

Abstract
Relatively few catalytic systems are able to control the stereochemistry of electronically excited organic intermediates. Here we report the discovery that a chiral Lewis acid complex can catalyze triplet energy transfer from an electronically excited photosensitizer. This strategy is applied to asymmetric [2+2] photocycloadditions of 2′-hydroxychalcones using tris(bipyridyl) ruthenium(II) as a sensitizer. A variety of electrochemical, computational, and spectroscopic data rule out substrate activation via photoinduced electron transfer and instead support a mechanism in which Lewis acid coordination dramatically lowers the triplet energy of the chalcone substrate. We expect that this approach will enable chemists to more broadly apply their detailed understanding of chiral Lewis acid catalysis to stereocontrol in reactions of electronically excited states.
The ability to control the stereochemistry of organic reactions is a defining characteristic of contemporary synthetic chemistry. Because access to structurally well-defined organic molecules is important for progress in fields of research ranging from drug discovery to materials science, numerous chiral catalysts have been developed to control the enantioselectivity of a wide range of mechanistically diverse organic transformations. Photochemical reactions, however, have long proven to be challenging to conduct in an enantioselective fashion, particularly using substoichiometric stereocontrolling catalysts.(1,2) This represents a fundamental gap in synthetic methodology because the reactivity of electronically excited organic molecules is distinctive and often impossible to replicate using non-photochemical techniques.(3,4)
Recently, a renewed interest in synthetic applications of photoinduced electron transfer(5) has resulted in the development of several strategies for performing highly enantioselective photocatalytic reactions. In general, these protocols have involved the photochemical generation of reactive intermediates using an organic or transition metal photoredox catalyst; subsequently, a second chiral Lewis acid,(6) Brønsted acid,(7,8) or organic catalyst(9,10) then influences the stereochemistry of ensuing reactions and affords highly enantioenriched products. The key bond-forming events in all of these photoredox reactions necessarily occur via photogenerated reactive intermediates in their electronic ground-state configurations. Although photoactivation provides a particularly convenient strategy for the generation of the open-shell radical and radical ion intermediates involved in these reactions, similar reactive intermediates can be generated via a variety of non-photochemical means.(11) Stereochemical control over the distinctive reactivity of excited-state organic intermediates remains a largely unsolved challenge.
The difficulty of controlling electronically excited organic molecules using exogenous chiral catalysts is commonly attributed to their characteristically short lifetimes, which arise from fast unimolecular vibrational and emissive deactivation pathways that are not available to ground-state reactive intermediates. Highly enantioselective catalytic reactions of photoexcited organic substrates have only recently been reported, using either chiral hydrogen-bonding organic sensitizers(12–16) or, more recently, chiral Lewis acid catalysts.(17–20)
We were particularly intrigued by this latter advance because Lewis acid catalysis offers a mature, well-understood platform for asymmetric synthesis. The ability to generalize a chiral Lewis acid strategy for excited state photoreactions would offer a powerful tool for organic photochemistry. Bach’s seminal results in this area have demonstrated that chiral oxazaborolidines can be used to mediate asymmetric [2+2] photocycloadditions of enones (17–20) (Figure 1A). These impressive proof-of-principle studies nevertheless rely upon subtle changes in the absorption properties of the substrate that arise upon coordination to a Lewis acid.(21) High levels of enantiomeric excess (ee) are only feasible if the catalyst–substrate complex can be photoexcited preferentially over the unbound, achiral substrate in order to minimize the participation of racemic background reactions. Because this approach requires a delicate balancing of the singlet excited state properties of both the free substrate and the chiral Lewis acid-substrate complex,(22) the range of substrates that have been shown to provide high enantioselectivies to date using Bach’s chiral oxazaborolidine strategy has proven to be somewhat narrowly constrained to cyclic enones.
Figure 1. Enantioselective Catalysis Involving Excited State Organic Intermediates.
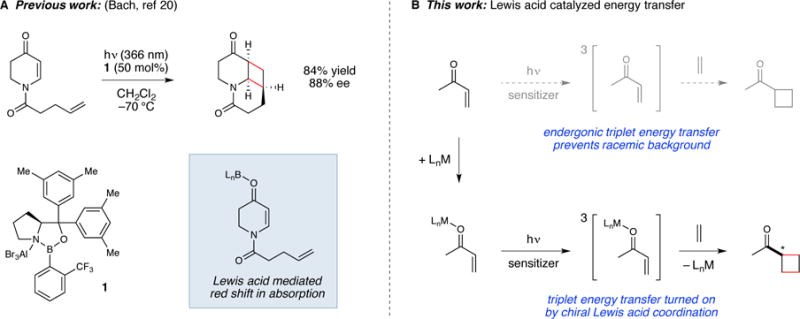
(A) Prior example of enantioselective Lewis acid catalyzed photoreaction involving direct photoexcitation of a chiral catalyst–substrate complex. (B) Design plan for Lewis acid catalysis of a triplet sensitization reaction.
We envisioned an alternative approach in which a chiral Lewis acid would serve as a catalyst for triplet energy transfer from a racemic triplet sensitizer. The coordination of Lewis acids to enones can significantly perturb the energy of their singlet excited states, a phenomenon first documented by Frederick Lewis several decades ago.(23,24) The effect of Lewis acid coordination on triplet energies, to the best of our knowledge, has not been the subject of similarly detailed exploration. We hypothesized that if coordination of a Lewis acid to an enone could produce a bathochromic shift in the energy of its singlet excited state, the same interaction might exert an analogous effect on its triplet state energy as well. If so, it should be possible to design a system in which triplet energy transfer from an appropriate sensitizer would become thermodynamically feasible only when a substrate enone is bound to a chiral Lewis acid co-catalyst (Figure 1B).
In previous work, we have shown that chiral Lewis acids are effective in controlling the stereochemical course of reactions initiated by photoinduced electron transfer from an electronically excited Ru*(bpy)32+ photocatalyst.(6) Among the most substantial benefits of this two-catalyst strategy is the ability to modify the structure of a chiral Lewis acid for optimal stereocontrol without deleteriously impacting the desirable photophysical properties of the sensitizer. This flexibility opens photochemical synthesis to the wide range of privileged Lewis acid scaffolds known to be highly effective in other enantioselective reactions, and we have subsequently shown that the same strategy can be applied to a number of other reactions exploiting chiral Lewis acid catalyzed photoredox activation.(25,26) We propose that this concept, when applied to the more difficult problem of catalytic energy transfer, can have similarly broad ramifications.
The feasibility of Lewis acid catalyzed triplet energy transfer was initially tested by examining the effect of exogenous Lewis acids on photocatalytic reactions of 2′-hydroxychalcone (2) (Table 1). This substrate provides an ideal model system because the 2′-hydroxyaryl ketone moiety is predisposed towards association with a variety of Lewis acidic metals and because the photochemical properties of 2 have been thoroughly characterized. In particular, 2 has been experimentally shown to possess a triplet state 54 kcal/mol higher in energy than its closed-shell singlet ground state.(27) This triplet energy (ET) is well outside of the range that should be reasonably accessible using Ru(bpy)32+ as a triplet sensitizer (ET = 46 kcal/mol).(28) Indeed, an experiment in which 2 and diene 3 were irradiated with visible light in the presence of Ru(bpy)3(PF6)2 showed minimal evidence of productive photoreaction (entry 1). On the other hand, when the same reaction was conducted in the presence of various oxophilic Lewis acid additives, we were delighted to observe the formation of [2+2] cycloadduct 4 in good to moderate yields (entries 2–5). Control experiments indicated that the cycloaddition occurs only in the presence of both the photocatalyst and Lewis acid co-catalyst, as no cycloadduct is observed when Ru(bpy)32+ is omitted or when the reaction is conducted in the dark (entries 6 and 7). Given the strict dependence on the presence of a Lewis acid, we speculated that a highly enantioselective reaction might result from the use of a chiral Lewis acid complex. After investigating several classes of chiral ligands, we were pleased to find that Sc(III) PyBox complexes generally worked well as chiral Lewis acid co-catalysts, and tBu-PyBox (7) in particular provided promising ee (entries 8–10). Subsequent optimization of standard reaction variables(29) afforded our optimized conditions (entry 11), which produced cyclobutane 4 in high yields and excellent enantioselectivity. Finally, we conducted an experiment using a 2:1 mixture of E and Z 2′-hydroxychalcone as the substrate and found that the geometry of the alkene had no discernable impact on the stereoselectivity of the reaction, consistent with a stepwise triplet cycloaddition (entry 12).
Table 1.
Optimization and control studies for Lewis acid catalyzed enantioselective cycloadditions of 2′-hydroxychalcone.
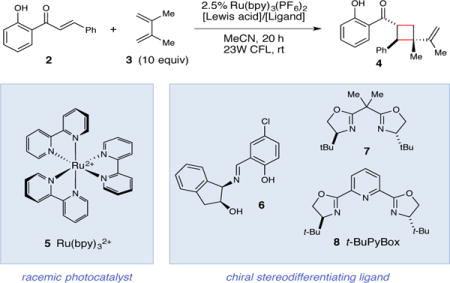
| |||||
|---|---|---|---|---|---|
| Entry | Lewis acid | Ligand | Note | Yield* | ee† |
| 1 | none | none | – | <5% | – |
| 2 | Al(OTf)3 (1 equiv) | none | – | 70% | – |
| 3 | Ti(OEt)4 (1 equiv) | none | – | 29% | – |
| 4 | Y(OTf)3 (1 equiv) | none | – | 15% | – |
| 5 | Sc(OTf)3 (1 equiv) | none | – | 88% | – |
| 6 | Sc(OTf)3 (1 equiv) | none | No Ru(bpy)3(PF6)2 | <5% | – |
| 7 | Sc(OTf)3 (1 equiv) | none | No light | <5% | – |
| 8 | Sc(OTf)3 (20 mol%) | 6 (30 mol%) | – | 52% | 0% |
| 9 | Sc(OTf)3 (20 mol%) | 7 (30 mol%) | – | 64% | 0% |
| 10 | Sc(OTf)3 (20 mol%) | 8 (30 mol%) | – | 75% | 74% |
| 11 | Sc(OTf)3 (10 mol%) | 8 (15 mol%) | 3:1 i-PrOAc:MeCN | 80% | 93% |
| 12‡ | Sc(OTf)3 (10 mol%) | 8 (15 mol%) | 3:1 i-PrOAc:MeCN | 89% | 93% |
Yields were determined by 1H NMR using phenanthrene as a calibrated internal standard.
Enantiomer ratios were determined by chiral SFC analysis. rt, room temperature.
Reaction conducted using a 2:1 ratio of Z and E 2′-hydroxychalcone as the starting substrate.
This reaction represents a rare example of an enantioselective catalytic intermolecular photocycloaddition(16) and, to the best of our knowledge, the only example of a highly enantioselective cycloaddition of an acyclic excited-state enone. Studies exploring the synthetic scope of this transformation are outlined in Figure 2.(30) Variation of the β-aryl moiety of the chalcone is well tolerated. Substrates with ortho, meta, and para substituents on this ring provide excellent ee and good yields (9–11). Chalcones with electron-rich β-aryl groups participate in an uncatalyzed background cycloaddition under CFL irradiation, and therefore require irradiation with a monochromatic blue LED (λmax = 450 nm) light source for optimal enantioselectivities (12–14).31 Heteroaryl groups are also easily accommodated using this method (14). Substitution of the hydroxyphenyl moiety is tolerated (15–16) so long as this substituent does not interfere with the putative binding site for the chiral Lewis acid. Finally, a range of substituted and unsubstituted dienes also provide high ee’s and good yields (17–19), and unsymmetrical dienes offer good levels of regioselectivity (18–19).
Figure 2. Scope of the enantioselective catalytic [2+2] cycloaddition of 2′-hydroxychalcones.
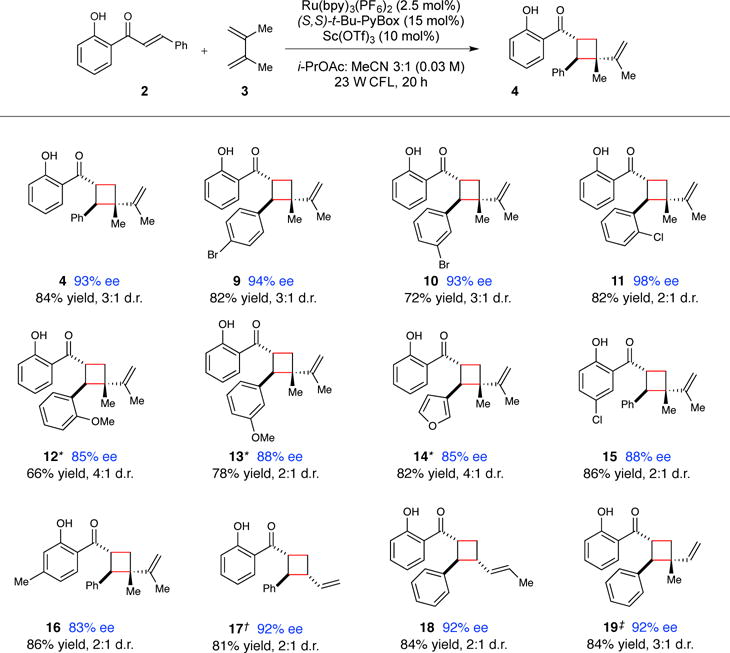
Data reflect the averaged isolated yields from two reproducible experiments. Diastereomer ratios were determined by 1H NMR analysis of the unpurified reaction mixture. Enantiomer ratios were determined using chiral SFC or HPLC analysis. See supplementary material for details. * Irradiation was conducted using a blue LED lamp instead of a 23 W CFL bulb, 2 h irradiation time. † 40 h irradiation time. ‡Isolated as a 6:1 mixture of regioisomers.
Several lines of evidence support the hypothesis that this transformation involves substrate activation via energy transfer rather than photoredox catalysis. First, Porco has reported that redox activation of 2′-hydroxychalcones affords [4+2] cycloadducts upon reaction with dienes,(32,33) rather than the [2+2] cycloadducts produced in the photocatalytic protocol we have developed. No traces of similar [4+2] Diels–Alder products were observed under our optimized photocatalytic conditions. We also independently investigated the possibility that this reactivity was the result of redox activation. Electrochemical characterization rules out photocatalytic one-electron oxidation as a mechanism of activation: no oxidation features below +1.60 V vs saturated calomel electrode (SCE) are observable in the cyclic voltammogram of 2, either in the presence or absence of Sc(OTf)3, indicating that Ru*(bpy)32+ is too weak an oxidant (*Eox = +0.77 V) to activate the chalcone substrate this way. We did, however, observe a reduction feature with a half-wave potential of –1.2 V (vs. SCE) in the cyclic voltammogram of 2 that shifts to –0.47 V in the presence of Sc(OTf)3. Although photoreduction of the Sc•2 complex by Ru*(bpy)32+ (*Ered = –0.81 V vs. SCE) cannot be ruled out on the basis of these electrochemical data, we found that a variety of substantially less reducing Ru photocatalysts also successfully mediate this cycloaddition. For example, in an experiment replacing Ru(bpy)3(PF6)2 with its much more electron-deficient analogue Ru(deeb)3(PF6)2 (20), from which photoreduction of the Sc•2 complex would be endergonic (*Ered = –0.42 V),(34) we nevertheless observed formation of cycloadduct 2 in 67% yield and similar enantioselectivity (Figure 3A).
Figure 3. Differentiation of electron transfer and energy transfer pathways.
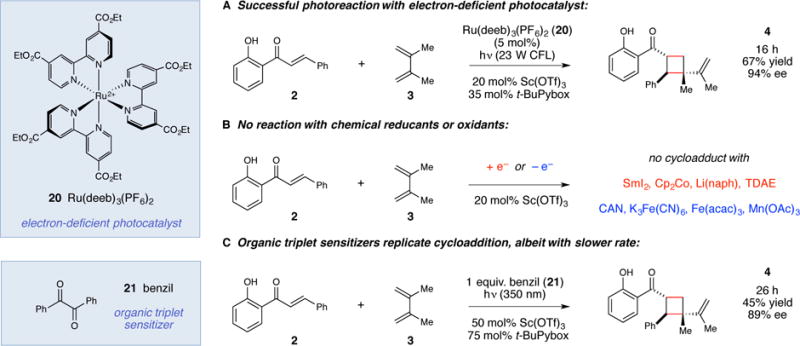
(A) Successful photocycloaddition using an electron-deficient photocatalyst rules out a mechanism involving initial enone photoreduction. (B) Experiments using chemical redox reagents fail to produce [2+2] cycloadducts. (C) UV-activated triplet sensitizer replicates reactivity with similar ee. For full details see the Supplementary Materials.
Consistent with these results, attempts to replicate the [2+2] reaction using Sc(OTf)3 in combination with a range of standard chemical one-electron reductants failed to produce any [2+2] products (Figure 3B). Similar experiments using chemical one-electron oxidants also did not afford any photocycloadducts. On the other hand, if an energy transfer process is indeed relevant to this Lewis acid catalyzed cycloaddition, it should be feasible to promote the reaction using alternative triplet sensitizers. Indeed, when the cycloaddition of 2′-hydroxychalcone 2 and diene 3 was performed under 350 nm irradiation in the presence of benzil (21, ET = ~54 kcal/mol),(35) we observed modest yields of the desired [2+2] cycloaddition product but comparable enantioselectivity to the Ru(bpy)3(PF6)2 catalyzed reaction (Figure 3C). Because of the high sensitivity of asymmetric catalysis to changes in mechanism,(36) we interpret this result as strong corroborating evidence for Lewis acid catalyzed triplet sensitization as the operative pathway.
Finally, we investigated the hypothesis that the Lewis acid co-catalyst in this energy transfer process serves to lower the triplet energy of the hydroxychalcone substrate. The S0–T1 gap for free 2 was computationally investigated (B3LYP/6-311+G(2d,p)). These calculations gave a triplet energy of 51 kcal/mol (Figure 4A), in reasonably good agreement with the reported experimental value of 54 kcal/mol.(27) The analogous computation on the Sc(III) complex of 2, however, suggested that the energy of the lowest optimized triplet state would be dramatically lowered to 32 kcal/mol.(37) This 20 kcal/mol bathochromic shift in triplet energy upon coordination to a Lewis acid would lie easily within a range where triplet energy transfer from Ru*(bpy)32+ would be exergonic, in good accord with our design plan. The magnitude of the effect suggested by calculation, however, was surprisingly large. To validate these computational results, we next investigated the emissive properties of 2 at near-IR wavelengths corresponding to the predicted triplet energy. In the absence of Sc(OTf)3, there was no observable emission signal at wavelengths longer than 800 nm. However, when the emission study was conducted in the presence of added Sc(OTf)3, we observed a feature at 876 nm, corresponding to an excited state energy of 33 kcal/mol, in excellent agreement with the computational prediction (Figure 4B). Moreover, the emission is partially quenched in the presence of oxygen, consistent with emission from a triplet state.
Figure 4. Computational and experimental evidence for a Lewis acid promoted decrease in triplet energy.
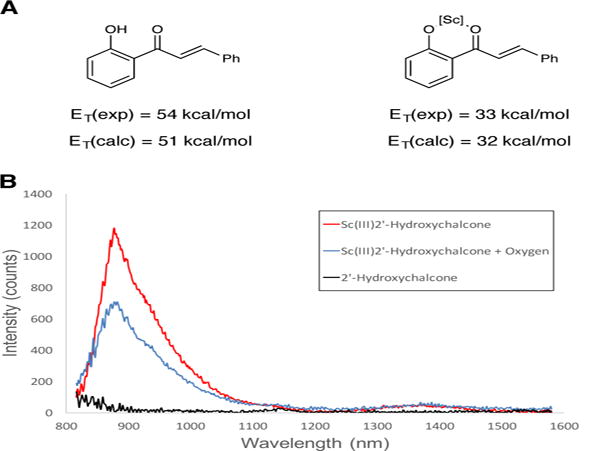
(A) Experimental and calculated S0–T1 gaps for 2′-hydroxychalcone 2 and its Sc(III) complex. (B) Experimental near-IR emission data for 2′-hydroxychalcone 2 in the absence (black) and presence (red) of Sc(OTf)3. The emission is partially quenched in the presence of oxygen (blue).
Collectively, these studies reveal a previously unrecognized effect of Lewis acid coordination on the excited states of organic substrates. We have found that complexation of 2′-hydroxychalcones with Sc(III) results in a dramatic decrease in the energy of the triplet state. Current work in our laboratory is aimed at investigating the applicability of this strategy to other Lewis basic organic substrates and other transformations, which we hope will enable a flexible and robust strategy for catalytic enantiocontrol in a broad range of organic triplet-state reactions.
Supplementary Material
Acknowledgments
We gratefully acknowledge experimental assistance from Prof. Michael Arnold and Michael Shea in obtaining near-IR luminescence spectra. We also thank Prof. Jennifer Schomaker and Minsoo Ju for access to chiral HPLC instrumentation. Experimental details for all studies reported in this paper are described in the Supplementary Materials. Funding for this project was provided by the NIH (GM09888). The computational resources were supported in part by NSF (CHE-0840494). T.B acknowledges support from an NIH Chemical Biology Interface Training grant (T32 GM008505). T.B. conceived of this project, conducted the optimization studies described in the Supplementary Materials, and performed the mechanistic studies. Z.M. collected the data reported in Table 1 and Figure 2 and conducted the cyclic voltammetry studies. D.B. and T.B. performed the computational studies. I.G. collected and analyzed X-ray crystallographic data, which are available free of charge from the Cambridge Crystallographic Data Centre under accession number CCDC 1507886.
Footnotes
All authors contributed to the writing and editing of the manuscript.
SUPPLEMENTARY MATERIALS
Materials and Methods
References (37–51)
Spectral and Chromatographic Data
References and Notes
- 1.Inoue Y. Asymmetric Photochemical Reactions in Solution. Chem Rev. 1992;92:741–770. [Google Scholar]
- 2.Brimioulle R, Lenhart D, Maturi MM, Bach T. Enantioselective Catalysis of Photochemical Reactions. Angew Chem Int Ed. 2015;54:3872–3890. doi: 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem Rev. 2008;108:1052–1103. doi: 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]
- 4.Bach T, Hehn JP. Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew Chem Int Ed. 2011;50:1000–1046. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- 5.Prier CK, Rankic DA, MacMillan DWC. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du J, Skubi KL, Schultz DM, Yoon TP. A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science. 2014;344:392–396. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J Am Chem Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- 8.Uraguchi D, Kinoshita N, Kizu T, Ooi T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J Am Chem Soc. 2015;137:13768–13771. doi: 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]
- 9.Nicewicz D, MacMillan DWC. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science. 2008;322:77–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiRocco DA, Rovis T. Catalytic Asymmetric Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. J Am Chem Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studer A, Curran DP. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew Chem Int Ed. 2015;55:58–102. doi: 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- 12.Muller C, Bauer A, Bach T. Light-Driven Enantioselective Organocatalysis. Angew Chem Int Ed. 2009;48:6640–6642. doi: 10.1002/anie.200901603. [DOI] [PubMed] [Google Scholar]
- 13.Alonso R, Bach T. A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2+2] Photocycloaddition Reactions Induced by Visible Light. Angew Chem Int Ed. 2014;53:4368–4371. doi: 10.1002/anie.201310997. [DOI] [PubMed] [Google Scholar]
- 14.Maturi M, Bach T. Enantioselective Catalysis of the Intermolecular [2+2] Photocycloaddition between 2-Pyridones and Acetylenedicarboxylates. Angew Chem Int Ed. 2014;53:7661–7664. doi: 10.1002/anie.201403885. [DOI] [PubMed] [Google Scholar]
- 15.Vallavoju N, Selvakumar S, Jockusch S, Sibi MP, Sivaguru J. Enantioselective Organophotocatalysis Mediated by Atropisomeric Thiourea Derivatives. Angew Chem Int Ed. 2014;53:5604–5608. doi: 10.1002/anie.201310940. [DOI] [PubMed] [Google Scholar]
- 16.Tröster A, Alonso R, Bauer A, Bach T. Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H) Quinolones Induced by Visible Light Irradiation. J Am Chem Soc. 2016;138:7808–7811. doi: 10.1021/jacs.6b03221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo H, Herdtweck E, Bach T. Enantioselective Lewis Acid Catalysis in Intramolecular [2+2] Photocycloaddition Reactions of Coumarins. Angew, Chem Int Ed. 2010;49:7782–7785. doi: 10.1002/anie.201003619. [DOI] [PubMed] [Google Scholar]
- 18.Brimioulle R, Bach T. [2+2] Photocycloaddition of 3-Alkenyloxy-2-cycloalkenones: Enantioselective Lewis Acid Catalysis and Ring Expansion. Angew Chem Int Ed. 2014;53:12921–12924. doi: 10.1002/anie.201407832. [DOI] [PubMed] [Google Scholar]
- 19.Brimioulle R, Guo H, Bach T. Enantioselective Intramolecular [2+2] Photocycloaddition Reactions of 4-Substituted Coumarins Catalyzed by a Chiral Lewis Acid. Chem Eur J. 2012;18:7552–7560. doi: 10.1002/chem.201104032. [DOI] [PubMed] [Google Scholar]
- 20.Brimioulle R, Bach T. Enantioselective Lewis Acid Catalysis of Intramolecular Enone Photocycloaddition Reactions. Science. 2013;342:840–843. doi: 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]
- 21.Brimioulle R, Bauer A, Bach T. Enantioselective Lewis Acid Catalysis in Intramolecular [2+2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J Am Chem Soc. 2015;137:5170–5176. doi: 10.1021/jacs.5b01740. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Cao X, Chen X, Fang W, Dolg M. Regulatory Mechanism of the Enantioselective Intramolecular Enone [2+2] Photocycloaddition Reaction Mediated by a Chiral Lewis Acid Catalyst Containing Heavy Atoms. Angew Chem Int Ed. 2015;54:14295–14298. doi: 10.1002/anie.201505931. [DOI] [PubMed] [Google Scholar]
- 23.Lewis FD, Howard DK, Oxman JD. Lewis Acid Catalysis of Coumarin Photodimerization. J Am Chem Soc. 1983;105:3344–3345. [Google Scholar]
- 24.Lewis FD, Barancyk SV. Lewis Acid Catalysis of Photochemical Reactions. 8. Photodimerization and Cross-Cycloaddition of Coumarin. J Am Chem Soc. 1989;111:8653–8661. [Google Scholar]
- 25.Ruiz Espelt L, McPherson IS, Wiensch EM, Yoon TP. Enantioselective Conjugate Additions of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J Am Chem Soc. 2015;137:2452–2455. doi: 10.1021/ja512746q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amador AG, Sherbrook EM, Yoon TP. Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J Am Chem Soc. 2016;138:4722–4725. doi: 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norikane Y, Itoh H, Arai T. Photochemistry of 2′-Hydroxychalcone. One-way Cis-Trans Photoisomerization Induced by Adiabatic Intramolecular Hydrogen Atom Transfer. J Phys Chem A. 2002;106:2766–2776. [Google Scholar]
- 28.Kalyanasundaram K. Photophysics, Photochemistry and Solar Energy Conversion with Tris(bipyridyl)ruthenium(II) and Its Analogues. Coord Chem Rev. 1982;46:159–244. [Google Scholar]
- 29.See Supplementary Material for more details.
- 30.The absolute configuration of the cycloadducts was determined by Riley oxidation of compound 9 to a crystalline derivative, which was unambiguously confirmed by single-crystal X-ray crystallography. See Supplementary Material for details.
- 31.The nature of this background process is currently poorly understood, and is the subject of ongoing studies.
- 32.Cong H, Ledbetter D, Rowe GT, Caradonna JP, Porco JA. Electron Transfer-Initiated Diels– Alder Cycloadditions of 2′-Hydroxychalcones. J Am Chem Soc. 2008;130:9214–9215. doi: 10.1021/ja803094u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cong H, Becker C, Elliott S, Grinstaff MW, Porco JA. Silver Nanoparticle-Catalyzed Diels– Alder Cycloadditions of 2′-Hydroxychalcones. J Am Chem Soc. 2010;132:7514–7518. doi: 10.1021/ja102482b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliott CM, Freitag RA, Blaney DD. Electrochemistry, Spectroelectrochemistry, and Photochemistry of a Series of New Covalently Linked Tris(2,2-bipyridine)ruthenium(II)/Diquat Complexes. J Am Chem Soc. 1985;107:4647–4655. [Google Scholar]
- 35.Herkstroeter WG, Lamola AA, Hammond GS. Mechanisms of Photochemical Reactions in Solution. XXVIII. Values of Triplet Excitation Energies of Selected Sensitizers. J Am Chem Soc. 1964;86:4537–4540. [Google Scholar]
- 36.Jacobsen EN, Zhang W, Guier ML. Electronic Tuning of Asymmetric Catalysts. J Am Chem Soc. 1991;113:6704–6706. [Google Scholar]
- 37.See Supplementary Material for details on these computational studies.
- 38.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 39.Cornejo A, Fraile JM, García JI, Gil MJ, Martínez-Merino V, Mayoral JA, Pires E, Villalba I. An efficient and general one-pot method for the synthesis of chiral bis(oxazoline) and pyridine bis(oxazoline) ligands. Synlett. 2005:2321–2324. [Google Scholar]
- 40.Ischay MA, Lu Z, Yoon TP. [2+2] Cycloadditions by Oxidative Visible Light Photocatalysis. J Am Chem Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minatti A, Zheng X, Buchwald SL. Synthesis of Chiral 3-Substituted Indanones via an Enantioselective Reductive-Heck Reaction. J Org Chem. 2007;72:9253–9258. doi: 10.1021/jo701741y. [DOI] [PubMed] [Google Scholar]
- 42.Furstner A, Gastner T. Total Synthesis of Cristatic Acid. Org Lett. 2000;2:2467–2470. doi: 10.1021/ol0061236. [DOI] [PubMed] [Google Scholar]
- 43.Bruker-AXS. APEX3. Madison; Wisconsin, USA: 2015. Version 2015.9-0. [Google Scholar]
- 44.Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J Appl Cryst. 2015;48:3–10. doi: 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheldrick GM. XPREP. Georg-August-Universität Göttingen; Göttingen, Germany: 2013b. Version 2013/1. [Google Scholar]
- 46.Sheldrick GM. The SHELX homepage. 2013a http://shelx.uni-ac.gwdg.de/SHELX/
- 47.Sheldrick GM. SHELXT– Integrated space-group and crystal-structure determination. Acta Cryst A. 2015a;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheldrick GM. Crystal structure refinement with SHELXL. Acta Cryst C. 2015b;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr. 2009;42:339–341. [Google Scholar]
- 50.Guzei IA, Programs IA. Gn. University of Wisconsin-Madison; Madison, Wisconsin, USA: 2007–2013. [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Jr, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox JD. Gaussian 09, Revision D.01. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.