

Abstract
Sialylation of glycoproteins and glycolipids is catalyzed by sialyltransferases in the Golgi of mammalian cells, whereby sialic acid residues are added at the nonreducing ends of oligosaccharides. Because sialylated glycans play critical roles in a number of human physio‐pathological processes, the past two decades have witnessed the development of modified sialic acid derivatives for a better understanding of sialic acid biology and for the development of new therapeutic targets. However, nothing is known about how individual mammalian sialyltransferases tolerate and behave towards these unnatural CMP‐sialic acid donors. In this study, we devised several approaches to investigate the donor specificity of the human β‐d‐galactoside sialyltransferases ST6Gal I and ST3Gal I by using two CMP‐sialic acids: CMP‐Neu5Ac, and CMP‐Neu5N‐(4pentynoyl)neuraminic acid (CMP‐SiaNAl), an unnatural CMP‐sialic acid donor with an extended and functionalized N‐acyl moiety.
Keywords: CMP-sialic acid, glycoengineering, selective exo enzymatic labeling, sialylation, sialyltransferases
Introduction
Sialic acids (Sias) are nine‐carbon electro‐negatively charged monosaccharides commonly found on deuterostome cell‐surface glycoconjugates.1 Because of their position, charge, and structural diversity, they are of particular importance in modulating a variety of cellular recognition events.2 In human tissue, the most common is N‐acetyl‐neuraminic acid (5‐acetamido‐3,5‐dideoxy‐d‐glycero‐d‐galacto‐nonulopyranos‐1‐onic acid, Neu5Ac). In vivo, it is synthesized from N‐acetyl mannosamine (ManNAc) or N‐acetyl glucosamine (GlcNAc) along a complex metabolic pathway.3 Indeed, sialic acids metabolism requires a variety of enzymes at different subcellular sites of mammalian cells; among these are the nuclear CMP‐Neu5Ac synthase (CSS or CMAS), the cytosolic UDP‐GlcNAc 2‐epimerase/N‐acetylmannosamine kinase (GNE), the cytosolic cytidine monophosphate‐N‐acetylneuraminic acid hydroxylase (CMAH), the Golgi CMP‐Neu5Ac transporter (SLC35A1), 20 Golgi sialyltransferases (STs), and four sialidases (Neus).3 With the advent of chemical biology, new metabolic glycoengineering methodologies, MOE (metabolic oligosaccharide engineering) or MGE (metabolic glycoengineering), have arisen to study sialic acid metabolic pathways in eukaryotes. These ground‐breaking strategies, pioneered by the groups of Reutter4 and Bertozzi5 hijack cell metabolism by the introduction of an unnatural monosaccharide bearing a chemical modification. From this starting point, two main approaches have emerged. First, chemical modifications of ManNAc, such as elongation of the N‐acyl side chain of peracetylated monosaccharides (largely used by the Reutter and Hortskorte groups), has led to the discovery of important and unexpected functions of the N‐acyl side chain of sialic acids, such as modulation of virus binding, stimulation of neural cell growth, and activation of T lymphocytes.6 The second approach, initially developed by the Bertozzi group, consists of specific covalent ligation of a specific probe to a chemical reporter, through universal bioorthogonal chemical reactions (click chemistry).7 Many bioorthogonal strategies have been reported, such as Staudinger–Bertozzi,8 copper catalyzed alkyne–azide cycloaddition (CuAAC),9 strain‐promoted alkyne–azide cycloaddition (SPAAC),10 and Diels–Alder reactions with inverse electronic demand (DAinv).11 These fast‐growing strategies reflect the development of chemical tools for biological issues, and the associated new instruments for bio‐imaging are essential to shed light on the metabolic pathways of sialic acid.
However, these MOE approaches are hindered by the inefficient use of monosaccharide analogues and somewhat limited by variability among cell lines in incorporating these unnatural sialic acids. To overcome these limitations, fluorescent and photoactivable cytidine‐5′‐monophospho‐sialic acids (CMP‐Sias)12 and CMP‐Neu5N‐acyl derivatives,13 which can be covalently attached to asialoglycoconjugates, were synthesized. As these CMP‐Sias analogues are not membrane‐permeable (thus limiting their use in living cells), selective exo‐enzymatic labeling (SEEL) for the eukaryotic cell surface was developed by using exogenous recombinant rat β‐galactoside (α2,6‐sialyltransferase ST6Gal I) and C‐5 or C‐9 azido‐modified CMP‐Sias to label cell‐surface N‐linked glycans.14 This approach was successfully used to achieve profiling of the cell‐surface glycoproteome.15 Moreover, a neuraminidase‐coupled SEEL strategy with specific sialyltransferases led to the efficient labeling and detection of sialylated N‐ and O‐glycosylproteins by mass spectrometry.16 Thus, the main advantage of SEEL over MOE is selective labeling of glycans on the cell surface without interfering with endogenous enzymes of the sialic acid pathway. In addition, SEEL offers a promising new strategy for the study of recombinant sialyltransferases and in particular of their tolerance toward different donor/acceptor substrates directly at the cell surface.
Sialyltransferases are key evolutionarily conserved enzymes involved in the final steps of the sialic acid pathway; they catalyze the transfer of sialic acid residues from an activated CMP‐Sia donor onto galactose (Gal), N‐acetylgalactosamine (GalNAc), or another sialic acid residue of glycoproteins or glycolipids.17 The twenty identified human sialyltransferases are classified into four families: ST6Gal, ST3Gal, ST8Sia, and ST6GalNAc (named according to the monosaccharide acceptor and glycosidic linkage formed).18 Molecular cloning and biochemical characterization are available for a limited number of mammalian sialyltransferases, and despite the increasing number of crystal structures of these enzymes,19 our understanding of their structure/activity relationships is still very limited. Several studies have underscored their exquisite acceptor substrate specificity, likely relying on specific sequence motifs (“sialylmotifs”)20 and their redundant activity in vivo.
A few studies have reported the sialylation kinetics of mammalian sialyltransferases with various exogenous glycoprotein substrates.21 However, almost nothing is known about how individual mammalian sialyltransferases behave towards CMP‐Sias donors and how these glycosyltransferases tolerate natural and unnatural sialic acids. It is interesting to note that the evolutionarily related mammalian polysialyltransferases ST8Sia II and ST8Sia IV22 differentially use N‐acyl derivatives of Neu5Ac, thus suggesting diverse donor substrate affinities.6c, 23 In order to address this question, we focused on the most widely known human glycoprotein β‐d‐galactoside α2,3/6‐sialyltransferases: ST6Gal I and ST3Gal I.24 These two enzymes catalyze the formation of α‐2–6 or α‐2–3 glycosidic linkages on the terminal Gal of type‐II (Galβ1,4GlcNAc) and type‐III (Galβ1,3GalNAc) disaccharides found on N‐glycans and O‐glycans, respectively. Each enzyme was released into the cell culture medium of transiently transfected HEK293 cells as truncated recombinant proteins, and used either for in vitro assays or with living cells to quantitatively evaluate the ability to use N‐acyl CMP‐Sia derivatives. For this purpose, CMP‐Neu5Ac and CMP‐N‐(4‐pentynoyl)neuraminic acid (CMP‐SiaNAl) bearing an extended N‐acyl group functionalized with alkyne group were synthesized as previously described.25
Results and Discussion
Chemical synthesis of CMP‐Sia derivatives: CMP‐SiaNAl, CMP‐Neu5Ac
To evaluate the donor substrate tolerance of sialyltransferases, we synthesized two activated CMP‐Sia donors (Scheme 1), one natural (CMP‐Neu5Ac) and one alkynyl derivative (CMP‐N‐(4‐pentynoyl)neuraminic acid or CMP‐SiaNAl) by using readily available CMP‐sialic acid synthase (E.C. 2.7.7.43) from Neisseria meningitidis as recently described.25 Each substrate was directly used with both ST6Gal I and ST3Gal I. The sialic acid residue was transferred by each sialyltransferase onto its preferred acceptor (Scheme 1).
Scheme 1.
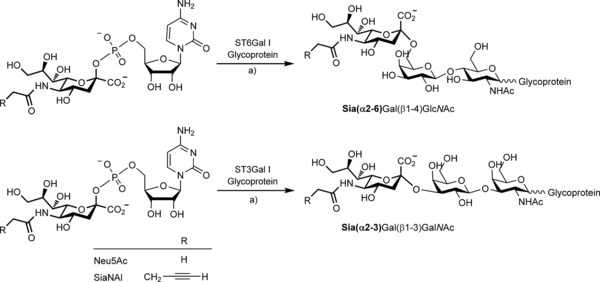
The two activated sialic acid donors used for selective sialylation of N‐ and O‐glycosylproteins. Left: CMP‐N‐acetylneuraminic acid (CMP‐Neu5Ac left) and the alkyne derived CMP‐N‐(4‐pentynoyl)neuraminic acid (CMP‐SiaNAl). Right: resulting sialylated products formed after enzymatic activity of ST6Gal I or ST3Gal I, with transferred sialic acid shown in bold. a) Cacodylate buffer (pH 6.2), 6 h, 37 °C.
Production of recombinant soluble active human α2,3/6sialyltransferases (Δ56ST3Gal I and Δ56ST6Gal I)
Recombinant expression of mammalian sialyltransferases in suitable amounts and quality is difficult, and few commercial sources of human sialyltransferases are available. As N‐terminal truncation of sialyltransferases impacts enzyme production and activity,26 we generated soluble active forms of the human ST3Gal I and ST6Gal I enzymes lacking the first 56 amino acids of the open reading frame (cytoplasmic tail and transmembrane and stem domains). The enzymes were named Δ56ST3Gal I and Δ56ST6Gal I, respectively. A FLAG‐tag and a preprotrypsin signal peptide were introduced into the recombinant chimeric proteins for efficient secretion of an N‐terminal FLAG‐tagged recombinant enzyme into the culture medium of transiently transfected human embryonic kidney cells (HEK293). Thirty‐six hours post‐transfection, we detected the secreted FLAG‐tagged enzyme by western blotting with an anti‐FLAG antibody (Figure 1). Δ56ST3Gal I and Δ56ST6Gal I migrated as multiple bands with an apparent molecular masses of 40–50 and 54 kDa, respectively corresponding to glycosylated protein isoforms as assessed by PNGase F treatment (data not shown). Low levels of Δ56ST6Gal I were consistently obtained, whereas Δ56ST3Gal I expressed at much higher levels in the cell culture medium of transfected HEK293 cells.
Figure 1.
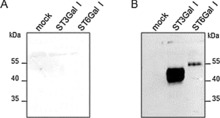
Production of recombinant soluble human α2,3/6‐sialyltransferases Δ56ST6Gal I and Δ56ST3Gal I in the cell culture medium of HEK293 transfected cells. Truncated isoforms (cytoplasmic tail and transmembrane domain deleted; 56 amino acid residues) were N‐terminally tagged with FLAG. These and an empty construct (mock) were transiently expressed in HEK293 cells. After 36 h, 30 μL of HEK293 cell culture medium was subjected to SDS‐PAGE (8 %) under reducing conditions and transferred to a nitrocellulose membrane. A) The membrane was stained with red ponceau, and B) western blotting was carried out with Bio M2 anti‐FLAG mAb (1/1000 e). Positions of the high‐range pre‐stained standards (Bio‐Rad Lab) are indicated. These data are representatives of several experiments and show that Δ56ST3Gal I is expressed at much higher levels than Δ56ST6Gal I in the culture medium, as evaluated from ImageJ analysis of the anti‐FLAG signal.
Reported kinetic data for human sialyltransferases are sparse and vary significantly for recombinant enzymes. In this study, we performed enzymatic reactions for crude extracts of Δ56ST3Gal I and Δ56ST6Gal I secreted in the HEK293 cell culture medium with freshly prepared CMP‐Neu5Ac and radiolabeled CMP‐[14C]Neu5Ac as donor substrate,25 and 60 μg of desialylated fetuin as an acceptor substrate. Fetal bovine fetuin contains three N‐glycans and three O‐glycans,27 and is commonly used as an acceptor substrate for both ST3Gal I and ST6Gal I. HEK293 cells transiently transfected with an empty pFLAG plasmid (mock) were used as a negative control, as this cell culture medium did not show any FLAG‐tagged protein or sialyltransferase activity (Figures 1 and 2). Sialylated product formation was monitored over time for each recombinant enzyme. The rate of sialylated fetuin formation remained constant for at least 5 to 6 h with either Δ56ST6Gal I or Δ56ST3Gal I (Figure 2 A). We also noticed that asialofetuin was sialylated much faster by Δ56ST3Gal I than by Δ56ST6Gal I: the formation of α2,3‐sialylated product reached a maximum after 6 h incubation, whereas the formation of α2,6‐sialylated product kept on increasing up to 15 h (Figure 2 A). We checked that our recombinant enzymes had negligible transfer activity onto potential targets in the medium (data not shown). We then incubated each enzyme for 6 h with various concentrations of CMP‐Neu5Ac (1 to 100 μm; Figure 2 B). Apparent kinetic parameters were obtained by nonlinear regression analysis of the Michaelis–Menten equation in Graph Pad. The experiment was carried out three times, and the apparent K M values were determined for the donor substrate CMP‐Neu5Ac as 2.79 μm (for Δ56ST6Gal I) and 41.23 μm (for Δ56ST3Gal I; Table 1). These values are consistent with those previously published.26b, 26d, 28
Figure 2.
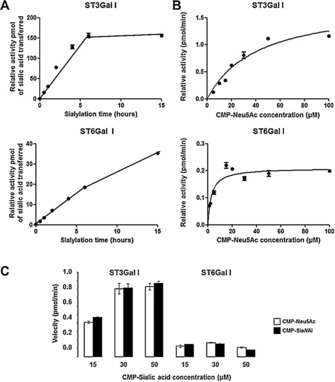
Determination of apparent kinetic parameters for Δ56ST6Gal I and Δ56ST3Gal I towards the natural donor substrate CMP‐Neu5Ac. Enzymatic reactions were performed with 10 μL of the culture medium of transfected HEK293 cells containing recombinant hΔ56ST3Gal I or hΔ56ST6Gal I (or no enzyme, mock) and 60 μg of asialofetuin (acceptor). A) Activity of enzymes for various incubation times with 20 μm CMP‐Neu5Ac (50 000 DPM/13.6 μm CMP‐[14C]Neu5Ac and 6.4 μm cold CMP‐Neu5Ac) donor substrate. Proteins were precipitated, filtered on glass‐fiber filters, and radioactivity was counted. B) Apparent kinetic parameters for Δ56ST6Gal I and Δ56ST3Gal I calculated for CMP‐Neu5Ac. Incubations with 10 μL of transfected culture medium were carried out for 6 h with various concentrations of donor substrate CMP‐Neu5Ac (50 000 DPM/ 13.6 μm CMP‐[14C]Neu5Ac and various amount of cold CMP‐Neu5Ac) and 60 μg of asialofetuin. Proteins were precipitated, filtered on glass‐fiber filters, and radioactivity was counted. The background signal (obtained with mock) was subtracted. The data are mean±SE (n=3) and were fitted to the Michaelis–Menten equation (fitted values±SE in Table 1). C) Enzymatic activity towards the two CMP‐Sias donors. Incubations of 10 μL of the culture medium of transfected HEK293 cells containing Δ56ST3Gal I (left) or Δ56ST6Gal I (right) were carried out for 6 h with various concentrations (1–50 μm) of freshly prepared CMP‐Neu5Ac or CMP‐SiaNAl and 50 000 DPM of radiolabeled CMP‐[14C]Neu5Ac (tracer) and 60 μg of asialofetuin. Specific [14C]Neu5Ac incorporation was calculated as described in the Experimental Section. Data are mean±SE (n=3).
Table 1.
Apparent K M and relative V max for Neu5Ac with 10 μL of Δ56ST3Gal I or Δ56ST6Gal I in the cell culture medium of transfected HEK293.
| V max [pmol min−1] | K M [μm] | V max/K M [a] | |
|---|---|---|---|
| Δ56ST6Gal I | 0.209±0.008 | 2.79±0.53 | 0.079 |
| Δ56ST3Gal I | 1.773±0.159 | 41.23±7.64 | 0.043 |
[a] Relative substrate activity.
We achieved immunoaffinity purification of each transfected cell medium (mock, ST6Gal I, and ST3Gal I) by using the M2 monoclonal antibody (anti‐FLAG mAb) bound to a protein G–Sepharose resin, with several washing steps and elution of the bound enzyme (with a 3×FLAG peptide). We could not obtain sufficient amount of purified ST6Gal I to perform kinetic studies. Kinetic parameters established for purified ST3Gal I were found to be similar to those obtained with crude enzyme, so we used the crude extracts rather than purified enzyme (data not shown).
Interestingly, incubations of Δ56ST3Gal I or Δ56ST6Gal I with various concentrations (1–50 μm) of freshly prepared CMP‐Neu5Ac or CMP‐SiaNAl and 50 000 DPM radiolabeled CMP‐[14C]Neu5Ac (as a tracer) showed no difference (Figure 2 C). These data further suggested that Δ56ST3Gal I and Δ56ST6Gal I are likely promiscuous with respect to the CMP‐SiaNAl donor carrying an extended N‐acyl group in the C5 position, as previously suggested.4
Characterization of the α2,3/6‐sialyltransferase activities towards the unnatural donor substrate CMP‐SiaNAl
We developed an in vitro sialyltransferase assay based on desialylated glycoproteins as acceptor substrates for probing Δ56ST3Gal I and Δ56ST6Gal I sialyltransferase activities towards the unnatural sialic acid donor substrate CMP‐SiaNAl. Sialylation reactions were carried out with the cell culture medium of transfected HEK293 cells containing either Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock) with 100 μm CMP‐SiaNAl and 50 μg of acceptor substrate (fetuin, asialofetuin, or α‐1‐acid glycoprotein). Alpha‐1‐acid glycoprotein (also known as orosomucoid) contains five N‐linked oligosaccharides, which are predominantly tri‐ and tetra‐antennary structures with LacNAc structures,29 and it is used as a specific substrate acceptor for ST6Gal I. Following incubation, chemical ligation of azido‐biotin to the transferred alkyne‐sialic acid was achieved by CuAAC. Resialylated glycoproteins covalently linked to biotin were separated by 8 % SDS‐PAGE, transferred to nitrocellulose, and detected by western blotting with anti‐biotin antibody. Several bands resulting from the Δ56ST6Gal I activity on asialofetuin and asialoorosomucoid could be detected (Figure 3 A), whereas no signal was detected when using fetuin as an acceptor. Similarly, Δ56ST3Gal I was active only on asialofetuin and not on fetuin or asialoorosomucoid. As expected, PNGase F treatment, which eliminates N‐glycans from resialylated fetuin, showed that Δ56ST6Gal I transfers alkyne‐sialic acid only on N‐glycans; Δ56ST3Gal I activity was resistant to PNGase F treatment, thus indicating that Δ56ST3Gal I likely transfers alkyne‐sialic acid on O‐glycans of asialofetuin (Figure 3 B).
Figure 3.
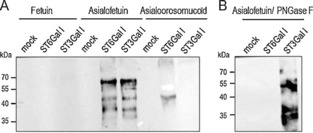
Enzymatic activity of Δ56ST6Gal I and Δ56ST3Gal I with CMP‐SiaNAl as a donor. A) Enzymatic reactions were performed for 4 h with 8.5 μL of the culture medium of transfected HEK293 cells containing either no enzyme (mock) or recombinant Δ56ST6Gal I (ST6Gal I) or Δ56ST3Gal I (ST3Gal I) and 50 μg of native fetuin, or desialylated fetuin (asialofetuin), or asialoorosomucoid (acceptor), and 100 μm freshly prepared CMP‐SiaNAl (conditions as described in Experimental Section). Covalent ligation of the SiaNAlkyne moiety with azido‐biotin was carried out by click chemistry. Re‐sialylated proteins were separated by SDS‐PAGE (8 %) and visualized by western blotting with peroxidase‐conjugated anti‐biotin antibody (16 ng mL−1). B) Specificity of Δ56ST3Gal I or Δ56ST6Gal I was assessed by PNGase F treatment of 20 μg of resialylated fetuin prior to click chemistry and western blot analysis. Then, covalent ligation of the SiaNAlkyne moiety with azido‐biotin and SDS PAGE were carried out. The signal previously detected on resialylated fetuin disappears upon PNGase F treatment of samples sialylated with Δ56ST6Gal I, whereas the signal remains for fetuin samples resialylated with Δ56ST3Gal I.
To assess the efficiency of SiaNAl transfer, we incubated each recombinant enzyme with various concentrations of CMP‐SiaNAl (2 to 200 μm) for 6 h with 200 μg of asialofetuin as an acceptor (Figure 4). Transferred SiaNAl was released from resialylated fetuin by acid hydrolysis and labeled with 1,2‐diamino‐4,5‐methylenedioxybenzene (DMB). Sensitive and specific detection of DMB‐coupled sialic acid was performed by LC‐MS in multiple reaction monitoring (MRM)‐MS3 mode. Apparent kinetic parameters were obtained by nonlinear regression analysis of the Michaelis–Menten equation in Graph Pad (Figure 4 B). The experiment was carried out three times, and the apparent K M values for the enzymes were determined for the donor substrate CMP‐SiaNAl as 6.55 μm (for Δ56ST6Gal I; Table 2) and 49.03 μm (for Δ56ST3Gal I; Table 2). The apparent K M values calculated for CMP‐Sia donors were relatively unaffected, thus leading to the conclusion that Δ56ST3Gal I and Δ56ST6Gal I efficiently and selectively transfer alkyne‐sialic acid on O‐glycans and N‐glycans, respectively, despite the extended N‐acyl chain of CMP‐SiaNAl.
Figure 4.
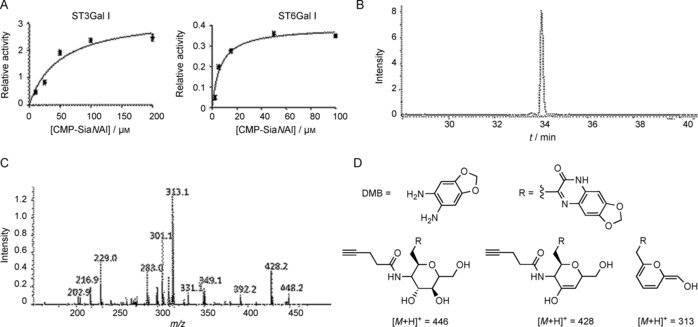
Determination of apparent kinetic parameters for Δ56ST6Gal I and Δ56ST3Gal I towards the unnatural donor substrate CMP‐SiaNAl. Incubations in 6.5 μL of culture medium of transfected HEK293 cells containing recombinant Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock) were carried out for 6 h with various concentrations of CMP‐SiaNAl (donor substrate) and 200 μg of asialofetuin. Sialic acids from each reaction were released, derivatized with DMB and subjected to LC‐MS3 analysis to quantify SiaNAl. A) The background signal (mock) was subtracted, and data were fitted to the Michaelis–Menten equation; apparent kinetic parameters for Δ56ST6Gal I and Δ56ST3Gal I were calculated for CMP‐SiaNAl (Table 2). B) Quantification was based on LC‐MS signal intensity from MRM‐MS3 analysis of the DMB‐SiaNAl specific [M+H]+ product at m/z 446. C) MS3 fragmentation pattern of [M+H]+ ion at m/z 446; D) Major fragmentation products.
Table 2.
Apparent K M and relative V max for SiaNAl with 6.5 μL of Δ56ST3Gal I or Δ56ST6Gal I in the cell culture medium of transfected HEK293.
| V max [pmol min−1] | K M [μm] | V max/K M [a] | |
|---|---|---|---|
| Δ56ST6Gal I | 0.390±0.016 | 6.55±1.07 | 0.059 |
| Δ56ST3Gal I | 3.275±0.249 | 49.03±9.91 | 0.067 |
[a] Relative substrate activity.
Cell‐based assays of α2,3/6‐sialyltransferase activity
By using a SEEL approach, Δ56ST3Gal I or Δ56ST6Gal I in the cell culture medium of transfected HEK293 cells was used to sialylate cell‐surface O‐glycans or N‐glycans with the two freshly prepared CMP‐Sia derivatives, CMP‐SiaNAl and CMP‐Neu5Ac. Lec2 CHO cells, which are deficient in the Golgi CMP‐sialic acid transporter SLC35A1,30 were chosen to assess Δ56ST3Gal I and Δ56ST6Gal I activities in vivo, as these cells express almost no sialic acid on the cell surface. Cell‐based methods including flow cytometry analysis of cell surface sialylation and confocal microscopy were used to evaluate sialyltransferase activity of the recombinant enzymes with labeled lectins (Figure 5). Detection of sialic acid binding was achieved by using SNA‐lectin (Sambucus nigra agglutinin, which mainly recognize α2,6‐linked sialic acid of N‐glycans) and MAA‐lectin (Maackia amurensis agglutinin II, MAL‐II, which is specific for α2,3‐linked sialic acid of O‐glycans).31 We checked that CMP‐Sia donors alone or cell culture medium containing no enzyme (mock) had no sialylation activity on Lec2 CHO cells (Figure 5). The cell‐surface O‐glycans of the cells could not be readily labeled by Δ56ST3Gal I with either CMP‐Neu5Ac or CMP‐SiaNAl substrates, thus suggesting either 1) too‐low donor substrate concentrations for efficient Δ56ST3Gal I activity, or 2) too‐low acceptor substrate concentration or accessibility on Lec2 CHO cells (as suggested previously by glycomics profiling studies of these cells).32 Suitable incubation conditions for Δ56ST3Gal I are under study to establish efficient sialylation of O‐glycans on Lec2 CHO cells. However, using Δ56ST6Gal I crude extract and the two CMP‐Sia donor substrates, we could obtain efficient and specific sialylation of Lec2 CHO N‐glycans (Figure 5 A). Interestingly, we noticed higher fluorescence intensity when using the unnatural CMP‐SiaNAl donor compared to CMP‐Neu5Ac, regardless of concentration (Figure 5 B), thus further suggesting either more‐efficient transfer of SiaNAl on cell‐surface glycoconjugates or better recognition of the extended N‐acyl group of CMP‐SiaNAl by the SNA‐lectin, as previously reported for the siglec‐1 recognition of N‐acyl‐substituted sialic acid.33
Figure 5.
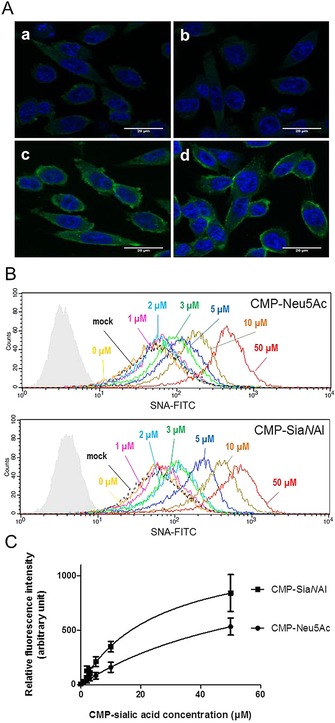
Δ56ST6Gal I activity in vivo using CHO Lec2 cells. A) Confocal microscopy images show labeling of cells using Δ56ST6Gal I. HEK293 cell culture medium (200 μL) expressing recombinant Δ56ST6Gal I enzyme (b, c, d) or no enzyme (a, mock) incubated for 2 h with CHO Lec2 cells and 20 μm CMP‐Neu5Ac (c) or CMP‐SiaNAl (d). CHO Lec2 cells were fixed in 4 % paraformaldehyde and treated with FITC‐SNA lectin, (1/75 e). Nuclei are stained with DAPI (blue). B) Flow cytometry analysis of CHO Lec2 cells after exogenous labeling with 300 μL of medium from HEK293 cells expressing Δ56ST6Gal I (or no enzyme, mock) and CMP‐Neu5Ac or CMP‐SiaNAl (0–50 μm, as indicated). Incubations were carried out for 2 h, and sialylated glycoconjugates were detected with FITC‐SNA lectin (1/100 e) and analyzed by flow cytometry. Autofluorescence control is shown in pale gray. No variation in fluorescence intensity was detected between CHO Lec2 cells incubated with no enzyme (dashed line, mock) or with enzyme but no CMP‐Sia donor (yellow, 0 μm). Significant variation in fluorescence was detected in CHO Lec2 cells incubated with cell culture medium expressing Δ56ST6Gal I and various amount of CMP‐Sias. Data are from one of three replicated experiments. C) Variation in fluorescence intensity (±SD) according to concentration of CMP‐Sias obtained in three independent experiments. These data indicate better detection of the transferred SiaNAl by the FITC‐SNA lectin.
Conclusion
As a first step towards deciphering the specificity of different monosialyltransferases in relation to the N‐acyl side chain of N‐acylneuraminic acid (sialic acid), we used two β‐galactoside α2,3/6‐sialyltransferases, ST6Gal I and ST3Gal I (specific for N‐glycans and O‐glycans, respectively) and two CMP‐Sia donors, the natural donor CMP‐Neu5Ac and CMP‐SiaNAl (with an engineered N‐acyl side chain). We synthesized both CMP‐Sia donors and produced soluble and active enzymes directly in the cell culture medium of transiently transfected HEK293 cells. These could be efficiently used in in vitro and in vivo approaches. We showed that both freshly prepared donor substrates were efficiently and selectively used to obtain α2,6‐sialylation of N‐glycans and α2,3‐sialylation of O‐glycans. For the first time, we investigated and compared the relative activities of these two human sialyltransferases towards natural and unnatural sialic acids. Additionally, we developed a versatile workflow for a better understanding of sialylation processes. This toolbox not only provides new methods for fundamental studies of sialyltransferases, but also paves the way for better control of cell‐surface glycoengineering, which is instrumental to control the behavior of cells and modulate biological recognition phenomena.
Experimental Section
General: Chemicals and reagents were purchased from Sigma–Aldrich, TCI (Zwijndrecht, Belgium), and Carbosynth (Compton, UK) and were used without further purification. The natural sialic acid analogue N‐Acetylneuraminic acid (Neu5Ac) was purchased from Carbosynth; the alkyne derivative N‐(4‐pentynoyl)neuraminic acid (SiaNAl) was synthesized and purified as previously described.25, 34 Inorganic pyrophosphatase from Saccharomyces cerevisiae (EC 3.6.1.1) and CMP‐sialic acid synthetase (CSS) from N. meningitidis group B (EC 2.7.7.43) were from Sigma–Aldrich. CMP‐[14C]Neu5Ac (55 mCi mmol−; 0.1 mCi mL−) was purchased from American Radiolabeled Chemicals (St. Louis, MO).
Cytidine‐5′‐monophospho‐N‐acetylneuraminic acid (CMP‐Neu5Ac) and cytidine‐5′‐monophospho‐N‐(4‐pentynoyl)neuraminic acid (CMP‐SiaNAl) synthesis: Activated sialic acids CMP‐Neu5Ac and CMP‐SiaNAl were synthesized and characterized by 31P NMR according to an optimized protocol recently reported.25 The obtained solutions were cooled to 4 °C and used in a subsequent enzymatic sialylation assay with no further purification, or aliquoted and kept at −80 °C.
Cloning and sialyltransferases constructions; expression of soluble fusion sialyltransferases: cDNAs encoding a truncated form of human Galβ1,3GalNAc α2,3‐sialyltransferase (ST3Gal I) and Galβ1,4GlcNAc α2,6‐sialyltransferase (ST6Gal I) lacking the first 56 N‐terminal amino acid residues were amplified by PCR by using primers (Eurogentec, Angers, France): Sense ST3Gal I: 5′‐AAAAA GCTTA GGCCT TGCAC CTGCA CCCAC TG‐3′, antisense ST3Gal I: 5′‐AAAGG TACCC TTCAC TGCGT CATCT CCCCT TG‐3′ (hΔ56ST3Gal I), Sense ST6Gal I (5′‐AAAAA GCTTG GGTCT GATTC CCAGT CTG‐3′), and antisense ST6Gal I (5′‐AAAGG ATCCT TAGCA GTGAA TGGTC CGGAA G‐3′). Both sense primers contained a HindIII site, and antisense primers contained a KpnI (ST3Gal I) or BamHI site (ST6Gal I) for subsequent cloning into expression vector p3xFLAG‐CMV‐9 (Sigma–Aldrich) as previously described.26d Amplified fragments were extracted from an 0.8 % agarose gel, subcloned into the vector, and the final construct was checked by enzymatic digestion and sequencing.
HEK293 cells (ATCC CRL‐1573) were maintained in DMEM medium (Lonza, Basel, Switzerland) complemented with fetal calf serum (10 %; Lonza) in 6‐wells plates at 37 °C under 5 % CO2. Confluent cells (70 %) were transiently transfected by using lipofectamine reagent (Invitrogen/Thermo Fisher Scientific) with ultraMEM (Lonza), by following the instruction of the manufacturer. After 36 h, the cell culture medium was collected, centrifuged to eliminate cell debris, and used as the crude enzyme fraction.
Sialyltransferase assays using radiolabeled CMP‐[14C]Neu5Ac: Assays were carried out essentially as previously described.35 Fetuin (Sigma–Aldrich) was desialylated by using TFA (0.1 m) for 2 h at 80 °C, and free sialic acid was eliminated by using a spectra/por3 dialysis membrane (MWCO 3500, 18 mm flat width; Spectrum Laboratories, Rancho Dominguez, CA) for 24 h. The contents were transferred to a glass tube and lyophilized. Desialylated fetuin was suspended (20 mg mL−1) in water. Asialoorosomucoid (Sigma–Aldrich) was prepared similarly. Asialofetuin was the acceptor substrate for both ST6Gal I and ST3Gal I enzymatic activity; asialoorosomucoid was the acceptor substrate solely for ST6Gal I. A standard reaction mixture (30 μL) contained cacodylate buffer (0.04 m, pH 6.2), MnCl2 (4 mm), Triton CF‐54 (0.08 %; Sigma–Aldrich), CMP‐Sia (20 μm: CMP‐[14C]Neu5Ac (22.5 nCi, 50 000 DPM, 13.6 μm) and cold CMP‐Neu5Ac (6.4 μm)), desialylated fetuin (2 mg mL−1) and enzyme (10 μL). The sialylation reaction was performed with the cell culture medium of transfected HEK293 cells expressing either Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock), and the mixture was incubated at 37 °C for 6 h. The reaction was stopped by precipitation of glycoproteins with phosphotungstic acid (PTA; 1 mL, 5 % in 2 n HCl) followed by filtration on Whatman GF/A glass microfiber filters. Radiolabeled sialic acid transfer onto glycoproteins was quantified by liquid scintillation counting in UltimaGold (3 mL; PerkinElmer) with a Hidex 300 SL counter.
For kinetic analysis, incubations were performed with donor substrate CMP‐Neu5Ac (1–100 μm) and medium (10 μL) of transfected HEK293 cells expressing either Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock). As the enzyme source was the crude enzyme fraction, the kinetic parameter K M and V max are apparent constants and are named app K M and app V max. These values were calculated by fitting the data to the Michaelis–Menten equation that describes enzyme activity in Prism 5 (GraphPad, La Jolla, CA).
SDS‐PAGE and western blot analysis: In order to check the production of sialyltransferases from the transfected HEK293 cells, culture medium (30 μL) was boiled with 4× Laemmli solution (10 μL; Tris⋅HCl (235 mm, pH 6.8), SDS (8 %), glycerol (40 %), 2‐mercaptoethanol (10 %), Bromophenol Blue (0.01 %)) for 10 min at 95 °C, then separated in an 8 % polyacrylamide gel and transferred to a nitrocellulose membrane. Ponceau staining was used as a loading control. Then, the membrane was saturated in blocking buffer (non‐fat milk (5 %) in TBST with Tween 20 (0.05 %)) for an hour and incubated overnight at 4 °C with anti‐flag antibody (4 μg mL−1; Sigma–Aldrich). After three TBST washing, the membrane was incubated with peroxidase‐conjugated anti‐mouse IgG antibody (1/10 000; GE Healthcare). The signal was detected by using the chemiluminescent reagent ECL 2 (Thermo Fisher Scientific) and a chemiluminescence fusion camera (Vilber Lourmat, Marne‐la‐Vallée, France).
To visualize enzymatic activity of the recombinant sialyltransferases Δ56ST3Gal I or Δ56ST6Gal I on the various glycoprotein acceptors using the unnatural sialic acid donor CMP‐SiaNAl, we set up a western blot approach. Sialylation reactions were performed with 8.5 μL of the culture medium of transfected HEK293 cells expressing either recombinant Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock) for 4 h with freshly prepared CMP‐SiaNAl (100 μm) and glycoprotein acceptor (50 μg; fetuin, asialofetuin, asialoorosomucoid or no exogenous acceptor). Following sialylation reaction, click chemistry was used to covalently link the alkyne moiety with azide‐PEG3‐biotin (Sigma–Aldrich). Briefly, sialylated glycoprotein (20 μg) was incubated for 1 h at 25 °C with CuSO4 (11.25 μm), 2‐(4‐((bis((1‐tert‐butyl‐1H‐1,2,3‐triazol‐4‐yl)methyl)amino)methyl)‐1H‐1,2,3‐triazol‐1‐yl)acetic acid (77.5 μm), azido‐biotin (6.96 μm; Sigma–Aldrich), and sodium ascorbate (61.88 μm) in PBS (20 μL). After click chemistry, the glycoprotein (20 μg) was boiled for 5 min in Laemmli solution. Samples were then resolved in an 8 % polyacrylamide gel, transferred to a nitrocellulose membrane, and ponceau staining was performed to check transfer and control loading. The membrane was saturated in blocking buffer for 1 h, then incubated for 1 h with peroxidase‐conjugated IgG fraction monoclonal mouse anti‐biotin (16 ng mL−1; Jackson Immunoresearch). After three TBST washing, the signal was detected by chemiluminescent reagent (SuperSignal West Femto Maximum Sensitivity Substrate; Thermo Fisher Scientific) and a chemiluminescence fusion camera (Vilber Lourmat).
To assess the specificity of the recombinant sialyltransferase (Δ56ST3Gal I or Δ56ST6Gal I), PNGase F (Roche) treatment of resialylated fetuin was carried out prior to click chemistry and western blot analysis. Resialylated glycoprotein (20 μg) was incubated in phosphate buffer (50 mm, 21 μL) and boiled (100 °C) for 10 min. After cooling, PNGase F (1 U) was added and incubated 4 h at 37 °C. After treatment, nine volumes of absolute ethanol were used to precipitate proteins.
Quantification of sialic acid (SiaNAl) transfer by micro‐LC/ESI‐MRM‐MS3 analysis: Sialyltransferase assays were performed essentially as described above with desialylated fetuin CMP‐SiaNAl (200 μg) at the indicated concentration in cacodylate buffer (30 μL) supplemented with cell culture medium (6.5 μL) containing either Δ56ST3Gal I or Δ56ST6Gal I (or mock). The reaction mixture was incubated for 6 h at 37 °C and stopped by addition of PTA (1 mL, 5 % in 2 n HCl). Following overnight precipitation at −20 °C, resialylated proteins were washed three times with TCA (10 %). The glycosidic linkage between sialic acid and acceptor monosaccharide was hydrolyzed with TFA (100 μL, 0.1 m), then the mixture was incubated for 2 h at 80 °C, and dried in a 5301 concentrator (Eppendorf). Derivation of sialic acid was performed for 2 h at 50 °C in the dark with DMB (32 μg), β‐mercaptoethanol (0.5 m), Na2S2O4 (9 mm), TFA (5 mm, 40 μL). Samples were stored at −20 °C before analyzing.
Quantitative analyses were performed in positive ion mode on an amaZon speed ETD ion trap mass spectrometer (Bruker Daltonics) equipped with a standard ESI source and controlled by Hystar software (ver. 3.2). The identification of MS2 fragment ions was based on previously published analyses of DMB‐coupled sialic acid by MS/MS.36 DMB‐coupled sialic acid separation was achieved on Prominence LC‐20AB micro LC system (Shimadzu). Samples were diluted fivefold in formic acid (0.1 %), and dilutions (5 μL) were applied to a Luna 3 μm analytical column (C18 (2), 100 Å, 150×1 mm; Phenomenex) with isocratic elution in acetonitrile/MeOH/water (4:6:90, v/v/v) at 60 μL min−1. Multiple reaction monitoring (MRM) of MS3 was used for DMB‐coupled Sia quantification (ion spray voltage 4500 V, dry gas flow rate 8 L min−1, 200 °C). Absolute quantifications were calculated by comparing ion intensities to references established for all sialic acid‐DMB derivatives.
Cell‐based analyses: FACS analysis and confocal microscopy: CHO‐Lec2 cells (ATCC CRL‐1736) were grown in DMEM with FCS (10 %) in a 24‐well plate to 70 % confluence. Cells were incubated for 2 h at 37 °C with CMP‐Neu5Ac or CMP‐SiaNAl (0–50 μm) in cell‐culture medium (200 μL) containing either Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock). After cell washing with Dulbecco's phosphate‐buffered saline (DPBS), cells were detached with EDTA (5 mm) and placed in a 96‐well plate. After centrifugation (260 g, 5 min), fluorescein isothiocyanate‐conjugated Sambucus nigra agglutinin (1 μg, 2 mg mL−1, FITC‐SNA lectin; Vector Laboratories) in PBS/ BSA (1 %; 50 μL) was incubated with cells for 1 h at 4 °C. Alternatively, of biotin‐conjugated Maackia amurensis lectin II (1 μg, 1 mg mL−1, MAL II, Vector Laboratories) in a PBS/ BSA (1 %; 50 μL) was incubated with cells for 1 h at 4 °C; after washing with PBS, FITC‐conjugated streptavidin (0.1 μg, 1 mg/mL, DyLight 488 Streptavidin; Vector Laboratories) was added to PBS/ BSA (1 %; 50 μL) and incubated for 1 h at 4 °C. Finally, after three washing with PBS, cells were transferred to 5 mL polystyrene round‐bottom tubes (BD Falcon), and fluorescence was quantified with a FACSCalibur cytometer (Becton Dickinson).
For confocal microscopy, CHO Lec2 cells were grown on coverslips in DMEM with FCS (10 %) in a 24‐well plate to 70 % confluency. After DPBS washings, cells were incubated with cell culture medium (200 μL) containing either Δ56ST3Gal I or Δ56ST6Gal I (or no enzyme, mock) and CMP‐Neu5Ac or CMP‐SiaNAl (20 μm) for 2 h. Then cells were washed, fixed with paraformaldehyde (4 %) for 20 min, then incubated with PBS/ BSA (1 %) and saponin (0.075 %) for 1 h at room temperature. Cells were incubated with SNA lectin (1.33 μg) in PBS/BSA (1 %; 50 μL) for 1 hour at room temperature and washed three times with PBS. DAPI (1/200 in PBS/BSA (1 %)) was incubated with cells for 20 min at room temperature. Finally, after three washing with PBS, coverslips were mounted on glass slides with mounting medium (Dako). Fluorescence was detected through an inverted Zeiss 700 confocal microscope.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank Elodie Richard and Dr. Christian Slomianny of the BICel‐Campus Lille1 facility for access to instruments and technical advices. We are indebted the Plateforme d'Analyse des Glycoconjugués (PAGés, http://plateforme‐pages.univ‐lille1.fr) and to the Research Federation FRABio (Univ.Lille, CNRS, FR 3688, FRABio, Biochimie Structurale et Fonctionnelle des assemblages Biomoléculaires) for providing the scientific and technical environment conducive to achieving this work. We are extremely grateful to Anne‐Marie Mir for her excellent technical assistance using radiolabeled compounds. We greatly acknowledge Dr. Bernadette Coddeville, Dr. Mathieu Carpentier, Dr. Agnès Denys and Dr. Fabrice Allain for helpful discussions.
M. Noel, P.-A. Gilormini, V. Cogez, N. Yamakawa, D. Vicogne, C. Lion, C. Biot, Y. Guérardel, A. Harduin-Lepers, ChemBioChem 2017, 18, 1251.
References
- 1. Varki A., Glycobiology 1992, 2, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schauer R., Curr. Opin. Struct. Biol. 2009, 19, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Y., Chen X., Appl. Microbiol. Biotechnol. 2012, 94, 887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kayser H., Zeitler R., Kannicht C., Grunow D., Nuck R., Reutter W., J. Biol. Chem. 1992, 267, 16934–16938. [PubMed] [Google Scholar]
- 5. Mahal L. K., Yarema K. J., Bertozzi C. R., Science 1997, 276, 1125–1128. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Keppler O. T., Horstkorte R., Pawlita M., Schmidt C., Reutter W., Glycobiology 2001, 11, 11R–18R; [DOI] [PubMed] [Google Scholar]
- 6b. Nischan N., Kohler J. J., Glycobiology 2016, 12, e1006010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Wratil P. R., Horstkorte R., Reutter W., Angew. Chem. Int. Ed. 2016, 55, 9482–9512; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9632–9665. [Google Scholar]
- 7. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 8. Saxon E., Luchansky S. J., Hang H. C., Yu C., Lee S. C., Bertozzi C. R., J. Am. Chem. Soc. 2002, 124, 14893–14902. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Meldal M., Tornoe C. W., Chem. Rev. 2008, 108, 2952–3015; [DOI] [PubMed] [Google Scholar]
- 9b. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711. [Google Scholar]
- 10. Agard N. J., Prescher J. A., Bertozzi C. R., J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- 11. Haun J. B., Devaraj N. K., Hilderbrand S. A., Lee H., Weissleder R., Nat. Nanotechnol. 2010, 5, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brossmer R., Gross H. J., Methods Enzymol. 1994, 247, 177–193. [DOI] [PubMed] [Google Scholar]
- 13. Wolf S., Warnecke S., Ehrit J., Freiberger F., Gerardy-Schahn R., Meier C., ChemBioChem 2012, 13, 2605–2615. [DOI] [PubMed] [Google Scholar]
- 14. Mbua N. E., Li X., Flanagan-Steet H. R., Meng L., Aoki K., Moremen K. W., Wolfert M. A., Steet R., Boons G.-J., Angew. Chem. Int. Ed. 2013, 52, 13012–13015; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13250–13253. [Google Scholar]
- 15. Sun T., Yu S.-H., Zhao P., Meng L., Moremen K. W., Wells L., Steet R., Boons G.-J., J. Am. Chem. Soc. 2016, 138, 11575–11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu S.-H., Zhao P., Sun T., Gao Z., Moremen K. W., Boons G.-J., Wells L., Steet R., J. Biol. Chem. 2016, 291, 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Harduin-Lepers A., Glycobiol. Insights 2010, 2, 29–61; [Google Scholar]
- 17b. Harduin-Lepers A., Mollicone R., Delannoy P., Oriol R., Glycobiology 2005, 15, 805–817; [DOI] [PubMed] [Google Scholar]
- 17c. Harduin-Lepers A., Vallejo-Ruiz V., Krzewinski-Recchi M.-A., Samyn-Petit B., Julien S., Delannoy P., Biochimie 2001, 83, 727–737. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Harduin-Lepers A. in Sialobiology: Structure, Biosynthesis and Function. Sialic Acid Glycoconjugates in Health and Diseases (Eds.: J. Tiralongo, I. Martinez-Duncker), Bentham, 2013, pp. 139–187; [Google Scholar]
- 18b. Tsuji S., Datta A. K., Paulson J. C., Glycobiology 1996, 6, v–vii. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Kuhn B., Benz J., Greif M., Engel A. M., Sobek H., Rudolph M. G., Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1826–1838; [DOI] [PubMed] [Google Scholar]
- 19b. Meng L., Forouhar F., Thieker D., Gao Z., Ramiah A., Moniz H., Xiang Y., Seetharaman J., Milaninia S., Su M., Bridger R., Veillon L., Azadi P., Kornhaber G., Wells L., Montelione G. T., Woods R. J., Tong L., Moremen K. W., J. Biol. Chem. 2013, 288, 34680–34698; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Rao F. V., Rich J. R., Rakić B., Buddai S., Schwartz M. F., Johnson K., Bowe C., Wakarchuk W. W., Defrees S., Withers S. G., Strynadka N. C. J., Nat. Struct. Mol. Biol. 2009, 16, 1186–1188; [DOI] [PubMed] [Google Scholar]
- 19d. Volkers G., Worrall L. J., Kwan D. H., Yu C.-C., Baumann L., Lameignere E., Wasney G. A., Scott N. E., Wakarchuk W., Foster L. J., Withers S. G., Strynadka N. C. J., Nat. Struct. Mol. Biol. 2015, 22, 627–635. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Audry M., Jeanneau C., Imberty A., Harduin-Lepers A., Delannoy P., Breton C., Glycobiology 2011, 21, 716–726; [DOI] [PubMed] [Google Scholar]
- 20b. Datta A. K., Curr. Drug Targets 2009, 10, 483–498; [DOI] [PubMed] [Google Scholar]
- 20c. Harduin-Lepers A., Recchi M.-A., Delannoy P., Glycobiology 1995, 5, 741–758; [DOI] [PubMed] [Google Scholar]
- 20d. Rohfritsch P. F., Joosten J. A. F., Krzewinski-Recchi M.-A., Harduin-Lepers A., Laporte B., Juliant S., Cerutti M., Delannoy P., Vliegenthart J. F. G., Kamerling J. P., Biochim. Biophys. Acta Gen. Subj. 2006, 1760, 685–692; [DOI] [PubMed] [Google Scholar]
- 20e. Takashima S., Biosci. Biotechnol. Biochem. 2008, 72, 1155–1167. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Gupta R., Matta K. L., Neelamegham S., Biochem. Biophys. Res. Commun. 2016, 469, 606–612; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Kono M., Ohyama Y., Lee Y.-C., Hamamoto T., Kojima N., Tsuji S., Glycobiology 1997, 7, 469–479; [DOI] [PubMed] [Google Scholar]
- 21c. Williams M. A., Kitagawa H., Datta A. K., Paulson J. C., Jamieson J. C., Glycoconjugate J. 1995, 12, 755–761. [DOI] [PubMed] [Google Scholar]
- 22. Harduin-Lepers A., Petit D., Mollicone R., Delannoy P., Petit J.-M., Oriol R., BMC Evol. Biol. 2008, 8, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horstkorte R., Muhlenhoff M., Reutter W., Nöhring S., Zimmermann-Kordmann M., Gerardy-Schahn R., Exp. Cell Res. 2004, 298, 268–274. [DOI] [PubMed] [Google Scholar]
- 24. Teppa R. E., Petit D., Plechakova O., Cogez V., Harduin-Lepers A., Int. J. Mol. Sci. 2016, 17, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilormini P. A., Lion C., Noel M., Krzewinski-Recchi M.-A., Harduin-Lepers A., Guérardel Y., Biot C., Glycobiology 2016, 26, 1151–1156. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Chen C., Ma J., Lazic A., Backovic M., Colley K. J., J. Biol. Chem. 2000, 275, 13819–13826; [DOI] [PubMed] [Google Scholar]
- 26b. Legaigneur P., Breton C., El Battari A., Guillemot J. C., Auge C., Malissard M., Berger E. G., Ronin C., J. Biol. Chem. 2001, 276, 21608–21617; [DOI] [PubMed] [Google Scholar]
- 26c. Luley-Goedl C., Czabany T., Longus K., Schmolzer K., Zitzenbacher S., Ribitsch D., Schwab H., Nidetzky B., J. Biotechnol. 2016, 235, 54–60; [DOI] [PubMed] [Google Scholar]
- 26d. Vallejo-Ruiz V., Haque R., Mir A.-M., Schwientek T., Mandel U., Cacan R., Delannoy P., Harduin-Lepers A., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2001, 1549, 161–173. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Baenziger J. U., Fiete D., J. Biol. Chem. 1979, 254, 789–795; [PubMed] [Google Scholar]
- 27b. Spiro R. G., Bhoyroo V. D., J. Biol. Chem. 1974, 249, 5704–5717. [PubMed] [Google Scholar]
- 28.
- 28a. Datta A. K., Paulson J. C., J. Biol. Chem. 1995, 270, 1497–1500; [DOI] [PubMed] [Google Scholar]
- 28b. Lee K.-Y., Kim H. G., Hwang M. R., Chae J. I., Yang J. M., Lee Y. C., Choo Y. K., Lee Y. I., Lee S.-S., Do S.-I., J. Biol. Chem. 2002, 277, 49341–49351. [DOI] [PubMed] [Google Scholar]
- 29. Imre T., Kremmer T., Héberger K., Molnár-Szöllősi É., Ludányi K., Pócsfalvi G., Malorni A., Drahos L., Vékey K., J. Proteomics 2008, 71, 186–197. [DOI] [PubMed] [Google Scholar]
- 30. Stanley P., Siminovitch L., Somatic Cell Genet. 1977, 3, 391–405. [DOI] [PubMed] [Google Scholar]
- 31. Geisler C., Jarvis D. L., Glycobiology 2011, 21, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. North S. J., Huang H.-H., Sundaram S., Jang-Lee J., Etienne A. T., Trollope A., Chalabi S., Dell A., Stanley P., Haslam S. M., J. Biol. Chem. 2010, 285, 5759–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Erikson E., Wratil P. R., Frank M., Ambiel I., Pahnke K., Pino M., Azadi P., Izquierdo-Useros N., Martinez-Picado J., Meier C., Schnaar R. L., Crocker P. R., Reutter W., Keppler O. T., J. Biol. Chem. 2015, 290, 27345–27359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilormini P. A., Lion C., Vicogne D., Levade T., Potelle S., Mariller C., Guérardel Y., Biot C., Foulquier F., Chem. Commun. 2016, 52, 2318–2321. [DOI] [PubMed] [Google Scholar]
- 35. Harduin-Lepers A., Stokes D. C., Steelant W. F. A., Samyn-Petit B., Krzewinski-Recchi M.-A., Vallejo-Ruiz V., Zanetta J.-P., Augé C., Delannoy P., Biochem. J. 2000, [PMC free article] [PubMed] [Google Scholar]; 352, 37–48. [Google Scholar]
- 36.
- 36a. Baumann A.-M., Bakkers M. J. G., Buettner F. F. R., Hartmann M., Grove M., Langereis M. A., de Groot R. J., Mühlenhoff M., Nat. Commun. 2015, 6, 7673; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Klein A., Diaz S., Ferreira I., Lamblin G., Roussel P., Manzi A. E., Glycobiology 1997, 7, 421–432. [DOI] [PubMed] [Google Scholar]