

Abstract
The recently rapid increase of obesity and type 2 diabetes mellitus has caused great burden to our society. A positive association between type 2 diabetes and risk of colorectal cancer has been reported by increasing epidemiological studies. The molecular mechanism of this connection remains elusive. However, type 2 diabetes may result in abnormal carbohydrate and lipid metabolism, high levels of circulating insulin, insulin growth factor-1, and adipocytokines, as well as chronic inflammation. All these factors could lead to the alteration of energy sensing pathways such as the AMP activated kinase (PRKA), mechanistic (mammalian) target of rapamycin (mTOR), SIRT1, and autophagy signaling pathways. The resulted impaired SIRT1 and autophagy signaling pathway could increase the risk of gene mutation and cancer genesis by decreasing genetic stability and DNA mismatch repair. The dysregulated mTOR and PRKA pathway could remodel cell metabolism during the growth and metastasis of cancer in order for the cancer cell to survive the unfavorable microenvironment such as hypoxia and low blood supply. Moreover, these pathways may coupling metabolic and epigenetic alterations that is central to oncogenic transformation. Further researches including molecular pathologic epidemiologic studies are warranted to better address the precise links between these two important diseases.
Keywords: PRKA, Biomarker, Carcinoma, Colon, Energy balance, Molecular pathologic epidemiology
1. Preface
Type 2 diabetes mellitus (hereafter, referred to as “type 2 diabetes”) and colorectal cancer are both common diseases that cause great burden to our society. Moreover, convincing evidence indicates that type 2 diabetes is associated with an elevated risk for colorectal cancer [1, 2]. Epidemiological studies have also proved that in colorectal cancer patients, diabetic patients have a worse prognosis and higher mortality than those without diabetes. Type 2 diabetes and colorectal cancer also share many risk factors such as aging, obesity, sedentary life style, diet, and smoking. Therefore, studying the underlying mechanisms would provide insights into the prevention and treatment strategy of cancer in diabetic patients.
2. Epidemiological association between type 2 diabetes and colorectal cancer
The International Diabetes Federation (IDF) reports that the number of diabetic patients will increase from 415 million in 2015 to 642 million by 2040 [3]; while the World Health Organization (WHO) -projected global incidence of cancer will increase from 14 million in 2012 to 22 million in 2032 [4]. In parallel, a strong association between colorectal cancer and diabetes has been supported by increasing epidemiological studies.
2.1 Type 2 diabetes increases the risk of colorectal cancer
Compared to the general population, type 2 diabetes increases the risk of colorectal cancer by up to three times [5, 6]. In the meta-analysis by Sun and Yu [7], which included 11 case–control and 28 cohort studies, diabetes is associated with a 29% increase of colorectal cancer [95% confidence index (CI): 1.23–1.35]. This effects persist even after adjustment for body mass index (BMI) and physical activity in the Nurses’ Health Study and Cancer Prevention Study [relative risk (RR) = 1.49 and 1.24, respectively] [8]. Even in young patients at 40–49 years, a dramatic increase of colorectal adenomas in diabetic patients has been reported [odds ratio (OR) = 3.1; 95% CI: 1.5–6.4]: these patients have similar incidence of adenomas as those who are 50–59 years old without diabetes [9] . Recently, the link between DM and colorectal cancer has been recognized in American Diabetes Association (ADA) guideline which suggests that patients with diabetes should receive age- and sex-appropriate cancer screenings and should also reduce their cancer risk factors which are modifiable such as smoking, obesity, and physical inactivity [10].
2.2 Type 2 diabetes aggravates the prognosis of colorectal cancer
Type 2 diabetes also increases the cancer specific mortality and overall mortality of colorectal cancer [11, 12]. In a meta-analysis including 26 observational studies in patients with colorectal cancer [13] it is found that diabetic patients have a 17% increased risk of all-cause mortality and a 12% increased risk of cancer-specific mortality, compared to those without diabetes. The disease-free survival and overall survival of the CRC patients were also associated with blood glucose levels in a multivariate analysis [14]. Results from one pathologic analysis has found that patients with diabetes have deeper tumor invasion, greater lymphovascular invasion, and higher TNM staging compared to those without diabetes [15]. Finally, one genetic research has found that some genes, which are associated with metabolic syndrome such as APOA2, APOC1, APOD and ABCA1, predict disease-free survival in stage II colorectal cancer patients [16]. Therefore, genetic factors as well as environmental factors contribute to the poorer prognosis of colorectal cancer in diabetic patients compared to the general population.
3. Energy sensing pathways and colorectal cancer
The molecular mechanism by which type 2 diabetes increases the incidence or aggravates the prognosis of colorectal cancer is not clear. However, nutrient-sensing pathways which coupling energy metabolism to signals of cell growth and survival are often dysregulated in diabetes, and are important contributors to cancers for diabetic patients.
3.1 PRKA (AMPK) signaling pathway
PRKA is the primary energy sensor in the cell, and actively participates in the control of cell metabolism and several cellular processes including proliferation and apoptosis [17, 18]. In mammals, PRKA is activated by increased AMP : ATP or ADP : ATP ratios. Any cellular process that either decreases ATP levels or increases AMP concentrations (such as fasting or exercise) can activate PRKA, whereas high nutrition (such as in obesity or diabetes) can inactivate PRKA (Figure 1).
Figure 1.
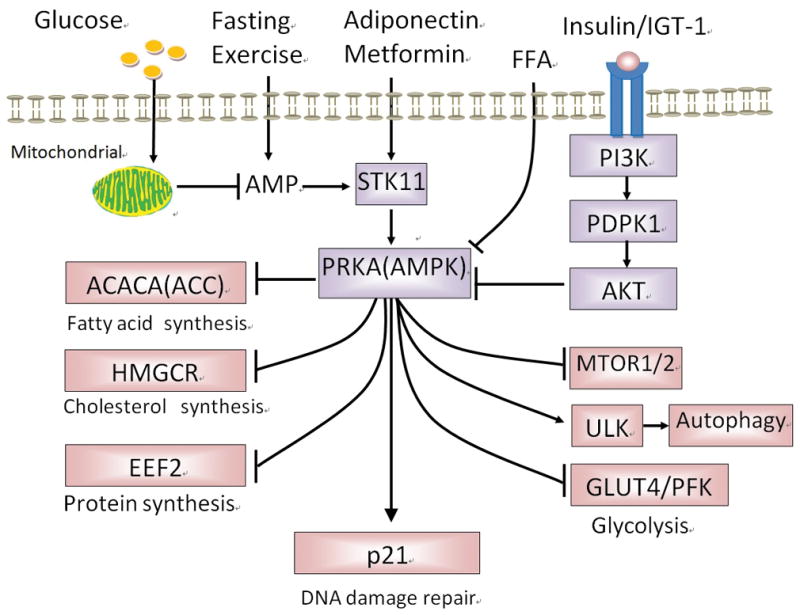
Roles of AMPK in cancer in type 2 diabetes. Under nutrition deficiency state, such as fasting, exercise, glucose deprivation, increased AMP activates AMPK which then switches off the synthesis of protein, fatty acid and cholesterol, but switches on the glycolysis and autophagy in order for the cell to survive in energy insufficient. However, in type 2 diabetes, AMPK is inhibited by high levels of plasma glucose, insulin and IGF1, which may promote anabolism to meet the increasing demanding of cancer cell growth. CaMMKK:calmodulin-dependent protein kinase 1 alpha; AICAR: 5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide; GLUT4: Glucose transporter 4; PFK-2: phosphofructokinase-2; eEF-2: eukaryotic translation elongation factor 2; STK11 (LKB1: Liver kinase B1); AMPK: AMP-activated protein kinase; ACC: acetyl-CoA carboxylase; IGF1: insulin growth factor-1; PI3K: phosphatidylinositol 3-kinase; PDPK1: phosphoinositide-dependent kinase-1; AKT: proteinase kinase B; mTOR: mechanistic (mammalian) target of rapamycin; ULK: Unc-51-Like Kinases; HMG-CoA:3-hydroxy-3-methylglutaryl-coenzyme A
PRKA activation promotes a metabolic and proliferative phenotype unfavorable to cancer cell growth, and as such is thought to negatively impact tumorigenesis [19]. There is a close association between PRKA and colorectal cancer. Germ-line mutations in STK11 (PRKA activator) are linked to Peutz-Jegher syndrome characterized by predisposition to gastrointestinal polyps and malignant tumors [20, 21], where colorectal cancer is the most common cancer [11]. In another study, it is found that low phosphorylated PRKA levels are associated with worse overall survival in a cohort of metastatic colorectal cancer patients treated with chemotherapy plus bevacizumab [22].
3.2 mTOR signaling pathway
mTOR is another important energy sensor in cell which may function dependently and also independently on PRKA. mTOR has two distinct multimolecular complexes differ in composition, substrate specificity, and mechanism of growth regulation. mTORC1 is a nutrient and energy sensor responding to changes in growth factor, amino acid and nutrient levels. Growth factors (such as insulin and IGF-1) are strong activators of mTORC1 acting through AKT signaling. AKT phosphorylates TSC2 and dissociates it from TSC1, which then activate mTORC1 [23, 24]. High nutrition will lead to mTORC1 activation because of the loss of PRKA inhibition [25]. Withdrawal of glucose, amino acids, or oxygen leads to rapid suppression of mTORC1 activity through PRKA dependent mechanism. Amino acids, particularly leucine and arginine, also activate mTORC1 through unknown mechanism [26]. Figure 2.
Figure 2.

Roles of mTOR signaling pathway in cancer in type 2 diabetes. mTOR signaling pathway is a key energy sensing pathway. In type 2 diabetes, the high level of nutrition (glucose, FFA and amino acid) as well as increased level of ROS, adipokines, insulin and IGF1, all contribute to the activation of mTOR. The activated mTOR will then inhibit autophagy, promote the synthesis of protein and fatty acid, up-regulate glycolysis, and stimulate angiogenesis, which is helpful for the cancer cell growth and metastasis. GLUT1: Glucose transporter 1; PRKA: AMPK, AMP-activated protein kinase; IGF1: insulin growth factor-1; PI3K: Phosphatidylinositol 3-kinase; PDPK1: phosphoinositide-dependent kinase-1; AKT: proteinase kinase B; mTOR: mammalian target of rapamycin; ULK: Unc-51-Like kinases; IKBKB: IKKkappa, I kappa B kinase; ROS: Reactive oxygen species; HIF1A: HIF-1alpha, Hypoxia-inducible factor-1 alpha; VEGFA: VEGF, Vascular endothelial growth factor; LDH: Lactate dehydrogenase; SREBF: SREBP, sterol regulatory element-binding proteins; FASN: Fatty acid synthase.
Strong evidences supporting the tumorigenesis role of mTOR come from the positive associations between familial cancer syndromes and mutations of mTOR regulators such as TSC1-TSC2, STK11 and PTEN [27]. Moreover, sporadic mutation or deregulation of mTOR upstream regulators (such as PI3K, AKT and PTEN) and downstream effectors (such as EIF4EBP1) are frequently reported to be associated with human cancer [28, 29]. High mTORC level, rictor expression and mTORC downstream effectors are also associated with poor prognosis of colorectal cancer patients [30, 31].
3.3 SIRT1 signaling pathway
SIRT1 is another important energy sensing protein which couples the cellular metabolic status to chromatin structure and regulation of gene expression by deacetylating histones, transcription factors, and transcription co-factors, and is associated with numerous cellular signaling pathways that include senescence [32, 33], apoptosis [34], DNA damage repair [35], and autophagy [36]. Upon glucose starvation or other metabolic changes associated with caloric restriction, elevation of NAD+/NADP+ ratio activates SIRT1, which then acts on its downstream targets including: (1) TP53. TP53 is one of the best-defined target proteins of the SIRT1 deacetylase. SIRT1 deacetylates and inactivates TP53, thereby exerting an antiapoptotic effect [37]. (2) ATM and histone deacetylase 1 (HDAC1). SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability [38]. (3) Liver X receptor proteins (LXRP). SIRT1 can also deacetylate and activate LXRP, thereby facilitating cholesterol efflux from the cell [39]. (4) Peroxisome proliferator-activated receptor γ coactivator 1α (PPARGC1A, also PGC-1α). PPARGC1A is another important modulator of metabolic pathways. SIRT1-dependent deacetylation of PPARGC1A enhances its ability to cooperate with transcription factors such as PPARG (peroxisome proliferator-activated receptor γ) which in turn induces expression of genes involved in fatty acid oxidation and gluconeogenesis [40, 41]. (5) HIF1A. SIRT1 deacetylates and inactivates HIF1A, thus inhibits the expression of genes targeted by HIF1A in certain tumors [42]. (6) NFKB1. SIRT1 inactivation leads to G1-phase arrest via NFKB1/CCND1 (cyclin D1) signaling [43].
It is proved that heterozygous deletion of SIRT1 accelerates tumor formation in TP53 +/−knock-out mice [44], and overexpression of the protein reduces intestinal tumor formation in adenomatous polyposis coli (Apc) min/+ mice [45]. In colorectal cancer patients, SIRT1 shows lower nuclear expression in tumor samples than in normal tissue samples; Patients with high nuclear expression of SIRT1 showed better survival and a lower chance of tumor recurrence [46]. In patients with colorectal adenocarcinoma, SIRT1 overexpression is observed in approximately 25% of stage I/II/III tumors but rarely in advanced stage IV tumors and approximately 30% of carcinomas shows lower SIRT1 expression than normal tissues [47]. Moreover, SIRT1 protein expression is gradually decreased during the normal-adenoma-adenocarcinoma-metastasis sequence in colorectal cancers [48]. Figure 3.
Figure 3.

Roles of SIRT1 signaling pathway in cancer in type 2 diabetes. SIRT1 signaling pathway is an important energy sensing pathway bridging energy metabolism to DNA stability. In type 2 diabetes, the high level of glucose and FFA will reduce the level of NAD+/NADH, which then decrease SIRT1 activity. The decreased SIRT1 activity loses its stimulation of fatty acid metabolism and its ability to maintain genomic stability, as well as its inhibition of inflammation and angiogenesis. All of those alterations may initiate the formation and growth of cancer cell. FFA: free fatty acid; PRKA: AMPK, AMP-activated protein kinase; mTOR: mammalian target of rapamycin; HIF1A: Hypoxia-inducible factor; VEGFA: VEGF, Vascular endothelial growth factor; NAD: Nicotinamide adenine dinucleotide; SIRT1: NAD-dependent deacetylase sirtuin-1; STK11: LKB1, liver kinase B1; PPARGC1A: PCG-1alpha, peroxisome proliferator-activated receptor gamma coactivator-1 α; LXR: liver X receptor; NFKB1: NF-kappaB, Nuclear factor-KappaB; HDAC1: histone deacetylase 1
However, an increase in SIRT1 expression has also been reported in many types of cancer including colorectal cancer [49], and high level of SIRT1 serves as an independent marker of poor prognosis for cancer patients [50]. Recently, it is found that intestine-specific SIRT1 heterozygous mice have enhanced intestinal tumor formation, whereas intestine-specific SIRT1 homozygous knockout mice have reduced development of colon cancer; Furthermore, SIRT1 reduction, but not deletion, is associated with human colorectal tumors, and colorectal cancer patients with low protein expression of SIRT1 have a poor prognosis [51]. Therefore, SIRT1 may function as a double-edged sword in colorectal cancer, but the consensus is that SIRT1 is a DNA stability guardian and possesses anticancer effects during the initiation of cancer.
3.4 Autophagy-related pathway
Autophagy is a cell process whereby proteins and organelles are recycled via lysosomal degradation to maintain cellular homoeostasis; however, autophagy can also be induced in response to various stresses including nutrient and oxygen deprivation [52]. Therefore, autophagy functions as another type of energy sensor. A major regulator of autophagy is mTOR pathway. The down regulation of mTOR under cellular stress triggers autophagy [53], and mTOR inhibitors, including rapamycin, have also been shown to induce autophagy [54]. The activation of PRKA will also repress mTOR and initiate autophagy [55]. A recent study found that PRKA can directly phosphorylate ULK1, which is required for mitochondrial homeostasis and cell survival during starvation [56]. It is also suggested that amino acid inhibit autophagy by inactivating RAF1/MEK/ERK1/2 pathway [57].
Autophagy has been found to inhibit inflammation [58], mitochondrial dysfunction [59] and genome instability [60] which are known promoters of cancer initiation (Figure 4). Autophagy is important in the switch from normal to malignant colorectal cells. UVRAG is a key autophagy effector and a guardian of chromosomal stability; one recent study discovered that a frame shift mutation leading to shortened form of UVRAG protein, which counteracts most of the tumor-suppressor functions of wild-type UVRAG and promotes tumorigenesis, epithelial-to-mesenchymal transition, and metastasis of colorectal cancer [61, 62]. Other mutations of autophagy related genes (ATG5, ATG2B) were also reported to be associated with colorectal cancer [63, 64]. Besides, BECN1 (Beclin 1) protein expression is found negatively related to liver and distant metastasis of colorectal cancer, and also positively associated with good prognosis in patients with colorectal cancer [65, 66]. Figure 4.
Figure 4.

Roles of Autophagy signaling pathway in cancer in type 2 diabetes. Autophagy has been found to limit inflammation, mitochondrial dysfunction and genome instability which are known promoters of cancer initiation. In type 2 diabetes, the high level of nutrition (glucose, FFA and amino acid) as well as increased level of ROS, insulin and IGF1, all contribute to the inhibition of autophagy, which may play key role in the initiation of cancer of type 2 diabetes. PRKA: AMP-activated protein kinase; IGF1: insulin growth factor-1; PI3K: Phosphatidylinositol 3-kinase; PDPK1: phosphoinositide-dependent kinase-1; AKT: proteinase kinase B; mTOR: mammalian target of rapamycin; ULK: Unc-51-Like kinases; IKBKB: I kappa B kinase; ROS: Reactive oxygen species; HIF1A: Hypoxia-inducible factor; VEGFA: VEGF, Vascular endothelial growth factor; LDH: Lactate dehydrogenase; SREBF: Sterol regulatory element-binding proteins; FASN: Fatty acid synthase; TSC: Tuberous sclerosis complex; BECN1: Beclin 1
However, autophagy has paradoxical and complex functions in cancer development. Increased autophagy was also found correlated with poor prognosis in colorectal cancer. The consensus is that autophagy possesses an anticancer effect at least in the initiation of tumors. At this stage, autophagy promotion may help preventing cancer.
4. Roles of energy sensing pathways in colorectal cancer in type 2 diabetes
As were discussed above, energy sensing pathways play important roles in coupling energy metabolism and DNA stability, and dysfunction of these pathways are closely associated with colorectal cancer. In type 2 diabetes, high level of glucose, FFA, insulin and IGF-1 will inactivate PRKA pathway which then lead to the activation of mTOR pathway and the inhibition of autophagy pathway both in colorectal epithelial tissues and in pancreatic tissues [67–75]. However, abnormal intestinal bacteria in type 2 diabetes may also inhibit PRKA activity through the modulation of metabolic products such as butyrate [76]. Additionally, high level of pro-inflammatory cytokines such as tumor necrosis factor-α and leptin increase mTORC1, whereas low level of adiponectin decreases mTOR level; Adiponectin also decreases the proliferation of intestinal epithelial cells in rats fed with high fat diet [77, 78]. Moreover, in type 2 diabetes, activated IκB kinase β (IKBKB) caused by hyperglycemia increases the activation of mTOR by phosphorylating TSC1 or interacting with racor [79, 80]. Besides, increased level of NAD+ because of high glucose or lipids will inhibit SIRT1 pathway which also contributes to the activation of mTORC pathway and the inhibition of autophagy pathway [81].
All of these dysregulated pathways may then increase the risk of carcinogenesis by decreasing genetic stability and DNA mismatch repair, or remodeling cell metabolism which is hepful for cancer cell to survive the unfavorable microenvironment such as hypoxia and low blood supply, or promoting protein synthesis, nucleotide synthesis and lipids synthesis which will favor the rapid proliferation of cancer cell (Figure 5).
Figure 5.

Alteration and interaction of energy sensing pathways in type 2 diabetes and their association with colorectal cancer. The energy sensing pathways are dysregulated in type 2 diabetes because of high glucose, insulin and IGF-1 level. In the mean time, these dysregulated pathways may then increase the risk of carcinogenesis by decreasing genetic stability and DNA mismatch repair to promote cancer genesis, or remodeling cell metabolism to promote protein synthesis, nucleotide synthesis and lipids synthesis which will favor the rapid proliferation of cancer cell. AMPK: AMP-activated protein kinase; IGF-1: insulin growth factor-1; mTORC1: mammalian target of rapamycin complex 1.
4.1 Effects on reprogramming metabolic pathways to boost nutrient needs for cancer progress
PRKA inhibits cancer cell progression by down-regulating the metabolic pathways that support uncontrolled proliferation of cancer cells. The Warburg effect is a key hallmark of cancer, whereas PRKA opposes Warburg effects by suppressing anabolic signals and promoting signals of oxidative phosphorylation [82, 83]. PRKA inhibition promotes a metabolic shift toward the Warburg effect in both non-transformed and malignant cells, indicating that this enzyme is a negative regulator of glycolysis and suppressor of tumor development through regulation of metabolic pathways that support uncontrolled proliferation. Moreover, mTORC1 promotes angiogenesis by activating hypoxia-inducible factor 1α (HIF1A, also HIF1α) to promote angiogenesis and glycolysis to sustain tumor growth and infiltration [84].
4.2 Effects on macromolecules synthesis to support rapid cancer cell proliferation
It is found that PRKA decreases the de novo synthesis of fatty acids and cholesterol through directly phosphorylating and inhibiting acetyl CoA carboxylase (ACACA, also ACC) and sterol regulatory element binding transcription factor 1 (SREBF-1, also SREBP-1) [85], which promotes cancer growth [86]. The decreased level of PRKA in type 2 diabetes may therefore lose its inhibition on protein synthesis, fatty acid and cholesterol synthesis and then promote cancer cell growth [87].
Inhibited PRKA could also promote the growth of cancer cells by activating autophagy through unc-51 like autophagy activating kinase 1 (ULK1) [88]. Recently, researchers have found that TRB3, which is closely associated with diabetes related cancer, interacts with autophagic receptor SQSTM1 (p62) and hinders SQSTM1 to bind to LC3 and ubiquitinated substrates, and therefore produces potent antitumour efficacies against tumor growth and metastasis [89].
Activated mTORC1 will directly phosphorylates the translational regulators eukaryotic translation initiation factor 4E (EIF4E) binding protein 1 (EIF4EBP1) and RPS6 kinase 1 (RPS6KB1, also S6K1), which, in turn, promote protein synthesis [90]. mTORC1 also promotes lipid biogenesis to sustain the rapid proliferation of cancer cells through SREBF1, SREBF2 and fatty acid synthase (FASN) [91]. In diet induced obesity/prediabetic mice, mTOR inhibitor rapamycin significantly reduced pancreatic tumor growth through inhibiting mTOR-regulated growth and survival signaling [92].
4.3 Effects on DNA stability and cancer initiation
Increasing evidences have also proved that PRKA may function as a house-keeper of DNA stability and play roles in DNA damage response pathway. It is found that in cancer cells, PRKA is rapidly phosphorylated and activated in response to radiation in order to survive the cancer [93, 94]; PRKA also functions as downstream of ATM which is a key mediator of the DNA damage response [95]. It is found that AMPKα1 deletion down-regulates cyclin-dependent kinase inhibitor p21 which play important roles in decreasing DNA double-strand breaks (DSB) [96]. Recently, it is found that in renal proximal tubular mouse cells exposed to high glucose as well as in kidney of db/db mice, activation PRKA by AICAR increases 8-oxoG-DNA glycosylase (OGG1) proteins and reduces oxidative DNA damage [97].
Several studies in mouse support the contention that SIRT1 improves genetic stability and suppresses tumor growth [98, 99]. Cockayne syndrome (CS) is an accelerated aging disorder characterized by progressive neurodegeneration caused by mutations in genes encoding the DNA repair proteins CS group A or B (CSA or CSB). Recent study has found that NAD(+) supplementation can rescue CS-associated phenotypes by activating SIRT1 [100]. In another study, it is proved that cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1; restored SIRT1 deacetylase activity can sensitize prematurely senescent cancer cells for apoptosis [101].
5 The anti-cancer effects of hypoglycemic agents depend on energy sensing pathways
Metformin has apparent role in decreasing colorectal cancer risk in type 2 diabetes [102, 103]. Moreover, in a meta-analysis it is found that metformin therapy reduce the risk of all cause of death by 44% and the risk of cancer specific death by 34% in colorectal cancer patients compared to those in non-users [104]. In another study, it is showed that the administration of low-dose metformin for 1 year to patients without diabetes was safe. Low-dose metformin reduced the prevalence and number of metachronous adenomas or polyps after polypectomy [105]. It is proved recently that metformin lowers blood glucose levels by inhibiting hepatic glucose production through intestinal PRKA but not hepatic PRKA dependent pathway [106]. In another study it is found that administration of metformin to P. obesus with insulin resistance and type 2 diabetes led to an up-regulation of intestinal PRKA signaling pathway and a reduction in ACC activity as well as improved blood lipids [70]. These data suggest that PRKA fulfills key functions in metabolic processes in the small intestine. It is also found that metformin suppresses colonic epithelial proliferation via the activation of PRKA [107]. Yang et al. have found that reduced cancer risk associated with metformin use is most evident in patients with T2DM who have low levels of HDL cholesterol [108]. Since HDL can also activate PRKA [109], therefore their results strongly supports the hypothesis that dysregulation of the PRKA pathway is a key feature linking T2DM and cancer.
Resveratrol, a phytoalexin present in few plant species, has demonstrated beneficial antidiabetic effects in animals and humans [110–112]. The beneficial effects of resveratrol occurred via mechanisms such as anti-inflammation, antioxidation, antihyperglycaemia, etc. The molecular targets of resveratrol include SIRT1, AMPK and autophagy [113–115]. It is found that resveratrol suppressed proliferation and invasion of two different human colorectal cancer cells in a dose-dependent manner, and interestingly, this was accompanied with a significant decrease in Ki-67 expression in SIRT1 dependent manner [116].
Thiazolidinediones (TZDs) is another important hypoglycemic agents which possess anti-cancer effects. One study has found that compared with other hypoglycemic agents, TZDs decreased cancer risk (including colorectal cancer) to 40%–50% in type 2 diabetic patients and this effect was dose-dependent [117]. In a meta-analysis, it is also found that TZDs is associated with decreased risk of colorectal cancer [118]. The anti-cancer effects of TZDs appear mainly to be independent of their PPARγ agonist activity but the molecular mechanisms involved in the anticancer action are not yet well understood. However, Wei et al. have declared that the anti-cancer effects of TZDs may be explained by its energy restriction effects including the transient induction of SIRT1 and the activation of AMPK [119]. Because of this, TZDs is considered as a new energy restriction-mimetic agent similar to resveratrol and 2-Deoxy-D-glucose which are also used as anti-cancer agents.
6. Prospects
In the future, it will be increasingly important to facilitate a seamless synergism between basic science, and clinical and population-based research. Ample evidence suggests that colorectal cancer represents a heterogenous group of neoplasms with differing sets of tumor molecular alterations [120–124]. Towards achievement of that goal, further studies based on molecular pathological epidemiology (MPE) are needed for us to uncover the underlying mechanism of the link between type 2 diabetes and colorectal cancer in large populations, and to strategize preventive measures of colorectal cancer incidence and mortality [125, 126]. The concept of MPE has gained considerable recognitions in the recent literature [127–134]. The MPE paradigm has been topics in international meetings [135–142]. In 2008, we have found that the adverse prognostic effect of obesity is present in patients with FASN-positive colon cancers, but not in patients with FASN-negative colon cancers [143]. Our subsequent investigations have found that a number of other tumor molecular changes interact with prediagnosis BMI to modify tumor aggressiveness [144–146]. Therefore, we have opened a new direction of MPE research to investigate interactive effects of dietary or lifestyle exposures and tumoral molecular features on tumor behavior (prognosis or clinical outcome), so that one can attribute the effects of dietary or lifestyle variables to a specific molecular subtype of cancer [126]. In future MPE projects, type 2 diabetes can be examined in relation to specific subtypes of colorectal cancer in analyses of colorectal cancer incidence and mortality. As another example, metformin, as a commonly used hypoglycemic agent for type 2 diabetes, has been proved possess strong anti-cancer effects in many studies [102, 147, 148]; however, in other studies, it is proved that metformin has no effect or even increase the risk of cancer in type 2 diabetes[149, 150]. Therefore, further MPE studies are needed for us to find the connection between type 2 diabetes, metformin use, and cancer, which may be helpful in the diagnosis, prevention and treatment of type 2 diabetes and cancer.
In conclusion, energy sensing pathway may bridge type 2 diabetes and colorectal cancer. Further studies including MPE studies are needed to address the links between these two diseases, which may bring great promise in the coming era of “precise medicine”.
Acknowledgments
This work was supported by grants from the U.S.A. National Institutes of Health (NIH) [K07 CA190673 (to R.N.), R01 CA151993 and R35CA197735 (to S.O.)]; National Natural Science Foundation of China (81473472, 81200612); Tianjin Municipal Natural Science Foundation of China (13JCZDJC30500); Tianjin City High School Science & Technology Fund Planning Project (20102217).
Abbreviations
- ACACA
ACC, acetyl-CoA carboxylase
- AICAR
5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide
- AKT
Proteinase kinase B
- BECN1
Beclin 1
- BMI
Body mass index
- CAMKK2
Calmodulin-dependent protein kinase beta
- EEF2
Eukaryotic translation elongation factor 2
- FASN
Fatty acid synthase
- FFA
Free fatty acid
- GLUT1
Glucose transporter 1
- GLUT4
Glucose transporter 4
- HDAC1
Histone deacetylase 1
- HIF1A
HIF-1alpha, hypoxia-inducible factor-1 alpha
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A
- IGF1
Insulin growth factor-1
- IKBKB
IKKbeta, I kappa B kinase
- LDH
Lactate dehydrogenase
- LXR
Liver X receptor
- mTOR
Mammalian target of rapamycin
- NAD
Nicotinamide adenine dinucleotide
- NFKB1
NF-kappaB, nuclear factor-KappaB
- PDK1
Phosphoinositide-dependent kinase-1
- PFK2
Phosphofructokinase-2
- PI3K
Phosphatidylinositol 3-kinase
- PPARGC1A
PCG-1alpha, peroxisome proliferator-activated-bold gamma coactivator-1 α
- PRKA
AMPK, AMP-activated protein kinase
- PTEN
Phosphatase and tensin homolog
- ROS
Reactive oxygen species
- SIRT1
NAD-dependent deacetylase sirtuin-1
- SREBF1
SREBP-1, sterol regulatory element-binding proteins-1
- STK11
LKB1, liver kinase B1
- TSC
Tuberous sclerosis complex
- ULK
Unc-51-like kinases
- VEGFA
Vascular endothelial growth factor
Footnotes
Use of Standardized Official Symbols:
We use HUGO (Human Genome Organisation)-approved official symbols for genes and gene products, including PRKA; mTOR; PIK3CA; all of which are described at www.genenames.org.
References
- 1.Giovannucci E, Harlan DM, Archer MC, et al. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsilidis KK, Kasimis JC, Lopez DS, et al. Type 2 diabetes and cancer: umbrella review of meta-analyses of observational studies. BMJ. 2015;350:g7607. doi: 10.1136/bmj.g7607. [DOI] [PubMed] [Google Scholar]
- 3.Jaacks LM, Siegel KR, Gujral UP, Narayan KM. Type 2 diabetes: A 21st century epidemic. Best Pract Res Clin Endocrinol Metab. 2016;30:331–343. doi: 10.1016/j.beem.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Hu FB. The global implications of diabetes and cancer. Lancet. 2014;383:1947–1948. doi: 10.1016/S0140-6736(14)60886-2. [DOI] [PubMed] [Google Scholar]
- 5.La Vecchia C, D'Avanzo B, Negri E, Franceschi S. History of selected diseases and the risk of colorectal cancer. Eur J Cancer. 1991;27:582–586. doi: 10.1016/0277-5379(91)90223-z. [DOI] [PubMed] [Google Scholar]
- 6.van de Poll-Franse LV, Haak HR, Coebergh JW, et al. Disease-specific mortality among stage I–III colorectal cancer patients with diabetes: a large population-based analysis. Diabetologia. 2012;55:2163–2172. doi: 10.1007/s00125-012-2555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L, Yu S. Diabetes mellitus is an independent risk factor for colorectal cancer. Dig Dis Sci. 2012;57:1586–1597. doi: 10.1007/s10620-012-2059-x. [DOI] [PubMed] [Google Scholar]
- 8.Hu FB, Manson JE, Liu S, et al. Prospective study of adult onset diabetes mellitus (type 2) and risk of colorectal cancer in women. J Natl Cancer Inst. 1999;91:542–547. doi: 10.1093/jnci/91.6.542. [DOI] [PubMed] [Google Scholar]
- 9.Vu HT, Ufere N, Yan Y, et al. Diabetes mellitus increases risk for colorectal adenomas in younger patients. World J Gastroenterol. 2014;20:6946–6952. doi: 10.3748/wjg.v20.i22.6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American Diabetes A. 3. Foundations of Care and Comprehensive Medical Evaluation. Diabetes Care. 2016;39(Suppl 1):S23–35. doi: 10.2337/dc16-S006. [DOI] [PubMed] [Google Scholar]
- 11.Barone BB, Yeh HC, Snyder CF, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300:2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein KB, Snyder CF, Barone BB, et al. Colorectal cancer outcomes, recurrence, and complications in persons with and without diabetes mellitus: a systematic review and meta-analysis. Dig Dis Sci. 2010;55:1839–1851. doi: 10.1007/s10620-009-0944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mills KT, Bellows CF, Hoffman AE, et al. Diabetes mellitus and colorectal cancer prognosis: a meta-analysis. Dis Colon Rectum. 2013;56:1304–1319. doi: 10.1097/DCR.0b013e3182a479f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang IP, Tsai HL, Huang CW, et al. High blood sugar levels significantly impact the prognosis of colorectal cancer patients through down-regulation of microRNA-16 by targeting Myb and VEGFR2. Oncotarget. 2016;7:18837–18850. doi: 10.18632/oncotarget.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma A, Ng H, Kumar A, et al. Colorectal cancer: Histopathologic differences in tumor characteristics between patients with and without diabetes. Clin Colorectal Cancer. 2014;13:54–61. doi: 10.1016/j.clcc.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Vargas T, Moreno-Rubio J, Herranz J, et al. Genes associated with metabolic syndrome predict disease-free survival in stage II colorectal cancer patients. A novel link between metabolic dysregulation and colorectal cancer. Mol Oncol. 2014 doi: 10.1016/j.molonc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lettieri Barbato D, Baldelli S, Pagliei B, et al. Caloric Restriction and the Nutrient-Sensing PGC-1alpha in Mitochondrial Homeostasis: New Perspectives in Neurodegeneration. Int J Cell Biol. 2012;2012:759583. doi: 10.1155/2012/759583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6:457–470. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirwany NA, Zou MH. AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed) 2014;19:447–474. doi: 10.2741/4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 21.Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 22.Zulato E, Bergamo F, De Paoli A, et al. Prognostic significance of AMPK activation in advanced stage colorectal cancer treated with chemotherapy plus bevacizumab. Br J Cancer. 2014;111:25–32. doi: 10.1038/bjc.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alayev A, Holz MK. mTOR signaling for biological control and cancer. J Cell Physiol. 2013;228:1658–1664. doi: 10.1002/jcp.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. doi: 10.3389/fnmol.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 26.Smith EM, Finn SG, Tee AR, et al. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–18727. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 27.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin ME, Perez MI, Redondo C, et al. 4E binding protein 1 expression is inversely correlated to the progression of gastrointestinal cancers. Int J Biochem Cell Biol. 2000;32:633–642. doi: 10.1016/s1357-2725(00)00007-8. [DOI] [PubMed] [Google Scholar]
- 30.Sticz T, Molnar A, Mark A, et al. mTOR activity and its prognostic significance in human colorectal carcinoma depending on C1 and C2 complex-related protein expression. J Clin Pathol. 2016 doi: 10.1136/jclinpath-2016-203913. [DOI] [PubMed] [Google Scholar]
- 31.Bigagli E, De Filippo C, Castagnini C, et al. DNA copy number alterations, gene expression changes and disease-free survival in patients with colorectal cancer: a 10 year follow-up. Cell Oncol (Dordr) 2016;39:545–558. doi: 10.1007/s13402-016-0299-z. [DOI] [PubMed] [Google Scholar]
- 32.Cohen HY, Miller C, Bitterman KJ, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 33.Bordone L, Cohen D, Robinson A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 35.Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- 36.Nath KA. The role of Sirt1 in renal rejuvenation and resistance to stress. J Clin Invest. 2010;120:1026–1028. doi: 10.1172/JCI42184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 38.Dobbin MM, Madabhushi R, Pan L, et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16:1008–1015. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Zhang S, Blander G, et al. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 40.Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 41.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 42.Lim JH, Lee YM, Chun YS, et al. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38:864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 43.Yang Q, Wang B, Gao W, et al. SIRT1 is downregulated in gastric cancer and leads to G1-phase arrest via NF-kappaB/Cyclin D1 signaling. Mol Cancer Res. 2013;11:1497–1507. doi: 10.1158/1541-7786.MCR-13-0214. [DOI] [PubMed] [Google Scholar]
- 44.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Firestein R, Blander G, Michan S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benard A, Goossens-Beumer IJ, van Hoesel AQ, et al. Nuclear expression of histone deacetylases and their histone modifications predicts clinical outcome in colorectal cancer. Histopathology. 2015;66:270–282. doi: 10.1111/his.12534. [DOI] [PubMed] [Google Scholar]
- 47.Kabra N, Li Z, Chen L, et al. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem. 2009;284:18210–18217. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang SH, Min KW, Paik SS, Jang KS. Loss of SIRT1 histone deacetylase expression associates with tumour progression in colorectal adenocarcinoma. J Clin Pathol. 2012;65:735–739. doi: 10.1136/jclinpath-2012-200685. [DOI] [PubMed] [Google Scholar]
- 49.Chen X, Hokka D, Maniwa Y, et al. Sirt1 is a tumor promoter in lung adenocarcinoma. Oncol Lett. 2014;8:387–393. doi: 10.3892/ol.2014.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang K, Lyu L, Shen Z, et al. Overexpression of SIRT1 is a poor prognostic factor for advanced colorectal cancer. Chin Med J (Engl) 2014;127:2021–2024. [PubMed] [Google Scholar]
- 51.Ren NS, Ji M, Tokar EJ, et al. Haploinsufficiency of SIRT1 Enhances Glutamine Metabolism and Promotes Cancer Development. Curr Biol. 2017;27:483–494. doi: 10.1016/j.cub.2016.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 53.Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26:2694–2701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 54.Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang CS, Kim JJ, Lee HM, et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy. 2014;10:785–802. doi: 10.4161/auto.28072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez AM, Csibi A, Raibon A, et al. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012;113:695–710. doi: 10.1002/jcb.23399. [DOI] [PubMed] [Google Scholar]
- 57.Haussinger D, Reinehr R, Schliess F. The hepatocyte integrin system and cell volume sensing. Acta Physiol (Oxf) 2006;187:249–255. doi: 10.1111/j.1748-1716.2006.01542.x. [DOI] [PubMed] [Google Scholar]
- 58.Joven J, Guirro M, Marine-Casado R, et al. Autophagy is an inflammation-related defensive mechanism against disease. Adv Exp Med Biol. 2014;824:43–59. doi: 10.1007/978-3-319-07320-0_6. [DOI] [PubMed] [Google Scholar]
- 59.Lopez de Figueroa P, Lotz M, Blanco FJ, Carames B. Autophagy activation protects from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015 doi: 10.1002/art.39025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park JM, Tougeron D, Huang S, et al. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS One. 2014;9:e100819. doi: 10.1371/journal.pone.0100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He S, Liang C. Frameshift mutation of UVRAG: Switching a tumor suppressor to an oncogene in colorectal cancer. Autophagy. 2015;11:1939–1940. doi: 10.1080/15548627.2015.1086523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He S, Zhao Z, Yang Y, et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat Commun. 2015;6:7839. doi: 10.1038/ncomms8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kang MR, Kim MS, Oh JE, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217:702–706. doi: 10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- 64.An CH, Kim MS, Yoo NJ, et al. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract. 2011;207:433–437. doi: 10.1016/j.prp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 65.Zhang MY, Gou WF, Zhao S, et al. Beclin 1 expression is closely linked to colorectal carcinogenesis and distant metastasis of colorectal carcinoma. Int J Mol Sci. 2014;15:14372–14385. doi: 10.3390/ijms150814372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giatromanolaki A, Koukourakis MI, Harris AL, et al. Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J Clin Pathol. 2010;63:867–872. doi: 10.1136/jcp.2010.079525. [DOI] [PubMed] [Google Scholar]
- 67.Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3:340–351. doi: 10.1038/nrd1344. [DOI] [PubMed] [Google Scholar]
- 68.Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009;8:3679–3683. doi: 10.4161/cc.8.22.9905. [DOI] [PubMed] [Google Scholar]
- 69.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- 70.Harmel E, Grenier E, Bendjoudi Ouadda A, et al. AMPK in the small intestine in normal and pathophysiological conditions. Endocrinology. 2014;155:873–888. doi: 10.1210/en.2013-1750. [DOI] [PubMed] [Google Scholar]
- 71.Joly E, Roduit R, Peyot ML, et al. Glucose represses PPARalpha gene expression via AMP-activated protein kinase but not via p38 mitogen-activated protein kinase in the pancreatic beta-cell. J Diabetes. 2009;1:263–272. doi: 10.1111/j.1753-0407.2009.00043.x. [DOI] [PubMed] [Google Scholar]
- 72.Han J, Zhang L, Guo H, et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015;138:668–675. doi: 10.1016/j.ygyno.2015.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo Z, Zhang Y, Li F, et al. Resistin induces insulin resistance by both AMPK-dependent and AMPK-independent mechanisms in HepG2 cells. Endocrine. 2009;36:60–69. doi: 10.1007/s12020-009-9198-7. [DOI] [PubMed] [Google Scholar]
- 74.Moon HS, Chamberland JP, Aronis K, et al. Direct role of adiponectin and adiponectin receptors in endometrial cancer: in vitro and ex vivo studies in humans. Mol Cancer Ther. 2011;10:2234–2243. doi: 10.1158/1535-7163.MCT-11-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kwon B, Querfurth HW. Palmitate activates mTOR/p70S6K through AMPK inhibition and hypophosphorylation of raptor in skeletal muscle cells: Reversal by oleate is similar to metformin. Biochimie. 2015;118:141–150. doi: 10.1016/j.biochi.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 76.Donohoe DR, Wali A, Brylawski BP, Bultman SJ. Microbial regulation of glucose metabolism and cell-cycle progression in mammalian colonocytes. PLoS One. 2012;7:e46589. doi: 10.1371/journal.pone.0046589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang CH, Cao GF, Jiang Q, Yao J. TNF-alpha promotes human retinal pigment epithelial (RPE) cell migration by inducing matrix metallopeptidase 9 (MMP-9) expression through activation of Akt/mTORC1 signaling. Biochem Biophys Res Commun. 2012;425:33–38. doi: 10.1016/j.bbrc.2012.07.044. [DOI] [PubMed] [Google Scholar]
- 78.Fujisawa T, Endo H, Tomimoto A, et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut. 2008;57:1531–1538. doi: 10.1136/gut.2008.159293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 80.Xu Y, Lai E, Liu J, et al. IKK interacts with rictor and regulates mTORC2. Cell Signal. 2013;25:2239–2245. doi: 10.1016/j.cellsig.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 81.Hou X, Xu S, Maitland-Toolan KA, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smolkova K, Plecita-Hlavata L, Bellance N, et al. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43:950–968. doi: 10.1016/j.biocel.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 83.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 85.Jung EJ, Kwon SW, Jung BH, et al. Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver. J Lipid Res. 2011;52:1617–1625. doi: 10.1194/jlr.M015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zechner R, Kienesberger PC, Haemmerle G, et al. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res. 2009;50:3–21. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]
- 87.Leprivier G, Remke M, Rotblat B, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–1079. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rao-Bindal K, Zhou Z, Kleinerman ES. MS-275 sensitizes osteosarcoma cells to Fas ligand-induced cell death by increasing the localization of Fas in membrane lipid rafts. Cell Death Dis. 2012;3:e369. doi: 10.1038/cddis.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hua F, Li K, Yu JJ, et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat Commun. 2015;6:7951. doi: 10.1038/ncomms8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 91.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cifarelli V, Lashinger LM, Devlin KL, et al. Metformin and Rapamycin Reduce Pancreatic Cancer Growth in Obese Prediabetic Mice by Distinct MicroRNA-Regulated Mechanisms. Diabetes. 2015;64:1632–1642. doi: 10.2337/db14-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sanli T, Rashid A, Liu C, et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010;78:221–229. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 94.Ha BG, Park JE, Cho HJ, et al. Inhibitory effects of proton beam irradiation on integrin expression and signaling pathway in human colon carcinoma HT29 cells. Int J Oncol. 2015;46:2621–2628. doi: 10.3892/ijo.2015.2942. [DOI] [PubMed] [Google Scholar]
- 95.Storozhuk Y, Hopmans SN, Sanli T, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108:2021–2032. doi: 10.1038/bjc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu H, Zhou Y, Coughlan KA, et al. AMPKalpha1 deficiency promotes cellular proliferation and DNA damage via p21 reduction in mouse embryonic fibroblasts. Biochim Biophys Acta. 2015;1853:65–73. doi: 10.1016/j.bbamcr.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Habib SL, Yadav A, Kidane D, et al. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle. 2016;15:3048–3059. doi: 10.1080/15384101.2016.1231259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh SK, Williams CA, Klarmann K, et al. Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. J Exp Med. 2013;210:987–1001. doi: 10.1084/jem.20121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gonfloni S, Iannizzotto V, Maiani E, et al. P53 and Sirt1: routes of metabolism and genome stability. Biochem Pharmacol. 2014;92:149–156. doi: 10.1016/j.bcp.2014.08.034. [DOI] [PubMed] [Google Scholar]
- 100.Guarente L. Linking DNA damage, NAD(+)/SIRT1, and aging. Cell Metab. 2014;20:706–707. doi: 10.1016/j.cmet.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 101.Back JH, Rezvani HR, Zhu Y, et al. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. J Biol Chem. 2011;286:19100–19108. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cardel M, Jensen SM, Pottegard A, et al. Long-term use of metformin and colorectal cancer risk in type II diabetics: a population-based case-control study. Cancer Med. 2014;3:1458–1466. doi: 10.1002/cam4.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cho YH, Ko BM, Kim SH, et al. Does metformin affect the incidence of colonic polyps and adenomas in patients with type 2 diabetes mellitus? Intestinal Res. 2014;12:139–145. doi: 10.5217/ir.2014.12.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mei ZB, Zhang ZJ, Liu CY, et al. Survival benefits of metformin for colorectal cancer patients with diabetes: a systematic review and meta-analysis. PLoS One. 2014;9:e91818. doi: 10.1371/journal.pone.0091818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Higurashi T, Hosono K, Takahashi H, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17:475–483. doi: 10.1016/S1470-2045(15)00565-3. [DOI] [PubMed] [Google Scholar]
- 106.Duca FA, Cote CD, Rasmussen BA, et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat Med. 2015;21:506–511. doi: 10.1038/nm.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hosono K, Endo H, Takahashi H, et al. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol Carcinog. 2010;49:662–671. doi: 10.1002/mc.20637. [DOI] [PubMed] [Google Scholar]
- 108.Yang X, Lee HM, Chan JC. Drug-subphenotype interactions for cancer in type 2 diabetes mellitus. Nat Rev Endocrinol. 2015;11:372–379. doi: 10.1038/nrendo.2015.37. [DOI] [PubMed] [Google Scholar]
- 109.Kimura T, Tomura H, Sato K, et al. Mechanism and role of high density lipoprotein-induced activation of AMP-activated protein kinase in endothelial cells. J Biol Chem. 2010;285:4387–4397. doi: 10.1074/jbc.M109.043869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Knop FK, Konings E, Timmers S, et al. Thirty days of resveratrol supplementation does not affect postprandial incretin hormone responses, but suppresses postprandial glucagon in obese subjects. Diabet Med. 2013;30:1214–1218. doi: 10.1111/dme.12231. [DOI] [PubMed] [Google Scholar]
- 111.Bhatt JK, Thomas S, Nanjan MJ. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr Res. 2012;32:537–541. doi: 10.1016/j.nutres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 112.Brasnyo P, Molnar GA, Mohas M, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106:383–389. doi: 10.1017/S0007114511000316. [DOI] [PubMed] [Google Scholar]
- 113.Wang B, Yang Q, Sun YY, et al. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med. 2014;18:1599–1611. doi: 10.1111/jcmm.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yar AS, Menevse S, Alp E. The effects of resveratrol on cyclooxygenase-1 and -2, nuclear factor kappa beta, matrix metalloproteinase-9, and sirtuin 1 mRNA expression in hearts of streptozotocin-induced diabetic rats. Genet Mol Res. 2011;10:2962–2975. doi: 10.4238/2011.November.29.7. [DOI] [PubMed] [Google Scholar]
- 115.Chang CC, Chang CY, Wu YT, et al. Resveratrol retards progression of diabetic nephropathy through modulations of oxidative stress, proinflammatory cytokines, and AMP-activated protein kinase. J Biomed Sci. 2011;18:47. doi: 10.1186/1423-0127-18-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Buhrmann C, Shayan P, Popper B, et al. Sirt1 Is Required for Resveratrol-Mediated Chemopreventive Effects in Colorectal Cancer Cells. Nutrients. 2016;8:145. doi: 10.3390/nu8030145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin HC, Hsu YT, Kachingwe BH, et al. Dose effect of thiazolidinedione on cancer risk in type 2 diabetes mellitus patients: a six-year population-based cohort study. J Clin Pharm Ther. 2014;39:354–360. doi: 10.1111/jcpt.12151. [DOI] [PubMed] [Google Scholar]
- 118.Bosetti C, Rosato V, Buniato D, et al. Cancer risk for patients using thiazolidinediones for type 2 diabetes: a meta-analysis. Oncologist. 2013;18:148–156. doi: 10.1634/theoncologist.2012-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wei S, Kulp SK, Chen CS. Energy restriction as an antitumor target of thiazolidinediones. J Biol Chem. 2010;285:9780–9791. doi: 10.1074/jbc.M109.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dienstmann R, Vermeulen L, Guinney J, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017 doi: 10.1038/nrc.2017.24. [DOI] [PubMed] [Google Scholar]
- 121.Kudryavtseva AV, Lipatova AV, Zaretsky AR, et al. Important molecular genetic markers of colorectal cancer. Oncotarget. 2016;7:53959–53983. doi: 10.18632/oncotarget.9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kocarnik JM, Shiovitz S, Phipps AI. Molecular phenotypes of colorectal cancer and potential clinical applications. Gastroenterol Rep (Oxf) 2015;3:269–276. doi: 10.1093/gastro/gov046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013;5:676–713. doi: 10.3390/cancers5020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013;14:16365–16385. doi: 10.3390/ijms140816365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ogino S, Stampfer M. Lifestyle factors and microsatellite instability in colorectal cancer: the evolving field of molecular pathological epidemiology. J Natl Cancer Inst. 2010;102:365–367. doi: 10.1093/jnci/djq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ogino S, Chan AT, Fuchs CS, Giovannucci E. Molecular pathological epidemiology of colorectal neoplasia: an emerging transdisciplinary and interdisciplinary field. Gut. 2011;60:397–411. doi: 10.1136/gut.2010.217182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rescigno T, Micolucci L, Tecce MF, Capasso A. Bioactive Nutrients and Nutrigenomics in Age-Related Diseases. Molecules. 2017;22 doi: 10.3390/molecules22010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weng M, Wu D, Yang C, et al. Noncoding RNAs in the development, diagnosis, and prognosis of colorectal cancer. Transl Res. 2016 doi: 10.1016/j.trsl.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 129.Patil H, Saxena SG, Barrow CJ, et al. Chasing the personalized medicine dream through biomarker validation in colorectal cancer. Drug Discov Today. 2017;22:111–119. doi: 10.1016/j.drudis.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 130.Lewis C, McQuaid S, Hamilton PW, et al. Building a 'Repository of Science': The importance of integrating biobanks within molecular pathology programmes. Eur J Cancer. 2016;67:191–199. doi: 10.1016/j.ejca.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 131.Alnabulsi A, Murray GI. Integrative analysis of the colorectal cancer proteome: potential clinical impact. Expert Rev Proteomics. 2016:1–11. doi: 10.1080/14789450.2016.1233062. [DOI] [PubMed] [Google Scholar]
- 132.Zhou L, Wang K, Li Q, et al. Clinical proteomics-driven precision medicine for targeted cancer therapy: current overview and future perspectives. Expert Rev Proteomics. 2016;13:367–381. doi: 10.1586/14789450.2016.1159959. [DOI] [PubMed] [Google Scholar]
- 133.Campbell PT, Newton CC, Newcomb PA, et al. Association between body mass index and mortality for colorectal cancer survivors: overall and by tumor molecular phenotype. Cancer Epidemiol Biomarkers Prev. 2015;24:1229–1238. doi: 10.1158/1055-9965.EPI-15-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kuipers EJ, Grady WM, Lieberman D, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065. doi: 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Campbell PT, Rebbeck TR, Nishihara R, et al. Proceedings of the third international molecular pathological epidemiology (MPE) meeting. Cancer Causes Control. 2017 doi: 10.1007/s10552-016-0845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Blomain ES, Waldman SA. Does obesity promote the development of colorectal cancer? Expert Rev Anticancer Ther. 2016;16:465–467. doi: 10.1586/14737140.2016.1162102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ogino S, Nishihara R, VanderWeele TJ, et al. Review Article: The Role of Molecular Pathological Epidemiology in the Study of Neoplastic and Non-neoplastic Diseases in the Era of Precision Medicine. Epidemiology. 2016;27:602–611. doi: 10.1097/EDE.0000000000000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martinez-Useros J, Garcia-Foncillas J. Obesity and colorectal cancer: molecular features of adipose tissue. J Transl Med. 2016;14:21. doi: 10.1186/s12967-016-0772-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sylvetsky AC, Nandagopal R, Nguyen TT, et al. Buddy Study: Partners for better health in adolescents with type 2 diabetes. World J Diabetes. 2015;6:1355–1362. doi: 10.4239/wjd.v6.i18.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ogino S, Campbell PT, Nishihara R, et al. Proceedings of the second international molecular pathological epidemiology (MPE) meeting. Cancer Causes Control. 2015;26:959–972. doi: 10.1007/s10552-015-0596-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Epplein M, Bostick RM, Mu L, et al. Challenges and opportunities in international molecular cancer prevention research: An ASPO Molecular Epidemiology and the Environment and International Cancer Prevention Interest Groups Report. Cancer Epidemiol Biomarkers Prev. 2014;23:2613–2617. doi: 10.1158/1055-9965.EPI-14-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kuller LH, Bracken MB, Ogino S, et al. The role of epidemiology in the era of molecular epidemiology and genomics: Summary of the 2013 AJE-sponsored Society of Epidemiologic Research Symposium. Am J Epidemiol. 2013;178:1350–1354. doi: 10.1093/aje/kwt239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ogino S, Nosho K, Meyerhardt JA, et al. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J Clin Oncol. 2008;26:5713–5720. doi: 10.1200/JCO.2008.18.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ogino S, Nosho K, Baba Y, et al. A cohort study of STMN1 expression in colorectal cancer: body mass index and prognosis. Am J Gastroenterol. 2009;104:2047–2056. doi: 10.1038/ajg.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ogino S, Shima K, Nosho K, et al. A cohort study of p27 localization in colon cancer, body mass index, and patient survival. Cancer Epidemiol Biomarkers Prev. 2009;18:1849–1858. doi: 10.1158/1055-9965.EPI-09-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ogino S, Nosho K, Shima K, et al. p21 expression in colon cancer and modifying effects of patient age and body mass index on prognosis. Cancer Epidemiol Biomarkers Prev. 2009;18:2513–2521. doi: 10.1158/1055-9965.EPI-09-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sehdev A, Shih YC, Vekhter B, et al. Metformin for primary colorectal cancer prevention in patients with diabetes: A case-control study in a US population. Cancer. 2014 doi: 10.1002/cncr.29165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh S, Singh H, Singh PP, et al. Antidiabetic medications and the risk of colorectal cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2013;22:2258–2268. doi: 10.1158/1055-9965.EPI-13-0429. [DOI] [PubMed] [Google Scholar]
- 149.Bodmer M, Becker C, Meier C, et al. Use of metformin is not associated with a decreased risk of colorectal cancer: a case-control analysis. Cancer Epidemiol Biomarkers Prev. 2012;21:280–286. doi: 10.1158/1055-9965.EPI-11-0992-T. [DOI] [PubMed] [Google Scholar]
- 150.Sakoda LC, Ferrara A, Achacoso NS, et al. Metformin use and lung cancer risk in patients with diabetes. Cancer Prev Res (Phila) 2015;8:174–179. doi: 10.1158/1940-6207.CAPR-14-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]