under some selection relative to other protein‐coding genes (Emblem et al., 2014). The patterns (gained, lost, or truncated) exhibited by these noncanonical ORFs are consistent with transposon‐like elements (Winckler, Szafranski, & Glöckner, 2005). The ORFs detected in this study showed the same characteristics as the ORFs above, so they may play similar roles. Transposable elements can influence neighboring genes by altering splicing and polyadenylation patterns, or by functioning as enhancers or promoters (Girard & Freeling, 1999; Slotkin & Martienssen, 2007; Waterland & Jirtle, 2003). They have been demonstrated to play essential roles in the host response to stress and in facilitating the adaptation of populations (Blot, 1994; Casacuberta & González, 2013; Chénais, Caruso, Hiard, & Casse, 2012). Considering the common features of the species that carry noncanonical ORFs and the particular characteristics of the environment in which they live, the ORFs identified in this study may play important roles in adjusting mitochondrial energy metabolism.
3.3. Genome composition and skewness
In metazoan mitogenomes, the frequency of each nucleotide utilized varies among different taxa. The mitogenome of metazoans usually has a strand‐specific bias in nucleotide composition (Alexandre, Nelly, & Jean, 2005). The A + T content of the mitogenome is extremely high in insects and nematodes and lower in vertebrates (Saccone, Giorgi, Gissi, Pesole, & Reyes, 1999). In the anthozoan mitogenome, the A + T content ranges from 54.9% to 68.1% (Brugler & France, 2007; Foox et al., 2016). The nucleotide composition of the H‐strand in Bolocera sp. was biased toward A and T, and the overall A + T content was 60.51%. Similar results have also been observed in other actiniarian mitogenomes (Foox et al., 2016; Zhang & Zhu, 2016). As is commonly found in metazoan mitogenomes, the A + T content varied in different regions (Table 2). The CR‐like element presented the highest A + T content (66.16%), and the lowest content was found in the 2 tRNA genes (49.65%).
Table 2.
Genomic characteristics of the mitogenome of Bolocera sp
| Species | GenBank accession NO. | H‐strand | 13 energy pathway protein‐coding genesa | lrRNA gene | srRNA gene | 2 tRNA genes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | A + T (%) | AT‐skew | GC‐skew | NO. of amino acid | A + T (%) | Length (bp) | A + T (%) | Length (bp) | A + T (%) | Length (bp) | A + T (%) | ||
| Bolocera sp. | KU507297 | 19,463 | 60.51 | −0.127 | 0.108 | 4,023 | 61.25 | 2,216 | 58.35 | 1,055 | 54.50 | 141 | 49.65 |
| Bolocera tuediae | HG423145 | 19,143 | 60.28 | −0.123 | 0.109 | 4,023 | 61.32 | 2,209 | 58.22 | 1,082 | 54.71 | 141 | 49.65 |
| Aiptasia pulchella | HG423148 | 19,790 | 62.43 | −0.104 | 0.110 | 4,090 | 62.76 | 2,178 | 60.42 | 1,065 | 57.00 | 141 | 47.52 |
| Alicia sansibarensis | KR051001 | 19,575 | 61.01 | −0.126 | 0.110 | 3,893 | 61.55 | 2,200 | 59.73 | 1,074 | 57.36 | 141 | 48.23 |
| Antholoba achates | KR051002 | 17,816 | 61.89 | −0.130 | 0.122 | 4,023 | 62.68 | 2,084 | 59.21 | 1,056 | 53.69 | 141 | 50.36 |
| Halcampoides purpurea | KR051003 | 18,038 | 57.88 | −0.108 | 0.083 | 3,933 | 58.67 | 2,192 | 57.48 | 1,082 | 55.27 | 141 | 51.06 |
| Hormathia digitata | HG423146 | 18,754 | 61.79 | −0.132 | 0.113 | 4,033 | 62.81 | 2,189 | 59.21 | 1,082 | 55.45 | 141 | 49.65 |
| Isosicyonis striata | KR051006 | 19,001 | 60.28 | −0.121 | 0.108 | 3,984 | 61.30 | 2,212 | 58.27 | 1,055 | 54.50 | 141 | 51.77 |
| Metridium senile | HG423143 | 17,444 | 61.86 | −0.129 | 0.112 | 3,953 | 62.67 | 2,188 | 59.37 | 1,082 | 55.27 | 141 | 51.06 |
| Nematostella sp. | DQ643835 | 16,389 | 60.86 | −0.117 | 0.090 | 3,945 | 61.42 | 602 | 56.81 | 693 | 57.29 | 141 | 49.65 |
| Sagartia ornata | KR051008 | 17,446 | 62.21 | −0.126 | 0.114 | 3,962 | 63.11 | 2,201 | 59.29 | 1,082 | 55.73 | 141 | 51.06 |
| Urticina eques | HG423144 | 20,458 | 59.32 | −0.118 | 0.094 | 3,956 | 60.47 | 2,214 | 57.50 | 1,057 | 53.93 | 141 | 49.65 |
The heg gene and two ORFs do not counted in the 13 energy pathway protein‐coding genes.
The AT skew was negative (−0.127), whereas the GC skew was positive (0.108) in Bolocera sp. In all sequenced actiniarian mitogenomes, the trends were the same, and the AT‐skew and GC‐skew values were similar (Table 2). This result indicated that actiniarian mitogenomes favor Ts and Gs. Similar nucleotide skew patterns have also been observed in the mitogenomes of other hexacorallian subclasses (Brugler & France, 2007; van Oppen et al., 2000).
3.4. Protein‐coding genes
In this study, 16 protein‐coding genes (13 energy pathway protein‐coding genes, a heg gene, and two unknown ORFs) were identified (Table 1). All of these genes were coded by the same strand (the H‐strand) and transcribed in the same direction. In metazoans, most of the mitochondrial protein‐coding genes start with an ATN codon (Liao et al., 2010; Ma et al., 2014; Wang, Chao, Fang, & Yu, 2016). In the present study, except for the inserted heg gene, which is considered to be a selfish genetic element (Edgell, 2009), all of the genes were initiated by typical ATG codons. This pattern of initiation codon usage has also been observed in other basal metazoans (Beagley et al., 1998; Boore & Brown, 1995; Pont‐Kingdon et al., 1998).
In metazoan mitogenomes, stop codons are frequently incomplete (Clary & Wolstenholme, 1985; Okimoto, Macfarlane, & Wolstenholme, 1990) and are presumed to be completed by post transcriptional polyadenylation (Ojala, Montoya, & Attardi, 1981). However, in this study, all of the protein‐coding genes were terminated by complete TAA (11) and TAG (5) termination codons. This situation has also been observed in some sequenced cnidaria (Flot & Tillier, 2007; Kayal & Lavrov, 2008). The typical initiation codons and completed termination codons indicated a near standard genetic code, which is rare in metazoan mitogenomes. This result indicated that Bolocera sp. might retain some features of its cnidarian ancestor.
No significant difference in codon usage was detected among the sequenced actiniarians, and the A + T contents of the 13 energy pathway protein‐coding genes were 61.25% in Bolocera sp. In the 13 protein‐coding genes, UUA (Leu, 7.21%), UUU (Phe, 6.21%), and AUU (Ile, 4.56%) were the most frequently utilized codons in Bolocera sp., and the third position of the codons showed relatively high percentage of A and T bases (A, 26.58% and T, 38.06%). These features reflected codon usage with A and T biases at the third codon position, which is similar to the biases observed in most metazoans (Liao et al., 2010; Ma et al., 2014; Miller, Murphy, Burridge, & Austin, 2005; Wang et al., 2016).
3.5. Transfer RNA genes
In most cnidarian mitogenomes, only two tRNAs (tRNATrp and tRNAMet) were detected (except for octocorallians, in which only one tRNAMet was detected) (Beagley et al., 1998; Beaton, Roge, & Cavalier‐Smith, 1998; Kayal & Lavrov, 2008). A study by Beagley and Wolstenholme (2013) showed that nuclear DNA‐encoded functional tRNAs were detected in mitochondria, and the missing tRNAs are believed to be encoded in the nucleus and later imported into the mitochondrion (Schneider & Maréchal‐Drouard, 2000). In this study, the mitogenome of Bolocera sp. contained two tRNAs (tRNATrp and tRNAMet) (Figure 3). Both tRNAs could fold into a clover‐leaf secondary structure, and the anticodon usage was identical to most of the observed sea anemone species. One mismatched base pair (C‐A) was detected in tRNA Met. Interestingly, the unmatched base pair occurred on the amino acid acceptor arm. Such stem mismatches seem to be a common phenomenon for mitochondrial tRNAs in many species (Jiang et al., 2013; Liao et al., 2010; Miller et al., 2005; Wang et al., 2016) and are probably corrected by a post RNA‐editing mechanism (Lavrov, Brown, & Boore, 2000).
Figure 3.
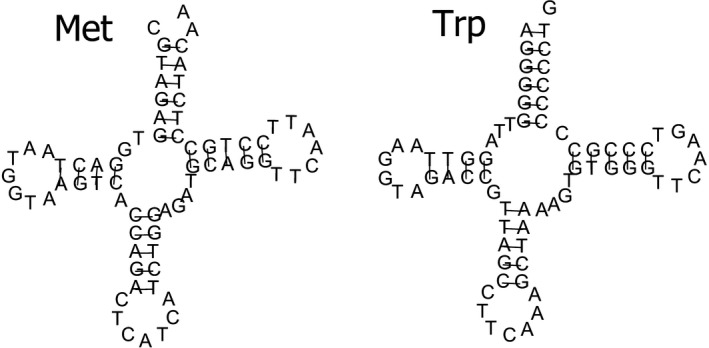
Inferred secondary structure of tRNAs in mitogenome of Bolocera sp.
3.6. Noncoding regions
A total of 22 noncoding regions (ranging from 4 to 789 bp) were identified in the Bolocera sp. mitogenome. The longest noncoding region (789 bp) was located between the ND3 gene and the second exon of the ND5 gene. The nucleotide content of the 789‐bp noncoding region was 238 As (30.16%), 284 Ts (36.00%), 130 Cs (16.48%), and 137 Gs (17.36%). The A + T content (66.16%) of the 789‐bp noncoding region was higher than that of other regions in the mitogenome. In addition, several special TTTT and AAA repeats and T + A‐rich regions were observed (Figure 4).
Figure 4.

The CR‐like sequences of Bolocera sp. The T + A‐rich regions were underlined, and the “G(A)nT” motifs were marked with box
The CR in the mitogenome is essential for transcription and replication in vertebrates (Fernández‐Silva, Enriquez, & Montoya, 2003). As it was not constrained in the same way as the protein‐coding genes, the mitochondrial CR is usually considered to be the most variable portion of the mitogenome (Marshall & Baker, 1997), showing the highest variation in the whole mitogenome (Aquadro & Greenberg, 1983). The structure of the CR has been intensively investigated in vertebrates, but not in invertebrates. Generally, in vertebrates, the mitochondrial CR is divided into three domains, including the extended terminal associated sequences (ETAS), the central conserved sequence blocks (CSB‐F, CSB‐E, and CSB‐D), and the conserved sequence blocks (CSB‐1, CSB‐2, and CSB‐3) (Pesole, Gissi, Chirico, & Saccone, 1999; Sbisà, Tanzariello, Reyes, Pesole, & Saccone, 1997). However, the nucleotide sequence, length, and number of each motif all vary considerably among vertebrate classes and even within a class (Rand, 1993; Ruokonen & Kvist, 2002; Shaffer & McKnight, 1996). In invertebrates, especially in anthozoans, similar CR structures are not clearly defined, but some similar features have been observed. In A. tenuis, the candidate mitochondrial CR contains repetitive elements and has the potential to form the typical secondary structures of vertebrate D–loops (van Oppen et al., 2000). In Pocillopora, the candidate mitochondrial CR exhibits three characteristics: large size, variability in nucleotide composition, and tandemly arranged repeated sequences (Flot & Tillier, 2007). With the exception of the repeated sequences, the rest of these characteristics were all observed in the 789‐bp noncoding region identified here. In addition, the special CR “G(A)nT” motif that is present in S. gregaria and C. parallelus (Zhang, Szymura, & Hewitt, 1995) was also observed in the 789‐bp noncoding region (Figure 4). In all organisms except primates, A + T > G + C in the CR domains (Sbisà et al., 1997), which was also observed in the 789‐bp noncoding region. Replication of mitogenome has been shown to initiate near hairpin structures (Clayton, 2000). In Drosophila, the origin of replication is located near a conserved stem‐loop structure (Saito, Tamura, & Aotsuka, 2005). In this study, the secondary structure of the 789‐bp noncoding fragment presented several similar stem‐loop structures (Figure 5), which is a characteristic feature of O L in vertebrates (Clayton, 1991). In short, while no repetitive elements were observed, the 789‐bp noncoding region exhibited typical CR structures observed in other invertebrates. Considering the primitiveness of sea anemones in evolution, there must be some primitive features maintained. Therefore, we concluded that the 789‐bp noncoding sequence was a candidate CR (that we defined as “CR‐like”) that has not been reported in other actiniarians. This would be a unique and/or primitive mitogenomic feature for Bolocera sp., which is in agreement with the CR differences observed within invertebrates. As no other obvious adjusting elements were detected in the Bolocera sp. mitogenome, this special CR‐like element may play an important role in regulating the transcription and replication of the mitogenome in the extreme environment of the deep sea.
Figure 5.
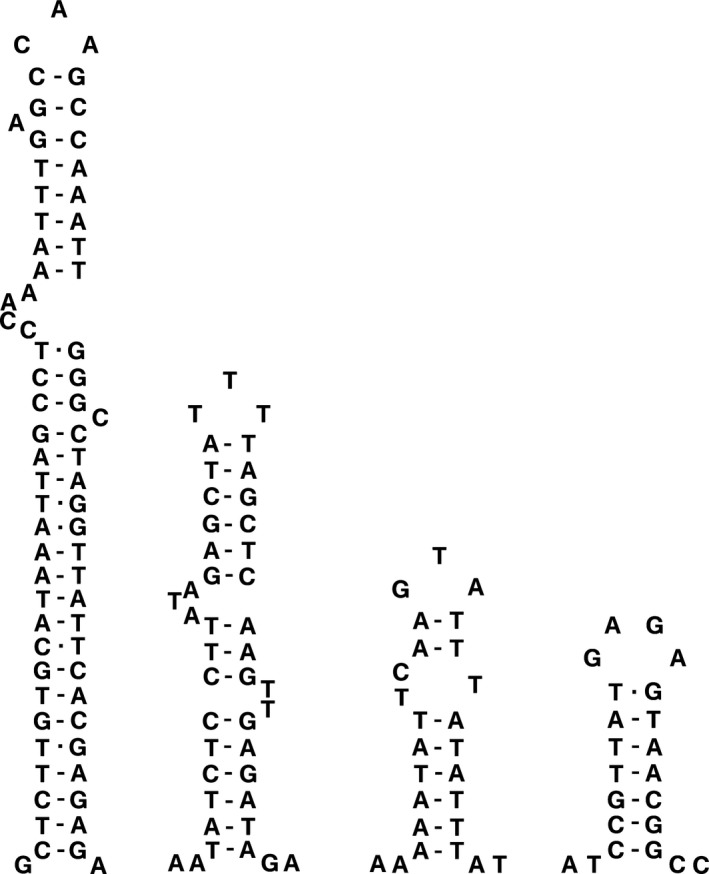
The potential stem‐loop structures in the 789‐bp noncoding “CR‐like” sequence of Bolocera sp.
3.7. Phylogenetic analyses
To investigate the relationships among anthozoan, we performed phylogenetic analysis based on the nucleotide datasets of the 13 mitochondrial energy pathway protein‐coding genes (Figure 6). All of the topologies showed a high support value. The phylogenetic tree of Anthozoa in this study showed that Bolocera sp. was clustered in the Actiniaria clade and had the closest relationship with Bolocera tuediae. The Actiniaria represented a monophyletic group and together with the sister groups Zoanthidea, Antipatharia, and Scleractinia clustered with the Hexacorallia group, which supported the classification of anthozoan (Daly, Fautin, & Cappola, 2003). Based on seven mitochondrial protein‐coding genes, Brugler and France (2007) found that zoanthid was at the base of the hexacoral clade, and the antipatharian clade had high support as a sister‐taxon to the scleractinian clade. However, in this study, based on the 13 protein‐coding genes, the antipatharians were a sister‐group to zoanthids, with a closer relationship to actiniarians, while scleractinians were located at the base of the hexacoral clade, which was identical to results obtained based on 18 S rRNA (Berntson, France, & Mullineaux, 1999). These findings also corroborated earlier studies of sea anemone phylogenetic relationships based on short mitochondrial and nucleotide sequences (Daly, Chaudhuri, Gusmão, & Rodríguez, 2008; Emblem et al., 2014).
Figure 6.
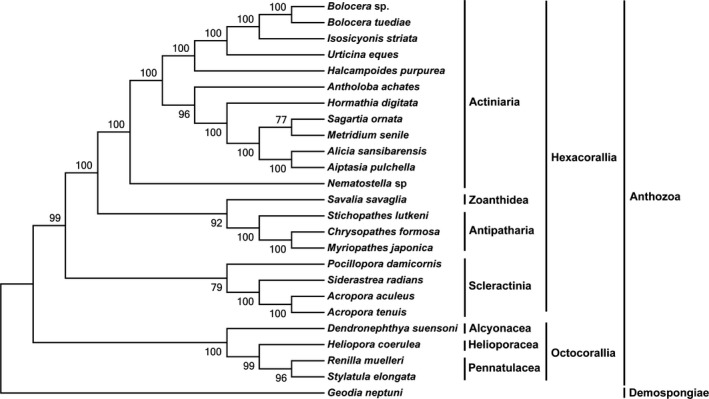
Phylogenetic tree of species of Anthozoa based on ML analysis of the nucleotide datasets. Geodia neptuni was selected as outgroup. Bootstrap support values are shown on the nodes
3.8. Positive selection analysis
The selective pressures imposed on the mitogenomes of sea anemones were evaluated using CODEML from the PAML package (Table 3). Two different tree‐building methods were used because the CODEML likelihood analysis is sensitive to tree topology. There were no significantly different ω ratios between branches of genus Bolocera and other species when we set the genus Bolocera as a foreground branch using the two‐ratio model (p > .05). In the analyses of individual genes, we found that three positive selection sites (31 L and 42 N in ATP6, 570 S in ND5) showed BEB values >0.95 using branch‐site models.
Table 3.
Selective pressure analyses of the mitochondrial genes of sea anemones
| Gene | Branch‐site model | Model compared | 2△lnL | LRT p‐value | Positive sites | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | ln L | Estimates of parameters | |||||||||
| ATP6 | Model A | −3,368.2077 | Site class | 0.0000 | 1.0000 | 2a | 2b | Model A versus Model A null | 2.1423 | .1433 |
31 L 0.994**
42 N 0.991** |
| Proportion | 0.0000 | 0.0000 | 0.9291 | 0.0709 | |||||||
| Background ω | 0.0719 | 1.0000 | 0.0719 | 1.0000 | |||||||
| Foreground ω | 0.0719 | 1.0000 | 999.0000 | 999.0000 | |||||||
| Model A null | −3,369.2788 | ||||||||||
| ND5 | Model A | −9,573.8530 | Site class | 0.0000 | 1.0000 | 2a | 2b | Model A versus Model A null | 0.3829 | .5360 | 570 S 0.978* |
| Proportion | 0.0000 | 0.0000 | 0.9074 | 0.0926 | |||||||
| Background ω | 0.0815 | 1.0000 | 0.0815 | 1.0000 | |||||||
| Foreground ω | 0.0815 | 1.0000 | 26.8046 | 26.8046 | |||||||
| Model A null | −9,574.0445 | ||||||||||
| Trees | Branch model | Model compared | 2△ln L | LRT p‐value | ||
|---|---|---|---|---|---|---|
| Model | ln L | Estimates of parameters | ||||
| NJ | Model 1 | −58,898.9152 | Model 1 versus Model 0 | 477.4924 | .0000 | |
| Two‐ratio | −59,137.6137 | ω0=0.0968 ω1=0.0795 | Two‐ratio versus Model 0 | 0.0954 | .9534 | |
| Model 0 | −59,137.6614 | ω=0.0968 | ||||
| ML | Model 1 | −58,898.6539 | Model 1 versus Model 0 | 478.0150 | .0000 | |
| Two‐ratio | −59,137.6137 | ω0=0.0968 ω1=0.0796 | Two‐ratio versus Model 0 | 0.0954 | .9534 | |
| Model 0 | −59,137.6614 | ω=0.0968 | ||||
*posterior probability >95%; **posterior probability >99%.
The NADH dehydrogenase complex, which likely functions as a proton pump (da Fonseca et al., 2008) and influences metabolic performance (Hassanin et al., 2009), has been considered important in the adaptive evolution of the mammalian mitogenome (da Fonseca et al., 2008; Xu et al., 2007). ND2 and ND6 were found to be under positive selection pressure in a mitogenome analysis of Chinese snub‐nosed monkeys, which is suggestive of adaptive changes related to high altitude and cold weather stress (Yu et al., 2011). ND6 was found to be under positive selection pressure in Tibetan horses living at high altitude (Ning et al., 2010). ATP synthase is directly associated with the produce of ATP (Mishmar et al., 2003; Weiss, Friedrich, Hofhaus, & Preis, 1991; Zhou, Shen, Irwin, Shen, & Zhang, 2014). It has been suggested that variation in ATPase proteins could result in significant variation in mitochondrial adaptation to different environments (Mishmar et al., 2003; Wallace, 2007). Members of the Caprini tribe that live in high‐altitude mountain regions show higher levels of adaptive evolution in the ATP synthase complex (Hassanin et al., 2009). The positive selection of ATP6 and ND5 observed in the present study could help us to better understand the adaptation of organisms to the deep‐sea environment.
4. CONCLUSION
This study characterized the complete mitogenome of a deep‐sea benthos species, Bolocera sp., which was speculated to be a new species of Bolocera. The study provided the following conclusions about deep‐sea organisms: (1) These organisms have monophyletic genome characteristics similar to those of shallow sea organisms: The basic gene content, order, and orientation of the species were identical to most of those reported homologous species, and phylogenetic analyses indicated that Bolocera sp. is closely related to Bolocera tuediae and belongs to the Actiniidae family. (2) Several genes experienced positive selection: Residues 31 L and 42 N in ATP6 and 570 S in ND5 were inferred to be positively selected sites for the branch of Bolocera sp. and B. tuediae, which may indicate that the genes were under positive selection pressure. (3) Novel genetic structures appeared: Some novel/unique gene features were observed in the mitogenomes of deep‐sea organisms compared with those of shallow sea species. In the mitogenome of Bolocera sp., two transposon‐like noncanonical ORFs and a CR‐like structure were detected. These novel genetic structures of Bolocera sp. may provide some clues regarding the adaptation to deep‐sea conditions. This study may shed light on the mitogenomic adaptation of sea anemones that inhabit the deep‐sea environment.
CONFLICT OF INTEREST
The authors report no conflict of interests. The authors alone are responsible for the content and the writing of the article.
ACKNOWLEDGMENTS
This work was financially supported by the Special Fund for Strategic Pilot Technology Chinese Academy of Sciences (Grant No. XDA11030203‐2), the National Science Fund for Excellent Young Scholars (Grant No. 41322038); the National Natural Science Foundation of China (Grant/Award Number: 41576145, 41606170); and GuangDong Oceanic and Fisheries Science and Technology Foundation of (A201601D03).
Zhang B, Zhang Y‐H, Wang X, Zhang H‐X, Lin Q. The mitochondrial genome of a sea anemone Bolocera sp. exhibits novel genetic structures potentially involved in adaptation to the deep‐sea environment. Ecol Evol. 2017;7:4951–4962. https://doi.org/10.1002/ece3.3067
Funding information
This research was supported by Special Fund for Strategic Pilot Technology Chinese Academy of Sciences (Grant/Award Number: XDA11030203‐2); National Science Fund for Excellent Young Scholars (Grant/Award Number: 41322038); the National Natural Science Foundation of China (Grant/Award Number: 41576145, 41606170); and Guangdong Oceanic and Fisheries Science and Technology Foundation (A201601D03)
REFERENCES
- Alexandre, H. , Nelly, L. , & Jean, D. (2005). Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Systematic Biology, 54, 277–298. [DOI] [PubMed] [Google Scholar]
- Aquadro, C. F. , & Greenberg, B. D. (1983). Human mitochondrial DNA variation and evolution: Analysis of nucleotide sequences from seven individuals. Genetics, 103, 287–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagley, C. T. , Okada, N. A. , & Wolstenholme, D. R. (1996). Two mitochondrial group I introns in a metazoan, the sea anemone Metridium senile: One intron contains genes for subunits 1 and 3 of NADH dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America, 93, 5619–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagley, C. T. , Okimoto, R. , & Wolstenholme, D. R. (1998). The mitochondrial genome of the sea anemone Metridium senile (Cnidaria): Introns, a paucity of tRNA genes, and a near‐standard genetic code. Genetics, 148, 1091–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagley, C. T. , & Wolstenholme, D. R. (2013). Characterization and localization of mitochondrial DNA‐encoded tRNAs and nuclear DNA‐encoded tRNAs in the sea anemone Metridium senile . Current Genetics, 59, 139–152. [DOI] [PubMed] [Google Scholar]
- Beaton, M. J. , Roge, A. J. , & Cavalier‐Smith, T. (1998). Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: Conserved gene order among octocorals. Journal of Molecular Evolution, 47(6), 697–708. [DOI] [PubMed] [Google Scholar]
- Bernt, M. , Braband, A. , Schierwater, B. , & Stadler, P. F. (2013). Genetic aspects of mitochondrial genome evolution. Molecular Phylogenetics and Evolution, 69, 328–338. [DOI] [PubMed] [Google Scholar]
- Berntson, E. A. , France, S. C. , & Mullineaux, L. S. (1999). Phylogenetic relationships within the class Anthozoa (Phylum Cnidaria) based on nuclear 18S rDNA sequences. Molecular Phylogenetics and Evolution, 13, 417–433. [DOI] [PubMed] [Google Scholar]
- Blot, M. (1994). Transposable elements and adaptation of host bacteria. Genetica, 93(1–3), 5–12. [DOI] [PubMed] [Google Scholar]
- Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Research, 27, 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore, J. L. , & Brown, W. M. (1995). Complete sequence of the mitochondrial DNA of the annelid worm Lumbricus terrestris . Genetics, 141(1), 305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, V. A. , Brooke, A. , Fordyce, J. A. , & McCracken, G. F. (2011). Genetic analysis of populations of the threatened bat Pteropus mariannus . Conservation Genetics, 12, 933–941. [Google Scholar]
- Brugler, M. R. , & France, S. C. (2007). The complete mitochondrial genome of the black coral Chrysopathes formosa (Cnidaria: Anthozoa: Antipatharia) supports classification of antipatharians within the subclass Hexacorallia. Molecular Phylogenetics and Evolution, 42, 776–788. [DOI] [PubMed] [Google Scholar]
- Casacuberta, E. , & González, J. (2013). The impact of transposable elements in environmental adaptation. Molecular Ecology, 22(6), 1503–1517. [DOI] [PubMed] [Google Scholar]
- Chénais, B. , Caruso, A. , Hiard, S. , & Casse, N. (2012). The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene, 509(1), 7–15. [DOI] [PubMed] [Google Scholar]
- Clary, D. O. , & Wolstenholme, D. R. (1985). The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. Journal of Molecular Evolution, 22(3), 252–271. [DOI] [PubMed] [Google Scholar]
- Clayton, D. A. (1991). Replication and transcription of vertebrate mitochondrial DNA. Annual Review of Cell Biology., 7, 453–478. [DOI] [PubMed] [Google Scholar]
- Clayton, D. A. (2000). Transcription and replication of mitochondrial DNA. Human Reproduction, 15(Suppl 2), 11–17. [DOI] [PubMed] [Google Scholar]
- Daly, M. , Chaudhuri, A. , Gusmão, L. , & Rodríguez, E. (2008). Phylogenetic relationships among sea anemones (Cnidaria: Anthozoa: Actiniaria). Molecular Phylogenetics and Evolution, 48, 292–301. [DOI] [PubMed] [Google Scholar]
- Daly, M. , Fautin, D. , & Cappola, V. A. (2003). Systematics of the Hexacorallia (Cnidaria: Anthozoa). Zoological Journal of the Linnean Society, 139(3), 419–437. [Google Scholar]
- Edgell, D. R. (2009). Selfish DNA: Homing endonucleases find a home. Current Biology, 19, 115–117. [DOI] [PubMed] [Google Scholar]
- Emblem, Å. , Karlsen, B. O. , Evertsen, J. , & Johansen, S. D. (2011). Mitogenome rearrangement in the cold‐water scleractinian coral Lophelia pertusa (Cnidaria, Anthozoa) involves a long‐term evolving group I intron. Molecular Phylogenetics and Evolution, 61, 495–503. [DOI] [PubMed] [Google Scholar]
- Emblem, Å. , Okkenhaug, S. , Weiss, E. S. , Denver, D. R. , Karlsen, B. O. , Moum, T. , & Johansen, S. D. (2014). Sea anemones possess dynamic mitogenome structures. Molecular Phylogenetics and Evolution, 75, 184–193. [DOI] [PubMed] [Google Scholar]
- Etter, R. J. , Rex, M. A. , Chase, M. C. , & Quattro, J. M. (1999). A genetic dimension to deep‐sea biodiversity. Deep Sea Research Part I: Oceanographic Research Papers, 46, 1095–1099. [Google Scholar]
- Feng, Y. , Li, Q. , Kong, L. , & Zheng, X. (2011). DNA barcoding and phylogenetic analysis of Pectinidae (Mollusca: Bivalvia) based on mitochondrial COI and 16S rRNA genes. Molecular Biology Reports, 38, 291–299. [DOI] [PubMed] [Google Scholar]
- Fernández‐Silva, P. , Enriquez, J. A. , & Montoya, J. (2003). Replication and transcription of mammalian mitochondrial DNA. Experimental Physiology, 88, 41–56. [DOI] [PubMed] [Google Scholar]
- Flot, J. F. , & Tillier, S. (2007). The mitogenome of Pocillopora (Cnidaria: Scleractinia) contains two variable regions: The putative D‐loop and a novel ORF of unknown function. Gene, 401, 80–87. [DOI] [PubMed] [Google Scholar]
- da Fonseca, R. R. , Johnson, W. E. , O'Brien, S. J. , Ramos, M. J. , & Antunes, A. (2008). The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics, 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foox, J. , Brugler, M. , Siddall, M. E. , & Rodríguez, E. (2016). Multiplexed pyrosequencing of nine sea anemone (Cnidaria: Anthozoa: Hexacorallia: Actiniaria) mitogenomes. Mitochondrial DNA. Part A, DNA mapping, sequencing, and analysis, 27, 2826–2832. [DOI] [PubMed] [Google Scholar]
- Girard, L. , & Freeling, M. (1999). Regulatory changes as a consequence of transposon insertion. Genesis, 25(4), 291–296. [DOI] [PubMed] [Google Scholar]
- Green, D. R. , & Reed, J. C. (1998). Mitochondria and apoptosis. Science, 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Gvoždík, V. , Moravec, J. , Klütsch, C. , & Kotlik, P. (2010). Phylogeography of the middle eastern tree frogs (Hyla, Hylidae, Amphibia) as inferred from nuclear and mitochondrial DNA variation, with a description of a new species. Molecular Phylogenetics and Evolution, 55, 1146–1166. [DOI] [PubMed] [Google Scholar]
- Hassanin, A. , Ropiquet, A. , Couloux, A. , & Cruaud, C. (2009). Evolution of the mitochondrial genome in mammals living at high altitude: New insights from a study of the tribe Caprini (Bovidae, Antilopinae). Journal of Molecular Evolution, 68, 293–310. [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Wang, G. , Tan, S. , Gong, S. , Yang, M. , Peng, Q. , … Zou, F. (2013). The complete mitochondrial genome sequence analysis of Tibetan argali (Ovis ammon hodgsoni): Implications of Tibetan argali and Gansu argali as the same subspecies. Gene, 521, 24–31. [DOI] [PubMed] [Google Scholar]
- Johansen, S. D. , Emblem, Å. , Karlsen, B. O. , Okkenhaug, S. , Hansen, H. , Moum, T. , … Seternes, O. M. (2010). Approaching marine bioprospecting in hexacorals by RNA deep sequencing. New Biotechnology, 27, 267–275. [DOI] [PubMed] [Google Scholar]
- Kayal, E. , & Lavrov, D. V. (2008). The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene, 410(1), 177–186. [DOI] [PubMed] [Google Scholar]
- Keskin, E. , & Can, A. (2009). Phylogenetic relationships among four species and a sub‐species of Mullidae (Actinopterygii; Perciformes) based on mitochondrial cytochrome B, 12S rRNA and cytochrome oxidase II genes. Biochemical Systematics and Ecology, 37, 653–661. [Google Scholar]
- Lavrov, D. V. , Brown, W. M. , & Boore, J. L. (2000). A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus . Proceedings of the National Academy of Sciences of the United States of America, 97, 13738–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, R. H. , Shore, G. D. , Brenneman, R. A. , Engberg, S. E. , Sitzmann, B. D. , Bailey, C. A. , … Louis, E. E. Jr (2010). Complete sequence and gene organization of the mitochondrial genome for Hubbard's sportive lemur (Lepilemur hubbardorum). Gene, 464, 44–49. [DOI] [PubMed] [Google Scholar]
- Liao, F. , Wang, L. , Wu, S. , Li, Y. P. , Zhao, L. , Huang, G. M. , … Li, M. G. (2010). The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). International Journal of Biological Sciences [Electronic Resource], 6(2), 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T. M. , & Eddy, S. R. (1997). tRNAscan‐SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Gao, W. , Gao, Y. , Tang, S. , Huang, Q. , Tan, X. , … Huang, T. (2008). Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion, 8, 352–357. [DOI] [PubMed] [Google Scholar]
- Ma, H. , Ma, C. , Li, C. , Lu, J. , Zou, X. , Gong, Y. , … Xia, L. (2014). First mitochondrial genome for the red crab (Charybdis feriata) with implication of phylogenomics and population genetics. Scientific Reports, 5, 11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. , Ma, C. , Li, X. , Xu, Z. , Feng, N. , & Ma, L. (2013). The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene, 519, 120–127. [DOI] [PubMed] [Google Scholar]
- Marshall, H. D. , & Baker, A. J. (1997). Structural conservation and variation in the mitochondrial control region of fringilline finches (Fringilla spp.) and the greenfinch (Carduelis chloris). Molecular Biology and Evolution, 14, 173–184. [DOI] [PubMed] [Google Scholar]
- Medina, M. , Collins, A. G. , Takaoka, T. L. , Kuehl, J. V. , & Boore, J. L. (2006). Naked corals: Skeleton loss in scleractinia. Proceedings of the National Academy of Sciences of the United States of America, 103(24), 9096–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, A. D. , Murphy, N. P. , Burridge, C. P. , & Austin, C. M. (2005). Complete mitochondrial DNA sequences of the decapod crustaceans Pseudocarcinus gigas (Menippidae) and Macrobrachium rosenbergii (Palaemonidae). Marine Biotechnology, 7, 339–349. [DOI] [PubMed] [Google Scholar]
- Mishmar, D. , Ruiz‐Pesinia, E. , Golik, P. , Macaulay, V. , Clark, A. G. , Hosseini, S. , … Wallace, D. C. (2003). Natural selection shaped regional mtDNA variation in humans. PNAS, 100, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer, D. D. , & Ferguson‐Miller, S. (2003). Mitochondria: Releasing power for life and unleashing the machineries of death. Cell, 112, 481–490. [DOI] [PubMed] [Google Scholar]
- Nielsen, H. , & Johansen, S. D. (2009). Group I introns: Moving in new directions. RNA Biology, 6, 375–383. [DOI] [PubMed] [Google Scholar]
- Ning, T. , Xiao, H. , Li, J. , Hua, S. , & Zhang, Y. P. (2010). Adaptive evolution of the mitochondrial ND6 gene in the domestic horse. Genetics and Molecular Research, 9, 144–150. [DOI] [PubMed] [Google Scholar]
- Ojala, D. , Montoya, J. , & Attardi, G. (1981). tRNA punctuation model of RNA processing in human mitochondrial. Nature, 290(5806), 470–474. [DOI] [PubMed] [Google Scholar]
- Okimoto, R. , Macfarlane, J. L. , & Wolstenholme, D. R. (1990). Evidence for the frequent use of TTG as the translation initiation codon of mitochondrial protein genes in the nematodes, Ascaris suum and Caenorhabditis elegans . Nucleic Acids Research, 18(20), 6113–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oppen, M. J. , Catmull, J. , McDonald, B. J. , Hislop, N. R. , Hagerman, P. J. , & Miller, D. J. (2000). The mitochondrial genome of Acropora tenuis (Cnidaria: Scleractinia) contains a large group I intron and a candidate control region. Journal of Molecular Evolution, 55, 1–13. [DOI] [PubMed] [Google Scholar]
- Osigus, H. J. , Eitel, M. , Bernt, M. , Donath, A. , & Schierwater, B. (2013). Mitogenomics at the base of metazoa. Molecular Phylogenetics and Evolution, 69(2), 339–351. [DOI] [PubMed] [Google Scholar]
- Perna, N. T. , & Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution, 41, 353–358. [DOI] [PubMed] [Google Scholar]
- Pesole, G. , Gissi, C. , Chirico, A. D. , & Saccone, C. (1999). Nucleotide substitution rate of mammalian mitochondrial genomes. Journal of Molecular Evolution, 48, 427–434. [DOI] [PubMed] [Google Scholar]
- Pont‐Kingdon, G. , Okada, N. A. , Macfarlane, J. L. , Beagley, C. T. , Watkins‐Sims, C. D. , Cavalier‐Smith, T. , … Wolstenholme, D. R. (1998). Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial muts: A possible case of gene transfer from the nucleus to the mitochondrion. Journal of Molecular Evolution, 46(4), 419–431. [DOI] [PubMed] [Google Scholar]
- Posada, D. , & Crandall, K. A. (1998). MODELTEST: Testing the model of DNA substitution. Bioinformatics, 14, 817–818. [DOI] [PubMed] [Google Scholar]
- Putnam, N. H. , Srivastava, M. , Hellsten, U. , Dirks, B. , Chapman, J. , Salamov, A. , … Shapiro, H. , et al. (2007). Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science, 317(5834), 86–94. [DOI] [PubMed] [Google Scholar]
- Rand, D. M. (1993). Endotherms, ectotherms, and mitochondrial genome‐size variation. Journal of Molecular Evolution, 37, 281–295. [DOI] [PubMed] [Google Scholar]
- Rex, M. A. (1981). Community structure in the deep‐sea benthos. Annual Review of Ecology and Systematics, 12, 331–353. [Google Scholar]
- Ruokonen, M. , & Kvist, L. (2002). Structure and evolution of the avian mitochondrial control region. Molecular Phylogenetics and Evolution, 23, 422–432. [DOI] [PubMed] [Google Scholar]
- Saccone, C. , Giorgi, C. D. , Gissi, C. , Pesole, G. , & Reyes, A. (1999). Evolutionary genomics in metazoa: The mitochondrial DNA as a model system. Gene, 238(1), 195–209. [DOI] [PubMed] [Google Scholar]
- Saito, S. , Tamura, K. , & Aotsuka, T. (2005). Replication origin of mitochondrial DNA in insects. Genetics, 171(4), 1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, H. L. , & Hessler, R. R. (1969). Ecology of the deep‐sea benthos. Science, 163, 1419–1424. [DOI] [PubMed] [Google Scholar]
- Sbisà, E. , Tanzariello, F. , Reyes, A. , Pesole, G. , & Saccone, C. (1997). Mammalian mitochondrial D‐loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene, 205, 125–140. [DOI] [PubMed] [Google Scholar]
- Schneider, A. , & Maréchal‐Drouard, L. (2000). Mitochondrial tRNA import: Are there distinct mechanisms? Trends in Cell Biology, 10(12), 509–513. [DOI] [PubMed] [Google Scholar]
- Shaffer, H. B. , & McKnight, M. L. (1996). The polytypic species revisited: Genetic differentiation and molecular phylogenetics of the tiger salamander Ambystoma tigrinum (Amphibian: Caudata) complex. Evolution, 50, 417–433. [DOI] [PubMed] [Google Scholar]
- Shearer, T. L. , Van Oppen, M. J. H. , Romano, S. L. , & Wörheide, G. (2002). Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Molecular Ecology, 11, 2475–2487. [DOI] [PubMed] [Google Scholar]
- Slotkin, R. K. , & Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nature Reviews Genetics, 8(4), 272–285. [DOI] [PubMed] [Google Scholar]
- Wallace, D. C. (2007). Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annual Review of Biochemistry, 76, 781–821. [DOI] [PubMed] [Google Scholar]
- Wang, Z. L. , Chao, L. , Fang, W. Y. , & Yu, X. P. (2016). The complete mitochondrial genome of two Tetragnatha spiders (Araneae: Tetragnathidae): Severe truncation of tRNAs and novel gene rearrangements in araneae. International Journal of Biological Sciences [Electronic Resource], 12(1), 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland, R. A. , & Jirtle, R. L. (2003). Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Molecular and Cellular Biology, 23(15), 5293–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, H. , Friedrich, T. , Hofhaus, G. , & Preis, D. (1991). The respiratory‐chain NADH dehydrogenase (complex I) of mitochondria. European Journal of Biochemistry., 197, 563–576. [DOI] [PubMed] [Google Scholar]
- Winckler, T. , Szafranski, K. , & Glöckner, G. (2005). Transfer RNA gene‐targeted integration: An adaptation of retrotransposable elements to survive in the compact Dictyostelium discoideum genome. Cytogenetic and Genome Research, 110(1–4), 288–298. [DOI] [PubMed] [Google Scholar]
- Wolstenholme, D. R. (1992). Animal mitochondrial DNA: Structure and evolution. International Review of Cytology, 141(6), 173–216. [DOI] [PubMed] [Google Scholar]
- Xu, S. , Luosang, J. , Hua, S. , He, J. , Ciren, A. , Wang, W. , … Zheng, X. (2007). High altitude adaptation and phylogenetic analysis of Tibetan horse based on the mitochondrial genome. Journal of Genetics and Genomics, 34, 720–729. [DOI] [PubMed] [Google Scholar]
- Yu, L. , Wang, X. P. , Ting, N. , & Zhang, Y. P. (2011). Mitogenomic analysis of Chinese snub‐nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high‐altitude adaptation. Mitochondrion, 11, 497–503. [DOI] [PubMed] [Google Scholar]
- Zhang, D. X. , Szymura, J. M. , & Hewitt, G. M. (1995). Evolution and structural conservation of the control region of insect mitochondrial DNA. Journal of Molecular Evolution, 40, 382–391. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , & Zhu, Q. (2016). Complete mitochondrial genome of the sea anemone, Anthopleura midori (Actiniaria: Actiniidae). Mitochondrial DNA. Part A, DNA Mapping, Sequencing, and Analysis, 1–2. [DOI] [PubMed] [Google Scholar]
- Zhou, T. , Shen, X. , Irwin, D. M. , Shen, Y. , & Zhang, Y. (2014). Mitogenomic analyses propose positive selection in mitochondrial genes for high‐altitude adaptation in galliform birds. Mitochondrion, 18, 70–75. [DOI] [PubMed] [Google Scholar]