

Abstract
We previously communicated that high α-selectivity that can be achieved in intramolecular glycosylations using a rigid bisphenol A template supplemented with linkers of various lengths. Herein, we present our investigation of the mechanistic aspects of the templated synthesis that helped to design an improved template-linker combination. We demonstrate that bisphenol A as the template in combination with phthaloyl linker allows for superior stereoselectivity and yields in glycosylations. Several mechanistic studies explore origins of the enhanced stereoselectivity and yields achieved using the phthaloyl linker.
Graphical Abstract
INTRODUCTION
An issue of controlling the stereoselectivity of glycosylations has been approached in a variety of modes.1,2 Among these, intramolecular approaches occupy an important niche among other methods available.3–6 The basis of the concept is that the two glycosylation components, glycosyl donor and acceptor, are tethered together using a suitable linker. The purpose of this tethering is to achieve an efficient facial selectivity due to steric or geometric constraints and forces.
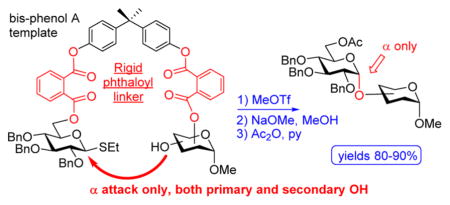
Recently, we introduced a type of templated oligosaccharide synthesis,7 a method where glycosylations were performed using the general conventions of the “molecular clamp” concept.8–16 Bisphenol A (BPA) was used as the template, and succinoyl, glutaryl, or phthaloyl linkers were used to tether glycosyl donors and acceptors together. The general outline of the templated synthesis is shown in Scheme 1. The templated synthesis differs from the general molecular clamping by allowing for glycosylation of different hydroxyl groups, not only those adjacent to the tether and allows for connecting multiple building blocks for oligosaccharide synthesis. If the synthesis of a disaccharide is targeted, a glycosyl donor equipped with linker 1 is connected to a glycosyl acceptor bearing linker N via a template. The resulting donor–acceptor tethered pair is then subjected to glycosylation, and the disaccharide is cleaved off of the template. In cases when the synthesis of an oligo- or even a polysaccharide is attempted, a series of building blocks equipped with various linkers are connected via a template in a sequential manner (Scheme 1). In principle, the connection can be performed as a polymerization if all building blocks are the same (or a copolymerization if repetitive sequences are attempted). The tethered donor–acceptor network is then subjected to glycosylation. Finally, the resulting oligosaccharide is cleaved from the template.
Scheme 1.

Molecular Clamping and Templated Oligosaccharide Synthesis
Our preliminary study dedicated to varying the linkers resulted in the development of a new concept that we named templated oligosaccharide synthesis. A range of disaccharides were obtained in good yields and with high stereoselectivity.7 We also demonstrated the possibility of extending the template to the synthesis of a trisaccharide, which was also obtained with complete α-stereoselectivity for both glycosylation steps.7 Described herein is a continuation of this study with the focus on dedicated mechanistic studies to reveal the driving forces of the templated synthesis and further improve the yields and stereoselectivities.
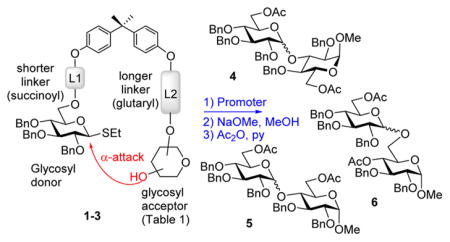
RESULTS AND DISCUSSION
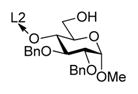
Having learned from the previous work by Fairbanks and coworkers,17,18 Warriner,19 and our own experimentation with flexible peptide-based templates,7 we anticipated that the rigidity of the template should be essential for the stereoselection. With this consideration in mind, we selected bisphenol A (BPA) as the rigid template. The preliminary results indicate that the rigidity of the template is essential for the stereoselection. The first series of conjugates 1–37 were designed to deliver the glycosyl acceptor from the bottom face (linker L1 is shorter than L2, succinoyl vs glutaryl, respectively). The results of this study are summarized in Table 1. When per-benzylated donor tethered with 3-OH acceptor 1 was activated with NIS/TfOH followed by the ester bond cleavage under Zemplen conditions (NaOMe) and standard acetylation (Ac2O/pyridine), disaccharide 47 was isolated in 60% yield (entry 1). The most encouraging outcome of this test reaction is that glycoside 4 was obtained with complete α-selectivity, and no traces of the β-linked diastereomer could be detected. In spite of this promising result, it also became apparent that NIS/TfOH is too powerful an activator for this system, as reflected by a modest yield of disaccharide 4 and relatively high rate of hydrolysis of the leaving group, as judged by the presence of the hemiacetal (1-OH derivative) isolated in 28% yield. Therefore, subsequent reactions have been conducted using MeOTf, a milder activator for thioglycosides.20 Thus, when the activation was performed in the presence of MeOTf at 0 °C, (1 → 3)-linked disaccharide 4 was isolated in a higher 73% yield (entry 2) and retains complete α-selectivity. The better results led us to conclude that the donor and acceptor were not held in close proximity to each other, but the linkers are playing a direct role in approach of the acceptor toward the forming oxacarbenium ion. When the promoter is stronger, increased intermolecular reaction with H2O acting as the nucleophile is the predominating mechanism. With the use of MeOTf, the system has an opportunity for the donor–acceptor pair to rearrange to form the α-linked macrocycle.
Table 1.
Template is Designed to Deliver the Nucleophile from the α-Face (L2 is Longer than L1)
| ||||
|---|---|---|---|---|
| entry | acceptor | promotera | time | product (yield, α/β ratio) |
| 1 |
 1 |
NIS/TfOH | 5 min | 4 (60%, α-only) |
| 2 |
1 |
MeOTf | 18h | 4 (73%, α-only) |
| 3 |
 2 |
MeOTf | 18h | 5 (81%, α-only) |
| 4 |
 3 |
MeOTf | 20 h | 6 (63%, 9.2/1) |
Performed in 1,2-dichloroethane in the presence of molecular sieves 4 Å at rt (NIS/TfOH) or 3 Å at 0 °C (MeOTf).
With optimized reaction conditions, the protocol was applied to glycosylation of tethered 4-OH acceptor 2, and disaccharide 57 was obtained in 81% yield and complete α-selectivity (entry 3). Glycosylation of tethered 6-OH acceptor 3 provided disaccharide 67 in 63% yield. Again, the preference was given to the formation of α-linked product, although the presence of the other diastereomer was also evident (α/β = 9.2/1, entry 4).
It is possible that the compromised stereoselectivity in this case is related to the fact that primary 6-hydroxyl group is more flexible and can reach out both from the bottom and from the top faces of the activated donor (oxacarbenium ion).21 A second possibility is that the primary 6-hydroxyl group is a less hindered nucleophile compared to the secondary acceptors. This might increase the number of approach vectors toward the oxacarbenium ion.
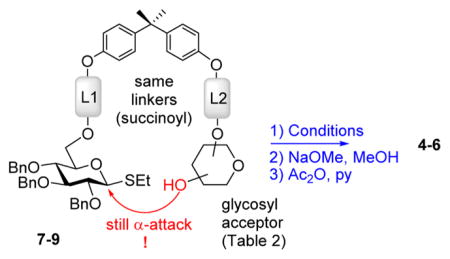
Having achieved promising stereoselectivity with the L2 longer than the L1 model, we were curious to investigate the stereoselectivity of glycosylations with spacers of the equal length (L1 = L2). For this purpose, we obtained a template with succinoyl linkers for both the glycosyl donor and acceptor attachment (7–9). As summarized in Table 2, complete α-stereoselectivity was still maintained in all reactions involving glycosylations of secondary glycosyl acceptors 7 and 87 (entries 1–4). Herein, we also tested the use of dimethyl(thiomethyl)sulfonium triflate (DMTST),22 another popular promoter for glycosidation of thioglycosides23 (entry 2). Nevertheless, the most consistent results and best yields have been achieved with MeOTf (entries 3–5). Once again, glycosylation of the primary glycosyl acceptor 9 provided only moderate stereoselectivity (α/β = 6.3/1, entry 5).
Table 2.
Template with Identical Linkers (L1 = L2 = Succinoyl) Still Provides Excellent α-Stereoselectivity
| ||||
|---|---|---|---|---|
| entry | acceptor | conditions | time | product (yield, α/β ratio) |
| 1 |
7 |
NIS/TfOH, MS4 Å CH2C12, −78 °C |
4h | 4 (71%, α-only) |
| 2 |
7 |
DMTST, MS 3Å 1,2-DCE, −30°C |
8h | 4 (68%, α-only) |
| 3 |
7 |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 16h | 4 (79%, α-only) |
| 4 |
8 |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 18h | 5 (76%, α-only) |
| 5 |
9 |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 20 h | 6 (78%, 6.3/1) |
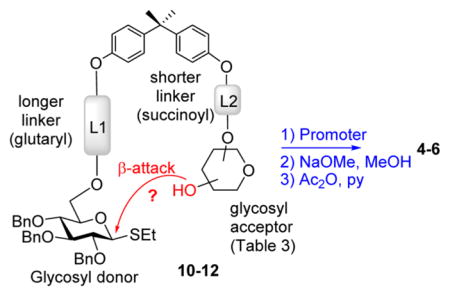
With good reaction yields and excellent α-stereoselectivity achieved in most template-mediated glycosylations, we also investigated a template wherein the glycosyl acceptor would be expected to be delivered from the top (β-) face. For this purpose, the glycosyl donor was attached via a linker L1 (glutaryl) longer than that of the glycosyl acceptor (L2, succinoyl) for compounds 10, 11,7 and 12. In this case, however, practically no selectivity was achieved. Results summarized in Table 3 clearly show that the synthesis of β-linked derivatives could not be accomplished using the longer linker L1. In all glycosylations attempted, α-linked disaccharides 4–6 were still formed as major products albeit with rather poor selectivity (entries 1–5).
Table 3.
Template is Designed to Deliver the Nucleophile from the β-Face (L1 is Longer Than L2)
| ||||
|---|---|---|---|---|
| entry | acceptor | promotera | time | product (yield, α/β ratio) |
| 1 |
10 |
NIS/TfOH | 5 min | 4 (70%, 1.2/1) |
| 2 |
10 |
MeOTf | 15h | 4 (83%, 3.0/1) |
| 3 |
11 |
NIS/TfOHb | 4h | 5 (69%, 2.1/1) |
| 4 |
11 |
MeOTf | 20 h | 5 (71%, 2.8/1) |
| 5 |
12 |
MeOTf | 24 h | 6 (71%, 7.9/1) |
Performed in 1,2-dichloroethane in the presence of molecular sieves 4 Å at rt (NIS/TfOH) or 3 Å at 0 °C (MeOTf).
Performed in CH2Cl2 at −78 °C.
Overall, we determined that the rigidity of BPA backbone structure creates a suitable environment for generating glycosidic linkages with superior stereoselectivity compared to those previously seen with peptide-based templates.17–19 Complete stereoselectivity was achieved in the synthesis of disaccharides derived from secondary glycosyl acceptors, whereas the (1 → 6)-linked disaccharide was produced with lower selectivity (up to α/β = 9/1).
It is possible that the compromised stereoselectivity is related to the ability of the more flexible primary hydroxyl group to reach out to both face of the activated donor (oxacarbenium ion intermediate). It became evident that the length of linkers may also have an effect on stereoselectivity, but the fact that the linkers of the same length still allowed for excellent α-stereoselectivity should help to reduce the number of options and focus our subsequent studies on the properties of the linker rather than its length. Hence, we began looking at the mechanistic aspects of the templated oligosaccharide synthesis so as to gain practical insights into the development of more effective linkers and further improve yields and stereoselectivity.
It occurred to us that using flexible succinoyl or glutaryl linkers may not be optimal for the effective positioning of the two coupling counterparts in a close proximity to ensure the effectiveness of this approach. This was investigated by setting up a simple test experiment wherein two glycosyl acceptors, tethered compound 17 and “free-floating” acceptor 13,24 were set to compete with the tethered donor. As illustrated in Scheme 2A, this simple competition experiment resulted in the preferential formation of the cross-coupling product 15 (51% yield, α/β = 3.0/1) rather than the tethered disaccharide 14. The latter was obtained in a lower yield of 20% albeit complete α-selectivity. In our opinion, this result serves as an indication that using a flexible spacer attachment is perhaps not the most ideal approach for the overall concept of the molecular clamping.
Scheme 2.

Competitive Glycosylation of Tethered Glucosides 1 and 16 vs Free-Floating Acceptor 13
The fact that the acceptor moiety is distanced from the anomeric center of the glycosyl donor is perhaps the major reason for relatively modest yields and relaxed stereoselectivity with primary hydroxyl groups.7 Hence, a further search was focused on more rigid spacer systems. Certainly, geometrical constraints should lead to the enhanced diastereocontrol by maintaining the reacting centers at proper orientation. The flexible linkers allow glycosylation of hydroxyl groups at remote locations from the tethering point. This distanced our templated approach from the traditional molecular clamping concept, wherein glycosylation was mainly possible at the adjacent position due to the high rigidity of the donor–acceptor pairs. Therefore, both the alignment and reactivity of tethered glycosyl donor/acceptor pairs would be important factors to consider in more rigid systems.
Bearing these considerations in mind, we investigated a more rigid phthaloyl linker with the following two anticipations. First, the enhanced rigidity would provide a more stringent acceptor delivery mode and hence help improve the stereoselectivity outcome for primary glycosyl acceptors. Second, the free rotation around a number of linkages in such BPA-phthaloyl template-linker combinations would still offer enough flexibility to glycosylate the hydroxyl group at remote positions. To investigate these, we obtained the tethered compound 16, which was subjected to the competition experiment with the free-floating acceptor 13. As illustrated in Scheme 2B, this experiment resulted in the preferential formation of the tethered disaccharide 17, which was isolated in 52% yield and with exclusive α-selectivity. The cross-coupling product 18 was also formed but in a lower yield (30%) in comparison to that recorded for the experiment with the flexible linkers (51%).
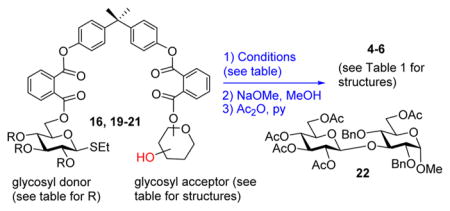
In our opinion, this result serves as a clear proof that a more rigid spacer attachment allows for the reaction components, glycosyl donor and acceptor, to be in close proximity while still maintaining complete α-selectivity and sufficient flexibility to glycosylate the remote hydroxyl groups. Encouraged by this preliminary result, we conducted the individual experiment with the per-benzylated donor tethered with the 3-OH acceptor 16. When compound 16 was activated with MeOTf followed by the ester bond cleavage under Zemplen conditions (MeONa) and standard acetylation (Ac2O/pyridine), disaccharide 4 was isolated in 71% yield and with complete stereoselectivity (entry 1, Table 4). No traces of the β-linked diastereomer could be detected.
Table 4.
Investigation of the Phthaloyl Linker in the BPA-Templated Glycosylations
| ||||
|---|---|---|---|---|
| entry | donor-acceptor | conditions | time | product (yield, α/β ratio) |
| 1 |
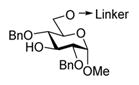 16, R = Bn |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 16 h | 4 (71%, α-only) |
| 2 |
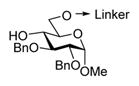 19, R = Bn |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 22 h | 5 (90%, α-only) |
| 3 | 19 | NIS/TfOH, MS 4Å, CH2C12, −78 °C | 2h | 5 (75%, α-only) |
| 4 |
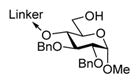 20, R = Bn |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 18 h | 6 (89%, α-only) |
| 5 |
21, R = Bz |
MeOTf, MS 3Å, 1,2-DCE, 0°C | 40 h | 22 (84%, β-only) |
| 6 | 21 | NIS/TfOH, MS 4Å, 1,2-DCE, 0°C | 10 min | 22 (75%, β-only) |
| 7 | 19 | MeOTf, MS 3Å, MeCN, 0 °C → rt | 24 h | 5 (69%, α-only) |
When essentially the same protocol was applied to glycosylation of the tethered 4-OH acceptor 19,7 disaccharide 5 was obtained in 90% yield and with complete α-selectivity (entry 2). In our previous study, we deemed NIS/TfOH too powerful an activator for the templated synthesis using flexible linkers.7 Also here, NIS/TfOH-promoted synthesis of disaccharide 5 was rather swift (2 h at −78 °C), but the increased rate was translated into the decreased yield of 75% (entry 3). Therefore, many subsequent reactions have been conducted using MeOTf, a milder activator for thioglyco-sides.20 MeOTf-promoted synthesis of the (1 → 6)-linked disaccharide 6 from the tethered donor–acceptor 207 also resulted in an excellent yield of 89% (entry 4). Even more importantly, complete α-selectivity in glycosylation of the primary alcohol was now obtained using this tethering approach.
Having achieved excellent yields and complete stereo-selectivities it all syntheses of α-linked disaccharides, we were curious to see whether essentially the same approach could be used for the synthesis of β-linked disaccharides. For this purpose, we obtained the benzoylated glycosyl donor that was tethered with the 3-OH acceptor 21.7 MeOTf-promoted glycosylation was rather sluggish (40 h) perhaps due to the disarmed nature of per-benzoylated donor or due to the hindrance caused by the acyloxonium ion used herein.25
Nevertheless, the reaction smoothly progressed, and disaccharide 227 was obtained in 84% yield with complete β-stereoselectivity (entry 5). The rate of this coupling could be significantly enhanced in the presence of NIS/TfOH (10 min), but the isolated yield of disaccharide 22 was reduced to 75% (entry 6). Again, the β-linked product was formed exclusively. Interestingly, when the benzylated donor tethered to 4-OH acceptor 19 was glycosylated in MeCN, a reaction solvent that is known to enhance β-selectivity,26 only the α-linked disaccharide 5 was obtained (68%, entry 7). This result implies that the effect of the intramolecular tethering on the stereoselectivity of glycosylation is stronger than that of solvent effects.
In general, the effect of the steric bulkiness of a substituent at C-6 is known to be beneficial for the formation of α-D-glucosides.27 This effect is attributed to shielding (steric or electronic) of the top face of the ring and hence favoring the nucleophilic attack from the opposite, bottom face. We wondered whether the steric bulk of the tethered glycosyl donors may contribute to the high α-stereoselectivity achieved in these reactions. This turned our attention to investigating whether it is the rigidity of the tethered structure rather than the effect of steric bulkiness at C-6 that is driving these glycosylations toward the α-linked products. To delineate between these two possible effects, glycosyl donor 23 and acceptor 24 (Scheme 3), both bearing bulky phenylphthaloyl substituents at C-6, were obtained. Glycosidation of donor 23 with acceptor 24 was performed using MeOTf as a promoter in 1,2-dichloroethane. The resulting disaccharide 25 was isolated in 84% yield, but the stereoselectivity was low (α/β = 2.8/1). In comparison to the intramolecular glycosylation of tethered donor–acceptor pairs, we can conclude that the steric bulkiness of the protecting group at C-6 in this case did not influence the stereoselectivity as much as tethering of the two components did.
Scheme 3.

Investigation of the Effect of Steric Bulkiness at C-6 on Stereoselectivity
Interestingly, the influence of acetonitrile as the reaction solvent was more notable in this case. Disaccharide 25 was obtained in 88% yield with a slightly reversed stereoselectivity (α/β = 1/1.2, Scheme 3). In our opinion, this may also serve as an indication that the steric bulkiness at C-6 has a minor contribution into the stereoselectivity achieved in tethered systems that were not influenced at all by the effect of acetonitrile.
Previously, excellent α-stereoselectivity was achieved with glycosyl donors equipped with a 6-O-phthaloyl linker attached to a bulky p-phenylbenzyl group.16 To investigate whether a phthaloyl linker connected to bisphenol A can have any effect on stereoselectivity of glycosylation, we obtained conjugate 26 equipped with a TBDMS-protected bisphenol A phthaloyl protecting group at C-6. Couplings of conjugate 26 with glycosyl acceptors 2728 and 2829 were practically non-stereoselective, and the respective disaccharides 29 and 30 were obtained in average yields and poor stereoselectivities (Scheme 3). This result indicates that the rigid bisphenol A template by itself has no stereodirecting impact on templated glycosylations.
Recently, Manabe, Ito, and their co-workers determined that glycosides carrying cyclic protecting groups may be prone to the β- to α-anomerization.30 This anomerization proceeds via the endocyclic mechanistic pathway. It is affected in the presence of a mild Lewis acid and it favored by the inner strain caused by the fused rings and required as the promoter. We were curious to investigate whether our tethered disaccharides’ selectivity was due to this endocyclic cleavage/anomerization pathway. In principle, that would also explain high α-stereoselectivity observed in all templated reactions. For the purpose of investigating the postglycosylational isomerization, we obtained a β-linked macrocyclic compound 31 (Scheme 4) and examined its anomerization. These reactions were first attempted in the presence of boron trifluoride etherate (BF3-OEt2) as a Lewis acid.30 No anomerization occurred over three days at room temperature; in fact, the starting material 31 could be recovered quantitatively. In addition, to mimic our actual glycosylation reaction conditions, we also investigated a MeOTf-mediated anomerization of compound 31. However, no anomerization took place under these reaction conditions, ruling out this possible explanation for the excellent α-stereoselectivity achieved in templated glycosylations.
Scheme 4.
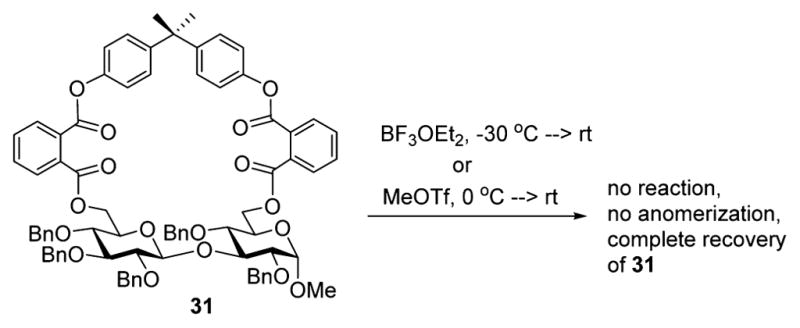
Investigation of a Possibility of the Endocyclic Cleavage Leading to Anomerization
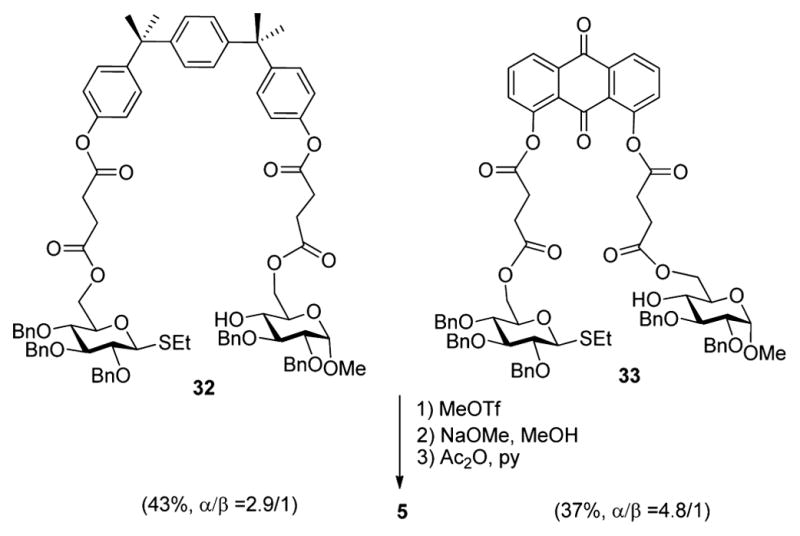
Upon seeing the effects of linker rigidity on the selectivity of glycosylation, we turned toward modifying the template rigidity. Two alternative template molecules, bisphenol P and anthraquinone, were chosen. In the case of bisphenol P-based conjugate 32, the extra aromatic functionality adds flexibility and increases the distance between donor and acceptor. As a result, a fair yield and poor stereoselectivity were observed in glycosidation of 32 (Scheme 5). It is possible that the tendency of bisphenol P to adopt the favored trans-conformation wherein the two hydroxyl groups are placed opposite of each other has further contributed in the decreased outcome in comparison to that of the BPA-based reactions. In the case of the anthraquinone-based conjugate 33, the placement of the hydroxyl groups on anthraquinone seems ideal, both acceptor and donor are facing each other. Hence, the reaction counterparts should be in closer proximity with each other as compared to those in bisphenol A. Nevertheless, template 33 also produced fair yields and selectivities, indicating that our initial choice of BPA as the template seems the most advantageous for the systems chosen.
Scheme 5.
Investigation of Other Related Templates
CONCLUSIONS
Overall, on the basis of the results of the mechanistic studies described herein, we would like to emphasize that the rigid bisphenol A template and phthaloyl linkers permit highly stereoselective glycoside bond formation. Efficient intra-molecular glycosylation with glycosyl donors equipped with a nonparticipating benzyl group at C-2 led to the exclusive formation α-linked disaccharides. Complete α-selectivity was obtained even with primary glycosyl acceptors that gave lowers stereoselectivity in our previous studies with flexible linkers. Extended studies revealed that it is indeed the tethering that offers the stereodirecting effect in α-glycosylations rather than steric bulkiness of C-6 substituents. We also demonstrated that β-linked glycosides can be efficiently formed with the aid of a participatory effect of the neighboring ester group. Further development of this methodology and its application to oligo- and polysaccharide synthesis in currently underway in our laboratory.
EXPERIMENTAL SECTION
General Experimental
The reactions were performed using commercial reagents, and the ACS grade solvents used for reactions were purified and dried in accordance with standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh), and reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 and 1,2-dichloromethane (DCE) were distilled from CaH2 directly prior to application. Molecular sieves (3 Å), used for reactions, were crushed and activated in vacuo at 390 °C for 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured using a polarimeter. 1H NMR spectra were recorded at 300, 500, or 600 MHz. 13C NMR spectra were recorded at 75, 125, or 150 MHz. The 1H NMR chemical shifts are referenced to the signal of the residual CHCl3 (δH = 7.27 ppm) for solutions in CDCl3. The 13C NMR chemical shifts are referenced to the central signal of CDCl3 (δC = 77.23 ppm) for solutions in CDCl3. HRMS determinations were made with the use of a mass spectrometer with FAB ionization and ion-trap detection.
General Procedure for Introducing the Succinoyl Linker
Succinic anhydride (402 mg, 4.02 mmol) was added to a solution of a partially protected derivative (1.34 mmol) in dry pyridine (5.0 mL), and the resulting mixture was stirred under argon for 16 h at rt. After that, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in dichloromethane (~100 mL) and washed with water (3 × 20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the respective succinoylated compounds.
Ethyl 2,3,4-Tri-O-benzyl-6-O-(3-hydroxycarbonylpropanoyl)-1-thio-β-D-glucopyranoside (35)
The title compound was obtained from ethyl 2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (34)31 as described previously,7 and its analytical data were the same as those reported previously.32
Methyl 2,3-Di-O-benzyl-6-O-(3-hydroxycarbonylpropanoyl)-α-D-glucopyranoside (37)
The title compound was obtained from methyl 2,3-di-O-benzyl-α-D-glucopyranoside (36)33 as described previously,7 and its analytical data for were the same as those reported previously.34
Methyl 2,4-Di-O-benzyl-6-O-(3-hydroxycarbonylpropanoyl)-α-D-glucopyranoside (39)
The title compound was obtained from methyl 2,4-di-O-benzyl-α-D-glucopyranoside (38)28 as described previously,7 and its analytical data for were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-4-O-(3-hydroxycarbonylpropanoyl)-6-O-triphenylmethyl-α-D-glucopyranoside (41)
Succinic anhydride (350 mg, 3.50 mmol) was added to a solution of methyl 2,3-di-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (40,35 539 mg, 0.88 mmol) in dry pyridine (3.0 mL), and the resulting mixture was stirred under argon for 16 h at rt. After that, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in dichloromethane (~100 mL) and washed with water (3 × 20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compounds in 60% yield (375 mg, 0.52 mmol) as a white amorphous solid. Analytical data for 41: Rf = 0.40 (ethyl acetate/toluene, 2/3, v/v); [α]D25 + 6.5 (c = 1, CHCl3); 1H NMR: δ, 2.04–2.13, 2.20–2.36 (2m, 6H, CH2CH2COOH), 3.04 (dd, 1H, J5,6a = 2.0 Hz, J6a,6b = 10.1 Hz, H-6a), 3.10 (dd, 1H, J5,6b = 6.0 Hz, H-6b), 3.47 (s, 3H, OCH3), 3.60 (dd, 1H, J2,3 = 9.5 Hz, H-2), 3.81 (m, 1H, H-5), 3.90 (dd, 1H, J3,4 = 9.4 Hz, H-3), 4.73 (dd, 2H, 2J = 11.3 Hz, CH2Ph), 4.62–4.76 (m, 3H, J1,2 = 3.5 Hz, H-1, CH2Ph), 4.95 (dd, 1H, J4,5 = 9.6 Hz, H-4), 7.14–7.42 (m, 25H, aromatic), 8.53 (d, 1H, J = 4.4 Hz, COOH) ppm; 13C NMR (75 MHz): δ 28.8, 28.9, 55.2, 62.6, 69.1, 70.8, 73.5, 75.4, 79.4, 79.8, 86.6, 98.0, 124.3, 127.0 (×2), 127.7, 127.8 (×5), 128.0, 128.1 (×4), 128.4 (×2), 128.5 (×3), 128.8 (×5), 137.6, 138.1, 138.5, 143.7 (×2), 147.9, 170.8, 176.3 ppm; HR-FAB MS [M + Na]+ calcd for C44H44NaO9 739.2883, found 739.2882.
General Procedure for Introducing Glutaryl Linker
Glutaric anhydride (459 mg, 4.02 mmol) was added to a solution of a partially protected derivative (1.34 mmol) in dry pyridine (5.0 mL), and the resulting mixture was stirred under argon for 16 h at rt. After that, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in dichloromethane (~100 mL) and washed with water (3 × 20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the respective glutarated compounds.
Ethyl 2,3,4-Tri-O-benzyl-6-O-(4-hydroxycarbonylbutanoyl)-1-thio-β-D-glucopyranoside (42)
The title compound was obtained from 34 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-6-O-(4-hydroxycarbonylbutanoyl)-α-D-glucopyranoside (43)
The title compound was obtained from 36 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,4-Di-O-benzyl-6-O-(4-hydroxycarbonylbutanoyl)-α-D-glucopyranoside (44)
The title compound was obtained from 38 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-4-O-(4-hydroxycarbonylbutanoyl)-6-O-triphenylmethyl-α-D-glucopyranoside (45)
The title compound was obtained from 40 as described previously,7 and its analytical data were the same as those reported previously.7
General Procedure for Introducing the Phthaloyl Linker
4-N,N-Dimethylaminopyridine (DMAP, 82 mg, 0.67 mmol) and phthalic anhydride (794 mg, 5.36 mmol) were added to a solution of a partially protected derivative (1.34 mmol) in dry pyridine (5.0 mL), and the resulting mixture was stirred under argon for 24 h at 50 °C. After that, the volatiles were removed under reduced pressure, and the residue was dissolved in dichloromethane (~100 mL) and washed with water (3 × 20 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane, 1/1, v/v) to afford respective compounds.
Ethyl 2,3,4-Tri-O-benzyl-6-O-(2-hydroxycarbonylbenzoyl)-1-thio-β-D-glucopyranoside (46)
The title compound was obtained from 34 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-6-O-(2-hydroxycarbonylbenzoyl)-α-D-glucopyranoside (47)
The title compound was obtained from 36 as described previously,7 and its analytical data were same as reported previously.7
Methyl 2,4-Di-O-benzyl-6-O-(2-hydroxycarbonylbenzoyl)-α-D-glucopyranoside (48)
The title compound was obtained from 38 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-4-O-(2-hydroxycarbonylbenzoyl)-6-O-tri-phenylmethyl-α-D-glucopyranoside (49)
The title compound was obtained from 40 as described previously,7 and its analytical data were the same as those reported previously.7
Ethyl 2,3,4-Tri-O-benzoyl-6-O-(2-hydroxycarbonylbenzoyl)-1-thio-β-D-glucopyranoside (51)
The title compound was obtained from 5036 as described previously,7 and its analytical data were the same as those reported previously.7
General Procedure for the Coupling of Linker to 4,4′-Bisphenol A
A solution of N,N′-dicyclohexylcarbodiimide (DCC, 0.27 mmol) in dry dichloromethane (1.0 mL) was added dropwise to a solution of a linker-containing sugar derivative (0.11 mmol) and 2,2-bis(4-hydroxyphenyl)propane (BPA, 0.22 mmol) in dry dichloromethane (1.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/1, v/v) to afford the respected BPA-containing compounds.
Methyl 2,3-Di-O-benzyl-6-O-(α,α,α-p-hydroxyphenyldimethyltolyl succinate)-α-D-glucopyranoside (52)
The title compound was obtained from 37 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,4-Di-O-benzyl-6-O-(α,α,α-p-hydroxyphenyldimethyltolyl succinate)-α-D-glucopyranoside (53)
A solution of N,N′-dicyclohexylcarbodiimide (DCC, 108 mg, 0.522 mmol) in dry dichloromethane (1.0 mL) was added dropwise (over 5 min) to a solution of 39 (124 mg, 0.261 mmol) and 2,2-bis(4-hydroxyphenyl)-propane (BPA, 90 mg, 0.392 mmol) in dry dichloromethane (3.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/1, v/v) to afford the title compound in 74% yield (58.7 mg, 0.086 mmol) as a colorless syrup. Analytical data for 53: Rf = 0.53 (ethyl acetate/toluene, 2/3, v/v); [α]D24 + 20.0 (c = 0.8, CHCl3); 1H NMR: δ 1.53 (s, 6H, C(CH3)2), 2.60–2.67, 2.73–2.80 (2m, 4H, COCH2CH2CO), 3.22 (s, 3H, OCH3), 3.29 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.33 (dd, 1H, J4,5 = 9.7 Hz, H-4), 3.71 (m, 1H, H-5), 4.02 (dd, 1H, J3,4 = 9.1 Hz, H-3), 4.25 (m, 2H, H-6a, 6b), 4.51 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.58 (m, 2H, CH2Ph), 4.68 (dd, 2H, 2J = 11.2 Hz, CH2Ph), 6.64–7.28 (m, 18H, aromatic) ppm; 13C NMR (75 MHz): δ 25.0, 25.7, 29.2, 29.5, 29.6, 29.9, 31.2, 33.9, 42.3, 49.6, 55.4, 63.7, 68.4, 73.3, 73.8, 74.7, 79.8, 97.5, 115.0, 120.9, 121.0, 127.9, 128.0 (×2), 128.1, 128.3 (×3), 128.4 (×2), 128.6, 128.8, 138.3, 142.7, 148.2, 148.5, 148.7, 153.7, 171.1, 172.0 ppm; HR-FAB MS [M + Na]+ calcd for C40H44NaO10 707.2832, found 707.2838.
Methyl 2,3-Di-O-benzyl-4-O-(α,α,α-p-hydroxyphenyldimethyltolyl succinate)-6-O-triphenylmethyl-α-D-glucopyranoside (54)
A solution of N,N′-dicyclohexylcarbodiimide (DCC, 230 mg, 1.12 mmol) in dry dichloromethane (3.0 mL) was added dropwise (over 5 min) to a solution of 41 (400 mg, 0.558 mmol) and 2,2-bis(4-hydroxyphenyl)propane (BPA, 230 mg, 0.837 mmol) in dry dichloromethane (6.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/1, v/v) to afford the title compound in 59% yield (307 mg, 0.33 mmol) as a colorless syrup. Analytical data for 54: Rf = 0.65 (ethyl acetate/toluene, 1/4, v/v); [α]D24 + 6.7 (c = 1, CHCl3); 1H NMR: δ 1.65 (s, 6H, C(CH3)2), 2.18–2.36, 2.45–2.50 (2m, 4H, COCH2CH2CO), 3.08 (m, 2H, H-6a, 6b), 3.49 (s, 3H, OCH3), 3.61 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.82 (m, 1H, H-5), 3.89 (dd, 1H, J3,4 = 9.5 Hz, H-3), 4.68 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.73 (dd, 2H, 2J = 11.5 Hz, CH2Ph), 4.75 (dd, 2H, 2J = 12.1 Hz, CH2Ph), 4.95 (dd, 1H, J4,5 = 9.5 Hz, H-4), 6.73–7.42 (m, 33H, aromatic) ppm; 13C NMR(75 MHz): δ 29.1, 29.3, 31.2 (×2), 42.3, 55.4, 62.8, 69.2, 71.0, 73.7, 75.6, 79.6, 80.0, 86.7, 98.2, 115.0 (×3), 120.9 (×3), 127.2 (×3), 127.8, 127.9 (×5), 128.2 (×3), 128.3 (×3), 128.4, 128.5 (×3), 128.7 (×3), 128.9 (×4), 129.2, 138.2, 138.7, 142.9, 143.9 (×3), 148.5, 148.7, 153.6, 170.7, 170.8 ppm; HR-FAB MS [M + Na]+ calcd for C59H58NaO10 949.3928, found 949.3929.
Methyl 2,3-Di-O-benzyl-6-O-(α,α,α-p-hydroxyphenyldimethyltolyl glutarate)-α-D-glucopyranoside (55)
The title compound was obtained from 43 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,4-Di-O-benzyl-6-O-(α,α,α-p-hydroxyphenyldimethyltolyl glutarate)-α-D-glucopyranoside (56)
The title compound was obtained from 44 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-4-O-(α,α,α-p-hydroxyphenyldimethyltolyl glutarate)-6-O-triphenylmethyl-α-D-glucopyranoside (57)
The title compound was obtained from 45 as described previously,7 and its analytical data were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-6-O-(α,α,α-(4-hydroxyphenyl)-dimethyltolyl phthalate)-α-D-glucopyranoside (58)
The title compound was obtained from 47 as described previously,7 and its analytical data for were the same as those reported previously.7
Methyl 2,4-Di-O-benzyl-6-O-(α,α,α-(4-hydroxyphenyl)-dimethyltolyl phthalate)-α-D-glucopyranoside (59)
The title compound was obtained from 48 as described previously,7 and its analytical data for were the same as those reported previously.7
Methyl 2,3-Di-O-benzyl-4-O-(α,α,α-(4-hydroxyphenyl)-dimethyltolyl phthalate)-6-O-triphenylmethyl-α-D-glucopyranoside (60)
The title compound was obtained from 49 as described previously,7 and its analytical data for were the same as those reported previously.7
General Procedure for the Synthesis of Tethered Donor–Acceptor Pairs
A solution of N,N′-dicyclohexylcarbodiimide (0.22 mmol) and 4-dimethylaminopyridine (0.033 mmol) in dry dichloromethane (1.0 mL) was added dropwise to a stirring solution of a BPA-containing conjugate (0.11 mmol) and a linker-containing counterpart (0.13 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford the respective tethered compounds. To yield tethered 6-OH compounds 3, 9, 12, and 20, the respective crude mixtures were dissolved in dichloromethane (2.0 mL); a 10% soln. of trifluoroacetic acid in wet dichloromethane (1.5 mL) was added dropwise, and the resulting mixture was stirred for 1 h at rt. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (~10 mL), sat. aq NaHCO3 (2 × 10 mL) and water (~10 mL). Organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford tethered compound 3, 9, 12, or 20, respectively
Tethered Compound 1
The title compound was obtained from 35 and 56 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 2
The title compound was obtained from 35 and 55 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 3
The title compound was obtained from 35 and 57 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 7
A solution of DCC (29 mg, 0.14 mmol) and DMAP (2.6 mg, 0.021 mmol) in dry dichloromethane (1.0 mL) was added dropwise (5 min) to a stirring solution of 53 (48 mg, 0.071 mmol) and 35 (50 mg, 0.085 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford tethered compound 7 in 76% yield (67 mg, 0.054 mmol) as a colorless syrup. Analytical data for 7: Rf = 0.53 (ethyl acetate/hexane, 1/1, v/v); [α]D24 + 5.6 (c = 1, CHCl3); 1H NMR: δ 1.26 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.58 (s, 6H, C(CH3)2), 2.63–2.71 (m, 6H, SCH2 CH3, COCH2 CH2 CO), 2.76–2.82 (m, 4H, COCH2CH2CO), 3.26 (s, 3H, OCH3), 3.28–3.42 (m, 3H, H-2, 2′, 4), 3.42–3.52 (m, 2H, H-4′, 5′), 3.65 (dd, 1H, J3′,4′ = 8.7 Hz, H-3′), 3.74 (m, 1H, H-5), 4.04 (dd, 1H, J3,4 = 9.2 Hz, H-3), 4.17–4.35 (m, 4H, H-6a, 6b, 6a′, 6b′), 4.42 (d, 1H, J1′,2′ = 9.8 Hz, H-1′), 4.50–4.62 (m, 5H, H-1, 2 × CH2Ph), 4.66–4.92 (m, 6H, 3 × CH2Ph), 6.91–7.34 (m, 33H, aromatic) ppm; 13C NMR (75 MHz): δ 15.3, 25.4, 29.2, 29.4 (×2), 31.1 (×2), 42.7 (×2), 55.4 (×2), 63.7, 63.9, 68.4, 73.3, 73.8, 74.7, 75.3, 75.7, 76.0, 77.1, 77.8, 79.8, 81.9, 85.4, 86.8, 97.6, 121.0 (×4), 127.9 (×5), 128.0 (×6), 128.1 (×2), 128.3 (×4), 128.4 (×3), 128.5 (×2), 128.6 (×5), 128.7 (×2), 128.8 (×2), 137.9, 138.1, 138.3, 138.5, 148.1 (×2), 148.7, 171.0 (×2), 172.0 (×2) ppm; HR-FAB MS [M + Na]+ calcd for C73H80NaO17S 1283.5014, found 1283.5012.
Tethered Compound 8
The title compound was obtained from 35 and 52 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 9
A solution of DCC (35 mg, 0.17 mmol) and DMAP (3 mg, 0.026 mmol) in dry dichloromethane (2.0 mL) was added dropwise (5 min) to a stirring solution of 35 (61 mg, 0.103 mmol) and 54 (68 mg, 0.074 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The crude mixture was dissolved in dichloromethane (2.0 mL); 10% soln. of trifluoroacetic acid in wet dichloromethane (1.5 mL) was added dropwise (1 min), and the resulting mixture was stirred for 1 h at rt. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (~10 mL), sat. aq NaHCO3 (2 × 10 mL), and water (~10 mL). Organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford tethered compound 9 in 69% yield (65 mg, 0.051 mmol, over 2 steps) as a colorless syrup. Analytical data for 9: Rf = 0.33 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 3.8 (c = 0.2, CHCl3); 1H NMR: δ 1.30 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.64 (s, 6H, C(CH3)2), 2.66–2.78 (m, 6H, SCH2CH3, COCH2CH2CO), 2.78–2.90 (m, 4H, COCH2CH2CO), 3.37 (s, 3H, OCH3), 3.43 (dd, 1H, J2′,3′ = 9.5 Hz, H-2′), 3.47–3.62 (m, 6H, H-2, 4′, 5, 5′, 6a′, 6b′), 3.67 (dd, 1H, J3′,4′ = 8.7 Hz, H-3′), 4.00 (dd, 1H, J3,4 = 9.4 Hz, H-3), 4.25 (dd, 1H, J5,6a = 4.6 Hz, J6a,6b = 11.9 Hz, H-6a), 4.38 (dd, 1H, H-6b), 4.47 (d, 1H, J1′,2′ = 9.8 Hz, H-1′), 4.55–4.74 (m, 5H, H-1, 2 × CH2Ph), 4.77–4.96 (m, 7H, H-4, 3 × CH2Ph), 6.95–7.35 (m, 33H, aromatic) ppm; 13C NMR (75 MHz): δ 15.3, 25.4, 29.1, 29.2, 29.4, 31.1, 42.7, 55.6, 61.2, 63.9, 71.2, 73.7, 75.3, 75.6, 75.7, 76.0, 77.1, 77.4, 77.7, 79.1, 79.7, 81.9, 85.4, 86.8, 98.5, 121.0 (×2), 121.1 (×2), 127.8, 127.9 (×3), 128.0 (×6), 128.1, 128.2 (×2), 128.3 (×2), 128.4 (×2), 128.5 (×2), 128.6 (×4), 128.7 (×7), 137.8, 138.1, 138.5, 138.9, 148.0, 148.2, 148.6, 148.7, 171.0 (×2), 172.0, 172.7 ppm; HR-FAB MS [M + Na]+ calcd for C73H80NaO17S 1283.5014, found 1283.5010.
Tethered Compound 10
A solution of DCC (55 mg, 0.27 mmol) and DMAP (4.9 mg, 0.004 mmol) in dry dichloromethane (2.0 mL) was added dropwise (5 min) to a stirring solution of 53 (108 mg, 0.132 mmol) and 42 (78 mg, 0.075 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford tethered compound 10 in 80% yield (170.1 mg, 0.171 mmol) as a colorless syrup. Analytical data for 10: Rf = 0.55 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 29.2 (c = 2, CHCl3); 1H NMR: δ 1.26 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.58 (s, 6H, C(CH3)2), 1.99 (m, 2H, COCH2 CH2 CH2 CO), 2.39, 2.57 (2dd, 4H, COCH2 CH2 CH2 CO), 2.65–2.82 (m, 6H, SCH2 CH3, COCH2CH2CO), 3.26 (s, 3H, OCH3), 3.27–3.32 (m, 2H, H-2, 4), 3.35 (dd, 1H, J2′,3′ = 10.1 Hz, H-2′), 3.52–3.56 (m, 2H, H-4′, 5′), 3.72 (dd, 1H, J3′,4′ = 8.5 Hz, H-3′), 3.80 (m, 1H, H-5), 4.04 (dd, 1H, J3,4 = 9.3 Hz, H-3), 4.13 (dd, 1H, J5′,6a′ = 4.5 Hz, J6a′,6b′ = 12.0 Hz, H-6a′), 4.22–4.38 (m, 3H, H-6a, 6b, 6b′), 4.42 (d, 1H, J1′,2′ = 9.8 Hz, H-1′), 4.51–4.65 (m, 5H, H-1, 2 × CH2Ph), 4.68–4.88 (m, 6H, 3 × CH2Ph), 6.95–7.31 (m, 33H, aromatic) ppm; 13C NMR (75 MHz): δ 15.4, 20.3, 25.2, 29.2, 29.5, 31.2, 33.3, 33.6, 42.7 (×2), 55.5, 63.6, 63.7, 68.5, 73.3, 73.4, 74.8, 75.4, 75.8, 76.0, 77.2, 77.3, 77.9, 79.8, 82.0, 85.4, 86.9, 97.6, 121.2 (×3), 128.0 (×3), 128.1 (×2), 128.2 (×2), 128.3 (×2), 128.4 (×3), 128.5 (×6), 128.6 (×2), 128.7 (×3), 128.8 (×2), 128.9 (×5), 130.2 (×2), 137.9, 138.1, 138.4, 138.6, 148.1, 148.2, 148.8, 171.1, 171.7, 172.1, 172.7 ppm; HR-FAB MS [M + Na]+ calcd for C74H82NaO17S 1297.5170, found 1297.5175.
Tethered Compound 11
The title compound was obtained from 35 and 52 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 12
A solution of DCC (36 mg, 0.176 mmol) and DMAP (3.2 mg, 0.026 mmol) in dry dichloromethane (5.0 mL) was added dropwise (5 min) to a stirring solution of 42 (63.2 mg, 0.11 mmol) and 54 (81 mg, 0.09 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The crude mixture was dissolved in dichloromethane (2.0 mL); 10% soln. of trifluoroacetic acid in wet dichloromethane (1.5 mL) was added dropwise (1 min), and the resulting mixture was stirred for 1 h at rt. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (~10 mL), sat. aq NaHCO3 (2 × 10 mL), and water (~10 mL). Organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford tethered compound 12 in 66% yield (87.3 mg, 0.132 mmol, over 2 steps) as a colorless syrup. Analytical data for 12: Rf = 0.33 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 11.2 (c = 2, CHCl3); 1H-n.m.r: δ 1.34 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.68 (s, 6H, C(CH3)2), 2.07 (m, 2H, COCH2CH2CH2CO), 2.45–2.51 (m, 4H, COCH2CH2CH2CO), 2.63–2.68 (m, 4H, COCH2CH2CO), 2.68–2.80 (m, 2H, SCH2CH3), 3.40 (s, 3H, OCH3), 3.47 (dd, 1H, J2′,3′ = 9.2 Hz, H-2′), 3.52–3.66 (m, 6H, H-2, 4′, 5, 5′, 6a′, 6b′), 3.73 (dd, 1H, J3′,4′ = 8.7 Hz, H-3′), 4.03 (dd, 1H, J3,4 = 9.5 Hz, H-3), 4.22 (dd, 1H, J5,6a = 4.5 Hz, J6a,6b = 11.9 Hz, H-6a), 4.44 (dd, 1H, H-6b), 4.50 (d, 1H, J1′,2′ = 9.8 Hz, H-1′), 4.60–4.76 (m, 5H, H-1, 2 × CH2Ph), 4.77–5.00 (m, 7H, H-4, 3 × CH2Ph), 6.96–7.02 (m, 4H, aromatic), 7.21–7.41 (m, 29H, aromatic) ppm; 13C NMR(75 MHz): δ 15.3, 20.2, 25.3, 29.0, 29.2, 31.1, 33.2, 33.4, 42.6, 55.6, 61.1, 63.5, 69.6, 71.1, 73.7, 75.3, 75.6, 75.7, 75.9, 77.0, 77.4, 77.8, 79.0, 79.6, 81.8, 85.3, 86.7, 98.4, 120.9 (×2), 121.1 (×2), 127.8, 127.9 (×3), 128.0 (×6), 128.1, 128.2 (×4), 128.4 (×2), 128.5 (×4), 128.6 (×3), 128.7 (×4), 137.7, 138.0 (×2), 138.4, 138.8, 148.0, 148.1, 148.5, 148.6, 171.0, 171.7, 172.7 (×2) ppm; HR-FAB MS [M + Na]+ calcd for C74H82NaO17S 1297.5170, found 1297.5160.
Tethered Compound 16
A solution of DCC (73 mg, 0.35 mmol) and DMAP (6.4 mg, 0.021 mmol) in dry dichloromethane (1.0 mL) was added dropwise (5 min) to a stirring solution of 59 (130 mg, 0.177 mmol) and 46 (136 mg, 0.213 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford tethered compound 16 in 81% yield (183 mg, 0.136 mmol) as a colorless syrup. Analytical data for 16: Rf = 0.63 (ethyl acetate/toluene, 3/7, v/v); [α]D25 + 35.0 (c = 0.5, CHCl3); 1H NMR: δ 1.16 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.57 (s, 6H, C(CH3)2), 2.61 (m, 2H, SCH2CH3), 3.20 (s, 3H, OCH3), 3.27 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.37 (m, 2H, J2′,3′ = 8.5 Hz, J4,5 = 9.8 Hz, H-2′, 4), 3.46–3.57 (m, 2H, H-4′, 5′), 3.63 (dd, 1H, J3′,4′ = 8.3 Hz, H-3′), 3.79 (m, 1H, J5,6a = 3.0 Hz, H-5), 4.02 (dd, 1H, J3,4 = 9.1 Hz, H-3), 4.33–4.57 (m, 10H, J1,2 = 3.4 Hz, H-1, 1′, 6a, 6b, 6a′, 6b′, 2 × CH2Ph), 4.61–4.88 (m, 6H, 3 × CH2Ph), 7.06–7.79 (m, 41H, aromatic) ppm; 13C NMR (75 MHz): δ 15.3, 25.1, 31.2, 42.7, 55.4, 64.5, 65.7, 68.4, 73.2, 73.8, 74.7, 75.3, 75.7, 76.0, 77.0, 77.4, 77.9, 79.7, 81.8, 85.2, 86.7, 97.4, 121.1 (×4), 127.9 (×4), 128.0 (×3), 128.1 (×5), 128.2 (×3), 128.3 (×5), 128.5 (×3), 128.6 (×6), 128.7 (×5), 128.8 (×2), 129.3 (×2), 129.4, 131.5, 131.6, 131.7, 131.9, 132.1, 132.2, 137.7, 138.0 (×2), 138.1, 148.2, 148.9, 166.4, 166.5, 166.8, 167.0 ppm; HR-FAB MS [M + Na]+ calcd for C81H80NaO17S 1379.5013, found 1379.5010.
Tethered Compound 19
The title compound was obtained from 46 and 58 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 20
The title compound was obtained from 46 and 60 as described previously,7 and its analytical data were the same as those reported previously.7
Tethered Compound 21
The title compound was obtained from 51 and 59 as described previously,7 and its analytical data were the same as those reported previously.7
Synthesis of Disaccharides 4–6 and 22
Typical NIS/TfOH-Promoted Glycosylation
A mixture of a donor–acceptor conjugate (0.032 mmol) and freshly activated molecular sieves (4 Å, 120 mg) in 1,2-dichloroethane (1.0 mL) was stirred under argon for 16 h at rt. The mixture was cooled to −78 °C; NIS (0.07 mmol) and TfOH (0.007 mmol) were added, and the resulting mixture was stirred at −78 °C until the disappearance of the starting material as indicated by TLC. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with 20% aq Na2S2O3 (~10 mL) and water (3× 10 mL). The organic phase was separated, dried over MgSO4, concentrated in vacuo, and dried. The residue was dissolved in dry methanol (1.0 mL); 1N soln. of NaOMe in MeOH (0.5 mL) was added, and the resulting mixture was stirred for 2–16 h at rt. After that, the reaction mixture was neutralized with Dowex (H+), and the resin was filtered off and washed successively with MeOH. The combined filtrate was concentrated in vacuo. The residue was dissolved in pyridine (1.0 mL); acetic anhydride (0.2 mL) was added dropwise, and the resulting mixture was stirred for 16 h at rt. After that, the reaction was quenched with methanol (~2.0 mL), and the volatiles were evaporated in vacuo. The residue was coevaporated with toluene, and the residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the respective disaccharide.
Typical MeOTf-Promoted Glycosylation
A mixture of a donor–acceptor conjugate (0.020 mmol) and freshly activated molecular sieves (3 Å, 100 mg) in 1,2-dichloroethane (0.5 mL) was stirred under argon for 16 h at rt. The mixture was cooled to 0 °C; MeOTf (0.06 mmol) was added, and the reaction mixture was stirred at 0 °C until the disappearance of the starting material, as indicated by TLC. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with sat aq NaHCO3 (~10 mL) and water (3 ×10 mL). The organic phase was separated, dried over MgSO4, concentrated in vacuo, and dried. The residue was dissolved in dry methanol (1.0 mL); 1N soln. of NaOMe in MeOH (0.5 mL) was added, and the resulting mixture was stirred for 2–16 h at rt. After that, the reaction mixture was neutralized with Dowex (H+), and the resin was filtered off and washed successively with MeOH. The combined filtrate was concentrated in vacuo. The residue was dissolved in pyridine (1.0 mL); acetic anhydride (0.2 mL) was added dropwise, and the resulting mixture was stirred for 16 h at rt. After that, the reaction was quenched with methanol (~2.0 mL), and the volatiles were evaporated in vacuo. The residue was coevaporated with toluene, and the residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the respective disaccharide.
Typical DMTST-Promoted Glycosylation
A mixture of a donor–acceptor conjugate (0.023 mmol) and freshly activated molecular sieves (3 Å, 90 mg) in 1,2-dichloroethane (0.5 mL) was stirred under argon for 16 h at rt. The mixture was cooled to −30 °C; DMTST (0.07 mmol) was added, and the resulting mixture was stirred at −30 °C until the disappearance of the starting material, as indicated by TLC. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with sat aq NaHCO3 (~10 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, concentrated in vacuo, and dried. The residue was dissolved in dry methanol (1.0 mL); 1N soln. of NaOMe in MeOH (0.5 mL) was added, and the resulting mixture was stirred for 2–16 h at rt. After that, the reaction mixture was neutralized with Dowex (H+), and the resin was filtered off and washed successively with MeOH. The combined filtrate was concentrated in vacuo. The residue was dissolved in pyridine (1.0 mL); acetic anhydride (0.2 mL) was added dropwise, and the resulting mixture was stirred for 16 h at rt. After that, the reaction was quenched with methanol (~2.0 mL), and the volatiles were evaporated in vacuo. The residue was coevaporated with toluene, and the residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the respective disaccharide.
Methyl 3-O-(6-O-Acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-6-O-acetyl-2,4-di-O-benzyl-α-D-glucopyranoside (4)
The title compound was obtained from various precursors (see Tables 1–4), and its analytical data were the same as those reported previously.7
Methyl 4-O-(6-O-acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-6-O-acetyl-2,3-di-O-benzyl-α-D-glucopyranoside (5)
The title compound was obtained from various precursors (see Tables 1–4), and its analytical data were the same as those reported previously.7
Methyl 6-O-(6-O-Acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-4-O-acetyl-2,3-di-O-benzyl-α-D-glucopyranoside (6)
The title compound was obtained from various precursors (see Tables 1–4), and its analytical data were the same as those reported previously.7
Methyl 6-O-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-6-O-acetyl-2,4-di-O-benzyl-α-D-glucopyranoside (22)
The title compound was obtained from precursor 21, and its analytical data were the same as those reported previously.7
Competition Experiments
General Procedure
A mixture of the tethered compound 1 or 16 (0.019 mmol), acceptor 1324 (0.016 mmol), and freshly activated molecular sieves (3 Å, 100 mg) in 1,2-dichloroethane (0.5 mL) was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C; MeOTf (0.06 mmol) was added, and the resulting mixture was stirred for 18 h at 0 °C. After that, the solid was filtered off through a pad of Celite and washed successively with dichloromethane. The combined filtrate (~30 mL) was washed with sat. aq NaHCO3 (~10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/5, v/v) to afford respective compounds.
Methyl 2,4,6-Tri-O-benzyl-α-D-glucopyranoside (13)
The title compound was synthesized according to the reported procedure, and its analytical data were essentially the same as those reported previously.37
Macrocyclic Disaccharide 14
The title compound was obtained from compound 1 in 20% yield (α only) as a clear film. Analytical data for 14: Rf = 0.63 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 15.6 (c = 1, CHCl3); 1H NMR: δ 1.59 (s, 6H, C(CH3)2), 2.60–2.88 (m, 8H, 2 × COCH2CH2CO), 3.27 (s, 3H, OCH3), 3.42–3.58 (m, 4H, H-2, 2′, 4′, 5′), 3.84–3.90 (m, 2H, H-4, 5), 3.97 (dd, 1H, J3′,4′ = 9.3 Hz, H-3′), 4.22–4.36 (m, 4H, H-3, 6a, 6a′, 6b′), 4.49–4.87 (m, 10H, H-1, 6b, 4 × CH2Ph), 4.88 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 5.42 (d, 1H, J1′,2′ = 3.6 Hz, H-1′), 6.84–7.34 (m, 33H, aromatic) ppm; 13C NMR (75 MHz): δ 29.5, 29.6, 29.7, 29.7, 29.9, 31.0, 42.6 (×2), 55.2, 68.5, 71.6, 73.2, 73.3, 74.9, 75.7, 77.3 (×2), 77.4 (×2), 78.3, 79.6, 79.8, 82.3, 96.4, 97.3, 121.0 (×2), 121.3 (×2), 127.7 (×5), 128.0 (×6), 128.1 (×3), 128.2 (×3), 128.4 (×2), 128.5 (×4), 128.6 (×2), 128.7 (×2), 128.8 (×2), 137.5, 137.8, 138.1, 138.6, 138.9, 148.0, 148.2, 148.6, 148.9, 170.8 (×2), 171.9, 172.0 ppm; HR-FAB MS [M + Na]+ calcd for C71H74NaO17 1221.4824, found 1221.4866.
Compound 15
The title compound was isolated as a colorless foam from the completion of the reaction between compound 1 and 13 in 51% yield (α/β = 3.0/1). Selected analytical data for α-15: Rf = 0.49 (ethyl acetate/toluene, 1/5, v/v); 1H NMR: δ 1.25 (s, 6H, 2 ×CH3), 2.35–2.84 (m, 8H, 2 × –CH2CH2–), 3.29–3.31 (m, 6H, 2 ×OCH3), 3.32–3.44 (m, 4H, H-2, 2″, 4, 4″), 3.47–3.81 (m, 5H, H-2′, 3′, 4′, 5′, 5″), 4.02–4.12 (m, 4H, H-3, 3″, 5, 6a′), 4.20–4.48 (m, 6H, H-6a, 6b, 3′, 6b′, 6a″, 6b″), 4.51–4.74 (m, 13H, H-1′, 1″, 51/2 CH2Ph), 4.81–4.93 (m, 5H, 21/2 CH2Ph), 5.54 (d, 1H, J1,2 = 3.4 Hz, H-1), 6.92–7.36 (m, 48H, aromatic) ppm; 13C NMR (150 MHz): δ 31.6, 31.8, 33.6, 45.1, 57.7, 57.9, 66.0, 66.1, 70.8, 71.1, 71.4, 72.2, 75.6, 75.8, 76.0, 76.2 (×2), 76.3, 77.2, 77.5, 78.2, 78.9, 79.6, 80.5, 81.0, 81.2, 82.1, 82.2, 84.9, 99.7, 100.0, 100.1, 123.4 (×2), 123.5 (×2), 129.3 (×2), 129.9, 130.1, 130.2, 130.3 (×2), 130.4 (x 8), 130.5 (×4), 130.6 (×2), 130.7 (×3), 130.8, 130.9 (×4), 131.0 (x 8), 131.1 (x 8), 131.2 (×2), 131.4 (×2), 140.3, 140.4, 140.5, 140.7, 140.9, 141.0, 141.2, 150.4, 150.6, 151.1, 151.2, 173.5, 173.6, 174.5 ppm, HR-FAB MS [M + Na]+ calcd for C99H106NaO23 1685.7023, found 1685.7056.
Macrocyclic Disaccharide 17
The title compound was obtained as a clear film from compound 16 in 52% yield (α only). Analytical data for 17: Rf = 0.63 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 65.8 (c = 1, CHCl3); 1H NMR: δ 1.62, 1.72 (2s, 6H, C(CH3)2), 3.31 (s, 3H, OCH3), 3.53 (dd, 1H, J2,3 = 9.1 Hz, H-2), 3.55–3.62 (m, 2H, H-2, 4), 3.68 (dd, 1H, J3′,4′ = 9.7 Hz, H-4′), 4.02–4.14 (m, 3H, H-5, 3′, 6a), 4.28 (dd, 1H, H-3), 4.29 (dd, 1H, 2J = 10.7 Hz, 1/2 CH2Ph), 4.42 (dd, 2H, 2J = 10.2 Hz, CH2Ph), 4.51 (m, 7H, H-5′,6a′, 6b′, CH2Ph), 4.87–5.01 (m, 4H, H-6b, 11/2 CH2Ph), 5.52 (d, 1H, J1,2 = 3.5 Hz, H-1), 7.05–8.00 (m, 41H, aromatic) ppm; 13C NMR (75 MHz): δ 31.5, 31.6, 42.9, 55.4, 64.3, 65.6, 67.1, 28.6, 72.2, 72.5, 73.2, 73.4, 75.2, 75.7, 77.4, 78.2, 79.7, 79.8,82.2, 96.3, 96.9, 120.5 (×2), 121.4 (×2), 127.8 (×2), 127.9 (×4), 128.0 (×6), 128.3 (×3), 128.4 (×2), 128.5 (×3), 128. Six (×4), 128.7 (×2), 129.0, 129.2, 129.5, 129.9, 130.2, 131.1, 131.2, 131.5, 131.6, 131.9 (×2), 132.1, 133.1, 137.2, 137.4, 137.9, 138.6, 138.8, 147.9, 148.3, 148.9, 149.0, 166.1, 166.3, 167.6, 167.7 ppm; HR-FAB MS [M + Na]+ calcd for C79H74NaO17 1317.4824, found 1317.4828.
Compound 18
The title compound was isolated as a colorless syrup from the completion reaction between 16 and 13 in 30% yield (α/β = 1.6/1). Selected analytical data for α-18: Rf = 0.38 (ethyl acetate/toluene, 1/5, v/v); 1H NMR (500 MHz): δ 1.57 (s, 6H, C(CH3)2), 3.35 (s, 3H, OCH3), 3.44 (dd, 1H, J2′,3′ = 9.8 Hz, H-2′), 3.50 (dd, 1H, H-4′), 3.80 (dd, 1H, H-3′), 3.95 (m, 1H, H-5′), 5.58 (d, 1H, J1′,2′ = 3.5 Hz, H-1′) ppm; 13C NMR (125 MHz): δ 96.6, 96.8, 97.3 ppm; HR-FAB MS [M + Na]+ calcd for C105H102NaO22 1737.6760, found 1737.6740.
Investigation of the Effect of the Steric Bulkiness at C-6
Ethyl 2,3,4-Tri-O-benzyl-6-O-(o-phenyloxycarbonyl)benzoyl-1-thio-β-D-glucopyranoside (23)
A solution of N,N′-dicyclohexylcarbodiimide (34 mg, 0.17 mmol) in dry CH2Cl2 (1.0 mL) was added dropwise to a solution of 46 (64 mg, 0.10 mmol) and phenol (7.8 mg, 0.083 mmol) in dry CH2Cl2 (1.0 mL) at 0 °C. The resulting mixture was stirred for 2 h, and during this time, the temperature was allowed to gradually increase to rt. After that, the reaction mixture was diluted with CH2Cl2 (~30 mL) and washed with water (3× 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexanes, 1/1, v/v) to afford the title compound in 68% yield (40 mg, 0.057 mmol) as a white amorphous solid. Analytical data for 23: Rf = 0.87 (ethyl acetate/toluene, 3/7, v/v); [α]D29 + 8.6 (c = 1, CHCl3); 1H NMR: δ 1.25 (t, 3H, J = 7.5 Hz, SCH2CH3), 2.73 (m, 2H, SCH2CH3), 3.44 (dd, 1H, J2,3 = 8.9 Hz, H-2), 3.54–3.62 (m, 2H, H-4, 5), 3.71 (dd, 1H, J3,4 = 8.6 Hz, H-3), 4.45 (dd, 1H, J6a,6b = 5.19), 4.47 (d, 1H, J1,2 = 8.9, H-1), 4.56–4.93 (m, 7H, H-6b, 3 × CH2Ph), 7.21–7.90 (24H, aromatic); 13C NMR (75 MHz): δ 15.3, 25.2, 64.7, 75.3, 75.7, 76.0, 77.1, 77.9, 81.8, 85.2, 86.7, 121.7 (×2), 126.2, 128.0 (×2), 128.1 (×2), 128.3 (×2), 128.5 (×2), 128.6 (×2), 128. Seven (×5), 129.4 (×2), 129.7 (×2), 131.6, 131.7, 131.8, 132.2, 137.7, 138.0, 138.4, 151.0, 166.9 ppm; HR-FAB MS [M + Na]+ calcd for C43H42O8SNa 741.2498, found 741.2499.
Methyl 2,3-Di-O-benzyl-6-O-(o-phenyloxycarbonyl)benzoyl-α-D-glucopyranoside (24)
The title compound was prepared from 47 as described for the synthesis of 23 in 83% as a colorless syrup. Analytical data for 24: Rf = 0.52 (ethyl acetate/hexane, 1/1, v/v); [α]D29 + 48.1 (c = 1, CHCl3); 1H NMR: δ 3.18 (s, 3H, OCH3), 3.23 (dd, 1H, J2,3 = 9.5 Hz, H-2), 3.34 (dd, 1H, J3,4 = 8.8 Hz, J4,5 = 9.8 Hz, H-4), 3.36 (m, 1H, H-5), 3.99 (dd, 1H, H-3), 4.44 (m, 3H, J1,2 = 3.5 Hz, H-1, 6a, 6b), 4.52 (m, 3H, 11/2 CH2Ph), 4.76 (d, 1H, 2J = 11.1 Hz, 1/2 CH2Ph), 7.11–7.79 (m, 19H, aromatic) ppm; 13C NMR (75 MHz): δ 55.3, 64.4, 68.3, 73.1, 73.7, 74.6, 77.3, 79.6, 97.3, 121.6 (×2), 126.1, 127.9, 128.1 (×2), 128.2 (×3), 128.5 (×2), 128.7 (×2), 129.2, 129.3, 129.5 (×2), 131.5 (×2), 131.8, 132.0, 137.9, 138.1, 150.9, 166.2, 166.9 ppm; HR-FAB MS [M + Na]+ calcd for C35H34O9Na 621.2101, found 621.2093.
Methyl O-(2,3,4-Tri-O-benzyl-6-O-(o-phenyloxycarbonyl)-benzoyl-D-glucopyranosyl)-(1 → 4)-2,3-di-O-benzyl-6-O-(o-phenyloxycarbonyl)benzoyl-α-D-glucopyranoside (25)
A mixture of 23 (21.2 mg, 0.030 mmol), 24 (16.2 mg, 0.027 mmol), and freshly activated molecular sieves (3 Å, 120 mg) in 1,2-dichloroethane (10 mL) was stirred under argon for 16 h at rt. The mixture was cooled to 0 °C; MeOTf (10.1 μL, 0.09 mmol) was added, and the resulting mixture was stirred under argon for 16 h at 0 °C. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichlorolethane. The combined filtrate (~30 mL) was washed with sat. aq NaHCO3 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound (28.4 mg, 84%, α/β = 2.5/1). Selected analytical data for α-25: Rf = 0.28 (ethyl acetate/hexane, 3/7, v/v); 1H NMR: δ 3.25 (s, 3H, OCH3), 3.40–3.60 (m, 5H, J2,3 = 9.2 Hz, H-2, 2′, 4, 4′, 5′), 3.93 (m, 1H, H-5), 4.04 (dd, 1H, H-3), 4.21–4.65 (m, 12H, H-1′, 3′, 6a, 6b, 6a′, 6b′, 3 × CH2Ph), 4.77–4.98 (m, 4H, 2 × CH2Ph), 7.04–7.86 (m, 43H, aromatic) ppm; 13C NMR (75 MHz): δ 30.0, 55.0, 68.1, 68.9, 73.4, 75.1, 75.7, 77.3, 78.1, 78.7, 78.9, 79.8, 82.3, 97.2, 97.3, 126.0, 126.1, 127.2, 127.5, 127.6, 127.7, 128.9, 128.0, 128.1, 128.3 (×2), 128.4, 128.5 (×2), 129.1, 129.2, 129.5, 131.4, 131.6, 131.8, 131.9, 132.0, 137.6, 137.3, 137.4, 138.2, 138.5, 150.9, 166.2, 166.3, 166.4, 166.9 ppm; HR-FAB MS [M + Na]+ calcd for C76H70NaO17 1277.4511, found 1277.4504.
Ethyl 2,3,4-Tri-O-benzyl-1-thio-6-O-(α,α,α-(4-t-butyldimethylsilyloxyphenyl)dimethyltolyl phthalate)-β-D-glucopyranoside (26)
A solution of N,N′-dicyclohexylcarbodiimide (154 mg, 0.74 mmol) and 4-dimethylaminopyridine (9.0 mg, 0.074 mmol) in dry CH2Cl2 (8.0 mL) was added dropwise to a solution of 46 (200 mg, 0.37 mmol) and α,α,α-(4-t-butyldimethylsilyloxyphenyl)dimethyl-p-cresol38 (383 mg, 1.12 mmol) in dry CH2Cl2 (8.0 mL) at 0 °C. The resulting mixture was stirred for 2 h, and during this time, the temperature was allowed to gradually increase to rt. After that, the reaction mixture was diluted with CH2Cl2 (~30 mL) and washed with water (3× 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexanes, 1/1, v/v) to afford the title compound in 72% yield (256 mg, 0.278 mmol) as a white amorphous solid. Analytical data for 26: Rf = 0.62 (ethyl acetate/hexane, 2/3, v/v); [α]D22 + 5.0 (c = 1, CHCl3); 1H NMR: δ 0.00 (s, 6H, Si(CH3)2), 0.79 (s, 9H, SiC(CH3)3), 1.07 (t, 3H, SCH2CH3), 1.39, 1.45 (2s, 6H, C(CH3)2), 2.53 (m, 2H, SCH2CH3), 3.26 (dd, 1H, J2,3 = 9.5 Hz, H-2), 3.35–3.45 (m, 2H, H-4, 5), 3.52 (dd, 1H, J3,4 = 8.5 Hz, H-3), 4.23–4.30 (m, 2H, H-1, 6a), 4.42 (dd, 1H, J5,6b = 1.5 Hz, H-6b), 4.52 (dd, 2H, 2J = 10.8 Hz, CH2Ph), 4.64 (dd, 2H, 2J = 10.2 Hz, CH2Ph), 4.71 (dd, 2H, 2J = 10.9 Hz, CH2Ph), 6.54–7.67 (m, 27H, aromatic) ppm; 13C NMR (75 MHz): δ −4.33, 1.08, 15.1, 18.2, 25.0, 25.7 (×3), 31.0, 31.1, 42.2, 64.6, 75.2, 75.6, 75.9, 76.9, 77.8, 81.7, 85.1, 86.6, 119.4, 120.8, 127.8 (×4), 127.9 (×2), 128.0 (×2), 128.2 (×2), 128.4 (×2), 128.5 (×5), 129.2, 129.3, 131.4, 131.5, 131.7, 132.1, 137.6, 137.9, 138.3, 143.0, 148.6, 148.8, 153.5, 166.4, 166.8 ppm; HR-FAB MS [M + Na]+ calcd for C58H66NaO9SSi 989.4095, found 989.4092.
Methyl 2,3,4-Tri-O-benzyl-α-D-glucopyranoside (27)
The title compound was synthesized according to the reported procedure, and its analytical data were essentially the same as those reported previously.37
Methyl 2,3,6-Tri-O-benzyl-α-D-glucopyranoside (28)
The title compound was synthesized according to the reported procedure, and its analytical data were essentially the same as those reported previously.37
Methyl O-[2,3,4-Tri-O-benzyl-6-O-(α,α,α-(4-t-butyldimethylsilyloxyphenyl)dimethyltolyl phthalate)-D-glucopyranosyl]-(1 → 6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (29)
A mixture of 26 (35 mg, 0.036 mmol), 27 (25 mg, 0.054 mmol), and freshly activated molecular sieves (3 Å, 100 mg) in 1,2-dichloroethane (1.0 mL) was stirred under argon for 16 h at rt. The mixture was cooled to 0 °C; MeOTf (17.9 mg, 0.108 mmol) was added, and the reaction mixture was stirred at 0 °C until the disappearance of the starting material, as indicated by TLC. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with sat aq NaHCO3 (~10 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, concentrated in vacuo, and dried. The residue was dissolved in dry methanol (1.0 mL); 1N soln. of NaOMe in MeOH (0.5 mL) was added, and the resulting mixture was stirred for 2–16 h at rt. After that, the reaction mixture was neutralized with Dowex (H+), and the resin was filtered off and washed successively with MeOH. The combined filtrate was concentrated in vacuo. The residue was dissolved in pyridine (1.0 mL); acetic anhydride (0.2 mL) was added dropwise, and the resulting mixture was stirred for 16 h at rt. After that, the reaction was quenched with methanol (~2.0 mL), and the volatiles were evaporated in vacuo. The residue was coevaporated with toluene, and the residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound in 57% yield (28 mg, 0.020 mmol, α/β = 1.7/1) as a white amorphous solid. Selected analytical data for α-29: Rf = 0.54 (ethyl acetate/hexane, 3/10, v/v); 1H NMR: δ 3.15 (s, 3H, OCH3), 3.20 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.27–3.52 (m, 4H, H-2, 2′, 4, 4′), 3.71–3.83 (m, 2H, H-3, 3′), 4.35 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.72 (d, 1H, H-1′) ppm; 13C NMR (75 MHz): δ 96.9, 97.9 ppm; HR-FAB MS [M + Na]+ calcd for C84H92NaO15Si 1391.6103, found 1391.6108.
Methyl O-[2,3,4-Tri-O-benzyl-6-O-(α,α,α-(4-t-butyldimethylsilyloxyphenyl)dimethyltolyl phthalate)-D-glucopyranosyl]-(1 → 4)-2,3,6-tri-O-benzyl-α-D-glucopyranoside (30)
The title compound was prepared as described for the synthesis of 29 from 26 (35 mg, 0.035 mmol) and 28 (25 mg, 0.072 mmol) in 61% yield (26.7 mg, 0.021 mmol) as a white amorphous solid. Selected analytical data for α-30: Rf = 0.55 (ethyl acetate/hexane, 3/10, v/v); 1H NMR: δ 3.18 (s, 3H, OCH3), 3.23–3.43 (m, 4H, H-2, 2′, 4, 4′), 3.62–3.73 (m, 2H, H-5, 6a), 3.74–3.80 (dd, 1H, H-3′), 3.85–3.91 (m, 2H, H-3, 6b), 4.42 (d, 1H, H-1), 5.40 (d, 1H, J1′2′ = 3.6 Hz, H-1′) ppm; 13C NMR (150 MHz): δ 96.6, 97.9 ppm; HR-FAB MS [M + Na]+ calcd for C84H92NaO15Si 1391.6103, found 1391.6110.
Synthesis of Compound 31 for Investigating a Possibility of Anomerization
Methyl 2,4-Di-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (61)
The title compound was prepared as previously reported.39 Analytical data for 61: Rf = 0.67 (ethyl acetate/toluene, 1/1, v/v); [α]D27 + 26.5 (c = 1, CHCl3); 1H NMR: δ 2.50 (d, 1H, J = 2.2 Hz, OH), 3.24 (dd, 1H, J5,6a = 3.2 Hz, J6a,6b = 10.1 Hz, H-6a), 3.47 (s, 3H, OCH3), 3.51–3.70 (m, 3H, J2,3 = 11.0, J4,5 = 10.0 Hz, H-2, 4, 6b), 3.83 (m, 1H, H-5), 4.10 (ddd, 1H, H-3), 4.39 (d, 1H, 2J = 10.9 Hz, 1/2 CH2Ph), 4.72 (d, 1H, 2J = 9.6 Hz, 1/2 CH2Ph), 4.80 (s, 2H, CH2Ph), 4.83 (d, 1H, J1,2 = 3.5 Hz, H-1), 7.01–7.54 (m, 25H, aromatic); 13C NMR (75 MHz): δ 55.0, 62.9, 69.9, 73.1, 73.8, 74.6, 78.1, 79.9, 97.4, 127.0, 127.7, 127.9, 128.1 (×9), 128.3 (×4), 128.7 (×3), 128.9 (×3), 138.2 (×6), 138.2, 144.0 (×3) ppm; HR-FAB MS [M + Na]+ calcd for C40H40O6Na 639.2722, found 639.2717.
Methyl O-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-(1 → 3)-2,4-di-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (63)
A mixture of ethyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside40 (62, 124 mg, 0.316 mmol), 61 (162 mg, 0.263 mmol), and freshly activated molecular sieves (3 Å, 360 mg) in 1,2-dichloroethane (4.0 mL) was stirred under argon for 2 h at rt. MeOTf (71 μL, 0.631 mmol) was added, and the resulting mixture was stirred for 4 h at rt. After that, the solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~50 mL) was washed with water (10 mL), sat. aq NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate–hexanes gradient elution) to afford the title compound (160 mg, 64%) as a white amorphous solid. Analytical data for 63: Rf = 0.63 (ethyl acetate/hexane, 3/10, v/v); [α]D27 + 9.5 (c = 1, CHCl3); 1H NMR: δ2.00, 2.07 (×2), 2.16 (4s, 12H, 4 × COCH3), 3.18 (dd, 1H, J5,6a = 4.4 Hz, J6a,6b = 9.9 Hz, H-6a), 3.45–3.52 (m, 5H, H-6b, 4, OCH3), 3.60 (dd, 1H, J2,3 = 9.63 Hz, H-2), 3.67 (dd, 1H, H-5′), 3.85 (dd, 1H, J5,6b = 4.3 Hz, H-5), 4.37 (dd, 1H, J5′,6a′ = 2.0 Hz, J6a′,6b′ = 12.4 Hz, H-6a′), 4.24–4.36 (m, 3H, H-3, 6b′, 1/2 CH2Ph), 4.60 (d, J = 11.4 Hz, 1/2 CH2Ph), 4.72 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.82–4.92 (m, 2H, CH2Ph), 5.09–5.28 (m, 4H, J4′,5′ = 10.3 Hz, H-1′, 2′, 3′, 4′), 7.00–7.48 (m, 25H, aromatic) ppm; 13C NMR (75 MHz): δ 20.7 (×3), 21.0, 54.9, 62.0, 63.1, 68.4, 69.9, 71.7, 72.2, 73.3, 73.7, 74.6, 76.3, 79.2, 81.2, 86.4, 97.1, 100.4, 127.0 (×3), 127.5, 127.8 (×4), 128.1 (×3), 128.3 (×6), 128.4, 128.8 (×7), 137.8, 138.2, 144.0 (×3), 169.5, 169.6, 170.3, 170.8 ppm; HR-FAB MS [M + Na]+ calcd for C54H58NaO15 969.3673, found 969.3673.
Methyl O-(2,3,4-Tri-O-benzyl-β-D-glucopyranosyl)-(1 → 3)-2,4-di-O-benzyl-α-D-glucopyranoside (64)
A solution of NaOMe in methanol (1M, ~1.0 mL) was added dropwise to a solution of 63 (150 mg, 0.22 mmol) in methanol (3.0 mL) until pH ~9, and the resulting mixture was kept for 1 h at rt. After that, Dowex (H+) was added until pH ~7, and the resin was filtered off and washed successively with methanol. The combined filtrate (~30 mL) was concentrated in vacuo and dried. The residue (117 mg, 0.218 mmol) was dissolved in pyridine (5.0 mL); triphenylmethyl chloride (243 mg, 0.872 mmol) was added, and the resulting mixture was stirred for 24 h at rt. After that, the volatiles were removed under the reduced pressure, and the residue was coevaporated with toluene and dried. The residual solid (210 mg, 206 mmol) was dissolved in dry DMF (3.0 mL) and benzyl bromide (0.1 mL, 0.93 mmol). The resulting solution was added dropwise over a period of 15 min to a stirring mixture of NaH (60% in mineral oil, 50 mg, 1.23 mmol) in DMF (3.0 mL) at 0 °C. The reaction mixture was then allowed to warm to rt and stirred for 16 h at rt. After that, the reaction mixture was poured on crushed ice and stirred until cessation of H2 evolution. The mixture was then extracted with ethyl acetate/diethyl ether (3 × 15 mL, 1/1, v/v), and the combined organic phase was washed with water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue (229 mg, 0.205 mmol) was dissolved in CH2Cl2 (10 mL); trifluoroacetic acid (0.3 mL) and water (100 μL) were added, and the resulting mixture was stirred for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~25 mL) and washed with water (10 mL), sat. aq NaHCO3 (2 × 10 mL), and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to obtain the title compound in 78% overall yield (0.138 mg, 0.171 mmol) as a colorless syrup. Analytical data for 64: Rf = 0.48 (ethyl acetate/toluene, 1/1, v/v); [α]D22 + 37.2 (c = 1, CHCl3); 1H NMR: δ 1.57 (br. s, 2H, OH), 3.19–3.23 (m, 4H, H-6a, OCH3), 3.34–3.71 (m, 10H, H-2, 2′, 3′, 4, 4′, 5, 5′, 6a′, 6b, 6b′) 4.24 (dd, 1H, J3–4 = 9.1, H-3), 4.29 (d, 1H, 2J = 11.7, 1/2 CH2Ph), 4.36 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.51–6.50 (m, 3H, 11/2 CH2Ph), 4.75–4.65 (m, 7H, H-1′, 3 × CH2Ph), 7.13–7.36 (m, 25H, aromatic) ppm; 13C NMR (75 MHz): δ 55.2, 61.7, 61.9, 70.3, 73.8, 74.7, 74.8, 75.1, 75.2, 75.7, 75.9, 77.8, 78.0, 81.1, 83.3, 84.8, 97.9, 102.5, 127.7 (×2), 127.9, 128.0, 128.1 (×2), 128.4 (×5), 128.5, 128.6 (×2), 129.1, 138.0, 138.1, 138.4, 138.6, 138.8 ppm; HR-FAB MS [M + Na]+ calcd for C48H54NaO11 829.3564, found 829.3535.
Methyl O-[2,3,4-Tri-O-benzyl-6-O-(o-hydroxycarbonyl)benzoyl-β-D-glucopyranosyl]-(1 → 3)-2,4-di-O-benzyl-6-O-(o-hydroxy-carbonyl)benzoyl-α-D-glucopyranoside (65)
The title compound was prepared from 64 (55 mg, 0.068 mmol), phthalic anhydride (2 ×61 mg, 2 × 0.818 mmol), and 4-(N,N-dimethylamino)pyridine (2 × 4.2 mg, 0.068 mmol) in accordance with the general procedure for introducing the phthaloyl linker in 85% as a colorless syrup. Analytical data for 65: Rf = 0.25 (methanol/dichloromethane, 1/9, v/v); [α]D26 + 2.8 (c = 1, CH3Cl); 1H NMR: δ 3.21 (s, 3H, OCH3), 3.31–3.42 (m, 4H, H-2, 2′, 4, 4′), 3.63–3.66 (m, 2H, H-3, 5′), 4.21–4.57 (m, 10H, H-1, 3, 5′, 6a, 6b, 6a′, 6b′, 2 × CH2Ph), 4.67–4.98 (m, 7H, H-1′, 3 × CH2Ph) 7.10–7.90 (m, 33H, aromatic); 13C NMR (75 MHz): δ 55.3, 65.2, 66.0 (×2), 67.6, 72.8, 73.7, 75.1, 75.3, 75.8, 76.0, 78.8, 79.3, 81.2, 83.0, 84.9, 97.3, 102.3, 127.7, 127.8, 127.9 (×4), 128.0, 128.1 (×4), 128.4 (×6), 128.5 (×6), 128.7 (×3), 128.8, 129.0 (×2), 129.2, 129.6 (×3), 130.5, 130.6, 131.3, 131.6, 132.1, 132.2, 133.8, 135.9, 137.6, 137.9, 138.3, 138.6 ppm; HR-FAB MS [M + Na]+ calcd for C64H62NaO17 1125.3885, found 1125.3896.
Macrocyclic Disaccharide 31
The title compound was prepared in accordance with the general procedure for the introduction of BPA linker from 65 (0.46 mg, 0.042 mmol) in 75% yield (41 mg, 0.032 mmol) as a colorless syrup. Analytical data for 31: Rf = 0.26 (ethyl acetate/dichloromethane, 1/9, v/v); [α]D26 + 18.2 (c = 1, CHCl3); 1H NMR: δ 1.25 (s, 6H, C(CH3)2), 3.30 (s, 3H, OCH3), 3.43–3.49 (m, 2H, H-2, 4), 3.52 (dd, 1H, J2′,3′ = 8.5 Hz, H-2′), 3.63 (br. s, 2H, H-4, 5), 3.73 (dd, 1H, J2,3 = 7.5 Hz, H-3), 3.90 (m, 1H, H-5′), 4.35 (d, 1H, 2J = 11.6 Hz, 1/2 CH2Ph), 4.41 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 4.44–4.66 (m, 8H, H-1, 6a, 6b, 6a′, 6b′, 11/2 CH2Ph), 4.87 (m, 2H, CH2Ph), 4.93 (d, 1H, 2J = 11.8 Hz, 1/2 CH2Ph), 4.99–5.09 (m, 2H, CH2Ph), 5.14 (d, 1H, J1′,2′ = 7.7 Hz, H-1′), 7.02–7.36 (m, 41H, aromatic) ppm; 13C NMR (125 MHz): δ 32.4, 45.0 (×2), 57.9, 67.4, 68.3, 70.9, 75.1, 76.3, 77.8, 77.9, 78.2, 78.6, 80.7, 83.8, 85.7, 87.5, 100.1, 105.3, 110.0, 130.3 (×3), 130.4 (×2), 130.5 (×8), 130.7(×3), 130.9 (x10), 131.0 (x19), 131.1 (×9), 131.7 (×2), 140.5, 141.2, 156.3, 156.4 ppm; HR-FAB MS [M + Na]+ calcd for C79H74NaO17 1317.4824, found 1317.4827.
Attempt to Anomerize 31 in the Presence of BF3-Et2O
A mixture of 31 (5.5 mg, 4.24 μmol) and freshly activated molecular sieves (3 Å, 20 mg) in 1,2-dichloroethane (0.5 mL) was stirred under argon for 2 h at rt. The mixture was cooled to −30 °C; BF3-Et2O (1 μL, 7.7 μmol) was added, and the reaction mixture was stirred at −30 °C for 7 h. After that, the reaction mixture was allowed to gradually warm to rt and stirred for additional 72 h. The solid was filtered off through a pad of Celite and rinsed successively with dichloromethane. The combined filtrate (~30 mL) was washed with sat aq NaHCO3 (~10 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, concentrated in vacuo, and dried. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford 31 quantitatively.
Attempt to Anomerize 31 in the Presence of MeOTf
The title reaction was performed as described in the typical procedure for MeOTf-promoted glycosylation (method B). No anomerization was detected.
Investigation of Other Templates
Methyl 2,3-Di-O-benzyl-6-O-(α,α′-bis(4-hydroxyphenyl)-1,4-diisopropylbenzene succinate)-α-D-glucopyranoside (66)
A solution of N,N′-dicyclohexylcarbodiimide (DCC, 164 mg, 0.80 mmol) in dry dichloromethane (2.0 mL) was added dropwise (over 5 min) to a solution of 37 (126 mg, 0.27 mmol) and 2,2-bis(4-hydroxyphenyl)-1,4-diisopropylbenzene (138 mg, 0.40 mmol) in dry dichloromethane (4.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 4 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/1, v/v) to afford the title compound in 79% yield (168 mg, 0.21 mmol) as a colorless syrup. Analytical data for 66: Rf = 0.53 (ethyl acetate/toluene, 2/3, v/v); [α]D24 + 21.9 (c = 0.8, CHCl3); 1H NMR: δ 1.68 (s, 12H, 2 × C(CH3)2), 2.11 (s, 1H, PhOH), 2.69–3.02 (m, 4H, COCH2CH2CO), 3.42 (s, 3H, OCH3), 3.49–3.58 (m, 2H, H-2, 4), 3.80–3.89 (m, 2H, J5,6a = 3.5 Hz, H-3, 5), 4.34 (d, 1H, J6a,6b = 12.0 Hz, H-6a), 4.51 (dd, 1H, H-6b), 4.65–5.06 (m, 5H, H-1, 2 × CH2Ph), 6.75–7.78 (m, 22H, aromatic) ppm; 13C NMR (75 MHz): δ 114.8 (×2), 120.8 (×2), 126.3 (×2), 126.4 (×2), 127.9 (×2), 128.0 (×3), 128.1, 128.2 (×4), 128.6 (×2), 128.7 (×2), 138.0, 138.6, 142.3, 147.3, 148.2, 148.4, 148.5, 153.6, 171.1, 172.5 ppm; HR-FAB MS [M + Na]+ calcd for C49H54NaO10 825.3615, found 825.3614.
Tethered Compound 32
A solution of DCC (38 mg, 0.186 mmol) and DMAP (2.6 mg, 0.021 mmol) in dry dichloromethane (2.0 mL) was added dropwise (5 min) to a stirring solution of 66 (50 mg, 0.062 mmol) and 35 (55 mg, 0.093 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford tethered compound 32 in 88% yield (76 mg, 0.055 mmol) as a colorless syrup. Analytical data for 32: Rf = 0.48 (ethyl acetate/toluene, 3/7, v/v); [α]D24 + 3.0 (c = 1, CHCl3); 1H NMR: δ 1.48 (t, 3H, J = 7.4 Hz, SCH2CH3), 1.81 (s, 12H, C(CH3)2), 2.85–2.98 (m, 6H, SCH 2 CH3, COCH2 CH2 CO), 3.00–3.06 (m, 4H, COCH2CH2CO), 3.55 (s, 3H, OCH3), 3.58–3.72 (m, 5H, H-2, 2′, 4, 4′, 5′), 3.86–3.96 (m, 3H, H-3, 3′, 5), 4.40–4.43 (m, 2H, H-6a′, 6b′), 4.54–4.66 (m, 3H, H-1′, 6a, 6b), 4.12 (d, 1H, J1,2 = 3.1 Hz, H-1), 4.74–5.19 (m, 10H, 5 × CH2Ph), 7.12–7.54 (m, 37H, aromatic) ppm; 13C NMR (75 MHz): δ 15.3, 25.3, 29.1, 29.3, 30.9, 42.4, 55.5, 63.7, 69.3, 70.0, 73.4, 75.2, 75.7, 75.9, 77.0, 77.4, 79.6, 81.2, 81.8, 85.3, 86.7, 98.3, 120.8, 126.5 (×4), 127.9 (×6), 128.1 (×3), 128.2 (×3), 128.3 (×5), 128.5 (×3), 128.6 (×3), 128.6 (×8), 128.8 (×3), 137.7, 137.9, 138.1, 138.4, 138.7, 147.6 (×2), 148.4 (×3), 171.1 (×2), 172.0, 172.5 ppm; HR-FAB MS [M+Na]+ calcd for C82H90NaO17S 1401.5796, found 1401.5795.
Methyl 2,3-Di-O-benzyl-6-O-[(8-hydroxy-9,10-dioxo-9,10-dihydroanthracen-1-yl) succinate]-α-D-glucopyranoside (67)
A solution of N,N′-dicyclohexylcarbodiimide (DCC, 87 mg, 0.42 mmol) in dry dichloromethane (2.0 mL) was added dropwise (over 5 min) to a solution of 37 (100 mg, 0.21 mmol) and 1,8-dihydroxyanthraquinone (76 mg, 0.32 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 4 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/toluene, 1/1, v/v) to afford the title compound in 84% yield (124 mg, 0.17 mmol) as a colorless syrup. Analytical data for 67: Rf = 0.36 (ethyl acetate/hexanes, 1/1, v/v); [α]D24 + 1.8 (c = 1, CHCl3); 1H NMR: δ 2.81 (t, 2H, J = 6.8 Hz, COCH2CH2CO), 3.08 (t, 2H, J = 6.9 Hz, COCH2CH2CO), 3.32 (s, 3H, OCH3), 3.37 (t, 1H, J4,5 = 9.7 Hz, H-4), 3.42, (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.68 (m, 1H, H-5), 3.71 (dd, 1H, J3,4 = 9.1, H-3), 4.24 (dd, 1H, J6a,6b = 12.1 Hz, J5,6a = 2.1 Hz, H-6a), 4.41(dd, 1H, J5,6b = 4.7 Hz, H-6b) 4.39 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.54–4.92 (m, 4H, 2 × CH2Ph), 7.09–8.20 (m, 16H, aromatic) ppm; 13C NMR (75 MHz): δ 29.0, 29.4, 55.4, 63.7, 69.9, 73.3, 75.6, 79.58, 81.2, 98.3, 116.6, 119.5, 124.6, 124.9, 125.4, 126.1, 128.0, 128.1 (×2), 128.2, 128.3 (×2), 128.6 (×2), 128.7, 129.1, 130.3, 132.7, 135.3, 135.6, 136.8, 138.0, 138.7, 150.4, 162.7, 170.9, 172.5, 181.8, 188.0 ppm; HR-FAB MS [M + Na]+ calcd for C39H36NaO12 719.2104, found 719.2102.
Tethered Compound 33
A solution of DCC (80 mg, 0.39 mmol) and DMAP (4.7 mg, 0.04 mmol) in dry dichloromethane (2.0 mL) was added dropwise (5 min) to a stirring solution of 67 (135 mg, 0.19 mmol) and 35 (173 mg, 0.29 mmol) in dry dichloromethane (2.0 mL) at 0 °C. The resulting mixture was allowed to warm to rt over 2 h. After that, the reaction mixture was diluted with dichloromethane (~30 mL) and washed with water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford tethered compound 33 in 81% yield (218 mg, 0.158 mmol) as a clear yellow syrup. Analytical data for 33: Rf = 0.50 (ethyl acetate/toluene, 1/9, v/v); [α]D23 + 6.0 (c = 1, CHCl3); 1H NMR: δ 1.31 (t, 3H, J = 7.4 Hz, SCH2CH3), 2.35–2.53 (m, 4H, COCH2CH2CO), 2.73 (m, 2H, SCH2CH3), 2.71–2.87 (m, 4H, COCH2CH2CO), 3.37 (s, 3H, OCH3), 3.41 (dd, 1H, J2′,3′ = 8.9 Hz, H-2′), 3.50 (m, 2H, H-2, 4′), 3.56 (dd, 1H, J5,6a = 9.6 Hz, H-5), 3.68 (dd, 1H, J3′,4′ = 8.6 Hz, H-3′), 3.85 (m, 1H, H-5), 3.92 (dd, 1H, J4′,5′ = 9.4 Hz, H-4′), 4.08–4.37 (m, 4H, H-6a, 6b, 6a′, 6b′), 4.45 (d, 1H, J1′,2′ = 9.8 Hz, H-1′), 4.55 (d, 1H, J1,2 = 6.9 Hz, H-1), 4.58–4.96 (m, 10H, 5 × CH2Ph), 5.00 (dd, 1H, J4,5 = 9.7 Hz, H-4), 7.16–7.81 (m, 31H, aromatic) ppm; 13C NMR (75 MHz): δ 15.3, 25.4, 28.9, 29.0 (×2), 29.5, 55.6, 62.8 (×2), 63.8, 67.7, 70.1, 73.7, 75.2, 75.5, 75.7, 75.9, 77.7, 79.2, 79.7, 81.8, 85.7, 86.7, 98.3, 116.7, 119.5, 124.7, 125.0, 126.1, 127.8, 127.9 (×2), 128.0, 128.1 (×4), 128.2 (×4), 128.3 (×3), 128.4 (×2), 128.5 (×4), 128.6 (×4), 130.5, 132.8, 135.4, 135.7, 136.8, 137.8, 138.0 (×2), 138.4, 138.6, 150.5, 162.7, 171.0, 171.2, 171.9 (×2), 181.9, 188.0 ppm; HR-FAB MS [M+Na]+ calcd for C72H72NaO19S 1295.4286, found 1295.4265
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences (Grant GM111835). We thank Dr. Rensheng Luo (UM, St. Louis) for help with acquiring spectral data using the 600 MHz NMR spectrometer that was purchased thanks to the NSF (Award CHE-0959360). Dr. Winter and Mr. Kramer (UM, St. Louis) are thanked for HRMS determinations.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b02151.
1H and 13C NMR spectra for all new compounds (PDF)
References
- 1.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 2.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 3.Jung KH, Muller M, Schmidt RR. Chem Rev. 2000;100:4423–4442. doi: 10.1021/cr990307k. [DOI] [PubMed] [Google Scholar]
- 4.Madsen J, Bols M. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, NY: 2000. pp. 449–466. [Google Scholar]
- 5.Ishiwata A, Lee YJ, Ito Y. Org Biomol Chem. 2010;8:3596–3608. doi: 10.1039/c004281a. [DOI] [PubMed] [Google Scholar]
- 6.Ziegler T. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2008. pp. 469–496. [Google Scholar]
- 7.Pornsuriyasak P, Jia XG, Kaeothip S, Demchenko AV. Org Lett. 2016;18:2316–2319. doi: 10.1021/acs.orglett.6b01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegler T, Lau R. Tetrahedron Lett. 1995;36:1417–1420. [Google Scholar]
- 9.Lau R, Schule G, Schwaneberg U, Ziegler T. Liebigs Ann. 1995;1995:1745–1754. [Google Scholar]
- 10.Ziegler T, Ritter A, Hurttlen J. Tetrahedron Lett. 1997;38:3715–3718. [Google Scholar]
- 11.Ziegler T, Lemanski G. Eur J Org Chem. 1998;1998:163–170. [Google Scholar]
- 12.Lemanski G, Ziegler T. Helv Chim Acta. 2000;83:2655–2675. [Google Scholar]
- 13.Ziegler T, Lemanski G, Hurttlen J. Tetrahedron Lett. 2001;42:569–572. [Google Scholar]
- 14.Huchel U, Schmidt RR. Tetrahedron Lett. 1998;39:7693–7694. [Google Scholar]
- 15.Muller M, Huchel U, Geyer A, Schmidt RR. J Org Chem. 1999;64:6190–6201. [Google Scholar]
- 16.Wakao M, Fukase K, Kusumoto S. Synlett. 1999;1999:1911–1914. [Google Scholar]
- 17.Tennant-Eyles RJ, Davis BG, Fairbanks AJ. Chem Commun. 1999:7–1038. [Google Scholar]
- 18.Tennant-Eyles RJ, Davis BG, Fairbanks AJ. Tetrahedron: Asymmetry. 2000;11:231–243. [Google Scholar]
- 19.Burt J, Dean T, Warriner S. Chem Commun. 2004:454–455. doi: 10.1039/b313571c. [DOI] [PubMed] [Google Scholar]
- 20.Lonn H. J Carbohydr Chem. 1987;6:301–306. [Google Scholar]
- 21.Whitfield DM. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 22.Ravenscroft M, Roberts RMG, Tillett JG. J Chem Soc, Perkin Trans 2. 1982:9–1972. [Google Scholar]
- 23.Andersson F, Fugedi P, Garegg PJ, Nashed M. Tetrahedron Lett. 1986;27:3919–3922. [Google Scholar]
- 24.Koto S, Takebe Y, Zen S. Bull Chem Soc Jpn. 1972;45:291–293. [Google Scholar]
- 25.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068–6070. [Google Scholar]
- 26.De Meo C, Farris M, Ginder N, Gulley B, Priyadarshani U, Woods M. Eur J Org Chem. 2008;2008:3673–3677. [Google Scholar]
- 27.Nigudkar SS, Demchenko AV. Chem Sci. 2015;6:2687–2704. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuester JM, Dyong I. Justus Liebigs Ann Chem. 1975;1975:2179–2189. [Google Scholar]
- 29.Garegg PJ, Hultberg H. Carbohydr Res. 1981;93:C10–C11. [Google Scholar]
- 30.Satoh H, Manabe S, Ito Y, Lüthi HP, Laino T, Hutter J. J Am Chem Soc. 2011;133:5610–5619. doi: 10.1021/ja201024a. [DOI] [PubMed] [Google Scholar]
- 31.Ray AK, Roy N. Carbohydr Res. 1990;196:95–100. doi: 10.1016/0008-6215(90)84108-7. [DOI] [PubMed] [Google Scholar]
- 32.Agoston K, Kroger L, Agoston A, Dekany G, Thiem J. Carbohydr Res. 2009;344:1428–1433. doi: 10.1016/j.carres.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Elchert B, Li J, Wang J, Hui Y, Rai R, Ptak R, Ward P, Takemoto JY, Bensaci M, Chang CWT. J Org Chem. 2004;69:1513–1523. doi: 10.1021/jo035290r. [DOI] [PubMed] [Google Scholar]
- 34.Kaeothip S, Paranjape G, Terrill SE, Bongat AFG, Udan MLD, Kamkhachorn T, Johnson HL, Nichols MR, Demchenko AV. RSC Adv. 2011;1:83–92. [Google Scholar]
- 35.Borsuk K, Kazimierski A, Solecka J, Urbanczyk-Lipkowska Z, Chmielewski M. Carbohydr Res. 2002;337:2005–2015. doi: 10.1016/s0008-6215(02)00139-8. [DOI] [PubMed] [Google Scholar]
- 36.Veeneman GH, van Boom JH. Tetrahedron Lett. 1990;31:275–278. [Google Scholar]
- 37.Ranade SC, Kaeothip S, Demchenko AV. Org Lett. 2010;12:5628–5631. doi: 10.1021/ol1023079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Meulenaer B, Baert K, Lanckriet H, Van Hoed V, Huyghebaert A. J Agric Food Chem. 2002;50:5273–5282. doi: 10.1021/jf0202739. [DOI] [PubMed] [Google Scholar]
- 39.Koto S, Inada S, Yoshida T, Toyama M, Zen S. Can J Chem. 1981;59 [Google Scholar]
- 40.Parameswar AR, Imamura A, Demchenko AV. Carbohydr Chem: Proven Synth Methods. 2012;1:187–196. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.