

Abstract
Interactions between cardiac myoglobin (Mb), nitrite, and nitric oxide (NO) are vital in regulating O2 storage, transport, and NO homeostasis. Production of NO through the reduction of endogenous myocardial nitrite by deoxygenated myoglobin has been shown to significantly reduce myocardial infarction damage and ischemic injury. We developed a mathematical model for a cardiac arteriole and surrounding myocardium to examine the hypothesis that myoglobin switches functions from being a strong NO scavenger to an NO producer via the deoxymyoglobin nitrite reductase pathway. Our results predict that under ischemic conditions of flow, blood oxygen level, and tissue pH, deoxyMb nitrite reduction significantly elevates tissue and smooth muscle cell NO. The size of the effect is consistent at different flow rates, increases with decreasing blood oxygen and tissue pH and, in extreme pathophysiological conditions, NO can even be elevated above the normoxic levels. Our simulations suggest that cardiac deoxyMb nitrite reduction is a plausible mechanism for preserving or enhancing NO levels using endogenous nitrite despite the rate-limiting O2 levels for endothelial NO production. This NO could then be responsible for mitigating deleterious effects under ischemic conditions.
Keywords: myoglobin nitrite reductase, myocardial infarction, mass transport, arteriole model
1. Introduction
Myoglobin (Mb) is an intracellular oxygen binding protein that has a variety of important functions in O2 storage and delivery. Mb has been shown to substantially contribute to nitric oxide (NO) – a powerful paracrine vasodilator - homeostasis in the heart (Flögel et al., 2010). In normoxia, Mb is a strong scavenger of NO. However, in hypoxic conditions, deoxygenated myoglobin (deoxyMb) has been shown to enzymatically reduce nitrite to NO. It is hypothesized that this is one of several nitrite reduction pathways in the body that provides a mechanism for the body to generate NO during conditions when oxygen-dependent endothelial nitric oxide synthase (eNOS) production falls (Faassen et al., 2010; Hendgen-Cotta et al., 2010).
Myoglobin nitrite reduction has been shown to reduce ischemic damage, mitigate ischemia/reperfusion injury, and reduce myocardial infarction size through NO release in animal and human heart studies (Hendgen-Cotta et al., 2008; Rassaf et al., 2007; Shiva et al., 2007; Totzeck et al., 2012a). The ability of myocardial Mb to effectively produce NO from nitrite in vivo has been challenged by its high affinity to oxygen, which reduces the availability of deoxyMb. However, endogenous nitrite, relatively high in tissue, has been shown to be sufficient to reduce ischemic damage, although the mechanism by which NO does this is not fully understood (Flögel et al., 2010; Totzeck et al., 2012b). Myoglobin deficient animals have been shown to be more susceptible to ischemic damage (Flögel et al., 2010; Rassaf et al., 2007), and myoglobin deficient animals with overexpressed inducible NOS have developed cardiac hypertrophy, ventricular dilatation, and fibrosis (Wunderlich et al., 2003). Due to experimental limitations, there are relatively few physiological human studies on cardiac Mb nitrite reduction (Ormerod et al., 2011; Webb et al., 2004). However, in vivo and in vitro studies have reported therapeutically beneficial uses of myocardial nitrite in treating ischemia through NO release in animals such as rats and mice, (Baker et al., 2007; Rassaf et al., 2014; Shiva et al., 2007; Tiravanti et al., 2004; Zweier et al., 2010) horses (Totzeck et al., 2012b), and fish (Pedersen et al., 2010). It is thought that Mb scavenges NO during normoxic conditions to maintain homeostasis and prevent deleterious transient NO effects, but releases NO during ischemic conditions such as low flow, low blood PO2, and low pH in order to inhibit cardiac respiration, reduce reactive oxygen species generation, and vasodilate cardiac vessels (Hendgen-Cotta et al., 2008; Rassaf et al., 2007).
Previous mathematical models have considered the effect of myoglobin as a scavenger on NO transport in microcirculatory vessels and the effects of myoglobin, nitrite, and NO reactions (Buerk, 2001; Kavdia et al., 2002; Kavdia and Popel, 2004; Tsoukias, 2008), but have not quantified the relative importance of deoxymyoglobin releasing NO from tissue nitrite in ischemic conditions relative to Mb scavenging of NO during normal oxygen and flow. The NO producing capabilities of cardiac Mb have previously been estimated using simple reaction kinetics calculations (Kim-Shapiro and Gladwin, 2014), without considering the effect of mass transport inside an arteriole environment. We have previously modeled (Liu et al., 2016) the effect of the deoxyhemoglobin nitrite reductase pathway on blood nitrite, but the deoxymyoglobin pathway is active on tissue nitrite and has different reaction kinetics. There is a need for mathematical models to analyze this scavenger and producer mechanism of myoglobin to explain the experimental effectiveness of cardiac tissue nitrite in releasing NO under ischemic conditions. The goal of this study is to determine the effectiveness of deoxyMb nitrite reductase during ischemia compared to its NO scavenging behavior under normoxic conditions. For this purpose, we developed a mathematical model for a cardiac arteriole and surrounding tissue to investigate the effect of deoxyMb nitrite reduction of endogenous nitrite in releasing NO during ischemia.
2. Model Development
2.1. Description of mathematical model
NO and O2 transport were considered in a cardiac arteriole and surrounding myocardial tissue, modeled as a cylindrical vessel with surrounding tissue. Coupled nonlinear partial differential equations were written in cylindrical coordinates and solved for steady-state conditions by finite element numerical methods using the software COMSOL v5.1 (COMSOL, Inc., Burlington, MA), similar to our previous approach (Buerk et al., 2011; Liu et al., 2016). The model has concentric cylindrical layers: (i) red blood cell (RBC) core, (ii) a RBC-free plasma layer, (iii) endothelium, (iv) vascular wall, and (v) perivascular cardiac tissue (Fig. 1). All model layers were assumed to have homogenous properties with uniform species reactions within.
Figure 1.
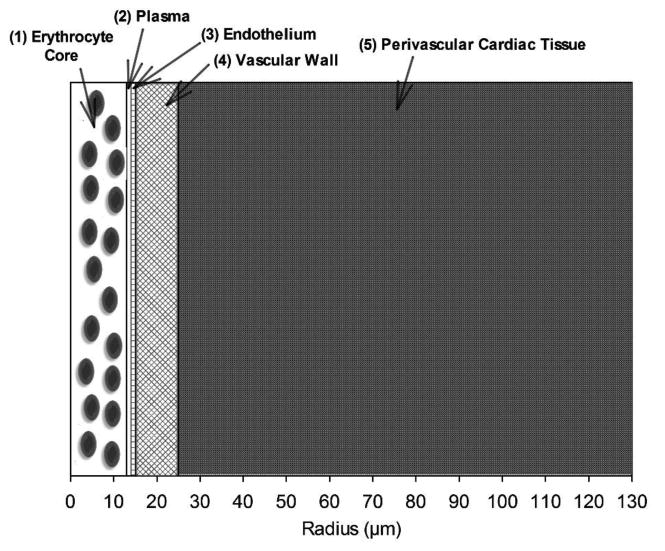
Schematic diagram of a radially concentric cylindrical model of a small cardiac arteriole and perivascular tissue. The layer radii are: (1) 13 μm, (2) 1 μm, (3) 1 μm, (4) 10 μm, and (5) 105 μm.
Concentration gradients were assumed to be axisymmetric and axial diffusion was assumed to be negligible. Blood flow was assumed to be well-developed and laminar. At steady state, the governing mass transport equation therefore simplifies to Eq. (1):
| (1) |
where D, C, and R are the diffusivity, concentration, and reactions of the species, i, corresponding to NO and O2. The axial flow velocity vz is given by Poiseuille flow. Table 1 lists the dimensions and physical constants in the model.
Table 1. Physical Parameters and Rate Constants.
| Parameter | Value(s) | References |
|---|---|---|
| Vessel geometry | ||
| Length | 300 μm | (Buerk et al., 2003) |
| Vessel Diameter | 28 μm | (Buerk et al., 2003) |
| Blood (0 < r < r1) | 13 μm | (Buerk et al., 2003) |
| Plasma (r1 < r < r2) | 1 μm | (Buerk et al., 2003) |
| Endothelium (r2 < r < r3) | 1 μm | (Buerk et al., 2003) |
| Vascular Wall (r3 < r < r4) | 10 μm | (Buerk et al., 2003) |
| Tissue (r4 < r < r5) | 105 μm | (Buerk et al., 2003) |
| Diffusion coefficients | ||
| NO | 3300 μm2 s-1 | (Buerk et al., 2003) |
| O2 | 2800 μm2 s1 | (Buerk et al., 2003) |
| Oxygen transport | ||
| Solubility coefficient | 1.34 μM Torr1 | (Buerk et al., 2003) |
| Myoglobin P | 2.31 Torr | (Curtis et al., 2012) |
| Blood PO2 | 0.1-120 Torr | See text |
| NO scavenging | ||
| Hemoglobin (λb) | 382.5 s-1 | (Buerk, 2009; Carlsen and Comroe, 1958) |
| Tissue (λt) | 5 s-1 | (Buerk, 2009) |
| MbO2 (λMb) | 3.7 × 107 M-1 s-1 | (Flögel et al., 2010) |
| Maximum O2 consumption: Qmax | ||
| Vascular wall | 1 μM s-1 | (Buerk, 2009) |
| Tissue | 20 μM s-1 | (Buerk, 2009) |
| eNOS production | ||
| Maximum RNOmax | 385.07 μM s-1 | See text |
| K for O -dependent production | 4.7 Torr | (Buerk et al., 2003) |
| Shear equation term (τref) | 24 dyne cm-2 | (Chen et al., 2011) |
| Blood flow | ||
| Centerline vmax | 10-1000 μm s-1 | (Buerk, 2009) |
| Dynamic viscosity (μ) | 2.3243 mPa s | (Buerk et al., 2011) |
| Myocardium | ||
| Nitrite | 20 μM | (Faassen et al., 2010; Hendgen-Cotta et al., 2008) |
| pH | 5.5-6.85 | (Buerk et al., 2003; Zweier et al., 2010, 1999) |
| Mb | 0.5 mM | (Masuda et al., 2008; Sylven et al., 1984) |
| Myoglobin nitrite reduction rate (kMb) | 12.4 M-1 s-1 | (Shiva et al., 2007) |
2.2. Nitric oxide
Coupled NO-O2 transport models have been developed previously by us (Chen, 2006; Lamkin-Kennard et al., 2004; Liu et al., 2016) and others (Kavdia et al., 2002; Tsoukias, 2008) and our approach here is similar. Endothelial nitric oxide synthase (eNOS) in layer 3 was assumed to uniformly produce NO with a linear shear stress dependence and O2-dependent Michaelis-Menten kinetics (Eq. 2). All other substrates and co-factors were assumed to be in abundant supply.
| (2) |
where Km is the Michaelis-Menten constant and τref is the reference value used by Chen et al. (Chen et al., 2011) RNOmax was calculated so that at baseline flow (centerline velocity = 1000 μm s-1), NO production matches the solely O2-dependent Michaelis-Menten values used by Buerk (Buerk, 2001). Shear stress was calculated as a function of flow using the laminar pipe flow wall shear stress equation (Eq. 3):
| (3) |
where μb - the dynamic viscosity of blood - was calculated using the modified microvessel viscosity equation by Pries et al. (Pries et al., 1994) NO is scavenged by hemoglobin in layer 1 and by oxymyoglobin in layer 5 with first order rate constants λb and λMb, respectively. The Hb scavenging constant λb has been estimated around 100-750 s-1 at a systemic hematocrit of 45%, and our chosen value of 382.5 s-1 falls within these values (Buerk, 2009; Carlsen and Comroe, 1958; Tsoukias and Popel, 2003). In layers 4 and 5, NO reacts with soluble guanylyl cyclase (sGC) with first order scavenging rate constant λt. NO is also produced by enzymatic myoglobin reduction of endogenous cardiac nitrite (described below).
2.3. Oxygen
RBC-rich layer 1 was assumed to have uniform, constant O2 delivery which was independently varied from severe anoxia to typical normoxia levels (0.1-120 Torr). The lower end of this range represents levels of oxygen representative of extreme ischemic events such as acute myocardial infarction, where flow and O2 are nearly entirely restricted, compared to more mild physiological hypoxia levels around 20 Torr (Faassen et al., 2010). The amount of O2 consumed by eNOS is twice that of NO produced. Oxygen consumption in the vascular wall and tissue was reversibly inhibited through inhibition of cytochrome oxidase by NO in a modified Michaelis-Menten equation (Eqs. 4 and 5) as modeled by Buerk (Buerk, 2001).
| (4) |
| (5) |
where Km* is assumed to be 1 Torr for these simulations.
2.4. Myoglobin and Mb nitrite reductase
Myoglobin and nitrite were assumed to be uniformly present and constant in the perivascular cardiac tissue (layer 5), and quantified with experimentally reported values for healthy adult humans (Table 1). Cardiac tissue nitrite levels vary depending on factors such as dietary habits and lifestyle (e.g. tobacco consumption, exercise), but have been shown to be around 20 μM (Faassen et al., 2010; Hendgen-Cotta et al., 2008). The oxymyoglobin saturation curve was defined using Eq. (6):
| (6) |
where P50 is 2.31 Torr.
Oxygenated myoglobin (MbO2) scavenges NO as in Eq. (7) with second order rate constant λMb (Ascenzi and Brunori, 2001).
| (7) |
Deoxygenated myoglobin reduces nitrite as a function of nitrite and deoxyMb concentration with second order rate constant kMb. This rate constant has been experimentally reported in the range of 12.0-12.86 M-1 s-1 at 37°C and pH = 7.4, changing ten-fold with each unit pH change (Gladwin and Kim-Shapiro, 2008; Hendgen-Cotta et al., 2010; Shiva et al., 2007). This nitrite reduction behavior is quantified as Eq. (8):
| (8) |
where kMb is 12.4 M-1 s-1. Postischemic cardiac tissue pH levels have been measured as low as 5.5, from a normal pH around 7-7.4 (Buerk et al., 2003; Zweier et al., 2010, 1999). All other secondary reactions between myoglobin, NO, and other nitroso species are assumed to be negligible as experimental evidence has shown that Eqs. 7 and 8 are the primary reactions affecting functional NO from nitrite reduction (Rassaf et al., 2007; Shiva et al., 2007).
2.5. Boundary conditions
Zero mass fluxes were assumed for all species at the center of the blood vessel (r = 0) and at the outermost boundary of layer 5 (r5) in Eq. (9):
| (9) |
The mass flux of a species exiting one layer is assumed to equal the mass flux entering the adjacent layer e.g. for the lumen boundary (r2) in Eq. (10):
| (10) |
2.6. Numerical methods
The model was solved in COMSOL v5.1 at steady state with relative accuracy of 1 × 10-6 to predict concentration profiles across the arteriolar domain (0 < r < 130 μm) and averaged across the smooth muscle cell (SMC) region (layer 4) and perivascular tissue region (layer 5) at the arteriole midline (z = 150 μm). Simulations were performed with and without tissue nitrite present in layer 5 while varying blood PO2, blood flow rates, and tissue pH to determine the effect of deoxyMb nitrite reductase in producing NO during ischemic conditions. Simulations done at zero blood oxygen and flow were not feasible due to solver limitations, so 0.1 Torr and 1% centerline vmax (10 μm s-1) were used to simulate almost complete ischemic blockage.
3. Results
Ischemia leads to a deficiency in blood PO2, which monotonically decreases tissue PO2, averaged across layer 5 (Fig. 2A). A decrease in blood PO from normoxic levels (around 120 Torr) to near anoxic levels (0.1 Torr) leads to a decrease in average tissue PO2 from 50.1 to 0.1 Torr, respectively (Fig. 2A inset). This change in tissue PO2 affects the saturation percentage of perivascular tissue myoglobin as in Eq. 6 (Fig. 2B).
Figure 2.
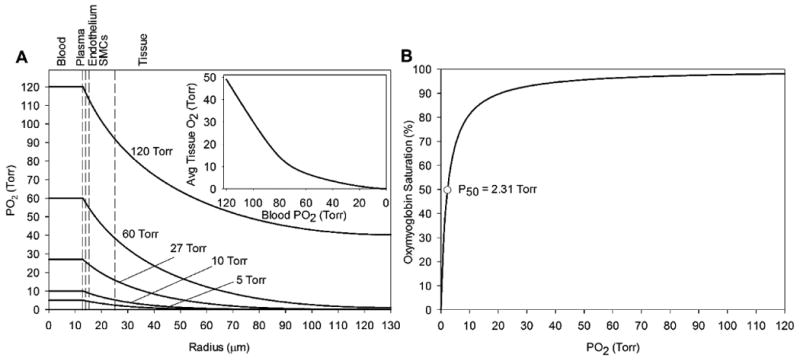
Effect of blood PO2 changes on (A) PO2 profiles across the arteriolar radius and (B) myoglobin oxygen binding. (Inset) Average tissue PO2 on the perivascular tissue region (25 < r < 130 μm) for various blood PO2 concentrations. The vertical dashed lines mark boundaries between the five radial model layers labeled above the graph.
Without the contribution of tissue nitrite, ischemic decreases in blood PO2 (and tissue PO2) lead to decreases in NO across the computational domain (Fig. 3). A decrease in blood PO2 from 120 to 1 Torr leads to a decrease in peak NO at the endothelium-SMC boundary (r = 15 μm) from 45.6 to 8.28 nM, respectively. Tissue NO is <5 nM at all blood oxygen values without tissue nitrite due to strong myoglobin scavenging.
Figure 3.
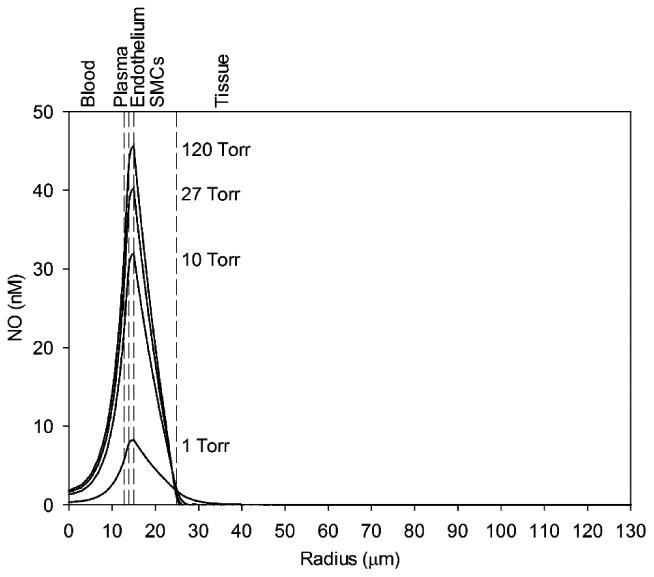
NO concentration profiles across the computational domain for various blood PO2 concentrations (indicated next to the corresponding peak) without cardiac nitrite. The vertical dashed lines mark boundaries between the five radial model layers labeled above the graph.
Ischemic decreases in flow also lead to changes in NO availability. Without tissue nitrite reduction, decreasing flow and blood PO2 down to near zero values (0.1 Torr, 1% flow) both lead to a decrease in averaged SMC NO in the 15 < r < 25 μm region (Fig. 4). Changes in tissue pH from ischemia have little effect on the shear- and O2- dependent NO produced by eNOS in layer 3.
Figure 4.

Average NO concentration in the smooth muscle cell region (15 < r < 25 μm) as a function of blood PO2 concentrations without cardiac nitrite for various blood flow rates. Blood flow is varied from maximum centerline velocities of 1000 to 10 μm s-1. There is no significant difference between different tissue pH values on these data.
With the added effect of tissue nitrite reduction by deoxyMb, there is a significant increase in NO, increasing in effect at lower blood PO2 (1 vs. 10 Torr) and lower pH (Figs. 5A and 5B, respectively). The increase in NO is greatest in magnitude at the tissue and SMC regions but relatively minor in the blood and plasma layers. The effect is significant at the SMC region for all flow values and stronger at the lower limit of pH = 5.5 (Fig. 6B) versus pH = 6.85 (Fig. 6A). There was little effect on the PO2 profiles with the incorporation of tissue nitrite reduction; variations in average tissue PO2 between no nitrite and nitrite for all flow and pH cases were <1 Torr (not shown).
Figure 5.
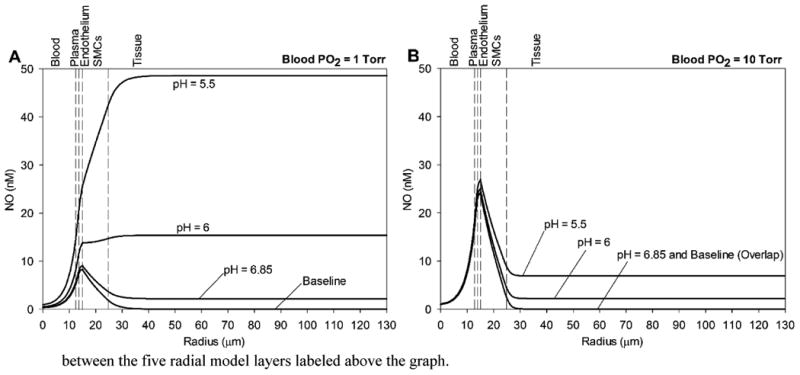
Effect of deoxyMb nitrite reductase on NO concentrations across the computational domain at low blood PO2 values (A) 1 Torr and (B) 10 Torr at pH values of 6.85, 6, and 5.5. The baseline NO profile is the condition with zero tissue nitrite (as in Fig. 3). The vertical dashed lines mark boundaries between the five radial model layers labeled above the graph.
Figure 6.
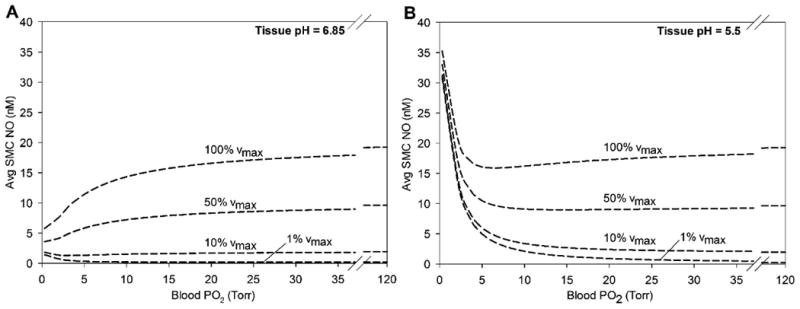
Effect of myocardial tissue nitrite reductase on average NO concentration in the smooth muscle cell region (15 < r < 25 μm) as a function of blood PO2 for tissue pH values of (A) 6.85 and (B) 5.5. The higher section of blood PO2 is omitted because the change in average SMC NO between blood PO2 values of 35 and 120 Torr is <1 nM.
The absolute elevation of both average SMC NO (Fig. 7A) and average tissue NO (Fig. 7B) by tissue nitrite is consistent at different flow values, but relatively higher at low flow because of the decrease in NO availability at lower shear stress. The elevation in NO is also higher in the tissue region than the SMC region, and higher as tissue pH decreases. Table 2 summarizes the changes in NO between the no nitrite and nitrite cases for various ischemic conditions at baseline flow.
Figure 7.
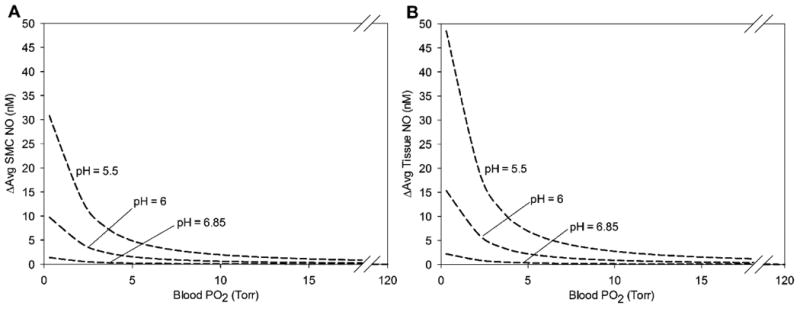
Effect of myocardial deoxyMb nitrite reductase on average NO concentration as a function of blood PO2 compared against the baseline no nitrite values in (A) the smooth muscle region (15 < r < 25 μm) and (B) the perivascular tissue region (25 < r < 130 μm) for tissue pH values of 6.85, 6, and 5.5. There is no significant difference in these data at different flow values. The higher section of blood PO2 is omitted because the average SMC NO between blood PO2 values of 20 and 120 Torr is <1 nM.
Table 2. Effect of Deoxymyoglobin Nitrite Reduction on NO at SMC and Tissue Regions.
| Tissue pH | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Blood PO2 (Torr) | Baseline NO (nM) | 5.5 | 6 | 6.85 | ||
| Avg. SMC | 1 | 4.41 | ΔNO (nM) | +30.9 | +9.75 | +1.38 |
| 5 | 11.2 | +4.85 | +1.53 | +0.22 | ||
| 10 | 14.3 | +1.95 | +0.62 | +0.09 | ||
| 27 | 17.3 | +0.47 | +0.15 | +0.02 | ||
|
| ||||||
| Avg. Tissue | 1 | 0.02 | ΔNO (nM) | +48.5 | +15.4 | +2.19 |
| 5 | 0.01 | +6.90 | +1.73 | +0.31 | ||
| 10 | 0* | +2.72 | +0.87 | +0.12 | ||
| 27 | 0* | +0.65 | +0.19 | +0.03 | ||
NO concentrations below 1 pM are reported as zero
A sensitivity analysis for the model parameters was conducted on six key parameters to examine the effect of small variations (± 5%) on average NO concentration in the SMC region with the effect of tissue nitrite reductase at blood PO2 = 1 Torr and baseline flow and pH (Fig. 8). The sensitivity was relatively low for dynamic viscosity of blood μb and cardiac Mb concentration. There was a significant positive correlation for tissue nitrite concentration, deoxyMb nitrite reductase rate constant kMb, and the Mb P50. There was a significant negative correlation with oxyMb NO scavenging rate λMb.
Figure 8.
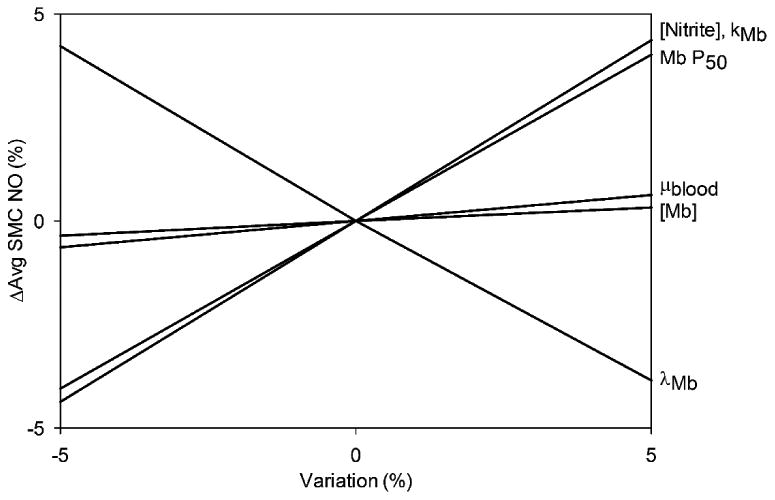
Sensitivity analysis showing the effect of minor variations (± 5%) in model parameters (listed at right) on relative differences in average NO in the smooth muscle cell region (15 < r < 25 μm) for blood PO2 = 1 Torr and baseline flow and pH.
4.1. Discussion
The results of our simulations suggest that in conditions of cardiac ischemia resulting in low blood PO2, low flow, and low pH, reduction of endogenous nitrite by deoxymyoglobin can significantly elevate both tissue and smooth muscle cell NO. Our computational model is the first attempt to explicitly model the interplay between myoglobin scavenging of NO at normoxia and its opposing role of producing NO from tissue nitrite under certain conditions. As blood PO2 falls from normoxic to hypoxic and near anoxic levels (Fig. 2A), tissue PO2 levels can drop to levels inside the range of the P50 of Mb (Fig. 2B), where the concentration of deoxyMb rapidly increases. This in turn leads to a significant increase in deoxyMb nitrite reduction, while simultaneously decreasing the scavenging NO by MbO2.
Similarly, decreases in flow and blood PO2 comparable to levels found during extreme cardiac ischemia, such as during a myocardial infarction, lead to significant shortages in NO production by eNOS (Figs. 3 and 4) (Gladwin and Kim-Shapiro, 2008; Rassaf et al., 2007). At extreme levels of ischemia (1% vmax, 1 Torr), there is almost zero NO across the arteriolar radius when there is no tissue nitrite. These results are consistent with previous mathematical models studying the effect of flow and blood oxygen levels on NO availability without compensatory nitrite reduction pathways (Chen, 2006; Tachtsidis et al., 2011; Tsoukias, 2008). Additionally, the presence of Mb in perivascular cardiac tissue at strongly scavenges NO; there is <1 nM NO in the tissue region at all conditions in our modeled cardiac arteriole at normoxia (Fig. 3) due to the high rate constant of the Mb-NO reaction (Eq. 7). Previous models show a similar reduction in NO in vascular SMC due to myoglobin scavenging with almost complete NO scavenging at the SMC-tissue border, but they did not model any recovery via deoxyMb nitrite reductase (Kavdia, 2006; Kavdia and Popel, 2004). It has been postulated that cardiac muscle Mb serves to protect the region from transient increases in cytosolic NO from nitric oxide synthase in the endothelium, the sarcoplasmic reticulum, and the mitochondria by inactivating excess NO (Blomberg et al., 2004; Flögel et al., 2001). In contrast, Mb will switch to becoming an NO producer at extremely low oxygenation and flow conditions, when O2-dependent eNOS production is limited.
When the effect of deoxyMb nitrite reduction of endogenous tissue nitrite is considered, our simulations predict that low blood flow and PO2 conditions result in significantly elevated NO across the endothelium, SMC, and tissue regions, in increasing order of magnitude (Fig. 5). This NO elevation effect is consistent at different flow values, but relatively more powerful at low flow conditions because of the lower baseline NO from shear-stress-dependent production (Fig. 6). Both blood PO2 and flow rates will decrease during ischemia. Similarly, lowering pH values from normal (6.85) to severely ischemic heart values will significantly increase the elevation of nitrite-derived NO (Fig. 7). The dependence of Mb nitrite reduction on pH has been experimentally shown to be very high (10-fold change in reaction rate for every unit pH change) and another factor in the large elevation of tissue and SMC NO following cardiac ischemia (Shiva et al., 2007; Totzeck et al., 2012a; Trochu et al., 2000). With the combination of very low flow, blood PO2, and tissue pH that could occur during severe ischemia (e.g. 10 μm s-1, 1 Torr, 5.5 tissue pH), NO elevations in blood and tissue can reach the 30-50 nM range, which is comparable to or even surpasses the steady state predictions at normal, non-ischemic conditions (Table 2).
These results are in agreement with experimental evidence showing that nitrite reduction by deoxyMb is a significant producer of NO in cardiac muscle in ischemic and hypoxic conditions (Rassaf et al., 2007; Shiva et al., 2007; Webb et al., 2004). Functionally significant levels (>5 nM) of NO produced by deoxyMb are detected with subendocardium oxygen levels around 2-5 Torr (Shiva et al., 2007), and NO production measured indirectly through NO-heme accumulation was appreciable only with hypoxia and levels of nitrite application similar to our simulation concentrations (20 μM) (Rassaf et al., 2007). Direct reporting of NO generation rates by deoxyMb during ischemia is rare due to experimental difficulties, but estimates from analysis of human and mice Mb are around 1-5 nM s-1 (Totzeck et al., 2012b; Zweier et al., 2010). In addition, Mb nitrite production of NO has been shown to significantly decrease myocardial infarction size and reduce ischemic injury or ischemia/reperfusion injury in animal experiments (Hendgen-Cotta et al., 2008; Shiva et al., 2007; Totzeck et al., 2012b; Webb et al., 2004). It is not exactly understood how this released NO reduces ischemic damage or mitigates ischemia-reperfusion injury. It has been hypothesized that the release of NO by deoxyMb during ischemia could downregulate cardiac energy consumption via reduced contractility and oxygen consumption. Additionally, the released NO can serve to reduce reactive oxygen species generation or inhibit adhesion molecule expression and platelet aggregation (Gladwin and Kim-Shapiro, 2008; Hendgen-Cotta et al., 2008; Lamkin-Kennard et al., 2004; Rassaf et al., 2007). Nitrite-derived NO from deoxyMb has been demonstrated to inhibit cellular respiration in myocardial infarctions, but it is not yet understood exactly how NO escapes autocapture by secondary reactions with myoglobin species such as deoxyMb and metMb (Hendgen-Cotta et al., 2008; Webb et al., 2004). It has been proposed that the proximity of nitrite-derived NO to mitochondria (specifically, cytochrome c oxidase binding to inhibit respiration) allows this NO to escape autocapture (Shiva et al., 2007; Totzeck et al., 2012a), or that in vivo conditions result in scavenging rates of NO by these secondary reactions that are sufficiently low to allow escape (Blomberg et al., 2004; Gladwin and Kim-Shapiro, 2008). This elevation of NO leads to downregulation of cardiac contractile function and energy metabolism (Hendgen-Cotta et al., 2010). This effect, known as short-term hibernation, leads to a dampening of high energy phosphates and oxygen consumption, and can potentially restore myocardial energy balance (Faassen et al., 2010; Heusch et al., 2005). Without an elevation of NO, ischemic conditions would lead to a depletion of NO which could exacerbate the condition by causing (or enhancing) vasoconstriction (Heusch et al., 2005; Trochu et al., 2000).
4.2. Model limitations
There are limitations to our model approach that could affect our predictions of NO bioavailability. We have assumed that the primary reactions involving Mb and NO are the MbO2 scavenging reaction (Eq. 7) and the deoxyMb nitrite reduction reaction (Eq. 8). There are second-order reactions involving NO and other nitrogen oxide species and different states of Mb (oxyMb, metMb, and deoxyMb) that could affect nitrite-NO interactions which we have not included in our model, but should have less significant effects than the deoxyMb nitrite reduction pathway (Eich et al., 1996; Flögel et al., 2010). There is uncertainty regarding the various parameters reported by experimental sources used in this model. Myoglobin concentrations and reaction rates have been primarily estimated in animal models due to experimental limitations in human studies (Rassaf et al., 2007; Totzeck et al., 2012a, 2012b). Human Mb is unique because it has Cys110, which has been shown to form S-nitroso-myoglobin (S-NO-Mb) during nitrite reduction and extend the lifetime of nitrite-derived NO (Rayner et al., 2005; Witting et al., 2001). This pathway has been hypothesized to enhance the transport of NO produced by cardiac Mb but since it has not been fully elucidated, is not present in this model. Since released NO is scavenged by Mb but might be preserved by S-NO-Mb, our model provides a worst case scenario for Mb nitrite reduction during ischemia, and may underestimate the effect in humans. The sensitivity analysis (Fig. 8) shows that tissue nitrite concentration, Mb nitrite reduction reaction rate, MbO2 scavenging of NO, and Mb P50 have a strong influence on the released NO from tissue nitrite. All of these factors impact the O2 saturation of myoglobin, which significantly affects nitrite-released NO.
We have also constructed our model with independent flow and blood PO2 input parameters, but these two conditions would be linked in vivo. We (Chen, 2006; Chen et al., 2011; Lamkin-Kennard et al., 2004) and others (Kavdia et al., 2002; Tsoukias et al., 2004) have examined the dynamics of red blood cell profiles, flow, and blood oxygen in the microcirculation through mathematical modeling, and incorporating these mechanisms is an area of potential future study. Models of oxygen transport in more complex microcirculatory networks, which could more fully characterize the interactions of microvascular hypoxia, blood flow, and O2 transport, have been reviewed by Goldman (Goldman, 2008). However, NO transport and hypoxic nitrite production in these networks has not been as extensively modeled. We have previously studied coupled oxygen and NO transport in paired arteriole-venule (Chen et al., 2007) and branching microcirculation environments (Chen et al., 2011), and plan to incorporate Mb nitrite reduction to extend these models. Infused nitrite into the bloodstream releasing NO has also been shown to have cytoprotective and vasodilatory effects, although the mechanism is not fully understood (Liu et al., 2015; Lundberg et al., 2008). Our model also does not simulate O2 consumption as a function of contractile activity, which has been postulated to be part of the cytoprotective effects of cardiac nitrite reduction (Flögel et al., 2010; Heusch et al., 2005). Recently, low-dosages (physiological levels) of nitrite infused into the bloodstream have been shown to reduce the effects of myocardial infarction, which is a phenomena that could be added to the model for future study (Ingram et al., 2013). In addition, reactive oxygen and nitrogen species have a significant effect on ischemic damage and ischemia/reperfusion injury, and mathematical modeling of the role of Mb nitrite reduction in these complex reactions is another area of potential study (Kavdia, 2006; Powers et al., 2014). There are also numerous other nitrite reductase pathways that have been identified as significantly active under hypoxic or ischemic conditions, and would likely add to the magnitude of the effects predicted here. These pathways include proteins such as xanthine oxidoreductase, cytochrome P450, and aldehyde oxidase (Faassen et al., 2010; Li et al., 2008). These other pathways have been shown to be effective in producing nitrite-derived NO in different organs and muscle vessels of the body depending on their enzyme compositions, including the heart (Zweier et al., 2010). Modeling these other tissue nitrite mechanisms is an area of potential future analysis. However, the deoxyMb nitrite reduction pathway alone has been shown be significant in elevating NO (Totzeck et al., 2012b).
4.3. Conclusion
Our mathematical model illustrates that while cardiac myoglobin serves as a strong scavenger of NO during normal conditions of flow or blood PO2, the deoxyMb nitrite reduction pathway can have a significant physiological effect on NO production during ischemic conditions. This effect is most pronounced at the lowest blood PO2 and tissue pH conditions and in some cases results in SMC and tissue NO above the normoxic levels. NO elevation is consistent at different flow rates, but relatively higher at lower flow due to the decreased baseline NO. Since we have demonstrated that cardiac tissue nitrite-derived NO can be significant, an important area of potential future study is modeling the in vivo observation that nitrite-derived NO can reduce cellular respiration and cardiac tissue oxygen consumption. Nevertheless, our results support experimental observations of the effectiveness of myoglobin nitrite reductase in releasing NO in the cardiac muscle during ischemic events and provide insight into mechanisms by which myocardial nitrite reduces myocardial infarction injury.
Highlights.
In normoxia, oxygenated Mb is a strong scavenger of NO to maintain homeostasis.
During ischemia, deoxygenated Mb can significantly generate NO from tissue nitrite.
NO elevation is highest at the lowest blood PO2 and tissue pH conditions.
This effect is consistent at different flow rates, but relatively higher at low flow.
This released NO could be responsible for mitigating deleterious ischemic effects.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute Grant U01HL116256.
Footnotes
Disclosures: We wish to confirm that there are no conflicts of interest, financial or otherwise, associated with this publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ascenzi P, Brunori M. Myoglobin: a pseudo-enzymatic scavenger of nitric oxide. Biochem Mol Biol Educ. 2001;29:183–185. doi: 10.1111/j.1539-3429.2001.tb00116.x. [DOI] [Google Scholar]
- Baker JE, Su J, Fu X, Hsu A, Gross GJ, Tweddell JS, Hogg N. Nitrite confers protection against myocardial infarction: Role of xanthine oxidoreductase, NADPH oxidase and KATP channels. J Mol Cell Cardiol. 2007;43:437–444. doi: 10.1016/j.yjmcc.2007.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg LM, Blomberg MRA, Siegbahn PEM. A theoretical study of myoglobin working as a nitric oxide scavenger. J Biol Inorg Chem. 2004;9:923–35. doi: 10.1007/s00775-004-0585-5. [DOI] [PubMed] [Google Scholar]
- Buerk DG. Mathematical modeling of the interaction between oxygen, nitric oxide and superoxide. Adv Exp Med Biol. 2009;645:7–12. doi: 10.1007/978-0-387-85998-9_2. [DOI] [PubMed] [Google Scholar]
- Buerk DG. Can We Model Nitric Oxide Biotransport? A Survey of Mathematical Models for a Simple Diatomic Molecule with Surprisingly Complex Biological Activities. Annu Rev Biomed Eng. 2001;3:109–143. doi: 10.1146/annurev.bioeng.3.1.109. [DOI] [PubMed] [Google Scholar]
- Buerk DG, Barbee KA, Jaron D. Modeling O2-dependent effects of nitrite reductase activity in blood and tissue on coupled NO and O2 transport around arterioles. Adv Exp Med Biol. 2011;701:271–276. doi: 10.1007/978-1-4419-7756-4. [DOI] [PubMed] [Google Scholar]
- Buerk DG, Barbee KA, Jaron D. Nitric oxide signaling in the microcirculation. Crit Rev Biomed Eng. 2011;39:397–433. doi: 10.1615/critrevbiomedeng.v39.i5.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerk DG, Lamkin-Kennard K, Jaron D. Modeling the influence of superoxide dismutase on superoxide and nitric oxide interactions, including reversible inhibition of oxygen consumption. Free Radic Biol Med. 2003;34:1488–1503. doi: 10.1016/S0891-5849(03)00178-3. [DOI] [PubMed] [Google Scholar]
- Carlsen E, Comroe JH. The rate of uptake of carbon monoxide and of nitric oxide by normal human erythrocytes and experimentally produced spherocytes. J Gen Physiol. 1958;42:83–107. doi: 10.1085/jgp.42.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. The influence of radial RBC distribution, blood velocity profiles, and glycocalyx on coupled NO/O2 transport. J Appl Physiol. 2006;100:482–492. doi: 10.1152/japplphysiol.00633.2005. [DOI] [PubMed] [Google Scholar]
- Chen X, Buerk DG, Barbee KA, Jaron D. A model of NO/O2 transport in capillary-perfused tissue containing an arteriole and venule pair. Ann Biomed Eng. 2007;35:517–529. doi: 10.1007/s10439-006-9236-z. [DOI] [PubMed] [Google Scholar]
- Chen X, Buerk DG, Barbee KA, Kirby P, Jaron D. 3D network model of NO transport in tissue. Med Biol Eng Comput. 2011;49:633–647. doi: 10.1007/s11517-011-0758-7. [DOI] [PubMed] [Google Scholar]
- Curtis E, Hsu LL, Noguchi AC, Geary L, Shiva S. Oxygen regulates tissue nitrite metabolism. Antioxid Redox Signal. 2012;17:951–61. doi: 10.1089/ars.2011.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eich RF, Li T, Lemon DD, Doherty DH, Curry SR, Aitken JF, Mathews AJ, Johnson KA, Smith RD, Phillips GN, Olson JS. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry. 1996;35:6976–83. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- Faassen EE Van, Bahrami S, Feelisch M, Hogg N, Kelm M, Kozlov AV, Li H, Lundberg JO, Mason R, Rassaf T, Samouilov A, Slama-schwok A, Shiva S. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2010;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flögel U, Fago A, Rassaf T. Keeping the heart in balance: the functional interactions of myoglobin with nitrogen oxides. J Exp Biol. 2010;213:2726–33. doi: 10.1242/jeb.041681. [DOI] [PubMed] [Google Scholar]
- Flögel U, Merx MW, Godecke A, Decking UKM, Schrader J, Go A, Decking UKM. Myoglobin: a scavenger of bioactive NO. Proc Natl Acad Sci USA. 2001;98:735–740. doi: 10.1073/pnas.011460298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin MT, Kim-Shapiro DB. The functional nitrite reductase activity of the heme-globins. Blood. 2008;112:2636–2647. doi: 10.1182/blood-2008-01-115261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D. Theoretical models of microvascular oxygen transport to tissue. Microcirculation. 2008;15:795–811. doi: 10.1080/10739680801938289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendgen-Cotta UB, Kelm M, Rassaf T. A highlight of myoglobin diversity: The nitrite reductase activity during myocardial ischemia-reperfusion. Nitric Oxide. 2010;22:75–82. doi: 10.1016/j.niox.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2008;105:10256–61. doi: 10.1073/pnas.0801336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol. 2005;288:H984–99. doi: 10.1152/ajpheart.01109.2004. [DOI] [PubMed] [Google Scholar]
- Ingram TE, Fraser AG, Bleasdale RA, Ellins EA, Margulescu AD, Halcox JP, James PE. Low-dose sodium nitrite attenuates myocardial ischemia and vascular ischemia-reperfusion injury in human models. J Am Coll Cardiol. 2013;61:2534–2541. doi: 10.1016/j.jacc.2013.03.050. [DOI] [PubMed] [Google Scholar]
- Kavdia M. A Computational Model for Free Radicals Transport in the Microcirculation. Antioxid Redox Signal. 2006;8:1103–1111. doi: 10.1089/ars.2006.8.1103. [DOI] [PubMed] [Google Scholar]
- Kavdia M, Popel AS. Contribution of nNOS- and eNOS-derived NO to microvascular smooth muscle NO exposure. J Appl Physiol. 2004;97:293–301. doi: 10.1152/japplphysiol.00049.2004. [DOI] [PubMed] [Google Scholar]
- Kavdia M, Tsoukias NM, Popel AAS. Model of nitric oxide diffusion in an arteriole: impact of hemoglobin-based blood substitutes. Am J Physiol Heart Circ Physiol. 2002;282:2245–2253. doi: 10.1152/ajpheart.00972.2001. [DOI] [PubMed] [Google Scholar]
- Kim-Shapiro DB, Gladwin MT. Mechanisms of nitrite bioactivation. Nitric Oxide. 2014;38:58–68. doi: 10.1016/j.niox.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkin-Kennard KA, Buerk DG, Jaron D. Interactions between NO and O2 in the microcirculation: a mathematical analysis. Microvasc Res. 2004;68:38–50. doi: 10.1016/j.mvr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Lamkin-Kennard Ka, Jaron D, Buerk DG. Impact of the Fåhraeus effect on NO and O2 biotransport: a computer model. Microcirculation. 2004;11:337–349. doi: 10.1080/10739680490437496. [DOI] [PubMed] [Google Scholar]
- Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: Critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem. 2008;283:17855–17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wajih N, Liu X, Basu S, Janes J, Marvel M, Keggi C, Helms CC, Lee AN, Belanger AM, Diz DI, Laurienti PJ, Caudell DL, Wang J, Gladwin MT, Kim-Shapiro DB. Mechanisms of human erythrocytic bioactivation of nitrite. J Biol Chem. 2015;290:1281–94. doi: 10.1074/jbc.M114.609222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Buerk DG, Barbee KA, Jaron D. A mathematical model for the role of N2O3 in enhancing nitric oxide bioavailability following nitrite infusion. Nitric Oxide. 2016;60:1–9. doi: 10.1016/j.niox.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466-c2. [DOI] [PubMed] [Google Scholar]
- Masuda K, Truscott K, Lin PC, Kreutzer U, Chung Y, Sriram R, Jue T. Determination of myoglobin concentration in blood-perfused tissue. Eur J Appl Physiol. 2008;104:41–48. doi: 10.1007/s00421-008-0775-x. [DOI] [PubMed] [Google Scholar]
- Ormerod JOM, Ashrafian H, Maher AR, Arif S, Steeples V, Born GVR, Egginton S, Feelisch M, Watkins H, Frenneaux MP. The role of vascular myoglobin in nitrite-mediated blood vessel relaxation. Cardiovasc Res. 2011;89:560–565. doi: 10.1093/cvr/cvq299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen CL, Faggiano S, Helbo S, Gesser H, Fago A. Roles of nitric oxide, nitrite and myoglobin on myocardial efficiency in trout (Oncorhynchus mykiss) and goldfish (Carassius auratus): implications for hypoxia tolerance. J Exp Biol. 2010;213:2755–62. doi: 10.1242/jeb.041624. [DOI] [PubMed] [Google Scholar]
- Powers SK, Ji LL, Kavazis AN, Jackson MJ. Reactive Oxygen Species: Impact on Skeletal Muscle. Crit Rev Biomed Eng. 2014;1:941–69. doi: 10.1002/cphy.c100054.REACTIVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pries AR, Secomb TW, Gessner T, Sperandio MB, Gross JF, Gaehtgens P. Resistance to blood flow in microvessels in vivo. Circ Res. 1994;75:904–15. doi: 10.1161/01.RES.75.5.904. [DOI] [PubMed] [Google Scholar]
- Rassaf T, Ferdinandy P, Schulz R. Nitrite in organ protection. Br J Pharmacol. 2014;171:1–11. doi: 10.1111/bph.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Flögel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Nitrite reductase function of deoxymyoglobin: Oxygen sensor and regulator of cardiac energetics and function. Circ Res. 2007;100:1749–1754. doi: 10.1161/CIRCRESAHA.107.152488. [DOI] [PubMed] [Google Scholar]
- Rayner BS, Wu BJ, Raftery M, Stocker R, Witting PK. Human S-nitroso oxymyoglobin is a store of vasoactive nitric oxide. J Biol Chem. 2005;280:9985–9993. doi: 10.1074/jbc.M410564200. [DOI] [PubMed] [Google Scholar]
- Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654–61. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- Sylven C, Jansson E, Book K. Myoglobin content in human skeletal muscle and myocardium: relation to fibre size and oxidative capacity. Cardiovasc Res. 1984;18:443–446. doi: 10.1093/cvr/18.7.443. [DOI] [PubMed] [Google Scholar]
- Tachtsidis I, Tisdall MM, Pritchard C, Leung TS, Elwell CE, Smith M. Oxygen Transport to Tissue XXXII. Oxyg. Transp. to tissue. Advances in Experimental Medicine and Biology. 2011;701:9–14. doi: 10.1007/978-1-4419-7756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiravanti E, Samouilov A, Zweier JL. Nitrosyl-heme complexes are formed in the ischemic heart: Evidence of nitrite-derived nitric oxide formation, storage, and signaling in post-ischemic tissues. J Biol Chem. 2004;279:11065–11073. doi: 10.1074/jbc.M311908200. [DOI] [PubMed] [Google Scholar]
- Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ, Semmler D, Shiva S, Williams D, Kipar A, Gladwin MT, Schrader J, Kelm M, Cossins AR, Rassaf T. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation. 2012a;126:325–34. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totzeck M, Hendgen-Cotta UB, Rammos C, Petrescu AM, Meyer C, Balzer J, Kelm M, Rassaf T. Assessment of the functional diversity of human myoglobin. Nitric Oxide. 2012b;26:211–6. doi: 10.1016/j.niox.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Trochu JN, Bouhour JB, Kaley G, Hintze TH. Role of Endothelium-Derived Nitric Oxide in the Regulation of Cardiac Oxygen Metabolism : Implications in Health and Disease. Circ Res. 2000;87:1108–1117. doi: 10.1161/01.RES.87.12.1108. [DOI] [PubMed] [Google Scholar]
- Tsoukias NM. Nitric oxide bioavailability in the microcirculation: insights from mathematical models. Microcirculation. 2008;15:813–834. doi: 10.1080/10739680802010070. [DOI] [PubMed] [Google Scholar]
- Tsoukias NM, Kavdia M, Popel AS. A theoretical model of nitric oxide transport in arterioles: frequency- vs. amplitude-dependent control of cGMP formation. Am J Physiol Heart Circ Physiol. 2004;286:H1043–56. doi: 10.1152/ajpheart.00525.2003. [DOI] [PubMed] [Google Scholar]
- Tsoukias NM, Popel AS. A model of nitric oxide capillary exchange. Microcirculation. 2003;10:479–95. doi: 10.1038/sj.mn.7800210. [DOI] [PubMed] [Google Scholar]
- Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci U S A. 2004;101:13683–8. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witting PK, Douglas DJ, Mauk AG. Reaction of human myoglobin and nitric oxide. Heme iron or protein sulfhydryl (s) nitrosation dependence on the absence or presence of oxygen. J Biol Chem. 2001;276:3991–8. doi: 10.1074/jbc.M005758200. [DOI] [PubMed] [Google Scholar]
- Wunderlich C, Flögel U, Gödecke A, Heger J, Schrader J. Acute Inhibition of Myoglobin Impairs Contractility and Energy State of iNOS-Overexpressing Hearts. Circ Res. 2003;92:1352–1358. doi: 10.1161/01.RES.0000079026.70629.E5. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Li H, Samouilov A, Liu X. Mechanisms of nitrite reduction to nitric oxide in the heart and vessel wall. Nitric Oxide. 2010;22:83–90. doi: 10.1016/j.niox.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL, Samouilov A, Kuppusamy P. Non-enzymatic nitric oxide synthesis in biological systems. Biochim Biophys Acta. 1999;1411:250–262. doi: 10.1016/S0005-2728(99)00018-3. [DOI] [PubMed] [Google Scholar]