

Abstract
Sepsis can induce an overwhelming systemic inflammatory response, resulting in organ damage and death. Suppressor of cytokine signaling 1 (SOCS1) negatively regulates signaling by cytokine receptors and Toll-like receptors (TLRs). However, the cellular targets and molecular mechanisms for SOCS1 activity during polymicrobial sepsis are unknown. To address this, we utilized a cecal ligation and puncture (CLP) model for sepsis; C57BL/6 mice subjected to CLP were then treated with a peptide (iKIR) that binds the SOCS1 kinase inhibitory region (KIR) and blocks its activity. Treatment with iKIR increased CLP-induced mortality, bacterial burden, and inflammatory cytokine production. Myeloid cell–specific SOCS1 deletion (Socs1Δmyel) mice were also more susceptible to sepsis, demonstrating increased mortality, higher bacterial loads, and elevated inflammatory cytokines, compared with Socs1fl littermate controls. These effects were accompanied by macrophage metabolic reprograming, as evidenced by increased lactic acid production and elevated expression of the glycolytic enzymes hexokinase, lactate dehydrogenase A, and glucose transporter 1 in septic Socs1Δmyel mice. Upregulation was dependent on the STAT3/HIF-1α/glycolysis axis, and blocking glycolysis ameliorated increased susceptibility to sepsis in iKIR-treated CLP mice. These results reveal a role of SOCS1 as a regulator of metabolic reprograming that prevents overwhelming inflammatory response and organ damage during sepsis.
Keywords: Immunology, Metabolism
Suppressor of cytokine signaling 1 (SOCS1) regulates metabolic reprograming during sepsis by inhibiting glycolysis in macrophages and neutrophils.
Introduction
Sepsis is characterized by life-threatening organ and tissue damage, which results from a dysregulated host immune response to disseminated infection (1). The recognition of different microbial components, such as pathogen-associated molecular patterns (PAMPs) and host-derived damage-associated molecular patterns (DAMPs), induces a number of signaling programs, culminating in the activation of transcription factors, such STATs, activator protein 1 (AP-1), and NF-κB, among others (2). Activation of these pathways results in production of proinflammatory cytokines, including IL-1β, TNF-α, and IL-6. Ultimately, the recognition of abundant PAMPs and DAMPs in sepsis resulting from severe systemic infection leads to cytokine storm, tissue damage, organ dysfunction, and even death (3–5).
SOCS1 is a protein that negatively regulates cytokine receptors, TLR signaling, and NF-κB activation. The SOCS1 protein has three functional domains: a kinase inhibitory region (KIR), an SH2 domain, and a SOCS box. The KIR domain functions as a pseudo-substrate that inhibits JAK2-mediated STAT1 and STAT3 activation (6, 7). The SH2 domain binds directly to the activation loop of JAK2 or apoptosis signal–regulating kinase 1 (ASK1), thereby blocking STAT or MAPK activation, respectively. The SOCS box targets proteins such as JAK2, myeloid differentiation primary-response gene 88 (MyD88) adaptor-like protein (MAL), and ASK1 for degradation by the ubiquitin-proteasome pathway through recruitment of the E3 ubiquitin ligase scaffold Cullin 5 and other components of the E3 ubiquitin ligase complex (6, 8, 9).
TLR ligands, such as LPS and CpG, are potent inducers of inflammation, as well as SOCS1 expression (10, 11). In turn, as noted above, SOCS1 blocks the activity of several signaling components of the TLR/NF-κB pathway, thus providing a mechanism to downregulate potentially harmful inflammation. Mechanistically, SOCS1 may inhibit the NF-κB pathway by binding to the p65 subunit and facilitating its degradation via the ubiquitin-proteasome system (12) or by blocking MAL-dependent p65 phosphorylation (9). We have further shown that SOCS1 also inhibits expression of MyD88 and prevents TLR- and IL-1 receptor–mediated NF-κB activation (13). Consistent with these observations, SOCS1-knockout mice show increased production of proinflammatory cytokines, such as TNF-α, IL-12, and IFN-γ, in response to TLR ligands in vitro (14, 15) and in vivo (16, 17). The precise role of SOCS1 in bacterial sepsis, however, remains undetermined.
Metabolic reprograming is associated with macrophage polarization, during which glycolysis (typically induced by LPS/IFN-γ) is a driver of the proinflammatory phenotype, and oxidative phosphorylation (induced by IL-4/IL-13) triggers macrophage anti-inflammatory and pro-resolution programs (18–21). Enhanced glycolysis (Warburg effect) is necessary for eliciting rapid and potent phagocytic and antimicrobial responses (18), and increased levels of glycolysis have also been associated with production of proinflammatory cytokines in response to TLR ligands. During LPS-induced endotoxic shock, increased glycolysis is observed in peritoneal macrophages, and this is accompanied by elevated levels of another critical component of aerobic glycolysis, pyruvate kinase M2 (PKM2). However, blocking glycolytic flux with a competitive inhibitor of hexokinase (the first enzyme in the glycolytic pathway) decreases IL-1β production, and mice treated with shikonin, an inhibitor of PKM2, show decreases in both glycolysis and IL-1β production, which protects against lethal endotoxic shock and sepsis (22).
HIF-1α is the primary molecule responsible for transcriptional control of glycolytic metabolism (23). Although stability of the HIF-1α protein is controlled by hypoxia and succinate, Hif-1α mRNA expression is tightly regulated by STAT3 (24). This suggests the possibility that SOCS1 may act as an upstream mediator of STAT3-regulated HIF-1α activation, although this has not been shown. Further, although it is somewhat expected that SOCS1 can inhibit macrophage glycolysis and the production of proinflammatory cytokines, the specific regulatory steps involved in SOCS1-mediated regulation of glycolysis during sepsis are unknown.
Here, we investigated the role of SOCS1 in a mouse model of polymicrobial sepsis and found that this protein acts as an endogenous brake for glycolysis by inhibiting the transcriptional upregulation of glycolytic enzymes in macrophages. This, in turn, dampens the systemic inflammatory response, culminating in both reduced tissue damage and improved survival. Our data further suggest that an improved understanding of the activity of known pleiotropic molecular brakes during the overwhelming inflammatory response occurring during sepsis might provide clues to new therapeutic opportunities to control cytokine storm and organ damage resulting from this disease.
Results
Pharmacological inhibition of SOCS1 impairs recovery from CLP-induced sepsis.
Although SOCS1 plays a significant role as an endogenous brake on endotoxin-mediated cytokine production in vivo, the specific role of SOCS1 in bacterial sepsis remains to be determined. Therefore, we initially determined whether Socs1 mRNA expression is altered during sepsis in both humans and mice. Our data show increased Socs1 mRNA expression in blood leukocytes from pediatric septic patients in comparison to normal controls. Likewise, peritoneal cells from mice with polymicrobial sepsis induced by cecal ligation and puncture (CLP) also showed increased Socs1 mRNA expression, as compared with cells from sham-operated mice (Figure 1, A and B).
Figure 1. Inhibition of SOCS1 increases bacterial burden and organ damage during sepsis.

(A) SOCS1 mRNA expression levels in the blood of septic pediatric patients and normal controls, as determined by qPCR (septic shock, n = 180 and normal controls, n = 52); ANOVA, and corrections for multiple comparisons were performed using a Benjamini-Hochberg false discovery rate of 5%. (B) Socs1 mRNA expression levels in C57BL/6 mouse peritoneal cells 18 hours after cecal ligation and puncture–induced (CLP-induced) sepsis, as determined by qPCR (n = 8 mice/group, t test, Mann-Whitney U test); *P < 0.05 vs. sham-operated mice or normal controls. (C) Survival rates for C57BL/6 mice treated with inhibitor of the kinase inhibitory region (iKIR) or scrambled peptide control prior to receiving moderate CLP. Survival was monitored for 9 days (n = 10 mice/group, log-rank [Mantel-Cox] test). Bacterial burden in the (D) blood and (E) peritoneal cavity (PC) was determined 18 hours after CLP (n = 4–5 mice/group, unpaired t test, Mann-Whitney U test). (F) Survival rates for of Socs1Δmyel and Socs1fl septic mice. Survival was monitored for 9 days (n = 13 mice/group, log-rank [Mantel-Cox] test). Inset: Immunoblot of hCD4 confirming Cre recombination in peritoneal cells from Socs1Δmyel mice and no recombination in Socs1fl (control) mice. (G) Bacterial burden in the blood of Socs1Δmyel and Socs1fl septic mice, 18 hours after CLP surgery (n = 7–9 mice/group, t test, Mann-Whitney U test). (H) Bioluminescent methicillin–resistant Staphylococcus aureus (MRSA) load was determined using the in vivo animal imaging (IVIS) detection system in the peritoneal cavity of mice treated with iKIR at 24 hours and 1 hour before infection. (I) Representative diffuse light imaging tomography (DLIT) MRSA CT overlay of mice treated with iKIR or peptide control and infected with bioluminescent MRSA for 24 hours (n = 5-6 mice/group, unpaired t test). (J) Bioluminescent MRSA infection in the kidney 24 hours after infection. (K) Representative DLIT MRSA CT overlays from the kidneys of mice treated with iKIR or peptide control and infected with bioluminescent MRSA for 24 hours (n = 5–6 mice/group, t test, Mann-Whitney U test). Scatter plot shows individual values, mean, and SEM; *P < 0.05, septic vs. control or naive group.
We next determined the ability of our KIR inhibitor peptide (iKIR) to inhibit SOCS1 activity by measuring STAT1 phosphorylation in macrophages pretreated with iKIR prior to challenge with methicillin-resistant Staphylococcus aureus (MRSA). We observed increased STAT1 activation in macrophages treated with iKIR, as compared to macrophages treated with control peptide. From these data, we conclude that iKIR blocks SOCS1 activity and increases STAT1 phosphorylation in vitro (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.92530DS1). To determine whether SOCS1 inhibits the inflammatory response and reduces mortality during sepsis, we treated mice with iKIR and evaluated survival after CLP surgery. We found that iKIR-treated septic mice showed increased mortality (Figure 1C) and an elevated bacterial burden in both the blood and peritoneal cavity (Figure 1, D and E) in comparison to septic mice treated with control peptide. These effects on mortality and bacterial load were accompanied by increased liver, kidney, and cardiac damage, as shown by enhanced production of aspartate transaminase, urea, and creatine kinase–MB (CK-MB), respectively (Supplemental Figure 2, A–C). To determine whether this increased bacterial load mediates organ injury, we administered antibiotic (ertapenem) to iKIR-treated mice 6 hours after the induction of sepsis and then twice a day for 3 days. Our results show that although antibiotic treatment greatly restored survival in septic mice treated with the scrambled peptide control, iKIR + antibiotic–treated mice showed decreased survival, indicating that SOCS1 blockage drives inflammation-mediated organ damage and mortality in septic mice (Supplemental Figure 3). Together these results show that pharmacological inhibition of SOCS1 increases organ damage during sepsis and suggest this could be responsible for the enhanced lethality that is observed in these animals.
Role of myeloid-specific SOCS1 during polymicrobial sepsis.
Macrophages and neutrophils are essential for mounting an appropriate immune response and for the early control of invading pathogens during sepsis (25). Here, we observed that iKIR-treated septic mice exhibited decreased numbers and frequency of neutrophils in the peritoneal cavity (Supplemental Figure 4, A and B). To further study the role of SOCS1-expressing myeloid cells in sepsis, we crossed Socs1fl and LysMcre/cre mice to generate progeny that are Socs1-deficient specifically in myeloid cells (Socs1Δmyel mice). Peritoneal cells from Socs1Δmyel and Socs1fl mice were then challenged with MRSA, and STAT1 phosphorylation was determined, in order to confirm the macrophage-specific Socs1 deletion. Indeed, we found that macrophages from Socs1Δmyel mice were hyperresponsive to MRSA, as evidenced by higher levels of STAT1 phosphorylation in these animals, as compared with littermate control Socs1fl mice (Supplemental Figure 1B). SOCS1 deletion was further confirmed by the presence of human CD4 (hCD4) in macrophages from Socs1Δmyel mice (Figure 1F, inset). When either female or male Socs1Δmyel mice were subjected to CLP, we also observed increased mortality and a substantial increase in bacteremia in comparison to septic WT littermates (Figure 1, F and G). Collectively, these results further suggest that in myeloid cells, SOCS1 acts as an endogenous brake that improves animal survival by limiting organ damage, and possibly enhancing bacterial clearance during polymicrobial sepsis.
SOCS1 prevents methicillin-resistant S.
aureus–induced sepsis. Although CLP is the gold standard animal model for sepsis, it is characterized by an intense acute inflammatory response, and because of the polymicrobial nature of the infection, the immune response could be unique to this model. Therefore, we determined whether blocking SOCS1 will also influence the outcome of sepsis induced by MRSA, a common cause of this condition (26–29). We observed that mice pretreated with iKIR, followed by MRSA-induced peritonitis with bioluminescent S. aureus, exhibited increased bacterial dissemination 24 hours after infection, as compared with controls (Figure 1, H and I). Because we observed higher bioluminescent signal in the area of the kidney, we removed the kidneys from iKIR-treated and control mice and imaged the isolated organs. We found that kidneys from mice treated with iKIR, but not the control peptide, showed robust bioluminescence in the kidney, indicating the presence of MRSA (Figure 1, J and K). These data indicate that SOCS1 exhibits protective effects in different models of bacterial sepsis.
Increased inflammatory response in iKIR-treated animals.
To determine whether SOCS1 inhibits a systemic inflammatory response during sepsis, we measured two cytokines, IL-6 and TNF-α, which are associated with inflammation-related organ damage, in serum from iKIR-treated and control septic mice. We found that iKIR-treated septic mice showed higher systemic cytokine levels than septic mice treated with the peptide control (Figure 2, A and B). Similar results were also observed in Socs1Δmyel septic mice compared with Socs1fl septic littermates (Figure 2, C and D). Several studies have shown that the physiopathology observed in SOCS1-knockout mice depends on IFN-γ production (30–32). Additionally, IFN-γ plays a detrimental role in polymicrobial sepsis (33). Therefore, we evaluated whether pharmacological inhibition of SOCS1 would lead to elevated levels of IFN-γ. Our data show that although CLP did not induce high levels of IFN-γ in mice treated with the scrambled peptide control, iKIR treatment significantly increased IFN-γ production in the peritoneal cavity, but not in the blood, of septic mice (Supplemental Figure 5). Collectively, these results show that SOCS1 acts to downregulate a cytokine associated with systemic inflammation during polymicrobial sepsis.
Figure 2. Pharmacological inhibition of SOCS1 or SOCS1 deficiency increases the systemic proinflammatory response in septic mice.
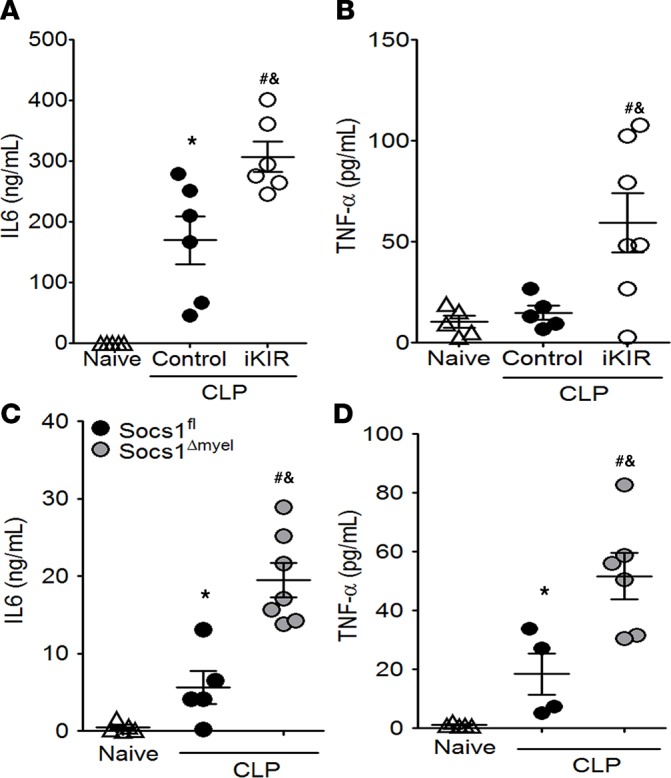
Quantification of IL-6 and TNF-α levels in serum from mice treated with inhibitor of the kinase inhibitory region (iKIR) or control peptide (A and B), and from Socs1Δmyel and Socs1fl mice (C and D) 18 hours after the onset of sepsis. Scatter plot shows individual values, mean, and SEM. *P < 0.05, control septic mice vs. naive, or Socs1fl septic mice vs. naive; #P < 0.05, iKIR-treated septic mice vs. naive, or Socs1Δmyel septic mice vs. naive; &P < 0.05 iKIR-treated septic mice vs. control-treated septic mice, or Socs1Δmyel septic mice vs. Socs1fl septic mice. In all circumstances, n = 4–7 mice/group and 1-way ANOVA followed by Bonferroni correction.
SOCS1 prevents sepsis-induced lung injury.
Systemic infection resulting from bacterial sepsis can affect a number of organs, including the lung (34). We therefore hypothesized that SOCS1 inhibition during sepsis may promote lung damage. To test this, we first measured levels of Socs1 mRNA in lung homogenates and observed increased expression at 18 hours after CLP (Figure 3A). We next inhibited SOCS1 by pretreating with iKIR prior to CLP and found that three common measurements of pulmonary inflammation — alveolar edema, capillary congestion, and neutrophil recruitment (as determined by myeloperoxidase [MPO] activity, a biochemical marker of neutrophil infiltration; refs. 35–38) — were increased in iKIR-treated septic mice, as compared with control septic mice (Figure 3, B–D). Myeloid-specific SOCS1 deletion also resulted in increased neutrophil migration to the lung and enhanced pulmonary inflammation, as evidenced from immunohistological analysis of tissues with an anti-Ly6G antibody, in septic mice (Figure 3, D and E). We further observed increased production of the neutrophil chemokine KC and production of IL-6 in iKIR-treated septic mice (Figure 3, F and G). These data show that SOCS1 can protect septic mice from lung injury and inflammation induced during sepsis.
Figure 3. SOCS1 inhibits lung injury during sepsis.

(A) Socs1 expression in the lung of septic mice was quantified 18 hours after cecal ligation and puncture (CLP) (n = 8–9 mice/group, unpaired t test). (B) Quantification of lung volume in inhibitor of the kinase inhibitory region–treated (iKIR-treated) septic mice 18 hours after CLP by microCT analysis (n = 6–7 mice/group, unpaired t test). (C) Histological analysis with H&E staining of lung tissue from Socs1Δmyel and Socs1fl septic and their respective naive mice (original magnification, ×100). (D) Detection of Ly6G+ neutrophils in Socs1Δmyel and Socs1fl mice, 18 hours after the onset of sepsis. Original magnifications, ×100 and ×400. (E) Pulmonary myeloperoxidase (MPO) activity in the lung of iKIR-treated septic mice. (F) KC and (G) IL-6 production in the lung of iKIR-treated septic mice. E: n = 4 mice/group, t test, Mann-Whitney U test. F: n = 4–5 mice/group, unpaired t test. Scatter plot shows individual values, mean, and SEM. *P < 0.05, iKIR-treated vs. control or naive mice.
SOCS1 targets STAT3 to prevent an overwhelming immune response during sepsis.
SOCS1 is known to inhibit STAT1 and STAT3 activation (7). However, phospho-STAT1 was not detected in septic mice 18 hours after CLP surgery (data not shown). Based on this, we speculated that SOCS1 negatively regulates STAT3, increasing the anti-inflammatory milieu and improving animal survival and host defense. Initially, we observed that both pharmacologic and genetic SOCS1 inhibition in septic mice increased Stat3 expression in peritoneal cells relative to that in septic control mice (Figure 4, A and B). Enhanced Stat3 expression was also observed in the lung of iKIR-treated septic mice (data not shown). Additionally, we found enhanced STAT3 phosphorylation in lung tissue from iKIR-treated septic mice, as compared with control peptide–treated septic mice (Figure 4C). To determine whether STAT3 activation drives increased lethality in SOCS1-deficient mice, we further treated iKIR-treated mice with a STAT3 inhibitor (STATTIC) (39) at 1 hour and 24 hours following the onset of sepsis. We observed that inhibition of STAT3 activation attenuated mortality and reduced bacterial burden in the peritoneal cavity of iKIR-treated septic mice in comparison to controls not given the STAT3 inhibitor (Figure 4, D and E). Collectively, these results suggest that SOCS1 exhibits a protective effect in sepsis by decreasing bacterial burden and lethality in septic mice, which is likely mediated through inhibition of excessive STAT3 activation and reduced proinflammatory cytokine production.
Figure 4. SOCS1 mediates protective effects during sepsis via blockage of STAT3 activation.

Levels of Stat3 mRNA expression 18 hours after onset of sepsis as quantified by qPCR in peritoneal cells from inhibitor of the kinase inhibitory region–treated (iKIR-treated) septic mice (A) or Socs1Δmyel septic mice (B). A: n = 3 mice/group, 1-way ANOVA followed by Bonferroni correction; B: n = 9–10 mice/group, t test. (C) Top: Lung cells were harvested 18 hours after induction of sepsis in mice treated with iKIR or control peptide and subjected to immunoblotting for determination of total STAT3 and phosphorylated STAT3 (Tyr705) expression. Bottom: Histogram showing mean densitometric analysis of immunoblots (n = 3–4 mice/group, 1-way ANOVA followed by Bonferroni correction). (D) Survival rates of iKIR-treated septic mice that were treated (or not) with STATTIC at 24 hours and 1 hour before cecal ligation and puncture (CLP) and once daily for 3 days after CLP (n = 7–8 mice/group, log-rank [Mantel-Cox] test). (E) Bacterial load in mice treated as in D. CFU determined 18 hours after CLP (n = 3–4 mice/group, 1-way ANOVA followed by Bonferroni correction). Scatter plot shows individual values, mean, and SEM. *P < 0.05, control septic mice vs. naive, Socs1Δmyel septic mice vs. Socs1fl; #P < 0.05, iKIR-treated septic mice vs. naive; %P < 0.05, STATTIC- and iKIR-treated septic mice vs. iKIR-treated septic mice; &P < 0.05, iKIR-treated septic mice vs. control septic mice.
SOCS1 inhibits glycolysis-associated gene expression in septic mice.
STAT3 is known to drive HIF-1α promoter activation and expression (24), and in turn, HIF-1α promotes expression of enzymes involved in glycolysis during Th17 differentiation (23). We found that animals treated with iKIR prior to the onset of sepsis showed higher levels of Hif-1α mRNA expression than mice treated with the peptide control (Figure 5A). This increased Hif-1α mRNA in iKIR-treated septic mice was accompanied by elevated mRNA expression of Glut1 (glucose transporter 1), which increases glucose uptake (40), and of Ldha (lactate dehydrogenase A) (Figure 5B), which enhances lactate production (41) (Figure 5C). An enhanced expression of glycolysis-associated genes was also observed in the lung of iKIR-treated septic mice (data not shown). Lactate is the final product of glycolysis, and lactate production can be used as an indicator of increased glycolytic flux (18, 22). Consistent with our mRNA expression data, we observed increased accumulation of lactate in the peritoneal cavity of iKIR-treated septic mice (Figure 5C). Hexokinase 1 (Hk1) is a key enzyme in glycolysis; this protein phosphorylates glucose, enabling it to be metabolized through the glycolytic pathway (42). We found that iKIR-treated septic mice exhibited increased Hk1 mRNA and protein expression (Figure 5, D and E). Further, elevated expression of glycolytic genes was also observed in septic Socs1Δmyel mice, relative to septic Socs1fl littermate controls (Figure 5, F and G). Collectively, these results indicate that SOCS1 functions to inhibit metabolic reprogramming during sepsis.
Figure 5. SOCS1 amplifies HIF-1α–mediated glycolysis during sepsis.

Septic mice treated with inhibitor of the kinase inhibitory region (iKIR) or control peptide were sacrificed 18 hours after cecal ligation and puncture (CLP), and mRNA expression levels of (A) Hif-1α (n = 3 mice/group, 1-way ANOVA followed by Bonferroni correction) or (B) Glut1 and Ldha were quantified in peritoneal cells by qPCR (n = 3–7 mice/group, 1-way ANOVA followed by Bonferroni correction). (C) Lactate levels in peritoneal exudate (n = 3–7 mice/group, 1-way ANOVA followed by Bonferroni). (D) Hk1 mRNA expression as determined by qPCR in peritoneal cells from mice treated as in A (n = 4–6 mice/group, t test, Mann-Whitney U test). (E) Lung cells were harvested 18 hours after CLP in mice treated with iKIR or control peptide and subjected to immunoblotting for determination of hexokinase expression. Numbers: Mean densitometric analysis of the bands from immunoblots (n = 3–4 mice/group, 1-way ANOVA followed by Bonferroni correction). Expression of (F) Hif-1α, (G) Glut1, and Ldha mRNA transcripts was determined by qPCR in peritoneal cells from Socs1Δmyel or Socs1fl septic mice. F: n = 4–7 mice/group, t test, Mann-Whitney U test. G: Glut1: n = 6–9 mice/group, t test, Mann-Whitney U test. Ldha: n = 4–13 mice/group, t test, Mann-Whitney U test. (H and I) Peritoneal macrophages from Socs1Δmyel or Socs1fl were stimulated with 100 ng/ml LPS for 24 hours, and ChIP assays were performed using anti-STAT3 (H), anti–HIF-1α (I), or isotype control IgG antibodies. Pulled-down DNA was subjected to qPCR amplification using specific primers against the promoter for Hif-1α (H) or Hk1 (I). H and I: n = 3 mice/group, 1-way ANOVA followed by Bonferroni correction. Scatter plot shows individual values, mean, and SEM. *P < 0.05, control septic mice vs. naive; #P<0.05, iKIR-treated septic mice vs. naïve; &P<0.05, iKIR-treated septic mice vs. control-treated septic mice, or Socs1Δmyel vs. Socs1fl septic mice. Experiments in vitro: *P < 0.05, LPS-Socs1fl vs. media-Socs1fl macrophages; #P < 0.05, LPS-Socs1Δmyel vs. media-Socs1Δmyel macrophages; &P < 0.05, LPS-Socs1fl vs. LPS-Socs1Δmyel macrophages. PC, peritoneal cavity.
STAT3-induced Hif-1α expression in LPS-challenged Socs1Δmyel macrophages.
To gain further insight into whether SOCS1 negatively regulates STAT3 binding to the Hif1α promoter and HIF-1α binding to the Hk1 promoter, we carried out a ChIP assay. We found that STAT3 binding to the Hif-1α promoter was increased in LPS-stimulated Socs1Δmyel macrophages in comparison to LPS-stimulated Socs1fl control macrophages (Figure 5H). We also found that HIF-1α binding to the Hk1 promoter was increased in LPS-stimulated Socs1Δmyel macrophages in comparison to LPS-stimulated Socs1fl littermate control macrophages (Figure 5I). Together these results indicate that SOCS1 prevents macrophage metabolic reprograming by blocking the STAT3/HIF-1α axis during sepsis.
SOCS1 inhibits glycolysis to prevent an exacerbated proinflammatory response.
LPS induces metabolic reprograming, which results in a switch from oxidative phosphorylation to aerobic glycolysis in macrophages and dendritic cells (18). To determine whether aberrant glycolysis is an important cause of enhanced mortality in SOCS1-deficient animals, we treated mice with the competitive hexokinase inhibitor 2-deoxyglucose (2-DG) once a day for 4 days and 1 hour before CLP. The animals also received iKIR or control peptide 24 hours and 1 hour before CLP. We found that lactate production was decreased following 2-DG administration in iKIR-treated septic mice compared with control septic mice treated only with iKIR (Figure 6A). Administration of 2-DG to iKIR-treated septic mice also restored resistance to sepsis, as evidenced by reduced bacterial burden in the blood and peritoneal cavity, and decreased levels of IL-1β production, as compared with control septic mice treated only with iKIR (Figure 6, B and C); however, no changes in TNF-α levels were observed (Figure 6D). Collectively, these results indicate that, during sepsis, SOCS1 blocks activation of the STAT3/HIF-1α axis and the glycolysis pathway, effectively reducing the proinflammatory response and potentially preventing sepsis-associated organ damage.
Figure 6. Increased glycolysis is responsible for iKIR-mediated animal mortality during sepsis.

Mice were treated with the competitive hexokinase inhibitor 2-deoxyglucose (2-DG; 0.5 g/kg, i.p.) daily for 4 days and 1 hour before cecal ligation and puncture (CLP). The animals were also treated with iKIR (inhibitor of the kinase inhibitory region), 24 hours and 1 hour before surgery. (A) Lactate levels in peritoneal exudate (n = 5–7 mice/group, 1-way ANOVA followed by Bonferroni correction). (B) Bacterial loads were determined in blood and peritoneal exudate 18 hours after CLP (n = 4–9 mice/group, 1-way ANOVA followed by Bonferroni correction). Levels of IL-1β (C) and TNF-α (D) were quantified in peritoneal exudate (n = 4–5 mice/group, 1-way ANOVA followed by Bonferroni correction). Scatter plot shows individual values, mean, and SEM. *P < 0.05, control-treated septic mice vs. naive; &P<0.05, iKIR vs. control-treated septic mice; %P<0.05, iKIR and 2-DG vs. iKIR-treated septic mice; #P<0.05, iKIR-septic mice vs. naive mice. PC, peritoneal cavity.
Discussion
Activation of pathogen recognition receptors triggers sequential events that drive production of inflammatory mediators (e.g., TNF-α, IL-6, IL-1β, KC, and MIP-2), as well the as the synthesis of microbicidal molecules, such as proteins and lipids involved in the generation of reactive oxygen species and reactive nitrogen species (RNS) (2, 43). Although the inflammatory response is required to control infection, when left unchecked, it can be harmful, driving tissue damage, organ failure, and death (43–45). During sepsis, the initial inflammatory events are important determinants of the outcome of disease. Therefore, identifying molecular brakes that can act to dial down the inflammatory response could provide critical therapeutic targets for preventing organ damage and morbidities associated with sepsis.
SOCS1 is a pleiotropic inhibitor of the inflammatory response, and can prevent activation of pathogen recognition receptors, cytokine receptors, and growth factors (15–17, 46–48). SOCS1 is known to act at different steps in the TLR pathway, and SOCS1-deficient mice are hypersensitive to endotoxemia (16, 17). Indeed, SOCS1 deficiency in myeloid cells leads to higher production of proinflammatory cytokines during IFN-γ stimulation (49). However, the possible role of SOCS1 as a relevant negative regulator involved in systemic infection is relatively understudied. Here, we found that (a) SOCS1 expression is increased in peritoneal cells of septic humans and mice; (b) blockage of the KIR domain before onset of sepsis increases animal mortality, bacterial load, and serum cytokine levels; (c) SOCS1 expression in myeloid cells leads to impaired bacterial clearance and exaggerated cytokine production during sepsis; (d) SOCS1 targets STAT3, but not STAT1, to reduce animal mortality and bacterial load; (e) SOCS1 induces metabolic reprograming via the STAT3/HIF-1α–mediated expression of genes involved in glycolysis; and (f) SOCS-mediated inhibition of glycolysis reverses the increased bacterial loads and enhanced inflammatory cytokine production observed in iKIR-treated septic mice. Thus, our results provide insights into the mechanisms whereby SOCS1 inhibits the inflammatory response and improves bacterial clearance and host survival in two models of bacterial sepsis.
The role of SOCS1 in the control of the proinflammatory response and its potential therapeutic value has been demonstrated for various inflammatory diseases (50, 51). Specifically, treatment with SOCS1 mimetic peptides, analogous to KIR, or a tyrosine kinase inhibitor peptide (Tkip) protects mice from developing murine encephalomyelitis and from the increased inflammatory response observed in keratinocytes stimulated with LPS (52–54). These results illustrate the potential for developing strategies based on SOCS1 in therapeutics targeting the pathogenesis of inflammatory disease.
Previous studies have shown that SOCS1 deficiency increases susceptibility of mice to endotoxic shock (16, 17). For example, Chung et al. demonstrated that whole-body SOCS1-heterozygous-knockout mice are more susceptible to polymicrobial sepsis, as compared with WT mice (55). However, these authors did not observe any change in SOCS1 expression in blood cells, peritoneal leukocytes, lung, or spleen (55). In contrast, we found increased expression of SOCS1 in both peritoneal and lung cells from septic mice. We speculate that these conflicting results could be due to differences in sepsis severity and/or the time points studied. Indeed, alterations in SOCS1 expression have been associated with disease severity for several inflammatory disorders (48, 56–58). To further determine whether SOCS1 expression is increased in patients with sepsis, we employed transcriptomic analysis and found elevated levels of SOCS1 in peripheral blood cells from septic pediatric patients as compared with healthy subjects. However, more studies are necessary to determine whether differences in SOCS1 expression correlate with severity of sepsis.
To investigate whether endogenously produced SOCS1 limits the inflammatory response and improves animal survival during sepsis, we employed two different approaches: (a) we pretreated mice with a peptide that block the KIR domain of the SOCS1 protein and (b) utilized mutant mice containing myeloid cells deficient for SOCS1. In both instances, loss of SOCS1 drives an exaggerated inflammatory response, poor host defense, and organ injury. Structurally, SOCS1 is composed of three main domains: KIR, SH2, and SOCS box (6, 11). Although here we studied the role of KIR domain in the pathogenesis of sepsis, the role of the other SOCS1 domains remains to be determined (53, 54). We focused our present efforts on deciphering the role of the KIR domain because it has been implicated in the pathogenesis of other inflammatory diseases (6). Further, previous studies have shown that SOCS1-KIR inhibition is sufficient to limit both STAT3 and STAT1 activation and to inhibit production of proinflammatory cytokines (6). Critically, we found that both pharmacologic blockage of the KIR domain or SOCS1 deletion in myeloid cells drives an aberrant systemic inflammatory response (higher IL-6 and TNF-α) and, as noted previously, increased levels of nitric oxide (data not shown) in septic mice, as compared with control septic animals. This exaggerated cytokine production is accompanied by a deficiency in bacterial clearance and increased organ damage (kidney, heart, liver, and lung). To elucidate whether the increased lethality observed in iKIR-treated mice was due to aberrant inflammation or excessive bacterial growth, we treated iKIR-challenged mice with antibiotic (ertapenem) every 12 hours for 3 days, and animal survival was determined over time. We observed that although antibiotic treatment increases animal survival in mice treated with scrambled control peptide, antibiotic did not improve survival in iKIR-treated mice (Supplemental Figure 3). These data suggest that cytokine-mediated organ damage drives animal mortality in the absence of SOCS1.
We, and others, have shown that neutrophil recruitment to the peritoneal cavity is essential to control infection (51, 59, 60). Here, we found that although iKIR treatment inhibits neutrophil recruitment to the site of infection, neutrophil accumulation could be detected in the lungs, indicating increased pulmonary inflammation. The mechanisms involved in this SOCS1-mediated differential neutrophil recruitment to the site of infection and lung will be investigated in the near future. In addition, we plan to determine whether SOCS1 modulates antimicrobial effector functions in phagocytes. Both STAT1 and STAT3 are activated by LPS to induce cytokine production (13, 61, 62). While STAT1 plays a deleterious role in polymicrobial sepsis, deletion of STAT3 in myeloid cells leads to higher mortality in polymicrobial sepsis, and loss of STAT3 is associated with enhanced systemic cytokine production and organ damage (63). Here, our data show that in the absence of SOCS1, STAT3, but not STAT1, is highly activated via phosphorylation. Further, treatment of mice with a STAT3 blocking peptide, STATTIC (39, 64, 65), reduces both mortality and bacterial burden in iKIR-treated septic mice. STAT3 is activated by IL-6 (66). Given that we observe increased STAT3 and IL-6 during sepsis in SOCS1-deficient mice, it remains to be determined whether increased IL-6 production is responsible for STAT3 activation, or whether STAT3 activation is the driver of IL-6 production in this model.
STAT3 is known to be involved in expression of HIF-1α, a master regulator of key glycolytic enzymes (24, 41, 67). Glycolysis is an essential pathway for immune cell function; it is required for the production of proinflammatory cytokines, phagocytosis, proliferation, and activation during infection (67, 68). Immune cells are particularly influenced by aberrant glycolysis, which can drive exaggerated proinflammatory cytokine production, macrophage polarization, and inflammasome activation (21, 69–71). Increased glycolysis is also associated with different pathogenic conditions, such as encephalomyelitis, arthritis, and diabetes (19, 21, 24). TLR4 is the most highly studied immune receptor involved in metabolic reprograming in macrophages and dendritic cells. TLR4 activation enables cells to generate ATP that is required for different effector functions, including phagocytosis, bacterial killing, production of inflammatory cytokines, and antigen presentation (19, 72). In macrophages, the protein PKM2 mediates enhanced glycolysis by interacting with HIF-1α and inducing expression of glycolytic enzymes, such as HK-2, LDHA, and pyruvate dehydrogenase kinase 1 (PDK-1), which, in turn, are regulated by HIF-1α (22). It is uncertain, however, how these glycolytic enzymes activate cytokine production. A recent study revealed that HK1, another glycolytic enzyme, activates the NLPR3 inflammasome to promote IL-1β processing (71). This enzyme is also an intracellular receptor that recognizes endogenous PAMPs during infection (70). Given that SOCS1 negatively regulates transcriptional programs involved in HIF-1α–induced HK1 expression in macrophages, it seems likely that SOCS1 also negatively regulates HK1 signaling during PAMP recognition. Furthermore, while we focused our work on transcriptional changes associated with lack of SOCS1, we could not rule out possible effects of SOCS1 on enzyme activity, given that SOCS1 can inhibit the activation in different enzymes, such as IKK and Src kinases (73–75). Therefore, SOCS1 could also directly interfere with different components of the glycolytic pathway. However, the possibility of such effects will be addressed in the near future.
Glycolysis is a well-ordered event, and regulators that control the expression and/or activation of glycolytic enzymes would provide attractive targets to control inflammatory diseases and the development of systemic inflammatory responses. Consistent with this, Yang et al. showed that pharmacological inhibition of PKM2 renders mice more resistant to lethal sepsis. This enhanced resistance was associated with reductions in both glycolysis and IL-1β production (22, 76). These data, combined with our observations here, suggest that targeting aerobic glycolysis might represent a viable therapeutic strategy for treatment of inflammatory diseases. Indeed, we found that SOCS1 blockage increases expression of enzymes involved in metabolic reprograming, resulting in increased glycolysis, and that prevention of HK activation (by treating mice with 2-DG) restores bacterial clearance and cytokine production in Socs1Δmyel septic mice. In addition to SOCS1, other negative regulators of metabolic reprogramming that are important for controlling inflammatory disease have been described. The orphan nuclear receptor ERR-α is both a regulator of energy metabolism and a negative regulator of inflammation (77). The question of whether SOCS1 associates with ERR-α and/or other inhibitors of glycolysis, however, remains to be determined.
In summary, our results identify SOCS1 as a regulator of glycolysis metabolism during polymicrobial sepsis and further reveal the mechanisms by which SOCS1 prevents an exacerbated proinflammatory response. This SOCS1-mediated regulatory program prevents excessive damage to the host and death. Our findings further suggest that targeting components in this pathway might provide a potential therapeutic strategy for the treatment of systemic inflammatory response syndrome.
Methods
Mice.
WT C57BL/6 mice were obtained from the Jackson Laboratory. C57BL/6 Socs1fl mice were obtained from Warren Alexander (Walter and Eliza Hall Institute, Parkville, Victoria, Australia) (78), and these were mated with C57BL/6 LysMcre/cre mice from the Jackson Laboratory and maintained at either the Laboratory Animal Resource Center (LARC), Indiana University School of Medicine, or the Division of Animal Care (DAC), Vanderbilt University Medical Center. Mice were genotyped before each experiment for detection of hCD4 as a reporter for cre-mediated recombination, as previously described (78). Mice carrying the SOCS1 deletion specifically in myeloid cells were denoted Socs1Δmyel. Socs1Δmyel and Socs1fl mice were crossed for at least 7–10 generations in our facility.
Sepsis induction.
Sepsis was induced by CLP, as previously described (79), using a 21 gauge needle to induce moderate sepsis. After CLP, mice were not given pain medicine (NSAIDs) before or after sepsis because this class of drugs is known to increase SOCS1 expression and therefore could skew our findings. Mice were given 1 ml s.c. buffered saline as fluid resuscitation therapy. The survival of the animals was evaluated 6 hours after CLP and twice per day, every day, during the 9 days after surgery. All procedures described here were approved by both the Vanderbilt University Medical Center and Indiana University School of Medicine, and when septic mice were in distress (abnormal posture or movement, weight loss, ocular discharge, ataxia, tremor, hypothermia, and cyanosis) for more than 48 hours, they were euthanized according to guidelines established by the IACUC at Vanderbilt University Medical Center and Indiana University School of Medicine. Peritoneal lavage was performed, and blood and lung were harvested 18 hours after surgery to measure cytokines, gene expression, and bacterial burden in septic mice.
Transcriptomic data.
Socs1 mRNA expression data were extracted from an existing microarray-based transcriptomic database, and the data have been deposited in the NCBI’s Gene Expression Omnibus (GEO GSE66099). Briefly, the samples were obtained from children with septic shock (n = 180) and normal controls (n = 52), and the RNA transcripts used for microarray analyses were derived from whole blood obtained within the first 24 hours of meeting criteria for septic shock.
Peptides and treatments.
The iKIR (DTHFRTFRSHSDYRR) and scrambled-iKIR control peptides (DTHFARTFARSHSDYRRI) were obtained from GenScript. A lipophilic palmitoyl group was added to the N-terminus for cell penetration (80). Peptides were dissolved in ultrapure water and then in PBS prior to use in vivo. Mice were treated with 50 μg of either the iKIR peptide or the scrambled-iKIR control (53) 24 hours and 1 hour before CLP via i.p. injection.
Mice were treated with STATTIC, a well-known inhibitor of STAT3 activation, 24 hours and 1 hour before CLP and once per day over the following 3 days (10 mg/kg, i.p.) (39, 65). The competitive hexokinase inhibitor 2-DG was diluted in PBS and administered via i.p. injection (0.5 g/kg) daily for 4 days and 1 hour before CLP.
Peritonitis caused by methicillin-resistant Staphylococcus aureus (MRSA) and in vivo animal imaging (IVIS).
C57BL/6 mice were treated with iKIR 24 hours and 1 hour before bacterial infection. Bioluminescent MRSA (81) was then administered via i.p. injection (2 × 108 CFU) in 200 μl PBS, and 24 hours after infection, mice were induced with 4%–5% isoflurane (balance medical oxygen), transferred to an isothermal stage of the IVIS SpectrumCT (PerkinElmer), and maintained with 2%–3% isoflurane (balance medical oxygen). Planar (2D) bioluminescence images were collected with the following settings at each time point: 30-second integration; 24.4-mm field of view; no filtration; F stop 1; and binning factor 8. To permit diffuse light imaging tomography (DLIT) associated with bioluminescent emission, multispectral image series were collected with the following settings: 120-second integration; 13.1-mm field of view; no filtration; F stop 1; and binning factor 16 at 560, 580, 600, 620, and 640 nm. Immediately afterward, calibrated CT images were collected to permit surface and dense tissue modeling using the following parameters: 50-ms exposure; 720 projections; 1-mA tube current; 50-kVp tube voltage; 120-μm Cu filter; and binning factor 4, reconstructed using filtered back-projection methods, yielding a final isotropic resolution of 0.015 mm. In all cases, images were acquired, reconstructed, and fused using Living Image (v4.3.1.0.15890, PerkinElmer). For planar analysis, images were imported in to Living Image, the kidneys were segmented via semiautomated methods, and total photon flux (p/s) was quantified for all tissues. For DLIT/CT, images were imported to Analyze 12.0 (Analyze Direct) and segmented using threshold-based region growing, and lung volumes (V) were computed according to Equation 1:
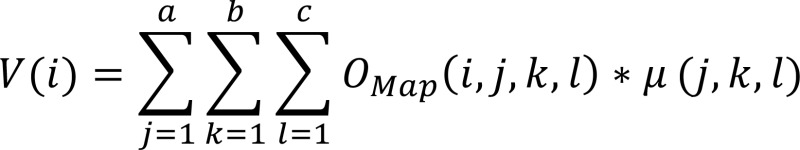
Equation 1
where i is the volume of the “i’th” region; j, k, and l are the “j’th,” “k’th,” and “l’th” object map indexes, respectively; a is the total number of row voxels; b is the total number of column voxels; c is the total number of slices; 0Map is object map; and μ is voxel dimension in millimeters.
Bacterial counts.
Peritoneal lavage fluid and blood were collected from septic mice 18 hours after surgery, as previously described (79). Serial dilutions were plated on Mueller-Hinton agar (Difco Laboratories), and plates were incubated for 18 hours at 37°C. For MRSA bacterial counts, peritoneal exudate was collected 24 hours after infection, and serial dilutions were plated on TSB agar. Colonies were counted after incubation overnight at 37°C. Results are expressed as log CFU/ml of blood or peritoneal exudate.
Microscopic determination of neutrophil migration during sepsis.
C57BL/6 mice were treated with iKIR or scrambled peptide and then sacrificed 18 hours after CLP surgery. Cells present in peritoneal cavity were harvested with PBS/EDTA solution, and total cell numbers were quantified using a Neubauer chamber. For differential counting, cells were concentrated on centrifuge slides by cytospin (300 g for 7 minutes), and stained with Diff-Quik, according to the manufacturer instructions (Merz & Dade).
MPO assay.
MPO activity in lung homogenates was determined 18 hours after CLP, as previously described (35–38, 82). Results are expressed as units of MPO activity per gram tissue.
Immunohistochemistry.
For histological analysis, slides from 5 μm of paraffin-embedded lung sections were treated with 10% hydrogen peroxide in distilled water to block endogenous peroxidase activity. Slides were blocked with PBS containing 8% serum. Sections were then incubated with Ly6G antibody (clone 1A8; BD Biosciences), followed by peroxidase-conjugated secondary antibody, color development, and hematoxylin counterstaining. Negative staining controls were generated by omitting the primary antibody from this procedure.
Cytokine and nitrite quantification.
Cytokine levels in blood and peritoneal exudate was measured by ELISA, according to the manufacturer’s instructions (BioLegend or R&D Systems). Nitrite was determined using the Griess reaction, as described in ref. 13.
Lactate activity assay.
Lactate levels in peritoneal exudate were quantified using a Lactate Colorimetric Assay Kit (Biovision), according to the manufacturer instructions.
Biochemical parameters.
Aspartate aminotransferase activity in serum from septic mice was determined in the Translation Core in the Center for Diabetes and Metabolic Disease at Indiana University School of Medicine. Renal dysfunction was evaluated by measuring serum levels of urea, and cardiac lesions were assessed by measuring levels of the isoenzyme CK-MB, according to the manufacturer instructions (Labtest).
Gene expression analysis.
Cells from the peritoneal cavity and total lung were harvested, and RNA extraction was performed using TRIzol reagent, according to the manufacturer’s instructions (Invitrogen). The RT2 First Strand Kit reverse transcription system (QIAGEN) was used for cDNA synthesis, and quantitative PCR (qPCR) was performed on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories). Relative gene expression was calculated using the comparative threshold cycle (Ct) and expressed relative to control or WT groups (ΔΔCt method). Primers for β-actin, Socs1, Stat3, Hif-1α, Glut-1, Ldha, Mct4, and Hk1 were purchased from Integrated DNA Technologies.
Immunoblotting.
Western blotting was performed as previously described (83). Membranes were incubated with the following primary antibodies: STAT3 (1:1,000; #9139, clone 124H6), phospho-STAT3 (Tyr705; 1:250; #9145, clone D3A7), STAT1 (#9172), phospho-STAT1 (Tyr701; 1:500; #9167, clone 58D6), HIF-1α (1:1,000; #14179, clone D2U3T), and HK1 (1:1,000; #2024, clone C35C4) (all from Cell Signaling Technology) or β-actin (1:20,000; # A5441, clone UC15, Sigma-Aldrich), and then washed and incubated with appropriate fluorophore-conjugated secondary antibodies (1:2,000, anti-rabbit IgG, HRP-linked antibody, #7074, from Cell Signaling Technology). Relative band intensities were quantified using ImageJ software (NIH), as previously described (13).
Peritoneal macrophage cell culture.
Macrophages from Socs1Δmyel mice were induced by injection with 2 ml 3% thioglycollate, and these were harvested 4 days after injection, as previously described (84). Macrophages were stimulated with 100 ng/ml LPS (Sigma-Aldrich), and 24 hours after stimulation, these were harvested for analysis by ChIP assay.
ChIP assay.
ChIP assays were performed using a SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) according to the manufacturer’s instructions (Cell Signaling Technology). Briefly, cells were fixed with formaldehyde and lysed; chromatin was then fragmented using micrococcal nuclease and sonicated (UP100H; Hielscher Ultrasonics) to obtain DNA fragments of approximately 150–900 bp. ChIP was performed using ChIP-validated antibodies to STAT3 (1:100; #9139) and HIF-1α (1:50; #14179) and ChIP-grade Protein G magnetic beads, all from Cell Signaling Technology. Chromatin was eluted from beads in elution buffer, according to the manufacturer’s instructions. DNA was purified using spin columns, and the samples were then subjected to qPCR as described above using the following primers: Hk1, forward, 5′-TGAACTCTGACAGGCTGTGG-3′ and reverse, 5′-AAACTAGGCTTCACAGA-3, and Hif1α, forward, 5′-GAGACTTCCCTTGTTGTGTAG-3′ and reverse, 5′-CTTACTCCTTAGCTGCGTGGC-3′ (24).
Histopathological examination.
Lung sections from septic and non-septic mice treated with iKIR or the scrambled peptide control were prepared as previously described (79), and sections (5 μm) were stained with H&E, as described previously (85). Sections of stained lung were imaged using an Infinity 1 camera (Lumenera) attached to a Nikon Eclipse Ci microscope.
Statistics.
Data were analyzed with StatSoft Inc. (2004) STATISTICA software, version 7. Data are expressed as mean ± SEM. Significance between two groups was estimated using the t test, 1-tailed for parametric data, and the Mann-Whitney U test for nonparametric data. Experiments with 3 or more groups were analyzed using 1-way ANOVA and corrected using the Bonferroni multiple comparison test and the Kruskal-Wallis test for nonparametric data. Survival curves are expressed as percent survival and were analyzed by a log-rank (Mantel-Cox) test; P < 0.05 was considered significant. The normalized transcriptomic data used from people with septic shock and healthy individuals were analyzed using GeneSpring GX 7.3 software (Agilent Technologies). All signal intensity–based data were used after robust multi-array average (RMA) normalization, which specifically suppresses all but the most significant variation among lower-intensity probe sets (86). All chips representing septic shock samples were then normalized to the respective median values of controls on a per-gene basis. Differences in mRNA abundance between study groups were determined using ANOVA, and corrections for multiple comparisons were performed using a Benjamini-Hochberg false discovery rate of 5%.
Study approval.
The animals were maintained in the Laboratory Animal Resource Center (LARC) at Indiana University School of Medicine (IUSM) (protocols 10500 and 10886) or at the Division of Animal Care (DAC) at Vanderbilt University Medical Center (protocol M1600154). In all cases, we adhered to specific guidelines for the use of experimental animals, as outlined in the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). Details of the human study protocol were previously published, and the consent form for the human transcriptomic analysis allows for a secondary analysis of clinical and biological data (87).
Author contributions
ARPA, JCAF, CHS, and SB conceived and designed the experiments. ARPA, SB, SS, SW, NGB, and BPM performed the experiments. ARPA, JCAF, CHS, SB, HW, and PRT analyzed the data. ARPA and CHS wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (HL-103777 and R01HL124159-01 to CHS; T32AI060519 to SB; GM099773 and GM108025 to HW); an American Lung Association Senior Research Training Fellowship (RT-349159) to NGB; and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and São Paulo Research Foundation (FAPESP), under grant agreements 2011/19670-0 (Projeto Temático) and 2013/08216-2 (Center for Research in Inflammatory Disease), and the CAPES Programa de Doutorado Sanduíche no Exterior (PDSE 99999.003387/2015-01) to ARPA. This study was also funded, in part, by an Indiana Institute for Biomedical Imaging Sciences Advanced Imaging Research Technology Development (AIRTD) grant to PRT. Analyte measures were completed in the Translation Core of the Center for Diabetes and Metabolic Diseases at Indiana University School of Medicine (1P30DK097512 R. Mirmira [PI]).
Footnotes
CHS’s present address is: Division of Infectious Diseases, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(13):e92530. doi:10.1172/jci.insight.92530.
Contributor Information
Annie Rocio Piñeros Alvarez, Email: anniep93@gmail.com.
Nicole Glosson-Byers, Email: nlglosson@yahoo.com.
Stephanie Brandt, Email: brandt@umail.iu.edu.
Soujuan Wang, Email: suewang@iupui.edu.
Hector Wong, Email: hector.wong@cchmc.org.
Sarah Sturgeon, Email: sarah.m.sturgeon@vanderbilt.edu.
References
- 1.Singer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. doi: 10.1038/nrdp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335(6071):936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54(2):281–288. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jamieson AM, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340(6137):1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed CM, Larkin J, Johnson HM. SOCS1 Mimetics and Antagonists: A Complementary Approach to Positive and Negative Regulation of Immune Function. Front Immunol. 2015;6:183. doi: 10.3389/fimmu.2015.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi R, et al. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J Exp Med. 2011;208(10):2055–2067. doi: 10.1084/jem.20110428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujimoto M, Naka T. SOCS1, a Negative Regulator of Cytokine Signals and TLR Responses, in Human Liver Diseases. Gastroenterol Res Pract. 2010:470468. doi: 10.1155/2010/470468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mansell A, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7(2):148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 10.Mostecki J, Showalter BM, Rothman PB. Early growth response-1 regulates lipopolysaccharide-induced suppressor of cytokine signaling-1 transcription. J Biol Chem. 2005;280(4):2596–2605. doi: 10.1074/jbc.M408938200. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 12.Ryo A, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12(6):1413–1426. doi: 10.1016/S1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 13.Serezani CH, Lewis C, Jancar S, Peters-Golden M. Leukotriene B4 amplifies NF-κB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J Clin Invest. 2011;121(2):671–682. doi: 10.1172/JCI43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. 2004;279(52):54702–54707. doi: 10.1074/jbc.M411043200. [DOI] [PubMed] [Google Scholar]
- 15.Sachithanandan N, et al. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes. 2011;60(8):2023–2031. doi: 10.2337/db11-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinjyo I, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17(5):583–591. doi: 10.1016/S1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa R, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17(5):677–687. doi: 10.1016/S1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez-Prados JC, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 20.Vats D, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang EV, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castaldo ET, Yang EY. Severe sepsis attributable to community-associated methicillin-resistant Staphylococcus aureus: an emerging fatal problem. Am Surg. 2007;73(7):684–687. [PubMed] [Google Scholar]
- 27.Sharma-Kuinkel BK, Zhang Y, Yan Q, Ahn SH, Fowler VG. Host gene expression profiling and in vivo cytokine studies to characterize the role of linezolid and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA) murine sepsis model. PLoS One. 2013;8(4):e60463. doi: 10.1371/journal.pone.0060463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakker M, Park JS, Carey V, Lee JC. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun. 1998;66(11):5183–5189. doi: 10.1128/iai.66.11.5183-5189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HK, Missiakas D, Schneewind O. Mouse models for infectious diseases caused by Staphylococcus aureus. J Immunol Methods. 2014;410:88–99. doi: 10.1016/j.jim.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander WS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98(5):597–608. doi: 10.1016/S0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 31.Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS. Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1) Proc Natl Acad Sci USA. 2002;99(2):943–948. doi: 10.1073/pnas.022628499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metcalf D, Di Rago L, Mifsud S, Hartley L, Alexander WS. The development of fatal myocarditis and polymyositis in mice heterozygous for IFN-gamma and lacking the SOCS-1 gene. Proc Natl Acad Sci USA. 2000;97(16):9174–9179. doi: 10.1073/pnas.160255197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romero CR, et al. The role of interferon-γ in the pathogenesis of acute intra-abdominal sepsis. J Leukoc Biol. 2010;88(4):725–735. doi: 10.1189/jlb.0509307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34(3):129–136. doi: 10.1016/j.it.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czaikoski PG, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. 2016;11(2):e0148142. doi: 10.1371/journal.pone.0148142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sônego F, et al. MyD88-, but not Nod1- and/or Nod2-deficient mice, show increased susceptibility to polymicrobial sepsis due to impaired local inflammatory response. PLoS One. 2014;9(8):e103734. doi: 10.1371/journal.pone.0103734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andonegui G, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111(7):1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pulli B, et al. Measuring myeloperoxidase activity in biological samples. PLoS One. 2013;8(7):e67976. doi: 10.1371/journal.pone.0067976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 40.Freemerman AJ, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289(11):7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206(Pt 12):2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 43.Sônego F, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. doi: 10.3389/fimmu.2016.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peña G, Cai B, Deitch EA, Ulloa L. JAK2 inhibition prevents innate immune responses and rescues animals from sepsis. J Mol Med. 2010;88(8):851–859. doi: 10.1007/s00109-010-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park JH, et al. Annexin A5 increases survival in murine sepsis model by inhibiting HMGB1-mediated pro-inflammation and coagulation. Mol Med. 2016;22:424–436. doi: 10.2119/molmed.2016.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carow B, et al. Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves Mycobacterium tuberculosis control in an interferon-gamma (IFN-gamma)-dependent manner. J Biol Chem. 2011;286(30):26873–26887. doi: 10.1074/jbc.M111.238287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fenner JE, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. 2006;7(1):33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida T, et al. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med. 2004;199(12):1701–1707. doi: 10.1084/jem.20031675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chong MM, Metcalf D, Jamieson E, Alexander WS, Kay TW. Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood. 2005;106(5):1668–1675. doi: 10.1182/blood-2004-08-3049. [DOI] [PubMed] [Google Scholar]
- 50.Baig MS, et al. NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J Exp Med. 2015;212(10):1725–1738. doi: 10.1084/jem.20140654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamim CF, Silva JS, Fortes ZB, Oliveira MA, Ferreira SH, Cunha FQ. Inhibition of leukocyte rolling by nitric oxide during sepsis leads to reduced migration of active microbicidal neutrophils. Infect Immun. 2002;70(7):3602–3610. doi: 10.1128/IAI.70.7.3602-3610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol. 2005;175(8):5077–5086. doi: 10.4049/jimmunol.175.8.5077. [DOI] [PubMed] [Google Scholar]
- 53.Jager LD, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol. 2011;232(1-2):108–118. doi: 10.1016/j.jneuroim.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madonna S, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol. 2013;43(7):1883–1895. doi: 10.1002/eji.201343370. [DOI] [PubMed] [Google Scholar]
- 55.Chung CS, Chen Y, Grutkoski PS, Doughty L, Ayala A. SOCS-1 is a central mediator of steroid-increased thymocyte apoptosis and decreased survival following sepsis. Apoptosis. 2007;12(7):1143–1153. doi: 10.1007/s10495-007-0059-7. [DOI] [PubMed] [Google Scholar]
- 56.Chinen T, et al. Suppressor of cytokine signaling-1 regulates inflammatory bowel disease in which both IFNgamma and IL-4 are involved. Gastroenterology. 2006;130(2):373–388. doi: 10.1053/j.gastro.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 57.Ramírez-Vélez G, et al. Constitutive phosphorylation of interferon receptor A-associated signaling proteins in systemic lupus erythematosus. PLoS One. 2012;7(7):e41414. doi: 10.1371/journal.pone.0041414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pathak S, et al. MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp Mol Med. 2015;47:e164. doi: 10.1038/emm.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trevelin SC, et al. Toll-like receptor 9 activation in neutrophils impairs chemotaxis and reduces sepsis outcome. Crit Care Med. 2012;40(9):2631–2637. doi: 10.1097/CCM.0b013e318258fb70. [DOI] [PubMed] [Google Scholar]
- 60.Alves-Filho JC, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010;16(6):708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 61.Greenhill CJ, et al. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol. 2011;186(2):1199–1208. doi: 10.4049/jimmunol.1002971. [DOI] [PubMed] [Google Scholar]
- 62.Peña G, et al. Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. Eur J Immunol. 2010;40(9):2580–2589. doi: 10.1002/eji.201040540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsukawa A, Takeda K, Kudo S, Maeda T, Kagayama M, Akira S. Aberrant inflammation and lethality to septic peritonitis in mice lacking STAT3 in macrophages and neutrophils. J Immunol. 2003;171(11):6198–6205. doi: 10.4049/jimmunol.171.11.6198. [DOI] [PubMed] [Google Scholar]
- 64.Okada S, Han S, Patel ES, Yang LJ, Chang LJ. STAT3 signaling contributes to the high effector activities of interleukin-15-derived dendritic cells. Immunol Cell Biol. 2015;93(5):461–471. doi: 10.1038/icb.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han Z, et al. Inhibition of STAT3 signaling targets both tumor-initiating and differentiated cell populations in prostate cancer. Oncotarget. 2014;5(18):8416–8428. doi: 10.18632/oncotarget.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.da Silva CG, et al. A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals. Hepatology. 2013;57(5):2014–2025. doi: 10.1002/hep.26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galván-Peña S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shirai T, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213(3):337–354. doi: 10.1084/jem.20150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolf AJ, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell. 2016;166(3):624–636. doi: 10.1016/j.cell.2016.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moon JS, et al. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015;12(1):102–115. doi: 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linossi EM, Nicholson SE. Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev. 2015;266(1):123–133. doi: 10.1111/imr.12305. [DOI] [PubMed] [Google Scholar]
- 74.Dimitriou ID, Clemenza L, Scotter AJ, Chen G, Guerra FM, Rottapel R. Putting out the fire: coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol Rev. 2008;224:265–283. doi: 10.1111/j.1600-065X.2008.00659.x. [DOI] [PubMed] [Google Scholar]
- 75.Wormald S, Hilton DJ. The negative regulatory roles of suppressor of cytokine signaling proteins in myeloid signaling pathways. Curr Opin Hematol. 2007;14(1):9–15. doi: 10.1097/00062752-200701000-00004. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Z, et al. Plumbagin Protects Mice from Lethal Sepsis by Modulating Immunometabolism Upstream of PKM2. Mol Med. 2016;22:162–172. doi: 10.2119/molmed.2015.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab. 2008;19(8):269–276. doi: 10.1016/j.tem.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chong MM, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity. 2003;18(4):475–487. doi: 10.1016/S1074-7613(03)00078-5. [DOI] [PubMed] [Google Scholar]
- 79.Ferreira AE, et al. PPAR-γ/IL-10 axis inhibits MyD88 expression and ameliorates murine polymicrobial sepsis. J Immunol. 2014;192(5):2357–2365. doi: 10.4049/jimmunol.1302375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waiboci LW, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol. 2007;178(8):5058–5068. doi: 10.4049/jimmunol.178.8.5058. [DOI] [PubMed] [Google Scholar]
- 81.Guo Y, Ramos RI, Cho JS, Donegan NP, Cheung AL, Miller LS. In vivo bioluminescence imaging to evaluate systemic and topical antibiotics against community-acquired methicillin-resistant Staphylococcus aureus-infected skin wounds in mice. Antimicrob Agents Chemother. 2013;57(2):855–863. doi: 10.1128/AAC.01003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alves-Filho JC, de Freitas A, Russo M, Cunha FQ. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit Care Med. 2006;34(2):461–470. doi: 10.1097/01.CCM.0000198527.71819.E1. [DOI] [PubMed] [Google Scholar]
- 83.Kim SH, Serezani CH, Okunishi K, Zaslona Z, Aronoff DM, Peters-Golden M. Distinct protein kinase A anchoring proteins direct prostaglandin E2 modulation of Toll-like receptor signaling in alveolar macrophages. J Biol Chem. 2011;286(11):8875–8883. doi: 10.1074/jbc.M110.187815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Z, et al. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PLoS One. 2015;10(2):e0115855. doi: 10.1371/journal.pone.0115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin EL, et al. Phosphoinositide-3 kinase gamma activity contributes to sepsis and organ damage by altering neutrophil recruitment. Am J Respir Crit Care Med. 2010;182(6):762–773. doi: 10.1164/rccm.201001-0088OC. [DOI] [PubMed] [Google Scholar]
- 86.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 87.Wong HR, et al. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics. 2007;30(2):146–155. doi: 10.1152/physiolgenomics.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.