

Introduction
Although the foundations of mass spectrometry-based lipidomics have been practiced for over 30 years, recent technological advances in ionization modalities in conjunction with robust increases in mass accuracy and resolution have greatly accelerated the emergence, growth and importance of the field of lipidomics. Moreover, advances in the separation sciences, bioinformatic strategies and the availability of robust databases have been synergistically integrated into modern lipidomic technologies leading to unprecedented improvements in the depth, penetrance and precision of lipidomic analyses and identification of their biological and mechanistic significance. The purpose of this “opinion” article is to briefly review the evolution of lipidomics, critique the platforms that have evolved and identify areas that are likely to emerge in the years to come. Through seamlessly integrating a rich repertoire of mass spectrometric, chemical and bioinformatic strategies, the chemical identities and quantities of tens of thousands to hundreds of thousands of different lipid molecular species and their metabolic alterations during physiologic or pathophysiologic perturbations can be obtained. Thus, the field of lipidomics which already has a distinguished history of exciting new discoveries in many disease states holds unparalleled potential to identify the pleiotropic roles of lipids in health and disease at the chemical level.
The Dawn of Mass Spectrometry-based Lipidomics
Over thirty years ago, we began our adventures in lipidomics exploring the hypothesis that alterations in myocardial membrane lipid constituents result in changes in membrane molecular dynamics and surface charge that precipitate myocardial electrophysiologic dysfunction, hemodynamic failure and sudden death. In the late 1970s, we discovered that negatively charged liposomes and changes in membrane molecular dynamics were potent activators of myocardial phospholipases [1]. However, the methods for identification of alterations in lipid content and composition were severely limited by the difficulties of the laborious techniques used for analysis of the majority of cellular lipids accompanied by corresponding inaccuracies in lipid identification and quantitation. Accordingly, we began the development of lipidomic strategies employing sequential straight and reversed phase HPLC with quantitation by GC-MS to identify alterations in lipid molecular species during cardiac ischemia which had the potential to identify the membrane alterations responsible for lethal ventricular arrhythmias, heart failure and sudden death [2]. At that time, it was clear that a new field needed to be developed which could accurately identify the alterations in cellular membrane constituents to mechanistically understand the roles of lipids in health and disease. In the early 1980s, fast atom bombardment mass spectrometry (FABMS) employing a beam of high-energy Xe atoms focused on a host matrix of glycerol containing peptide guests was developed [3]. We adapted this soft ionization method for lipids resulting in the ionization of the intact phospholipid parent ions with minimal fragmentation. We were astounded when this technology identified plasmalogens (which contain a vinyl ether linkage at the sn-1 position) as the major phospholipid constituents of myocardial sarcolemma [4]. Moreover, ethanolamine glycerophospholipids in sarcolemma were predominantly plasmalogens containing arachidonic acid at the sn-2 position (Fig 1) [4]. These results had profound implications for myocardial membrane physical properties and lipid-mediated signal transduction. Subsequent studies using FABMS analysis of sarcoplasmic reticulum and mitochondria demonstrated the abundance of arachidonic acid containing plasmalogens in sarcoplasmic reticulum while mitochondria were comprised predominantly of diacyl phospholipids containing linoleic acid and arachidonic acid [5]. These results demonstrated the profound differences in the lipidome in specific subcellular organelles and their non-uniform spatial distribution in the cell. Critical to the success of these initial lipidomic experiments was the use of HPLC to separate lipid classes followed by soft ionization by FAB to volatize intact phospholipid molecular ions in the absence of significant fragmentation. Collectively, the use of HPLC followed by mass spectrometry identified the class, subclass, molecular species and regiospecificity of ~80 phospholipids in canine myocardium many of which were spatially localized in specialized subcellular compartments [4, 5]. The unanticipated predominance of plasmalogens containing almost exclusively arachidonic acid at the sn-2 position in myocardial sarcolemma focused our attention on the chemical, biophysical and signaling properties of plasmalogens and their downstream metabolites. Furthermore, this early technology provided a robust platform for identifying the molecular species of lipids in different cell types and tissues during physiologic or pathophysiologic perturbations. Through the use of these technologies, we identified a diverse array of phospholipid molecular species in human myocardium [6], synaptic vesicles [7] and electrophysiologically active cells [8]. However, the limited sensitivity of FAB ionization prevented its application to large-scale lipidomic analyses.
Figure 1. Plasmalogens Are the Major Storage Depot of Arachidonic Acid in Sarcolemma.
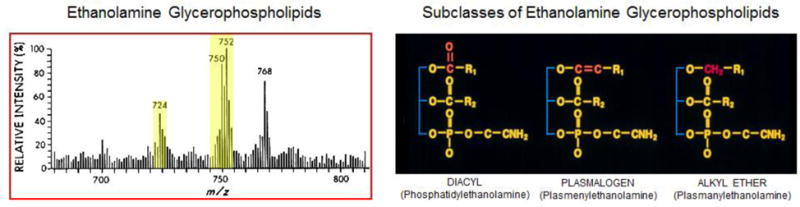
Left panel, fast atom bombardment mass spectrometry of sarcolemmal ethanolamine glycerophospholipids performed using highly purified sarcolemma prior to HPLC purification of ethanolamine glycerophospholipids. Approximately 500 pg of ethanolamine glycerophospholipids were dissolved in CHCl3,/CH3OH prior to obtaining fast atom bombardment mass spectra using a Xenon beam to ionize lipids as guests in a glycerol matrix, on a copper probe using a commercially available gas chromatography detector. Right panel, chemical structures of subclasses of ethanolamine diacyl, plasmalogen and alkyl ether glycerophospholipids.
(Biochemistry 1984 Jan3; 23(1):158–165. 0006-2960/84/0423-0158$01.50/0 1984 American Chemical Society.)
Electrospray Ionization Mass Spectrometry of Phospholipids Greatly Extends the Sensitivity, Specificity and Power of Lipidomics
In 1989, John Fenn identified the amazing power of electrospray ionization to affect the soft ionization of charged peptides, which led to his sharing the Nobel Prize in Chemistry in 2002 [9]. Reasoning that electrospray ionization of zwitterionic phospholipids was likely chemically related to the ESI-mediated soft ionization of peptides, in the early 1990s we were able to directly inject organic extracts of biologic tissues or fluids into an electrospray ion source resulting in a 1000 fold increase in sensitivity in comparison to FAB. This was accompanied by a robust increase in the identification of a large repertoire of lipid molecular species from multiple different classes in either the positive or negative ion mode [10]. Of course, knowing the m/z of a phospholipid molecular species only provides boundary conditions for its polar head group, aliphatic chain composition and regiospecificity. Accordingly, we directly injected an organic extract of bovine heart ethanolamine or choline glycerolipids into a triple quadrupole mass spectrometer using Q1 as a mass analyzer to select the peak of interest, Q2 as a collision chamber for the selected peaks and Q3 as product ion mass analyzer. Analysis of parent ion mass and product ion masses led to the identification of ~150 lipid molecular species in erythrocyte plasma membranes directly from organic extracts within minutes [11]. During this era, we determined that the ionization efficiency of an analyte is highly dependent on the magnitude of the dipole in the lipid analyte and, if present, its adduct ion. Thus, we could selectively ionize different classes of zwitterionic, negatively and positively charged phospholipids by multiplexing the charge, pH of the infusion solution or adduct ion. In general, charged or zwitterionic lipids have high ionization efficiencies while non-polar lipids typically require either an adduct ion for direct ionization or derivatization with a suitable group to effect ionization. Moreover, we demonstrated that within the range of typical fatty acyl compositions in each class and subclass, the molecular ion intensities were closely correlated with the abundance of the molecular species in the sample after accounting for its isotopologue distribution. Thus, by adding multiple internal standards of similar chain length and class during the extraction procedure, the quantitative analysis of a myriad of molecular species from multiple different lipid classes and subclasses in a high throughput manner was possible. Furthermore, we demonstrated that the position of the aliphatic chain in sn-1 vs. sn-2 lysolipids could be identified by the fragmentation patterns of the molecular ions [12] which was a simple solution to a long-standing problem in lipid chemistry. This approach, now known as shotgun lipidomics, was successful in identifying alterations in lipid composition in platelets after thrombin activation [13], acylcarnitine accumulation during myocardial ischemia [14], the importance of arachidonic acid-induced changes in currents in electrophysiologically active cells [8] and the alterations in lipid content in diabetic myocardium [15].
Multi-Dimensional Mass Spectrometry-based Shotgun Lipidomics (MDMS-SL)
During the initial development of shotgun-lipidomics, assignments were made manually by the mass of the parent ion and the identity and relative abundance of its product ions which identified the chemical components of the molecular ion of interest (i.e., backbone, polar head group, aliphatic chains, regiospecificity and its adduct ions). During this period, the overwhelming abundance of many thousands of lipid metabolites was recognized and it soon became apparent that the resolution of this complexity would be highly beneficial. We reasoned that the electrical propensity of a lipid to ionize in acidic or basic conditions could be utilized for selective ionization of specific lipid classes in the ESI ion source based upon the pH of the infusion solution. Through multiplexing the pH of the infusion solution and its adduct ions, the spectral complexity of molecular ions was greatly reduced in a process now known as intrasource separation. Furthermore, we demonstrated fragmentation patterns could be utilized to determine multiple aspects of lipid structure and regiospecificity [12]. Since cellular lipids are typically compromised of linear combinations of aliphatic chains and polar head groups linked to either a glycerol or sphingosine backbone, the use of intrasource separation can selectively ionize lipids with distinct electrical propensities by adjusting the pH or addition of selected adduct ions just prior to injection into the mass spectrometer thus reducing spectral complexity. Thus, the first step of Multi-Dimensional Mass Spectrometry-based Shotgun Lipidomics (MDMS-SL) was collection of spectra at neutral pH in the negative ion mode (e.g., for PS,PI,CL,PA). The second step of MDMS-SL was the addition of small amounts of LiOH just prior to infusion into the ESI ion source to facilitate ionization of ethanolamine molecular species in the negative ion mode which were not ionized under neutral conditions. In the third step of MDMS-SL, the spectrometer was switched to the positive ion mode leading to the ionization of choline phospholipids, acylcarnitines, lysolipids, Li+-adducted triglycerides and other non-polar lipids. Through altering the pH of the infusion solution, using multiple fragmentation energies and the judicious choice of adduct ions and powerful bioinformatic analyses MDMS-SL was a powerful plaform for lipidomic analyses. The details of the development and utilization of MDMS-SL have been reviewed in detail previously [16, 17].
Importantly, in 1997 Brugger and coworkers developed methods for precursor ion scanning (PIS) and neutral loss scanning (NLS) of lipids [18] which we quickly recognized as powerful tools that could be immediately integrated into our intrasource separation and fragmentation strategies. Through combining intrasource separation and multiple PIS and NLS of salient aliphatic chains and head groups, the molecular structures of prominent lipids were readily determined directly from extracts of biologic tissues or fluids. The power of multiplexing ionization conditions, product ion analysis in conjunction with multiple PIS and NLS with changes in collision energies, and the identification of regiospecific fragmentation patterns was remarkably successful in creating a simple platform for high throughput lipidomic analysis. We also point out that Ekroos and Shevchenko used multiple precursor ion scanning to identify molecular species of phospholipids by direct infusion [19].
As an example of the power of this MDMS-SL technology to identify the chemical mechanisms underlying human disease, we show a 2-dimensional positive ion array of the molecular ions from control and diabetic mouse myocardium analyzed by MDMS-SL (Fig 2). Two features of this array are apparent. First, in diabetic myocardium (shown on the right) there is a fourfold increase in the mass of triglycerides shown by comparing the MS1 scan in comparison to control myocardium (left). Second, neutral loss scanning indicated dramatic alterations in the arachidonic acid content in triglycerides in the diabetic mice (note the change in the neutral loss pattern from control vs. diabetic mice in molecular species containing arachidonic acid, NL 304.3 (20:4)) [20]. Since the molecular species content of triglycerides reflects the metabolic history of a cell, these results demonstrate the increased incorporation of arachidonic acid (which is normally predominantly present in cellular phospholipids) into the cellular triglyceride pool in conjunction with an increase in acylcarnitines. Further studies demonstrated a striking decrease in cardiolipin content and molecular species distribution [20]. The most straightforward explanation for these findings was that diabetes activated myocardial phospholipases resulting in alterations in cardiolipin content and molecular species distribution precipitating mitochondrial dysfunction. The resultant mitochondrial dysfunction attenuated the processing of fatty acids leading to accumulation of triglycerides that were enriched in arachidonic acid released by activated phospholipases. Thus, MDMS-SL technology identified the activation of phospholipases leading to mitochondrial bioenergetic dysfunction, lipid accumulation, and maladaptive changes in cellular signaling that collectively result in diabetic cardiomyopathy.
Figure 2. Positive Ion 2D Mass Spectra from Control and Diabetic Mouse Myocardium.
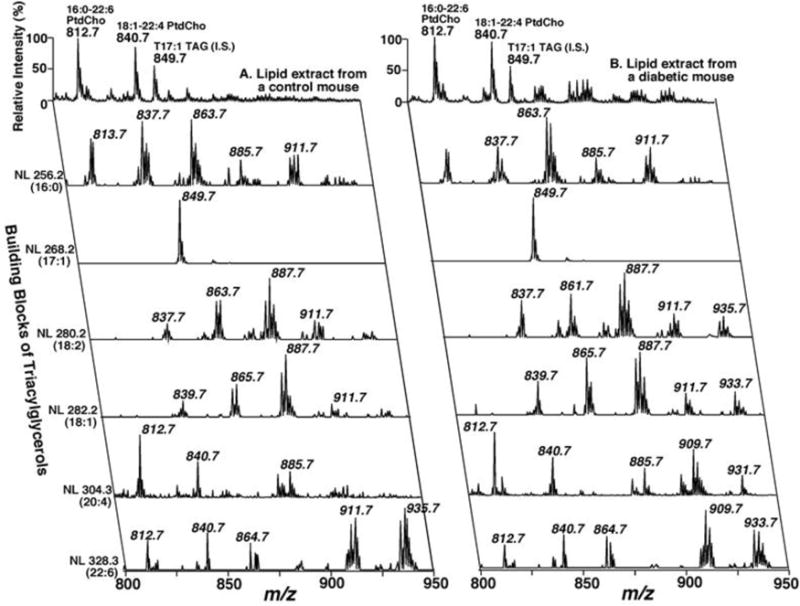
Triacylglycerol molecular species analyses by two-dimensional electrospray ionization mass spectrometry. The top trace is the molecular ion scan of myocardial lipid extracts from control or diabetic mice using Li+ as the adduct ion. The first-dimension spectra (top trace) were obtained in the positive-ion mode using intrasource separation. Next, neutral loss (NL) scanning of all naturally occurring aliphatic chains in triglycerides of myocardial chloroform extracts of control (left panel) or diabetic mice (right panel) were utilized to identify the molecular species assignments and quantify individual triacylglycerol molecular species by comparisons with selected internal standards. For tandem mass spectrometry in the positive-ion neutral loss (NL) mode, both the first and third quadrupoles were coordinately scanned with a mass difference (i.e., neutral loss) corresponding to the neutral loss of a nonesterified fatty acid from TAG molecular species. All mass spectral traces were displayed after normalization to the base peak in the individual spectrum.
(Biochemistry 2005 Nov24; 44:16684–16694, 10.1021/bi051908a)
Shotgun Lipidomic Enhancements
Over the decades that we have been using MDMS-SL, a wide range of derivatization procedures for targeting over 30 specific classes of lipids have been developed that result in increased signal-to-noise (S/N) by addition of highly efficient ionizable groups which can be engineered to result in a favorable mass shift or charge-switch derivatization through addition of moieties containing a permanent positive charge (Fig 3)). Additional sample handling techniques such as liquid/liquid partitioning have been particularly useful in the analysis of non-polar lipids allowing facile identification of diglycerides and monoglycerides (e.g.,[21, 22]) without α-hydroxy migration. We have also used Fmoc derivatization to markedly enhance the S/N in ethanolamine glycerolipids [7]. Noteworthy amongst recent derivatization enhancements for targeted analysis has been the use of charge-switch derivatization with aminomethyl phenyl pyridium (AMPP) and carbodiimide [23] developed by Gelb and coworkers which creates an amide with a permanent positive charge resulting in increases in S/N by ~50 fold for fatty acids. This method has provided improvements in the analysis of a wide variety of fatty acids and their oxidized derivatives through different strategies. For example, the localization of double bond isomers within the aliphatic chain can be accomplished by the use of AMPP derivatization [24]. This reagent also has far reaching advantages for the chiral analysis of oxylipins present in diminutive amounts using chiral chromatographic separation as discussed below.
Figure 3. Enhanced Shotgun Lipidomics Approaches.
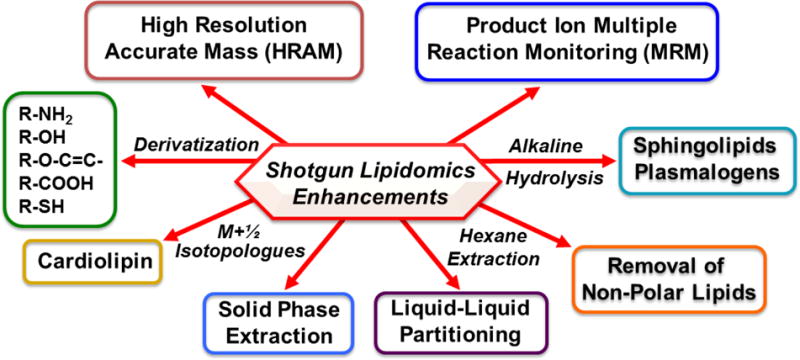
The analytic power of shotgun lipidomics can be extended through utilizing the unique chemical characteristics of specific lipid classes. These include the derivatization of lipid moieties to increase signal intensity and/or engender a mass shift to facilitate mass spectrometric analyses. For example, since cardiolipin is largely the only doubly negatively charged lipid in skeletal muscle and myocardium, the use of a M+1/2 isotopologue approach for cardiolipins successfully resolves their presence from neighboring groups. Other modifications including multiplexed extractions, liquid/liquid partitioning, liquid/solid partitioning (solid phase extraction), and/or alkaline hydrolysis to enrich for sphingolipids or ether lipids have also proven useful. In some tissues (e.g., adipose tissue), the removal of nonpolar lipids by hexane extraction is judicious prior to the subsequent analysis of polar lipid constituents. More recent enhancements include the use of MeOH and I2 for plasmalogen identification and the use of N-hydroxysuccinimide esters for phosphatidic acid analysis.
(Chem. Biol. 2011 Mar25; 18(3):284–291. http://dx.doi:10.1016/j.chembiol.2011.01.014 2011 Elsevier Ltd.)
Procedures we previously used for subclass identification (e.g., plasmalogen hydrolysis by acidic vapor) have largely been replaced by derivatization with methanol and I2 which is specific for addition to the plasmalogen vinyl ether linkage as developed by the Reid group [25]. This derivatization procedure is extremely effective, simple and has markedly advanced the throughput of MDMS-SL. A second strategy we applied to analysis of biologic tissues was in the identification and quantitation of CL molecular species. Since, CL is the overwhelmingly predominant class of lipids possessing a double negative charge in most cellular contexts, quantification of the isotopologue pattern using the M+1/2 for analysis was employed [26]. Continued progress in the ability to integrate novel chemical strategies for high throughput targeted analysis, in conjunction with improvements in ion mobility MS, FAIMS and other new technologies will facilitate the continued growth of MDMS-SL and its utility in the identification of novel metabolites, signaling pathways and the elucidation of the chemical mechanisms mediating diverse disease states.
High Mass Accuracy Shotgun Lipidomics
Perhaps one of the most important advances impacting direct infusion mass spectrometry has been the development of high-resolution high mass accuracy (HRAM) mass spectrometry with its extraordinary resolving power (>100,000) and mass accuracy (<2ppm). Shevchenko and coworkers have greatly extended the power of shotgun lipidomics through the use of HRAM mass spectrometry with polarity switching using direct infusion without sacrificing mass accuracy [27]. Similarly, in a recent account by Reid and coworkers, the advantages of an HRAM MS platform for shotgun lipidomics has been rigorously demonstrated [28]. The use of HRAM MS has further extended the power and throughput of shotgun lipidomics allowing identification of ~600 lipid molecular species of lipids from over 30 molecular classes in only 1 to 2 min of data acquisition [28].
Liquid Chromatography-Mass Spectrometry (LC-MS) and Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
The most widely used approach for identification of lipid molecular species and their quantitation employs the initial separation of lipid analytes by liquid chromatography on the bases of hydrophobicity (e.g., RPHPLC) or polarity, (e.g., ion exchange) with the column eluent injected directly into the mass spectrometer. Chromatography is a powerful dimension which has many benefits including the ability to structurally analyze phospholipids or non-polar lipids that are difficult or impossible to do so by direct infusion techniques alone. For example, HPLC can separate cis vs. trans olefins, enantiomers using a chiral chromatography matrix and discriminate species with identical elemental compositions from different phospholipid molecular classes based on retention times. Moreover, the use of liquid chromatography is well-suited to the analysis of lipids in the extremely low abundance regime which is not yet possible with shotgun lipidomics or other direct infusion technologies.
After acquiring LC-MS or LC-MS/MS data for each sample, the results are typically processed by using either targeted or untargeted methods. While targeted strategies are conceptually simpler and often performed by using standard vendor software, the analyses can be tedious and time consuming when the number of lipids being analyzed is large. An alternative approach for investigating hundreds to thousands of lipids unbiasedly is to apply untargeted data processing methods. These methods are global in scope, but it is common for researchers to focus only on those lipid species that have statistically significant changes in concentrations between experimental groups. The major challenge of applying untargeted approaches is a step in the workflow termed “correspondence determination”, which refers to recognizing signals across multiple samples as the same lipid species. Correspondence determination is complicated by experimental drift factors, such as column degradation or retention of moieties in the mobile phase that cause deviations in chromatographic retention times. The challenge of peak alignment is commonly addressed by using the Orbiwarp algorithm [29], which is implemented in the widely used XCMS open-source software program developed by Siuzdak and colleagues [30, 31]. A second challenge in untargeted analyses is that some detected signals do not correspond to real molecules but rather are a result of noise (e.g., random fluctuations in the baseline). The newest generations of software, such as “Warpgroup” developed by the Patti group, identify “consensus signals” that can be detected across all sample replicates and thus dramatically reduce informatic noise in data sets [32]. This leads to difficulties in quantitation without careful attention to the variety of complexes formed during ionization. For example, species can be detected as either the H+, Na+ or NH4+ adduct ions which each have to be measured (or related to appropriate internal standards) to quantify the analyte of interest. A recent untargeted study showed that some species in untargeted LC-MS/MS experiments can be detected as over 100 signals [33]. In contrast, in shotgun lipidomics, judicious choices of adduct ions (e.g., the oxytropic properties of LiCl) are exploited for charge remote fragmentation where only a single adduct ion needs to be considered. While direct infusion approaches contain an identical amount of ion suppression throughout the entire analysis, ion suppression varies widely during the gradients used for HPLC in the mobile phase as well as large differences in ion suppression at various points during peak elution where changes in concentration >100 fold may give rise to ion suppression. Finally, based on known solubility coefficients of monomer vs. micelle, in some studies the concentration of some of the analytes exceeds its monomeric solubility resulting in aggregation and resultant changes in elution times and ionization efficiencies. These factors notwithstanding, LC-MS/MS is the preferred method for lipidomic analyses in the extremely low abundance regime where the salutary aspects of increased analyte concentration during peak elution, differential elution times for cis vs. trans olefin isomers, identification of stereochemistry by chiral chromatography and discrimination of confounding mass overlaps are currently difficult or intractable problems for direct infusion approaches.
Identification and Quantitation of Lipids in the Extremely Low Abundance Regime
The most commonly used methods for quantitation of lipids in the extremely low abundance regime use LC-MS/MS approaches employing either selected ion monitoring (SIM) or multiple reaction monitoring (MRM) [34]. In the case of SIM, ion intensities are calculated from the m/z of known (anticipated) constituents in which the duty cycle of the mass spectrometer is focused on the targeted analyte. A common problem in SIM is the presence of additional analytes that are not separated by the column that are present under the targeted peak(s). These anomalous analytes which coelute with similar masses can only be detected by MS2 full mass scans and even then some structural and quantitative ambiguities remain. Although many investigations have utilized MRM with putative diagnostic transitions, it is clear that the analytic integrity of MRM depends on the fact that ions entering the precursor window (typically set at +/− 1Th) do not generate fragment ions within the window of product ions (typically +/− 1Th) reaching the detector. We and others, have observed multiple problems with LC and MRM where ions of analytes or contaminants of unknown origin have masses and transitions within the designated windows, but do not have accurate masses of the anticipated product ions and thus lead to high false discovery rates and/or inaccurate quantitation [35]. These observations demonstrate that a substantial lack of specificity and quantitative errors occur in many cases where MRMs are used without high mass accuracy examination of product ions. We have addressed this problem by trapping product ions and injecting them into the Orbitrap™ to verify their accurate masses to ensure that the anticipated product ion is measured without contamination of other isobaric ions not related to the molecular ion of interest (Fig 4) [35]. A similar strategy, of course, is readily used with qTOFs where high mass accuracy and high resolution of both the precursor and product ions can be directly obtained. Whatever method is chosen, it is clear that MRMs are subject to error unless additional constraints are applied for product ion identification and/or the use of multiple signature product ion transitions are utilized. This brings into question the accuracy of quantitation when only one transition is used without accurate mass determination of the anticipated product ion as has routinely been performed in the past. The use of high-resolution high mass accuracy analysis of product ions should minimize errors in this regard. Even then, structural ambiguities still remain (e.g. stereochemistry). Due to the ease of extraction of bis-allylic protons, it is also important to determine the stereochemistry of the analyte for the complete definition of the lipidome since oxidation can occur enzymatically (usually resulting in a single or a predominant stereoisomer) or non-enzymatically where a mixture of stereoisomers are present.
Figure 4. Selected Reaction Monitoring of the total ion current chromatograms of targeted oxidized aliphatic chains.
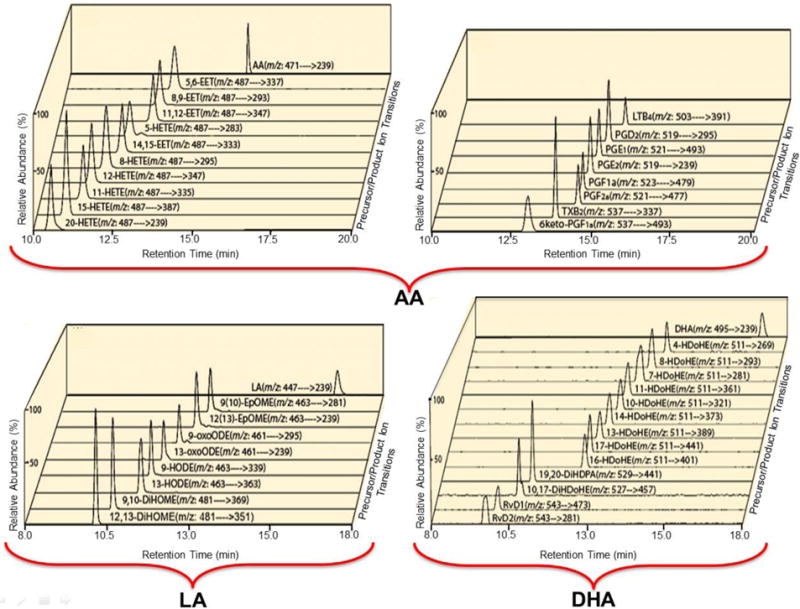
The carboxyl linkage of targeted oxidized aliphatic chains were charge-switch derivatized with aminomethyl phenyl pyridium (AMPP) by condensation with carbodiimide. The resultant charge-switch derivatized oxidized metabolites of linoleic (LA), arachidonic (AA) and docosahexaneoic (DHA) acids possessed ~30-80 fold increases in S/N due both to the switch to the positive ion mode in conjunction with the permanent positive charge in the pyridinium ring. The indicated fatty acid metabolite standards were derivatized with AMPP and measured by LC MS/MS via SRM in the positive ion mode following chromatographic separation using a C18 reverse phase HPLC column. Signature transitions from each of the analytes were used (as shown) and product ions were collected and reinjected into the Orbitrap™ to further substantiate the integrity of the SRM through accurate mass determination of their molecular identity. Representative SRM chromatograms for the standards are shown whose product ions contained the anticipated masses after reinjection into the Orbitrap™.
(Anal. Biochem. 2013 Jul11; 442:40-50. http://dx.doi.org/10.1016/j.ab.2013.06.014 2005 American Chemical Society)
Shotgun Lipidomics vs. Chromatography-based Analyses
Generally speaking, in analytical chemistry there are distinct attributes of different technologies to solve specific analytic problems with differing boundary conditions. For example, shotgun lipidomics is high throughput and extremely accurate for the quantitation of moderate abundance analytes, but is not suitable for studying analysis of analytes in the extremely low abundance regime. Of course this might change with the improvement of front-end technologies which act as de facto high resolution separation platforms such as ion mobility filters, field accelerated ion mobility or the development of other innovative approaches [36]. However, at the present time, the only accurate way to penetrate the extremely low abundance regime of the lipidome is through chromatography-based techniques which now have sophisticated bioinformatic software for peak alignment, accurate determination of the beginning and end of analyte elution and analysis of the multiple adduct ions formed during chromatography. For MRMs, accurate mass analysis of product ions has been shown to be necessary to minimize false discovery rates in the low abundance regime [35]. Alternatively, multiple diagnostic fragmentation patterns can also decrease the false discovery rate.
In practice, for moderately abundant lipids, direct infusion approaches are simple, take only minutes to run, and spectra can be directly compared after normalization to internal standards. Moreover, in MDMS-SL enabling derivatization reactions, spectral enhancements and HRAM mass spectrometry have extended the accuracy and confidence of quantitation by direct spectral comparisons that are preferable in many targeted as well as untargeted mass spectrometric investigations.
Reverse Lipidomics for Discovery of Novel Signaling Pathways
In order to further penetrate the extremely low abundance regime, it is necessary to have pre-knowledge of the molecular mass, signature transitions and chromatographic elution properties of unknown molecules in connected through a metabolic network. To this end, we have described an approach where purified enzymes convert substrates to products oftentimes resulting in previously unknown lipids. Candidate novel natural products, produced in these purified systems often were not previously identified due to their low abundance in a sea of the complexity of hundreds of thousands of lipids. The resulting unknown analytes can be used to determine elution times, signature fragmentation patterns and HRAM MS of molecular and product ions (Fig 5). An example of this approach is the identification of enzyme catalyzed production of oxidized arachidonoyl-lysolipids using mass spectrometric parameters identified from in situ reactions with purified enzymes (e.g., COX-2, 15-lipoxygenase) and lysolipid substrates. Proof of the existence of the enzymatically generated products as bona fide natural products was enabled by using the identified HRAM mass spectrometric parameters, fragmentation patterns, and chiral chromatography to identify these novel metabolites in biologic systems thereby discovering previously unknown lipid signaling pathways [37]. This strategy is related to reverse engineering paradigms which have been used to solve previously intractable problems in other disciplines by similar strategies.
Figure 5. Reverse Lipidomics to Identify Novel COX-2 Reaction Products.
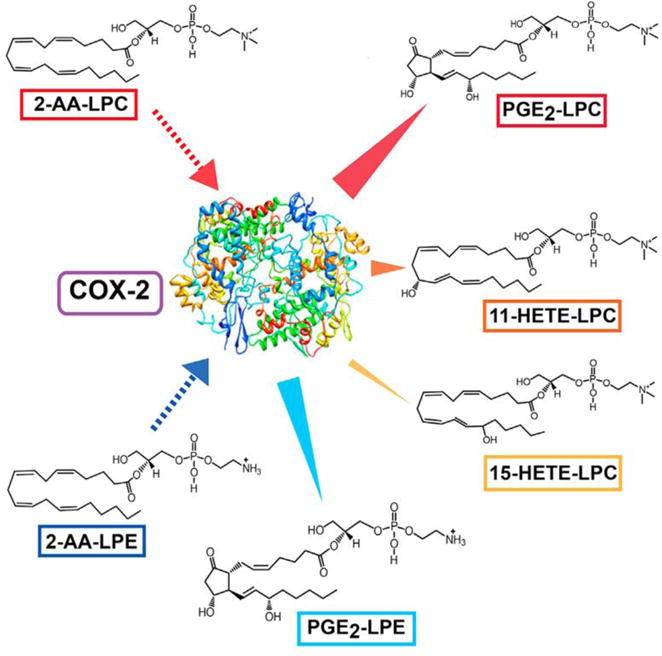
In the large majority of cases, identification of extremely low abundance metabolites requires pre-knowledge of the m/z of the precursor ion, a signature transition and the m/z of the product ion. One way to identify novel natural products is through parallel chemical mechanisms with computer docking of potential substrates and incubate purified enzymes to generate a multiplicity of products with pre-knowledge of their molecular masses and signature transitions. This, in favorable cases, results in a plethora of new enzyme generated products which are now well defined and are likely present in the lipidome if searched for by the appropriate MRMs with accurate mass analysis of product ions and confirmation of identical fragmentation patterns and identical chromatographic retention times. In the case shown above, we reasoned that COX-2 directly oxidizes 2-arachidonoyl-lysophospholipids to eicosanoid-lysolipids and identified the eicosanoid lysolipids emanating from COX-2 mediated catalysis in biologic samples.
(Cell Chem. Biol. 2016 Oct20; 23:1-11, http://dx.doi.org/10.1016/j.chembiol.2016.08.009 2016 Elsevier Ltd.)
The Inherent Limitations of MS-based Lipidomics: Challenges in Space
One obvious limitation of lipidomics is that although it can identify tens to hundreds of thousands of lipid metabolites from tissue, it cannot identify their cell type of origin since biologic tissues are comprised of multiple cell types. Furthermore, analysis of crude lipid extracts cannot determine the distribution of component lipids in subcellular organelles or their spatial juxtaposition to membrane compartments or membrane domains. Moreover, since the metabolic flux of metabolites varies greatly in individual subcellular compartments or membrane domains, their relationship to specific signaling events and/or communication between subcellular compartments cannot be directly accessed. Thus, when tissue is analyzed, lipids are mixed from multiple different cells and intracellular compartments which collectively both confound mechanistic interpretation as well as lose important information that is cell type or subcellular membrane compartment specific. Cell culture experiments can compare differences in specific cells, but cells in culture often have vastly different metabolic phenotypes and lipid compositions than those present in vivo. Furthermore, since each cell has multiple subcellular compartments whose lipids and metabolic flux are different, both in the resting state and after cellular activation, interpretation of data and mechanistic hypotheses must be made with caution. These factors notwithstanding, great progress has been made in identifying alterations in lipid composition and the mechanisms responsible for specific disease states and regulation of critical cellular processes. Through careful choice of tissues, stable isotope labels, and physiologic vs. pathophysiologic conditions, substantial information has already demonstrated the power of lipidomic analyses in mechanistically identifying alterations in cellular regulatory pathways, disease states, and responses to external perturbations.
Imaging mass spectrometry
To begin to resolve the critical issue of compartmentation in discrete regions of biologic tissues, cells and subcellular compartments, substantial efforts have focused on increasing the sensitivity, mass accuracy, resolving power and spatial resolution of imaging mass spectrometry (IMS). Historically, MALDI has been the primary platform for analysis of lipid metabolites and great advances in MALDI-based imaging have been made in the Caprioli group [38]. MALDI matrices with minimal ionization of low molecular weight constituents have demonstrated the power of MALDI for lipid analysis [39]. However, the resolution of MALDI is limited due to the size of the matrix crystals as well as the width of the laser beam needed to obtain adequate S/N. Using a variety of innovative approaches recent studies have demonstrated that MALDI can faithfully examine analytes at the cellular level with a spatial resolution of ~10μm. A second ionization methodology, DESI, was developed by the Cooks group [40]. One of the advantages of DESI is that it is a matrix-free approach that can be performed at ambient pressures to image lipids with ~10 μm resolution. The matrix-free imaging at ambient pressures has led to widespread use of this technology in tissue imaging of lipids [41]. A third IMS technology developed by Siuzdak and coworkers is Nanostructure-Initiator Mass Spectrometry (NIMS) which uses a silicon surface and a choice of initiators emailed ref (43). Through multiplexing various coatings on the silicon surface and different initiators a chemical pallette can be generated to measure a diverse group of compounds with extraordinary sensitivity [42]. A good example of the power of this approach is the identification of highly spatially localized analytes in the brain [43]. Its application to human disease was substantiated in identifying alterations in cholesterol metabolites and distribution in the Smith Lemli-Opitz syndrome [44].
Finally, secondary ion MS (SIMS) uses a high-energy ion beam to desorb and directly generate secondary ions from the surface. SIMS currently has the best resolution (~50nm) but the high energy of the ion beam typically results in extensive fragmentation of analytes. For example, the Winograd group has used high energy primary ion beams for the desorption of secondary ions that are analyzed by TOF MS. Through rastering the ion beam, an image of the chemical constituents at the surface is generated. In favorable cases, the lateral resolution and sensitivity can detect lipids at attomolar concentrations with resolution at the 50 nm scale [45].
The Inherent Limitations of MS-based Lipidomics: Challenges in Time
One previous difficulty has been interpretation of stable isotope labeling patterns since bioinformatic solutions to stable isotope flux were not previously available. Recently, the Patti group has developed a powerful bioinformatics approach designated 13CXCMS [32] to combine stable isotope labeling with untargeted metabolomic tools to unbiasedly track the fates of lipids. This bioinformatics approach is a highly effective tool to track the metabolic fate of lipids, alterations in substrate usage, the impact of individual enzymes in genetically engineered animals, alterations in metabolic flux after receptor stimulation and changes in metabolic flux in disease states. This enabling approach allows detailed analyses of the metabolic flux of specific molecules in total cells, but defining differences in turnover rates in specific subcellular organelles remains challenging. One approach is to measure the flux of lipids which are not compartmentalized or are confined to a single compartment. A variety of IMS approaches with stable isotopes will yield critical mechanistic information on the metabolic flux of discrete lipids in specialized subcellular compartments.
Concluding Remarks
The growth of lipidomics over the last 35 years has been remarkable allowing identification and quantitation of many thousands of lipids with a remarkable degree of reproducibility and precision. Many innovative approaches have been taken, the technology of the discipline has advanced beyond all expectations and the increasing roles of bioinformatics in data analysis have greatly accelerated progress in the field. The remaining issues of the coordinated regulation of lipid alterations in space and time are quite challenging, but given the advances in mass spectrometry-based lipidomics made over the last 35 years innovative approaches to further the understanding of alterations of the roles of lipids in health and disease are close at hand.
Supplementary Material
Highlights.
The foundations of mass spectrometry-based lipidomics have evolved over 4 decades.
The emergence of lipidomics has been catalyzed by development of new ionization techniques.
Advances in resolution and mass accuracy facilitated the growth of the field.
Lipidomic data analysis requires consideration of flux and spatial proximity.
Reverse lipidomics can identify unknown lipid signaling pathways.
Acknowledgments
This work was supported by NIH grants RO1HL118639 and RO1HL133178.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gross RW, Sobel BE. Augmentation of cardiac phospholipase activity induced with negative liposomes. Trans Assoc Am Physicians. 1979;92:136–147. [PubMed] [Google Scholar]
- 2.Gross RW, Sobel BE. Isocratic high-performance liquid chromatography separation of phosphoglycerides and lysophosphoglycerides. J Chromatogr. 1980;197:79–85. doi: 10.1016/s0021-9673(00)80538-5. [DOI] [PubMed] [Google Scholar]
- 3.Williams DH, Bojesen G, Auffret AD, Taylor LC. Study of ‘difficult peptides’ from Paracoccus cytochrome c-550 and a dolphin cytochrome c. Fast atom bombardment: a new method for molecular weight and sequence determination of peptides. FEBS Lett. 1981;128:37–39. doi: 10.1016/0014-5793(81)81073-3. [DOI] [PubMed] [Google Scholar]
- 4.Gross RW. High plasmalogen and arachidonic acid content of canine myocardial sarcolemma: a fast atom bombardment mass spectroscopic and gas chromatography-mass spectroscopic characterization. Biochemistry. 1984;23:158–165. doi: 10.1021/bi00296a026. [DOI] [PubMed] [Google Scholar]
- 5.Gross RW. Identification of plasmalogen as the major phospholipid constituent of cardiac sarcoplasmic reticulum. Biochemistry. 1985;24:1662–1668. doi: 10.1021/bi00328a014. [DOI] [PubMed] [Google Scholar]
- 6.Hazen SL, Hall CR, Ford DA, Gross RW. Isolation of a human myocardial cytosolic phospholipase A2 isoform. Fast atom bombardment mass spectroscopic and reverse-phase high pressure liquid chromatography identification of choline and ethanolamine glycerophospholipid substrates. J Clin Invest. 1993;91:2513–2522. doi: 10.1172/JCI116487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glaser PE, Gross RW. Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry. 1994;33:5805–5812. doi: 10.1021/bi00185a019. [DOI] [PubMed] [Google Scholar]
- 8.Gubitosi-Klug RA, Yu SP, Choi DW, Gross RW. Concomitant acceleration of the activation and inactivation kinetics of the human delayed rectifier K+ channel (Kv1.1) by Ca(2+)-independent phospholipase A2. J Biol Chem. 1995;270:2885–2888. doi: 10.1074/jbc.270.7.2885. [DOI] [PubMed] [Google Scholar]
- 9.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 10.Han X, Gross RW. Electrospray ionization mass spectroscopic analysis of human erythrocyte plasma membrane phospholipids. Proc Natl Acad Sci U S A. 1994;91:10635–10639. doi: 10.1073/pnas.91.22.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han X, Gross RW. Structural determination of picomole amounts of phospholipids via electrospray ionization tandem mass spectrometry. J Am Soc Mass Spectrom. 1995;6:1202–1210. doi: 10.1016/1044-0305(95)00568-4. [DOI] [PubMed] [Google Scholar]
- 12.Han X, Gross RW. Structural determination of lysophospholipid regioisomers by electrospray ionization tandem mass spectrometry. J Am Chem Soc. 1996;118:451–457. [Google Scholar]
- 13.Han X, Gubitosi-Klug RA, Collins BJ, Gross RW. Alterations in individual molecular species of human platelet phospholipids during thrombin stimulation: electrospray ionization mass spectrometry-facilitated identification of the boundary conditions for the magnitude and selectivity of thrombin-induced platelet phospholipid hydrolysis. Biochemistry. 1996;35:5822–5832. doi: 10.1021/bi952927v. [DOI] [PubMed] [Google Scholar]
- 14.Ford DA, Han X, Horner CC, Gross RW. Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry. 1996;35:7903–7909. doi: 10.1021/bi960552n. [DOI] [PubMed] [Google Scholar]
- 15.Han X, Abendschein DR, Kelley JG, Gross RW. Diabetes-induced changes in specific lipid molecular species in rat myocardium. Biochem J. 2000;352(Pt 1):79–89. [PMC free article] [PubMed] [Google Scholar]
- 16.Han X, Gross RW. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 17.Han X, Yang K, Gross RW. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev. 2012;31:134–178. doi: 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brugger B, Erben G, Sandhoff R, Wieland FT, Lehmann WD. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc Natl Acad Sci U S A. 1997;94:2339–2344. doi: 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ekroos K, Chernushevich IV, Simons K, Shevchenko A. Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal Chem. 2002;74:941–949. doi: 10.1021/ac015655c. [DOI] [PubMed] [Google Scholar]
- 20.Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44:16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]
- 21.Yang K, Jenkins CM, Dilthey B, Gross RW. Multidimensional mass spectrometry-based shotgun lipidomics analysis of vinyl ether diglycerides. Anal Bioanal Chem. 2015;407:5199–5210. doi: 10.1007/s00216-015-8640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang K, Dilthey BG, Gross RW. Shotgun Lipidomics Approach to Stabilize the Regiospecificity of Monoglycerides Using a Facile Low-Temperature Derivatization Enabling Their Definitive Identification and Quantitation. Anal Chem. 2016;88:9459–9468. doi: 10.1021/acs.analchem.6b01862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bollinger JG, Thompson W, Lai Y, Oslund RC, Hallstrand TS, Sadilek M, Turecek F, Gelb MH. Improved sensitivity mass spectrometric detection of eicosanoids by charge reversal derivatization. Anal Chem. 2010;82:6790–6796. doi: 10.1021/ac100720p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang K, Dilthey BG, Gross RW. Identification and quantitation of fatty acid double bond positional isomers: a shotgun lipidomics approach using charge-switch derivatization. Anal Chem. 2013;85:9742–9750. doi: 10.1021/ac402104u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fhaner CJ, Liu S, Zhou X, Reid GE. Functional group selective derivatization and gas-phase fragmentation reactions of plasmalogen glycerophospholipids. Mass Spectrom (Tokyo) 2013;2:S0015. doi: 10.5702/massspectrometry.S0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han X, Yang K, Yang J, Cheng H, Gross RW. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J Lipid Res. 2006;47:864–879. doi: 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuhmann K, Almeida R, Baumert M, Herzog R, Bornstein SR, Shevchenko A. Shotgun lipidomics on a LTQ Orbitrap mass spectrometer by successive switching between acquisition polarity modes. J Mass Spectrom. 2012;47:96–104. doi: 10.1002/jms.2031. [DOI] [PubMed] [Google Scholar]
- 28.Ryan E, Reid GE. Chemical Derivatization and Ultrahigh Resolution and Accurate Mass Spectrometry Strategies for “Shotgun” Lipidome Analysis. Acc Chem Res. 2016;49:1596–1604. doi: 10.1021/acs.accounts.6b00030. [DOI] [PubMed] [Google Scholar]
- 29.Prince JT, Marcotte EM. Chromatographic alignment of ESI-LC-MS proteomics data sets by ordered bijective interpolated warping. Anal Chem. 2006;78:6140–6152. doi: 10.1021/ac0605344. [DOI] [PubMed] [Google Scholar]
- 30.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahieu NG, Genenbacher JL, Patti GJ. A roadmap for the XCMS family of software solutions in metabolomics. Curr Opin Chem Biol. 2016;30:87–93. doi: 10.1016/j.cbpa.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahieu NG, Spalding JL, Patti GJ. Warpgroup: increased precision of metabolomic data processing by consensus integration bound analysis. Bioinformatics. 2016;32:268–275. doi: 10.1093/bioinformatics/btv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahieu NG, Spalding JL, Gelman SJ, Patti GJ. Defining and Detecting Complex Peak Relationships in Mass Spectral Data: The Mz.unity Algorithm. Anal Chem. 2016;88:9037–9046. doi: 10.1021/acs.analchem.6b01702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou W, She J, Tolstikov VV. A comprehensive workflow of mass spectrometry-based untargeted metabolomics in cancer metabolic biomarker discovery using human plasma and urine. Metabolites. 2013;3:787–819. doi: 10.3390/metabo3030787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Moon SH, Mancuso DJ, Jenkins CM, Guan S, Sims HF, Gross RW. Oxidized fatty acid analysis by charge-switch derivatization, selected reaction monitoring, and accurate mass quantitation. Anal Biochem. 2013;442:40–50. doi: 10.1016/j.ab.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng X, Renslow RS, Makola MM, Webb IK, Deng L, Thomas DG, Govind N, Ibrahim YM, Kabanda MM, Dubery IA, Heyman HM, Smith RD, Madala NE, Baker ES. Structural Elucidation of cis/trans Dicaffeoylquinic Acid Photoisomerization Using Ion Mobility Spectrometry-Mass Spectrometry. J Phys Chem Lett. 2017;8:1381–1388. doi: 10.1021/acs.jpclett.6b03015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Moon SH, Jenkins CM, Sims HF, Gross RW. Cyclooxygenase-2 Mediated Oxidation of 2-Arachidonoyl-Lysophospholipids Identifies Unknown Lipid Signaling Pathways. Cell Chem Biol. 2016;23:1217–1227. doi: 10.1016/j.chembiol.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reyzer ML, Caprioli RM. MALDI-MS-based imaging of small molecules and proteins in tissues. Curr Opin Chem Biol. 2007;11:29–35. doi: 10.1016/j.cbpa.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 39.Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis of cellular glycerophospholipids enabled by multiplexed solvent dependent analyte-matrix interactions. Anal Chem. 2008;80:7576–7585. doi: 10.1021/ac801200w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 41.Eberlin LS, Ferreira CR, Dill AL, Ifa DR, Cooks RG. Desorption electrospray ionization mass spectrometry for lipid characterization and biological tissue imaging. Biochim Biophys Acta. 2011;1811:946–960. doi: 10.1016/j.bbalip.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yanes O, Woo HK, Northen TR, Oppenheimer SR, Shriver L, Apon J, Estrada MN, Potchoiba MJ, Steenwyk R, Manchester M, Siuzdak G. Nanostructure initiator mass spectrometry: tissue imaging and direct biofluid analysis. Anal Chem. 2009;81:2969–2975. doi: 10.1021/ac802576q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanisevic J, Epstein AA, Kurczy ME, Benton PH, Uritboonthai W, Fox HS, Boska MD, Gendelman HE, Siuzdak G. Brain region mapping using global metabolomics. Chem Biol. 2014;21:1575–1584. doi: 10.1016/j.chembiol.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patti GJ, Shriver LP, Wassif CA, Woo HK, Uritboonthai W, Apon J, Manchester M, Porter FD, Siuzdak G. Nanostructure-initiator mass spectrometry (NIMS) imaging of brain cholesterol metabolites in Smith-Lemli-Opitz syndrome. Neuroscience. 2010;170:858–864. doi: 10.1016/j.neuroscience.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian H, Wucher A, Winograd N. Reducing the Matrix Effect in Organic Cluster SIMS Using Dynamic Reactive Ionization. J Am Soc Mass Spectrom. 2016;27:2014–2024. doi: 10.1007/s13361-016-1492-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.