

Abstract
Cyclobutenones, four-membered ketones bearing an unsaturated carbon–carbon double bond, and their structural sibling benzocyclobutenones, possess unique reactivity. Owing to their inherent high ring strain, such structures readily undergo ring opening under a variety of conditions, including thermolysis, photolysis, and transition metal catalysis, to afford reactive intermediates that can be trapped with nucleophiles, dienophiles, and unsaturated bonds. Their electron-deficient enone moieties are good electrophiles for facile nucleophilic addition. Such properties render cyclobutenones versatile synthons, serving as excellent coupling partners in a vast array of synthetically valuable transformations.
Keywords: cycloaddition, electrocyclic reactions, enones, homogeneous catalysis, strained molecules
Graphical Abstract
1. Introduction
Recent years have witnessed a dramatic increase in the use of four-membered ring compounds as building blocks for the rapid construction of complex molecules.[1] Owing to their inherent ring stain, such small ring structures can undergo ring-opening reactions triggered by myriad reaction conditions, allowing them to serve as coupling partners and core structures in a variety of transformations. Of the four-membered ring compounds, the properties of structures with saturated carbon–carbon bonds have been extensively reviewed, but four-membered ring structures bearing unsaturated carbon–carbon bonds, namely cyclobutenones and benzocyclobutenones, have received less attention.[2] Cyclobutenones and benzocyclobutenones can be conveniently prepared through [2+2] cycloadditions between alkynes or benzynes and ketenes or their equivalents,[2] although alternative methods, such as intramolecular nucleophilic addition,[3a] 4π-electrocyclic ring closure,[3b] and intramolecular transition metal-catalyzed cross-couplings,[3c–e] are also known. This Minireview focuses on representative transformations of cyclobutenones and benzocyclobutenones; examples of cyclobutenols, cyclobutediones, and squaric acid derivatives, as well as preparation of cyclobutenones and benzocyclobutenones,[1–3] will not be covered.
As an overview of the reactions of cyclobutenones and benzocyclobutenones (Scheme 1), early reports primarily involved photolysis and thermolysis, in which vinylketene is generally the reactive intermediate. Thus, cyclobutenones and benzocyclobutenones have been widely incorporated in transformations including [4+2]/[2+2] cycloadditions. In addition, given the nature of the conjugate enone moiety, both 1,2- and 1,4-nucleophilic additions have been investigated. Moreover, compared to cyclopentenones and cyclohexenones, cyclobutenones were recently found to be exceptional dienophiles for Diels–Alder reactions as such cycloadditions benefit from the release of ring strain. Finally, cyclobutenones and benzocyclobutenones have been frequently employed as substrates in C–C-activation reactions,[4] in which transition metals, including nickel, ruthenium, rhodium and cobalt, can selectively activate either C1–C2 or C1–C4 carbon–carbon bonds.
Scheme 1.

General reaction profiles for cyclobutenones.
2. Thermal Ring Opening
The electrocyclic reactions of cyclobutenes have been extensively studied and the stereoselectivity can be readily predicted. Terms like “conrotatory” and “disrotatory” are well defined and widely used to describe such transformations.[5] However, these terms are inadequate for describing the electrocyclic ring opening transformation of cyclobutenones and benzocyclobutenones, as there is only one carbon center carrying sp3 hybridization. In the ring opening of cyclobutenones, the terms “inward” and “outward” are frequently employed to describe the rotation: if a substituent rotates “inward”, it becomes cis to the ketene moiety, whereas it rotates “outward” to be trans to the ketene moiety (Scheme 2).[5] Houk and coworkers explored the effect of C4 substituents on the rotation barrier under thermal conditions and found that, although the substituent effects are less pronounced compared to cyclobutenes, electron donating groups prefer to rotate “outward”, whereas electron-withdrawing groups tend to rotate “inward”.[6] Such results can be attributed to the fact that, when rotating “outward”, the high-lying electron-donor orbital of the substituent can mix with the low lying σ* (LUMO) of the breaking bond, thus stabilizing the intermediate and lowering the activation energy.
Scheme 2.
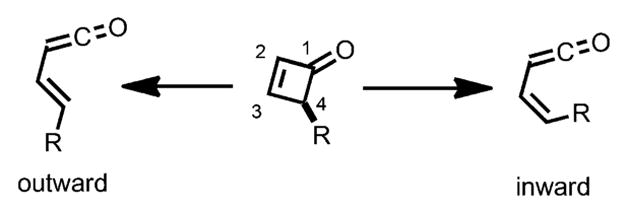
“Inward” and “outward” rotation.
In 1956, Jenny and Roberts studied the racemization of optically active 2,4-dichloro-3-phenylcyclobutenone 1 (Scheme 3).[7] They rationalized that a slowly formed vinylketene intermediate, which underwent rapid cyclization to give back the cyclobutenone, was responsible for the racemization. The putative vinylketene intermediate could either undergo rapid ring closure to furnish racemic 1 or react with acetic acid or acetate to give mixed anhydride 2.
Scheme 3.
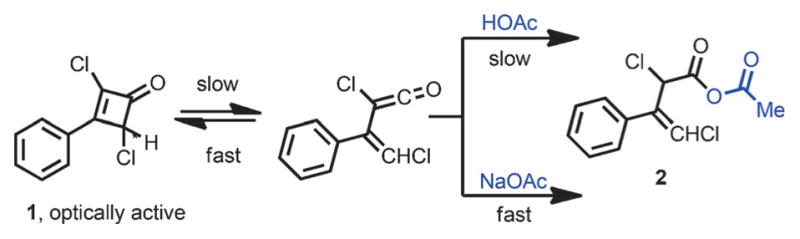
Preliminary observation for vinylketene intermediate.
In 1975, Mayr reported that 2,4,4-trimethylcyclobutenone 3 also underwent facile ring opening in boiling cyclohexane, and the vinylketene intermediate was trapped in situ by ethyl vinyl ether to afford cyclobutanone 4 (Scheme 4).[8] Huisgen and Mayr later reported that the vinylketene intermediate formed from cyclobutenone 5 can be intercepted by nucleophiles such as methanol and aniline, to give ester 6 or amide 7.[9]
Scheme 4.
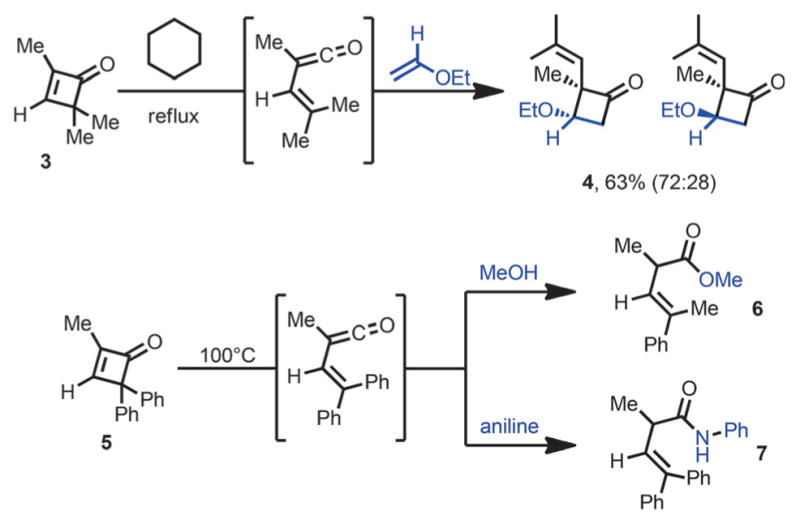
Vinylketene intermediates react with olefins and nucleophiles.
In 1977, Ficini et al. reported an unexpected rearrangement during the cycloaddition of ynamines with cyclobutenones (Scheme 5).[10] Unlike the cycloaddition of ynamines with cyclopentenones or cyclohexenones,[11] the reaction of ynamines with strained cyclobutenone 8 did not afford the [2+2] product 9. Instead, 3-amino-4-vinyl-cyclobutenones 10 were obtained in moderate yields. It was proposed that 10 came from intermediate 9, through a concerted rearrangement involving one π bond and two σ bonds, and at low temperature, the disrotatory ring-opening of 9 to form phenols is thermally forbidden. However, upon heating, 10 was converted to polysubstituted phenols 11. This transformation was proposed to involve thermal ring opening of the cyclobutenone followed by ring closure and aromatization.
Scheme 5.
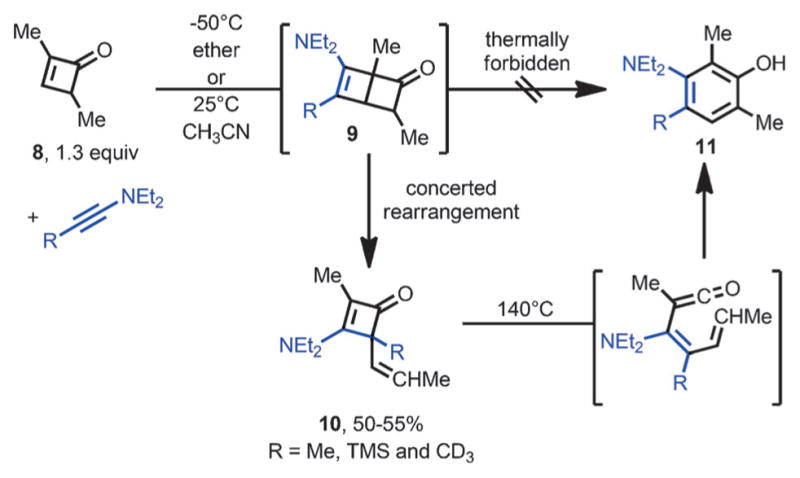
Ficini-type rearrangement with ynamines and cyclobutenones.
Eight-membered rings can also be accessed from cyclobutenones. In 1982, the Danheiser group developed a formal [4+4] annulation process to synthesize unsymmetrical highly functionalized cyclooctadienones (Scheme 6).[12] This reaction employed 1,3-dienes and vinylketenes as coupling partners, with cyclobutenones serving as the vinylketene precursor. Under the reaction conditions, the in situ generated vinylketene underwent a facile [2+2] cycloaddition to afford 2,3-divinylcyclobutanone 12, which upon heating gave cyclooctadienone 13 through a [3,3]-sigmatropic rearrangement.
Scheme 6.

Formation of cyclooctane derivatives by sigmatropic rearrangement.
The Danheiser group in 1984 reported an efficient approach to access highly substituted aromatic compounds from cyclobutenones and heteroatom (O, N or S)-substituted alkynes 14 via four successive pericyclic reactions (Scheme 7).[13] Upon heating above 80 °C, 4π-electrocyclic ring opening of cyclobutenone reversibly generated a vinylketene intermediate 15, which subsequently reacted with the electron-rich alkyne in a regiospecific [2+2] cycloaddition to give 4-vinyl cyclobutenones 16. As reported in Ficini’s work,[10] these 4-vinyl cyclobutenones can undergo a retro-4π/6π cyclization cascade to furnish highly substituted phenol derivatives.
Scheme 7.

Phenol formation by [2+2] cycloaddition of cyclobutenones with heterodisubstituted alkynes.
Later, Danheiser et al. also successfully applied this benzannulation method to the total synthesis of Mycophenolic Acid 19 (Scheme 8).[14] The key step in the synthesis involves the construction of pentasubstituted resorcinol by a cycloaddition cascade between cyclobutenone 17 and alkynyl ether 18.
Scheme 8.
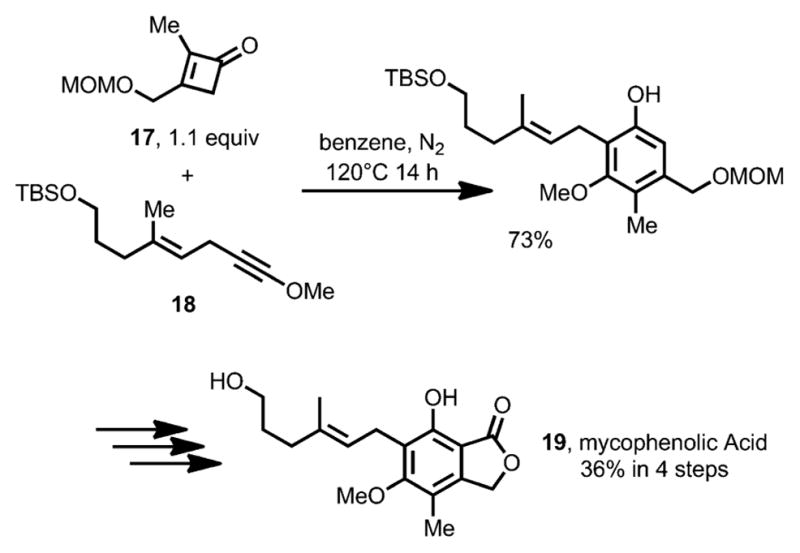
Total synthesis of mycophenolic acid. MOM=methoxymethyl acetal.
Although methoxy-substituted alkynes are excellent coupling partners for the benzannulation reaction with cyclobutenones, removal of the methyl group to generate free phenols is not trivial. In 1988, the groups of Kowalski[15] and Danheiser[16] independently developed an extended protocol using alkynyl silyl ethers 20 as substrates (Scheme 9). Under thermal conditions, the annulation proceeded with high yields to afford a variety of monosilyl-protected resorcinols. The silyl group can be smoothly removed under acidic conditions to give free phenols. By using this method, the Kowalski group accomplished an elegant synthesis of Δ-6-tetrahydrocannabinol 21. In both reports, 2,4-disubstituted cyclobutenones failed to furnish the desired annulation products.
Scheme 9.

Benzannulation with trialkylsiloxyalkynes as coupling partners. TIPS=triisopropylsilyl.
Although alkynyl ethers have been demonstrated as good coupling partners in benzannulation with cyclobutenones, the use of ynamines for phenol synthesis has been less common.[10] In Danheiser’s cyclobutenone benzannulation with N,N-dimethyl ynamines (Scheme 10),[13] allenes 22 were observed as byproducts. The formation of such allene products were suggested to have resulted from a zwitterionic intermediate 23.[17] This intermediate can either cyclize to provide the desired phenol product or afford allene 22 via an alkylideneoxete intermediate 24.
Scheme 10.
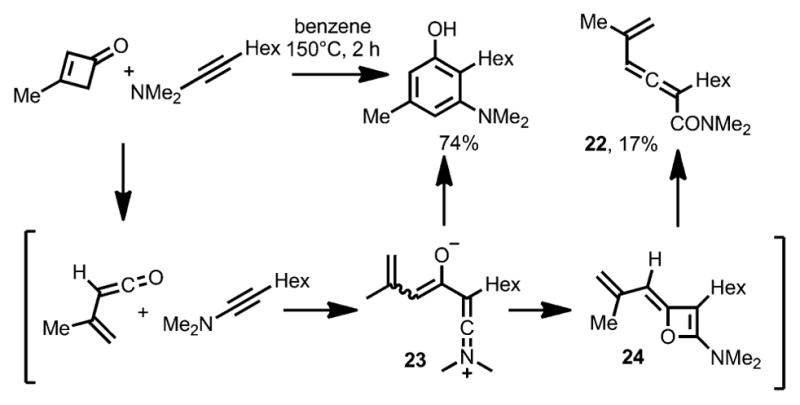
Allene side-product formation in Danheiser benzannulation.
Although the zwitterionic intermediate was able to give phenol products, the regioselectivity would be different from the electrocyclic reaction pathway (e.g., 25a and 25 b), as depicted in Scheme 11. Considering the unusual rearrangement reported by Ficini et al. (e.g, 25c),[10] the benzannulation of vinylketenes from cyclobutenones could provide three different isomers.
Scheme 11.
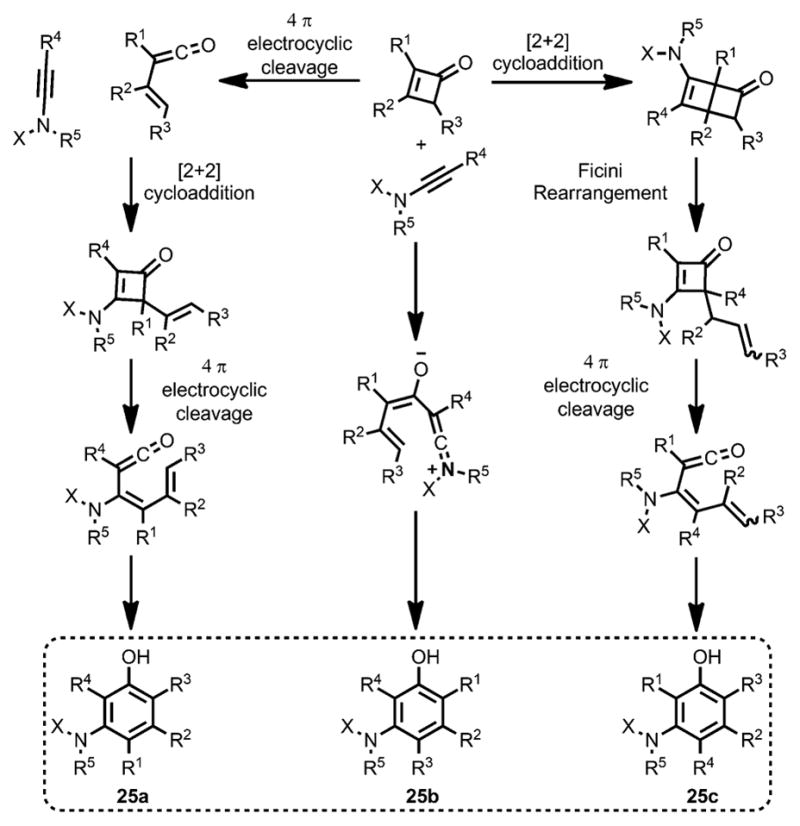
Three different regioisomers involved in the benzannulation.
To suppress these alternative regioisomeric products, less nucleophilic ynamides were employed in the benzannulation (Scheme 12).[18] Compared to ynamines, the ynamides favor the desired concerted [2+2] cycloaddition with the ketene rather than carbonyl addition. Under thermal conditions, the benzannulation between cyclobutenones 26 and ynamides 27 proceeded with high regioselectivity. By using this method, Danheiser and co-workers accomplished the formal synthesis of (+)-FR900482 (28, Scheme 13).
Scheme 12.
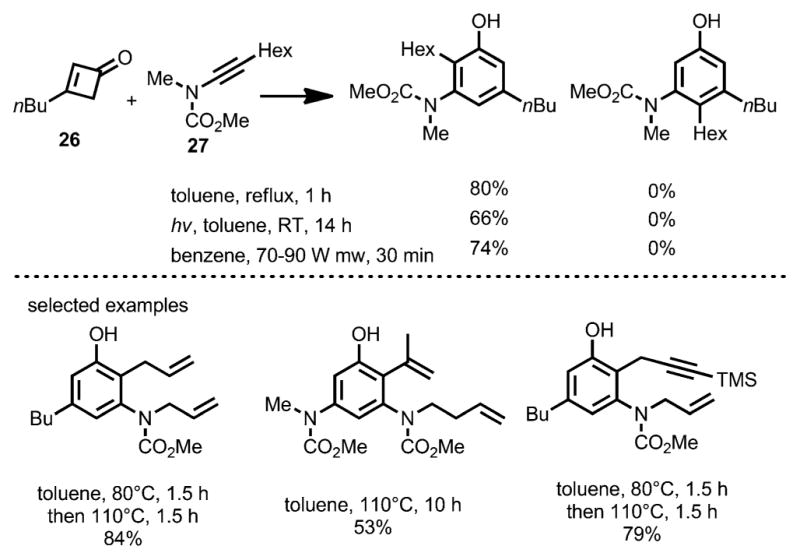
Ynamides as coupling partners for regioselective benzannulation.
Scheme 13.

Formal synthesis of (+)-FR900482. Teoc=2-(trimethylsilyl)ethoxycarbonyloxy, TBS=t-butyldimethylsilyl.
In addition to the intermolecular coupling between cyclobutenones and unsaturated π bonds, intramolecular cyclizations by electrocyclic ring opening of cyclobutenones have also been developed. In 1975, Mayr reported that cyclobutenones 29 were converted into the corresponding α-naphthols 30 quantitatively in refluxing cyclohexane (Scheme 14).[8] For cyclobutenone 31, bearing two different substituents at the C4 position, a mixture of 49 % of α-naphthol 32, 21 % of 33 and 30 % of 34 were isolated, indicating the formation of both cis-and trans-vinylketenes.
Scheme 14.
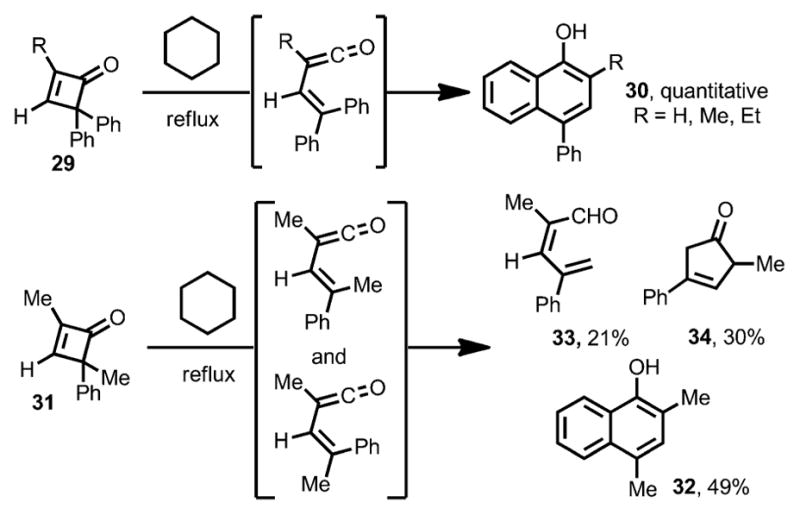
Intramolecular cyclization followed by thermal ring opening.
In 1985, Moore and co-workers developed the synthesis of polysubstituted benzoquinones through thermal rearrangement of 4-alkynylcyclobutenols 35 prepared from squaric acid derivatives (Scheme 15).[19] Upon 4π-electrocyclic ring opening, the resulting vinylketene intermediate 36 underwent ring closure to afford zwitterions 37 and/or 38, eventually leading to products 39 and/or 40. The selectivity was largely affected by the substituent: strong electron-withdrawing groups favored the formation of cyclopentenediones, whereas electron-donating groups facilitated the formation of benzoquinones.
Scheme 15.

Benzoquinone synthesis involving silyl or ally migration.
Interestingly, an allyl migration occurred when the trimethylsilyl group was replaced with an allyl group (e.g., 41).[20] By using aryl-substituted substrates (42, Scheme 16), aryl-fused benzoquinones were formed upon concomitant oxidation with Ag2O or CeIV. In all cases, the vinylketene intermediates were generated by an outward ring opening with regards to the oxygen substituent.
Scheme 16.
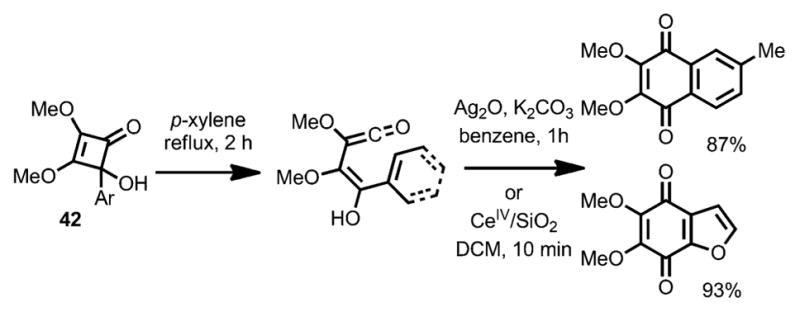
Benzoquinone synthesis involving intramolecular cyclization.
In 1988, Moore and Perri reported a detailed study on the substituent effect of the aryl group in the benzannulation reaction of 4-arylcyclobutenols (Scheme 17).[21] When cyclobutenones 43 and 44 were subjected to the reaction conditions, the same naphthalene-fused benzoquinone product was observed. Given that the 1-position of naphthalene is more reactive towards electrophilic substitution, this result indicated that some electrophilic character was required in the transition state of this reaction. In addition, the electrocyclic ring opening of cyclobutenone 45 was expected to give phenol 46. However, a mixture of quinones 47 and 48 was isolated instead, in which the ring closure of the vinylketene intermediate had selectively occurred at the carbon bearing the methoxy group followed by concomitant tautomerization or elimination of methanol.
Scheme 17.

Substituent effects on benzoquinone synthesis.
Moreover, although in most cases the hydroxy group favors outward rotation during ring opening, the presence of the strongly electron-rich aryl group (e.g., substrate 49) favors inward rotation, affording vinylketene intermediate 50, which led to the formation of γ-lactones 51 and 52 after workup with silica gel (Scheme 18).
Scheme 18.

Hydroxy group inward rotation in the presence of an electron-donating group.
In 1992, Liebeskind and co-workers developed a one-pot method for α-vinylation or arylation followed by benzannulation from 4-chlorocyclobutenone 53 (Scheme 19).[22] Through a Pd-catalyzed Stille coupling, a range of vinyl- and aryltin compounds were coupled with 53 to generate reactive cyclobutenone intermediate 54 in situ; subsequent thermal ring-opening and 6π-cyclization furnished the corresponding phenols 55 in high yields. Interestingly, when vinylzirconium 56 was employed, upon thermolysis, the reaction selectively afforded one of the two regioisomers 57 or 58, depending on the choice of catalyst system. For example, with 10 mol % [{Pd(allyl)Cl}2] and 20 mol % PPh3, phenol 57 was produced, whereas with 10 mol % [Pd(PPh3)4] and 40 mol % of PPh3, phenol 58 was produced. However, the reason for this selectivity is not yet clear.
Scheme 19.

Phenol synthesis by one-pot Pd-catalyzed cross-coupling/benzannulation sequence.
Later, syntheses of 1,4-quinone derivatives by thermal ring opening of cyclobutenones were extensively studied with substituents including a range of vinyl,[23,24] aryl, and heteroaryl[25] groups. However, in most cases, the formation of quinones required an oxidation step (Scheme 20).
Scheme 20.
Oxidation-free benzoquinone synthesis.
Aiming to avoid the oxidative modification, Harrowven and co-workers introduced a leaving group to the olefin moiety.[26] Under microwave conditions, cyclobutenone 59 afforded the desired quinone 60 in the absence of any oxidant (Scheme 21). However, an unusual phenomenon was observed: on increasing the electron density of the incorporated olefin moiety, carbocyclic compound 61, putatively through a carbonyl-ene reaction, became the major product. Interestingly, when 4-(o-styryl)cyclobutenones (e.g., 62, Scheme 22) were subjected to the reaction conditions with elevated temperature, benzobicyclo[3.2.1] structures 63 were afforded in high yields, which was proposed to involve a diradical intermediate 64.
Scheme 21.
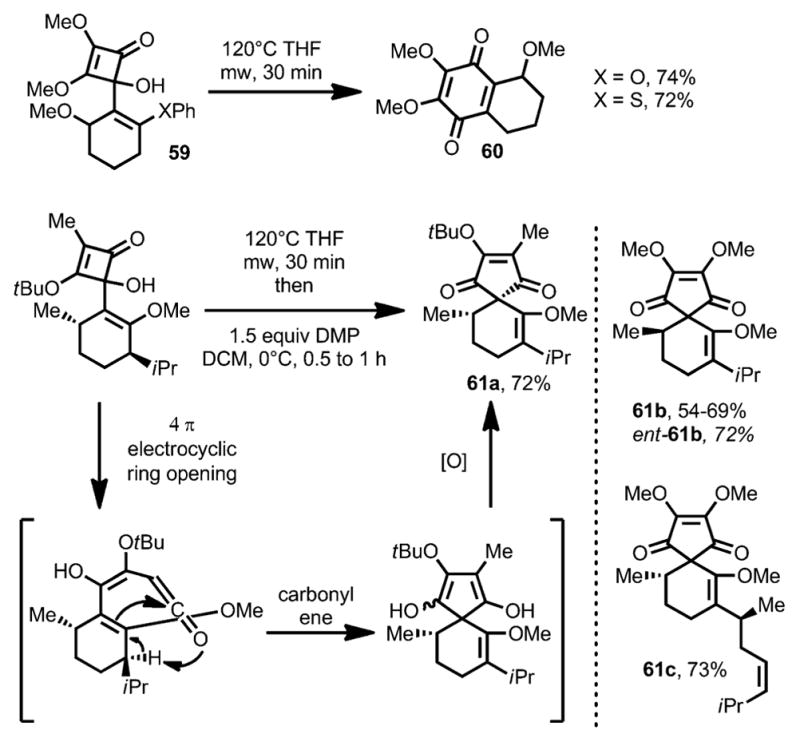
Substituent-controlled formation of carbocyclic ring vs. quinone derivatives.
Scheme 22.

Formation of benzobicyclo[3.2.1] structure via diradical intermediate.
Moore and co-workers later expanded this transformation to cyclobutenones bearing 4-allyl substituents (Scheme 23).[27] Upon 4π-electrocyclic ring opening, the resulting vinylketene intermediate 65 underwent a facile [2+2] cycloaddition to furnish the bicyclo[3.2.0]heptenones 66 as the final products. Substrates bearing free 4-OH (R=H), although they successfully underwent a similar ring expansion (e.g., 66g), were rather limited in substrate scope as a 2-phenyl substituent would lead to an unusual product 66 h. Substrate 66e, which undergoes facile deprotection to afford diketone 67, was used to unveil the mechanism of the reaction with 66h. Although it is stable under thermolysis in toluene, 67 was converted into tricycle 66h in the presence of a catalytic amount of acetic acid.
Scheme 23.

Bicyclo[3.2.0]heptenone formation from 3-allylcyclobutenones.
The vinylketene intermediate can also be intercepted intra-molecularly by a σ-nucleophile. In 2009, Shimizu and co-workers developed the synthesis of β-lactams through thermal ring opening of aminocyclobutenone 68.[28] Upon 4π-electrocyclic ring opening, the resulting amino vinylketene intermediate was attacked by the amine moiety to provide the β-lactams 69 in good yields. Although inherently favoring the cis isomer (3:1–4:1 without adding bases), the stereoselectivity can be well controlled: when 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was added, trans-69 was favored; in contrast, in the presence of 1,4-dimethylpiperazine, cis-69 was afforded exclusively. In 2014, the same group developed the enantioselective version of this transformation by using as the starting materials chiral aminocyclobutenones 68 (Scheme 24),[29] which were obtained from asymmetric reduction of the corresponding iminocyclobutenones 70 by using chiral phosphoric acid 71 as catalyst and dihydrobenzothiazole 72 as the reductant. Upon thermolysis with different bases, both trans- and cis-β-lactams were afforded in high yield and enantioselectivity.
Scheme 24.

Base-controlled chiral lactam formation through enantioselective reduction of iminocyclobutenones. PMP = p-methoxyphenyl.
3. Photolysis-Induced Ring Opening
In 1967, Baldwin and co-workers studied the ring opening of 2,4-dichloro-3-phenylcyclobutenone 73 (Scheme 25).[30,31] Under thermal conditions, E-β,γ-unsaturated ester 74 was provided; in contrast, under irradiation by a high-pressure mercury lamp, Z-β,γ-unsaturated ester 75 was afforded. The stereoselectivity was tentatively explained by the calculated excited state of the reaction. Based on Kikuchi’s work,[32] the n–π* singlet excited state could lead to a perpendicular diradical, which caused an inward roation.
Scheme 25.
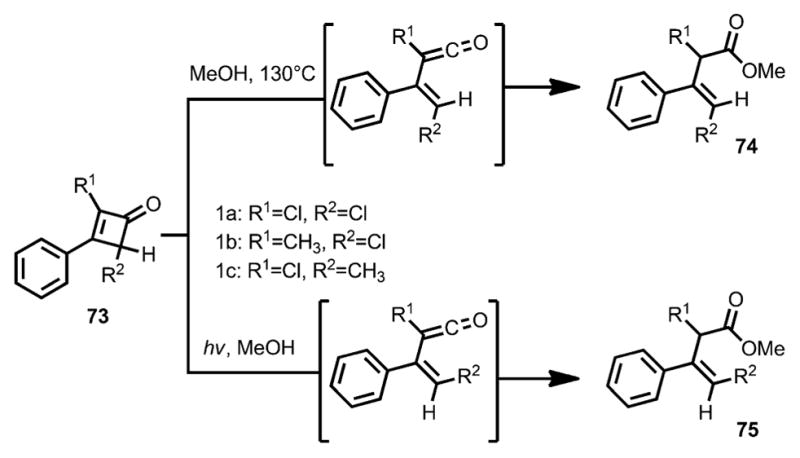
Inward and outward ring opening by thermolysis and photolysis.
The involvement of a vinylketene intermediate under photolysis conditions was observed by Chapman and Lassila by using infrared spectroscopy.[33] Upon irradiation with a high-pressure mercury lamp, 2,3,4,4-tetrachlorocyclobutenone 76 (Scheme 26) afforded a new carbonyl band at 2145 cm−1, which was believed to be a ketene carbonyl peak of 77.
Scheme 26.
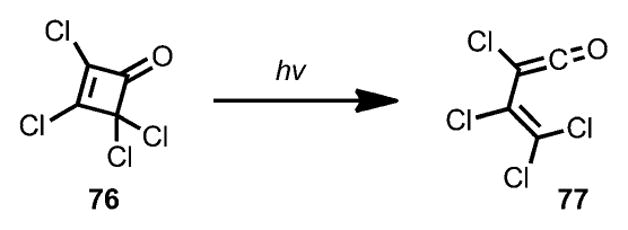
Formation of vinylketene under photolysis.
In 1974, Toda and Todo reported the photochemical ring-opening of methylenecyclobutenone 78 (Scheme 27).[34] Under UV irradiation, the ring opening of 78 afforded allene–ketene intermediate 79. Similar to other vinylketene intermediates, 79 can be intercepted intramolecularly by a hydroxy group, affording furanone 80 as the final product. With methanol as solvent, however, photolysis provided isomeric compound 81 as the product.[35] Control experiments showed that, although there was no conversion under the photolysis conditions, 80 could isomerize to 81 rapidly upon heating in the presence of a base.
Scheme 27.

Intramolecular nucleophilic attack after ring opening.
Later, the same group demonstrated that, upon ring-opening, the allene–ketene intermediate 82 could undergo a rapid [2+2] cycloaddition with benzophenone to afford an oxetanone intermediate, which eventually led to product 83 after a concomitant elimination of diphenylketene 84 (Scheme 28).[36] A similar transformation was realized under an oxygen atmosphere with elimination of carbon dioxide, albeit with a lower yield.
Scheme 28.
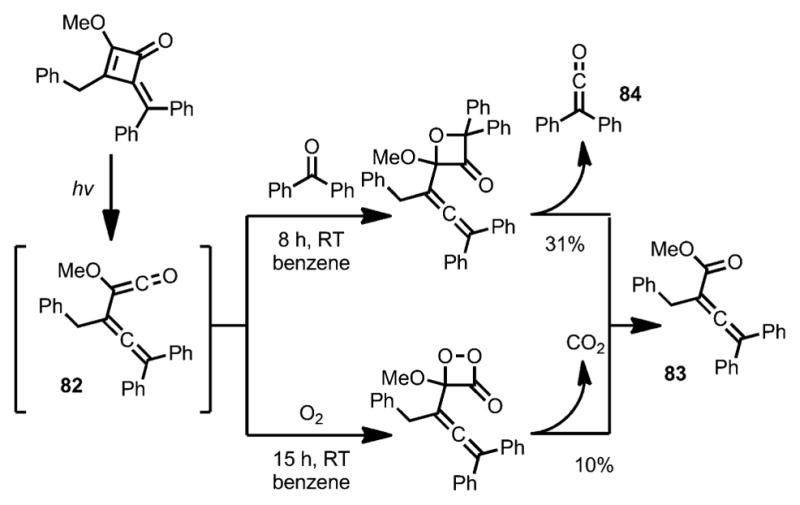
Ring opening with concomitant one-carbon loss.
In 1984, Schiess et al. reported the cycloaddition of benzocyclobutenone 85 (Scheme 29) with various dienophiles under flash photolysis conditions.[37] Upon UV irradiation in ether, benzocyclobutenone 85 underwent isomerization to vinylketene 86 with a lifetime of 170 ms at room temperature. Unlike simple vinylketenes, which typically react with electron-rich olefins, 86 acted as a diene and rapidly formed [4+2] cycloaddition products with diazo, activated carbonyl compounds or Michael acceptors, likely driven by forming aromatic ketones.
Scheme 29.
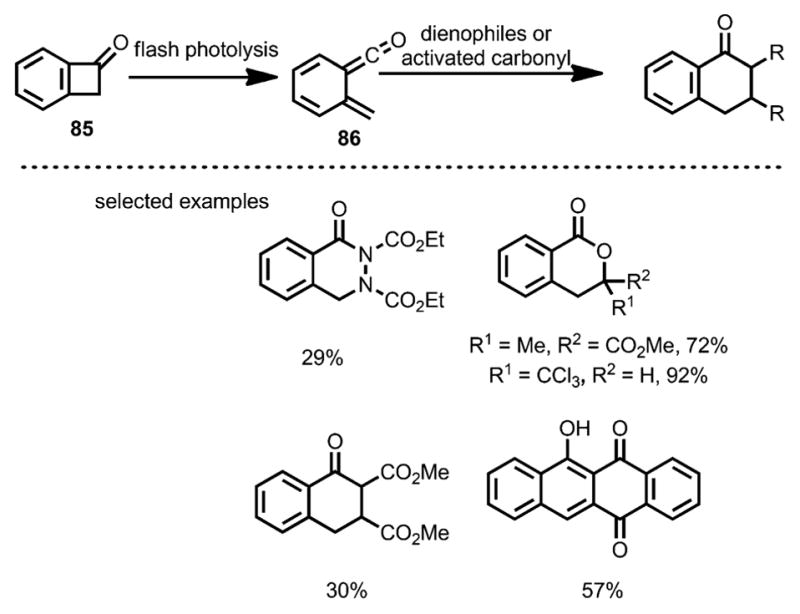
Reaction of benzocyclobutenones with activated carbonyl and dienophiles.
4. Nucleophile-Induced Ring Opening
Due to the α,β-unsaturated ketone moiety, cyclobutenones can be attacked by nucleophiles in a 1,2- or 1,4-addition fashion. The resulting alkoxide or enolate often triggers the subsequent ring opening of the four membered ring structures. In the 1950s, Roberts and co-workers studied the reaction of 3-phenyl-4,4-dichlorocyclobutenone 87 with hydroxide (Scheme 30).[38] 1,2-Addition of hydroxide to the carbonyl moiety in 87 initiated ring opening through retro-4π-cyclization, giving 4,4-dichloro-3-phenyl-2-butenoic acid 88 as a final product in good yield. A similar transformation in the presence of nBuOH was later reported by Hassner and Dillon, which eventually led to the formation of the corresponding ester.[39]
Scheme 30.
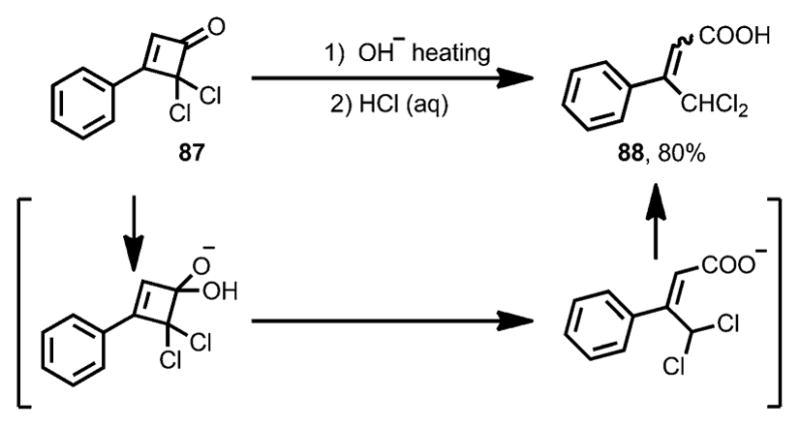
Early observation of nucleophile-induced ring opening.
In 2001, Murakami et al. reported the ring opening of cyclobutenones triggered by silyl anions (Scheme 31).[40] When 3-phenylcyclobutenone 89 was treated with a silylcuprate at −78 °C, the 1,4-addition product could be smoothly trapped by acetic anhydride to afford vinyl acetate 90, which, upon heating, afforded diene product 91 in an excellent yield as one stereoisomer. When silyllithium was used as a nucleophile, 89 underwent 1,2-addition to afford highly reactive intermediate 92, which, upon treatment with acetic anhydride at −78 °C, gave the ring-opening product 93 in excellent yield. Based on these results, the same group later developed an efficient approach for the stereoselective synthesis of highly functionalized 1,3-dienes from cyclobutenones.[41]
Scheme 31.

Silyl nucleophile induces stereoselective synthesis of 1,3-dienes.
In 2004, Magomedov et al. reported a cascade reaction sequence for the synthesis of heavily functionalized cyclohexanones through the ring opening of cyclobutenone 94, which was initiated by addition of vinyllithium reagent 98 (Scheme 32).[42] Upon 1,2-addition, alkoxide 95 underwent a charge-accelerated 4π conrotatory ring opening to generate triene 96, which, through 6π ring closure and isomerization, eventually resulted in cyclohexenone 97 in high yield as the trans isomer. Reaction intermediates, such as α,β-unsaturated ketone 99 and cyclobutenol 100, were successfully isolated by quenching the reaction at low temperature. As expected, treatment of cyclobutenol 100 with lithium diisopropylamide (LDA) provided cyclohexone 101 a in 94 % yield. Other pericyclic rearrangements upon 1,2-addition and ring opening can also occur under the reaction conditions, leading to more diverse products (Scheme 33).
Scheme 32.

Lithium salt induced electrocyclic reactions.
Scheme 33.
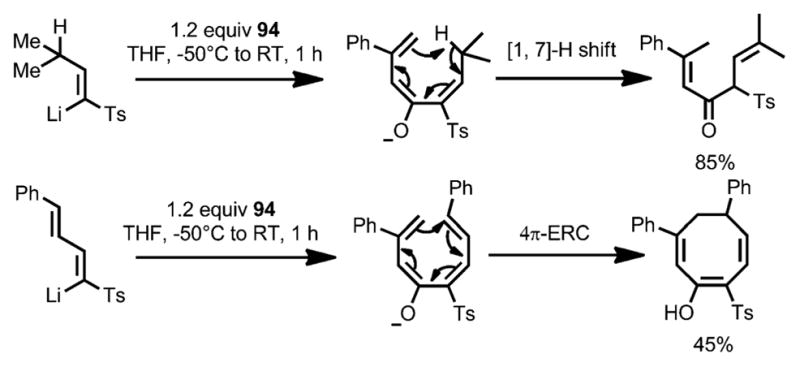
Pericyclic rearrangements involved in the ring opening. ERC=electrocyclic ring closure.
In 1993, Cammers-Goodwin reported the ring opening of cyclobutenones triggered by tributylphosphine in alcoholic solvents (Scheme 34).[43] When 3-phenylcyclobutenone 94 was treated with a catalytic amount of tributylphosphine in methanol, the ring-opening product 102 was afforded, albeit contaminated by approximately 1 % yield of the conjugated ester 103. Tributylphosphine was demonstrated to be crucial for the transformation as triethylamine failed to afford any desired product. Interestingly, the β,γ-unsaturated ester 102 can be converted into the conjugated product 103 upon gentle heating in alcohol with a base, indicating that the tributylphosphine catalyst led to the kinetic product. In contrast, under the same reaction condition, 3-pentylcyclobutenone gave methoxy-substituted cyclobutanone 104 in high yield. The ring-opening product 105 was afforded only when an increased concentration of tributylphosphine and higher temperature were applied. A mechanism was proposed whereby the reaction started with 1,4-addition of tributylphosphine to the cyclobutenone and concomitant protonation, followed by an SN1 displacement by methanol to afford cyclobutanone 104, or a Grob-type fragmentation to give the ring-opening product 102.
Scheme 34.

Tributylphosphine-catalyzed ring opening of cyclobutenones.
Inspired by the phosphine-catalyzed ring opening reaction, in 2015 Zhang and co-workers reported the enantioselective synthesis of spirocycles 108 through a formal [4+2] cycloaddition between cyclobutenones 106 and isatylidenemalononitriles 107, catalyzed by amino acid-derived chiral phosphines 109 (Scheme 35).[44] Compared with phosphine catalysts bearing a single or weak double H-bond donor (e.g., 109 a–e), phenylalanine-derived catalyst 109 f, with a strong double H-bond donor moiety, proved optimal, affording the cycloadduct in excellent yield and good enantioselectivity. NaI was proposed to provide a counterion that stabilized the zwitterionic intermediate. The substitution pattern of cyclobutenones only had a marginal effect on reactivity and enantioselectivity.
Scheme 35.

Chiral phosphine-catalyzed enantioselective spirocyclization. PMB=p-me-thoxybenzyl.
Unlike Cammers-Goodwin’s proposal (Scheme 34),[43] a 1,2-addition of the phosphine catalyst to the cyclobutenone was proposed as the initial step (Scheme 36). The following ring opening led to a 1,4-dipolar intermediate 110, which then underwent a stepwise addition/elimination sequence to generate the spiroannulation product.
Scheme 36.
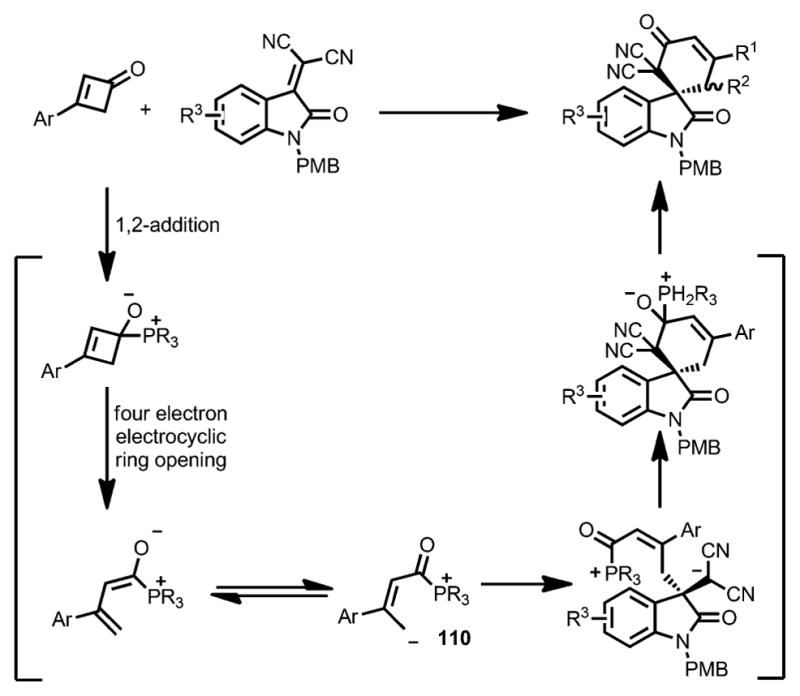
Mechanism for chiral phosphine-catalyzed enantioselective spirocyclization.
Such a formal [4+2] cycloaddition with cyclobutenones can also be realized with N-heterocyclic carbene (NHC) catalysts. In 2015, Chi and co-workers reported that a chiral NHC carbene (e.g., 115) catalyst effectively coupled imines 111 with cyclobutenones 112 in good yield and high enantioselectivity (Scheme 37).[45] As proposed, the enolate intermediate 113, formed after the 1,2-addition and concomitant ring opening, underwent a stepwise addition with imines to give lactam 114, bearing a quaternary carbon center as a single diastereomer. Both cyclic sulfonyl imines (e.g., 116) and tert-butyloxycarbonyl (Boc)-protected isatin-derived imines (e.g., 117) are suitable substrates. Cyclobutenones with a substituent at the 2-position showed low reactivity in this transformation.
Scheme 37.

Chiral N-heterocyclic carbene-catalyzed enantioselective lactam synthesis.
Later, Chi and co-workers also developed a formal [3+2] cycloaddition between 4-chlorocyclobutenones 118 and azomethine imines catalyzed by a chiral isothiourea catalyst 119 (Scheme 38).[46] Unlike their earlier work,[45] upon 1,2-addition and ring opening, the α-carbon of the enolate intermediate 120, instead of the γ-carbon, attacked the azomethine imines and eventually provided heterocycle 121. A variety of functional groups were well tolerated, giving high diastereo- and enantioselectivity, although bulky substrates lowered both the yields and enantioselectivities. For cyclobutenones bearing a proton or a methyl group at the C4 position or having 4,4-dichloro substituents, no cycloaddition was observed.
Scheme 38.

Chiral isothiourea-catalyzed formal [3+2] cycloaddition.
The nucleophile-initiated ring opening of benzocyclobutenones was first reported by Cava and Muth in 1960.[47a] Unlike monocyclic cyclobutenones, the cleavage of benzocyclobutenones could occur at either proximal or distal position, resulting in carboxylic acids 122 and/or 123 (Scheme 39).[47]
Scheme 39.
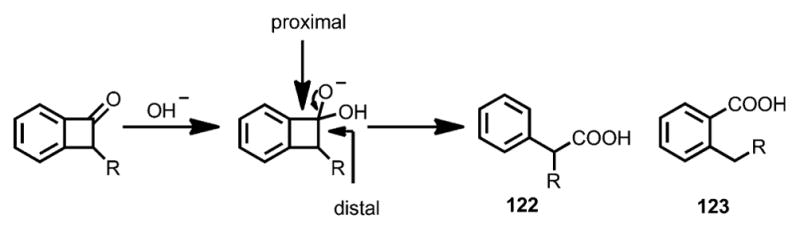
Early observation of nucleophile-induced ring opening of benzocyclobutenones.
In 1997, the Suzuki group reported that lithium tetramethylpiperidide (LiTMP) could attack the carbonyl moiety of benzocyclobutenone 124, leading to ring opening predominantly at the C1–C2 bond (Scheme 40).[48] The resulting anionic intermediate 125 reacted with benzaldehyde smoothly to afford alkoxide intermediate 126, which, upon intramolecular cyclization, resulted in lactone 127 in decent yields.
Scheme 40.
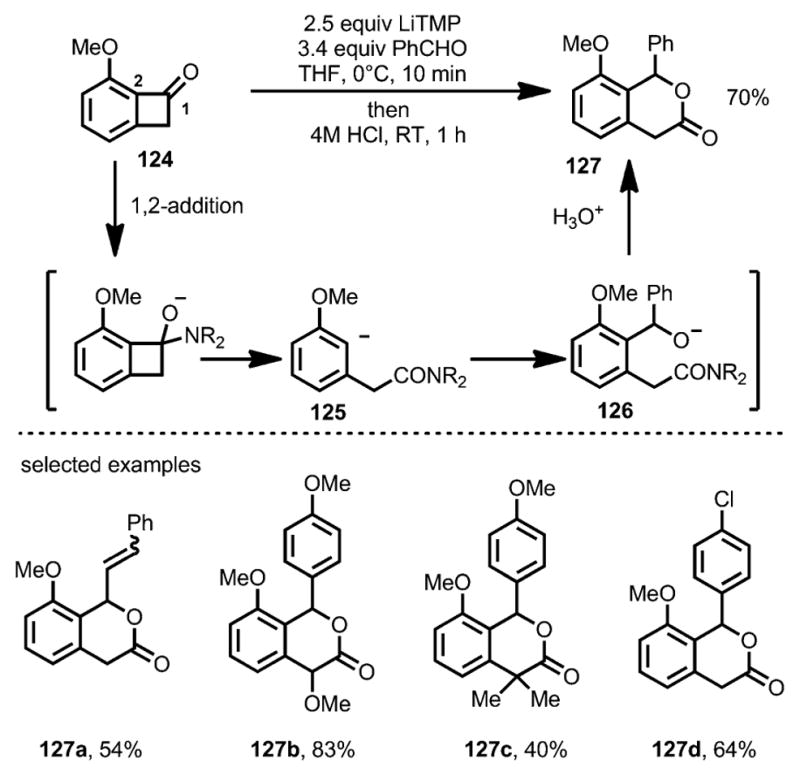
Lithium salt-induced ring opening trapped by aldehydes.
In 2011, the Aguilar and co-workers reported the coupling between benzocyclobutenone and Fischer carbene 128 (Scheme 41).[49] Upon addition with an alkyllithium reagent, benzocyclobutenone 129 underwent ring opening at the proximal position to afford oxy-ortho-quinodimethane 130, which attacks the Fischer carbene to provide intermediate 131. Followed by a 1,2-metal migration, the seven-membered ring oxonium intermediate 132 was attacked intramolecularly by the alkoxide and then protonation to give bridged compound 133 as the final product. However, yields were generally low for this reaction.
Scheme 41.
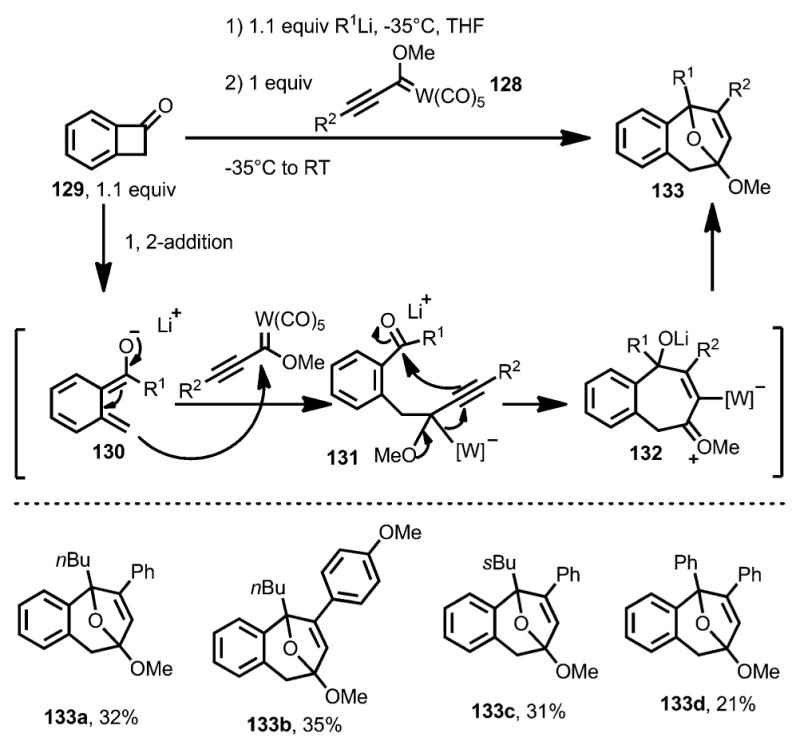
Lithium salt-induced ring opening trapped by Fischer carbenes.
In 2006, Matsuya et al. reported the rapid synthesis of 2,3-benzodiazepine derivatives 134 through a diazoanion-mediated benzocyclobutenone ring-opening transformation (Scheme 42).[50] Upon oxyanion-facilitated ring opening, an 8π-electrocyclization followed by isomerization led to 2,3-benzodiazepines as the final products. This method was later widely used in the syntheses of benzodiazepine[51] and unfused diazepine[52] derivatives.
Scheme 42.
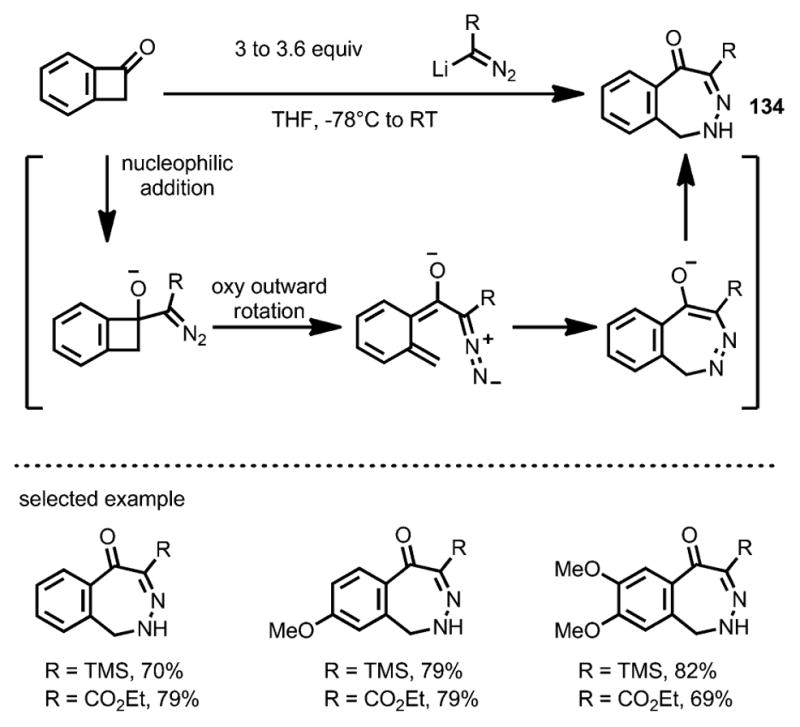
Diazoanion-induced ring opening and synthesis of 2,3-benzodiazepine derivatives.
5. Transition-Metal-Catalyzed Ring opening
Transition metal catalysis has emerged as a powerful tool in the functionalization of chemically inert bonds. Owing to their high ring strain, cyclobutenones are widely used in transition-metal-catalyzed carbon–carbon bond activation reactions.
5.1 Reactions involving stoichiometric transition metals
The first example of transition-metal insertion into cyclobutenones was reported by Liebeskind and co-workers in 1990 (Scheme 43).[53] Upon treatment with Wilkinson’s catalyst, 3-substituted and 2,3-disubstituted cyclobutenones smoothly produced rhodacyclopentenones 135 in good yields through ring-opening at the C1–C4 bond. However, 4-substituted cyclobutenones only afforded a mixture of complex products under the reaction condition, likely due to the steric hindrance.[54] Interestingly, benzocyclobutenones generally gave two regioisomeric rhodacyclopentenones 136 and 137, which can arise from cleavage of either the C1–C2 or C1–C8 bond. When an isolated mixture of 136 and 137 was heated at 130 °C, conversion into 137 was observed, indicating that rhodacycle 137 came from 136 under the reaction conditions and was the thermodynamic product. These rhodacyclopentenones 135–137 were found to be reluctant to react with alkynes.
Scheme 43.

The first example of transition-metal insertion into cyclobutenones.
In 2013, Yamashita, Nozaki, and Murakami studied the reactivity of a rhodium complex 138 containing an electron-rich pincer-type ligand towards C–C activation (Scheme 44).[55] Remarkably, the rhodium complex 139 underwent oxidative addition into the benzocyclobutenone C–C bond at room temperature with exclusive C1–C4 selectivity.
Scheme 44.
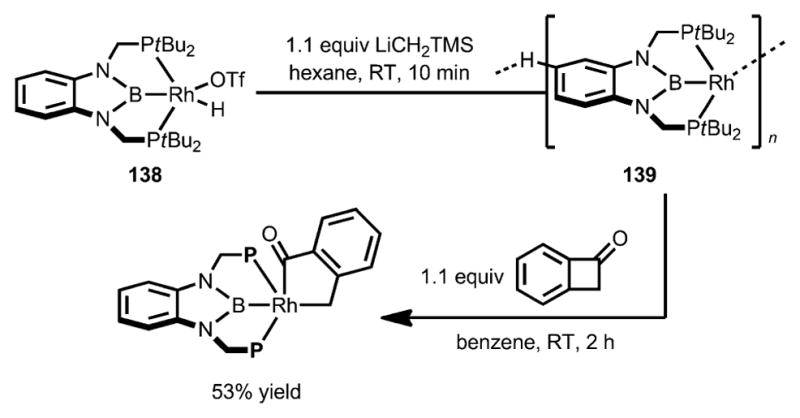
PBP pincer rhodium complex-enabled selective insertion of benzocyclobutenones.
The insertion of cobalt complexes was also investigated (Scheme 45).[54,56] Unlike [CoCl(PPh3)3] and [Co(η5-C5H5)(CO)2], only the more electron-rich cobalt precatalyst [Co(η5-C9H7)(PPh3)3] 140 was able to react with cyclobutenones to afford η4-vinylketene π-complexes 141. In sharp contrast to the reaction with rhodium (see above, Scheme 43), 3,4-disubstituted cyclobutenones afforded the η4-vinylketene products in higher yields as an E/Z isomeric mixture. Although the E isomers were kinetically favored, they could nevertheless be converted into their Z isomers with an elongated reaction time. These η4-vinylketene complexes (141) are spectroscopically different from the rhodacyclopentenones.[54] In particular, the stretching frequency of the carbonyl group of 141 is substantially higher, at 1756–1787 cm−1, whereas that of the rhodacyclopentenones appeared at around 1645 cm−1.
Scheme 45.
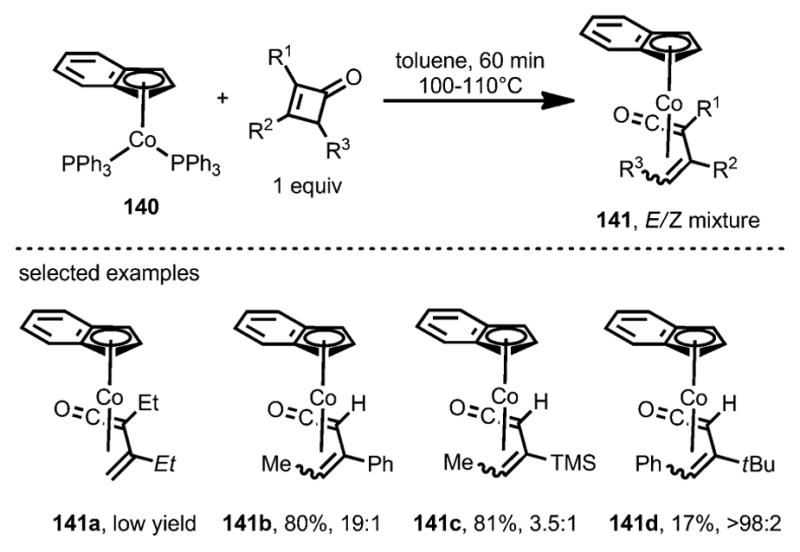
η4-Vinylketene complex formation through insertion of cobalt complexes into cyclobutenones.
Interestingly, 3-ethoxy-substituted cyclobutenone 142 gave cobaltacyclopentenone 143 as the only product (Scheme 46).[54] A phenyl-substituted cobaltacyclopentenone 144 was obtained when the π complex 141 e was subjected to a large excess of triphenylphosphine or to a CO atmosphere, although, in both cases, η4-vinylketene 141 e could be regenerated by heating. The treatment of benzocyclobutenone with [Co(η5-C9H7)(PPh3)3] also afforded similar cobaltacyclopentenones 145 and 146; however, no isomerization was observed when the isolated pure 145 and 146 were subjected to the reaction conditions.
Scheme 46.
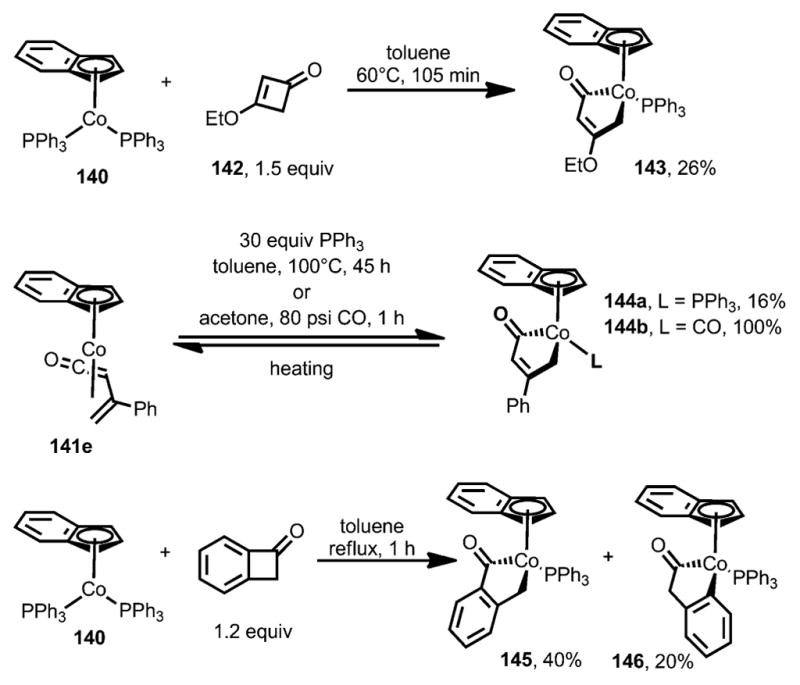
Cobaltacyclopentenone complex formation with cyclobutenones and benzocyclobutenones.
When superstoichiometric ZnCl2 was used as an additive, a new cobalt complex 147 was isolated with a nonconjugated enone moiety (Scheme 47). The exact reason remains unclear.
Scheme 47.
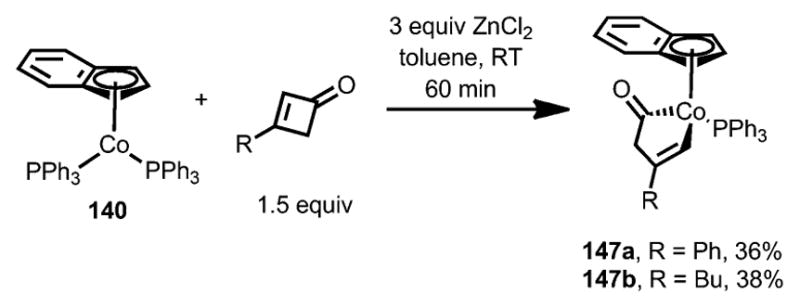
Formation of different cobaltacyclopentenones by addition of ZnCl2.
Three mechanisms were proposed regarding the formation of η4-vinylketene cobalt complexes.[54] Mechanism 1 (Scheme 48) involves thermolysis of cyclobutenones to form vinylketene intermediate 148, which was then trapped by the cobalt complex. This mechanism was excluded for two reasons. Firstly, cobalt insertion can happen at room temperature, whereas the thermolysis would be very slow. Secondly, the transformation can occur with isopropanol as solvent, in which the free vinylketene intermediate would be trapped rapidly by solvent instead of the cobalt species. Mechanism 2 is initiated by direct oxidative addition into the C1–C4 bond of cyclobutenones, followed by loss of PPh3 to give the η4-vinylketene cobalt complexes. However, the conversion from isolated 149 into η4-vinylketene cobalt complexes was very slow at 60 °C, whereas the insertion of cyclobutenone to form the η4–π complex happened smoothly at this temperature. Mechanism 3, which is the most consistent with experimental results, proceeds by pre-coordination of the cyclobutenone carbon–carbon double bond to [Co(η5-C9H7)(PPh3)3], followed by phosphine loss and subsequent ring opening.
Scheme 48.
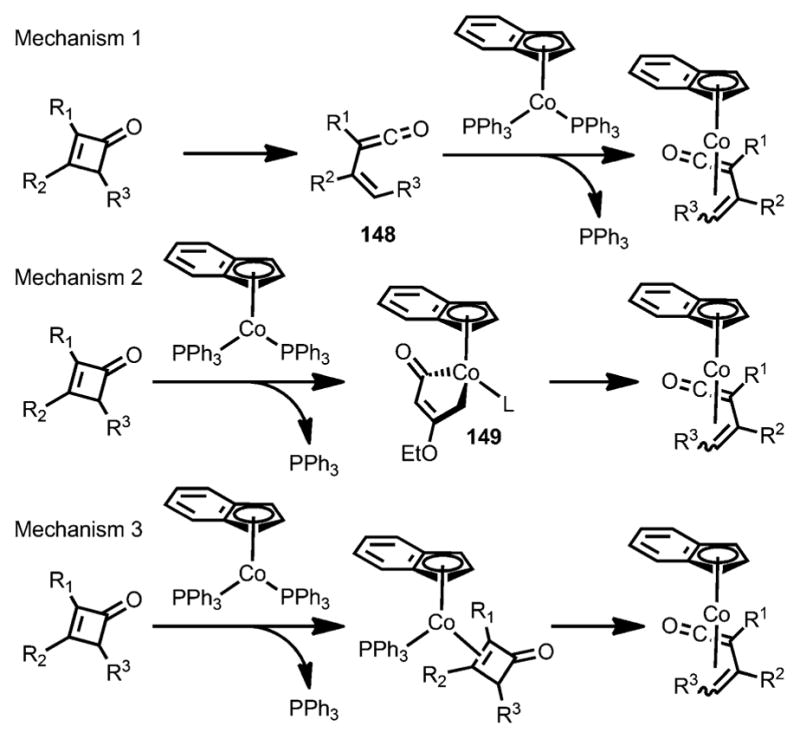
Three mechanisms proposed for the formation of η4-vinylketene complexes.
η4-Vinylketene cobalt complex 150 was found to react with a variety of alkynes to produce phenol derivatives (Scheme 49).[56] When unsymmetrical alkynes were used, a mixture of regioisomers was obtained. 1,5-Cyclooctadiene (cod) was employed as an additive to inhibit alkyne trimerization. However, 3,4-disubstituted η4-vinylketene cobalt complex 151 was only able to react with highly electron-deficient dimethyl acetylenedicarboxylate to provide a phenol derivative along with the cyclohexadienone complex 152.
Scheme 49.

Stoichiometric cobalt-enabled benzannulation reaction.
5.2 Reactions involving catalytic transition metals
In 1991, Liebeskind and co-workers reported the first catalytic phenol synthesis from cyclobutenones and alkynes (Scheme 50).[57] When the two coupling partners were subjected to a catalytic amount of [Ni(cod)2] at 0 °C, phenol products 153 were rapidly formed. Such a transformation was proposed to be initiated by a nickel-η4-vinylketene or nickelcyclopentenone intermediate followed by migratory insertion and reductive elimination sequence. Although both 3-substituted and 3,4-disubstituted cyclobutenones were suitable substrates, 2,3-disubstituted cyclobutenones failed to provide the desire products. The tolerance of a tributylstannyl substituent (e.g., 153 e) allows further functionalization of the product. In contrast to Danheiser’s benzannulation,[13] the nickel-catalyzed phenol synthesis can use non-activated alkynes and gives a different regioselectivity (e.g., ortho-153 vs para-153). The size of the alkyne substituents only had a marginal effect on the regioselectivity, whereas the heteroatom-containing substrates (e.g., 153 b and 153 d) showed an increased regioselectivity, which was proposed to come from a chelation effect (e.g., 154).
Scheme 50.

Nickel-catalyzed benzannulation of cyclobutenones and alkynes.
In 2011, Harrity and co-workers expanded the scope of the nickel-catalyzed benzannulation to alkynylboronates 155 (Scheme 51).[58] Unlike the rhodium catalyst, which provided cyclobutenone dimerization 156 as the major product, [Ni(cod)2] yielded the desired phenol boronic ester 157 in good yield and excellent regioselectivity, which allowed further derivatization through cross-coupling reactions. As this method was compatible with 3,4-disubstituted cyclobutenones, the regioselectivities of the coupling with 3-substituted cyclobutenones were predominantly determined by the substituents on the alkynylboronates. For example, sp3-based substituents showed high selectivity for the regioisomer where the pinacolboryl (Bpin) group was meta to the phenolic OH (e.g., 158 a), whereas sp2-based substituents resulted in a modest level of selectivity for the regioisomer where the Bpin group was ortho to the phenolic OH (e.g., 158 b).
Scheme 51.
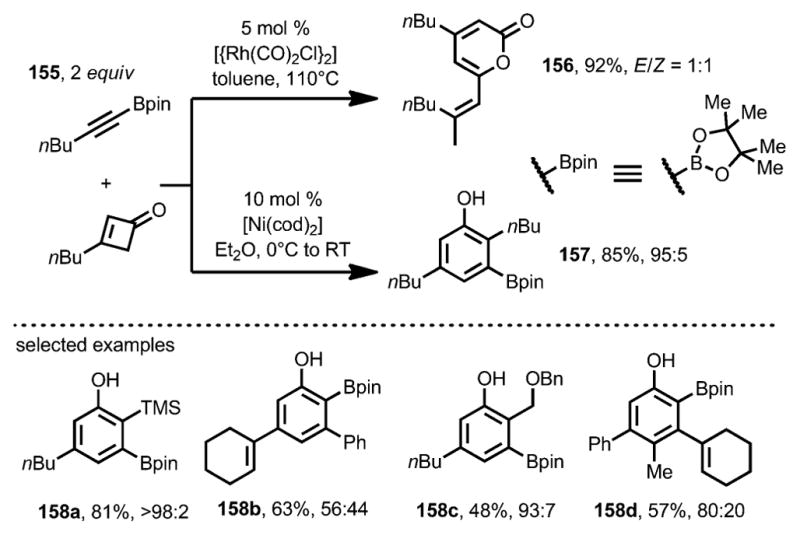
Nickel-catalyzed benzannulation with alkynylboronates.
In 2015, the same group reported a detailed study of the nickel-catalyzed benzannulation with alkynes bearing aryl substituents (Scheme 52).[59] When norbornadiene was added as a promoter to enhance the catalyst lifetime, various alkynes were compatible, giving heavily substituted phenols in good yields, and the regioselectivity was predominantly controlled by the substituents on the alkynes: an alkyl group tends to be incorporated ortho to the phenol OH moiety, whereas an aryl group prefers to stay at the meta position. Unlike Liebeskind’s work,[57] the presence of heteroatoms only had a marginal effect on the regioselectivity.
Scheme 52.
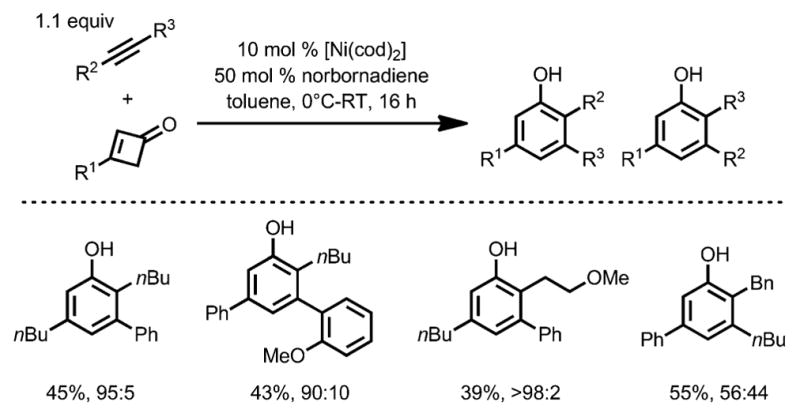
Regioselectivity in nickel-catalyzed benzannulation.
Two mechanisms were proposed for this benzannulation reaction (Scheme 53).[59] The first one is initiated by ring opening of the cyclobutenone to form either a metallocyclopentenone or a η4-vinylketene complex 159, which would then undergo migratory insertion and reductive elimination to afford the phenol product. The second reaction pathway was initiated by cyclometallation between the carbonyl group and the alkyne to form metallacycle 160. Similarly to the nickel-catalyzed cyclobutanone activation,[60] subsequent β-carbon elimination and reductive elimination would provide the phenol products. While both mechanisms are possible, the isolation of ester 161, when adding phenol to the standard reaction conditions, suggested involvement of a vinylketene-like intermediate.
Scheme 53.
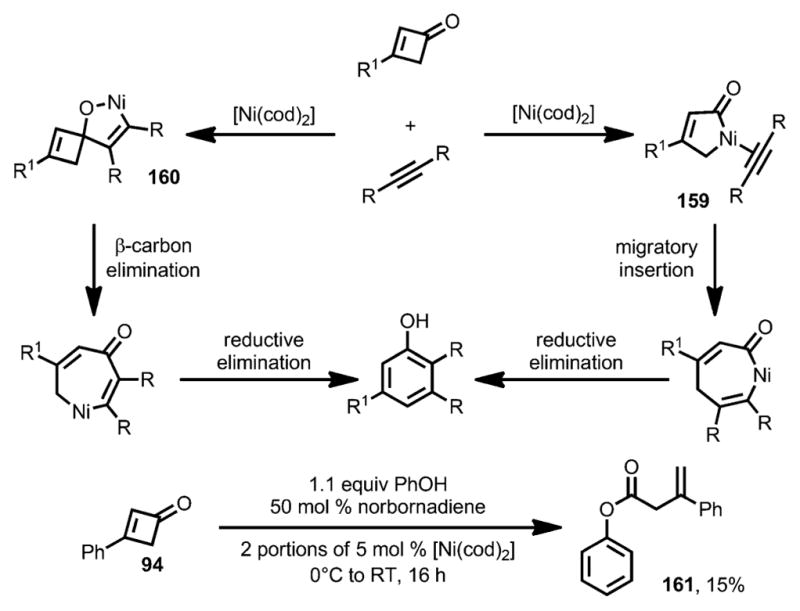
Mechanism for nickel-catalyzed benzannulation.
In 1993, Huffman and Liebeskind developed a transitionmetal-catalyzed synthesis of medium-sized rings from cyclopropyl and cyclobutyl-substituted cyclobutenones 162 (Scheme 54).[61] It was proposed that the rhodium or ruthenium first inserted into the C1–C4 bond of cyclobutenone 162, which was followed by β-carbon elimination and reductive elimination to give cycloheptadienone 163. Product 164 was also obtained, likely through an olefin isomerization from 163 by 1,5-hydrogen shift under thermal conditions.
Scheme 54.
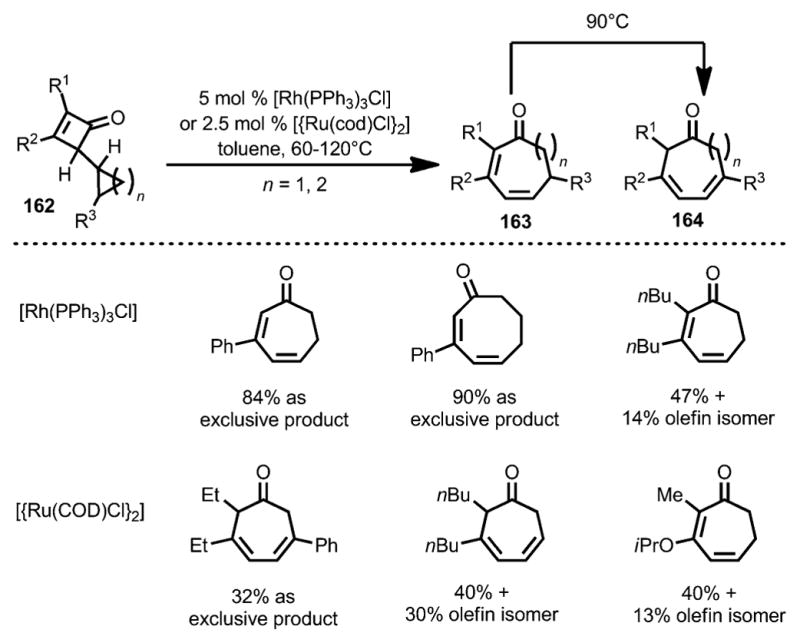
Rhodium-catalyzed medium-size ring synthesis.
In 2004, Kondo, Mitsudo, and co-workers developed a Rh/Ru-catalyzed dimerization of cyclobutenones (Scheme 55).[62] When cyclobutenone 165 was treated with a catalytic amount of [{RuCl2(CO)3}2] in refluxing toluene, 2-pyranone 166 was afforded as a mixture of E and Z isomers. However, use of [{Rh(CO)2Cl}2] provided the desired dimers in E configuration as the sole product.
Scheme 55.
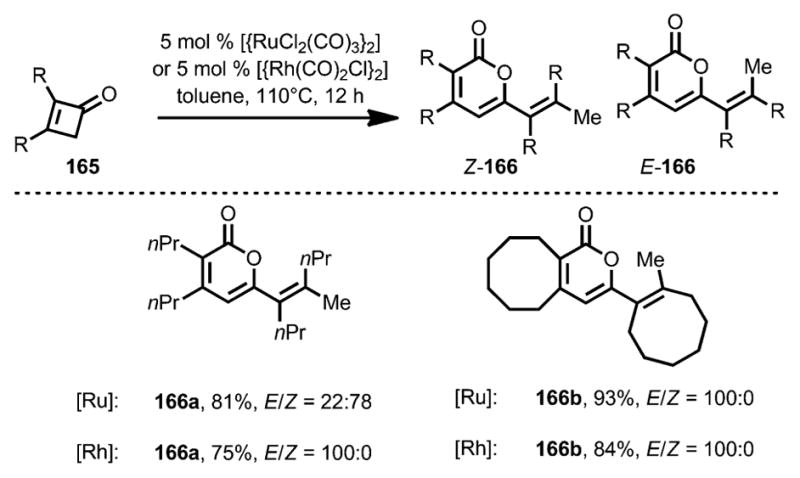
Dimerization of 2,3-disubstituted cyclobutenones.
In addition, with [{Rh(CO)2Cl}2] as the catalyst, the reaction between cyclobutenone and norbornene predominantly gave the decarbonylative coupling product 167 in good yield. However, under a CO atmosphere, the direct coupling occurred to give cyclohexenone 168 (Scheme 56). Interestingly, when 13CO was used, the corresponding 13C-labeled product (168 b) was formed, indicating a facile yet reversible decarbonylation/reinsertion process.
Scheme 56.
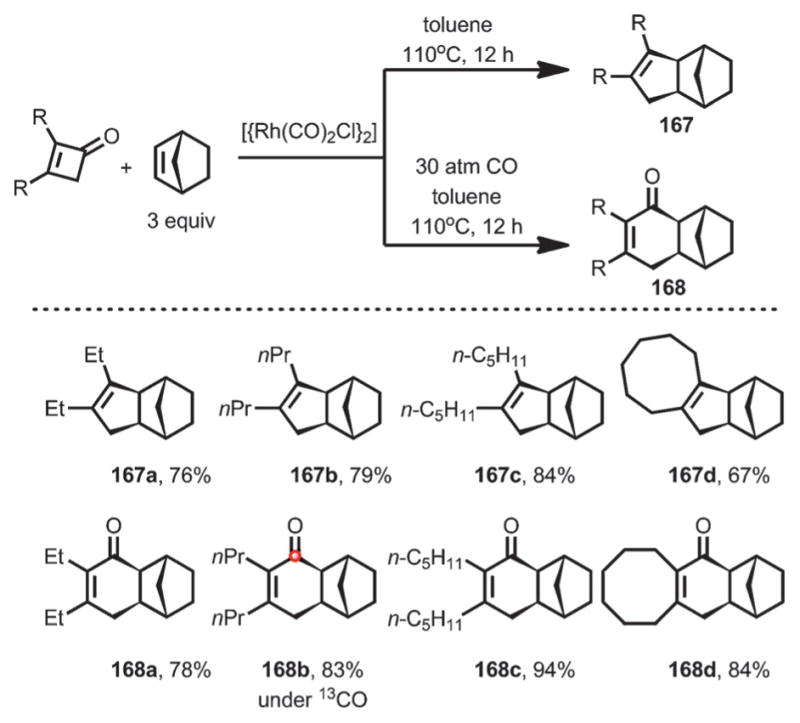
Direct/decarbonylative coupling between cyclobutenones and norbornene.
In 2007, Kondo and co-workers reported a new phenol synthesis from cyclobutenones and electron-deficient olefins 169 with the help of a rhodium catalyst (Scheme 57).[63] Tricyclohexylphosphine was employed as a ligand to increase the catalyst reactivity and suppress cyclobutenone dimerization. Although the precise mechanism remains unclear, it was proposed that, upon ring opening, the resulting η4-vinyl-ketene intermediate 170 underwent a Diels–Alder-type cyclization to afford cyclohexenone 171, which, upon dehydrogenation, led to the phenol products with high regioselectivity.
Scheme 57.
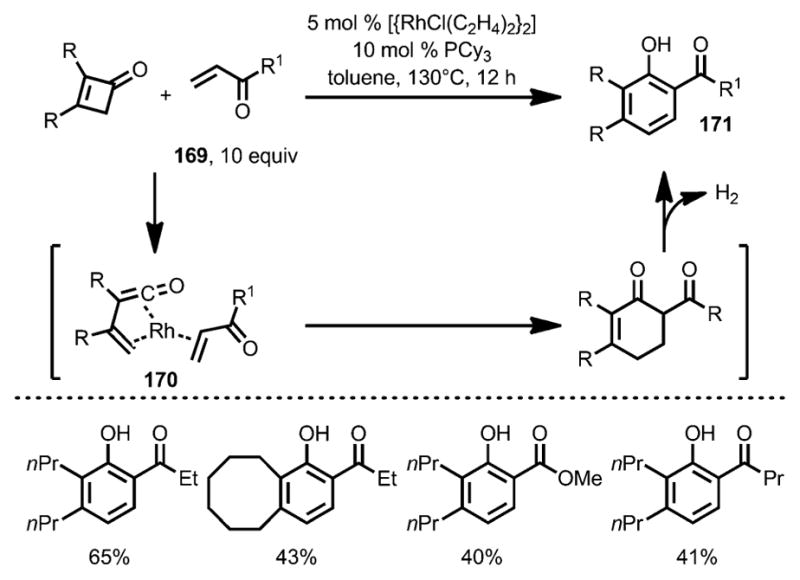
Phenol synthesis from cyclobutenones and electron-deficient alkenes.
In 2012, Xu and Dong developed a Rh-catalyzed intramolecular coupling between olefins and benzocyclobutenones, whereby insertion occurred selectively at the C1–C2 position (Scheme 58).[64] Upon treatment with [{Rh(cod)Cl}2] and 1,4-bis(diphenylphosphino)butane (dppb) as a bidentate phosphine ligand, benzocyclobutenone 172 tethered with an olefin moiety afforded the benzofused ring structure 173. Ligands with a larger bite angle were found to give higher yields. This method is compatible with a variety of mono- and 1,1-disubstituted alkenes. For more challenging substrates, such as those containing longer tethers, aryl substituents or trisubstituted alkenes, ZnCl2 was employed as a Lewis acid cocatalyst.
Scheme 58.
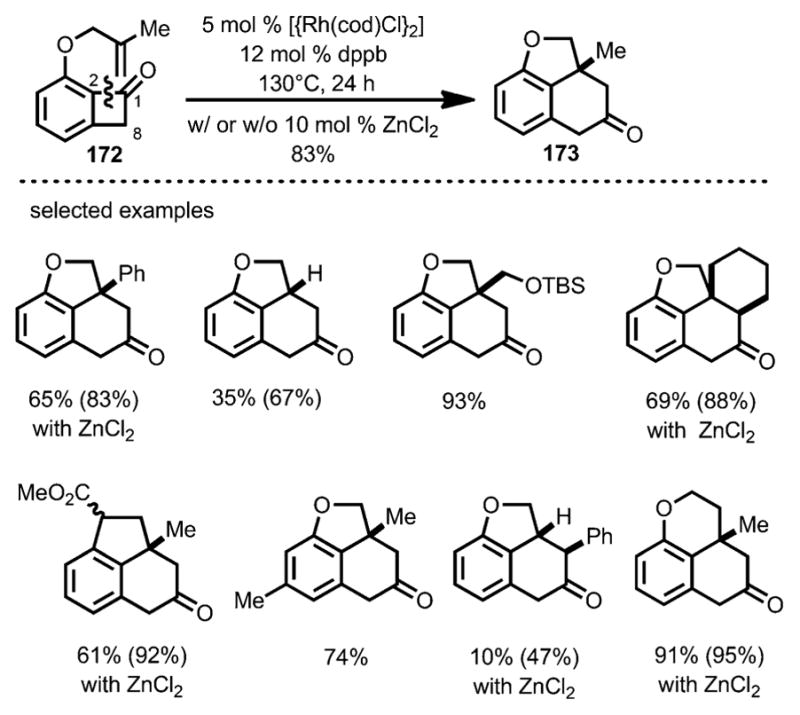
Rhodium-catalyzed carboacylation for the synthesis of benzofused ring systems. Values in parentheses refer to the yields based on recovered starting material.
The enantioselective version of this transformation was later reported by the same group (Scheme 59).[65] (R)-DTBM-SEG-PHOS and (R,R)-DIOP were found to be suitable chiral ligands to provide excellent enantioselectivity. The resulting products can be reduced to the corresponding saturated tricycles with four consecutive chiral centers (e.g., 174).
Scheme 59.

Enantioselective intramolecular carboacylation. THS=tetrabutylammonium hydrogen sulfate; TPAP=tetrapropylammonium perruthenate; NMO=N-methylmorpholine-N-oxide. Values in parentheses refer to the yields based on recovered starting material.
To better understand the mechanism and the ligand effect responsible for the observed high reactivity and enantioselectivity, Liu and co-workers undertook a detailed computational study of this rhodium-catalyzed intramolecular carboacylation reaction (Scheme 60).[66] According to the DFT calculations, instead of direct insertion into the C1–C2 bond of benzocyclobutenone (Scheme 60, path b), the cleavage is more likely to occur from cleavage of the C1–C8 bond (Scheme 60, path a), followed by a carbonyl migratory process with CO reinsertion being the rate-determining step (RDS). The increased reactivity of bidentate ligands with larger bite angles was attributed to release of the ligand–substrate repulsion in intermediate 175. The enantioselectivity was attributed to the steric repulsion between the equatorial aryl group of the chiral ligand and the C8-methylene of the benzocyclobutenone substrate.
Scheme 60.

Computationally derived proposal of the mechanism.
To enable insertion of more sterically hindered trisubstituted alkenes, such as 176, Xu and Dong developed a new catalyst system that employed electron-deficient [{Rh(CO)2Cl}2] and P(C6F5)3 (Scheme 61).[67] By using this method, the tetracyclic core of the proposed cycloinumakiol 177 was efficiently constructed. Ultimately, the syntheses of both diastereomers of the proposed structure were achieved, leading to the correct structural assignment of the natural product.
Scheme 61.
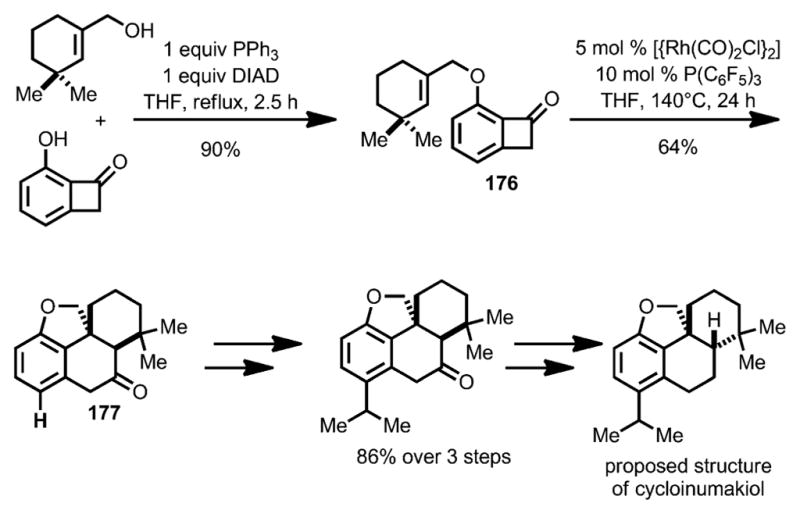
“Cut-and-sew”-strategy enabling total synthesis of proposed structure of cycloinumakiol. DIAD=diisopropyl azodicarboxylate.
In addition to forming fused rings, spirocycles can also be constructed from olefin-tethered benzocyclobutenones through properly choosing the catalyst system (Scheme 62).[68] It was anticipated that, after Rh insertion into the benzocyclobutenone C–C bond and subsequent migratory insertion into the olefin, whereas reductive elimination would lead to fused-ring products, β-H elimination and decarbonylation would afford the spirocycle 178.
Scheme 62.
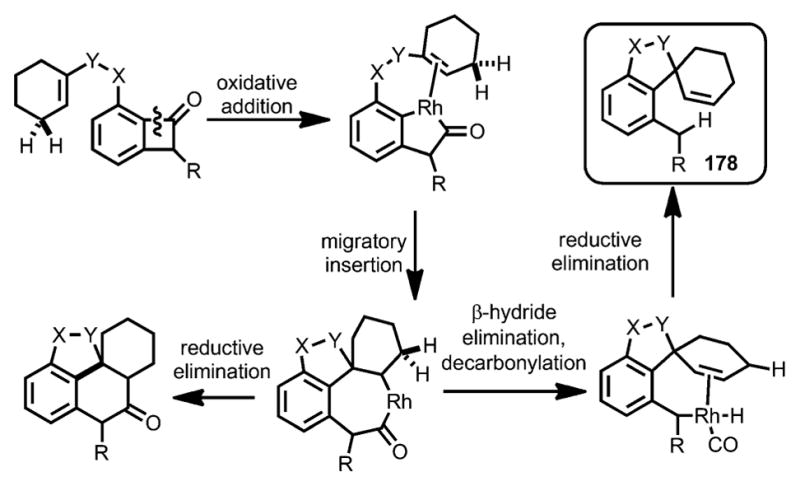
Rhodium-catalyzed decarbonylative spirocyclization.
When 10 mol % of P(C6F5)3 was used alongside 5 mol % [{Rh(CO)2Cl}2], a variety of cyclic trisubstituted olefins, as well as electron-deficient enone moieties, can react to give spirocycles 179 in decent yields, whereas in some cases the products were isolated as isomeric mixtures of alkenes (e.g., 179 b, d, g, and h; Scheme 63). Due to the near-neutral reaction conditions, this spirocyclization transformation tolerated functional groups including ketones, enamides, esters, dienes, benzyl and vinyl ethers, and even unprotected tertiary alcohols.
Scheme 63.

Substrate scope for decarbonylative spirocyclization.
The intramolecular coupling between alkynes and benzocyclobutenones was also reported by Dong and co-workers (Scheme 64).[69] The mechanism was proposed to be similar to the intramolecular alkene carboacylation.[64–66] The resulting enone moiety from the “cut and sew”” sequence underwent tautomerization to give β-naphthols as the final product. ZnCl2 was again found to be an effective Lewis acid cocatalyst to assist medium-sized ring formation (e.g., 180 d and 180 e).
Scheme 64.
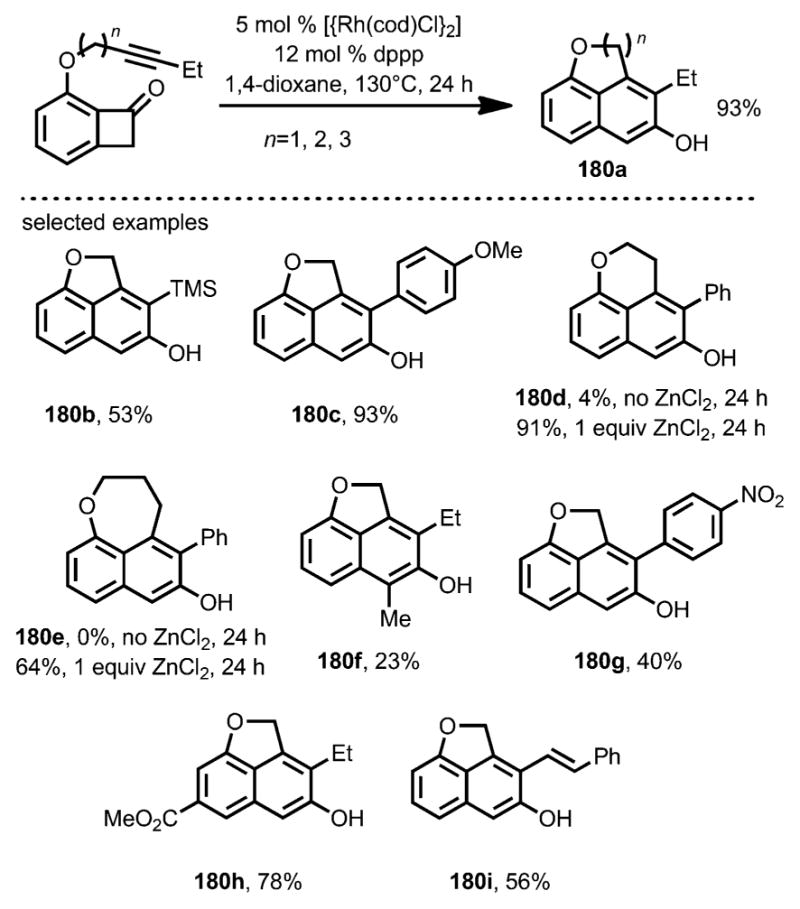
Catalytic direct intramolecular coupling of alkynyl benzocyclobutenone. Dppp=1,3-bis(diphenylphosphino)propane.
Interestingly, a decarbonylative coupling was realized by using DTBM-SEGPHOS as the ligand under argon flow, whereby tricyclic indenes (e.g., 181) were isolated as the products (Scheme 65). Thus, a divergent approach from alkyne-tethered benzocyclobutenones was realized (Scheme 66).
Scheme 65.
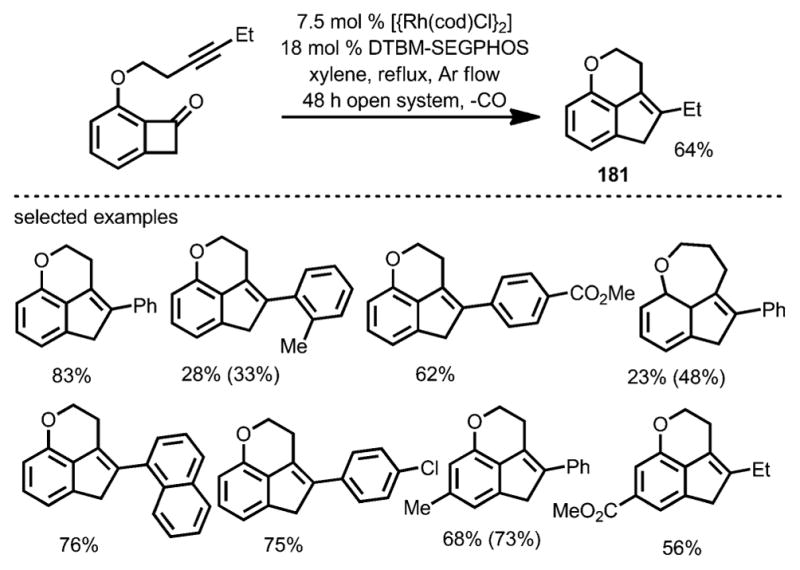
Catalytic decarbonylative intramolecular coupling of alkynyl benzocyclobutenone. Values in parentheses refer to the yields based on recovered starting material.
Scheme 66.
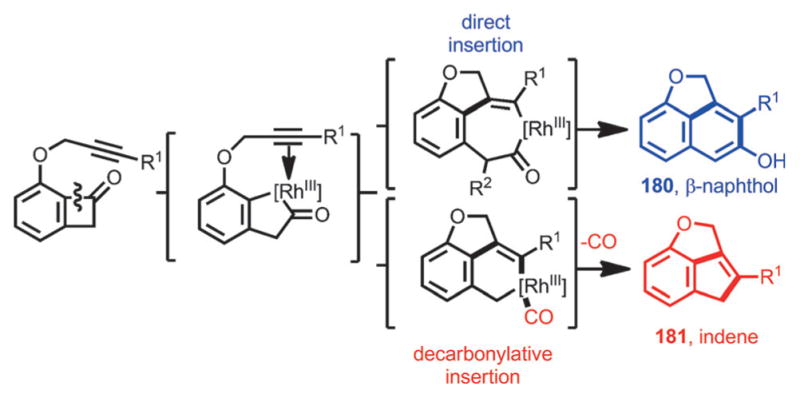
Rhodium-catalyzed divergent syntheses of fused β-naphthol and indene scaffolds.
Apart from coupling with nonpolar π bonds in substrates such as alkenes and alkynes, the Dong group later expanded the “cut and sew” transformation by utilizing more polar C=N bonds, for example, in oximes, as intramolecular coupling partners (Scheme 67).[70] The cationic [{Rh(cod)(CH3CN)2}]BF4 was found to be the optimal precatalyst. High enantioselectivity and decent yields were achieved by using a combination of two chiral ligands. This method provides an unusual way to prepare chiral fused lactams.
Scheme 67.

Enantioselective rhodium-catalyzed carboacylation of C=N bonds. SDP=7,7′-bis(diphenylphosphino)-2,2′,3,3′-tetrahydro-1,1′-spirobiindene.
In 2015, Martin and co-workers developed a nickel-catalyzed intermolecular coupling reaction between benzocyclobutenone and dienes or acetylenes (Scheme 68).[71] Upon treatment with [Ni(cod)2] as the precatalyst and tri(4-trifluoromethylphenyl)phosphine as the ligand, benzocyclobutenone 182 reacted smoothly with diene 183 to afford the [4+4] cyclization product 184 a in excellent yield, with a trace amount of formal [4+2] addition product 185. The coupling with unsymmetrical 1,3-dienes occurred in a regio- and diastereoselective fashion (e.g., 184 c and 184 i). For substrates having enolizable protons, an intramolecular transannular aldol reaction occurred under the reaction conditions, giving 186 as a single regioisomer and diastereoisomer. By using PPh3 as the ligand, the coupling between benzocyclobutenone and diphenylacetylene 187 occurred smoothly to give enone 188 in excellent yields. In all cases, no C1–C4 cleavage was observed.
Scheme 68.

Nickel-catalyzed intramolecular coupling with acetylenes and dienes.
In 2015, Dong and co-workers reported a rhodium-catalyzed decarbonylative coupling of 3-amino-4,4-disubstitutedcyclo-butenones 189 and tethered alkenes to access [3.1.0]bicyclic products 190 (Scheme 69).[72] Under thermal conditions, vinyl cyclobutanone product 191 was isolated, in the form of two conformational isomers, 191 a and 191 a′, presumably because the C3–N bond is incapable of free rotation. In contrast, when a more electron-deficient precatalyst [{Rh(CO)2Cl}2] was used, [3.1.0]bicyclic 190 was the predominant product. The transformation was proposed to be initiated by retro-4π electrocyclization of the cyclobutenone moiety through thermolysis. The resulting vinylketene intermediate 192 either undergoes [2+2] with the tethered alkene moiety to give the cyclobutanone product or a rhodium-catalyzed cyclometallation to give five-membered rhodacycle 193, which can undergo subsequent decarbonylative reductive elimination to form cyclopropane 190. When isolated cyclobutanones 191 a and 191 a′ were subjected to the [{Rh(CO)2Cl}2] conditions, no formation of cyclopropane 190 was observed, which excluded the possibility of forming cyclopropane 190 by decarbonylation of cyclobutanone 191. This transformation can tolerate different amide groups and tether linkages. Interestingly, when a 1,1-disubsituted olefin was used as the coupling partner, fused cyclobutanone was formed exclusively in high yield.
Scheme 69.

Rhodium-catalyzed formation of [3.1.0]bicyclic structures.
In 2015, by adding a cyclopropyl group to the benzocyclobutenone moieties, Yu and co-workers developed a rhodium-catalyzed [7+1] cycloaddition to form benzocyclooctenones (BCOs; Scheme 70).[73] Synthesized by 1,2-addition of cyclopropyl Grignard reagents to benzocyclobutenones, benzocyclobutene 194 underwent facile ring opening to form diene intermediate 195. Assisted by a rhodium catalyst, cyclometallation and then β-carbon elimination occurred to give intermediate 196, which, upon CO insertion, affored BCO 197 in good yield.
Scheme 70.

Rhodium-catalyzed [7+1] cycloaddtion. [Si]=trialkylsilyl.
Although most transition-metal-catalyzed cyclobutenone and benzocyclobutenone-mediated transformations involve coupling with unsaturated units, Dong and co-workers recently reported a reagent-free catalytic ring expansion of cyclobutenones and benzocyclobutenones bearing 3-alkyl substituents (Scheme 71).[74] As indicated in Liebeskind’s work,[53] the insertion of rhodium into the C1–C8 bond of benzocyclobutenones is kinetically favored. In the absence of coordination of unsaturated π moieties,[64–70] concomitant β-H elimination could occur after the oxidative addition to give the acylrhodium species 198, which, upon hydride re-insertion and reductive elimination, leads to ring-expanded five-membered ring products 199.
Scheme 71.
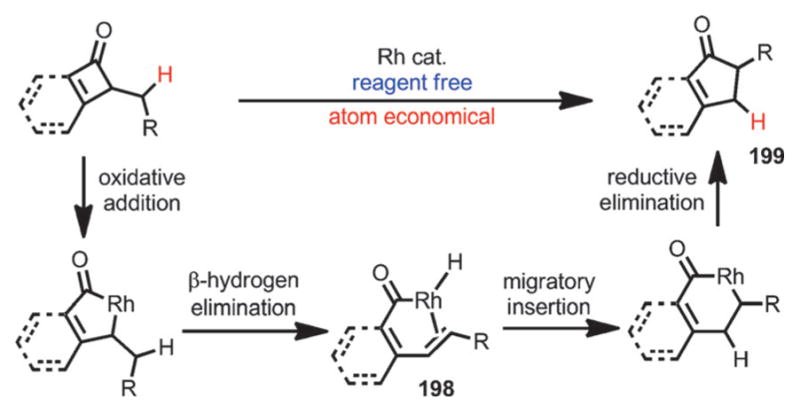
Rhodium-catalyzed ring expansion of cyclobutenones.
By properly choosing the Rh precatalyst and bidentate phosphine ligands, benzocyclobutenones and simple cyclobutenones both afforded the desired ring-expansion product, that is, 1-indanones and cyclopentenones, in good yields (Scheme 72). The involvement of a β-H elimination/migratory insertion/reductive elimination sequence was supported by deuterium labeling experiments and the isolation of ring-opening olefin products 200 and 201.
Scheme 72.

Substrate scope and mechanism study for the cyclobutenone ring expansion.
6. Diels–Alder Reactions
Besides having been widely investigated for ring-opening transformations, cyclobutenones also possess remarkable dienophilicity compared with cyclopentenones and cyclohexenones, which is promoted by the strain relief during the cycloaddition. Hence, cyclobutenones can efficiently participate in Diels–Alder reactions. The first Diels–Alder reaction that employed cyclobutenones as dienophiles was reported by Kelly and McNutt in 1975 (Scheme 73).[75] Treatment of 4,4-dimethylcyclobutenone 202 with 1,3-diphenylisobenzofuran 203 proceeded smoothly to afford adduct 204 in 62 % yield. BF3·OEt2 was used as a catalyst for the reaction between cyclopentadiene and 202, from which the exo adduct 205 was isolated as the major product.
Scheme 73.
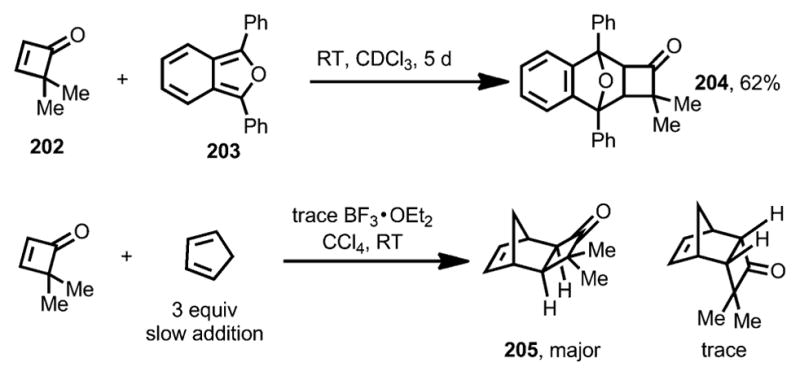
Diels–Alder cycloadditions of 4,4-dimethylcyclobutenone.
In 1991, Viehe, Sustmann, and co-workers reported the Diels–Alder reaction of 3-cyanocyclobutenone 206 (Scheme 74).[76] Cycloaddition with a variety of dienes proceeded with high efficiency at ambient temperature.
Scheme 74.

Diels–Alder cycloaddition involving 3-cyanocyclobutenone.
In 2010, Danishefsky and co-workers reported the first Diels–Alder cycloaddition that incorporated the unsubstituted parent structure of cyclobutenones 207 (Scheme 75).[77] A variety of dienes underwent cycloaddition rapidly and some reactions even occurred at −40 °C. When less reactive dienes, such as furan and 2,3-dimethylbutadiene, were used, ZnCl2 was added as a Lewis acid catalyst to promote the transformation.
Scheme 75.
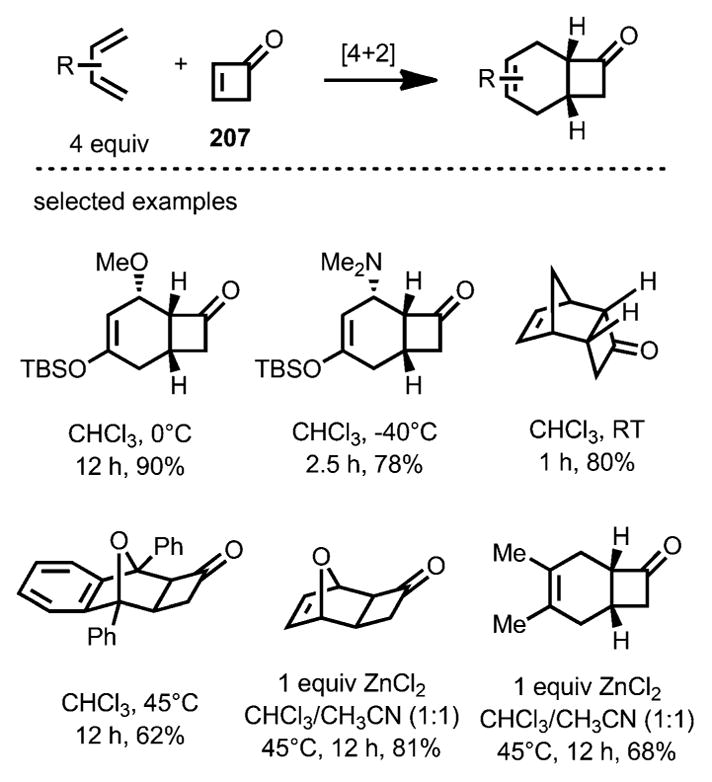
Diels–Alder cycloaddition incorporating parent cyclobutenone.
7. Conclusion and Outlook
The high yet predictable reactivities of cyclobutenones and benzocyclobutenones allow them to serve as versatile synthons, providing two, three or four carbon atoms in a variety of synthetic transformations. Their participation in such a large variety of reactions, including Diels–Alder reactions, [2+2] electrocyclizations, and cycloadditions with 1,3-dipolar species, has allowed their use in the synthesis of various structurally attractive and biologically important molecules. The recent development of transition-metal-catalyzed C–C bond activations of cyclobutenones and benzocyclobutenones has led to a renaissance of the field for new reactivity discovery. With the advent of more facile routes to cyclobutenones and benzocyclobutenones, it is expected that more synthetically useful applications of these intriguing molecules will be developed.
Acknowledgments
We thank NIGMS (R01GM109054) and the Welch Foundation (F 1781) for research grants. G.D. is a Searle Scholar and Sloan Fellow. Prof. Dr. M. C. Young is greatly acknowledged for editing and proof-reading the manuscript.
Biographies
 Peng-hao Chen received his BSc. degree in Chemistry from the University of Science and Technology of China (P. R. China) in 2012 under the supervision of Professor Liu-zhu Gong. He is currently a PhD candidate in organic chemistry under the supervision of Professor Guangbin Dong at the University of Texas at Austin (USA). His research is focused on transition-metal-catalyzed C–C bond activation.
Peng-hao Chen received his BSc. degree in Chemistry from the University of Science and Technology of China (P. R. China) in 2012 under the supervision of Professor Liu-zhu Gong. He is currently a PhD candidate in organic chemistry under the supervision of Professor Guangbin Dong at the University of Texas at Austin (USA). His research is focused on transition-metal-catalyzed C–C bond activation.
 Guangbin Dong received his BSc. degree from Peking University (P. R. China) and completed his PhD in chemistry from Stanford University (USA) with Professor Barry M. Trost, where he was a Larry Yung Stanford Graduate Fellow. In 2009, he began research with Prof. Robert H. Grubbs at California Institute of Technology (USA) as a Camille and Henry Dreyfus Environmental Chemistry Fellow. In 2011, he joined the Department of Chemistry and Biochemistry at the University of Texas at Austin (USA) as an Assistant Professor and a CPRIT Scholar. Currenty, he is a Professor of Chemistry at the University of Chicago. His research interests entail the development of powerful chemical tools for addressing questions of biological importance.
Guangbin Dong received his BSc. degree from Peking University (P. R. China) and completed his PhD in chemistry from Stanford University (USA) with Professor Barry M. Trost, where he was a Larry Yung Stanford Graduate Fellow. In 2009, he began research with Prof. Robert H. Grubbs at California Institute of Technology (USA) as a Camille and Henry Dreyfus Environmental Chemistry Fellow. In 2011, he joined the Department of Chemistry and Biochemistry at the University of Texas at Austin (USA) as an Assistant Professor and a CPRIT Scholar. Currenty, he is a Professor of Chemistry at the University of Chicago. His research interests entail the development of powerful chemical tools for addressing questions of biological importance.
References
- 1.For reviews on cyclobutanes, see: Namyslo JC, Kaufmann DE. Chem Rev. 2003;103:1485–1537. doi: 10.1021/cr010010y.Sadana AK, Saini RK, Billups WE. Chem Rev. 2003;103:1539–1602. doi: 10.1021/cr010022j.Nemoto H. Chem Pharm Bull. 2007;55:961–974. doi: 10.1248/cpb.55.961.Seiser T, Saget T, Tran DN, Cramer N. Angew Chem Int Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053.Angew Chem. 2011;123:7884–7896.
- 2.For reviews on cyclobutenones and benzocyclobutenones, see: Belluš D, Ernst B. Angew Chem Int Ed Engl. 1988;27:797–827.Angew Chem. 1988;100:820–850.Martin R, Flores-Gaspar A. Synthesis. 2013:563–580.Kondo T, Mitsudo T-A. Chem Lett. 2005;34:1462–1467.. The term “benzocyclobutanone” was occasionally used. However, due to the unsaturated nature of the benzene moeity and the similar reactivity as cyclobutenones, the term “benzocyclobutenone” is considered a more accurate description.
- 3.a) Aidhen IS, Ahuja JR. Tetrahedron Lett. 1992;33:5431–5432. [Google Scholar]; b) Zhao YL, Yang SC, Di CH, Han XD, Liu Q. Chem Commun. 2010;46:7614–7616. doi: 10.1039/c0cc02470h. [DOI] [PubMed] [Google Scholar]; c) Álvarez-Bercedo P, Flores-Gaspar A, Correa A, Martin R. J Am Chem Soc. 2010;132:466–467. doi: 10.1021/ja909811t. [DOI] [PubMed] [Google Scholar]; d) Martin R, Flores-Gaspar A. Org Synth. 2012;89:159–169. [Google Scholar]; e) Flores-Gaspar A, Gutiérrez-Bonet Á, Martin R. Org Lett. 2012;14:5234–5237. doi: 10.1021/ol3023819. [DOI] [PubMed] [Google Scholar]; f) Mehta G, Kotha S. Tetrahedron. 2001;57:625–659. [Google Scholar]; h) Liebman JF, Greenberg A. Chem Rev. 1976;76:311. [Google Scholar]
- 4.a) Souillart L, Cramer N. Chem Rev. 2015;115:9410–9464. doi: 10.1021/acs.chemrev.5b00138. [DOI] [PubMed] [Google Scholar]; b) Dong G, editor. Topics in Current Chemistry, Vol. 346: C–C Bond Activation. Springer; Heidelberg: 2014. [DOI] [PubMed] [Google Scholar]; c) Chen F, Wang T, Jiao N. Chem Rev. 2014;114:8613–8661. doi: 10.1021/cr400628s. [DOI] [PubMed] [Google Scholar]; d) Jones WD. Nature. 1993;364:676–677. [Google Scholar]; e) Murakami M, Ito Y. In: Activation of Unreactive Bonds and Organic Synthesis. Murai S, Alper H, Gossage RA, Grushin VV, Hidai M, Ito Y, Jones WD, Kakiuchi F, van Koten G, Lin YS, Mizobe Y, Murai S, Murakami M, Richmond TG, Sen A, Suginome M, Yamamoto A, editors. Vol. 3. Springer; Heidelberg: 1999. pp. 97–129. [Google Scholar]; f) Rybtchinski B, Milstein D. Angew Chem Int Ed. 1999;38:870–883. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; Angew Chem. 1999;111:918–932. [Google Scholar]; g) Perthuisot C, Edelbach BL, Zubris DL, Simhai N, Iverson CN, Müller C, Satoh T, Jones WD. J Mol Catal A. 2002;189:157–168. [Google Scholar]; h) van der Boom ME, Milstein D. Chem Rev. 2003;103:1759–1792. doi: 10.1021/cr960118r. [DOI] [PubMed] [Google Scholar]; i) Miura M, Satoh T. In: Palladium in Organic Synthesis. Tsuji J, editor. Vol. 14. Springer; Heidelberg: 2005. pp. 1–20. [Google Scholar]; j) Jun C-H, Park J-W. In: Directed Metallation. Chatani N, editor. Vol. 24. Springer; Heidelberg: 2007. pp. 117–143. [Google Scholar]; k) Cramer N, Seiser T. Synlett. 2011:449–460. [Google Scholar]; l) Murakami M, Matsuda T. Chem Commun. 2011;47:1100–1105. doi: 10.1039/c0cc02566f. [DOI] [PubMed] [Google Scholar]; m) Ruhland K. Eur J Org Chem. 2012:2683–2706. [Google Scholar]; n) Dermenci A, Coe JW, Dong G. Org Chem Front. 2014;1:567–581. doi: 10.1039/c4qo00053f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Moore HW, Decker OHW. Chem Rev. 1986;86:821–830. [Google Scholar]; b) Dolbier WR, Jr, Koroniak H, Houk KN, Sheu C. Acc Chem Res. 1996;29:471–477. [Google Scholar]
- 6.Niwayama S, Kallel EA, Sheu CM, Houk KN. J Org Chem. 1996;61:2517–2522. doi: 10.1021/jo950884i. [DOI] [PubMed] [Google Scholar]
- 7.Jenny EF, Roberts JD. J Am Chem Soc. 1956;78:2005–2009. [Google Scholar]
- 8.Mayr H. Angew Chem Int Ed Engl. 1975;14:500–501. [Google Scholar]; Angew Chem. 1975;87:491–492. [Google Scholar]
- 9.Huisgen R, Mayr H. J Chem Soc Chem Commun. 1976:55–56. [Google Scholar]
- 10.Ficini J, Falou S, Dangelo J. Tetrahedron Lett. 1977;18:1931–1934. [Google Scholar]
- 11.For a review on ynamines, see: Ficini J. Tetrahedron. 1976;32:1449–1486.
- 12.Danheiser RL, Gee SK, Sard H. J Am Chem Soc. 1982;104:7670–7672. [Google Scholar]
- 13.Danheiser RL, Gee SK. J Org Chem. 1984;49:1672–1674. [Google Scholar]
- 14.Danheiser RL, Gee SK, Perez JJ. J Am Chem Soc. 1986;108:806–810. [Google Scholar]
- 15.Kowalski CJ, Lal GS. J Am Chem Soc. 1988;110:3693–3695. [Google Scholar]
- 16.Danheiser RL, Nishida A, Savariar S, Trova MP. Tetrahedron Lett. 1988;29:4917–4920. [Google Scholar]
- 17.Delaunois M, Ghosez L. Angew Chem Int Ed Engl. 1969;8:72–73. [Google Scholar]; Angew Chem. 1969;81:33–34. [Google Scholar]
- 18.Mak XY, Crombie AL, Danheiser RL. J Org Chem. 2011;76:1852–1873. doi: 10.1021/jo2000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson JO, Nguyen NV, Foland LD, Moore HW. J Am Chem Soc. 1985;107:3392–3393. [Google Scholar]
- 20.Perri ST, Foland LD, Decker OHW, Moore HW. J Org Chem. 1986;51:3067–3068. [Google Scholar]
- 21.Moore HW, Perri ST. J Org Chem. 1988;53:996–1003. [Google Scholar]
- 22.Krysan DJ, Gurski A, Liebeskind LS. J Am Chem Soc. 1992;114:1412–1418. [Google Scholar]
- 23.a) Ezcurra JE, Karabelas K, Moore HW. Tetrahedron. 2005;61:275–286. [Google Scholar]; b) Tiedemann R, Turnbull P, Moore HW. J Org Chem. 1999;64:4030–4041. [Google Scholar]; c) Onofrey TJ, Gomez D, Winters M, Moore HW. J Org Chem. 1997;62:5658–5659. [Google Scholar]; d) Heerding JM, Moore HW. J Org Chem. 1991;56:4048–4050. [Google Scholar]; e) Enhsen A, Karabelas K, Heerding JM, Moore HW. J Org Chem. 1990;55:1177–1185. [Google Scholar]; f) Foland LD, Decker OHW, Moore HW. J Am Chem Soc. 1989;111:989–995. [Google Scholar]; g) Xu SL, Taing M, Moore HW. J Org Chem. 1991;56:6104–6109. [Google Scholar]; h) Xia H, Moore HW. J Org Chem. 1992;57:3765–3766. [Google Scholar]; i) Xu SL, Moore HW. J Org Chem. 1992;57:326–338. [Google Scholar]
- 24.a) Liu F, Liebeskind LS. J Org Chem. 1998;63:2835–2844. [Google Scholar]; b) Sun L, Liebeskind LS. J Org Chem. 1995;60:8194–8203. [Google Scholar]; c) Liebeskind LS, Granberg KL, Zhang J. J Org Chem. 1992;57:4345–4352. [Google Scholar]; d) Edwards JP, Krysan DJ, Liebeskind LS. J Am Chem Soc. 1993;115:9868–9869. [Google Scholar]; e) Gurski A, Liebeskind LS. J Am Chem Soc. 1993;115:6101–6108. [Google Scholar]; f) Kwan HL, Moore HW. Tetrahedron Lett. 1993;34:235–238. [Google Scholar]; g) Liebeskind LS, Wang J. J Org Chem. 1993;58:3550–3556. [Google Scholar]; h) Liebeskind LS, Bombrun A. J Org Chem. 1994;59:1149–1159. [Google Scholar]
- 25.a) Harrowven DC, Pascoe DD, Demurtas D, Bourne HO. Angew Chem Int Ed. 2005;44:1221–1222. doi: 10.1002/anie.200462268. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;117:1247–1248. [Google Scholar]; b) Peña-Cabrera E, Liebeskind LS. J Org Chem. 2002;67:1689–1691. doi: 10.1021/jo016034m. [DOI] [PubMed] [Google Scholar]; c) Perri ST, Dyke HJ, Moore HW. J Org Chem. 1989;54:2032–2034. [Google Scholar]
- 26.Harrowven DC, Pascoe DD, Guy IL. Angew Chem Int Ed. 2007;46:425–428. doi: 10.1002/anie.200603538. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2007;119:429–432. [Google Scholar]
- 27.a) Xu SL, Moore HW. J Org Chem. 1989;54:6018–6021. [Google Scholar]; b) Xu SL, Xia H, Moore HW. J Org Chem. 1991;56:6094–6103. [Google Scholar]
- 28.Hachiya I, Yoshitomi T, Yamaguchi Y, Shimizu M. Org Lett. 2009;11:3266–3268. doi: 10.1021/ol901192y. [DOI] [PubMed] [Google Scholar]
- 29.Hachiya I, Ito A, Shimizu M. Asian J Org Chem. 2014;3:614–618. [Google Scholar]
- 30.Baldwin JE, Mcdaniel C. J Am Chem Soc. 1967;89:1537–1537. [Google Scholar]
- 31.Baldwin JE, Mcdaniel MC. J Am Chem Soc. 1968;90:6118–6124. [Google Scholar]
- 32.Kikuchi O. Bull Chem Soc Jpn. 1982;55:1669. [Google Scholar]
- 33.Chapman OL, Lassila JD. J Am Chem Soc. 1968;90:2449–2450. [Google Scholar]
- 34.Toda F, Todo E. Chem Lett. 1974:1279–1280. [Google Scholar]
- 35.Toda F, Todo E. Bull Chem Soc Jpn. 1976;49:2503–2505. [Google Scholar]
- 36.Toda F, Todo Y, Todo E. Bull Chem Soc Jpn. 1976;49:2645–2646. [Google Scholar]
- 37.Schiess P, Eberle M, Huysfrancotte M, Wirz J. Tetrahedron Lett. 1984;25:2201–2204. [Google Scholar]
- 38.a) Roberts JD, Kline GB, Simmons HE. J Am Chem Soc. 1953;75:4765–4768. [Google Scholar]; b) Silversmith EF, Roberts JD. J Am Chem Soc. 1956;78:4023–4024. [Google Scholar]; c) Silversmith EF, Kitahara Y, Caserio MC, Roberts JD. J Am Chem Soc. 1958;80:5840–5845. [Google Scholar]
- 39.Hassner A, Dillon JL. J Org Chem. 1983;48:3382–3386. [Google Scholar]
- 40.Murakami M, Miyamoto Y, Ito Y. Angew Chem Int Ed. 2001;40:189–190. [PubMed] [Google Scholar]; Angew Chem. 2001;113:182–184. [Google Scholar]
- 41.Murakami M, Miyamoto Y, Ito Y. J Am Chem Soc. 2001;123:6441–6442. doi: 10.1021/ja010639i. [DOI] [PubMed] [Google Scholar]
- 42.Magomedov NA, Ruggiero PL, Tang Y. J Am Chem Soc. 2004;126:1624–1625. doi: 10.1021/ja0399066. [DOI] [PubMed] [Google Scholar]
- 43.Cammers-Goodwin A. J Org Chem. 1993;58:7619–7621. [Google Scholar]
- 44.Li Y, Su X, Zhou W, Li W, Zhang J. Chem Eur J. 2015;21:4224–4228. doi: 10.1002/chem.201406475. [DOI] [PubMed] [Google Scholar]
- 45.Li BS, Wang Y, Jin Z, Zheng P, Ganguly R, Chi YR. Nat Commun. 2015;6:6207. doi: 10.1038/ncomms7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B-S, Wang Y, Jin Z, Chi YR. Chem Sci. 2015;6:6008–6012. doi: 10.1039/c5sc01972a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.a) Cava MP, Muth K. J Am Chem Soc. 1960;82:652–654. [Google Scholar]; b) Bradley JC, Durst T. Can J Chem. 1995;73:1660–1665. [Google Scholar]; c) Gokhale A, Schiess P. Helv Chim Acta. 1998;81:251–267. [Google Scholar]
- 48.Matsumoto T, Hamura T, Kuriyama Y, Suzuki K. Tetrahedron Lett. 1997;38:8985–8988. [Google Scholar]
- 49.García-García P, Novillo C, Fernández-Rodríguez MA, Aguilar E. Chem Eur J. 2011;17:564–571. doi: 10.1002/chem.201002092. [DOI] [PubMed] [Google Scholar]
- 50.Matsuya Y, Ohsawa N, Nemoto H. J Am Chem Soc. 2006;128:13072–13073. doi: 10.1021/ja065277z. [DOI] [PubMed] [Google Scholar]
- 51.Matsuya Y, Katayanagi H, Ohdaira T, Wei ZL, Kondo T, Nemoto H. Org Lett. 2009;11:1361–1364. doi: 10.1021/ol900154x. [DOI] [PubMed] [Google Scholar]
- 52.Sugimoto K, Hayashi R, Nemoto H, Toyooka N, Matsuya Y. Org Lett. 2012;14:3510–3513. doi: 10.1021/ol301474g. [DOI] [PubMed] [Google Scholar]
- 53.Huffman MA, Liebeskind LS, Pennington WT. Organometallics. 1990;9:2194–2196. [Google Scholar]
- 54.Huffman MA, Liebeskind LS, Pennington WT. Organometallics. 1992;11:255–266. [Google Scholar]
- 55.Masuda Y, Hasegawa M, Yamashita M, Nozaki K, Ishida N, Murakami M. J Am Chem Soc. 2013;135:7142–7145. doi: 10.1021/ja403461f. [DOI] [PubMed] [Google Scholar]
- 56.Huffman MA, Liebeskind LS. J Am Chem Soc. 1990;112:8617–8618. [Google Scholar]
- 57.Huffman MA, Liebeskind LS. J Am Chem Soc. 1991;113:2771–2772. [Google Scholar]
- 58.Auvinet AL, Harrity JP. Angew Chem Int Ed. 2011;50:2769–2772. doi: 10.1002/anie.201007598. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:2821–2824. [Google Scholar]
- 59.Stalling T, Harker WR, Auvinet AL, Cornel EJ, Harrity JP. Chem Eur J. 2015;21:2701–2704. doi: 10.1002/chem.201405863. [DOI] [PubMed] [Google Scholar]
- 60.Murakami M, Ashida S, Matsuda T. J Am Chem Soc. 2005;127:6932–6933. doi: 10.1021/ja050674f. [DOI] [PubMed] [Google Scholar]
- 61.Huffman MA, Liebeskind LS. J Am Chem Soc. 1993;115:4895–4896. [Google Scholar]
- 62.Kondo T, Taguchi Y, Kaneko Y, Niimi M, Mitsudo TA. Angew Chem Int Ed. 2004;43:5369–5372. doi: 10.1002/anie.200461002. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2004;116:5483–5486. [Google Scholar]
- 63.Kondo T, Niimi M, Nomura M, Wada K, Mitsudo T-a. Tetrahedron Lett. 2007;48:2837–2839. [Google Scholar]
- 64.Xu T, Dong G. Angew Chem Int Ed. 2012;51:7567–7571. doi: 10.1002/anie.201202771. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:7685–7689. [Google Scholar]
- 65.Xu T, Ko HM, Savage NA, Dong G. J Am Chem Soc. 2012;134:20005–20008. doi: 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]
- 66.Lu G, Fang C, Xu T, Dong G, Liu P. J Am Chem Soc. 2015;137:8274–8283. doi: 10.1021/jacs.5b04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu T, Dong G. Angew Chem Int Ed. 2014;53:10733–10736. doi: 10.1002/anie.201404802. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:10909–10912. [Google Scholar]
- 68.Xu T, Savage NA, Dong G. Angew Chem Int Ed. 2014;53:1891–1895. doi: 10.1002/anie.201310149. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014;126:1922–1926. [Google Scholar]
- 69.Chen PH, Xu T, Dong G. Angew Chem Int Ed. 2014;53:1674–1678. doi: 10.1002/anie.201310100. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:1700–1704. [Google Scholar]
- 70.Deng L, Xu T, Li H, Dong G. J Am Chem Soc. 2016;138:369–374. doi: 10.1021/jacs.5b11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juliá-Hernández F, Ziadi A, Nishimura A, Martin R. Angew Chem Int Ed. 2015;54:9537–9541. doi: 10.1002/anie.201503461. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:9673–9677. [Google Scholar]
- 72.Zhou X, Zafar I, Dong G. Tetrahedron. 2015;71:4478–4483. doi: 10.1016/j.tet.2015.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.a) Fu XF, Xiang Y, Yu ZX. Chem Eur J. 2015;21:4242–4246. doi: 10.1002/chem.201405712. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Yu ZX. Acc Chem Res. 2015;48:2288–2296. doi: 10.1021/acs.accounts.5b00037. [DOI] [PubMed] [Google Scholar]
- 74.Chen P-H, Sieber J, Senanayake CH, Dong G. Chem Sci. 2015;6:5440–5445. doi: 10.1039/c5sc01875g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelly TR, Mcnutt RW. Tetrahedron Lett. 1975;16:285–288. [Google Scholar]
- 76.Bienfait B, Coppemotte G, Merenyi R, Viehe HG, Sicking W, Sustmann R. Tetrahedron. 1991;47:8167–8176. [Google Scholar]
- 77.a) Li X, Danishefsky SJ. J Am Chem Soc. 2010;132:11004–11005. doi: 10.1021/ja1056888. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pham HV, Paton RS, Ross AG, Danishefsky SJ, Houk KN. J Am Chem Soc. 2014;136:2397–2403. doi: 10.1021/ja410220w. [DOI] [PMC free article] [PubMed] [Google Scholar]