

Abstract
The negatively charged nonulose sialic acid (Sia) is essential for murine development in vivo. In order to elucidate the impact of sialylation on differentiation processes in the absence of maternal influences, we generated mouse embryonic stem cell (mESC) lines that lack CMP‐Sia synthetase (CMAS) and thereby the ability to activate Sia to CMP‐Sia. Loss of CMAS activity resulted in an asialo cell surface accompanied by an increase in glycoconjugates with terminal galactosyl and oligo‐LacNAc residues, as well as intracellular accumulation of free Sia. Remarkably, these changes did not impact intracellular metabolites or the morphology and transcriptome of pluripotent mESC lines. Moreover, the capacity of Cmas −/− mESCs for undirected differentiation into embryoid bodies, germ layer formation and even the generation of beating cardiomyocytes provides first and conclusive evidence that pluripotency and differentiation of mESC in vitro can proceed in the absence of (poly)sialoglycans.
Keywords: CMP-sialic acid synthase, differentiation, glycosylation, metabolism, sialic acids
Introduction
Every mammalian cell is coated by a glycocalyx, which consists of a multitude of glycans linked to proteins or lipids embedded in the cell surface. Glycans are potent regulators of numerous cellular functions. Interference with lipid glycosylation or incorrect N‐ or O‐linked glycosylation of proteins results in developmental defects and foetal loss.1 The negatively charged nine‐carbon sugar sialic acid (Sia) forms a terminal saccharide in most surface‐expressed and soluble glycoconjugates. Biosynthesis of Sia starts in the cytosol and is driven by the bi‐functional enzyme UDP‐N‐acetylglucosamine (UDP‐GlcNAc) 2‐epimerase/N‐acetylmannosamine (ManNAc) kinase (gene: Gne), which catalyses the epimerisation and subsequent phosphorylation of UDP‐GlcNAc to ManNAc‐6‐P (Scheme 1). Neu5Ac‐9‐phosphate synthase metabolises ManNAc‐6‐P and phosphoenolpyruvate (PEP) to N‐acetylneuraminic acid (Neu5Ac)‐9‐phosphate, which is dephosphorylated to the free monosaccharide Neu5Ac by Neu5Ac‐9‐phosphate phosphatase.
Scheme 1.
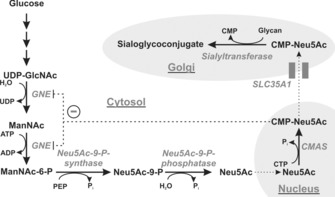
Biosynthesis of sialoglycoconjugates in vertebrates. Enzymes are italicised: UDP‐GlcNAc 2‐epimerase/ManNAc kinase (GNE), CMP‐sialic acid synthetase (CMAS), CMP‐sialic acid transporter (SLC35A1). Cell organelles are shaded in grey. CMP‐Neu5Ac is a feedback inhibitor of GNE (−).
Activation of Neu5Ac to its cytidine‐monophosphate diester is catalysed by the nuclear‐located CMP‐Sia synthase (CMAS, E.C. 2.7.7.43). CMP‐Neu5Ac is transported into the Golgi apparatus by the CMP‐Sia transporter (gene: Slc35a1), where Sia is transferred onto nascent glycoconjugates by sialyltransferases in α2,3‐ or α2,6‐linkage onto glycans terminated with either galactose (Gal) or N‐acetylgalactosamine (GalNAc), or in α2,8‐linkage to terminal Sia. Furthermore, polysialyltransferases can build Sia homopolymers in α2,8‐linkage (polySia) onto sialyl residues. In addition to its function as donor sugar, CMP‐Sia also serves as a feedback inhibitor for GNE and thus regulates de novo biosynthesis of Sia.2
At the cell surface Sia can serve as a receptor recognised by ligands such as lectins, but it can also mask underlying structures to impede the binding of, for example, Gal‐specific receptors.3 Aberrant sialylation has been associated with renal failure,4 myopathies,5 auto‐immune diseases6 and metastasis of cancer.7 Most importantly, mammalian development strictly depends on the integrity of the sialylation pathway. Blocking Sia de novo biosynthesis (e.g., mouse Gne knockout) is lethal around midgestation (E10),8 and interfering with 9‐O‐acetylation of Sia arrests embryonic development at the two‐cell stage.9 Despite its great influence on mammalian embryogenesis, the mechanisms by which Sia impedes embryonic development are still unclear.
Embryonic stem cells (ESCs) are useful to address in vitro the mechanisms of intrinsic early embryonic differentiation. ESCs can be obtained either from blastocysts10 or as induced pluripotent cells (iPSCs) from reprogrammed somatic cells.11 They can be differentiated in vitro into embryoid bodies (EBs), which reflect formation and development of the early embryo. Only a few studies have addressed the impact of sialylation on ESC biology, and different observations have been made between human and mouse ESCs. In human ESC lines hyposialylation induced by knockdown of ST6GAL1 (α2,6‐sialyltransferase) led to alterations in cellular pluripotency and impeded reprogramming of somatic cells;12 however, hyposialylation in murine ESC (mESC) following loss of GNE activity did not impact pluripotency.13 Nevertheless, in the latter, alterations in the expression of differentiation markers in Gne‐knockout mESC suggested a role for sialylation in differentiation.14 In these studies, either only α2,6‐Sia or the entire cell‐surface sialylation was reduced, thus possibly accounting for the divergent results. We therefore sought to analyse mESC development upon complete loss of sialylation. As CMP‐Sia is the donor substrate for all sialyltransferases, we interfered with CMP‐Sia biosynthesis by genetic ablation of CMAS in murine mESCs, and we demonstrated that the early steps in murine embryonic development in vitro proceed independently from sialoglycoconjugates.
Results
Generation and biochemical characterisation of CMASdeficient murine ESC
Genetic inactivation of Cmas in murine blastocysts was accomplished by excision of exon 4, which encodes residues essential for enzymatic activity (Figure 1 A),15 followed by confirmation of homologous integration (Figure 1 B). We used blastocysts 3.5 days after fertilisation (E3.5) from two heterozygous (Cmas +/−) matings to independently generate three wild‐type (WT, Cmas +/+), eight heterozygous (Cmas +/−) and four homozygous (Cmas −/−) mESC lines (genotyping described in Figure S1 in the Supporting Information). In order to confirm successful interference of Cmas expression, mRNA levels were monitored by quantitative PCR (qPCR, Figure 1 C). The expression of Cmas mRNA in Cmas +/− mESCs was clearly reduced compared to WT mESCs; in Cmas −/− mESCs, no Cmas mRNA signal was obtained (Figure 1 C). Consistently, CMAS protein expression was reduced in Cmas +/− and absent in Cmas −/− mESCs, thus indicating successful genetic inactivation (Figure 1 D).
Figure 1.
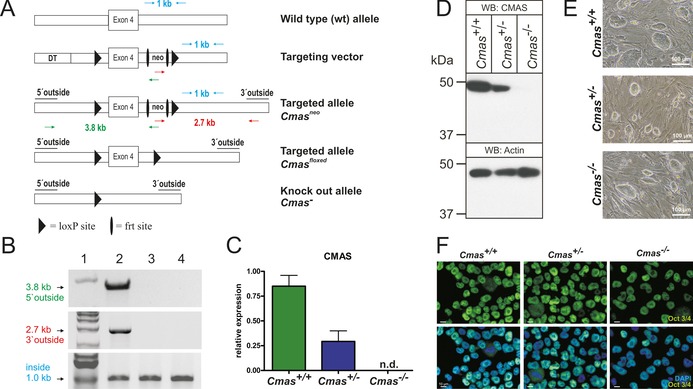
Targeting strategy and characterisation of CMAS‐deficient mESC. A) Cmas targeting strategy. Targeting vector with diphtheria toxin cassette (DT) to increase homologous recombination, frt‐flanked neomycin resistance cassette (neo), exon 4 and neo flanked by LoxP sites. Correct homologous integration of the targeting vector into ES cells was confirmed by the neo and a 5′ outside primer pair (green arrows; amplifying a 3.8 kb fragment) and by neo and a 3′ outside primer pair (red arrows; amplifying a 2.7 kb fragment). Inside primers were used as a control (light blue arrows; 1 kb fragment). Correctly targeted mESCs were used to generate mutant mice. The neo cassette was deleted by inter‐crosses with ACTFLPe mice, and exon 4 and the remaining frt site were deleted by crosses with Zp3‐cre mice, thereby resulting in the Cmas knock‐out allele. B) PCR analysis of homologous integration. A PCR product of 3.8 kb was amplified with neo‐ and 5′ outside primers (green in A), and a 2.7 kb product was amplified with neo‐ and 3′ outside primers (red in A). Both fragments occurred correctly only in the targeted Cmas neo mESC (lane 2), not in wild‐type mESC (lane 3) or in wild‐type mouse tail tissue (lane 4). A 1 kb PCR fragment from inside primers served as control for the PCR reaction (blue in A). 1 kb marker in lane 1. C) Quantitative PCR of Cmas expression from feeder‐free cultures of Cmas +/+, Cmas +/− and Cmas −/− mESCs (n=3 of one representative cell line with the respective genotype). n.d.=not detectable. D) Cell lysates of Cmas +/+, Cmas +/− and Cmas −/− mESC were separated by SDS‐PAGE, blotted and immunostained with anti‐CMAS antibody. The 48 kDa CMAS protein was detected in Cmas +/+ and Cmas +/− lysates but not in Cmas −/−. Anti‐actin staining was used as loading control. E) Morphology of Cmas +/+, Cmas +/− and Cmas −/− mESC cultured on MEFs supplemented with LIF. F) Indirect immunofluorescence staining of Oct3/4 in Cmas +/+, Cmas +/− and Cmas −/− mESC cultured feeder‐free with LIF supplementation. Representative results from one cell line per genotype are shown in D)–F).
When cultured on murine embryonic fibroblasts, the morphology of Cmas −/− mESC was indistinguishable from that of WT or Cmas +/−, with characteristic round stem‐cell colonies (Figure 1 E). Indirect immunofluorescence staining of the key pluripotency octamer binding transcription factor3/4 (Oct3/4, also known as Pou5f1) was performed to verify the mESC identity of the isolated cell lines. Oct3/4 was expressed with nuclear localisation in WT and in heterozygous and homozygous mutants (Figure 1 F). In order to evaluate if genetic ablation of CMAS affects pluripotency at the transcriptional level, expression of the pluripotency‐sustaining factors Oct3/4, SRY (sexdetermining region Y)‐box 2 (Sox2), and homeobox protein Nanog was investigated by qPCR. No significant genotypedependent differences were observed (Figure S2). Thus, we successfully generated pluripotent WT and hetero‐ and homozygous CMAS‐deficient mESC lines.
Genetic ablation of CMAS results in complete loss of cell‐surface sialylation with concomitant increase in LacNAc structures
Although there are some convincing arguments (and cell models) to support the hypothesis that CMAS is the only enzyme producing activated Sia as the donor sugar for sialyltransferases, a definitive proof has not been provided. With the new cell models we addressed this question by comparing cell‐surface sialylation of the isolated pluripotent mESC lines. Glycolipids were addressed by direct immunofluorescence analysis of the monosialotetrahexosylganglioside GM1 with FITC‐conjugated cholera toxin subunit B (CTXB; Figure 2 A). CTXB specifically binds to GM1 with high affinity, but does not recognise the asialo counterpart GA1.16 Binding of FITC‐conjugated CTXB was observed in WT and Cmas +/− mESC lines with a similar pattern, however, Cmas −/− mESCs did not show CTXB reactivity, thus revealing loss of the sialylated a‐series ganglioside GM1 (Figure 2 A).
Figure 2.
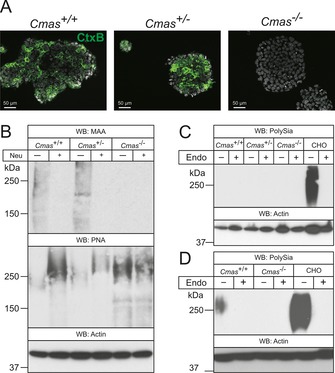
Cmas −/− mESC lack Sia on gangliosides and proteins. A) Direct immunofluorescence staining of the ganglioside GM1 with FITC‐conjugated CTXB in undifferentiated mESC lines. B) Whole‐cell lysates of Cmas +/+, Cmas +/− and Cmas −/− mESC were analysed before and after neuraminidase (Neu) treatment by SDS‐PAGE and western blotting. Detection of α2,3‐linked sialic acids was performed with Maackia amurensis agglutinin (MAA); galactose‐capped glycans were stained with peanut agglutinin (PNA). Cell lysates of C) undifferentiated Cmas +/+, Cmas +/− and Cmas −/− mESCs, and D) differentiated WT and CMAS‐depleted mESC were analysed with or without prior endoneuraminidase NF treatment (Endo) by SDS‐PAGE and western blotting. Detection of polysialic acid was performed with mAb 735. Anti‐actin staining was used as loading control. In order to maintain the pluripotent state, mESC lines were cultured feeder‐free with LIF supplementation. For undirected monolayer differentiation, mESC lines were cultivated for eleven days without LIF supplementation. Representative results from one cell‐line per genotype are shown.
Protein sialylation in mESC lines was investigated by western blotting and lectin analysis (Figure 2 B). Terminal α2,3‐linked Sia residues in WT and Cmas +/− mESC lysates stained with Maackia amurensis lectin (MAA) showed a similar pattern with similar intensity. Removal of terminal Sia by pretreatment with neuraminidase (Neu) abolished these signals and exposed the common penultimate disaccharide galactose‐β1,3‐N‐acetylgalactosamine, which is recognised by peanut agglutinin (PNA). In contrast, homozygous Cmas −/− mESCs were not stained by the Sia‐specific lectin MAA; however, increased PNA reactivity without pretreatment and equal reactivity with PNA before and after Neu treatment indicated increased asialo‐glycoproteins on the cell surface. Furthermore, polySia (a further indicator of surface sialylation) was only detected in differentiated WT mESCs and in control lysates of Chinese hamster ovary (CHO) cells, but not in the Cmas −/− mESC lines (Figure 2 C, D). Specificity of the polySia signal was controlled by endoneuraminidase NF (endoNF) treatment.
In order to confirm the asialo phenotype and to monitor global alterations of cell‐surface glycosylation, the N‐glycome was investigated by high performance multiplexed capillary gel electrophoresis with laser‐induced fluorescence detection (xCGE‐LIF).17, 18 This highly sensitive method allows detecting minor changes in the overall N‐glycosylation pattern. The identity of N‐glycans was verified by comprehensive exoglycosidase treatment and annotated according to our internal N‐glycan database (Figure S3 A). Identical N‐glycosylation patterns were observed in WT and Cmas +/− mESC lines, thus indicating sialylation at wild‐type level despite reduced Cmas expression in Cmas +/− mESCs (Figure S3 B). In contrast, α2,3‐ and α2,6‐sialylated N‐glycans were absent in Cmas −/− mESCs (1–6 in Figure 3 A), and sialidase treatment did not result in any change of the electropherogram of Cmas −/− mESC (Figure 3 B), thus supporting that Cmas −/− mESC are not able to sialylate glycans. Concurrently with the loss of sialylated N‐glycans in Cmas −/− mESCs, the signal intensities of N‐glycans capped with galactose increased (Figure 3 A, peaks 7–10), and oligo‐LacNAc bearing glycans appeared (Figure 3 C, 11–15).
Figure 3.
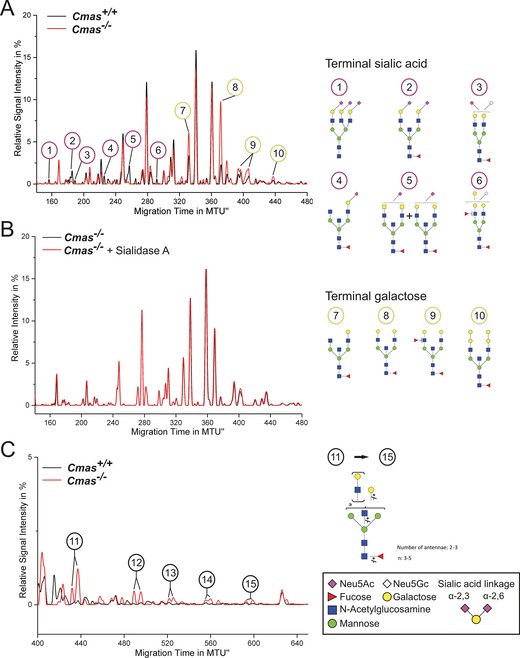
Loss of CMAS activity in mESCs entails increased exposure of galactose and oligo‐LacNAc residues at the cell surface. The y‐axis of normalised electropherogram is divided by the summed peak height of all quantifiable peaks (S/N≥9); relative signal intensity [%] of total peak height is plotted. A) and C) Differential xCGE‐LIF analysis of N‐glycans from undifferentiated Cmas +/+ and Cmas −/− mESCs: electropherogram regions from A) 140 to 480 normalised migration time units (MTU) and C) 400 to 650 MTU. B) Electropherogram of undifferentiated Cmas −/− mESCs before (black) and after sialidase treatment (red). N‐Glycan structures in xCGE‐LIF analyses are annotated: sialylated N‐glycans (1–6), galactose capped N‐ glycans (7–10) and oligo‐LAcNAc capped N‐glycans (11–15). Example N‐glycan structures 1 to 15 are depicted (right; detailed N‐glycan annotation in Figure S4). Activity and specificity of A. urefaciens sialidase was confirmed by treatment of bovine fetuin and subsequent xCGE‐LIF analysis of the well‐defined N‐glycans (Figure S3 A). Structures are presented following the Consortium for Functional Glycomics notation (www.functionalglycomics.org/glycomics/molecule/jsp/carbohydrate/carbMoleculeHome.jsp). Linkage positions of sialic acids are indicated by differing angles. All mESC lines were cultured feeder‐free with LIF supplementation to maintain the pluripotent state (Cmas +/+ n=3, Cmas +/− n=5, Cmas −/− n=4). Representative results from one cell line per genotype are shown.
Taken together, our data show that activation of Sia to CMP‐Sia exclusively relies on CMAS and is crucial for the biosynthesis of sialylated glycoconjugates. Furthermore, ablation of CMAS alters the mESC glycome by replacing usually sialylated N‐glycans with galactose and oligo‐LacNAc‐capped structures. As heterozygous (Cmas +/−) mESCs show a WT sialylation pattern we focused next on WT and Cmas −/− mESC lines.
CMAS activity is essential for maintaining cellular homeostasis of sialic acid biosynthesis
In order to analyse the consequences of Cmas knockout on intracellular metabolite concentration, we first quantified Sia levels by labelling with 1,2‐diamino‐4,5‐methylenedioxy‐benzene (DMB) and HPLC analysis.19 Different Sia derivatives, for example Neu5Ac, 5‐N‐glycolylneuraminic acid (Neu5Gc) and 5‐N‐acetyl‐9‐O‐acetylneuraminic acid, were used as standards (Figure 4 A, top). Neu5Ac appeared as the predominant Sia derivative in pluripotent WT and in Cmas −/− mESCs (Figure 4 A, bottom, dotted line). As expected, Cmas −/− mESCs exhibited substantial cytosolic accumulation of Neu5Ac (Figure 4 A, bottom, solid line). Peak quantification revealed an almost 30‐fold increase compared to WT (Figure 4 B). In order to confirm this metabolic effect in a second model we tested WT and CMAS −/− human embryonic kidney (HEK 293) cells (Figure 4 C): there was even greater accumulation (200‐fold increase of intracellular Neu5Ac). Although the absolute size of the Sia pool in the cytosol might be cell type‐ or species‐specific, our data are in perfect agreement with the described regulatory function of CMP‐Sia in de novo biosynthesis.
Figure 4.
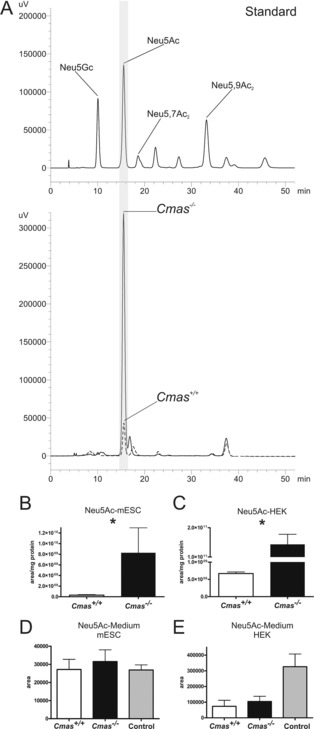
Cmas −/− mESC accumulate intracellular Neu5Ac. A) Reversed‐phase HPLC elution profile of the DMB‐labelled Sia derivatives. Sia standards (top) and samples obtained from intracellular metabolite extracts (bottom) of Cmas +/+ (‐ ‐ ‐ ‐) and Cmas −/− (—) mESCs. B) and C) Quantification of intracellular Neu5Ac by integration of peak areas from RP‐HPLC analysis of B) Cmas +/+ and Cmas −/− mESCs and C) CMAS +/+ and CMAS −/− HEK293 cells (peak area per mg protein; Student's t‐test * p<0.05, n=3 with three technical replicates per biological replicate). Generation and biochemical characterisation of CMAS −/− HEK293 cells in Figure S4. Quantification of free Neu5Ac in the cell culture supernatant of WT and CMAS‐deficient D) mESCs and E) HEK cells by integration of peak areas of DMB‐derivatised Sia from RP‐HPLC analyses (Student's t‐test * p<0.05, n=2). Cell‐free medium was used as control. All mESC lines were cultured feeder‐free with LIF supplementation to maintain the pluripotent state.
Notably, the Sia accumulation in our Cmas −/− cell lines is highly reminiscent of what has been described in sialuria patients, a clinical consequence of Gne mutations that prevent feedback inhibition by CMP‐Sia. Because sialuria patients excrete Neu5Ac in high concentration (up to 15 000 μmol per mmol creatinine; normal <74 μmol mmol−1),20 we analysed if Cmas −/− mESCs release Sia into the cell‐culture medium. After two days of cultivation, samples were taken from WT and knockout cells, and Sia concentrations were compared to that in fresh medium. To our surprise, there was no significant difference in Sia release between mutant and WT cells, either in ESCs (Figure 4 D) or HEK 293 cells (Figure 4 E). Remarkably, independent of HEK 293 genotype, these cells deprived Sia from the medium, an effect that needs further investigation.
Intracellular Neu5Ac accumulation alters neither associated metabolites nor intracellular glycosylation
Because of the massively elevated cytosolic Neu5Ac concentrations in Cmas‐knockout cells, we assumed that other metabolites and/or pathways would also be changed(Figure 5 A). Neu5Ac can be catabolised to ManNAc and pyruvate by the action of N‐acetylneuraminate pyruvate lyase.21 The key metabolite, pyruvate, can be used in the tricarboxylic acid (TCA) cycle or converted to lactate by lactate dehydrogenase (LDH). However, compared to the respective WT lines, neither intracellular nor secreted lactate levels were significantly altered in CMAS‐deficient mESC or HEK 293 cells (Figure 5 B, C). Similarly, no genotype‐related changes could be detected for the TCA cycle metabolites citrate, aconitate, α‐ketoglutarate, succinate, malate and fumarate (Figure 5 D–I).
Figure 5.
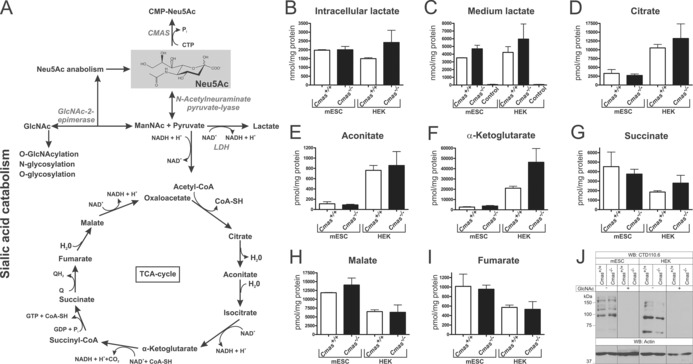
Increased levels of Neu5Ac affect neither related metabolite levels nor global O‐GlcNAcylation. A) Sialic acid anabolism and catabolism in mammals. Enzymes are italicised: CMP‐Sia synthetase (CMAS), lactate dehydrogenase (LDH). Quantification of B) intracellular and C) cell‐culture lactate levels from WT and CMAS‐deficient mESC and HEK metabolite extracts by HPLC‐MS/MS (nmol per mg protein). D–I) Quantification of intracellular TCA cycle metabolite levels from WT and CMAS‐depleted mESC and HEK metabolite extracts by HPLC‐MS/MS (pmol per mg protein). J) Analysis of global O‐GlcNAcylation. Whole‐cell lysates of WT and CMAS‐deficient mESC and HEK cells were analysed by SDS‐PAGE and western blotting with mAb CTD110.6. Specificity of antibody staining was controlled by preincubation of the primary antibody with N‐Acetylglucosamine (GlcNAc). Anti‐actin staining was used as loading control. All mESC lines were cultured feeder‐free with LIF supplementation to maintain the pluripotent state (Cmas +/+ mESC n=2, Cmas −/− mESC n=3, for all HEK cell lines n=3).
We then investigated the intracellular O‐GlcNAcylation outcome. This pathway was researched because we speculated that the elevated Sia levels might drive ManNAc epimerisation to GlcNAc by the action of GlcNAc‐2‐epimerase. GlcNAc in turn can be phosphorylated and activated to UDP‐GlcNAc, which is further utilised for N‐, O‐ and glucosaminoglycan (GAG) biosynthesis as well as intracellular O‐GlcNAcylation (Figure 5 A). In western blot analysis of whole‐cell lysates by using the O‐GlcNAc specific CTD110.6 antibody, highly similar staining patterns (including band intensities) were observed between Cmas −/− and WT cells (Figure 5 J), thus arguing against altered global O‐GlcNacylation in Cmas‐knockout cells. Thus, elevated cytosolic Neu5Ac levels influenced neither related metabolite levels nor intracellular glycosylation.
Sialylation is dispensable for differentiation into EBs and for onset of germ layer formation in mESCs
Next, we investigated at the transcriptome, protein and histologic levels if sialoglycans are a prerequisite for differentiation of mESCs towards the three germ lineages during undirected EB formation. In order to ensure uniform differentiation conditions, mESCs were seeded in a hanging drop culture, thus giving rise to one EB per drop and subsequent individual cultures thereof. We analysed the mRNA expression pattern of two undifferentiated WT and three Cmas −/− mESCs (time point 0, “LIF”) and resultant EBs after 2, 4 and 8 days of differentiation. The global gene expression profile was subjected to principal component analysis (PCA; Figure 6 A). This statistical procedure is a valuable tool to compare samples with large amounts of data, as it converts the data into a coordinate system, thereby reducing the dimensions of a dataset of multiple samples without losing useful information. The three principal components (PC1–3; x/y/z axes) cover 88 % of the variability in the whole data set obtained from the five cell lines; thus giving a very good overall impression of the analysed transcriptomes. The data points of the five undifferentiated mESC lines (LIF) are all very close together, thus indicating extremely high similarity of the transcriptomes. Although the distance between individual data points of the cell lines at a distinct time‐point increased during differentiation, the data points were still close together, and genotype‐specific differences were not observed. The only gene with a significantly altered mRNA expression level between WT and Cmas −/− was Cmas itself, which was absent in the knockout mESC lines (p value: 6.44×10−24, q value: 5.5×10−21). During the time course the data points formed a tilted C‐shaped pattern. This pattern is known from in vitro differentiated human ESC lines,22, 23 and indicates induction of developmental processes and a continuous differentiation of all mESC lines independent of the genotype. In order to verify correct exit from pluripotency and induction of germ layer formation, mRNA expression levels for selected pluripotency‐ and differentiation‐related genes were compared (Figure 6 B–E). The levels of the pluripotency markers (Oct3/4, Sox2, and Nanog) were similar in WT and Cmas −/− mESC lines at time zero, just as the changes during the time‐course of pluripotency exit and up to day 8 of differentiation (Figure 6 B). At day 4, expression of mesodermal marker genes closely linked to gastrulation (brachyury, fibroblast growth factor 8 (Fgf8) and Wnt3; Figure 6 C) peaked and declined again at day 8. The embryonic ectodermal markers of early epiblast development fibroblast growth factor 5 (Fgf5) and orthodenticle homeobox protein 2 (Otx2) rose at day 2 and declined at day 8 with a similar pattern for all genotypes, thus indicating differentiation of the embryonic ectoderm (Figure 6 D). Formation of the endodermal germ layer was monitored by expression of the definitive visceral endoderm markers forkhead box protein A2 (Foxa2) and Sox1724 (Figure 6 E). Foxa2 and Sox17 mRNA expression was induced at day 4 of differentiation with almost comparable intensity and time‐course in Cmas +/+ and Cmas −/− EBs. Thus, the mRNA expression patterns of endo‐, ecto‐ and mesoderm‐specific genes seems unimpaired in Cmas −/− EBs. In order to confirm germ‐layer formation in vitro, EBs were fixed and paraffin embedded after eight days of culture. WT EBs consist of an outer layer of primitive visceral endoderm surrounding an inner core of pluripotent cells. Further differentiation and cavity formation of WT EBs reflects germ‐layer formation and building of the proamniotic cavity in vivo. Immunohistological staining of EB sections of all genotypes exhibited a similar morphology, including cavity formation (Figure 6 F). The endodermal marker Disabled‐2 was expressed in epithelial cells surrounding the EB and lining the internal cavities in WT and Cmas −/− EBs. Similarly, the ectododermal marker Nestin (an intermediate filament protein) was present in EBs of both genotypes. In order to monitor the progress of mesodermal differentiation, expression of the cardiac muscle cell marker α‐smooth muscle actin (α‐SMA) was investigated. α‐SMA was expressed in all EBs at day 8 after LIF deprivation, thus confirming formation of mesodermal cardiac progenitor cells. Furthermore, rhythmic pulsing was observed in monolayer differentiation experiments of WT and Cmas −/− mESC after eleven days of cultivation, thus indicating the capability of WT and Cmas −/− ESCs to differentiate into functional cardiomyocytes (Supporting Movies S1 and S2).
Figure 6.
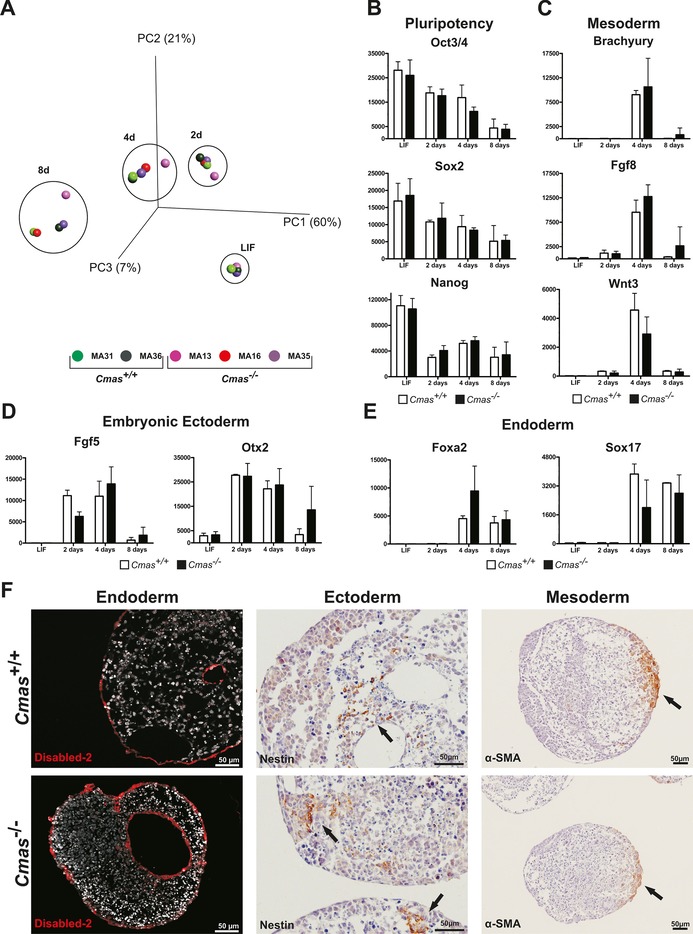
Loss of sialylation does not affect primary germ layer formation of mESCs in vitro. A) Principal component analysis of transcriptomic data from undifferentiated (LIF) mESC and after two, four and eight days of EB differentiation of Cmas +/+ (green, n=2) and Cmas −/− (red and purple, n=3) mESC lines. B)–E) mRNA expression quantified by normalised fluorescence intensity of B) pluripotency factors Oct3/4, Sox2 and Nanog; C) mesodermal markers Brachyury, Fgf8 and Wnt3; D) embryonic ectodermal markers Fgf5 and Otx2; and E) endodermal markers Foxa2 and Sox17 obtained from transcriptome array data. F) Indirect immunofluorescence and immuno‐histochemical analysis of sections from paraffin‐embedded Cmas +/+ and Cmas −/− EBs after eight days of differentiation. The capacity to form primary germ layers was detected by staining for the marker proteins Disabled‐2, Nestin and α‐smooth muscle actin (α‐SMA).
Discussion
The exposed terminal position of Sia on cell‐surface glycans predisposes the sugar as an important signalling component in cellular communication.7 Several pathologies caused by reduced synthesis of Sia and/or sialoglycoconjugates have been described in humans and in animal model system.4, 25, 26 Interestingly, mutations causing complete loss of Sia biosynthesis are not observed in humans and are embryonic‐lethal in mice.8 It thus seems to be a general rule that loss of Sia interferes with development and viability of vertebrates. The mechanisms by which Sia impacts embryonic development and differentiation are not well understood, but are of increasing interest with regard to the application of ESCs in regenerative medicine. We used mESCs as a valuable tool to address this question. In CMAS‐depleted mESCs and HEK 293 cells, we observed complete loss of sialylation on glycoconjugates at the cell surface, with intracellular accumulation of free Neu5Ac. Significantly, these experiments provide the first proof that CMAS is the only enzyme capable of activating Sia in mammalian cells and confirm earlier data demonstrating the role of CMP‐Sia as a feedback inhibitor of GNE.2
Our finding that intracellular Sia accumulation has no obvious influence either on major metabolic routes or on global O‐GlcNAcylation levels was unexpected. However, our data corroborate a recent investigation carried out with breast cancer cells,27 in which CMAS expression in 231MFP cells was reduced by shRNA. As in our study, changes in the levels of lactate or TCA‐cycle metabolites were not identified.27 Although metabolic changes have been observed to exert a marked influence on gene regulation and differentiation in mESCs,28 intracellular Sia accumulation does not seem to be critical for pluripotency and differentiation in mESCs.
At the cell surface, terminal Sia is linked to galactose (Gal) or N‐acetylgalactosamine (GalNAc) residues. Loss of Sia on Cmas −/− mESC was associated with an increase in Gal‐ and oligoLacNAc‐capped glycans. The latter observation was also described for HL‐60 cells (human promyelocytic leukemia) after treatment with the global sialyltransferase inhibitor 3F‐NeuAc.29 Hence, sialylation might serve as an end‐point signal of glycosylation for certain glycans; and, in the absence of the donor substrate CMP‐Sia, LacNAc structures get steadily elongated, most likely due to the action of competing glycosyltransferases, for example, β1,4‐galactosyl‐ and β1,3‐N‐acetylglucosaminyltransferases that add LacNAc repeats. Loss of Sia on glycoconjugates at the cell surface presumably has two consequences: it abolishes the binding of Sia‐specific lectins, which could lead to loss of function of, for example, Siglecs; and it could readily be accompanied by a gain in function of lectins preferring neutral glycans, for example, galectins.30 Thus, it seems likely that cell‐surface sialylation has an impact on pluripotency and/or differentiation in mESCs, as recently reported for human ESC and iPSC lines. Reduction of α2,6‐sialylation by down‐regulation of the sialyltransferase ST6GAL1 entails a decrease in the mRNA expression of pluripotency factor Oct3/4 (POU5F1) in hPSCs, and impedes the induction of pluripotency in somatic cells.12 Furthermore, neuraminidase treatment induced differentiation in human iPSCs derived from menstrual blood‐derived mesenchymal cells (iPS‐MBMC) as well as of human embryonic stem cells (hESC), thus suggesting that sialylation is involved in the maintenance of pluripotency.31 In stark contrast, murine asialo Cmas −/− ESCs did not exhibit any change in gene expression of the pluripotency network, thus suggesting that the maintenance of pluripotency in murine ESCs is not dependent on cell‐surface sialylation. These divergent results (between human and murine ESCs) might be attributable to species‐specific differences32 as well as to alterations in the differentiation states of mESC and hiPSC/hESC.33 Interestingly, in an mESC proteomic investigation, CMAS was identified as an interaction partner of the central pluripotency factor Oct3/4.34 However, as we show here, neither Oct3/4 mRNA expression level nor protein localisation was altered in Cmas −/−, compared to WT mESC.
In terms of differentiation capacity, different effects of cell‐surface sialylation have been reported for murine and human ESCs. Although polySia is essential for endodermal germ layer differentiation in hESC in vitro,35 mice with complete loss of polysialic acid are viable and fertile.36 Analysis of hyposialylated GNE‐deficient mESC in vitro also revealed a probable role of sialylation in differentiation.14 Although impaired in de novo Sia synthesis, GNE‐deficient mESC were able to retrieve free Sia by the lysosomal salvage pathway.37 Hence, the sialylation status of GNE‐deficient mESCs depends on the presence of sialoglycoconjugates and free Sia in the medium. Impaired differentiation and even the demise of EB from GNE‐deficient mESCs was overcome by culturing the cells in medium with high levels of Sia, thus suggesting that sialylation has an impact on processes regulating embryonic differentiation in vitro.14 In contrast, in Cmas −/− mESCs the exit from pluripotency and induction of differentiation into the three germ layers was not affected by sialylation. The observed discrepancy between Gne −/− and Cmas −/− mESCs might reflect different effects of hyposialo‐ and asialo‐cell surfaces for signalling and/or cell–cell interaction, respectively; or this might be attributable to genetic differences in the murine models.38 Moreover, a regulatory function of GNE (independent of its enzymatic activity) has also been postulated recently and must be considered.39 Strikingly, sialylation is dispensable for ecto‐, endo‐ and mesoderm formation: the three germ layers formed in vitro (Figure 6 F), thus confirming the data obtained in the comparative transcriptome and differentiation marker analysis of Cmas +/+ and Cmas −/− mESCs (as well as derived EBs). In fact, Cmas −/− mESCs gave rise to beating foci, thus clearly implying the capacity for cardiomyogenic differentiation. The data presented in this study leave no doubt that loss of CMAS (and the resulting sialylation capacity) has little if any impact on the early steps of murine ESC differentiation in vitro.
Sialylation is a ubiquitous, fundamental process in vertebrates, but it seems to be dispensable for pluripotency as well as for early differentiation of mESC. As sialylation is a relatively new “invention” in evolution, our data inspire the speculation that early developmental processes evolved in the absence of Sia and remained independent of sialylation in embryogenesis, whereas Sia became pivotal in the later stages of development. This idea is supported by the fact that in both human and mouse the degree of sialylation increases with differentiation, both in vitro and in vivo.40 However, with the observed discrepancies between human and murine ESCs, more data of sialylation‐impaired animal models from different species are required to fully understand the role of sialic acid in an evolutionary context.
Experimental Section
Mice: Mouse strains were obtained from the animal facility (ZTL) of Hannover Medical School. In order to inactivate the Cmas gene, a targeting vector was constructed in which exon 4 (encoding the active site of the protein) was flanked by loxP sites. An frt‐flanked neomycin resistance gene (neo) for positive selection was inserted into intron 4, and a diphtheria toxin gene for negative selection was inserted into intron 3. The entire vector was sequence‐verified and introduced into E14‐1 ESCs. Correct homologous integration of the construct was verified by PCR (Figure 1 A, B). A single integration event was confirmed by southern blot analysis. ES cells were injected into C57Bl/6J blastocysts. The neo cassette was deleted by crossing Cmas neo/neo mice with the Flp‐deleter strain SJL‐Tg(ACTFLPe)9205Dym/J. Resulting Cmas floxed mice were bred to homozygosity and crossed with the Cre‐deleter strain C57BL/6‐Tg(Zp3‐cre)93Knw/J. Upon further backcrosses with Bl/6 mice, heterozygous Cmas knockout mice (Cmas +/−) were obtained. These animals were crossed to NMRI, and intercrosses of Cmas +/− animals gave rise to Cmas −/− embryos. The targeting vector and resulting Cmas alleles are shown in Figure 1 A; Cmas mutants and their genotyping by PCR is in Figure S1 (primers are available upon request). Animals were hosted in the ZTL of the Hannover Medical School under specific pathogen‐free conditions. All animal experiments were carried out in compliance with German law for protection of animals and were approved by the local authorities (TV 33.12–42502‐04‐13/1312).
Cell culture: Generation of Cmas +/+, Cmas +/− and Cmas −/− murine embryonic stem cells (mESC) was performed as previously described.41 In brief, E3.5 blastocysts from heterozygous Cmas +/− matings were cultured on irradiated murine embryonic fibroblasts (MEF) in high‐glucose Dulbecco's modified Eagle's medium (DMEM, Biochrom, Cambridge, UK) supplemented with knockout serum replacement (KOSR, 15 %; Gibco), l‐glutamine (2 mm; Biochrom), 2‐mercaptoethanol (0.1 mm), leukaemia inhibitory factor (LIF, 1000 U mL−1; Merck Millipore), CHIR99021 (3 μm; kindly provided by Prof. Scheper, Institute of Technical Chemistry, Leibniz University, Hannover) and PD0325901 (1 μm; Sigma–Aldrich) at 37 °C under CO2 (5 %). After 6 to 9 days, the outgrowth of the inner cell mass was enzymatically disaggregated and further cultivated on MEFs in mESC medium (medium as above but with foetal calf serum (FCS, 15 %; Biochrom) and without KOSR, CHIR99021 or PD0325901). All experiments were carried out in mESC medium; depletion of MEFs was accomplished by dilution of mESC for at least three passages. For EB formation, feeder‐free pluripotent mESCs were trypsinised, counted and seeded on the lids of cell‐culture dishes (2000 cells, 25 μL per drop of LIF‐deprived mESC medium. After 48 h, homogenous EB formation was accomplished by transferring one EB per well to uncoated wells of 48‐well dishes (agitation at 75 rpm). In order to analyse the capacity of mESCs to form beating foci, mESCs were cultured as a monolayer without MEFs and deprived of LIF for 11 days. The medium was changed every day, and the occurrence of beating foci was monitored by bright field microscopy. Beating foci containing monolayer cultures of differentiated mESC were used for western blot analysis of polysialic acid expression upon differentiation.
HEK 293 cells (kindly provided by the Institute of Cell Biology, Hannover Medical School) were cultivated at 37 °C under CO2 (5 %) in DMEM/Ham's F‐12 medium (1:1; Biochrom) supplemented with FCS (10 %), l‐glutamine (2 mm) and penicillin/streptomycin (100 U mL−1). Cells were passaged by rinsing with fresh medium. CHO cells42 were similarly cultivated in medium supplemented with FCS (5 %) and sodium pyruvate (1 mm). Cells were passaged with trypsin (0.05 %)/EDTA (0.02 %).
Generation of HEK 293 CMAS −/− clones: HEK 293 CMAS −/− cells were generated by CRISPR/Cas9‐mediated genome editing with a guide RNA targeting residues 128 to 155 of CMAS exon 1 (5′‐GCAGC CCTAA TTCTG GCCCG‐3′). The target sequence was cloned into plasmid pX330‐U6‐Chimeric_BB‐CBh‐hSpCas9 (Addgene plasmid 42230 a kind gift from Feng Zhang).43 Wild‐type cells were cotransfected with plasmid pX330‐U6‐Chimeric_BB‐CBh‐hSpCas9‐hCMASgRNA and a vector containing a neomycin or puromycin resistance gene. After 24 h, the cells were selected with G418 or puromycin for 4–5 days, and single‐cell clones were obtained by limiting dilution. Clones were screened for insertions and deletion by PCR and sequencing of the products by using primers 5′‐AGGAA GATGG ACTCG GTGG‐3′ and 5′‐CCGGA TCTTG CACAT CCCT‐3′. Sequence alignment and analysis were performed with SnapGene (GSL Biotech, Chicago, IL).
Western blotting: Cells were lysed in RIPA buffer (supplemented with 1 μm PugNAc to analyze O‐GlcNAcylation); protein concentration was determined by a BCA Assay (Thermo Fisher Scientific). Sialidase and endosialidase treatment was performed by incubation with either neuraminidase from Arthrobacter urefaciens (20 U g−1 protein cell lysate; EY Laboratories, San Mateo, CA) or endoneuraminidase NF (4 mg g−1 protein cell lysate) for 1 h at 37 °C. Endoneuraminidase NF was purified as reported earlier.44 Protein (30 μg) was separated by 8 or 12 % SDS‐PAGE, transferred to PVDF membrane (or nitrocellulose for analysis of O‐GlcNAcylation) and incubated with primary antibody: in‐house made polyclonal rabbit anti‐CMAS antibody S59 (1:10 000);45 mouse anti‐actin antibody (1:100 000, Merck Millipore); in‐house made mouse anti‐PolySia antibody 735 (1:3000).46 After incubation with either anti‐rabbit HRP‐conjugated secondary antibody (1:15 000, Jackson ImmunoResearch Laboratories, West Grove, PA) or HRP‐conjugated goat anti‐mouse secondary antibody (1:15 000, Southern Biotech, Birmingham, AL), detection was performed with enhanced chemiluminescence. Lectin analysis (MAA, PNA) were performed with a DIG glycan differentiation kit (Roche), according to the manufacturer's instructions and detected with HRP‐conjugated anti‐digoxigenein antibody (1:5000, Roche) and enhanced chemiluminescence. O‐GlcNAcylation was detected with antibody CTD110.6 (1:1.000, Cell Signaling) before and after preincubation with GlcNAc (0.1 m) at 4 °C for 10 min and HRP‐conjugated mouse‐IgM secondary antibody (1:15.000, Jackson).
Indirect immunofluorescence and CtxB staining: Feeder‐free mESCs were seeded onto gelatin‐coated coverslips, and after two days of culture, were fixed with paraformaldehyde (4 %), permeabilised with Triton X‐100 (0.2 %), blocked with BSA (1 %) in PBS (blocking solution) and incubated with mouse anti‐Oct3/4 antibody (Santa Cruz Biotechnology) in blocking solution (1:50) for 1 h at RT. After three washes with PBS, Alexa Fluor 488‐conjugated anti‐mouse IgG2b (1:500; Molecular Probes) was applied in blocking solution (1 h, RT). For CTXB staining, fixed cells were incubated with FITC‐conjugated CTXB (1:200 in blocking solution; Sigma–Aldrich) for one hour. Samples were mounted in Vectashield (Vector Laboratories, Burlingame, CA) containing DAPI (to stain nuclei) and analysed with an AxioObserver.Z1 microscope (Zeiss) equipped with a Zeiss AxioCam MRm digital camera and an ApoTome.
Quantitative PCR: RNA preparation, cDNA synthesis and quantitative PCR analysis were as previously descibed46] with the exception that after TRIzol extraction, RNA was precipitated with isopropanol (500 μL per mL TRIzol), and the resulting pellet was washed in ethanol (1 mL, 75 %, v/v), air dried and dissolved in RNAse‐free water. The primers (Sigma–Aldrich) are listed in Table S1.
Multiplexed capillary gel electrophoresis with laser‐induced fluorescence detection (xCGE‐LIF): Cells were lysed in RIPA buffer (Tris (50 mm, pH 8) with NaCl (150 mm), NP‐40 (1 % (v/v); Roche), sodium deoxycholate (0.5 %, w/v; Sigma–Aldrich) and SDS (1 %, w/v; Serva Heidelberg, Germany)) supplemented with HALT protease inhibitor (Thermo Fisher Scientific) by pipetting up and down a few times followed by freezing at −80 °C overnight. Thawed cells were sonicated two times for one minute, followed by centrifuging (13 000×g, 15 min, 4 °C). Proteins were precipitated with a fourfold excess of acetone at −20 °C overnight. Further sample preparation and xCGE‐LIF glycoanalysis was as previously described.17 Briefly, protein pellets were fully dissolved in PBS containing SDS (2 %, w/v) and incubated for 10 min at 60 °C, then remaining SDS was neutralised by adding IGEPAL (8 %; Sigma–Aldrich) in PBS. N‐glycans were released from denatured and linearised plasma protein for 3 h at 37 °C by addition of peptide‐N‐glycosidase F (PNGase F; Sigma–Aldrich) in PBS. In order to obtain N‐glycosylation patterns by xCGE‐LIF, released N‐glycans were first labelled with 8‐aminopyrene‐1,3,6‐trisulfonic acid (APTS; Sigma–Aldrich), and excess label was removed by using a polyacrylamide‐based stationary phase in hydrophilic‐interaction‐chromatography (HILIC) mode. For this, Bio‐Gel P10 (200 μL, 100 mg mL−; Bio‐Rad) in MilliQ water/ethanol/acetonitrile (70:20:10, v/v/v) was applied to each well of a 0.45 μm GHP filter plate (Pall Corporation, New York, NY). The solvents were removed by using a vacuum manifold (Merck Millipore). All wells were prewashed with MilliQ water (3×200 μL), followed by equilibration with acetonitrile (80 %, 3×200 μL). APTS‐labelled samples were placed in wells containing Bio‐Gel P10 suspension and shaken (500 rpm) for 5 min to improve glycan binding. For purification, wells were washed with acetonitrile (80 %, 5×200 μL) containing triethylamine (100 mm; Sigma–Aldrich), adjusted to pH 8.5 with acetic acid (Sigma–Aldrich), then washed with acetonitrile (80 %, 3×200 μL) All washing steps were performed by addition of solution, incubation for 2 min and removal of solvent by vacuum. For elution, Bio‐Gel P10 (swollen size 100 μL) and MilliQ water (2×200 μL) were applied to each well followed by 5 min incubation with shaking (500 rpm). The eluates were removed by vacuum and collected in a 96‐well storage plate (Thermo Fisher Scientific). The combined eluates were stored at −20 °C. Purified APTS‐labelled N‐glycans were separated by capillary gel electrophoresis and monitored by laser‐induced fluorescence detection. glyXtool (glyXera, Magdeburg, Germany) was used for processing of xCGE‐LIF data, and for normalisation of migration times to an internal standard. N‐Glycan fingerprints were created from the electropherograms: signal intensity in relative fluorescence units (RFU) is plotted on the y‐axis against normalised migration time units (MTU) on the x‐axis. Because of the high reproducibility of normalised migration times, N‐glycan fingerprints of different samples can be compared. N‐glycan structural elucidation and peak annotation was made by using our in‐house N‐glycan database. For correct annotation of the glycans, several exoglycosidase digests (including chemical release of sialic acids by mild acid hydrolysis) were performed: Streptococcus pneumoniae α(2–3) sialidase (Prozyme, Hayward, CA), Clostridium perfringens α(2–3,6) sialidase (Prozyme), Arthrobacter ureafaciens α(2–3,6,8) sialidase (Prozyme), Xanthomonas α(1–3,4) fucosidase (QABio, San Mateo, USA), Xanthomonas manihotis α(1–2) fucosidase (New England Biolabs), bovine kidney α(1–2,3,4,6) fucosidase (Prozyme), X. manihotis β(1–3) galactosidase (New England Biolabs), Bacteroides fragilis β1‐4) galactosidase (New England Biolabs), Jack bean β(1–4,6) galactosidase (Prozyme), green coffee bean α(1–3,4,6) galactosidase (Prozyme), Streptococcus pneumoniae β(1–2,3,4,6)‐N‐acetylglucosaminidase (New England Biolabs), Jack bean α(1–2,3,6) mannosidase (Prozyme).
Neu5Ac quantification: Intracellular mESC metabolites were extracted from one confluent well of a 12‐well plate by using acetonitrile/methanol/water (2:2:1, v/v/v) as solvent. Samples were centrifuged (20 800 g, 10 min), and the supernatants were evaporated and dissolved in H2O. Protein concentration was determined b a BCA assay (Thermo Fisher Scientific). Extraction of metabolites from medium (50 μL) after two days of culture was accomplished as described for intracellular metabolites with acetonitrile/methanol (1:1) as extraction solvent. Free sialic acids were derivatised with DMB (as described by Hara et al.).48 Briefly, Sia was labelled for 2.5 h at 50 °C with DMB (8 mm; Dojindo Laboratories, Kumamoto, Japan) in propionic acid (1.5 m) containing 2‐mercaptoethanol (0.8 m) and sodium hydrosulfite (14.2 mm). DMB‐derivatised Sia was separated by using a Prominence CBM‐20A UFLC system (Shimadzu) equipped with a XSelect CSH C18 reversed‐phase HPLC column (4.6 μm×250 mm, 5.0 mm particle size; Waters). Isocratic elution was performed with acetonitrile/methanol/H2O (9:7:84, v/v/v) at a flow rate of 0.3 mL min−1, and fluorescence was monitored with a RF‐10A XL fluorescence detector (excitation 372 nm, emission 456 nm; Shimadzu). For the assignment of individual peaks, the DMB‐labelled Glyko Sialic Acid Reference Panel (Prozyme) or DMB‐derivatised bovine submaxillary mucin (Sigma–Aldrich) was used.
Metabolite analysis: Intracellular and medium metabolites were extracted as described for Neu5Ac quantification. For HPLC‐MS/MS analysis a SIL‐HTc liquid chromatography system (Shimadzu) with an inserted guard column and a Kinetex C18 column (Phenomenex) was coupled to an API4000 triple quadrupole mass spectrometer (Sciex, Framingham, MA) with electrospray ionisation, operated in negative ionisation mode. An isocratic gradient (water/methanol (97:3, v/v) containing formic acid (0.2 %)) was applied for chromatographic separation. Metabolite identification was done using multiple reaction monitoring with two mass transitions per analyte, of which one was used as quantifier. An internal standard (13C2‐citrate, Sigma–Aldrich) was added during the extraction procedure to each sample, and a calibration curve with customised standard substances ([13C2]2,4‐citrate: 492078, citrate: 251275, lactate: 71718, cis‐aconitate: A3412, α‐ketoglutarate: 75892, succinate: W327700, malate: 112577, fumarate: F1506; Sigma–Aldrich) at appropriate concentrations was applied for quantitation.
Immuno‐histochemistry: After eight days of differentiation, EBs were harvested, washed once in ice cold PBS and fixed in paraformaldehyde (4 %, v/v). Subsequently, EBs were washed again in PBS (30 min) and dehydrated by a graded ethanol series with subsequent paraffination. Paraffinised EBs were sliced into 3 μm sections with an RM 2265 microtome (Leica) and rehydrated. Sections were subsequently heated at 95 °C for 10 min in Target Retrieval Solution (Dako/Agilent Technologies), blocked with either BSA (1 %) or non‐fat dry milk (5 %) in PBS (blocking solution) and subsequently incubated with primary antibody in blocking solution over night at 4 °C: mouse anti‐Disabled‐2 (1:200, BD Transduction Laboratories), mouse anti‐actin α‐smooth muscle (Sigma–Aldrich), rabbit anti‐Nestin (1:100, Abcam). Secondary antibodies were either Alexa Fluor 555‐conjugated anti‐mouse IgG (Molecular Probes), visualised by fluorescence microscopy as described earlier, or HRP‐coupled anti‐rabbit IgG (Jackson ImmunoResearch) visualised by using DAB+ chromogen (Dako). Nucei were counterstained with vectashield or hematoxylin, respectively. Imaging was performed in an AxioObserver.Z1 microscope equipped with an AxioCam Mrm camera for fluorescence, or an AxioCam Mrc camera for bright‐field pictures of DAB‐stained tissue.
Transcriptomics: RNA was prepared by the Trizol method from pluripotent mESCs cultivated under feeder‐free conditions with LIF supplementation, as well as from EBs after 2, 4 and 8 days of differentiation. Total RNA (396 ng) was used for the synthesis of aminoallyl‐UTP‐modified (aaUTP) cRNA with the Quick Amp Labeling kit, No Dye (Agilent Technologies) according to the manufacturer's recommendations with ATP, CTP, GTP (2.5 mm each) and UTP, aaUTP (1.25 mm each) by the use of NTP Set and aminoallyl‐UTP (Thermo Fisher Scientific). aaUTP‐cRNA was labelled with Alexa Fluor 555 Reactive Dye (Life Technologies) as described in the Amino Allyl MessageAmp II Kit Manual (Life Technologies). Prior to the reverse transcription reaction, One‐Color Spike‐In Kit solution (0.5 μL of a 1:1000 stock; Agilent Technologies) was added to each of RNA sample (396 ng). Fluorescently labelled cRNA populations (500 ng each) were used for fragmentation, hybridisation and washing steps, as recommended in the One‐Color Microarray‐Based Gene Expression Analysis Protocol V5.7 (Agilent Technologies). Slides were scanned on a G2565CA Microarray Scanner (pixel resolution 3 μm, bit depth 20; Agilent Technologies) at the Research Core Unit Metabolomics at Hannover Medical School. Data extraction was performed with Feature Extraction Software V10.7.3.1 (Agilent Technologies) by using the extraction protocol file “GE1_107_Sep09.xml”, except that the Multiplicative detrending algorithm was inactivated. Statistical analyses were carried out in Qlucore Omics explorer (Scheelevägen, Sweden). A p value of 0.05 and a q value of 0.05 were applied to multi‐group comparisons between control and CMAS‐negative mESC and EBs in order to identify differentially expressed genes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
We are very grateful to Rita Gerardy‐Schahn for continuous support and fruitful discussions. Robert Lindner and Ortwin Naujok kindly provided us with fluorescently labelled CTXB and anti‐Nestin antibody, respectively. Microarray raw data used or referred to in this publication were generated by the Research Core Unit Transcriptomics (RCUT) of Hannover Medical School. Support concerning transcriptome processing and analyses given by Oliver Dittrich‐Breiholz was very helpful. We thank Kerstin Flächsig‐Schulz and Ulrike Bernard for laboratory expertise and documentation and Prof. Dr. Scheper (Institute of Technical Chemistry, Leibniz University of Hannover) for kindly providing CHIR 99021. Erdmann Rapp acknowledges support by the European Union under the projects “HighGlycan” (European Commission (EC) grant no. 278535) and “HTP‐GlycoMet” (European Commission (EC) grant no. 324400). For Samanta Cajic and Erdmann Rapp this work was supported by the German Federal Ministry of Education and Research (BMBF) under the project “Die Golgi Glykan Fabrik 2.0” (grant identifier 031A557). A.M.K. and B.W. were supported by grants from the Deutsche Forschungsgemeinschaft (DFG, WE 5585/1‐1 and MU 1849/2‐1).
M. Abeln, K. M. Borst, S. Cajic, H. Thiesler, E. Kats, I. Albers, M. Kuhn, V. Kaever, E. Rapp, A. Münster-Kühnel, B. Weinhold, ChemBioChem 2017, 18, 1305.
References
- 1. Ioffe E., Stanley P., Proc. Natl. Acad. Sci. USA 1994, 91, 728–732; [DOI] [PMC free article] [PubMed] [Google Scholar]; Xia L., Ju T., Westmuckett A., An G., Ivanciu L., McDaniel J. M., Lupu F., Cummings R. D., McEver R. P., J. Cell Biol. 2004, 164, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kornfeld S., Kornfeld R., Neufeld E. F., O'Brien P. J., Proc. Natl. Acad. Sci. USA 1964, 52, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhuo Y., Bellis S. L., J. Biol. Chem. 2011, 286, 5935–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weinhold B., Sellmeier M., Schaper W., Blume L., Philippens B., Kats E., Bernard U., Galuska S. P., Geyer H., Geyer R., Worthmann K., Schiffer M., Groos S., Gerardy-Schahn R., Münster-Kühnel A. K., J. Am. Soc. Nephrol. 2012, 23, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Celeste F. V., Vilboux T., Ciccone C., de Dios J. K., Malicdan M. C., Leoyklang P., McKew J. C., Gahl W. A., Carrillo-Carrasco N., Huizing M., Hum. Mutat. 2014, 35, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Müller J., Nitschke L., Nat. Rev. Rheumatol. 2014, 10, 422–428. [DOI] [PubMed] [Google Scholar]
- 7. Varki A., Trends Mol. Med. 2008, 14, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwarzkopf M., Knobeloch K.-P., Rohde E., Hinderlich S., Wiechens N., Lucka L., Horak I., Reutter W., Horstkorte R., Proc. Natl. Acad. Sci. USA 2002, 99, 5267–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Varki A., Hooshmand F., Diaz S., Varki N. M., Hedrick S. M., Cell 1991, 65, 65–74. [DOI] [PubMed] [Google Scholar]
- 10. Robertson E., Bradley A., Kuehn M., Evans M., Nature 1986, 323, 445–448. [DOI] [PubMed] [Google Scholar]
- 11. Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Cell 2007, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 12. Wang Y.-C., Stein J. W., Lynch C. L., Tran H. T., Lee C.-Y., Coleman R., Hatch A., Antontsev V. G., Chy H. S., O'Brien C. M., Murthy S. K., Laslett A. L., Peterson S. E., Loring J. F., Sci. Rep. 2015, 5, 13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weidemann W., Klukas C., Klein A., Simm A., Schreiber F., Horstkorte R., Glycobiology 2010, 20, 107–117. [DOI] [PubMed] [Google Scholar]
- 14. Weidemann W., Hering J., Bennmann D., Thate A., Horstkorte R., Int. J. Mol. Sci. 2013, 14, 20555–20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Münster A.-K., Weinhold B., Gotza B., Mühlenhoff M., Frosch M., Gerardy-Schahn R., J. Biol. Chem. 2002, 277, 19688–19696. [DOI] [PubMed] [Google Scholar]
- 16. Antes P., Schwarzmann G., Sandhoff K., Chem. Phys. Lipids 1992, 62, 269–280. [DOI] [PubMed] [Google Scholar]
- 17. Hennig R., Cajic S., Borowiak M., Hoffmann M., Kottler R., Reichl U., Rapp E., Biochim. Biophys. Acta 2016, 1860, 1728–1738. [DOI] [PubMed] [Google Scholar]
- 18. Huffman J. E., Pučić-Baković M., Klarić L., Hennig R., Selman M. H. J., Vučković F., Novokmet M., Krištić J., Borowiak M., Muth T., Polašek O., Razdorov G., Gornik O., Plomp R., Theodoratou E., Wright A. F., Rudan I., Hayward C., Campbell H., Deelder A. M., et al., Mol. Cell. Proteomics 2014, 13, 1598–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumann A.-M., Bakkers M. J. G., Buettner F. F. R., Hartmann M., Grove M., Langereis M. A., de Groot R. J., Mühlenhoff M., Nat. Commun. 2015, 6, 7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilcken B., Don N., Greenaway R., Hammond J., Sosula L., J. Inherited Metab. Dis. 1987, 10, 97–102. [DOI] [PubMed] [Google Scholar]
- 21. Schauer R., Sommer U., Krüger D., van Unen H., Traving C., Biosci. Rep. 1999, 19, 373–383. [DOI] [PubMed] [Google Scholar]
- 22. Blakeley P., Fogarty N. M. E., del Valle I., Wamaitha S. E., Hu T. X., Elder K., Snell P., Christie L., Robson P., Niakan K. K., Development 2015, 142, 3151–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu L.-F., Leng N., Zhang J., Hou Z., Mamott D., Vereide D. T., Choi J., Kendziorski C., Stewart R., Thomson J. A., Genome Biol. 2016, 17, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yasunaga M., Tada S., Torikai-Nishikawa S., Nakano Y., Okada M., Jakt L. M., Nishikawa S., Chiba T., Era T., Nishikawa S.-I., Nat. Biotechnol. 2005, 23, 1542–1550. [DOI] [PubMed] [Google Scholar]
- 25. van Karnebeek C. D. M., Bonafe L., Wen X.-Y., Tarailo-Graovac M., Balzano S., Royer-Bertrand B., Ashikov A., Garavelli L., Mammi I., Turolla L., Breen C., Donnai D., Cormier-Daire V., Heron D., Nishimura G., Uchikawa S., Campos-Xavier B., Rossi A., Hennet T., Brand-Arzamendi K., et al., Nat. Genet. 2016, 48, 777–784. [DOI] [PubMed] [Google Scholar]
- 26. Hinderlich S., Weidemann W., Yardeni T., Horstkorte R., Huizing M., Top. Curr. Chem. 2013, 366, 97–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kohnz R. A., Roberts L. S., DeTomaso D., Bideyan L., Yan P., Bandyopadhyay S., Goga A., Yosef N., Nomura D. K., ACS Chem. Biol. 2016, 11, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moussaieff A., Rouleau M., Kitsberg D., Cohen M., Levy G., Barasch D., Nemirovski A., Shen-Orr S., Laevsky I., Amit M., Bomze D., Elena-Herrmann B., Scherf T., Nissim-Rafinia M., Kempa S., Itskovitz-Eldor J., Meshorer E., Aberdam D., Nahmias Y., Cell Metab. 2015, 21, 392–402. [DOI] [PubMed] [Google Scholar]
- 29. Rillahan C. D., Antonopoulos A., Lefort C. T., Sonon R., Azadi P., Ley K., Dell A., Haslam S. M., Paulson J. C., Nat. Chem. Biol. 2012, 8, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macauley M. S., Crocker P. R., Paulson J. C., Nat. Rev. Immunol. 2014, 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alisson-Silva F., de Carvalho Rodrigues D., Vairo L., Asensi K. D., Vasconcelos-dos-Santos A., Mantuano N. R., Dias W. B., Rondinelli E., dos Santos Goldenberg R. C., Urmenyi T. P., Todeschini A. R., Glycobiology 2014, 24, 458–468. [DOI] [PubMed] [Google Scholar]
- 32. Rossant J., Development 2015, 142, 9–12. [DOI] [PubMed] [Google Scholar]
- 33. Davidson K. C., Mason E. A., Pera M. F., Development 2015, 142, 3090–3099. [DOI] [PubMed] [Google Scholar]
- 34. Ding J., Xu H., Faiola F., Ma'ayan A., Wang J., Cell Res. 2012, 22, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berger R. P., Sun Y. H., Kulik M., Lee J. K., Nairn A. V., Moremen K. W., Pierce M., Dalton S., Stem Cells 2016, 34, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weinhold B., Seidenfaden R., Röckle I., Mühlenhoff M., Schertzinger F., Conzelmann S., Marth J. D., Gerardy-Schahn R., Hildebrandt H., J. Biol. Chem. 2005, 280, 42971–42977. [DOI] [PubMed] [Google Scholar]
- 37. Weidemann W., Klukas C., Klein A., Simm A., Schreiber F., Horstkorte R., Glycobiology 2010, 20, 107–117. [DOI] [PubMed] [Google Scholar]
- 38. Zurita E., Chagoyen M., Cantero M., Alonso R., González-Neira A., López-Jiménez A., López-Moreno J. A., Landel C. P., Benitez J., Pazos F., Montoliu L., Transgenic Res. 2011, 20, 481–489. [DOI] [PubMed] [Google Scholar]
- 39. Kontou M., Weidemann W., Bauer C., Reutter W., Horstkorte R., Neuroreport 2008, 19, 1239–1242. [DOI] [PubMed] [Google Scholar]
- 40. Nairn A. V., Aoki K., dela Rosa M., Porterfield M., Lim J.-M., Kulik M., Pierce J. M., Wells L., Dalton S., Tiemeyer M., Moremen K. W., J. Biol. Chem. 2012, 287, 37835–37856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Czechanski A., Byers C., Greenstein I., Schrode N., Donahue L. R., Hadjantonakis A.-K., Reinholdt L. G., Nat. Protoc. 2014, 9, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eckhardt M., Mühlenhoff M., Bethe A., Koopman J., Frosch M., Gerardy-Schahn R., Nature 1995, 373, 715–718. [DOI] [PubMed] [Google Scholar]
- 43. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Science 2013, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schulz E. C., Schwarzer D., Frank M., Stummeyer K., Mühlenhoff M., Dickmanns A., Gerardy-Schahn R., Ficner R., J. Mol. Biol. 2010, 397, 341–351. [DOI] [PubMed] [Google Scholar]
- 45. Schaper W., Bentrop J., Ustinova J., Blume L., Kats E., Tiralongo J., Weinhold B., Bastmeyer M., Münster-Kühnel A.-K., J. Biol. Chem. 2012, 287, 13239–13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frosch M., Görgen I., Boulnois G. J., Timmis K. N., Bitter-Suermann D., Proc. Natl. Acad. Sci. USA 1985, 82, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thiesler C. T., Cajic S. S., Hoffmann D., Thiel C., van Diepen L., Hennig R., Sgodda M., Weiβmann R., Reichl U., Steinemann D., Diekmann U., Huber N. M. B., Oberbeck A., Cantz T., Kuss A. W., Körner C., Schambach A., Rapp E., Buettner F. F. R., Mol. Cell. Proteomics 2016, 15, 1435–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hara S., Yamaguchi M., Takemori Y., Furuhata K., Ogura H., Nakamura M., Anal. Biochem. 1989, 179, 162–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary