

Abstract
To date, over 100 small molecule oncology drugs have been approved by the US Food and Drug Administration. Due to the inherent heterogeneity of tumors, these small molecules are often administered in combination to prevent emergence of resistant cell subpopulations. Therefore, new combination strategies to overcome drug resistance in patients with advanced cancer are needed. In this study, we performed a systematic evaluation of the therapeutic activity of over 5,000 pairs of FDA-approved cancer drugs against a panel of 60 well-characterized human tumor cell lines (NCI-60) to uncover combinations with greater than additive growth-inhibitory activity. Screening results were compiled into a database, termed the NCI-ALMANAC (A Large Matrix of Anti Neoplastic Agent Combinations), publically available at https://dtp.cancer.gov/ncialmanac. Subsequent in vivo experiments in mouse xenograft models of human cancer confirmed combinations with greater than single-agent efficacy. Concomitant detection of mechanistic biomarkers for these combinations in vivo supported the initiation of two phase I clinical trials at the NCI to evaluate clofarabine with bortezomib and nilotinib with paclitaxel in patients with advanced cancer. Consequently, the hypothesis-generating NCI-ALMANAC web-based resource has demonstrated value in identifying promising combinations of approved drugs with potent anticancer activity for further mechanistic study and translation to clinical trials.
Keywords: cancer drug screening, combination therapy, chemotherapy, molecularly targeted therapy, apoptosis
Introduction
Despite recent advances in translational oncology (1), new therapies for cancer are urgently needed, as are new approaches to facilitate the rapid translation of promising preclinical data to clinical evaluation. Recognizing that combining anticancer drugs—whether cytotoxic or molecularly targeted—with distinct mechanisms of action is the approach most likely to overcome single-agent resistance and produce sustained clinical remissions (2,3), we determined that it would be of value to systematically screen pairwise combinations of 104 US Food and Drug Administration (FDA)-approved oncology drugs in the NCI-60 panel of human tumor cell lines. Our objective was to create a database, the NCI-ALMANAC (A Large Matrix of Anti Neoplastic Agent Combinations), that could be searched to identify combinations with greater antitumor activity than either agent alone. Promising combinations could then be selected for further evaluation in vitro or in human tumor xenografts models. By screening only approved drugs with proven activity and established safety profiles, combinations identified from the NCI-ALMANAC database offer potential for rapid translation into Investigational New Drug (IND)-exempt clinical trials.
The NCI-60 panel is one of many drug development resources that are, and will continue to be, supported by the NCI; each of the cell lines has been extensively characterized at the molecular level; exome sequence, mRNA expression, microRNA expression, protein quantification, protein modification, DNA methylation, enzyme activity, and metabolomics data are all publicly available (4,5). These datasets enable molecular data mining in the context of combination drug studies to assess the engagement of multiple potential targets and/or downstream markers of drug action, both in animal model systems and in patients during early phase clinical trials (6). They also support research into the multiple mechanisms driving resistance to cancer therapeutic agents, including mutation or amplification of the gene encoding the drug target, enhanced drug efflux or metabolism, activation of compensatory signaling networks that bypass the effects of target engagement, changes in DNA damage response or epigenetic pathways, or alterations in the tumor microenvironment (7).
Although a variety of active clinical trials are testing drug combinations based on accepted precepts of tumor cell biology, the preclinical hypotheses supporting many of these studies may be difficult to validate in patients without clearly defined mechanisms of action, resistance, and toxicity for each component of the combination, as well as for the combination itself (8). Indeed, the heterogeneity observed across individual tumors hints at the possibility of distinct co-occurring resistance mechanisms within a patient, such that different regions of the same malignancy may vary in their sensitivity (or resistance) to an individual agent at the time that treatment is initiated (9–11). Hence, developing new combination strategies to overcome drug resistance in patients with advanced cancer is a therapeutic imperative (8,12). Promising drug combinations may be discovered with synthetic lethal screens (13) utilizing siRNA or shRNA libraries, or CRISPR-Cas9 genome editing (14), to identify targets for commonly used anticancer drugs (15,16); however, in this study we utilized an unbiased, “hypothesis-free” phenotypic screening approach (8). The results of our comprehensive combination screening effort are available in the NCI-ALMANAC database, a web-based resource intended for hypothesis-generating assessments of oncology drugs combinations. Based on the outcome of this screening program, several promising drug combinations were examined in human tumor xenografts developed from NCI-60 cell lines. From our in vitro-to-in vivo translation effort, two promising drug combinations never before tested in human studies—clofarabine with bortezomib and paclitaxel with nilotinib—were chosen for clinical evaluation.
Materials and Methods
Cell Lines and Drug Screening
NCI-60 cell lines were obtained from the NCI Developmental Therapeutics Program Tumor Repository in the mid-1990s–early 2000s and distributed for drug screening and xenograft experiments in 2010–2012. For each lot of cells, the Repository performs Applied Biosystems AmpFLSTR® Identifiler® testing with PCR amplification to confirm consistency with the published Identifiler® STR profile (17) for the given cell line. Each cell line was tested for mycoplasma when it was accepted into the repository; routine mycoplasma testing of lots is not performed. Cells are kept in continuous culture at NCI for no more than 20 passages.
Oncology drugs were obtained through commercial sources. Experiments were performed at 3 locations (NCI’s Frederick National Laboratory for Cancer Research, SRI International, and University of Pittsburgh). At NCI, drug treatment was performed according to the standard NCI-60 testing protocol (https://dtp.cancer.gov/discovery_development/nci-60/default.htm), using 96-well plates and 2-day drug exposures; the endpoint is determination of Sulphorhodamine B levels in duplicate wells. For each drug pair, both single agents were tested at 5 concentrations. For drug combinations, one agent was tested at 5 concentrations together with one of 3 different concentrations of the second agent, generating 5×3 combination matrices. At SRI International and University of Pittsburgh, this protocol was modified: cells were plated in 384-well plates, fetal bovine serum was increased to 10%, CellTiter-Glo® was used as an endpoint, and drugs were tested at 3 concentrations as single agents and in 3×3 concentration matrices for combinations, in individual wells. Drug concentrations were chosen based on clinical relevance; where possible, concentrations were selected to be below the human peak plasma concentration (Cmax) at the FDA-approved clinical dose and also to yield measurable activity in our assay systems. In a few cases, one or more concentrations above the Cmax were selected to achieve some growth inhibition in vitro. Growth percent was calculated as described in the standard NCI-60 testing protocol. Inclusion of a time zero measurement in all experiments allowed for determination of cell killing and growth inhibition. Fifty-nine of the NCI-60 cell lines passed pre-defined quality control parameters for > 90% of the drug pairs. The SK-MEL-2 melanoma cell line was an outlier; data for only 65% of the drug pairs met quality control standards as the cells grew poorly in 384-well plates at both testing locations.
Development and Use of the NCI ComboScore
Determination of combination benefit (“ComboScore”) utilized a modification of Bliss independence (18), as follows: Let YiApBq be the growth fraction for the ith cell line exposed to the pth concentration of drug A and the qth concentration of drug B, defined as:
where T0 is the time zero endpoint measurement, is the endpoint measurement after 2 days, and is the endpoint measurement after 2 days for the control well. Define YiAp; YiBq as the growth fractions when only exposed to drug A or drug B, respectively. The expected growth fraction for the combination is:
where truncates the growth fraction at 100. The final combination score for the cell line and the drug combination is the sum of the differences in expected versus observed growth fractions:
These calculations set a ceiling of 100% growth, i.e., the expected combination growth percentage cannot be greater than the control. One screening site had a high incidence of “reversals,” where the growth percent increased as the concentration of one or both drugs was increased, i.e., apparent enhancement of growth by drug treatment. While the phenomenon of hormesis can be real, the frequency with which this was observed, at only one of the screening locations, argued against this being actual hormesis. All data for a drug pair/cell line were removed from the dataset if the growth percent increased by 50% or greater between any 2 adjacent doses of either drug. ComboScores are available for download at https://dtp.cancer.gov/ncialmanac.
Evaluation of Combination Therapies Registered with Clinicaltrials.gov
For initial analysis of the clinical status of all NCI-ALMANAC combinations, a drug pair was designated “clinically tested” if the two names were provided as an intervention in the same trial listed in the clinicaltrials.gov database (see Supplementary Methods for further detail).
Heat Map
JMP 11 (SAS institute) software was utilized for preparation of heat maps and statistical analyses. Two-way hierarchical clustering utilized the Ward method with data standardization and without imputation of missing values.
Human Tumor Xenografts
Animal experiments were performed at both SRI International and Frederick National Laboratory for Cancer Research. Both SRI International and Frederick National Laboratory are accredited by AAALAC International and follow the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council; 2011; National Academies Press; Washington, D.C.).
For mouse inoculation, tumor cells from the NCI-60 panel were used at the 4th to 6th in vitro passage of cryopreserved cell stocks. Cells (1×107 cells/0.1 mL/injection) were subcutaneously inoculated bilaterally into female athymic nu/nu NCr mice, and therapeutic studies were initiated upon reaching a target volume of 100 mm3. Sample sizes were n = 10–20 mice for vehicle-treated groups and n = 5–10 mice for single-agent- or combination-treated groups. Mice were treated with drug doses that had been demonstrated to be the MTD for the single agents and the combination in prior experiments by the NCI Developmental Therapeutics Program (see Supplementary Data for dosage regimens); in some studies, treatments at lower doses were also performed. For each mouse, the time from treatment initiation to tumor volume doubling was estimated using exponential growth interpolation when the event was between two time points. Mice for which tumor volume had not doubled by the end of study were considered censored at the last observation. The distribution of time-to-tumor-volume-doubling was estimated for each treatment cohort using the Kaplan-Meier method, and the log-rank test was used to determine significant differences between combination and single-agent treatment groups.
Biomarker Analyses of Human Tumor Xenografts
Analyses of apoptosis (19) and DNA damage response biomarkers (20,21) were performed as described previously (see Supplementary Methods for details).
Results
The NCI-ALMANAC Dataset and Web Tools
Pairwise combinations of 104 FDA-approved anticancer drugs (Supplementary Table S1) were screened in each of the cell lines of the NCI-60 panel to discover those with enhanced growth inhibition or cytotoxicity profiles compared to either single agent (Fig. 1A). Combination activity was reported as a “ComboScore” (22); positive ComboScores indicated greater than additive in vitro activity; negative scores indicated less-than-additive activity (see the Methods). A total of 5,232 drug pairs were evaluated in each of the cell lines; 304,549 experiments were performed to test each drug at either 9 or 15 combination dose points, for a total of 2,809,671 dose combinations. The NCI-ALMANAC is publicly available at https://dtp.cancer.gov/ncialmanac; the data can be visualized as a heat map summarizing the entire dataset, as a bar graph of the ComboScores for a particular drug pair in all cell lines, or as dose-response curves for one drug combination in a given cell line (Supplementary Fig. S1). Finally, users can obtain an overview of which drugs or mechanistic categories of drugs perform best in combination with a given drug of interest.
Figure 1. NCI-ALMANAC screen strategy and summary results.
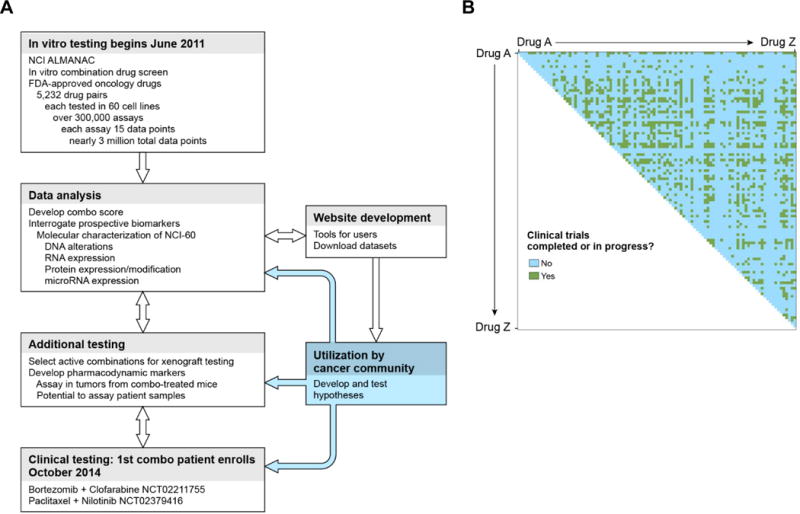
A, workflow diagram for the NCI-ALMANAC screen and follow-up preclinical and clinical studies. Double-headed arrows indicate the iterative flow of information between different stages of the project. B, NCI-ALMANAC reveals a large number of potentially active, clinically novel drug combinations (according to clinicaltrials.gov analysis). Data were downloaded from clinicaltrials.gov and processed to identify the cancer trials that had utilized the drug combinations examined in this study. Each drug is shown as both the abscissa and ordinate, alphabetically, with green indicating drug pairs that were identified as having been reported as tested in combination in a clinical trial and blue indicating pairs that have not.
We first researched what fraction of the NCI-ALMANAC drug pairs appeared on clinicaltrials.gov for an estimation of clinical experience (see Methods for details). At the start of this study in June 2011, nearly 75% of the > 5,000 pairs of FDA-approved drugs examined had not been reported to clinicaltrials.gov as being part of an ongoing or completed study (Fig. 1B). Next, to identify patterns in cellular sensitivity and combination activity, we conducted a two-dimensional hierarchical clustering analysis. The resulting heat map of ComboScores across cell lines and drug combinations revealed that most drug combinations have selective activity for a subset of cell lines, as indicated by the blue-red heterogeneity throughout most of the heat map, though some combinations did exhibit patterns of predominantly greater than additive (highly red) or predominantly less-than-additive (highly blue) activity across most of the NCI-60 cell lines (Fig. 2A and B). Additionally, except for leukemia cell lines, sensitivity to drug combinations appeared to be independent of cellular histology, as cell lines from the same source tissue did not cluster together.
Figure 2. In vitro activity of drug combinations in the NCI-60 panel.
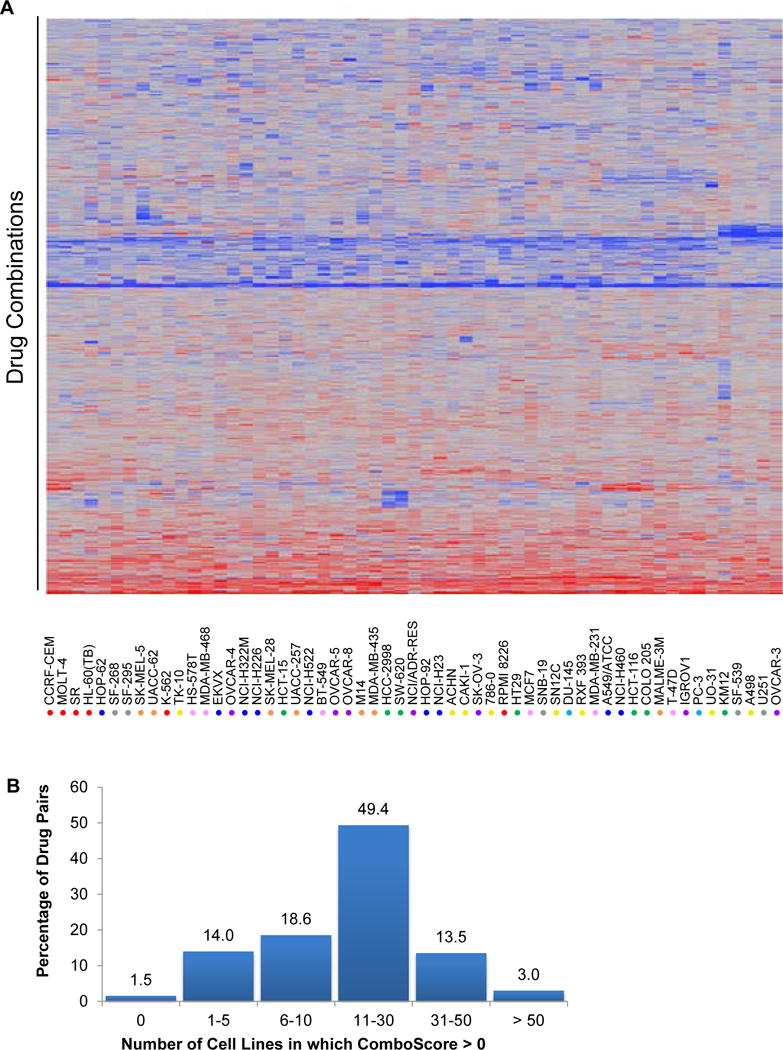
A, two-way hierarchical clustering was used to cluster ComboScores for 57 cell lines (the SK-MEL-2, SNB-75, and LOX-IMVI lines had many missing values and were excluded) and 4,629 drug combinations. Red denotes positive ComboScores (i.e., better-than-additive effects), and blue denotes negative values (i.e., less-than-additive effects). Tumor tissue derivations for the NCI-60 cell lines are indicated by colored circles: red = leukemia; green = colon; blue = non-small cell lung; gray = CNS; orange = melanoma; purple = ovarian; yellow = renal; turquoise = prostate; and pink = breast. B, the majority of drug pairs had in vitro activity in 11–30 cell lines of the NCI-60 panel, indicating that most drug pairs only have greater than additive activity in a subset of the NCI-60 cell lines.
Examination of Promising Anticancer Drug Combinations from the NCI-ALMANAC in Human Tumor Xenografts
Because it was not feasible to examine all drug combinations in follow-up animal model experiments, a subset of combinations with greater than additive activity was selected for in vivo analysis based on the ComboScore, the ability of relevant cell lines to grow as xenograft implants, and clinical investigator input regarding clinical utility. An initial study was performed using doses at or near the maximum tolerated dose (MTD) of each single agent to eliminate those combinations with excessive toxicity (requiring dose reduction >50%). For the remaining combinations, the more clinically pertinent criterion of “greater-than-single-agent” activity was applied to the follow-up xenograft analyses to identify combinations that could potentially confer additional efficacy in patients. We also differentiated between “novel” combinations (those that had not been previously tested together exclusively) and “non-novel” combinations (those tested together exclusively in a clinical trial arm), according to clinicaltrials.gov and the literature.
Various methods have been used to assess drug efficacy in xenograft studies, although to our knowledge, no method has been validated for comparing results of a high-throughput xenograft screen. For our purposes, we chose Kaplan-Meier and log-rank analyses of time-to-tumor-volume-doubling. Out of 13 “non-novel” combinations that were selected for xenograft analysis based on high in vitro ComboScores (Supplementary Table S2), 5 yielded greater-than-single-agent efficacy in at least one of the xenograft models tested. Furthermore, each of these 5 combinations has demonstrated appreciable efficacy (partial and/or complete responses) in clinical trials (Supplementary Table S3). Despite the small number of combinations examined, this analysis suggested that the NCI-ALMANAC results may be used as a starting point to select novel combinations for further study and potential clinical use.
Given these promising results, 20 novel combinations were evaluated for greater-than-single-agent efficacy in one or more xenograft models derived from NCI-60 cell lines, yielding a total of 44 unique “combination + model” experiments (in several cases, multiple schedules and/or doses were examined for each “combination + model” set; in these cases, only the best outcome was scored). The analysis revealed that 14 of the 44 experiments yielded significantly improved antitumor efficacy for the combination compared to both single-agents (P < 0.05; Fig. 3 and Supplementary Table S4). Additionally, a panel of xenograft experts designated experiments for which the tumor regression appeared superior for the combination compared to the respective single agents, despite lack of statistical significance in log-rank analysis of time-to-tumor-doubling; 7 such experiments were identified (Fig. 3, Supplementary Table S4). For the remaining experiments, the combination did not yield significantly improved efficacy compared to either single agent (Supplementary Table S4 and Fig. 3); in 2 experiments, this was due to substantial single-agent efficacy (i.e., ≤ 20% of mice treated with the single agent experienced a tumor doubling). Following this analysis, two of the combinations that exhibited significantly improved antitumor activity compared to their respective single agents were selected for further efficacy and mechanistic analysis.
Figure 3. Antitumor efficacy of NCI-ALMANAC-derived drug combinations in the tested NCI-60 xenograft models.
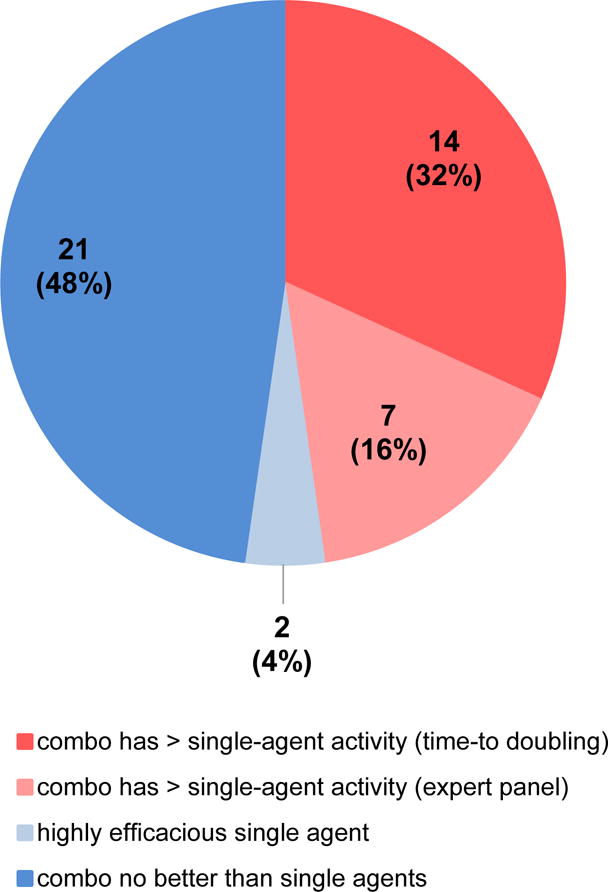
A total of 44 unique xenograft efficacy experiments, involving 20 different drug combinations that had not previously undergone clinical testing (according to clinicaltrials.gov), were performed using sets of drug combinations and NCI-60-derived models that demonstrated positive in vitro ComboScores. Experiments in which combination treatment yielded improved antitumor efficacy compared to both single-agent treatments were identified by Kaplan-Meier and log-rank analysis of time-to-tumor-volume-doubling (P < 0.05; dark pink) or by a panel of xenograft experts (light pink). Experiments with no greater-than-single-agent activity are shown in dark blue, while those for which one single agent was highly efficacious are shown in light blue. See Supplementary Table S4 for a complete list of xenograft experiments and results.
Clofarabine and Bortezomib
The purine nucleoside analog clofarabine and the proteasome inhibitor bortezomib play important roles in the treatment of hematopoietic malignancies; however, neither agent had previously been demonstrated to exhibit appreciable activity against human solid tumors (23,24). Based on greater than additive activity in vitro (Fig. 4A), we tested this combination in five solid tumor xenograft models and determined that the combination caused significant tumor regressions in two colon cancer models, HCT-116 (P < 0.01) and HT29 (P < 0.001), and in the non-small cell lung cancer model NCI-H522 (P < 0.01; Fig. 4B and Supplementary Table S4). In HCT-116 xenografts treated with bortezomib (0.75 mg/kg), clofarabine (60 mg/kg), or both agents in combination on a Q2Dx5 dosing schedule for two treatment cycles, the combination had significantly greater efficacy than either single agent (P < 0.01) as determined by log-rank analysis of the time-to-tumor-doubling (Supplementary Table S4); in a second experiment, a single treatment cycle at these doses yielded slightly greater-than-single-agent efficacy (P < 0.1; Fig. 4B and Supplementary Table S4). This combination was well tolerated, with no observed median body weight loss > 30% (data not shown). In contrast, although the clofarabine and bortezomib combination had greater than additive efficacy in the ovarian cancer cell line OVCAR-3 and the melanoma line M14 in vitro (Fig. 4A), neither single-agent nor combination treatment elicited antitumor effects in the corresponding xenograft models (Fig. 4C and Supplementary Table S4).
Figure 4. Therapeutic activity of the NCI-ALMANAC-derived combination of bortezomib and clofarabine.
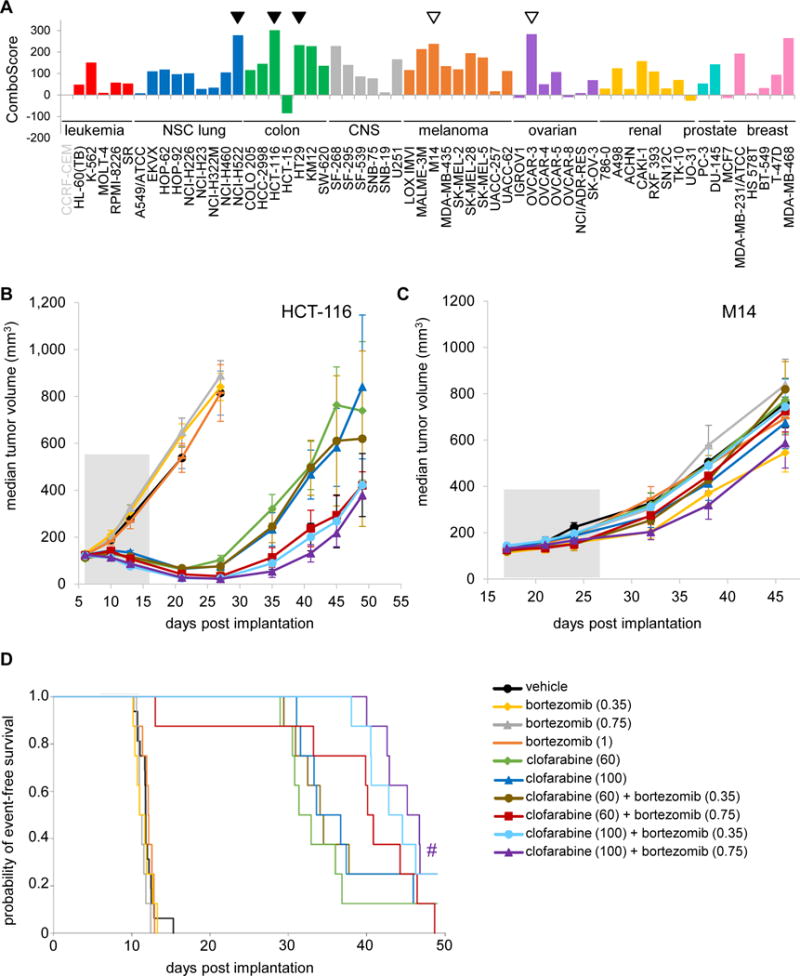
A, the combination of bortezomib and clofarabine in vitro yielded positive ComboScores across several cell types. Tumor tissue derivations for the NCI-60 cell lines are indicated by bar color: red = leukemia; green = colon; blue = non-small cell lung; gray = CNS; orange = melanoma; purple = ovarian; yellow = renal; turquoise = prostate; and pink = breast. A leukemia cell line for which no ComboScore was available for this combination is indicated by gray text. Models for which xenograft experiments revealed the presence or absence of in vivo greater-than-single agent activity are indicated by filled triangles or empty triangles, respectively. B and C, clofarabine-bortezomib combination treatment exhibits enhanced efficacy relative to the respective single-agent treatments in the human colon cancer HCT-116 xenograft model (B) but not in the M14 melanoma xenograft model (C). Median tumor volumes are shown for mice treated with vehicle, single-agent bortezomib (5×Q2D, intraperitoneal [IP] injection), single-agent clofarabine (5×Q2D, oral administration [PO]), or the combination (schedule and administration for each agent as indicated for the respective single-agent treatments); treatment commenced on day 6 for the HCT-116 experiment (B), and on day 17 for the M14 experiment (C) (dosing period indicated by gray-shaded area in both graphs). D, Kaplan-Meier curves for time-to-tumor doubling analysis of HCT-116 xenograft models treated with the clofarabine-bortezomib combination. The y-axis indicates probability of event-free survival, where an event is defined as one tumor doubling or drug-related death (the latter occurred for only one animal in the 60 mg/kg clofarabine + 0.75 mg/kg bortezomib-treated group). (#) denotes the combination-treated group exhibiting greater-than-single-agent time-to-tumor-doubling compared to both bortezomib-alone and clofarabine-alone groups at the corresponding doses (P < 0.1), as determined by log-rank tests. Doses (mg/kg) for each treatment group are indicated in the legend (n = 16 mice for the vehicle group, n = 8 mice for all other groups). Error bars indicate standard error of the median.
Because both bortezomib and clofarabine have been demonstrated to induce apoptosis in a variety of human tumor cell lines (25,26), we examined whether the activity of the combination in the treatment-responsive HCT-116 xenograft models might be explained by an enhanced modulation of apoptotic cascade proteins. To this end, tumor samples from one responsive (HCT-116) and one nonresponsive (M14) model were collected at various time points after treatment with clofarabine, bortezomib, or the combination, and tumor extracts were surveyed for changes in the apoptotic pathway using a 15-biomarker apoptosis multiplex panel [(19) and Supplementary Methods]. Based on results from a pilot time-course study (data not shown), we selected time points at 6 h and 24 h post-dose-5 for subsequent experiments.
In the responsive HCT-116 model, bortezomib alone had virtually no effect compared to vehicle on apoptosis markers at 6 h, while clofarabine alone significantly altered 3 of the 5 examined apoptosis markers (cleaved caspase 3 and mitochondrial/nuclear lamin-B and survivin) relative to vehicle (Fig. 5A). In contrast, compared to both vehicle and bortezomib alone, the bortezomib-clofarabine combination significant altered levels of all 5 of the measured apoptosis markers: cytosolic survivin, cleaved caspase 3, and mitochondrial/nuclear lamin-B, BAX, and survivin (Fig. 5A). These trends persisted at the 24 h post-treatment time point (Supplementary Fig. 2A). Additionally, Western blot analysis showed that treatment of HCT-116 xenograft models with clofarabine alone or with the combination yielded high levels of p53 upregulation/stabilization relative to vehicle control (Supplementary Fig. S2B). These results in the sensitive HCT-116 model contrasted with those observed for the nonresponsive M14 model, in which bortezomib-clofarabine combination treatment did not produce this apoptosis-associated biomarker signature (Fig. 5B and Supplementary Fig. S2C). Overall, in the sensitive HCT-116 xenograft model, the intrinsic apoptotic pathway was activated primarily by clofarabine, and the addition of bortezomib enhanced this activation.
Figure 5. Bortezomib-clofarabine combination treatment modulates markers of apoptosis and DNA damage in responsive, but not unresponsive, xenograft models.
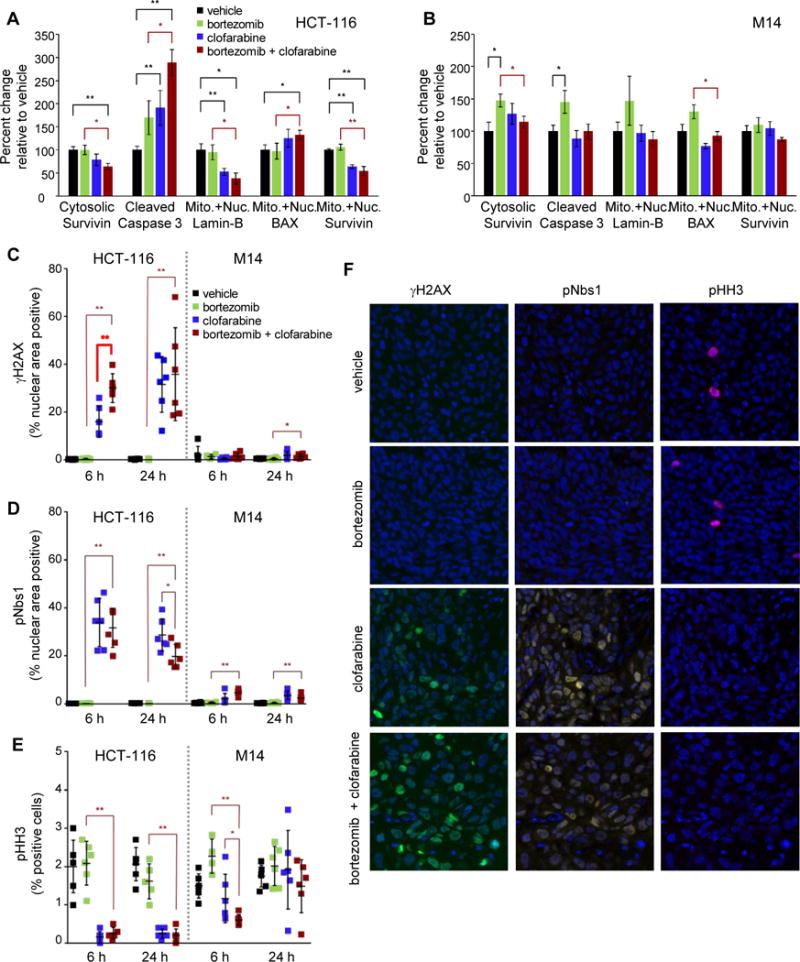
Mice bearing HCT-116 or M14 xenografts were treated with either vehicle, 0.75 mg/kg bortezomib (5×Q2D, IP), 60 mg/kg clofarabine (5×Q2D, PO), or the combination of bortezomib and clofarabine with the same dosage regimens. Tissues were harvested at 6 h and 24 h post dose 5 and processed into cell lysates (for apoptosis marker assays, A and B) or were formalin-fixed and paraffin-embedded for quantitative immunofluorescence analysis of DNA damage response and cell cycle markers (C–F). Apoptosis markers were measured in the therapeutically responsive HCT-116 (A) or nonresponsive M14 (B) xenograft models at 6 h following the day 5 dose. Error bars represent SEM; n = 6 mice per treatment group. Statistically significant differences between the combination and single-agent treatment groups are indicated in red, while those compared to vehicle are indicated in black (*P < 0.05, **P < 0.01; P-values derived from unpaired, nonparametric Mann-Whitney tests). One outlier data point for caspase-3 in the clofarabine HCT-116 group was removed. Effects of drug treatment on nuclear levels of γH2AX (C), pNbs1 (D), or pHH3-Ser10 (E) were examined in HCT-116 (left) and M14 (right) models at 6 h and 24 h following the day 5 dose. Representative images from the HCT-116 6 h group are shown (F). Error bars (C–E) represent SD (n = 6 mice per treatment group, except for the HCT-116 bortezomib 24 h group [n = 5 due to unsuitable tissue quality] and the M14 bortezomib 6 h group [n = 4]), and an average of 5,000 cells per mouse tumor were analyzed. Statistically significant differences between the combination and single-agent treatment groups are indicated (*P < 0.05, **P < 0.01); the sole statistically significant greater-than-single-agent difference between the HCT-116 clofarabine group and the combination group is bolded for emphasis (C). In the HCT-116 experiments (C–E, left), biomarker levels for both the clofarabine group and combination group were significantly different than vehicle at 6 h and 24 h time points (P < 0.01 vs. vehicle for all; P-values derived from nonparametric Mann-Whitney tests).
Clofarabine is known to induce DNA damage response signaling (27); therefore, we evaluated the effects of these treatments on markers of DNA damage by immunofluorescence analysis of tumor segments using a validated assay (20,21). The combination yielded a significantly greater induction of γH2AX compared to both bortezomib alone and clofarabine alone at the 6 h time point (Fig. 5C and F), the latter indicating a role for bortezomib in potentiating clofarabine-induced DNA damage. Such substantial changes in γH2AX levels were not observed in the nonresponsive M14 model (Fig. 5C, right). The DNA damage marker pNbs1 was significantly increased in the clofarabine- and combination-treated groups relative to the vehicle-treated group as early as 2 h (data not shown), at 6 h, and at 24 h in HCT-116, but not M14, models (Fig. 5D and F). Finally, the mitotic marker histone H3–pSer10 (pHH3) was lower in both the clofarabine-only and combination groups relative to vehicle at 6 and 24 h in the HCT-116 model (Fig. 5E and F) but was not consistently lower in M14 xenografts (Fig. 5E), suggesting that sustained cell cycle arrest was specific to the responsive model. As expected, treatment with bortezomib alone did not induce markers of DNA damage or cell cycle arrest (Fig. 5C–F). Together, these data provide mechanistic evidence that DNA damage, cell cycle arrest, and apoptosis contribute to the efficacy of the bortezomib-clofarabine combination in the responsive HCT-116 xenograft model.
Paclitaxel and Nilotinib
The combination of the microtubule-stabilizing agent paclitaxel and the BCR-Abl kinase inhibitor nilotinib also had not been explored clinically. All 6 hematopoietic tumor cell lines tested had positive ComboScores for the paclitaxel-nilotinib combination, and this drug combination gave positive ComboScores in three of the triple-negative breast cancer NCI-60 cell lines (Hs578T, MDA-MB-468, and BT-549), but negative scores in the two estrogen receptor-positive breast cancer lines (MCF7 and T-47D) (Fig. 6A). Based on these data, the effects of the nilotinib-paclitaxel combination on breast cancer xenografts derived from triple-negative cell lines were examined. Nilotinib-paclitaxel was well tolerated and highly efficacious in the MDA-MB-468 xenograft model; the combination (at either of the examined paclitaxel doses) was superior to both single agents (Fig. 6B and Supplementary Table S4). Strikingly, the combination treatment resulted in complete tumor regression, with no tumor regrowth observed for over 80 days following the end of therapy (day 134; Fig. 6B). Single-agent nilotinib had no significant effects on tumor growth compared to vehicle, and single-agent paclitaxel yielded only partial tumor control, with tumor regrowth commencing around day 65–75 (Fig. 6B). Hs578T and BT-549 grew poorly as xenografts and were therefore not assessable in vivo, so we instead examined two additional triple-negative models derived from non–NCI-60 cell lines, SUM-52 PE and SUM-149PT. In both of these models, the nilotinib-paclitaxel combination yielded greater-than-single-agent efficacy (Supplementary Table S4). An additional triple-negative NCI-60 cell line, MDA-MB-231, did not yield valid in vitro screen results for the nilotinib-paclitaxel combination due to a “reversal” profile (see Methods) and also did not yield greater-than-single-agent efficacy when tested as a xenograft model (Supplementary Table S4). Beyond breast cancer models, the nilotinib-paclitaxel combination also had greater-than-single-agent efficacy in the RXF 393 renal carcinoma model, in line with its greater than additive activity in the corresponding cell line in vitro (Supplementary Table S4). In contrast, the U251 cell line, where the combination was less efficacious in vitro (ComboScore of 21), yielded no greater-than-single-agent efficacy when tested as a xenograft model (Supplementary Table S4).
Figure 6. In vitro activity, in vivo efficacy, and mechanistic analysis of the NCI-ALMANAC-derived nilotinib-paclitaxel combination.
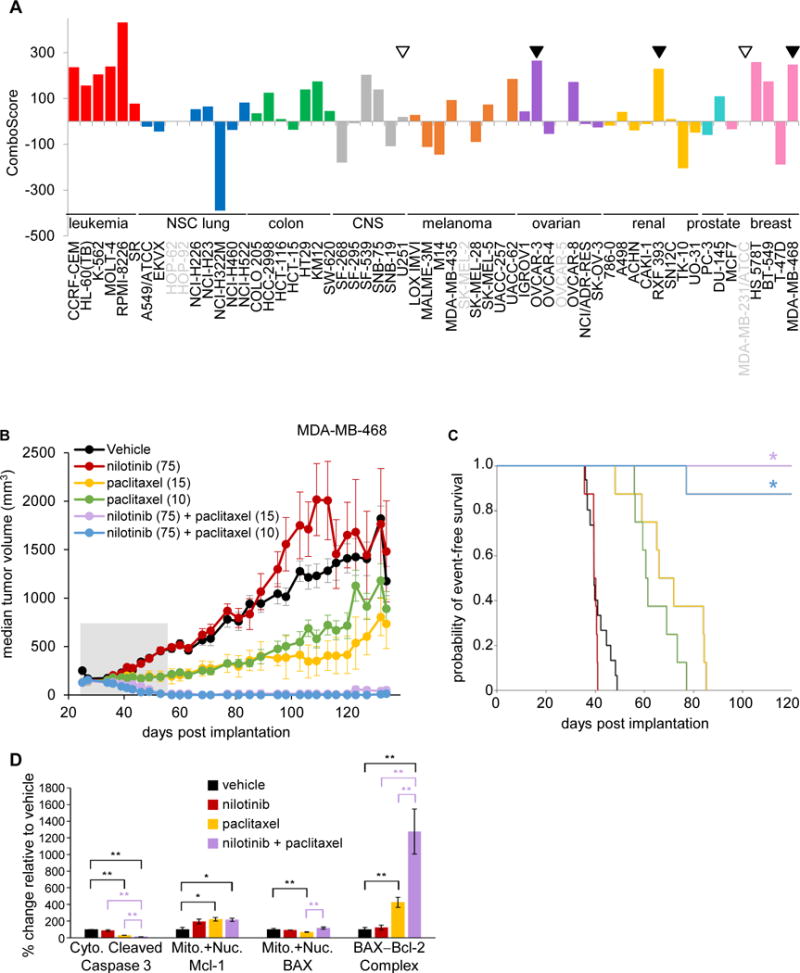
A, the combination of nilotinib and paclitaxel in vitro yielded positive ComboScores across several cell types. Tumor tissue derivations for the NCI-60 cell lines are indicated by bar color: red = leukemia; green = colon; blue = non-small cell lung; gray = CNS; orange = melanoma; purple = ovarian; yellow = renal; turquoise = prostate; and pink = breast. Cell lines for which no ComboScore data are available for this combination are indicated by gray text. Models for which xenograft experiments revealed the presence or absence of in vivo greater-than-single agent activity are indicated by filled triangles or empty triangles, respectively. B, nilotinib/paclitaxel combination treatment exhibits enhanced efficacy relative to the respective single-agent treatments in the MDA-MB-468 triple-negative breast cancer xenograft model. Median tumor volumes are shown for mice treated with vehicle, single-agent nilotinib (75 mg/kg QDx5, PO for 4 cycles beginning on days 25, 32, 39, and 46), single-agent paclitaxel (on one of two dose schedules for 2 cycles beginning on days 25 and 39: [1] 15 mg/kg, Q7Dx2, IV; or [2] 10 mg/kg, QDx3, IV); or [3] the respective combinations (schedule and administration as indicated for the respective single-agent treatments; dosing period indicated by gray-shaded area). n = 15 mice for vehicle group, and n = 7–8 mice for all other groups. Error bars indicate standard error of the median. C, Kaplan-Meier curves for time-to-tumor doubling analysis of MDA-MB-468 xenograft models treated with the nilotinib-paclitaxel combination. The y-axis indicates probability of event-free survival, where an event is defined as one tumor doubling. Asterisks denote combination-treated groups exhibiting significantly greater-than-single-agent time-to-tumor-doubling compared to both nilotinib-alone and paclitaxel-alone groups at the corresponding doses (*P < 0.001), as determined by log-rank tests (n = 16 mice for the vehicle group, n = 8 mice for all other groups). Treatment groups, and doses (mg/kg) for each, are indicated in the legend in (B). D, nilotinib-paclitaxel combination treatment does not induce apoptotic pathway biomarkers in therapeutically responsive MDA-MB-468 xenograft models. Mice were treated with either vehicle, single-agent nilotinib (75 mg/kg QDx19, PO), single-agent paclitaxel (15 mg/kg, Q7Dx3, IV), or the combination (schedule and administration as indicated for the respective single-agent treatments). Tissues were harvested on day 15 following the start of treatment and processed into cell lysates, which were then subjected to subcellular fractionation for measurement of the indicated apoptosis markers. Error bars represent SEM; n = 6 mice per treatment group. Statistically significant differences between the combination and single-agent treatment groups are indicated in purple, while those compared to vehicle are indicated in black (*P < 0.05, **P < 0.01); P-values derived from nonparametric Mann-Whitney tests.
To identify potential biomarkers of response to this combination, we first queried the NCI-60 molecular markers database to look for associations between in vitro ComboScore and drug-related molecular signatures. We found no significant correlations between nilotinib-paclitaxel ComboScores and either: 1) protein and RNA expression levels for the non-BCR-Abl kinases known to be inhibited by nilotinib (28), or 2) activity and RNA expression levels of the ABC family efflux pumps that have been reported to be inhibited by nilotinib (29–31) (Supplementary Fig. S3A). To further examine the possibility of nilotinib-mediated reduction in paclitaxel efflux via ABC transporter inhibition, we analyzed tumor paclitaxel levels in MDA-MB-468 xenograft models and found comparable tumor paclitaxel concentrations in animals treated with the nilotinib-paclitaxel combination and those treated with paclitaxel alone (Supplementary Fig. S3B), providing further evidence that the mechanism of action for this combination does not involve modulation of paclitaxel efflux.
Because apoptosis, amongst other forms of cell death, has been postulated to play a role in tumor cell killing by paclitaxel and/or nilotinib (32,33), we examined biomarkers of apoptosis in response to single-agent and combination treatment in the MDA-MB-468 breast cancer xenograft model. No increases in the apoptosis marker cleaved caspase-3 were observed in either single-agent or combination treatment groups compared to the vehicle control (Fig. 6D), supporting a non-apoptotic mechanism of antitumor activity for this drug pair. Corroborating this finding, levels of mitochondrial Mcl-1, associated with anti-apoptotic activity (34), were higher in the combination and single-agent treatment groups compared to vehicle (Fig. 6D). Though total levels of the canonically anti-apoptotic BAX–Bcl-2 complex were higher following nilotinib-paclitaxel combination treatment relative to the vehicle and single-agent treatments (Fig. 6D), subcellular fractionation experiments revealed that this was primarily due to an increase in ER-associated BAX–Bcl-2 (data not shown), which is involved in the perturbation of calcium homeostasis that occurs in both apoptosis and necroptosis (35). Together, these results suggest that the nilotinib-paclitaxel combination likely suppresses tumor growth through a caspase-3−independent mechanism of cell death.
Discussion
More than 100 small-molecule drugs have been approved for cancer therapy by the FDA (36), and are often used in combination with other agents to address the multiple molecular pathway aberrations, and consequent drug resistance, associated with many malignancies. However, a broad range of potential FDA-approved drug combinations has never been evaluated clinically. We determined that it would be valuable to systematically screen all possible two-drug combinations in vitro to identify novel pairs of FDA-approved drugs with greater in vitro growth inhibition than would be expected from the sum of their single agent activities. A number of combinations identified in the NCI-ALMANAC screen were further evaluated in animal models for greater-than-single-agent efficacy. Although combination experiments have been conducted before (16,37–39), to our knowledge, the NCI-ALMANAC is the most extensive anticancer drug combination screening effort for which the data and search tools are publicly available.
Our comprehensive in vitro screen identified 1,898 drug pairs with positive ComboScores in at least 1 cell line. Consequently, we performed in vivo testing of a small subset of drug pairs that have not, to our knowledge, been clinically tested (Supplementary Table S4). Agents were tested at or near their MTD to assess whether combination therapy could achieve efficacy superior to that of the single agents. Forty-eight percent of these novel, NCI-ALMANAC-derived combinations exhibited greater-than-single-agent activity in at least one model (Fig. 3), suggesting that the NCI-ALMANAC may be helpful in identifying additional combinations with clinical efficacy in the future.
The bortezomib-clofarabine combination conferred an antitumor activity advantage over single-agent clofarabine or bortezomib in the HCT-116 xenograft model (Fig. 4B). In view of the known induction of apoptosis in response to single-agent bortezomib (40) or clofarabine (27), we investigated the effects of this combination on apoptotic pathway markers. We found that the antitumor activity of the clofarabine-bortezomib combination was accompanied by several significant apoptosis-associated molecular changes relative to the single-agent- and/or vehicle-treated arms, including decreases in cytosolic and mitochondrial/nuclear survivin and mitochondrial/nuclear-associated lamin-B, as well as increases in caspase 3 activation and mitochondrial/nuclear-associated BAX (Fig. 5A). In contrast, for the nonresponsive M14 xenograft model, in which no antitumor efficacy advantage was observed for the combination, we observed no consistent alterations in levels of these apoptotic biomarkers, suggesting that the apoptotic pathway activation observed in the responsive HCT-116 model is associated with the antitumor efficacy of the clofarabine-bortezomib combination. The mechanism whereby clofarabine-bortezomib treatment reduces survivin levels in HCT-116 xenografts could be either p53-dependent (41), as indicated by our Western blot analysis (Supplementary Fig. S1E), or p53-independent (42). Regardless, the greater-than-single-agent activity of this combination has some precedent; previous in vitro experiments have demonstrated that bortezomib-induced cytotoxicity and pro-apoptotic signaling are enhanced by purine nucleoside analogs (43).
Because clofarabine is both a DNA damaging agent and a DNA synthesis inhibitor (27), we evaluated the role of DNA damage in the activity of the bortezomib-clofarabine combination. The combination and clofarabine alone both significantly increased levels of the DNA damage response markers γH2AX and pNbs1 and decreased levels of the cell proliferation marker pHH3 in HCT-116 xenografts compared to vehicle and single-agent bortezomib treatment (Fig. 5C–F); γH2AX levels were also significantly higher in the combination arm relative to the clofarabine-only arm at the 6 h time point (Fig. 5C). Hence, the ability of clofarabine to induce DNA damage (which could then trigger p53-mediated apoptosis) and/or directly alter apoptotic pathway components (27) might lower the apoptosis threshold in HCT-116 xenografts, rendering these tumors more susceptible to the cytotoxic effects of bortezomib-induced proteasome inhibition.
Though the pharmacodynamic effects of clofarabine and bortezomib converge on apoptosis, potential upstream interactions remain to be explored; for example, it is possible that bortezomib-mediated proteasome inhibition stabilizes a protein required for the uptake, recycling/stability, mechanism of action, or activating phosphorylation of clofarabine. A whole-proteome analysis in Jurkat cells revealed that bortezomib treatment yielded a reproducible 12–13–fold upregulation of nucleoside diphosphate kinase, which catalyzes the final step of clofarabine activation, as well as more modest upregulation of the clofarabine-activating enzyme deoxycytidine kinase and the clofarabine targets DNA polymerase-α and ε (27,44). Another possibility is that proteasome inhibition by bortezomib stabilizes DNA damage response proteins, thereby potentiating this response following its induction by clofarabine. Though these mechanistic details remain to be elucidated, the in vivo efficacy of the clofarabine-bortezomib combination, along with a prior NCI review of the relationship between efficacy in xenograft models and activity in human phase II trials (45) and the development of a validated multiplex assay for apoptotic pathway components (19), provided support for initiating a phase I trial of this drug pair in patients with advanced, refractory cancers (clinicaltrials.gov identifier: NCT02211755).
The novel NCI-ALMANAC-derived combination of nilotinib and paclitaxel exhibited greater-than-single-agent efficacy in our experiments with the MDA-MB-468 triple-negative breast cancer model and others. Previous studies have found that nilotinib enhances intracellular accumulation of paclitaxel and other agents in ABC transporter–overexpressing cell lines via inhibition of ABC transporter–mediated drug efflux (46,47). Furthermore, nilotinib-paclitaxel combination treatment has been shown to yield greater-than-single-agent efficacy in xenograft models overexpressing ABC transporters ABCB1 or ABCC10 (30). However, the lack of association between in vitro ABC transporter activity or expression and nilotinib-paclitaxel ComboScore (Supplementary Fig. S3A) suggests that ABC transporter inhibition may not account for the greater than additive activity of this combination that we observed. The comparable tumor paclitaxel concentrations in MDA-MB-468 xenograft models treated with paclitaxel alone versus those treated with the nilotinib-paclitaxel combination (Supplementary Fig. S3B) cast further doubt on this hypothesis for the enhanced activity of this combination. Another potential mechanism of action might be the known inhibition by nilotinib of the cytochrome P450s (including CYP2C8) involved in the metabolism of paclitaxel (48), which would increase systemic paclitaxel exposure; however, we also found comparable plasma paclitaxel concentrations in MDA-MB-468 xenograft models treated with paclitaxel alone versus the nilotinib-paclitaxel combination (data not shown), rendering this hypothesized mechanism of action also unlikely.
Recent studies have demonstrated that both paclitaxel and nilotinib induce a variety of overlapping apoptotic and necroptotic cell death mechanisms (32,33), that might contribute to the efficacy of this drug pair. Our analysis of apoptotic pharmacodynamic biomarkers revealed the absence of apoptosis-associated changes in cleaved caspase 3 or Mcl-1 levels following nilotinib-paclitaxel combination treatment in MDA-MB-468 xenografts, suggesting that an alternative mechanism of cell death may be responsible for the antitumor activity of this combination. Paclitaxel has previously been shown to induce both apoptosis and necrosis in a cell-cycle stage–specific manner (49), and a number of kinase inhibitors are known to induce necrosis (50), suggesting a potential role for nilotinib in shifting the apoptotic-necroptotic balance in paclitaxel-induced cell death. Regardless, the impressive in vitro activity and in vivo efficacy of this combination have motivated phase 1 evaluation of this drug pair (clinicaltrials.gov identifier: NCT02379416).
Despite the limitations inherent in the translation of in vitro activity into in vivo efficacy (due in part to the size of the screen, which precluded comprehensive investigation of the effect of different drug administration doses and schedules), and the fact that large-scale animal studies could not easily be conducted to validate all of our screening results, the NCI-ALMANAC generated interesting hypotheses that were further addressed with mechanistic biomarker profiling and xenograft efficacy studies. To explore drug combinations of interest identified in the NCI-ALMANAC screen, we enriched our xenograft testing for pairs that had not been studied previously in a clinical trial (as defined by searching clinicaltrials.gov). Because only FDA-approved small molecules were used in the NCI-ALMANAC screen, our results have the potential for rapid clinical translation. Thus, the NCI-ALMANAC should prove to be a valuable resource that will inform the development and selection of novel combinations of FDA-approved anticancer agents for future clinical trials.
Supplementary Material
Acknowledgments
The authors wish to thank the University of Pittsburgh and SRI International for acquisition of in vitro data, SRI for performing follow-up xenograft experiments, Dr. Karen Schweikart for excellent project management, Dr. Robert Kinders for helpful discussion regarding the biomarker analyses, Dr. Daniel Zaharevitz for construction of the NCI-ALMANAC website, and Drs. Mariam Konaté and Sarah Miller, Kelly Services, for editorial assistance in the preparation of this manuscript.
Financial Support: This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This research was supported [in part] by American Recovery and Reinvestment Act funds.
Footnotes
A Large Matrix of Anti-Neoplastic Agent Combinations
Disclosure of Potential Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Doroshow JH, Kummar S. Translational research in oncology-10 years of progress and future prospects. Nat Rev Clin Oncol. 2014;11(11):649–62. doi: 10.1038/nrclinonc.2014.158. [DOI] [PubMed] [Google Scholar]
- 2.Devita VT, Schein PS. Drugs in combination for treatment of cancer - rationale and results. N Engl J Med. 1973;288(19):998–1006. doi: 10.1056/NEJM197305102881905. [DOI] [PubMed] [Google Scholar]
- 3.Chen S-h, Lahav G. Two is better than one; toward a rational design of combinatorial therapy. Curr Opin Struct Biol. 2016;41:145–50. doi: 10.1016/j.sbi.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther. 2010;9(5):1451–60. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Institute NC. NCI-60 Human Tumor Cell Lines Screen. Aug 1; < https://dtp.cancer.gov/discovery_development/nci-60>. Accessed 2016 August 1.
- 6.Doroshow JH, Parchment RE. Oncologic phase 0 trials incorporating clinical pharmacodynamics: from concept to patient. Clin Cancer Res. 2008;14(12):3658–63. doi: 10.1158/1078-0432.CCR-07-4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–26. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Carragher NO, Brunton VG, Frame MC. Combining imaging and pathway profiling: an alternative approach to cancer drug discovery. Drug Discov Today. 2012;17(5–6):203–14. doi: 10.1016/j.drudis.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Alifrangis CC, McDermott U. Reading between the lines: understanding drug response in the post genomic era. Mol Oncol. 2014;8(6):1112–19. doi: 10.1016/j.molonc.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature genetics. 2014;46(3):225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21(6):1258–66. doi: 10.1158/1078-0432.CCR-14-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang LH, Pfister TD, Parchment RE, Kummar S, Rubinstein L, Evrard YA, et al. Monitoring drug-induced gammaH2AX as a pharmacodynamic biomarker in individual circulating tumor cells. Clin Cancer Res. 2010;16(3):1073–84. doi: 10.1158/1078-0432.CCR-09-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You J, Li Y, Fang N, Liu B, Zu L, Chang R, et al. MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One. 2014;9(3):e91827. doi: 10.1371/journal.pone.0091827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt SM, Gull M, Brandli AW. Engineering Xenopus embryos for phenotypic drug discovery screening. Advanced drug delivery reviews. 2014;69–70:225–46. doi: 10.1016/j.addr.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3(140):ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A. 2014;111(6):2349–54. doi: 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y, Chanock SJ, et al. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8(4):713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- 19.Srivastava AK, Jaganathan S, Stephen L, Hollingshead MG, Layhee A, Damour E, et al. Effect of a Smac mimetic (TL32711, Birinapant) on the apoptotic program and apoptosis biomarkers examined with validated multiplex immunoassays fit for clinical use. Clin Cancer Res. 2016;22(4):1000–10. doi: 10.1158/1078-0432.CCR-14-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(30):3409–15. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888) in combination with irinotecan in patients with advanced solid tumors. Clin Cancer Res. 2016;22(13):3227–37. doi: 10.1158/1078-0432.CCR-15-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drewinko B, Loo TL, Brown B, Gottlieb JA, Freireich EJ. Combination chemotherapy in vitro with adriamycin. Observations of additive, antagonistic, and synergistic effects when used in two-drug combinations on cultured human lymphoma cells. Cancer Biochem Biophys. 1976;1(4):187–95. [PubMed] [Google Scholar]
- 23.Kantarjian HM, Gandhi V, Kozuch P, Faderl S, Giles F, Cortes J, et al. Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21(6):1167–73. doi: 10.1200/JCO.2003.04.031. [DOI] [PubMed] [Google Scholar]
- 24.Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y, et al. Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future oncology (London, England) 2014;10(10):1795–807. doi: 10.2217/fon.14.30. [DOI] [PubMed] [Google Scholar]
- 25.Sayers TJ, Brooks AD, Koh CY, Ma WH, Seki N, Raziuddin A, et al. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102(1):303–10. doi: 10.1182/blood-2002-09-2975. [DOI] [PubMed] [Google Scholar]
- 26.Genini D, Adachi S, Chao Q, Rose DW, Carrera CJ, Cottam HB, et al. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96(10):3537–43. [PubMed] [Google Scholar]
- 27.Zhenchuk A, Lotfi K, Juliusson G, Albertioni F. Mechanisms of anti-cancer action and pharmacology of clofarabine. Biochem Pharmacol. 2009;78(11):1351–59. doi: 10.1016/j.bcp.2009.06.094. [DOI] [PubMed] [Google Scholar]
- 28.Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804(3):445–53. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Eadie LN, Hughes TP, White DL. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin Pharmacol Ther. 2014;95(3):294–306. doi: 10.1038/clpt.2013.208. [DOI] [PubMed] [Google Scholar]
- 30.Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013;328(2):307–17. doi: 10.1016/j.canlet.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villar VH, Vogler O, Martinez-Serra J, Ramos R, Calabuig-Farinas S, Gutierrez A, et al. Nilotinib counteracts P-glycoprotein-mediated multidrug resistance and synergizes the antitumoral effect of doxorubicin in soft tissue sarcomas. PLoS One. 2012;7(5):e37735. doi: 10.1371/journal.pone.0037735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1–2):61–68. doi: 10.1007/s11010-010-0638-8. [DOI] [PubMed] [Google Scholar]
- 33.Shaker ME, Ghani A, Shiha GE, Ibrahim TM, Mehal WZ. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim Biophys Acta. 2013;1833(8):1992–2003. doi: 10.1016/j.bbamcr.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G, Brady HJ. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem. 2006;281(9):5750–59. doi: 10.1074/jbc.M505688200. [DOI] [PubMed] [Google Scholar]
- 35.Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium. 2011;50(3):211–21. doi: 10.1016/j.ceca.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Oncology FADf. 2016 Aug 1; 1995–2016. < https://www.centerwatch.com/drug-information/fda-approved-drugs/therapeutic-area/12/oncology>. August 1, 2016.
- 37.Miller ML, Molinelli EJ, Nair JS, Sheikh T, Samy R, Jing X, et al. Drug synergy screen and network modeling in dedifferentiated liposarcoma identifies CDK4 and IGF1R as synergistic drug targets. Sci Signal. 2013;6(294):ra85. doi: 10.1126/scisignal.2004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt L, Kling T, Monsefi N, Olsson M, Hansson C, Baskaran S, et al. Comparative drug pair screening across multiple glioblastoma cell lines reveals novel drug-drug interactions. Neuro-oncology. 2013;15(11):1469–78. doi: 10.1093/neuonc/not111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roller DG, Axelrod M, Capaldo BJ, Jensen K, Mackey A, Weber MJ, et al. Synthetic lethal screening with small-molecule inhibitors provides a pathway to rational combination therapies for melanoma. Mol Cancer Ther. 2012;11(11):2505–15. doi: 10.1158/1535-7163.MCT-12-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milano A, Perri F, Caponigro F. The ubiquitin-proteasome system as a molecular target in solid tumors: an update on bortezomib. OncoTargets and therapy. 2009;2:171–8. doi: 10.2147/ott.s4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21(17):2613–22. doi: 10.1038/sj.onc.1205353. [DOI] [PubMed] [Google Scholar]
- 42.Bhat UG, Gartel AL. Differential sensitivity of human colon cancer cell lines to the nucleoside analogs ARC and DRB. Int J Cancer. 2008;122(6):1426–29. doi: 10.1002/ijc.23239. [DOI] [PubMed] [Google Scholar]
- 43.Duechler M, Linke A, Cebula B, Shehata M, Schwarzmeier JD, Robak T, et al. In vitro cytotoxic effect of proteasome inhibitor bortezomib in combination with purine nucleoside analogues on chronic lymphocytic leukaemia cells. European journal of haematology. 2005;74(5):407–17. doi: 10.1111/j.1600-0609.2004.00406.x. [DOI] [PubMed] [Google Scholar]
- 44.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10(7):634–7. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti J, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84(10):1424–31. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR, Jr, Chen X, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009;78(2):153–61. doi: 10.1016/j.bcp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, et al. Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10) PLoS One. 2009;4(10):e7520. doi: 10.1371/journal.pone.0007520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim MJ, Lee JW, Oh KS, Choi CS, Kim KH, Han WS, et al. The tyrosine kinase inhibitor nilotinib selectively inhibits CYP2C8 activities in human liver microsomes. Drug Metab Pharmacokinet. 2013;28(6):462–67. doi: 10.2133/dmpk.dmpk-13-rg-019. [DOI] [PubMed] [Google Scholar]
- 49.Liao PC, Lieu CH. Cell cycle specific induction of apoptosis and necrosis by paclitaxel in the leukemic U937 cells. Life sciences. 2005;76(14):1623–39. doi: 10.1016/j.lfs.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 50.Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY, Zaorsky NG, et al. Necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy. Oncotarget. 2016;7(35):57391–413. doi: 10.18632/oncotarget.10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.