

Abstract
Heme oxygenases are composed of two isozymes, Hmox1 and Hmox2, that catalyze the degradation of heme to carbon monoxide (CO), ferrous iron, and biliverdin, the latter of which is subsequently converted to bilirubin. While initially considered to be waste products, CO and biliverdin/bilirubin have been shown over the last 20 years to modulate key cellular processes, such as inflammation, cell proliferation, and apoptosis, as well as antioxidant defense. This shift in paradigm has led to the importance of heme oxygenases and their products in cell physiology now being well accepted. The identification of the two human cases thus far of heme oxygenase deficiency and the generation of mice deficient in Hmox1 or Hmox2 have reiterated a role for these enzymes in both normal cell function and disease pathogenesis, especially in the context of cardiovascular disease. This review covers the current knowledge on the function of both Hmox1 and Hmox2 at both a cellular and tissue level in the cardiovascular system. Initially, the roles of heme oxygenases in vascular health and the regulation of processes central to vascular diseases are outlined, followed by an evaluation of the role(s) of Hmox1 and Hmox2 in various diseases such as atherosclerosis, intimal hyperplasia, myocardial infarction, and angiogenesis. Finally, the therapeutic potential of heme oxygenases and their products are examined in a cardiovascular disease context, with a focus on how the knowledge we have gained on these enzymes may be capitalized in future clinical studies.
I. PERSPECTIVE
Since the generation of mice deficient in heme oxygenase-1 (Hmox1) and heme oxygenase-2 (Hmox2) nearly 20 years ago (426, 630), interest in the roles of these enzymes in normal physiology and disease pathology has bourgeoned. In particular, heme oxygenases have been implicated in vascular biology for close to two decades. Both isoforms of Hmox have been studied in the context of vascular tone, and much interest has focused on the role of Hmox1 in disease. In particular, the contribution of Hmox1 and the products formed during its enzymatic activity has been studied extensively in the context of vascular diseases, including atherosclerosis, ischemia/reperfusion (I/R) injury, and intimal hyperplasia. This review presents the current understanding of the roles of Hmox1 and Hmox2 in the vascular system, from their roles in normal physiology to the effect of Hmox1 expression in disease settings to the potential of modulating Hmox1 activity and/or its products, carbon monoxide and biliverdin/bilirubin, as novel therapies to treat vascular diseases. This review aims to highlight the complexity of Hmox-dependent vascular regulation, in particular the key and often-paradoxical role(s) that heme oxygenases play in the modulation of the vascular system.
II. INTRODUCTION
A. Heme Oxygenases
Heme oxygenases catalyze the regiospecific degradation of heme b (iron protoporphyrin IX) to carbon monoxide (CO), ferrous iron, and biliverdin IXα (397). Biliverdin IXα is then converted to bilirubin IXα by an NADPH-dependent biliverdin reductase (BVR) (537) (Figure 1). Heme oxygenases are evolutionarily highly conserved enzymes, and they have been identified in unicellular organisms including several bacterial (487, 646, 647) and yeast species (237, 418, 428). By comparison, BVR is less conserved. Heme oxygenase-mediated formation of biliverdin IXα consumes three molecules of oxygen per mole heme oxidized and seven electrons originating from NADPH and being supplied by cytochrome P-450 reductase (535). Notably, heme oxygenases use heme as both the prosthetic group and substrate of the enzyme (620).
FIGURE 1.
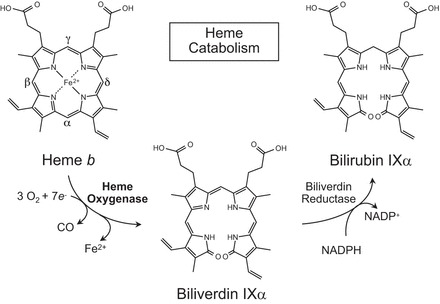
Schematic of the heme oxygenase reaction pathway. Heme oxygenases degrade heme b to sequentially yield carbon monoxide (CO), ferrous iron (Fe2+), and biliverdin IXα with the reaction requiring 3 mol of molecular oxygen and 7 electrons. In mammals, bilirubin IXα is subsequently reduced to bilirubin IXα by an NADPH-dependent biliverdin reductase.
In the first step of the reaction mechanism, ferric iron in the heme-heme oxygenase complex is reduced in an NADPH-dependent reaction (406, 623). Molecular O2 is bound to the complex as an oxyferrous intermediate that accepts a second electron from NADPH to form a ferric hydroperoxide intermediate (621, 624, 625). This intermediate hydroxylates the α-methine bridge carbon of the heme ring, forming hydroxy-heme (442, 592). The α-methine bridge carbon then becomes eliminated as CO resulting in the sequential formation of verdoheme and ferribiliverdin-IXα complex (BV-Fe III) (236, 457, 621, 625). Finally, ferribiliverdin-IXα is reduced resulting in the release of ferrous iron (Fe2+) and biliverdin-IXα (620, 623). Heme oxygenases exhibit a clear preference for heme b; however, a modest activity towards heme c and hematoheme is observed (268, 337, 626). Recently, a blue pigment named CV-bilin was identified in insects as a high-molecular-mass derivative of biliverdin-IXα that was likely formed from heme a (229), although evidence for mammalian heme oxygenase acting on heme a is currently not available.
In mammalian systems, two distinct enzymes make up the Hmox family: Hmox1 encoded by the gene HMOX1 and Hmox2 encoded by the gene HMOX2. In humans and rats, Hmox1 and Hmox2 are paralogs, sharing ∼43% sequence similarity in their amino acid sequences (98). Both proteins are characterized by a 24-amino acid sequence known as the “heme binding pocket” that allows for heme binding, with a conserved histidine imidazole residue (His-25 in Hmox1 and His-45 in Hmox2) acting as the heme iron ligand (198, 352). In addition to this, both proteins contain a hydrophobic region at the COOH terminus that acts as a membrane anchor (200, 351) and locates the proteins to the endoplasmic reticulum. Hmox2 contains heme regulatory domains that are absent from Hmox1 (350). These domains contain Cys residues that provide redox regulated binding sites for heme in addition to the proximal histidine residue required for enzymatic activity (138, 618). While both Hmox proteins use heme as substrate and cofactor, they differ in their physiological properties (Table 1). Biochemically, Hmox1 and Hmox2 have also been reported to have different Km values (0.24 ad 0.67 μM, respectively).
Table 1.
Functions of Hmox1 and Hmox2 in mammalian systems
| Hmox1 (Inducible) | Hmox2 (Constitutive) |
|---|---|
| Iron homeostasis | Oxygen sensing |
| Antioxidant defense | Antioxidant defense during changes in oxygen concentration |
| Regulation of inflammatory responses | Regulation of inflammatory responses |
| Wound healing | Wound healing |
| Regulation of cell proliferation | Regulation of cell proliferation |
| Angiogenesis | Neovascularization |
| Mitochondrial function | Neuroprotection |
| Erythropoiesis | Redox sensing |
| Regulation of innate and adaptive immunity |
1. Heme oxygenase-1
The expression of Hmox1 is induced in response to a variety of endogenous and exogenous stimuli including heat, heme, ultraviolet (UV) irradiation, lipopolysaccharide (LPS), growth factors, oxidative stress, hypoxia, cobalt protoporphyrin-IX (CoPP), iron starvation, and others (reviewed in Refs. 121, 450). There are differences in the molecular regulation of the human HMOX1 gene compared with the rodent HMOX1 genes (reviewed in Ref. 485). Hmox1 induction can be mediated by several transcription factors such as Nrf2, AP-1, Yin Yang 1 (YY1), and others (Figure 2). Conversely, Bach-1 (474) and JunD (183) repress Hmox1 expression, while metalloporphyrins such as tin protoporphyrin-IX (SnPP), zinc protoporphyrin-IX or manganese protoporphyrin-IX, tin, and chromium mesoporphyrins can act as competitive inhibitors of Hmox activity in vitro and in vivo. These inhibitors are used extensively in experimental studies, although they are not specific and light-sensitive compounds, so that care must be taken when using such compounds and interpreting data derived from their use.
FIGURE 2.
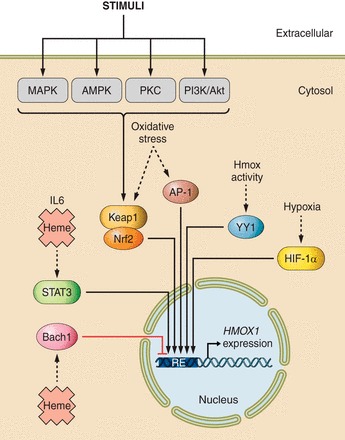
Signaling pathways and transcription factors modulating HMOX1 expression via different response elements (RE). Various extra- and intracellular stimuli activate the mitogen-activated protein kinases (MAPK), protein kinase C (PKC), 5'-AMP-activated protein kinase (AMPK), and phosphoinositide 3-kinase/RAC-alpha serine/threonine-protein kinase (PI3K/Akt) signaling pathways, leading to Hmox1 activation through the transcription factor Nrf2. Oxidative stress, including reactive nitrogen species (168, 373), also induce HMOX1 expression at least in part via Nrf2. Other transcription factors, including activator protein-1 (AP-1), signal transducer and activator of transcription 3 (STAT3), Yin Yang 1 (YY1), and hypoxia inducible factor-1α (HIF-1α) induce HMOX1 expression through various stimuli such as oxidative stress, hypoxia, heme, and interkeukin-6 (IL-6). Bach1 is a repressor of HMOX1 expression.
There is emerging evidence that Hmox1 gene expression may be regulated by microRNAs (miRNA) in multiple ways (reviewed in Ref. 328). Initial work identified two miRNAs to directly regulate HMOX1 expression via interaction with the HMOX1-3' untranslated region (29). More recent studies confirmed that Hmox1 expression can be modulated by miRNA in podocytes (609), osteoclasts (230), monkey kidney cells (604), adipose-derived mesenchymal stem cells (67), T-cells (639), and renal-cell carcinoma cells (150). There is also evidence that miRNA can affect Hmox1 expression indirectly by regulating the expression and activity of transcription factors such as Nrf2 (267) and Bach1 (186, 430). Finally, there is also data indicating that Hmox1 may regulate miRNA expression. Thus cells deleted of (261) or overexpressing Hmox1 (261, 312) display differential expression of a range of miRNA species, indicating that Hmox1 expression may affect the miRNA transcriptome.
Hmox1 is most highly expressed in tissues involved in the degradation of senescent red blood cells, such as the spleen, liver, and bone marrow where the enzyme is expressed in mononuclear phagocytes (535, 536). In most other tissue, Hmox1 is undetectable under basal conditions but can be induced rapidly by the stimuli outlined above. Both the 32 kDa Hmox1 and 36 kDa Hmox2 are anchored, via their COOH terminus, to the endoplasmic reticulum where they colocalize and interact with cytochrome P-450 reductase, an interaction required for maximal enzymatic activity (192, 314). In addition to the endoplasmic reticulum, Hmox1 has also been reported to reside in caveolae, mitochondria, and the nucleus (40, 238, 311, 494). Such alternative cellular localization appears to be associated with truncation of the COOH terminus of Hmox1 and loss of enzymatic activity (40, 311). Recent studies have also reported the detection of Hmox1 in plasma in several disease conditions (42, 629). While these observations suggest a role for Hmox1 in addition to heme breakdown (311), they also raise a number of currently unresolved issues, such as the identity of the signals/processes involved in alternative subcellular localization and the precise mode of action of Hmox1 if different to enzymatic degradation of heme.
2. Heme oxygenase-2
Hmox2 is a ∼36 kDa constitutively expressed protein (338, 544), with the highest expression of Hmox2 identified in the brain (519) and testis. Indeed, to date, only corticosteroids have been reported to induce Hmox2 (336, 437, 579). As indicated earlier, specific Cys residues contained in a cysteine-proline motif of the heme regulatory domains of Hmox2 act as a thiol/disulfide redox switch that regulates the Kd for heme, with the oxidized disulfide state of the enzyme having higher affinity for heme than the reduced state (435, 618). This redox switch has been implicated in the reported role of Hmox2 in oxygen sensing via the BKCa2+ channel in the carotid body (593). As BK channels detect low arterial Po2, they become acutely and reversibly inhibited, causing cell depolarization, subsequent voltage-gated Ca2+ influx that is ultimately transduced into increased ventilation. Similar to Hmox2, the BK channel is regulated by a thiol/disulfide redox switch that contains a CXXC motif that modulates heme binding (617). While the thiol/disulfide redox switch in the two proteins contains similar midpoint redox potentials, it has been proposed that they exert opposite albeit complementary effects on the affinity for heme (435). Thus, under normoxic conditions, sufficient O2 is present to poise the thiol/disulfide switches of Hmox2 and the BK channel in the disulfide state, where Hmox2 has high affinity and the BK channel has low affinity for heme. The resulting release of heme from the BK channel promotes the open state and high K+ transport, while degradation of heme by Hmox2 results in formation of CO. Conversely, under low O2 concentration and low affinity of Hmox2 for heme, the BK channel can bind available heme, poising the channel in the closed state.
Oxygen sensing by the carotid body requires CO as well as hydrogen sulfide (H2S) synthesis by cystathione-γ-lyase, and it has been suggested that Hmox2-derived CO works in concert with H2S to modulate O2 sensing (419, 628). Under normoxic conditions, Hmox2-derived CO stimulates protein kinase G-dependent phosphorylation of Ser377 in cystathione-γ-lyase, thereby inhibiting formation of H2S. In hypoxia, Hmox2-dependent formation of CO is attenuated, resulting in increased H2S that stimulates carotid body sensory nerve activity by inhibiting O2-sensitive K+ channels. These studies suggest variations in CO-regulated H2S as a fundamental mechanism in carotid body O2 sensing. It is noteworthy in this context that heme-dependent regulation of ion channels is not limited to the BK channel, but likely extends to other heme-binding ion channels (54). Thus, by regulating cellular heme concentrations as well as CO and H2S, Hmox2 (and in some cases perhaps also Hmox1) may contribute to the control of a number of complex biological processes.
B. Arterial Homeostasis
The vasculature refers to the arrangement of blood vessels in an organ composed of arteries, capillaries, and veins. In general, arteries provide an elastic conduit for the transport of oxygenated blood to the capillaries in target organs. Capillaries are the site of chemical exchanges between blood and tissue, whereas veins return the deoxygenated blood to the heart. Arterial homeostasis refers to the control of arterial tone and blood flow, arterial patency, as well as the response to arterial injury (459). This homeostasis is regulated by a complex interplay of neural factors mediated by the autonomic nervous system, humoral factors mediated by circulating mediators, local factors generated in the vessel wall, and autoregulatory mechanisms. Together, these systems ensure optimal perfusion of tissues and organs both at rest and during stress conditions.
1. Vessel structure
Arteries consist of three concentric layers: the tunica adventitia, the tunica media, and the tunica intima (Figure 3). The tunica adventitia is the outermost layer consisting mainly of fibroblasts and connective tissue and is supported by an external elastic lamina. The tunica media is positioned between the external and internal elastic laminae, and composed of vascular smooth muscle cells (VSMC) and elastic tissue. The VSMC are connected with tight junctions allowing for close mechanical coupling of cells for vessel constriction. Arterial VSMC maintain a basal tone, and the control of vessel diameter occurs through modulation of this tone. VSMC also play a crucial role in vascular remodeling after injury. Finally, the tunica intima is the innermost layer of a vessel comprised of a single layer of endothelial cells (EC) and supported by the internal elastic lamina.
FIGURE 3.
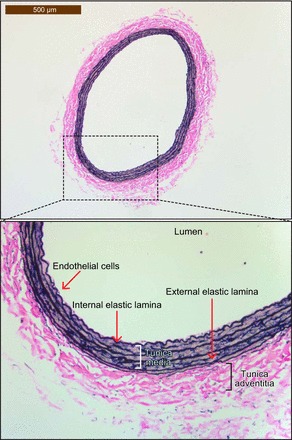
Cross-section of a hematoxylin and eosin-stained section of the left carotid artery from a Sprague-Dawley rat. The artery is lined by a single layer of endothelial cells (also referred to as the tunica intima) sitting on top of the internal elastic lamina. Following this is the tunica media composed of VSMC and elastic tissue. The final layer is the tunica adventitia that is comprised mainly of fibroblasts and connective tissue and that is supported by the external elastic lamina.
2. Neural regulation
Neural regulation of blood pressure, flow, and tone depends on the cardiovascular centers located in the medulla oblongata. Sympathetic neurons innervate the heart, blood vessel wall, adrenal glands, and kidneys, resulting in widespread control of cardiac and vascular function. These neurons use norepinephrine as a neurotransmitter to act on α-adrenergic receptors (α1 and α2) in blood vessels and β-adrenergic receptors (β1 and β2) in the heart and coronary arteries to modulate arteriolar resistance, venous capacitance, and cardiac function. Apart from the coronary arteries, most blood vessels are dominated by α-adrenergic receptors either α1-receptors (located on vascular postjunctional terminals) or α2-receptors (located on vascular prejunctional terminals). α-Adrenergic stimulation via α1- and α2-receptors leads to VSMC contraction and constriction of arteries. In addition, α2-receptors can reduce norepinephrine release, thereby exerting negative feedback regulation on the system.
In the coronary arteries β2-adrenergic receptors are the dominant adrenergic receptor, with stimulation leading to vasodilation. In contrast, β1-receptors are more prevalent in the heart where activation results in increased heart rate and contractility. Parasympathetic nerves that use acetylcholine to activate muscarinic receptors also innervate the vessel wall. Muscarinic receptors exert effects opposite to that of adrenergic stimulation. Specifically, acetylcholine-mediated stimulation of muscarinic receptors leads to a reduction of heart rate and vasodilation. Cholinergic vasodilation occurs via two distinct mechanisms. The first, more direct mechanism appears to be endothelium dependent, via synthesis of nitric oxide (NO) by endothelial nitric oxide synthase (eNOS). The second mechanism occurs through the stimulation of cholinergic receptors in the prejunction region of adrenergic neurons. Stimulation of cholinergic nerves inhibits the release of norepinephrine, thereby attenuating α-adrenergic stimulation and the resulting arterial constriction.
Neural regulation of blood pressure occurs via baroreceptors (mechanoreceptors) in blood vessels that sense and respond to the degree of stretch caused by blood flowing through the vessel. Changes in blood volume activate baroreceptors, which in turn trigger sympathetic or parasympathetic stimulation in vessels. The resulting vasoconstriction or dilation causes the normalization of blood pressure. Regulation of blood pressure also occurs via chemoreceptors in the carotid and aortic bodies, although to an overall lesser extent than regulation by baroreceptors. Chemoreceptors sense changes in blood gases, with rising CO2, falling O2, and/or falling pH indicating inadequate tissue perfusion. In such circumstances, chemoreceptors increase sympathetic stimulation, leading to increased blood pressure and blood oxygen concentrations (through increased ventilation) to enhance tissue blood supply.
3. Humoral
Endocrine control of vascular tone involves the release of the catecholamines epinephrine and norepinephrine. Circulating catecholamines affect vascular tone mainly through epinephrine generated in the adrenal medulla. Physiological levels of epinephrine result in vasodilation via stimulation of β-adrenergic receptors. Under conditions of stress, norepinephrine concentrations increase, and this may result in peripheral vasoconstriction via stimulation of α-adrenergic receptors. An important humoral mediator of vascular tone is the renin-angiotensin system (RAS), a hormone system that regulates fluid balance and blood pressure. In response to reduced renal blood flow, renin is released into the circulation. Renin catalyzes the conversion of angiotensinogen to angiotensin I, a peptide with limited biological activity. Angiotensin converting enzyme then converts angiotensin I to angiotensin II, a powerful vasoconstrictor that can act on VSMC in arteries, arterioles, and some veins. In addition, angiotensin II stimulates adrenal production of aldosterone, resulting in renal sodium absorption, an increase in plasma volume, and increased arterial pressure. The vasoconstriction exerted by angiotensin II significantly affects vascular tone and blood pressure. Circulating vasoactive factors such as antidiuretic hormone (vasopressin) and atrial natriuretic hormones also affect vessel tone and are stimulated by changes in vessel pressure.
4. Local
In addition to the neural and humoral control, blood flow and blood vessel perfusion and tone are also efficiently regulated at the local level, as a result of vessels responding to mechanical forces and chemical stimuli. The endothelium produces a variety of substances that regulate vascular tone and platelet aggregation in response to a range of physiological and pathological stimuli (for an extended list, see Table 2) These substances include derivatives of arachidonic acid, NO, and endothelin that are produced in response to stimuli such as increased shear stress in the vessel wall, hypoxia, thrombin, and neural mediators such as acetylcholine, epinephrine, and mediators of inflammation (Table 2). Endothelial cells also synthesize plasminogen activators and their inhibitors in response to stimuli associated with clot formation to regulate clot formation and dissolution.
Table 2.
Local mediators of vascular tone and vessel patency
| Pathway | Early Pathway Events | Downstream Signaling | Effects | Main Physiological Function | Reference Nos. | Evidence for Pathway Modulated by Hmox1 |
|---|---|---|---|---|---|---|
| Myogenic constriction | Activation of an unknown mechanosensor | Increase in phospholipase C, inositol 1,4,5-trisphosphate, protein kinase C, VSMC depolarization, VSMC [Ca2+]i, myosin light-chain Ca2+ sensitivity and actin polymerization | Constriction | Autoregulation of blood flow, capillary pressure stabilization | 178, 253, 366, 588 | 379 |
| Cytochrome P-450 metabolism of arachidonic acid | Formation of 20-hydroxyeicosatetraenoic acid, increase in protein kinase C, decrease in VSMC potassium channel opening, increase in VSMC [Ca2+]i | Constriction | Autoregulation of blood flow, capillary pressure stabilization | 270, 330 | 102 | |
| Cyclooxygenase metabolism of arachidonic acid | Production of prostacyclin and stimulation of IP receptor | Increase in adenylate cyclase, cAMP, protein kinase A activity, VSMC hyperpolarization and decrease in VSMC [Ca2+]i | Relaxation | Flow mediated dilation, endothelium-dependent, observed when other mediators are inhibited, renal blood flow regulation, platelet inhibition | 93, 94, 197, 256 | 299 |
| Production of PGE2 and stimulation of EP1, EP2, EP3 and EP4 receptors | EP2, EP4: Increase in adenylate cyclase, protein kinase A, K+ efflux, decrease in VSMC [Ca2+]i | EP2, EP4: relaxation | Renin secretion and systemic vascular tone | 4, 88, 431, 469, 527 | 47, 165 | |
| EP1, EP3: Increase in phospholipase C, inositol 1,4,5-trisphosphate and decrease in cAMP | EP1, EP3: constriction | |||||
| Production of thromboxane A2 and stimulation of TP receptors | Increase in phospholipase C, inositol 1,4,5-trisphosphate, VSMC [Ca2+]i, and myosin light-chain Ca2+ sensitivity, decrease in adenylate cyclase and cAMP, increase in phosphodiesterase expression, inhibition of endothelial hyperpolarization, increase in superoxide production | Constriction | Regulation of vascular tone, inhibition of EC-derived hyperpolarization, increase in reactive species, cross-reactivity with other prostanoid receptors under disease conditions | 191, 287, 355, 364, 378 | 471 | |
| Nitric oxide | sGC | Increase in cGMP, protein kinase G, receptor modulation, VSMC hyperpolarization; decrease in [Ca2+]i and myosin light-chain Ca2+ sensitivity | Relaxation | Blood pressure regulation, relaxation, inhibition of platelet aggregation, regulation of cardiac contractility | 44, 87, 443, 463, 526 | 216 |
| Carbon monoxide | sGC/BKca | Increase in; cGMP, protein kinase G and activation of BKca channels | Relaxation | Blood pressure regulation, relaxation | 147, 216, 379, 574, 587, 603 | |
| High CO concentration in skeletal muscle | Negative modulation of the NO pathway | Constriction | 203, 212 | |||
| EC-derived hyperpolarization of VSMC (not mediated by NO or cyclooxygenase-derived products) | K+ efflux through endothelial calcium-activated K+ channels | Activation of VSMC Kir and Na+-K+-ATPase channels, VSMC hyperpolarization leading to decreased VSMC [Ca2+]i | Relaxation | Regulation of vascular tone in resistance arteries, regulation of blood pressure, conduction of relaxation along arterial branches | 49, 97, 125, 253, 254, 479 | 298 |
| Electrical conductance through junctional proteins | Connexins mediate electrical conduction of hyperpolarization along EC and VSMC leading to decreased VSMC [Ca2+]i | Relaxation | Regulation of vascular tone of potential importance during functional antagonism of relaxation, conducted relaxation | 127, 345, 608 | ||
| Small molecule movement through myoendothelial gap junctions | Inositol 1,4,5-trisphosphate or Ca2+ movement through myoendothelial gap junctions, increase in EC [Ca2+]i, activation of eNOS and/or IKCa channels, subsequent activation of pathways described above | Relaxation | Regulation of vascular tone, VSMC negative feedback to counteract constriction | 274, 387, 460, 545 | ||
| Cytochrome P-450 metabolism of arachidonic acid | Epoxyeicosatrienoic acid act in: 1) autocrine manner raising EC [Ca2+]i through TRP receptors and subsequently activating Ca2+-activated K+ channels; and/or 2) paracrine action whereby epoxyeicosatrienoic acids diffuse to VSMC activating unidentified receptors, VSMC Ca2+- activated K+ channels and subsequent hyperpolarization and closure of voltage-gated Ca2+ channels | Relaxation | Regulation of vascular tone in response to cyclic stretch and shear flow and produced in inflammatory states | 58, 137, 190, 562 | 453, 454 | |
| Lipoxygenase metabolism of arachidonic acid | Formation of trihydroxyeicosatrienoic acids, hydroxyepoxyeicosatrienoic acids, and hydroxyeicosatetraenoic acids; VSMC hyperpolarization through K+ efflux | Relaxation | Regulation of vascular tone through hyperpolarization, roles in inflammation | 151–153, 638 | ||
| EC source of hydrogen peroxide | Oxidation of protein kinase G1α, activation of BKCa channels, VSMC hyperpolarization, decrease in VSMC [Ca2+]i | Relaxation | Proposed EC-derived hyperpolarizing factor. Mice with protein kinase G1α increased blood pressure despite normal response to NO | 53, 376, 429 | ||
| EC source of hydrogen sulfide | Formation of sulfide, nitrogen hybrid groups, enhanced NO donor activity, oxidation of protein kinase G1α, VSMC hyperpolarization, and decrease in [Ca2+]i | Relaxation | Proposed EC-derived hyperpolarizing factor | 95, 96, 300, 512 | ||
| EC source of bradykinin | Activation of B1 and B2 receptors, increase in EC [Ca2+]i, EC hyperpolarization, formation of epoxyeicosatrienoic acids, activation of BKCa channels | Relaxation | Inflammatory responses, relaxation and pain | 125, 589 | ||
| Endothelin-1 | ETA receptor | Increase in phospholipase C, inositol 1,4,5-trisphosphate, diacylglycerol, protein kinase C, and VSMC [Ca2+]i, increased superoxide production | Constriction | Regulation of vascular tone, more pronounced effects in age and disease | 105, 251, 284, 602 | |
| ETB Receptor | Increase in EC [Ca2+]i, increased prostacyclin and NO production | Relaxation | 105, 126, 632 | |||
| Angiotensin | Angiotensin type 1 receptor | Increase in phospholipases C, A2 and D; inositol 1,4,5-trisphosphate; diacylglycerol; VSMC [Ca2+]I; RhoA kinase activation. Stimulation of protein kinase C, BKca channel internalization, NADPH oxidase activation, increased superoxide production | Constriction | Regulation of vascular tone, electrolyte and blood volume and systemic blood pressure | 280, 356 | |
| Angiotensin type 2 receptor | Angiotensin II metabolism to angiotensin (1–7), III, and IV; increased NO release | Relaxation | 154, 155 | 189 |
In addition, autoregulatory mechanisms adjust blood flow to appropriate levels in response to changes in blood pressure. Metabolic and myogenic autoregulatory processes are active, independent of the endothelium and nerve stimulation. Metabolic stimuli such as changes in O2, adenosine, K+, CO2, H+, and lactate lead to alterations in vessel tone as an autoregulatory response, through the opening and closing of precapillary sphincters. Changes in blood flow also result in the myogenic response, where VSMC in the walls of arterioles and arteries stretch to protect against fluctuations in blood pressure and flow.
5. Vascular injury
To maintain vascular homeostasis, a balance between vascular injury and vascular repair must be maintained. Vascular injury can occur as a result of physiological and pathological events such as high shear stress and mechanical factors and elevated blood cholesterol. In addition, vascular injury occurs in response to interventional procedures such as percutaneous transluminal angioplasty and stenting. In response to acute vascular injury, three stages of repair occur. In the first stage, platelets adhere to the injured surface of the vessel and become activated. Subsequently, growth and chemotactic factors are released that recruit and activate VSMC and macrophages. Finally, activated VSMC and macrophages produce cytokines and growth factors that result in smooth muscle cell proliferation and matrix production. This remodeling process and recovery of endothelial integrity leaves the previously injured section of the vessel wall with a larger intimal layer than normal, a process known as intimal hyperplasia. The extent of this intimal hyperplasia is governed by the extent of vessel damage and the time required for regrowth of an intact endothelium, a process referred to as re-endothelialization. In situations of modest injury and rapid re-endothelialization, migration of VSMC to the intima is prevented with little or no intimal hyperplasia (522).
C. Vascular Diseases
In the 20th century, several noncommunicable diseases surpassed the number of communicable diseases, with cardiovascular diseases (CVD) leading the list (271). CVD remains the major global cause of mortality in addition to having a staggering effect on health care resource utilization (91, 271). Vascular diseases are defined by any pathological condition that affects the circulatory tree with ensuing loss of coordinated homeostatic function of different cell types that are responsible for ensuring adequate blood flow to all organs. While there are several entities of vascular diseases including but not limited to aneurysms, intimal hyperplasia, varicose veins, and vascular calcification, atherosclerosis is by far the most common cause of morbidity and mortality within this group. In an attempt to tackle this overwhelming clinical challenge, a great deal of research has been performed to identify risk factors and elucidate underlying pathophysiological and molecular pathways involved in this process (334). Evidence suggests that the development of atherosclerosis generally ensues over several decades, involves medium- and large-sized arteries, and constitutes a cascade of events that are fueled by endothelial dysfunction, inflammation, and proliferative events (131).
Atherosclerosis is also associated with a state of heightened oxidative stress, although it remains unclear whether, and if so how, the different reactive species and oxidative events cause the disease (504). Similarly, the presence of oxidative stress is not limited to atherosclerosis but is also part of several other vascular diseases, such as I/R and transplant arteriosclerosis. Therefore, enzymes with antioxidant properties may play beneficial roles in the formation of atherosclerotic lesions by minimizing oxidative stress and potentially associated oxidative injury. Following the discovery of the protective nature of the Hmox1 (384), extensive research has identified multiple pathways that are modulated in a beneficial manner in the context of atherosclerosis and other vascular diseases. The contribution of Hmox1 induction in vascular diseases is complex and involves different aspects that are mediated either by rapid removal of the toxic heme or generation of byproducts with potent antioxidant and anti-inflammatory properties or effects. A more detailed description of how Hmox1 is involved in vascular health and disease will be entailed in sections below.
III. HMOX IN VASCULAR HEALTH
A. Heme Oxygenases in the Normal Development of the Vascular System
To date, only two cases of human HMOX1 deficiency have been reported (434, 605) so that information on the human phenotype of this gene deficiency is limited. In both cases, however, similar phenotypes were observed characterized by generalized inflammation, nephropathy, asplenia, anemia, and tissue iron deposition. In addition, vascular injury was reported in both patients, suggesting the importance of Hmox1 in vascular health. Genetic approaches in murine models have been used extensively to characterize the role of Hmox1 in vascular and other systems, with the physiological importance of Hmox1 reinforced in the phenotype of HMOX1 gene knockout (Hmox1−/−) mice. Matings between Hmox1+/− mice do not yield the expected Mendelian ratio of offspring, with most HMOX1 gene null deletions being lethal (426, 427). This finding could explain the low penetrance of HMOX1 deficiency observed in humans. Hmox1−/− mice that are born exhibit phenotypes similar to the two human cases (Table 3), in particular signs of vascular injury and increased monocyte adhesion to vessel walls.
Table 3.
Comparison of human and mouse Hmox1 deficiency
| Phenotype | Human Cases | Hmox1 −/− Mice |
|---|---|---|
| Birth rate | Abortion, stillbirth | 5% |
| Development | Impaired | Impaired |
| Anemia | Observed | Observed |
| Red blood cells | Anisocytosis | Anisocytosis |
| Leukocytosis | Observed | Observed |
| Bone marrow hyperplasia | Observed | Not observed |
| Coagulation, thrombus | Observed | Not observed |
| Ferritin, heme | Elevated | Elevated |
| Reticuloendothelial system | Increased foam cells | Increased monocytes |
| Spleen | Asplenia | Enlarged |
| Fatty streaks/plaque | Observed | Not observed |
| Vascular injury | Observed | Observed |
| Glomerulonephritis | Observed | Not observed |
| Amyloid deposition | Observed | Not observed |
[From Kawashima et al. (227).]
The embryonic lethality seen in Hmox1−/− mice can be explained in part by Hmox1 affecting the development of the placental vasculature. Early studies showed that induction of placental Hmox1 expression augments placental blood flow while inhibition of Hmox1 increases resistance in the placenta (8, 327, 354). More recent studies indicate a critical role for Hmox1 in spiral artery remodeling (642). Uteroplacental blood flow increases dramatically during gestation and is facilitated by angiogenesis in the placenta, in particular the remodeling of the maternal uterine spiral artery system. In Hmox1+/− mice, malformations in the fetal placenta have been reported, with insufficient spiral artery remodeling and enlargement (596, 642). This insufficient remodeling of the spiral artery diminishes the number of uterine natural killer cells and angiogenic factors secreted by them at the fetomaternal interface (315, 642). Uterine natural killer cells regulate the remodeling of the maternal uterine vasculature via the production of pro-angiogenic factors such as vascular endothelial growth factor (VEGF), placental growth factor, and interferon-γ. Thus the expression of Hmox1 at the fetomaternal interface is important for optimal formation of the placental vasculature. HMOX1 may also increase placental blood flow by stimulating the release of corticotrophin (388), a pituitary gland hormone that stimulates the cortex of the adrenal gland to secrete its hormones, including corticosterone. In addition, Hmox1 has been reported to mediate the vascular effects of VEGF in chick embryo development, as judged by both the ability of VEGF to increase both Hmox1 expression and angiogenesis, and the Hmox1 inhibitor zinc mesoporphyrin to markedly attenuate angiogenesis (135).
It has also been proposed that decreased placental expression of Hmox1 contributes to the pathology of preeclampsia, with Hmox1 negatively regulating the release of the anti-angiogenic factors soluble fms-like tyrosine kinase-1 (sFlt1) and soluble endoglin (sEng) (99). Consistent with this contention, pregnant woman with pregnancy-induced hypertension and preeclampsia exhale less CO, an indirect measure of Hmox1 activity (26). Moreover, there is evidence from a small study suggesting that Hmox1 protein expression is decreased in placenta of women with preeclampsia (8). More recently, however, a comparatively larger study reported no evidence for decreased placental Hmox1 expression in preeclampsia (541). This study also did not observe placental Hmox1 expression to negatively regulate placental sFlt1 and sEng (541). Therefore, the role of Hmox1 in the pathology of preeclampsia requires clarification.
In contrast to Hmox1, Hmox2 is not implicated in the development of the vascular system, compared with wild-type littermates; Hmox2−/− mice develop indistinguishably into adulthood (425).
B. Heme Oxygenases and Hematopoiesis
There is now convincing evidence that Hmox1 is important in the regulation of the processes that bookend the life span of red blood cells (144). Poss and Tonegawa (426) first reported Hmox1-deficient adult mice to develop microcytic anemia associated with abnormally low serum iron, enlarged spleens and deposits of iron in the liver and kidney that contributed to macromolecular oxidative damage, tissue injury, and chronic inflammation. While the observed splenomegaly in Hmox1−/− mice appears at odds with the asplenia observed in the human case of HMOX1 deficiency (605), the splenic enlargement observed in young Hmox1−/− mice progresses to red pulp fibrosis, atrophy, and functional hyposplenism in animals older than 9 mo (260). Spleens in Hmox1−/− mice have significantly decreased numbers of macrophages (260) that allow iron recycling by returning iron from cleared red blood cells to the bone marrow for use in erythropoiesis (143). As a result of this decrease in splenic macrophages, body iron becomes redistributed from the spleen to hepatocytes and proximal tubular cells of the kidney instead of bone marrow cells (260, 426). This likely contributes to the microcytic anemia observed in Hmox1−/− mice (144). Following phagocytosis of senescent red blood cells, Hmox1 becomes induced in splenic macrophages where it plays an important role in both the iron recycling required for normal hematopoiesis and protecting the macrophages from the pro-oxidant heme released during red blood cell clearance.
Hmox1 also plays a significant role in the bone marrow, the primary organ responsible for erythropoiesis in adult mammals. During erythropoiesis, bone marrow macrophages interact with erythroid cells to form multicellular clusters termed erythroblastic islands. Within these erythroblastic islands, the macrophages supply the rapidly hemoglobinizing erythroblasts with iron and growth factors. Bone marrow macrophages also engulf and destroy nuclei expelled by erythroblasts in a process termed enucleation, leading to the release of anuclear reticulocytes into the circulation. Erythroblastic island macrophages in the bone marrow express Hmox1 (144), likely as the result of the macrophages engulfing and being exposed to the cytoplasmic hemoglobin contained in the erythroid nuclei expelled during enucleation. Compared with wild-type animals, Hmox1−/− mice display markedly lower numbers of erythroblastic islands with abnormal macrophage morphology (144). Moreover, bone marrow from Hmox1−/− mice contains fewer erythroblastic island macrophages and erythroid cells with abnormal adhesion molecule expression required for interaction of the two cell types (144). Hmox1 deficiency also dramatically decreases the frequency of macrophages expressing the phosphatidylserine receptor Tim4 in bone marrow and spleen, and this is associated with an increase in life span of circulating red blood cells (144). Overall, these findings suggest that macrophages and relevant receptors needed for steady-state erythropoiesis and red blood cell removal are depleted in Hmox1−/− mice, and that a prolonged lifespan of the erythrocytes minimizes the extent of anemia.
Similar to steady-state erythropoiesis, a deficiency in Hmox1 also leads to disrupted stress erythropoiesis. Thus bone marrow transfer from Hmox1+/− mice is ineffective in protecting lethally irradiated recipient mice, even though Hmox1+/− mice display a more rapid recovery to acute hematopoietic stress induced by 5-fluorouracil (61). Moreover, using a transplant model to induce stress conditions, Cao et al. (60) reported Hmox1 deletion to affect stress erythropoiesis through the retention of erythroblasts in the erythroblastic islands of the spleen.
C. Heme Oxygenases and Vascular Homeostasis
Under normal conditions, the endothelium maintains vascular tone and blood flow, and it limits vascular inflammation, intravascular thrombosis, and VSMC proliferation by synthesizing and releasing vasoactive substances such as NO. Both Hmox1 and Hmox2 can modulate vascular homeostasis in multiple ways. Of these, CO is thought to be primarily involved (see, however, Ref. 62), although some biological effects have also been attributed to bilirubin.
1. Regulation of vascular tone
Jones et al. (216) reported mesenteric and femoral arteries from Hmox1−/− mice to be slightly more sensitive to vasoconstrictors and to show a decrease in both endothelium-dependent (response to agents that act via eNOS/NO) and endothelium-independent relaxation (response to the NO-donor sodium nitroprusside) (216). The finding that Hmox1 deficiency is associated with attenuated endothelium-dependent relaxation was confirmed recently in aortic segments of littermate Hmox1+/+, Hmox1+/−, and Hmox1−/− mice (587). In that study, aortic segments of Hmox1+/+ mice were reported to express Hmox1 protein (587), as assessed by Western blotting using monoclonal anti-Hmox1 antibody (ADI-OSA-110 Enzo Life Sciences; Dr. Andreas Daiber, Johannes Gutenberg University, Mainz, Germany, personal communication). This observation is contrary to the general understanding that this stress protein is expressed only in “stressed” vasculature (71, 224). Irrespective, basal blood pressure is not different between nonstressed Hmox1+/+, Hmox1+/−, and Hmox1−/− mice (590). Mechanistic studies based on activators and inducers of soluble guanylate cyclase (sGC) suggest that in arteries of Hmox1−/− animals, the heme group of sGC is more oxidized and hence partly refractory to stimulation by NO compared with vessels from Hmox1+/+ mice (216). These findings suggest that Hmox1 affects vascular tone via its antioxidant activity, maintaining sGC in the reduced, NO-sensitive state.
Unlike Hmox1, Hmox2 is constitutively present in the vasculature, with cerebrovascular expression especially high. While endothelium-dependent and endothelium-independent relaxation of carotid arteries are not different between littermate Hmox2+/+, Hmox2+/−, and Hmox2−/− mice (224), CO derived from Hmox2 has been suggested to contribute to the maintenance of cerebral vessel studies based on studies using metalloporphryin-based inhibitors (reviewed in Ref. 415). Hmox2-derived CO formation by brain, isolated cerebral vessels, and cultured cerebral vascular endothelial cells increases in response to a variety of conditions and agents, including epileptic seizures, hypoxia, and glutamate (415). As indicated earlier, however, interpretation of metalloporphyrin-based inhibitor studies is complicated, as these chemicals modulate heme oxygenases indiscriminately, and they also affect other heme-containing enzymes, including NO synthase (325) and sGC (163).
Similar to NO, CO can modulate the activity of sGC and hence affect the cGMP signal transduction pathway (235). However, comparatively much higher concentrations of CO than NO are required to activate purified sGC in vitro (147, 510). Moreover, varying effects of CO on arterial relaxation have been reported depending on the experimental conditions studied. For example, exogenous, as well as endogenously formed, CO can promote endothelium-dependent constriction of isolated gracilis muscle arterioles (212). This vasoconstriction was converted into relaxation by NO synthase blockade and by l-arginine (212), consistent with CO promoting endothelium-dependent constriction by inhibiting endothelial NO formation (203). The overall impact of CO on vascular tone depends on the anatomical arrangement and the relative concentrations of heme oxygenases/CO and eNOS/NO present. Thus micromolar concentrations of CO cause constriction by inhibiting eNOS activity and decreasing NO (203), while lower concentrations of CO (≤100 nM) give rise to relaxation such as in liver sinusoids (157, 513). Vessel relaxation by CO may involve the release of NO from a large intracellular pool and, therefore, may mimic the vascular effects of NO (539).
In addition to regulating sGC, CO has also been reported to inhibit the production of the potent vasoconstrictor endothelin-1 (369). Moreover, studies by Suematsu and co-workers (367) have shown that Hmox2-derived CO enhances cerebral blood flow in hypoxia by regulating the hydrogen sulfide pathway (367). Under normoxic conditions, Hmox2 constitutively expressed in the brain gives rise to micromolar concentrations of CO that inhibit cystathionine β-synthase and hence formation of the vasodilator hydrogen sulfide (367). In hypoxia, Hmox2 senses the lower O2 concentration (see sect. IIA2) and forms less CO. This releases the constitutive inhibition of cystathionine β-synthase, allowing for synthesis of hydrogen sulfide that then mediates vasodilation of precapillary arterioles (367).
2. Regulation of vascular inflammation
A protective role of Hmox1 against vascular inflammation is implied by the observation that human HMOX1 deficiency is characterized by vascular inflammation (605). Similar to the human situation, Hmox1−/− mice have increased endothelial disruption and apoptosis and are prone to endothelial denudation, vascular injury, and intravascular thrombosis (546). Indeed, there is evidence for Hmox1-dependent anti-inflammatory activities to both the vasculature and circulating inflammatory cells. Pro-inflammatory stimuli including LPS induce the expression of Hmox1 in vascular EC, and this is important for the maintenance of vascular integrity during inflammation and the protection of EC from injury and apoptosis (reviewed in Refs. 450, 451). Compared with wild-type mice, aortic segments from Hmox1−/− mice also express higher levels of phagocyte-type NADPH oxidase 2 (NOX2) and the chemokine receptor CCR2 (587). Together, this promotes a pro-inflammatory milieu via increased formation of superoxide and entry of phagocytes into arteries. Indeed, when exposed to an inflammatory challenge, Hmox1−/− mice show enhanced vascular inflammation, characterized by increased aortic infiltration of CD11b+Ly6Chi monocytes and Ly6G+ neutrophils when compared with Hmox1+/+ mice (587).
Apoptosis of EC is a prominent feature in acute and chronic inflammation and is observed in numerous conditions, such as hyperoxia (408), I/R injury (408), and chronic graft rejection (15, 488). In vitro studies have shown that expression of Hmox1 prevents injury to EC mediated by activated phagocytes, hydrogen peroxide, or exposure to heme (2, 427, 610). Protection against hemoglobin and heme toxicity is important during vascular inflammation where hemolysis can occur and heme proteins may become denatured and release their heme moiety. Upon release into the circulation, hemoglobin and heme are usually bound by haptoglobin and hemopexin, respectively, i.e., acute phase proteins that aid the hepatic detoxification of hemoglobin and heme (for recent reviews, see Refs. 466, 565). However, these endogenous protective systems can be overwhelmed, with resulting incorporation of the cytotoxic heme or accumulation of iron derived from hemoglobin within the vascular endothelium, resulting in the expression of Hmox1 and/or ferritin in EC (21). Hmox1 serves to rapidly eliminate pro-oxidant heme and convert it into antioxidant bile pigments (500). Induction of Hmox1 activity is also associated with increased cellular iron efflux by regulating an iron pump, a process that limits the availability of redox-active iron and contributes to the inhibition of EC apoptosis (136).
The expression of Hmox1 can also decrease the pro-inflammatory properties and responses of immune phagocytes. For example, low concentrations of CO attenuate LPS-induced macrophage expression of the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), while at the same time increasing the anti-inflammatory cytokine interleukin-10 (IL-10) (407). Similarly, macrophages from Hmox1−/− mice exhibit increased pro-inflammatory cytokines compared with cells from wild-type animals (405). Moreover, using the murine macrophage cell line RAW 264.7, Taillé et al. (524) reported Hmox1 expression to inhibit NOX2 activity via decreasing cellular heme and downregulating cytochrome b558. Such anti-inflammatory protection by Hmox1 is also observed in humans. Thus, in circulating human monocytes of individuals with hypertension, mRNA concentrations of HMOX1 inversely correlate with those of CD14 (587), a classical marker of inflammatory monocytes. Similarly, the levels of CCR2, the receptor that binds monocyte chemoattractant protein-1 (MCP-1) and recruits monocytes to sites of inflammation, are increased in circulating monocytes of Hmox1−/− compared with Hmox1+/+ mice (587).
D. Heme Oxygenases and Endothelial Cell Function
In the presence of cardiovascular disease risk factors or following percutaneous coronary interventions and stenting, arterial EC become injured and lost. The repair of injured/lost EC and reestablishing of the physical continuity and function of the endothelial layer, a process referred to as re-endothelialization, is crucial for vascular health and disease. For example, areas that take longer to re-endothelialize following percutaneous coronary interventions have significantly more intimal thickening than areas that are rapidly covered with endothelium (173). The migration and proliferation of remaining resident EC, as well as the recruitment of circulating endothelial progenitor cells are important contributors to re-endothelialization, and Hmox1 promotes these repair processes.
1. Promotion of endothelial cell growth and re-endothelialization
Hmox1 regulates the proliferation of vascular cells in a cell-specific manner: it promotes the growth of EC (109) whereas it inhibits the proliferation of VSMC (Figure 4; see sect. IIIE1). In human EC, transduction with a retroviral vector encoding human HMOX1 revealed several differentially expressed genes encoding proteins modulating cell growth and cell cycle progression (3). These changes included HMOX1-induced increases in the expression of cyclin E and D, VEGF and its receptor I, and endothelial growth factor and hepatic-derived growth factor. In contrast, cyclin-dependent kinase inhibitors p21 and p27; cyclin-dependent kinases 2, 5, and 6; and MCP-1 were regulated down (3).
FIGURE 4.
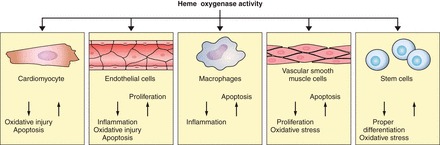
Hmox1 expression impacts on cardiomyocytes, vascular cells, and stem cells in a cell-type specific, in part opposing manner. In EC, increased Hmox1 expression promotes cell proliferation and decreases apoptosis inflammation and oxidative injury. Conversely, Hmox1 increases apoptosis and decreases the proliferation of VSMC. Hmox1 expression has been reported to increase apoptosis of macrophages and decrease inflammation. In various stem cells, Hmox1 expression has been associated with normal differentiation and protection against oxidative injury, while in cardiomyocytes, Hmox1 expression decreases apoptosis and oxidative stress.
Importantly, Hmox1 also promotes re-endothelialization following vascular injury, as demonstrated using a pharmacological inducer of Hmox1 in rabbits (272) or adenoviral overexpression of the HMOX1 gene in mice (309). In the former case, Hmox1 induction resulted in decreased intimal thickening (272), indicating that it translated into disease prevention. The enhanced re-endothelialization seen after viral HMOX1 overexpression could be recapitulated by exposing the mice to CO (250 ppm) (309), suggesting that Hmox1-derived CO may at least in part be responsible for the in vivo promotion of EC growth. Moreover, systemic Hmox1 induction leads to an increase in circulating progenitor cells (309) that contribute to re-endothelialization (598). This is consistent with bone marrow cells from Hmox1−/− mice generating fewer endothelial colony-forming cells than bone marrow from wild-type mice (598).
2. Protection against endothelial cell apoptosis
Similar to the cell-type specific growth effects discussed in the preceding section, overexpression of Hmox1 promotes and blocks apoptosis in VSMC (318) and endothelial cells (50), respectively. In the case of EC, Hmox1 inhibits apoptosis via formation of CO and activation of the p38 mitogen-activated protein kinase (MAPK) pathway (50, 490). The latter is achieved by Hmox1 directing signals from upstream kinases to the cytoprotective isoform of p38 MAPK, p38β, while enhancing the proteasomal degradation of p38α, the cytotoxic isoform of p38 MAPK (240, 486). Attenuation of EC apoptosis is also important for the prevention of thrombosis, and both CO and bilirubin rescue Hmox1−/− mice from accelerated thrombus formation via prevention of EC apoptosis, inactivation of platelets, and adaptive remodeling by bone marrow-derived progenitor cells (546).
3. Effects of heme oxygenase-2
Consistent with the notion that Hmox2 affects eNOS activity in vivo (see above), Bellner et al. (31) reported aortic endothelial cells from Hmox2−/− mice to contain lower concentrations of eNOS protein and phosphorylated eNOS compared with cells from wild-type mice. These authors also reported Hmox2 deficiency to result in “activated” EC, characterized by increased expression of gp91phox (NOX2) and the inflammatory cytokines IL-1β and interleukin-6 (IL-6) (31). Hmox2 has also been implicated in the responses of EC to hypoxia. Thus, in rats exposed to hypoxia, Hmox2 protein was reported to increase in aortic EC, and this was linked to a decrease in aortic contraction to phenylephrine (160). The authors suggested the existence of an unrecognized mechanism by which Hmox2 inhibits systemic reactivity in diseases that involve hypoxic conditions.
4. Protective effects of bilirubin
In addition to CO, bilirubin has also been reported to protect EC. In vitro work suggests that bilirubin protects against NADPH-mediated superoxide production (269), and bilirubin treatment can reduce oxidative stress induced by various stimuli (209). The role of heme oxygenases in regulating oxidative stress will be discussed in section IVB. While the cellular benefits of bilirubin are generally thought to reflect those of biliverdin reduction by BVR (209), there are examples where the biological activity observed with bilirubin is not recapitulated by biliverdin. For example, in human EC, TNF-α-mediated increase in E-selectin and vascular cell adhesion protein 1 is inhibited by increased Hmox1 activity or bilirubin treatment, whereas addition of biliverdin and CO had no effect (489). Finally, bilirubin has been reported to suppress interferon-γ-induced MHC class II expression on murine EC similarly to induction of Hmox1 (599), suggesting that the pigment modulates immune responses.
E. Heme Oxygenases and VSMC Function
Like EC, VSMC are essential for the normal functioning of the arterial vasculature. Through contraction and relaxation, VSMC alter the lumen diameter and hence blood supply to tissues. VSMC also proliferate and migrate during arterial remodeling in response to physiological conditions. The switch in VSMC from a synthetic to a proliferative state is also important in pathological situations such as atherosclerosis (sect. VA), intimal hyperplasia (sect. VB), and hypertension (sect. VF). Given this, much research has focused on understanding and modulating VSMC proliferation and migration.
1. Inhibition of VSMC growth
In contrast to the situation with EC, HMOX is a negative regulator of the growth of VSMC (Figure 5). Pharmacological inhibition of HMOX or addition of the CO trap hemoglobin potentiates the proliferation of VSMC in response to serum, angiotensin II, endothelin, and hypoxia (368, 421, 540). Similarly, Hmox1−/− mice are characterized by enhanced VSMC proliferation and DNA synthesis compared with wild-type animals (117). Conversely, induction of Hmox1 expression by hemin or overexpression of the HMOX1 gene induces VSMC apoptosis and blocks their growth in porcine and rat models (64, 107, 117, 318, 640). Many endogenous agents are known to induce Hmox1 in VSMC and to inhibit VSMC proliferation. These agents include NO (123, 171, 319), cAMP (122), platelet-derived growth factor (124), angiotensin II (204, 587), 15-deoxy-Δ12,14-prostaglandin J2 (306), and peroxisome proliferator-activator receptor (PPAR) ligands (265). In addition, numerous pharmacological agents modulate VSMC proliferation and migration via induction of Hmox1 (Table 4), and many are thought to represent novel mechanisms by which VSMC growth may be controlled in vivo.
FIGURE 5.

Schematic showing the mechanisms by which CO and bilirubin affect VSMC proliferation. CO has been reported to 1) activate soluble guanylate cyclase (sGC) leading to increased cGMP; 2) decrease cytokine-mediated apoptosis; 3) decrease NADPH oxidase 1 (Nox1) activity; 4) decrease T-type Ca2+ channel activity; 5) increase cell cycle progression through the p38 MAPK pathway; and 6) increase caveolin expression. Bilirubin has been reported to decrease cell cycle progression through hypophosphorylation of retinoblastoma protein (Rb) and by increasing intracellular Ca2+, leading to a decrease in the transcription factor Yin Yang 1 (YY1) and altered transcription of genes important for VSMC cell cycle control.
Table 4.
Pharmacological agents reported to modulate VSMC proliferation and migration through induction of Hmox1
| Pharmacological Agent | Agent Property | Model | Effect | Reference Nos. |
|---|---|---|---|---|
| Tunicamycin | Blocks N-linked glycosylation and induced ER stress | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation and migration | 619 |
| Naringenin | Flavanone | Rat aortic SMC | Inhibits TNF-α-induced proliferation and migration | 69 |
| Malabaricone C | Compound isolated from Myristica cinnamomea (nutmeg) | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation and migration | 276 |
| AVE 0991 | Angiotensin-(1–7) Mas receptor antagonist | Rat aortic SMC | Inhibits angiotensin II-induced VSMC proliferation and migration | 477 |
| Trans-caffeic acid phenethyl ester | Phenolic compound isolated from honeybee propolis | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation and migration | 447 |
| Resveratrol | Phenolic compound isolated from fruit such as grape skins | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation and migration | 243 |
| Celastrol | Pentacyclic tripterine isolated from the root of Tripterygium wilfordii | Rat aortic SMC | Inhibits angiotensin II-induced VSMC proliferation and migration | 627 |
| Neferine | Alkaloid from lotus seed embryos | Human umbilical vein SMC | Inhibits angiotensin II-induced VSMC proliferation and migration | 294 |
| Isoproterenol | Nonselective β-adrenoreceptor agonist | Rat aortic SMC | Inhibits angiotensin II-induced VSMC proliferation and migration | 242 |
| Aprotinin | Small bovine pancreatic trypsin inhibitor | VSMC from WT and spontaneous hypertensive rats | Inhibits VSMC proliferation | 80 |
| NS-398 | Pharmaceutical COX-2 inhibitor | Rat aortic SMC | Inhibits IL-1β-stimulated VSMC proliferation and migration | 79 |
| Curcumin | Principal curcuminoid of Curcuma longa (turmeric) | Rat aortic SMC | Inhibits 5% FBS-stimulated VSMC proliferation and migration | 412 |
| Paclitaxel | Chemotherapeutic | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation and migration | 78 |
| Rapamycin | Macrolide produced by Streptomyces hygroscopicus | Human pulmonary SMC | Inhibits PDGF-induced VSMC proliferation | 558 |
| Eupatolide | Sesquiterpene lactone isolated from the plant Inula britannica | Rat aortic SMC | Inhibits PDGF-induced VSMC proliferation | 245 |
| Valsartan | Angiotensin II receptor agonist | Rat aortic SMC | Inhibits balloon injury-induced VSMC proliferation | 296 |
| Dimethyl fumarate | Multiple sclerosis and psoriasis treatment | Rat carotid SMC | Inhibits balloon injury-induced VSMC proliferation | 400 |
| Simvastatin | HMG-CoA reductase inhibitor | Human aortic SMC; rat aortic SMC | Inhibits serum-stimulated VSMC proliferation | 277 |
| Rat pulmonary artery SMC | Inhibits serotonin-induced VSMC proliferation | 286 | ||
| Tricarbonyldichlororuthenium (II) dimer ([Ru(CO)3Cl2]2) | CO-releasing molecule | Human pulmonary artery SMC | Inhibits serum- induced VSMC proliferation | 495 |
| Sulfasalazine | Anti-inflammatory drug | Thoracic aortic SMC | Inhibits PDGF-induced VSMC proliferation | 244 |
| YS 49 | Analog of higenamine, a β2 adrenoreceptor agonist | Rat thoracic aorta SMC | Inhibits angiotensin II-induced VSMC proliferation and migration | 518 |
CO, carbon monoxide; ER, endoplasmic reticulum; IL-1β, interleukin-1β; mTOR, mechanistic target of rapamycin; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; PDGF, platelet-derived growth factor; SMC, smooth muscle cell; TNF-α, tumor necrosis factor-α; VSMC, vascular smooth muscle cells.
There is a considerable body of evidence suggesting that inhibition of VSMC proliferation by Hmox1 is mediated through the release of CO (368, 369, 421, 495). Several mechanisms have been proposed for this antiproliferative activity of CO, including an increase in cellular concentrations of cGMP (369), inhibition of NADPH oxidase 1 (NOX1) activity (444) and T-type Ca2+ channels (118), and activation of the p38 MAPK pathway (239). The study suggesting inhibition of T-type Ca2+ channels by CO employed patch-clamp recordings in different types of smooth muscle cells and showed relevant Ca2+ current and VSMC proliferation to be inhibited by blocking T-type channels (118). Activation of the p38β MAPK signaling pathway has been proposed as the mechanism by which exogenous CO (250 ppm) inhibits VSMC proliferation and intimal hyperplasia in rats following angioplasty-mediated injury of the left carotid artery (239). Exposure to CO led to increased expression of caveolin-1 and the cell cycle inhibitor kinase p21waf1/cip1 in injured arteries. As genetic depletion of caveolin-1 abolished the antiproliferative effect of CO, it was concluded that increased expression of caveolin-1 mediated the growth inhibitory properties of the gas (239). Together these data suggest that CO can modulate VSMC proliferation via multiple mechanisms, although care needs to be taken when attempting to extrapolate data obtained exclusively with exogenously administered CO to CO produced as a product of Hmox1 enzymatic activity.
Studies have also indicated that Hmox1 and the products of its enzymatic activity affect cell cycle progression by controlling the G1/S cell cycle transition to control VSMC proliferation. For example, induction of Hmox1 via hemin increases cells in G1 phase while it decreases cells in S or G2/M phase (291). Conversely, treatment with an Hmox1 inhibitor (tin-mesoporphyrin) decreases cells in G1 and increased cells in S or G2/M phase (291). Genetic overexpression of HMOX1 (117), CO administration (421), or treatment with bilirubin (422) arrests VSMC in the G1/S transition of the cell cycle. A recent in vitro study (509) suggested that bilirubin treatment inhibits VSMC growth by two distinct mechanisms. First, the pigment leads to an impaired phosphorylation and activation of the Raf/ERK/MAPK pathway resulting in hypophosphorylated retinoblastoma protein and suppression of cell cycle transition. Second, bilirubin increases intracellular Ca2+ that then leads to a decrease in the transcription factor YY1 and altered transcription of genes important for VSMC cell cycle control (509). Unfortunately, these studies employed bilirubin at a nonphysiologically high concentration (100 μM) so that their physiological relevance is unclear.
F. Hmox1 and the Regulation of Renal Hemodynamics and Blood Pressure
The regulation of blood pressure is a delicate process that is actively synchronized at multiple levels and involves mechanisms that work in a well-orchestrated manner. In this regard, the kidneys play a key role as important regulators of blood pressure. Hmox1 activity has been recognized to be a crucial component of blood pressure regulation. This notion was first suggested by reports that showed chemical induction of Hmox1 using tin was associated with prevention of development of hypertension in a genetic model of spontaneously hypertensive rats (455). The beneficial role of Hmox1 in this model of hypertension has been recapitulated using hemin as an inducer of Hmox1 expression (281, 343). Furthermore, the long-term effects of Hmox1 activation on modulation of blood pressure have been studied. Spontaneous hypertensive rats were administered hemin via subcutaneously implanted osmotic minipumps for three consecutive weeks (573). Remarkably, this treatment resulted in significant lowering of blood pressure, and intriguingly, these effects were sustained for a period of 9 mo after removal of the pumps (573).
The mechanism by which Hmox1 induction exerts its antihypertensive effects is attributed to a decrease in heme proteins such as cytochrome P-450 enzymes. This can lead to a decrease in the metabolism of arachidonic acid to 20-hydroxyeicosatetraenoic acid, a known vasoconstrictor with sodium retention properties (Table 2) (281, 343). In contrast, inhibition of heme oxygenase enzymatic activity has been shown to increase blood pressure and peripheral resistance of normotensive rats (215), a finding corroborated by others employing genetic manipulation of Hmox1. Accordingly, while overexpression of Hmox1 lowers blood pressure in hypertensive rats (452), Hmox1−/− mice demonstrate exaggerated blood pressure in a deoxycorticosterone acetate-induced systemic hypertension model (385). Moreover, while systolic blood pressure is not significantly different between Hmox1+/+ and Hmox1−/− mice at baseline, following a one kidney-one clip model of renovascular hypertension, deletion of Hmox1 led to a more severe development of hypertension that was also accompanied by higher mortality, serum creatinine concentrations, as well as cardiac hypertrophy (590). Others have reported that an increase in Hmox1 expression prevents hypertension induced in a model of renovascular hypertension with increased renin production (47). Moreover, Hmox1 overexpression suppresses angiotensin II-induced hypertension in rats (611).
Given the compelling evidence for Hmox1 mediating blood pressure, investigators have examined the role of the products of the heme oxygenase reaction in regulating this complex process. As a result, CO has emerged as a major component of this regulatory axis. The vasodilatory property of CO was described first in the pulmonary vasculature (521). Subsequently, numerous investigators have examined the antihypertensive actions of CO (reviewed in Refs. 275, 496). There are various pathways that have been proposed for CO to exert its antihypertensive effects. For example, CO is known to modulate neurotransmitter release (258) and the action potential of neurons in important nuclei that are responsible for central regulation of blood pressure in the brain, namely, the nucleus tractus solitarii (214). Importantly, inhibition of Hmox1 in this nucleus increases blood pressure while microinjection of CO into the ipsilateral nucleus reverses this finding in rats (214). These results have been validated further by injection of hematin to induce Hmox1 in the nucleus tractus solitarii (320). At the level of the vasculature, evidence also suggests involvement of the sGC/cGMP pathway (147) and high-conductance calcium-activated K+ channels in VSMC (575). There are other potential pathways that may be involved in CO-mediated regulation of blood pressure that include activation of phosphatidylinositol 3-kinase (PI3K) and RAC-alpha serine/threonine-protein kinase (Akt) as well as inhibition of the production of vasoconstrictive agents (e.g., endothelin-1) and are discussed in more detail elsewhere (496).
Bilirubin, a product of heme oxygenase activity, has also been reported to possess antihypertensive properties. The Gunn rat, an inherited model of severe unconjugated hyperbilirubinemia, is protected against hypertension induced by deoxycorticosterone acetate (385) and angiotensin II (423). These attributes have been confirmed in other, more moderate models of hyperbilirubinemia (556). Interestingly, a recent comprehensive analysis of National Health and Nutrition Examination Surveys (between 1999 and 2012) further corroborated the experimental findings in human subjects and revealed lower prevalence of hypertension in subjects with higher serum bilirubin concentrations (571).
While the majority of studies suggest that Hmox1 and products of its reaction possess antihypertensive properties, some contradicting studies have also been reported. For instance, Teran et al. (538) showed that chemical inhibition of Hmox activity in Dahl salt-sensitive rats lowered blood pressure in high salt treated animals. Furthermore, they suggested that CO leads to endothelial dysfunction that may contribute to the development of hypertension (538). Additionally, another study suggested that increased Hmox1-mediated generation of CO in obese Zucker rats leads to hypertension and arteriolar endothelial dysfunction, findings that were independent of metabolic parameters (213).
The kidneys also play an important role in chronic regulation of blood pressure that entails different pathways. Hence, renal hemodynamics and homeostasis of sodium are of paramount significance. Deficiency of Hmox1 has been demonstrated to result in perturbations of renal hemodynamics following ischemia (543). Another line of evidence suggests that Hmox1 is essential in preserving renal perfusion following angiotensin II administration, as its inhibition leads to increased renal vascular resistances and decreased renal blood flow (386). Induction of Hmox1 via hemin revealed important regulatory properties in the context of renal hemodynamics. These studies reported that induction of Hmox1 in rats is accompanied with higher renal blood flow, glomerular filtration rate, urinary flow and sodium excretion, while renal vascular resistance is significantly lower (46). Moreover, the plateau of the perfusion autoregulation of the kidneys is elevated and renal vascular changes in response to angiotensin II infusion are abrogated (46). Heme oxygenase-mediated CO release is also known to affect the vasoconstriction of afferent arterioles (439).
In addition, Hmox1 has been shown to directly regulate renal sodium excretion. In an elegant study, Li et al. (288) postulated that heme oxygenase activity with subsequent CO generation in the renal medulla may play a role in sodium handling and hence may have an overall effect on blood pressure. To prove this, they infused renal medulla of rats with chromium mesoporphyrin (an inhibitor of heme oxygenase activity) that markedly blunted pressure-dependent natriuresis (288). Such inhibition of heme oxygenase activity was also associated with significant elevation of mean arterial pressure in rats (288). In agreement with this premise, intravenous administration of hemin results in increased urinary CO concentrations accompanied with increased water and sodium excretion (445). Moreover, Hmox1 overexpression has been reported to inhibit superoxide generation in the kidney in response to angiotensin II (433). CO has been shown to exert similar effects (231) that may play an important role in the regulation of sodium reabsorption in this segment of the kidney.
As evident, there is a strong body of evidence to support the antihypertensive role of Hmox1. However, similar to the pathogenesis of hypertension, actions of Hmox1 on blood pressure modulation appear complex and involve various independent and overlapping pathways. Some of these potential pathways are illustrated in Figure 6.
FIGURE 6.
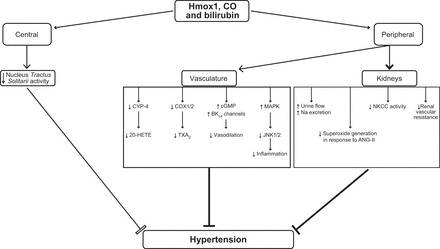
Potential pathways involved in hypertension that are modulated by Hmox1 and its products. Increased Hmox1 activity and CO both decrease the activity of nucleus tractus solitarii. CO activates MAPK, leading to inhibition of Jun amino-terminal kinases (JNK) 1/2 with subsequent mitigation of inflammation. CO can also activate sGC, leading to an increase in cGMP. This effect along with activation of high-conductance calcium-activated K+ channels (K-ca) leads to vasodilation. Additionally, CO inhibits cyclooxygenase-1 (COX) 1/2 and cytochrome P-450 (CYP-4) with subsequent reduction in thromboxane (TXA2) and 20-hydroxyeicosatetraenoic acid (20-HETE), respectively, both of which are vasoconstrictors and have sodium retention properties. Angiotensin II (ANG II)-mediated increase in superoxide generation with ensuing increased sodium reabsorption and renal vascular resistance is also known to be attenuated by HMOX1 induction and generation of CO and bilirubin. The activity of the Na+-K+-Cl− cotransporter (NKCC) in the thick ascending loop of Henle is also directly modulated by Hmox1 activity, leading to a decrease in Na+ absorption.
IV. REGULATION BY HEME OXYGENASES OF PROCESSES CONTRIBUTING TO VASCULAR DISEASES
As mentioned in section III, the protection afforded by heme oxygenases in the arterial vasculature arises from their multiple activities, including anti-inflammatory, antioxidant, and cell growth regulatory actions, although the exact mechanism(s) by which Hmox1/2 exert these effects remains only partially understood. Inducible Hmox1 is the more studied isoform in the context of vascular diseases and disease-linked processes. However, interest in the constitutively expressed Hmox2 in disease processes is increasing (31, 33), with currently available information also suggesting a role for this isozyme in inflammation and angiogenesis.
A. Inflammation
Hmox1 plays a critical role in acute and chronic inflammation. Hmox1−/− mice develop chronic inflammatory disease that progresses with age (426), and the first human case of Hmox1 deficiency was characterized by kidney and vascular inflammation (605). Although expression of Hmox1 can be induced in all tissues and cell types, the anti-inflammatory effects of Hmox1 appear to depend primarily on its role in myeloid cells (monocytes, macrophages, and dendritic cells) and EC. Willis et al. (594) first suggested the involvement of Hmox1 in inflammation in vivo using a carrageenan-induced model of pleurisy in rats. In that study, pharmacological modulation of Hmox1 resulted in striking effects on inflammation, with elevation of Hmox1 levels suppressing, and inhibition of Hmox1 increasing the inflammatory exudates (594). Since then numerous studies have reinforced the protective role of Hmox1 and Hmox2 in mammalian models of inflammatory disease (for a recent review of Hmox1 in inflammation, see Ref. 451).
1. Anti-inflammatory effects of Hmox1/CO
Overall, there is strong evidence for an anti-inflammatory action of CO gas and also CO-releasing molecules in preclinical animal models of CVD, inflammatory disorders, and organ transplantation (for a review and perspective of use of CO as a therapy for human diseases, see Refs. 252, 374). The anti-inflammatory benefits of CO were reported initially in a model of acute lung injury (408). Otterbein et al. (408) exposed rats to hyperoxia and observed concomitant treatment with CO gas (50-500 ppm) to significantly decrease both neutrophil influx into the airways and apoptosis of pulmonary cells compared with animals exposed to hyperoxia alone (408). As mentioned in section II, treatment with CO also decreases circulating concentrations of pro-inflammatory cytokines (TNF-α, IL-1β) in acute inflammation such as that provoked by administration of LPS (278, 370, 407). Several studies have reported additional anti-inflammatory actions of CO, including the inhibition of Toll-like receptor 2, 4, 5 and 9 signaling (380) and downregulation and inhibition of nuclear translocation of the proinflammatory factor high-mobility group box 1 (547). There are also studies reporting an association between CO and the activation of the transcription factors heat shock factor 1 (240) and immunoresponsive gene 1 (208).
What is less clear from the above studies is whether, and if so to what extent, the biological effects observed with exogenous CO reflect or even recapitulate the activities of CO arising from enzymatic degradation of heme by Hmox1/2. However, Hmox1 has been reported to be a downstream (278, 441) and upstream effector of IL-10, via activation of the p38 MAPK (407), the JNK signaling pathway, and the transcription factor AP-1 (370). In addition, there is evidence that Hmox1 induction through adiponectin (340), Bach1 deficiency (172), or hemin treatment (207, 390) modulates macrophage polarization towards an inflammation resolving M2 type, and promotes cell proliferation and wound healing. A recent study reported deletion of Hmox1 in the myeloid lineage to decrease the ability of myeloid progenitors to differentiate towards macrophages and that exposure of mice to CO gas in a model of marginal bone marrow transplantation improved donor myeloid cells expansion and differentiation (583).
2. Hmox1/CO and inflammasomes
Inflammasomes are intracellular multiprotein complexes that promote the cleavage of caspase-1 and subsequently lead to the secretion of pro-inflammatory cytokines such as IL-1β and the recruitment and activation of phagocytes in response to a variety of inflammatory triggers (247). Current research highlights the potential role of Hmox1 and CO in the regulation of the inflammasome dependent on NACHT, leucine-rich region, and pyrin domain-containing protein 3 (NLRP3). Specifically, hemin and other inducers of Hmox1 inhibit the activation of the NLRP3 inflammasome in models of liver (247) and lung injury (283, 326), thereby limiting the inflammatory response and damage. Recent work also suggests that macrophage-derived CO modulates the activation of the NLRP3 inflammasome and is a key modulator of macrophage defense (584). Wegiel et al. (584) suggested that CO derived from Hmox1 functions to “assess” the cellular environment. If bacteria are present, the CO formed leads to the activation of the inflammasome to drive pathogen clearance. A more recent study reported CO to inhibit the molecular interaction between the NLRP3 inflammasome and caspase-1 in bone marrow-derived macrophages exposed to LPS or ATP (221). CO also inhibited the decrease in mitochondrial membrane potential associated with the activation of the NLRP3 inflammasome, indicating that CO may negatively regulate the activation of the NLPR3 inflammasome by preventing mitochondrial dysfunction (221).
3. Effects of biliverdin on inflammatory responses
There is a growing body of work reporting the potential role of biliverdin as a negative modulator of inflammation. Administration of biliverdin decreases tissue inflammation and serum concentrations of pro-inflammatory cytokines, and increases anti-inflammatory cytokines in models of endotoxin-induced acute lung injury (462) and sepsis-induced inflammation (410). Similarly, treatment with biliverdin decreases neutrophil influx into the liver in a swine model of hepatic I/R (13), to ameliorate corneal inflammation in Hmox2−/− mice (472), and decreases LPS-induced expression of several proinflammatory cytokines in a human ex vivo model of inflammation (39).
Wegiel and colleagues (580, 582) proposed two possible signaling pathways by which biliverdin could attenuate inflammation. First, biliverdin may modulate IL-10 concentrations through a signaling cascade involving PI3K, activation of Akt, and downstream production of IL-10. Accordingly, binding of biliverdin to BVR on the cell surface of macrophages stimulates phosphorylation of BVR that allows it to bind PI3K and activate Akt, leading to IL-10 production (580). Second, biliverdin may affect the inflammatory response through inhibition of the transcription and expression of Toll-like receptor 4. Specifically, BVR becomes S-nitrosylated in response to biliverdin by eNOS-derived NO (582). This leads to the translocation of BVR to the nucleus, where it binds to the promoter of Toll-like receptor 4 and represses its expression. The pathways proposed by Wegiel and colleagues (580, 582) may help explain how biliverdin can influence cell function in addition to its role as an antioxidant (reviewed in sect. IVD). It is important to note, however, that the biliverdin concentrations used in these studies, i.e., 5–50 μM, are orders of magnitude higher than those present in the circulation in vivo.
It has also been proposed that the combined use of biliverdin and CO provides enhanced protection against injury as the two molecules may modulate different aspects of the inflammatory response. Thus, in a model of I/R injury in cardiac and renal transplantation, intraperitoneal administration of biliverdin (50 mg/kg) plus exposure to CO (20 ppm) increased recipient survival and decreased inflammation and tissue injury, while it increased the survival of recipients compared with monotherapy (CO or biliverdin alone) (382). Unfortunately, Nakao et al. (382) did not report circulating concentrations of biliverdin following intraperitoneal administration of the pigment, although they were likely in the low micromolar range judged by a recent study reporting the pharmacokinetics of biliverdin administered via this route (51). If so, intraperitoneal administration of biliverdin represents a pharmacological approach for the treatment of inflammatory conditions rather than reflecting endogenous formation of the pigment via the action of Hmox1/2.
4. Hmox2 and inflammation
Considerably less is known about the role of Hmox2 in inflammation. In a model of peritonitis, the number of inflammatory cells infiltrating the peritoneum was reported to be higher in Hmox2−/− mice compared with wild-type animals (472), suggesting that Hmox2 regulates the inflammatory response in some manner. With the use of a murine corneal injury model, Hmox2−/− mice also displayed aberrant wound healing and an exaggerated inflammatory response (32, 472). In that study, Hmox2−/− mice exhibited a delayed and more persistent response to corneal wound injury in terms of proinflammatory cytokines. Moreover, Hmox2−/− mice displayed increased neutrophil and macrophage infiltration compared with wild-type animals that was attenuated by treatment with biliverdin (32, 33). Compared with wild-type animals, peritoneal macrophages from Hmox2−/− mice display an altered more proinflammatory phenotype and decreased phagocytic activity (30). Finally, studies in cerebral microvascular EC indicate that overexpression of Hmox2 suppresses, while knockdown of Hmox1 increases, the expression of proinflammatory cytokines (68). Together, these results are consistent with Hmox2 minimizing persistent and chronic inflammation, although additional studies are required to substantiate such function of biliverdin.
B. Oxidative Stress
As indicated earlier, oxidative stress is implicated in a number of vascular diseases particularly atherosclerosis that has been the subject of previous extensive reviews (504), and hence will not be discussed further. The concept of “oxidative stress” in biological settings has been formulated in 1985 (483), and there has been considerable focus on the role of heme oxygenases, in particular Hmox1, as an adaptive mechanism to protect cells in situations of oxidative stress. Indeed, the early work of Keyse and Tyrrell (234) identified induction of Hmox1 as a cellular response to oxidative stress. Drawing from the significant body of work by Helmut Sies (480–484), oxidative stress has been defined initially as “the disturbance in the prooxidant-antioxidant balance in favor of the former,” and then redefined as “the imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage” (482). This definition underscores issues central to the accurate and appropriate studying of oxidative stress in biological systems. In particular, it reiterates that stress occurs from the imbalance of oxidants and antioxidants not just the presence of oxidants. This is important as oxidants, particularly hydrogen peroxide, play important physiological roles as signaling molecules and are not intrinsically deleterious. In addition, it highlights that oxidative challenge or loss of antioxidants per se does not establish oxidative stress nor is oxidative damage a necessary follow-on from oxidative challenge or loss of antioxidants. Adaptive cellular stress responses can compensate for or negate oxidative stress such that oxidative damage is not taking place. Hmox1 is part of this adaptive response and represents an evolutionarily conserved mechanism by which cells respond to various forms of stress, including oxidative stress.
1. Measuring oxidative stress
Measuring the occurrence of oxidative stress can principally be approached by determining changes in certain redox couple (e.g., oxidized/reduced glutathione, protein thiol redox state) or the concentration of reactive species (e.g., superoxide anion radical, hydrogen peroxide) and/or oxidized molecules (e.g., oxidized protein, lipid, DNA). Despite much advances in this field however, the accurate and specific measurement of oxidative stress in complex biological systems remains a challenge. Thus it is becoming increasingly clear that redox couples are highly regulated and subject to spatiotemporal changes (for a recent review, see Ref. 217). Similarly, the concentration of reactive species also changes in a spatiotemporal manner, adding to the challenge that reactive species are usually present at low concentrations and have short half-lives. There is a multitude of assays available to measure reactive species and that are based on the use of chemical and biological probes, electron spin resonance spectroscopy, and colorimetric assays, and many of these methods have been reviewed extensively (for the detection of reactive species in the vascular setting, see Refs. 111, 534). Importantly, all of these methods have limitations that need to be considered carefully when applied to complex biological systems. Probes such as dichlorodihydrofluorescein diacetate, dihydrorhodamine, and dihydroethidine are used most widely for evaluating the presence of oxidants as seen in the sections below. However, the use of these (and other fluorescent) probes has many caveats (reviewed in Refs. 332, 577, 595). In particular, such dyes are not selective and cannot be used to pinpoint changes in particular reactive species. For example, an increase in dichlorodihydrofluorescein diacetate-derived fluorescence alone does not provide evidence for an increase in reactive species. Therefore, the appropriate use of these probes must be carried out cautiously, with confounding issues factored into the experimental design and data analysis. Similarly, while markers of oxidative damage such as protein carbonyls, lipid oxidation products, protein, and DNA damage are often used in vivo, they are at best surrogate measurements of oxidative damage.
2. Hmox1 induction as a protective mechanism against oxidative stress
As indicated, early studies by Keyse and Tyrrell (233, 234) demonstrated that Hmox1 mRNA is induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, cadmium chloride, iodoacetamide, menadione, and sodium arsenite. These findings supported earlier studies (156, 182, 536) reporting the induction of Hmox1 in a variety of mammalian cells by chemical carcinogens, endotoxemia, hemin, thiol-reactive agents. Overall, these findings led to the proposal that induction of Hmox1 was part of the general response to oxidative stress, mediated primarily at the transcriptional level (232).
Hmox1-deficient cells have also provided key insights into the role of Hmox1 as a response to oxidative stress. Treatment of embryonic fibroblasts obtained from Hmox1−/− mice with hemin, hydrogen peroxide, the redox-cycling drug paraquat, or calcium chloride resulted in enhanced fluorescence derived from 2',7' dichlorodihydrofluorescein (a nonspecific measure of oxidative stress), and severely affected cell viability (427). While there are critical issues surrounding the use of 2',7' dichlorodihydrofluorescein as stated above, this initial finding led to the proposed role of Hmox1 as a mediator of stress resistance in vivo. More direct evidence in support of this proposal comes from a recent study reporting that red blood cells isolated freshly from nonstressed Hmox1−/− mice have a higher dimer-to-monomer ratio of peroxiredoxin 2 than corresponding cells from wild-type animals (144). Peroxiredoxin 2 is the major hydrogen peroxide-removing enzyme in red blood cells, and engagement of the enzyme in the metabolism of hydrogen peroxide results in an increase in its dimer-to-monomer ratio (27). Similar to these findings in mice, an Epstein-Barr virus-transformed lymphoblastoid cell line derived from the first human case of HMOX1 deficiency, displays high sensitivity to hemin with marked cell death compared with cells derived from control subjects that do not display damage under these conditions (605). Moreover, enhancement of cell death has recently also been reported in pluripotent stem cells deficient in Hmox1 and treated with an oxidant (308). Overall, these studies highlight the importance of Hmox1 in cell survival upon oxidant challenge.
Yeast cells lacking the yeast homolog of Hmox1 (Hmx1) also display enhanced sensitivity to oxidants (89), indicating that Hmox1-mediated protection against oxidant stress is an evolutionarily conserved mechanism. Deletion of HMX1 also led to the differential expression of 480 transcripts (89). In particular, decreased expression of genes encoding antioxidant enzymes such as γ-glutamylcysteine synthetase, glutathione peroxidase, catalase, and methionine sulfoxide reductase was observed that could explain the oxidant sensitivity of cells lacking Hmx1. There is increasing evidence that Hmox1 can localize to the nucleus and mediate cytoprotection (40, 89, 311). Interestingly, this ability of Hmox1 to protect cells may not require its enzymatic activity, but instead be mediated via activation of key oxidant-responsive transcription factors (40, 185, 311). Similarly, it has been suggested that Hmox1's ability to protect against oxidative stress may also not depend on the products derived from heme degradation, the roles of which are discussed below. In an early study, Epstein-Barr virus-transformed lymphoblastoid cells derived from a human case of HMOX1 deficiency transfected with H25A-Hmox1 which lacks enzymatic activity increased the expression of catalase, increased glutathione concentrations, and increased resistance to hydrogen peroxide treatment (185). This idea has been supported by Dennery and co-workers, who reported hypoxia to induce the formation of a COOH-terminal-truncated, enzymatically inactive form of Hmox1 to localize to the nucleus where it mediates the activation of transcription factors such as AP-1 and Nrf2 (40, 311). Perinuclear localization of Hmx1 was also observed in yeast cells exposed to hydrogen peroxide (89), indicating that nuclear targeting of Hmox1 may be an evolutionarily conserved mechanism by which cells respond to various stresses.
The exact mechanism by which a nuclear form of Hmox1 could affect gene transcription is still unknown. Direct DNA binding, binding to transcription factors, or transcription factor complexes have all been suggested as potential mechanism(s). In addition to the nucleus, translocation of Hmox1 has also been reported for other subcellular compartments, such as mitochondria (38) and caveolae (238). Mitochondria-localized Hmox1 has been reported to protect against apoptosis in gastric mucosal cells isolated from indomethacin-treated rats (38). These studies point to broader functions of Hmox1 in mediating cellular protection rather than through the actions of CO, biliverdin, and/or bilirubin.
3. Antioxidant protective activities of bile pigments
In terms of the mechanism by which Hmox1 alleviates oxidative stress, a focus has been placed on the antioxidant activities of biliverdin and bilirubin. As this topic has been reviewed previously (499), the following provides a short summary only. Both biliverdin and bilirubin are powerful oxidant scavengers in vitro (392, 393, 502, 503, 507, 508) (Table 5), although bilirubin has received more attention in terms of modulating oxidative stress. This is because in mammalian systems, biliverdin is rapidly converted to bilirubin so that the former usually does not accumulate in appreciable quantities. Several studies have shown the ability of bilirubin to inhibit various forms of macromolecule oxidation in vitro. For example, albumin-bound bilirubin can act synergistically with α-tocopherol (biologically the most active form of vitamin E) and consequently effectively inhibits low-density lipoprotein (LDL) lipid peroxidation (393) and possibly also inhibits membrane lipid oxidation. Bilirubin is also an effective inhibitor of protein oxidation (391, 392); a scavenger of hypochlorous acid (506), various reactive nitrogen species (225, 339), and chloramines (45); and an inhibitor of myeloperoxidase-mediated protein oxidation (45).
Table 5.
Reaction of (conjugated) bilirubin and biliverdin with different reactive oxygen and nitrogen species
| Reactive Species | Scavenging |
|---|---|
| Superoxide anion radical | No |
| Hydrogen peroxide, alkyl hydroperoxides | No |
| Singlet oxygen | Yes |
| Quinones | Yes |
| Peroxyl radicals | Yes |
| α-Tocopheroxyl radical | Yes |
| Mixed function oxidase/hydroxyl radical | Yes |
| Hypochlorous acid | Yes |
| Peroxynitrite | Yes |
| Nitroxyl radical | Yes |
[Adapted from Stocker (499).]
While biliverdin and bilirubin possess antioxidant activity in various in vitro systems, their antioxidant role in vivo is less clear. Several cell culture models have been used in an attempt to clarify the role of bilirubin as a cellular antioxidant. When added to culture medium at micromolar concentrations, bilirubin protects EC, VSMC, and cardiomyocytes from oxidant-induced toxicity (83, 372, 600). However, the supraphysiological concentrations of bilirubin used in these studies complicate their interpretation in a pathophysiological context. In a series of high-profile publications, Snyder and co-workers reported that even submicromolar concentrations of bilirubin protected cells in culture against the toxic effects of high micromolar concentrations of hydrogen peroxide (23, 115, 470). It was proposed that by acting as an antioxidant, bilirubin itself becomes oxidized to biliverdin and then recycled to bilirubin by BVR (470), with this bilirubin-biliverdin antioxidant cycle amplifying the antioxidant activity of bilirubin and providing cellular protection. However, this bilirubin-biliverdin recycling hypothesis of cellular antioxidant protection is highly improbable (353). This is because a prerequisite for this proposed antioxidant redox cycle is the quantitative (stoichiometric), or near quantitative, oxidation of bilirubin to biliverdin, yet there is no physiologically relevant chemical precedence in the literature for such quantitative oxidation of bilirubin to biliverdin (353). Moreover, decreasing BVR protein and activity in HeLa cells using RNA interference has been reported to be without effect on hydrogen peroxide-mediated cell death, just as BVR overexpression failed to enhance protection of cells against hydrogen peroxide-mediated damage, irrespective of whether bilirubin or biliverdin were added to the cells as substrate for the putative redox cycle (333). Therefore, the physiological importance and mechanism by which endogenously formed biliverdin/bilirubin provide antioxidant protection to cells remains to be established unambiguously.
4. Antioxidant activities of CO
CO has also been studied in the context of preventing oxidative damage though to a lesser extent than bilirubin. In contrast to bilirubin (and NO), CO is an inert molecule and does not directly engage in reactions with oxidants. Therefore, the reported antioxidant activity of CO is most likely due to indirect actions of the gaseous molecule. In one study, CO was reported to prevent ethanol-elicited hepatic oxidative damage and inflammatory stress through activation of the p38 MAPK pathway (295). In another study, CO was observed to block JNK phosphorylation and apoptosis induced by the redox-cycling and superoxide-generating drug menadione via decreased expression of the caspase-9, -6, and -3 (90). In addition to this, CO treatment was also shown to protect astrocytes against tert-butylhydroperoxide-induced apoptosis through a Bcl-2-dependent pathway that was associated with improved oxidative phosphorylation (11). The authors concluded that CO protects astrocytes against oxidative stress-induced apoptosis by improving metabolism, particularly mitochondrial oxidative phosphorylation. Similarly, CO has been suggested to protect against damage through modulating mitochondrial function in humans (440). In that study, CO inhalation during exercise increased expression of superoxide dismutase 2 and genes involved in mitochondrial biogenesis (440). Indeed, Piantadosi and co-workers (440) have shown that CO can regulate the expression of transcription factors that are activated in conditions of oxidative stress and/or are under redox control, and that this ultimately leads to the induction of genes involved in mitochondrial biogenesis, and mitochondria-specific and cellular repair mechanism (for a recent review, see Ref. 24). The link between Hmox1/CO, oxidative stress, and mitochondrial biogenesis has also been explored in a cardiovascular setting in relation to doxorubicin toxicity. Doxorubicin is a frequently used anthracycline anticancer drug whose clinical use is problematic due to associated toxic cardiomyopathy, mitochondrial damage, and apoptosis. In mice, CO exposure was shown to prevent doxorubicin-mediated cardiomyopathy through the induction of pathways controlling mitochondrial biogenesis, oxidative stress responses, and apoptosis (514). Moreover, cardiac-specific overexpression of Hmox1 protects mice against doxorubicin-induced cardiomyopathy and related mitochondrial derangement (193). These findings have led to the suggestion that overexpression of Hmox1 or CO treatment may play a novel role in protecting the heart from oxidative injury by regulating mitochondrial quality control.
5. Hmox2 and oxidative stress
Compared with Hmox1, Hmox2 has received less attention in the context of oxidative stress. Despite this however, Hmox2 has been indicated to be important in the response to oxidative stress in various cell types. Of note, Hmox2−/− mice display heightened sensitivity in a hyperoxia-induced model of lung injury, associated with increased protein carbonyl and decreased glutathione contents (108). These results were hypothesized to be due to accumulation of iron in the lungs of the stressed Hmox2−/− mice, with Hmox2 proposed to be important in iron turnover during oxidative stress. Similarly, inhibition of Hmox2 expression by small interfering RNA was reported to increase oxidative stress and apoptosis in EC exposed to hypoxia, conditions that led to a decrease in cellular Hmox1 (174). Together, these studies indicate that Hmox2 is important in maintaining cell viability and minimizing oxidative stress during changes in oxygen concentration, consistent with the proposed role of Hmox2 in oxygen sensing. Moreover, aortic EC isolated from Hmox2−/− mice express more gp91phox/Nox2 and display increased hydroethidine-derived fluorescence compared with EC from wild-type animals (31), suggesting that Hmox2-deficient EC experience greater oxidative stress in form of increased Nox2-derived superoxide.
C. Iron Metabolism
Iron is an essential element and an absolute necessity for all living organisms. Iron has an important ability to exist in two redox states and is able to readily accept and donate electrons interconverting between ferric (Fe3+) and ferrous (Fe2+) states (305). This characteristic substantiates its involvement as a cofactor in a multitude of diverse molecular and biological activities that range from being an essential component of hemoglobin and myoglobin to taking part in the electron transport chain through cytochromes in the mitochondria (305). The very property that makes iron indispensable for life also has the potential to pose a serious threat for the living organism. Once iron exceeds the metabolic needs of the cell, it may form a low-molecular-weight pool, otherwise referred to as the labile iron pool that can be readily involved in generation of potent oxidants with potential hazardous effects that can damage proteins, lipid membranes, and even nucleic acids (167). Given such deleterious potential and the fact that humans do not possess an effective pathway for excretion of iron, multiple other mechanisms have evolved to pedantically control the level of bioavailable iron both at systemic and cellular level (177).
1. Iron metabolism in atherosclerosis
The pro-oxidant nature of iron has been implicated in the pathogenesis of many vascular disorders (424, 585). While under physiological conditions free iron in circulation is almost nonexistent, heme can provide an alternative source of redox-active iron. In fact, compared with other sites in the body, the vascular endothelium is more prone to heme exposure due to the high content of heme in the form of hemoglobin within red blood cells. Red blood cells are susceptible to lysis that leads to the release of hemoglobin that may then become oxidized in the extracellular space. Oxidized hemoglobin (referred to as methemoglobin) contains the ferric form of iron that facilitates the release of heme moiety (52). The hazardous properties of free heme are well recognized (20). Within the circulation heme can cause toxicity to EC and also has the potential to oxidize LDL (20). Oxidation of LDL occurs in the process of atherosclerosis (504). Indeed, the most detailed studied aspect of iron within the context of vascular diseases is atherosclerosis. The initial “iron hypothesis” was proposed by Sullivan in 1981 where he suggested that the higher incidence of heart disease among men and postmenopausal women compared with premenopausal women may be linked to higher level of stored iron and hence recommended blood donation as a preventive measure (515). Failure of hormone replacement therapy to reduce the risk in postmenopausal women further supported this hypothesis (194). Given the enormity of morbidity and mortality associated with CVD and atherosclerosis, the hypothesis generated great enthusiasm among investigators and numerous studies in cellular and animal models as well as epidemiological and clinical studies have been conducted to further explore the “iron hypothesis” (557). Based on this hypothesis, patients with iron accumulation should have higher incidence of CVD. However, as discussed in more detail below, a direct causative role of iron in the pathogenesis of atherosclerosis remains to be validated despite rigorous research.
At the cellular level, there is overwhelming evidence that excess iron is detrimental given that it acts as a major catalyst for generation of potentially toxic oxidants (424). However, animal models of atherosclerosis have not provided convincing evidence to support such a role (499). For instance, while Lee et al. (279) reported substantial decrease in atherosclerotic lesions of apolipoprotein E gene-deficient (Apoe−/−) mice fed a low-iron diet, others (250) have reported that iron overload too mitigates atherosclerosis in Apoe−/− mice. Similarly, administration of heme decreases rather than increases atherosclerosis in LDL receptor gene knockout mice (202). Human studies have also stirred further controversy in this regard. For instance, several studies reported that when adjusted for other factors, body stores of iron (mainly estimated by measuring levels of serum iron, serum ferritin, and transferrin saturation rates) positively correlate with coronary artery disease and ensuing complications (6, 184, 273, 436, 458, 552). In contrast, there are a number of studies that have failed to find such an association (145, 341, 492). These controversial observations are also repeated by studying patients suffering from hemochromatosis wherein these patients do not display higher incidence of atherosclerosis (reviewed in Ref. 557).
Notably, iron is increased in atherosclerotic lesions compared with normal arterial walls (516). Several studies demonstrated that much of the iron within plaques is associated with macrophages and foam cells (557). Our understanding of macrophage plasticity and polarization status has markedly changed in the past few years. We are now aware that the iron handling machinery of the macrophages is heavily dependent on its polarization status (92, 148). The key role of macrophages in the process of atherogenesis has also been elegantly described by multiple studies (365). Deletion of macrophages using a CD-11b/diphtheria toxin receptor is protective against formation of atherosclerotic plaques (511). Another aspect of this pathway is the contribution of ferritin. Ferritin expression is increased in atherosclerotic plaques (414), and furthermore, new evidence suggests that ferritin may play a key role in activation and polarization of macrophages (43). Therefore, the observed increase in the expression of Hmox1 (572) and ferritin within the atherosclerotic lesions may be an adaptive response that is intended to minimize the progression of the disease. Taken together, while there is convincing evidence to support a deleterious effect for iron in the context of generation of oxidative stress, experimental findings to explore the role of iron in atherosclerosis have been inconclusive. This may be due to several factors, including the complex regulation of iron homeostasis that entails multiple proteins such as ferritin, and the potential effects that expression of these proteins may have during various stages of atherosclerosis. Hence, further investigation into the role of iron/ferritin induced macrophage activation and how that may modulate the initiation and progression of atherosclerosis merit consideration.
2. Iron metabolism in other CVD
Free iron is also a significant risk factor in I/R-mediated injury. The main source of iron during I/R is through cellular injury and release of intracellular heme from heme proteins. The injurious potential of iron in this setting has been highlighted by administration of iron chelators markedly reducing the level of tissue necrosis after reperfusion (175). Another clinically relevant area of iron-induced cardiovascular damage is restrictive (infiltrative) cardiomyopathy. This is an irreversible and devastating condition that is a frequent finding in patients suffering from iron overload states. This clinical entity results from accumulation of iron that can lead to stiffness of the walls of the ventricles resulting in significant resistance against normal filling with blood during diastole. In fact, patients with mutations in the human hemochromatosis protein (also known as HFE) that lead to systemic iron overload, have significantly higher risk of cardiomyopathy when compared with the age- and sex-matched population without mutations in human hemochromatosis protein (394).
D. Regulation of Growth of Vascular Cells
In addition to playing a role in vasculogenesis and angiogenesis during fetal development, Hmox1 has been implicated in adult neovascularization and angiogenesis following tissue injury such as ischemia and wounding or in diseases such as cancer. Following vascular injury, the ability to adequately revascularize is paramount in tissue healing. Data to date suggest that expression of Hmox1, likely via its products CO and biliverdin/bilirubin, influence angiogenesis through various mechanisms. A correlation between Hmox1 expression and angiogenesis has been suggested in various cancers (170, 396, 520, 542), with several human and murine tumors expressing high levels of Hmox1 (159, 335). Pharmacological inhibition of Hmox1 has been shown to reduce tumor growth in murine cancer models (132, 181, 525), leading to Hmox1 inhibitors such as pegylated or micellar zinc protoporphyrin (132, 133, 206) suggested as a potential chemotherapeutics.
1. Hmox1 and VEGF-mediated angiogenesis
The important role of Hmox1 in the growth of vascular cells was observed in early experiments where overexpression of Hmox1 increased coronary microvascular EC proliferation and formation of capillary-like structures in a two-dimensional Matrigel assay (109). Numerous studies subsequently underscored the role of Hmox1 in both vasculogenesis and angiogenesis through its effects on pro-angiogenic factors such as VEGF, stromal cell-derived factor 1 (SDF-1), and anti-angiogenic factors such as sFlt1 and sEng. Consistent with these observations, Hmox1−/− mice and cells treated with Hmox1 inhibitors show decreased production of VEGF (82), while the formation of sFlt1 and circulating concentrations of sEng are elevated compared with relevant controls (99, 100). Moreover, Hmox1 has been implicated in the synthesis of VEGF in EC (120, 218, 219). This points to the expression of Hmox1 and VEGF in EC resulting in a positive-feedback loop in which VEGF increases Hmox1 expression that in turn augments VEGF synthesis. Furthermore, aortic rings from Hmox1−/− mice are unable to form capillary sprouts ex vivo in response to the chemokine SDF-1 that itself activates Hmox1 expression (110). It has been hypothesized that CO mediates the effects of Hmox1 on VEGF and SDF-1, as the gaseous molecule itself elicits VEGF synthesis (76, 219). CO is also thought to enhance EC migration after injury by involving the regulation of the nuclear receptor Rev-erbα and chromatin remodeling (285). The authors suggested that CO represses the activity of the Rev-erbα-associated co-repressor histone deacetylase 3, thereby enhancing EC migration by enabling transcription of pro-angiogenic genes (such as vascular cell adhesion molecule 1) and inhibiting anti-angiogenic genes (e.g., thrombospondin). Indeed, it has been proposed that Hmox1 plays a unique role in modulating VEGF-driven and inflammation-independent angiogenesis: the enzyme increases VEGF-driven angiogenesis and at the same time inhibits leukocyte migration and inflammation at sites of injury (55, 56). As a corollary, these results suggest that Hmox1 deficiency may promote inflammation-driven angiogenesis. Additionally, it has been shown that activation of the Hmox1 via genetic modulation or chemical induction leads to a significant decrement in the concentration of certain micro-RNAs as well as an increase in SDF-1 (261). Moreover, SDF-1 was found to be a major mediator of Hmox1 activity (Figure 7) and such effects warrant further investigations in the context of angiogenesis.
FIGURE 7.
Interaction of stromal derived factor-1 (SDF-1), Hmox1, and endothelial nitric oxide synthase (eNOS) in response to vascular injury. Vascular injury, e.g., wounding or ischemia (110), results in the release of SDF-1 and subsequent induction of the phosphatidylinositol-4,5-bisphosphate 3-kinase/RAC-alpha serine/threonine-protein kinase/eNOS (PI3K/Akt/eNOS) pathway in bone marrow stromal cells. NO and Hmox1 (309, 598, 601) are critical for the mobilization of endothelial progenitor cells (EPC) from the bone marrow to the circulation. In circulating EPC, Hmox1 and eNOS release CO and NO, respectively, which induce phosphorylation of vasodilator-stimulated phosphoprotein (VASP) and its redistribution to the leading edge of the cells (282). This promotes migration and vascular repair. In addition, there is also evidence suggesting that Hmox1 regulates SDF-1 expression (309, 601).
2. Hmox2 and repair after injury
Using a mouse corneal injury model, Schwartzman and co-workers (32, 472) reported in vivo deletion of Hmox2 to markedly impair the inflammatory and reparative response of the cornea to injury, leading to unresolved corneal inflammation and chronic inflammatory complications, including ulceration, perforation, and exacerbated neovascularization (32, 472). Interestingly, Hmox2 deficiency was also reported to be associated with impaired injury-mediated Hmox1 induction (472), indicating a link between Hmox2 and Hmox1 induction. Mechanistic studies by the same group revealed that Hmox2 deletion transforms EC from a “normal” to an “activated” phenotype that is characterized by increases in inflammatory, oxidative, and angiogenic factors (31). Specifically, aortic EC derived from Hmox2−/− mice on a mixed C57BL/6 × 129/Sv genetic background showed higher expression of VEGF receptor 1, a marked angiogenic response, and increased expression of NOX2 compared with wild-type cells obtained from a separate line of mice (31). Moreover, treatment with biliverdin (10 μM) ameliorated the angiogenic activity of Hmox2−/− cells (31), suggesting that normal Hmox activity is essential for appropriate neovascularization post-injury. The delayed wound healing response caused by Hmox2 deficiency was confirmed recently in a cutaneous excisional wound model (324). In that study, however, Hmox2−/− mice demonstrated decreased vascularization and reduced collagen deposition independent from inflammation and Hmox1 expression (324). Rather, in these studies differences in the expression of the cytokine CXCL-11 were observed between Hmox2−/− and wild-type mice during wound repair (324). Overall, these studies provide strong evidence that Hmox2 plays an important role in normal wound healing response, including epithelial cell proliferation and neovascularization, although the underlying mechanisms remain to be confirmed, and this may require the use of littermate Hmox2−/− and Hmox2+/+ mice with more limited genetic variance.
V. HMOX IN VASCULAR DISEASE
A. Atherosclerosis
Atherosclerosis is a chronic disease of arteries that results in the formation of plaques or lesions in the arterial wall that contain intra- and extracellular lipid deposits, inflammatory cells, VSMC, and secreted products such as collagen and elastin (Figure 8) (302, 303). Atherosclerosis represents a state of heightened inflammation and oxidative stress, yet definitive evidence that inflammation and uncompensated oxidative stress, such as that leading to LDL oxidation, contribute to and cause human disease remains to be established (see Refs. 303, 504 for reviews). What is clear is that in early atherogenesis the monolayer of EC that lines the inner surface of arteries becomes functionally altered or “activated.” Such “activation” takes place predominantly at sites of the arterial tree where the hemodynamics of the pulsatile blood flow is such that the endothelium-protective laminar shear stress is interrupted. At a functional level, the “activated” endothelium is characterized by the expression of adhesion molecules that capture circulating leukocytes, increased endothelial dysfunction (generally defined as a decrease in bioavailable NO), increased endothelial permeability, and changes to the subendothelial extracellular matrix (303, 504). These changes lead to the entry and retention of LDL and inflammatory cells, mostly monocytes, within the arterial wall. Once resident within the artery, monocytes differentiate to macrophages that engulf lipoproteins and become lipid-laden cells, referred to as foam cells. Atherosclerotic lesions also contain VSMC that migrate from the media into the neointima, where they proliferate, secrete extracellular matrix, and form a fibrous cap that covers the lesion. Typically, the fibrous cap overlays areas rich in foam cells, some of which undergo apoptosis that results in the release and extracellular accumulation of lipid and cell debris, giving rise to the necrotic core of atherosclerotic plaques (303). Over time, such plaques cause clinical manifestations by limiting blood flow giving rise to ischemia, or by provoking the formation of a thrombus that can cause abrupt local interruption of blood flow. Thrombi are thought to commonly arise as a result of physical “rupture” of thin, collagen-poor fibrous caps with abundant macrophages in close proximity (303).
FIGURE 8.
Potential modulatory roles of Hmox1 in atherogenesis. Hmox1 expression in macrophages, foam cells, EC, and VSMC has been associated with decreases in EC injury; EC dysfunction; expression of the adhesion molecules vascular cell adhesion molecule 1 (VCAM-1) and intracellular adhesion molecule 1 (ICAM-1); release of the inflammatory cytokines monocyte chemoattractant protein-1 (MCP-1), IL-1β, TNF-α, and IL-6; and decrease in oxidative damage. In addition, increased Hmox1 expression has been reported to increase the stability of atherosclerotic lesions.
1. Hmox1 protects against experimental atherosclerosis
There is substantial evidence indicating that Hmox1 plays a protective role in atherosclerosis via multiple processes. Hmox1 expression was observed first in atherosclerotic lesions of Apoe−/− mice, and fatty streaks and fibrous plaques were noted in the aorta of a 6-yr-old boy with HMOX1 deficiency (227, 572). Prior to these important in vivo findings, it was shown that LDL oxidized by exposure to hemin plus hydrogen peroxide or by copper induces Hmox1 expression in EC and epithelial cells in vitro (5). In lesions of Apoe−/− mice, immunostaining of Hmox1 was detected in EC, macrophage-derived foam cells, and medial VSMC. In human lesions, Hmox1 was detected in intimal macrophages and medial VSMC. In both models, Hmox1 was not detected in sections of normal/healthy arteries. Following this early study, Hmox1 expression has been confirmed in macrophages, foam cells, EC, and VSMC of atherosclerotic lesions in animal models and humans (74, 201, 202, 297, 371, 383, 493). The expression of Hmox1 may have clinical relevance for atherosclerosis and CVD outcome in humans, as judged by genetic analyses. Thus polymorphisms that are known to affect HMOX1 promoter activity, such as (GT)n repeats (259, 263, 301, 417) and the T(-413)A single nucleotide polymorphism (59), have identified an association between decreased HMOX1 promoter activity and increased atherosclerotic disease progression in CVD patients.
There is strong evidence for Hmox1 protecting against the development of atherosclerosis in murine and rabbit models of hyperlipidemia-mediated disease. Pharmacological studies show that induction of Hmox1 expression attenuates atherosclerotic lesion formation in LDL-receptor gene knockout (Ldlr−/−) mice, while treatment of these mice with an Hmox1 inhibitor (tin protoporphyrin) increases lesion size (202). Similar results have been observed in Watanabe heritable hyperlipidemic rabbits (201) that lack a functional LDL receptor. Moreover, genetic manipulation of Hmox1 reiterates the findings from pharmacological studies. Specifically, hypercholesterolemic Apoe−/− mice develop larger lesions when also deficient in Hmox1 (614), while adenovirus-mediated overexpression of Hmox1 decreases lesion formation in Apoe−/− mice (220). As in these models, enzymatically active Hmox1 is expressed predominantly in lesion macrophages (383), Orozco et al. (405) examined whether macrophage Hmox1 expression constitutes a significant component of the protective role in atherosclerosis. They observed atherosclerotic lesions in lethally irradiated Ldlr−/− mice reconstituted with bone marrow from Hmox1−/− mice to exhibit greater macrophage content compared with animals receiving bone marrow from wild-type mice, although the size of the lesions were comparable. These findings suggest that Hmox1 expression in lesion macrophages decreases the pro-inflammatory milieu within the artery wall. In advanced (but not early) human atherosclerotic lesions, heme oxygenase activity is also detectable in EC, together with enhanced VEGF expression (371), although the functional implication of this is not clear.
2. Hmox1 and atherosclerotic plaque stability
In addition to Hmox1 modulating the progression of experimental atherosclerosis, Hmox1 has been implicated in modulating atherosclerotic plaque stability. As indicated earlier, the stability of atherosclerotic plaques is considered an important determinant of plaque rupture that gives rise to acute vascular occlusion and clinical sequelae. Using biopsy samples from patients with carotid artery disease, Cheng et al. (74) observed Hmox1 expression to correlate with features of plaque vulnerable to rupture, such as increased macrophages and lipid contents, and decreased VSMC and collagen contents. Likewise, in a mouse model of vulnerable plaque (Apoe−/− mice implanted with a carotid cast), Hmox1 expression was higher in lesions with a vulnerable phenotype compared with stable lesions (74). Induction of Hmox1 expression by treatment with copper protoporphyrin or adenoviral overexpression of Hmox1 increased intimal VSMC and fibrous cap thickness while it decreased the necrotic core-to-intima ratio and lipid accumulation in the lesion (74). These features of stable plaques were reversed in animals treated with the Hmox1 inhibitor zinc protoporphyrin (74). In a second animal study, vulnerable lesions were induced by local transfection of recombinant p53 adenovirus into rabbit plaques (290). As in the murine model, induction of Hmox1 (by hemin treatment) increased fibrous cap thickness and decreased the intima-to-media ratio, and prevented plaque rupture (290). While these studies support the notion that Hmox1 expression stabilizes plaque composition, directing lesions towards a more stable phenotype, the underlying mechanism remains to be elucidated. It is also important to note that to date, there is no single established animal model of plaque rupture so that care needs to be taken when attempting to extrapolate the above findings in model systems to the human situation.
3. Hmox1 and endothelial cell activation
In vitro studies show that pretreatment of vascular EC with heme arginate to induce Hmox1 attenuates endothelial dysfunction and the pro-inflammatory response to subsequent exposure to TNF-α, including the expression of vascular cell adhesion molecule 1, MCP-1, and macrophage stimulating factor (226). Interestingly, this protective activity was observed in cells treated with bilirubin but not with CO-releasing molecule 2 (226). Taking a systems-level approach, Romanoski et al. (446) reported HMOX1 expression in EC to be a critical modulator of cellular transcriptional response of human aortic EC to autoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine. Conversely, HMOX1 silencing increased induction of proinflammatory cytokines (446). The increase in Hmox1 induced by autoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine was also associated with increased expression of the genes encoding the LDL receptor and insulin-induced gene 1 (446), suggesting that endothelial Hmox1 may modulate sterol regulation on treatment with autoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine.
Further evidence for Hmox1 expression attenuating EC activation comes from the early study of Ishikawa et al. (202). They reported induction of Hmox1 in Lldr−/− mice to increase NO in a process that was inhibited by treatment of the animals with tin protoporphyrin (202). Consistent with these findings, Hmox1 induction has been observed to reverse impaired endothelium-dependent relaxation of thoracic aortic rings from Lldr−/− mice (226). These effects were reproduced by bilirubin (not CO pretreatment), suggesting a distinct role for bilirubin in modulating endothelial function in murine models of atherosclerosis.
4. Serum bilirubin and CVD
As alluded to, among the products arising during the degradation of heme, bilirubin has been most studied in the context of atherosclerosis and associated CVD. Numerous studies have reported mildly elevated concentrations of serum bilirubin to strongly associate with a decreased prevalence of chronic diseases, particularly CVD (for recent reviews, see Refs. 63, 566). Similarly, elevated serum total bilirubin associates with a decrease in inflammatory markers (10), suggestive of an anti-inflammatory activity alongside its known antioxidant activities (reviewed in Ref. 499). While these associations are consistent with the notion that bilirubin protects against CVD, they do not establish such a causal relationship. Importantly, recent genome-wide association studies and a conditional linkage study indicate that UGT1A1 (UDP-glucuronosyltransferase 1) rather than HMOX1/2 is the major gene controlling serum bilirubin concentrations in humans (reviewed in Ref. 310). UDP-glucuronosyltransferase 1 is found primarily in liver cells where bilirubin glucuronidation takes place. The resulting conjugated bilirubin is secreted from the liver into bile that becomes eventually excreted with solid waste via the intestinal tract. Therefore, a change in UDP-glucuronosyltransferase 1 activity will simultaneously (and inversely) change the concentration of bilirubin in serum and bile, and the latter could also impact on atherosclerosis and associated diseases. For example, bile and conjugated bilirubin are important regulators of dietary fat absorption and the gut microbiota (139), both of which impact on atherosclerosis (576). Within the circulation and the arterial vasculature, elevated concentrations of bilirubin may have beneficial anti-inflammatory activities. Thus biliverdin and bilirubin attenuate monocyte chemotaxis (199) and blunt TNF-α induced endothelial expression of adhesion molecule expression and neutrophil adhesion to EC (348).
B. Intimal Hyperplasia
Intimal hyperplasia arises from vascular injury subsequent to revascularization procedures such as angioplasty, stenting, as well as pulmonary hypertension, bypass grafting, and transplantation (see sect. IVC). The pathophysiology of intimal hyperplasia is complex (reviewed in Refs. 158, 363, 416). It involves leucocyte-endothelial interactions that trigger the coagulation cascade leading to the local formation of thrombus. This is followed by phenotypic changes of medial SMC causing their migration to, and proliferation in, the intima. The excessive intimal proliferation of VSMC and associated deposition of extracellular matrix in the lumen of the vessel wall gives rise to lesions and narrowing of the vessel lumen (or restenosis), reduced blood flow, and potentially vessel occlusion. In addition to intimal hyperplasia, arterial remodeling contributes significantly to the extent of restenosis. Clinically, intimal hyperplasia can limit the success of therapeutic interventions, and this has led to substantial interest in pathways or processes that may be targeted to protect vessels from intimal hyperplasia subsequent to injury.
1. Hmox1 and intimal hyperplasia
Hmox1 as a potential modulator of intimal hyperplasia was reported first in a rat balloon injury model in tandem with pharmacological induction and inhibition of Hmox1 by hemin and tin protoporphyrin, respectively (9). Treatment with hemin prior to and after balloon injury increased serum bilirubin and Hmox1 expression, while it decreased neointima formation, all of which could be reversed by treatment with tin protoporphyrin. The ability of increased Hmox1 to mitigate experimental neointima formation after balloon injury was subsequently reported in multiple studies using similar pharmacological agents (540, 549, 550). Consistent with the above findings, Hmox1−/− mice form larger lesions after femoral artery wire injury compared with wild-type mice (117). Furthermore, adenovirus-mediated overexpression of Hmox1 decreases the neointima-to-media area ratio in a rat carotid injury (548) and a porcine balloon-injury model (117). Finally, increased Hmox1 may also protect humans against restenosis as judged by an HMOX1 promoter microsatellite polymorphism study (130). Thus, 6 mo after balloon dilation in the femoropopliteal segment, the extent of restenosis was observed to be decreased in patients with short (GT)n repeat alleles (that normally lead to increased Hmox1 expression) compared with patients with long (GT)n repeat alleles (130).
Studies have also been performed to examine whether induction of Hmox1 could improve rates of in-stent stenosis. For instance, rabbit and rat iliac arteries were stented, and the role of Hmox1 activation via hemin or probucol administration was examined. Results showed that induction of Hmox1 was accompanied by a marked decrement in cellular processes that commonly lead to in-stent stenosis such as inflammation, apoptosis, and cellular proliferation, while at the same time it promoted in-stent re-endothelialization (195, 530). Moreover, CO donor application was able to mimic these findings highlighting the central role of CO in this regard (195). Also, the antiproliferative effects of drugs coating stents such rapamycin and paclitaxel have been attributed to Hmox1 (78, 558). Both of these drugs are able to stimulate Hmox1 induction in a dose-dependent manner, and more importantly, inhibition of heme oxygenase reverses their inhibitory effects on VSMC proliferation (78, 558).
Mechanistically, evidence points towards Hmox1 expression in VSMC as being important in the ability of Hmox1 to inhibit intimal hyperplasia. This is because VSMC from Hmox1−/− mice proliferate more rapidly when exposed to serum than VSMC from wild-type mice (409), while the disease-inhibitory activity of Hmox1 overexpression is associated with decreased VSMC proliferation (117). Moreover, in a rat carotid balloon-injury model, Hmox1 protein expression following injury was observed initially in the media and then in the intima where it remained (540), i.e., analogous to the spatiotemporal movement of VSMC after injury. Inhibition of intimal hyperplasia by pharmacological induction of Hmox1 with probucol increases VSMC expression of Hmox1 as well as YY1 (28), a transcription factor that regulates the expression of genes important for cell growth and differentiation. Cellular studies confirmed YY1 to be downstream of Hmox1 induction, with Hmox1-derived CO implicated as the mediator (28). Moreover, RNA interference knockdown of YY1 prevents probucol or adeno-Hmox1 from inhibiting VSMC proliferation in vitro and intimal hyperplasia in vivo (Figure 9) (28). Overall, these results support an important role for Hmox1 and YY1 in VSMC in the inhibition of intimal hyperplasia.
FIGURE 9.
Probucol (PB) inhibits injury-induced intimal hyperplasia, and this is prevented by Yin Yang 1 (YY-1) knockdown. Carotid arteries obtained from Zucker rats fed normal food without (Ctrl) or with 1% PB for 14 days before and 14 days after balloon injury and receiving YY1 siRNA or YY-1 scrambled RNA (scRNA) immediately before injury. Injury was assessed by intima-to-media- ratio. [From Beck et al. (28).]
2. Antiproliferative mechanisms of Hmox1 on VSMC
As we have learned in the preceding sections, expression of Hmox1 decreases VSMC proliferation, and this is thought to represent a key pathway in the inhibition of intimal hyperplasia by Hmox1. Mechanistically, Hmox1 has been reported to inhibit VSMC proliferation via several pathways, including effects on cell cycle progression, apoptosis, and p38 MAPK signaling through cGMP (409). The effect of Hmox1 on cGMP is thought to be independent of NO (409). Increased Hmox1 expression stimulates apoptosis while it inhibits DNA replication in medial cells (548), just as it attenuates DNA synthesis and cell proliferation in primary VSMC (117). Specifically, increased Hmox1 expression enhances G1/G0 arrest and VSMC from Hmox1−/− mice show increased G1/S transition compared with wild-type mice after wire injury, while the expression of the G1 cyclin-dependent kinase inhibitor p21Cip1 is decreased (117). It is possible that these findings relate to the observation that adenoviral overexpression or pharmacological induction of Hmox1 inhibits VSMC proliferation in vitro via induction of YY1 (28), as YY1 itself attenuates VSMC proliferation and intimal hyperplasia via modulation of p21Cip1 (461). In any case, it would be interesting to test whether VSMC-specific overexpression of Hmox1 and associated attenuation of VSMC growth is sufficient for inhibition of intimal hyperplasia in vivo.
3. Antiproliferative mechanisms of CO on VSMC
Among the Hmox1-derived products, CO has been identified as a potential driver of regulating VSMC proliferation. Thus CO administration (250 ppm) for 2 days prior to balloon injury has been reported to suppress intimal hyperplasia in rodent arterial-injury models (409). In that study, CO administration increased the expression of p21Cip1 in VSMC in vitro, although the suppression of intimal hyperplasia by CO was not dependent on changes to p21Cip1, an observation similar to that reported by Tulis et al. (551). However, Otterbein et al. (409) observed CO to increase cGMP, in line with previous observations that linked the antiproliferative effects of CO to cGMP in a distinct and NO-independent manner (540). Based on in vitro studies, Otterbein et al. (409) postulated that similar to NO, CO increases cGMP and that this leads to activation of p38 MAPK and cGMP-dependent protein kinases such as protein kinases G1α and G1β (409). Kim et al. (239) subsequently showed that the decrease in intimal hyperplasia observed in a rat carotid artery balloon injury model with CO treatment (250 ppm, 2 h before surgery or during the first 24 h post-surgery) depended on increased cGMP and stimulation of p38 MAPK. Moreover, CO increased the expression of caveolin-1 in neointimal lesions, and genetic depletion of caveolin-1 prevented the antiproliferative effects of CO (239). The authors concluded that CO-mediated activation of p38β MAPK increases caveolin-1, which then acts as a tumor suppressor protein mediating the growth-inhibitory properties of the gas. Infusing the external rat carotid artery with phosphate-buffered saline containing 875 μM CO for 30 min immediately after balloon injury, Tulis et al. (551) reported the gas-mediated decrease in neointima formation to be associated with decrease in the arterial expression of the G1 cyclins A and E, which are critical for cell entry and progression through S phase. The above studies suggest that exogenous CO inhibits intimal VSMC proliferation and hyperplasia likely by various mechanisms that may depend on the route and concentration of the administered gas. While these findings have potential therapeutic implications, the relationship between exogenously administered CO with that produced as a result of Hmox1/2 activity in the prevention of intimal hyperplasia, however, remains to be established.
4. Antiproliferative effects of biliverdin and bilirubin on VSMC
Several studies implicate biliverdin and bilirubin in the antiproliferative action of Hmox1 on VSMC proliferation following vascular injury (381, 401, 422). Compared with wild-type rats, hyperbilirubinemic Gunn rats show minimal intimal hyperplasia in response to injury, and intraperitoneal administration of biliverdin (50 μmol/kg) before balloon injury, or short local exposure before and after injury, significantly suppresses intimal hyperplasia (381, 401). Consistent with these findings, treatment of primary rat and mouse VSMC with biliverdin or bilirubin (10–500 μM) decreases their proliferation through cell cycle arrest and inhibition of the phosphorylation of retinoblastoma protein and decreased levels of cyclins A, D1, and E. In contrast to exogenous CO, bilirubin treatment was observed to inhibit p38 MAPK (401). Interestingly, in vitro treatment of VSMC with bilirubin (200 μM) was reported to decrease the expression of YY1 (401) that otherwise has been reported to decrease VSMC proliferation and intimal hyperplasia (28, 461). More recently, the effect of local bilirubin administration to the exposed adventitia after balloon injury was studied in a rat carotid artery balloon injury model. For this purpose, Peyton et al. (422) applied DSMO-dissolved bilirubin in pluronic gel to the exposed adventitia of the carotid artery. Such treatment significantly decreased neointima formation, increased the expression of cyclin A and D1, and arrested VSMC in G0/G1 without affecting ATP content or mitochondrial function (422).
The above studies show that both CO and bilirubin can mediate antiproliferative effects on VSMC proliferation in arterial injury models. It would be interesting to see the combined effects of CO and bilirubin on minimizing neointima formation, especially as modulation of endogenous Hmox1 expression in a therapeutic setting would increase the concentrations of both products.
5. Pharmacological modulation of VSMC by Hmox1 induction
Several drugs have been reported to decrease intimal hyperplasia following arterial injury via induction of Hmox1. For example, the lipid-lowering drug probucol (that inhibits restenosis in humans and is still used in Japan for the treatment of hyperlipidemia) and its monosuccinate ester succinobucol induce vascular Hmox1 expression, promote re-endothelialization, and inhibit intimal VSMC proliferation and hyperplasia (107, 272, 357, 597, 598). This inhibition of intimal hyperplasia is blocked completely by tin protoporphyrin (597), indicating that the effects of probucol/probucol derivatives are mediated through induction of Hmox1. Mechanistic studies employing Hmox1−/− mice and tin protoporphyrin suggest that bone marrow and circulating progenitor cells contribute to the observed re-endothelialization (598). This conclusion contrasts the findings of an earlier study be Kong et al. (257). These authors reported transplantation of Hmox1 overexpression endothelial progenitor cells to the site of injury did not enhance re-endothelialization or reduce intimal hyperplasia beyond that seen with control, GFP-expressing endothelial progenitor cells alone (257). Therefore, to establish whether endothelial progenitor Hmox1 is important in the prevention of intimal hyperplasia requires additional studies.
In addition to probucol and some of its derivatives, butylated hydroxanisole (317), sulfasalazine (244), the sesquiterpene lactone eupatolide (245), dimethylfumarate (400), valsartan (296), and epigallocetechin-3-gallate (316) have all been reported to attenuate experimental intimal hyperplasia after balloon injury by the induction of Hmox1. Clearly, Hmox1 induction by various pharmacological agents is of potential therapeutic interest to limit intimal hyperplasia, and other vascular diseases discussed in this review.
In addition to YY1 (28), there appear to be other proteins that act both up- and downstream of Hmox1 and that affect intimal hyperplasia. Thus overexpression of thymidine phosphorylase leads to expression of Hmox1 in vitro, and this may explain the decrease in VSMC migration and proliferation observed when thymidine phosphorylase is overexpressed in an in vivo model of arterial injury (293). In that study, thymidine phosphorylase-induced expression of Hmox1 increased the concentrations of p27KIP1 (293), suggesting that inhibition of VSMC proliferation was mediated by this member of cyclin-dependent kinase inhibitors. Another study reported induction of the peptidyl prolyl isomerase Pin1 to promote neointima formation after injury by downregulating Hmox1 expression (246).
C. Transplant Arteriosclerosis
The field of transplantation has benefited from major advances in the past several decades, and solid organ transplantation remains the only viable treatment for end-stage organ failure. Nevertheless, its attainment as a viable long-term curative option is still encumbered mainly by the development of transplant vasculopathy that is commonly referred to as transplant arteriosclerosis (TA). The characteristic findings of this predominant limiting condition affecting the long-term survival of the allograft involves severe and often diffuse intimal hyperplastic lesions that eventually lead to diminished blood flow to the allograft and thereby organ dysfunction (644). The pathogenesis of TA involves a chronic inflammatory state where the activated recipient's immune cells damage the endothelium of the graft. This is followed by release of cytokines that ultimately leads to migration and proliferation of VSMC with subsequent narrowing of the lumen and mitigation of perfusion (644). The clinical significance of TA has invoked numerous studies in search of a curative modality, to inhibit or at least delay the development and/or progression of TA. Accordingly, the potential application of Hmox1 in this clinical entity has been tested by multiple investigations.
One study found that the Allotrap peptide RDP58 therapy in an aortic allotransplant model provided inhibition of vascular intimal thickening, medial necrosis, and adventitial cellular inflammation (292). These findings were attributed to Hmox1 induction and decrement in production of TNF-α and inhibition of VSMC proliferation (292). Another study reported Hmox1 gene transfer to inhibit TA following aortic transplantation in rats, with the beneficial effects of Hmox1 attributed to inhibition of apoptosis and decreased expression of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) (116). Inhibition of leukocyte infiltration and proliferation of VSMC were reported in another model of rat aorta transplant where overexpression of Hmox1 was achieved via adenoviral gene transfer (48). More elaborate studies have provided evidence for the paramount role of CO in mediating the beneficial attributes of Hmox1 induction to alleviate TA. For instance, overexpression of Hmox1 via gene transfer and administration of methylene chloride (which releases CO after degradation) led to comparable protection, including a decrease in intimal thickness, leukocyte infiltration, cytokine production, and cell adhesion molecule expression in the transplanted aorta (66). Another study examined the role of inhaled CO in an aorta transplantation model. Exposure to CO (250 ppm) immediately following transplantation and for the subsequent 56 days abolished development of intimal hyperplasia as well as leukocyte infiltration compared with animals that were exposed to air (409). A protective role of CO in TA has been explored further, and it has been shown that donor Hmox1 expression attenuates intimal hyperplasia in recipient grafts (86). In addition, using CORM-3 as a CO donor, these beneficial effects were found to be dependent on the modulatory actions of CO on T-cells (86).
Other studies more specifically focused on the effects of Hmox1 on the immune system and its role in TA. One study showed that adoptive transfer of dendritic cells from Hmox1−/− mice prior to vascular allograft transplantation was associated with a marked infiltration of CD4+ T-cells as well as increased IgG deposition (75). Such outcomes following deletion of Hmox1 in dendritic cells were associated with an intensified degree of arteriosclerotic lesions. These results highlight the fundamental role of dendritic cells in transplant mediated immune responses and also reveal the regulatory role of Hmox1 as its deficiency leads to a state of augmented development of transplant vasculopathy.
The beneficial effects of other molecules, like the anti-inflammatory and immunosuppressive cytokine IL-10, in TA have also been attributed to Hmox1 expression. In a rat model of TA, another study found that the prevention of neointimal hyperplasia and immune cell infiltration by IL-10 depends on Hmox1 activity, because inhibition of heme oxygenase activity by tin protoporphyrin reversed the beneficial activities of adenoviral overexpression of IL-10 (70).
D. Myocardial Infarction
Myocardial infarction (MI) is an irreversible coagulative necrosis of the myocardium that is often a consequence of coronary artery disease and the number one cause of mortality in the Western world. The myocardium is the most reliant tissue on oxygen delivery and hence any interruptions in perfusion could have detrimental consequences that extend beyond the initial injury and carry significant risk of mortality and morbidity. Consequently, minimizing the extent of myocardial injury during an I/R event has a significant beneficial outcome. The advantageous role of Hmox1 and its products have been closely studied in this context. Several lines of evidence support the role of Hmox1 in protecting the myocardium. Hmox1 expression is increased robustly in porcine myocardium following I/R (475, 476). In contrast, absence of Hmox1 is associated with severe right ventricular dilation and infarction when mice are subjected to chronic (up to 7 wk) hypoxia (615). In another study, hearts from Hmox1+/− mice that contain ∼40% less Hmox1 protein than heart from Hmox1+/+ animals, were subjected to I/R ex vivo. Such decrease in Hmox1 content was accompanied with increased infarct size, creatine kinase release, reduced ventricular recovery, and increased oxidative stress (622). Furthermore, preconditioning (a strong cardioprotective approach) failed to protect hearts from Hmox1+/− mice (622), further highlighting the essential role of Hmox1 expression in minimizing the injurious effects of I/R.
The central role of Hmox1 in protecting the myocardium has also been demonstrated. Cardiac-specific overexpression of Hmox1 in mice is associated with significantly smaller areas of infarction following I/R as well as reduced inflammation and oxidative damage (616). Moreover, contractile function of the myocardium following ischemia is preserved in these mice in an Hmox1 dose-dependent manner (616). Other in vivo models support these encouraging results. For instance, delivery of a hypoxia-inducible Hmox1 expression vector under the ventricle-specific MLC2v promoter to the left ventricle three days prior to ischemia leads to reduction in infarct size, apoptosis and improvement in left ventricular function (528, 529). These findings were reiterated by a more remote gene delivery, namely, via quadriceps muscle transfection. Such remote delivery of either the Hmox1 transcriptional activator hypoxia-inducible factor-1α or the Hmox1 gene per se were shown to have marked cardioprotective effects both in ex vivo (101) and in vivo (37) models of MI. Notably, the cardioprotective effects of Hmox1 induced by gene transfer have been shown to be present even one year after the initial gene transfer (289). Furthermore, to date, no adverse functional consequences were observed with this long-term approach (289). Another cardioprotective role of Hmox1 is its potential ability to stabilize atherosclerotic plaques, thereby preventing plaque rupture and ensuing thrombosis and MI (see sect. IVA).
The Hmox1-mediated beneficial effects in I/R-induced myocardial injury extend to the downregulation of inflammatory changes. Such changes may exacerbate the initial injury through activation of several proinflammatory pathways that lead to infiltration of leukocytes and subsequent tissue injury exerted via release of cytokines and generation of reactive oxygen species. It has been demonstrated that while deficiency of Hmox1 leads to heightened postischemic inflammatory changes in murine hearts, delivery of Hmox1 significantly mitigates the inflammation in both murine and porcine hearts subjected to I/R (179).
1. Products of heme catabolism in myocardial infarction
The products formed during enzymatic heme catabolism have also been examined to further explore the mechanism(s) by which Hmox1 induction may confer cardioprotection. To this end, CO and bilirubin have been extensively studied. The CO releasing molecule CORM-3 was found to reduce infarct size when administered at the time of reperfusion (164). The same group reported that administration of CORM-3 24 h before coronary occlusion also leads to significant reduction in infarct size (497), suggesting that CO may function as a preconditioning mimetic. Administering CO via inhalation, others have observed that CO exerts its protection via modulation of p38 MAPK and Akt-eNOS pathway (146). Other studies have also confirmed the cardioprotective role of bilirubin (34, 84, 141) and conjugated bilirubin (19). These experimental findings support the clinical observations where mild elevation of serum bilirubin as in patients with Gilbert's syndrome (deficiency of the UGT1A1 enzyme resulting in unconjugated hyperbilirubinemia) has been found to be associated with decreased risk of coronary artery disease (see sect. IVA4).
As mentioned earlier, postinfarction complications constitute significant risk for morbidity and mortality. One of the most frequent and potentially lethal complications is ventricular arrhythmia that develops frequently following reperfusion. Importantly, Hmox1 expression is associated with protection against I/R-induced ventricular fibrillation, a protective response attributed to generation of CO (18, 555). These findings have been validated by inhibition of heme oxygenase activity as well as chemical induction of Hmox1 using N-tert-butyl-α-phenylnitrone (17). Another major complication of MI is the postinfarct ventricular remodeling. This is a process where the injured myocardium is replaced by fibrotic tissue and can eventually lead to derangements in contractility and congestive heart failure. Cardiac-specific overexpression of Hmox1 in a model of permanent coronary artery ligation improves survival and reduces left ventricular dilatation (568). In addition, these results could be recapitulated by utilizing CORM-3 as means of CO delivery (568). One study, however, reported worsening cardiac remodeling following induced MI in rats (361). In that study chronic inhalation of CO (3 wk of 500 ppm CO) led to increased infarct size and hypertrophic ventricular remodeling (361).
As discussed above, the cardioprotective effects of Hmox1 in MI are well established. Accordingly, they have led to a clinical trial to investigate its potential application following infarction induced cardiac injury NCT00483587.
E. Stroke
Cerebrovascular accident, commonly referred to as stroke, is a consequence of interruption of blood supply to the brain and is either ischemic or hemorrhagic in nature. Regardless of the nature of the cerebrovascular accident, detrimental complications can follow that underline the status of stroke as a leading cause of death and disability around the world. Hmox1 expression is induced during stroke. One of the earliest studies to demonstrate this used a middle cerebral artery ligation model, where Hmox1 was expressed robustly in EC within areas of infarction and in glia at the margins of infarctions (395). Interestingly, Hmox1 was also induced in glial cells in noninfarcted areas of the cortex inside as well as outside the middle cerebral artery distribution (395). This pattern of expression has been confirmed following injection of hemoglobin in cisterna magna that increased Hmox1 markedly in microglia (553).
1. Hmox1 in cerebral hemorrhage
Hmox1 is also induced in microglia throughout the brain following experimental subarachnoid hemorrhage (SAH) in rats (346). It was suggested that Hmox1 induction plays a beneficial role in ameliorating the vasospasm induced by hemoglobin. In fact, multiple studies have confirmed this notion, and their results underscore the significance of Hmox1 induction in attenuating vasospasm that frequently follows SAH. In one study Hmox1-expressing adenovirus was injected into the cisterna magna of rats in a model of SAH, and this significantly inhibited contraction of the basilar artery and it improved cerebral blood flow (404).
A number of free radical scavengers have been trialed for their ability to inhibit cerebral vasospasm in SAH. In this context, nicaraven (a synthetic radical scavenger with anti-vasospastic properties and used in patients with SAH) was reported to induce Hmox1 following SAH (478). Importantly, the inhibition of vasospasm by nicaraven was abrogated when Hmox1 induction was blocked by antisense oligodeoxynucleotides (478), indicating that the delayed cerebral vasospasm observed with nicaraven was mediated by Hmox1 induction. Utilizing a novel approach to achieve Hmox1 protein transduction into the cerebral arteries, Ogawa et al. (399) provided further evidence for Hmox1 overexpression to decrease vasospasm in the setting of SAH.
There have been conflictive findings with regard to the role of Hmox1 during intracerebral hemorrhage. In a model of intracerebral hemorrhage, selective overexpression of Hmox1 in astrocytes has been reported to decrease mortality, blood-brain barrier disruption, perihematomal cell injury, and neurological deficits (73). In apparent contrast, another study reported Hmox1 to exacerbate early brain injury in a similar model of intracerebral hemorrhage that was attributed mostly to excess iron being released from heme oxygenase-catalyzed heme degradation (569). More recently, Schallner et al. (467) investigated the role of microglia specific Hmox1 expression in SAH. Elegant studies using chemical inhibitors of Hmox activity as well as conditional deletion of Hmox1 in microglia lead to heightened neuronal cell death, vasospasm, impaired cognitive function, and clearance of cerebral blood burden following SAH (467). Importantly, CO inhalation was able to reverse these deleterious effects in the absence of Hmox1 expression. Furthermore, the authors reported significantly higher levels of Hmox1 activity in cerebrospinal fluid of patients with SAH compared with that in patients with unruptured cerebral aneurysms (467).
2. Hmox in ischemic stroke
A protective activity of Hmox1 has also been implicated in ischemic stroke. Shah et al. (473) observed Hmox1−/− mice to have substantially larger infarct size and neurologic deficit score compared with their wild-type littermates 7 days after occlusion of the middle cerebral artery. A similar protective effect was seen in wild-type mice treated with Ginkgo biloba extract, EGb 761, a popular and standardized natural extract, containing ginkgo-flavonol glycosides and terpene lactones (473). Interestingly, EGb 761 treatment had no beneficial effect in Hmox1−/− mice subjected to middle cerebral artery occlusion. Using a Hmox1 transgenic mice that overexpress Hmox1 under the control of the neuron-specific enolase promoter, another study confirmed the protective role of Hmox1 during ischemic stroke. Hmox1 overexpression decreased stroke volume and ischemic cerebral edema following middle cerebral artery occlusion, and this was related to increased neuronal concentrations of cGMP (413). Further evidence underscoring the protective role of the Hmox enzyme system in ischemic stroke is highlighted in Hmox2−/− mice. These mice demonstrate increased apoptosis around the infarct area following focal transient ischemia (114). Furthermore, cerebellar granule cultures of Hmox2−/− mice reveal augmented susceptibility to injury and apoptosis (114).
F. Pulmonary Hypertension
Pulmonary hypertension is a severe clinical condition that results in a proliferative vasculopathy of the pulmonary arteries with significant cellular proliferation, fibrosis, and vasoconstriction, leading to narrowing of the lumen with subsequent elevation in the pulmonary arterial pressure and right ventricular failure. The development of pulmonary hypertension is multifactorial and involves various pathways and cellular components that include EC, VSMC, inflammatory cells, as well as platelets and fibroblasts (211). The most common initiating factor in this context appears to be hypoxia that leads to pulmonary vasoconstriction with the ultimate consequence of VSMC proliferation and extracellular matrix deposition.
The protective role of Hmox1 in development and progression of pulmonary hypertension is based on several findings. Hmox1 is robustly induced in VSMC under hypoxic conditions in vitro (369). Moreover, chronic hypoxia also induces Hmox1 expression in lungs (643). In agreement with the premise of Hmox1 as a protective gene in pulmonary hypertension, it has been demonstrated that induction of Hmox1 via hemin treatment leads to mitigation of hypoxia-induced pulmonary hypertension and vascular remodeling in rats (81). Such treatment and induction of Hmox1 has also been shown to improve right ventricular systolic pressure, which correlates closely with the degree of severity of pulmonary hypertension (643). Other evidence supporting these findings has emerged from transgenic mice with lung-specific Hmox1 overexpression. It has been proposed that a hypoxic environment generates a proinflammatory milieu in the lung that precedes the vascular changes and ensuing pulmonary hypertension. To this end, overexpression of Hmox1 in lungs of transgenic mice blunted the pulmonary inflammation induced by hypoxia, and this was associated with marked improvement in parameters associated with pulmonary hypertension (359). In a rat model of monocrotaline-induced pulmonary hypertension, IL-10 gene transfer improved pulmonary artery pressure, weight ratio of right ventricle to left ventricle, mortality, as well as leukocyte infiltration and vascular cell proliferation (205). Importantly, these benefits of IL-10 were abrogated by treatment of the animals with tin protoporphyrin (205), suggesting protection by IL-10 was mediated by Hmox1 induction.
Further investigations have revealed the role of different products arising during heme oxygenase-mediated heme degradation in cardiopulmonary protection. The effects of CO and biliverdin have been tested in Hmox1−/− mice exposed to hypoxia. Treatment with CO gas (20–60 ppm) abolished hypoxia-induced right ventricular systolic pressure and arteriolar remodeling in both Hmox1−/− and wild-type mice, but was not able to rescue right ventricular failure and fibrosis (559). In contrast, biliverdin (50 μmol/kg) was able to mitigate right ventricular fibrosis and thrombus formation, whereas it had no effect on pulmonary hypertension-induced arteriolar remodeling (559). In three different models of rodent pulmonary hypertension, CO reversed established pulmonary hypertension as well as the morphological changes to the pulmonary vasculature to near normal (648).
In addition to the above-mentioned evidence, other investigators have shown that the beneficial effects of several drugs/molecules in pulmonary hypertension are mediated via Hmox1 induction. For example, in a monocrotaline-induced pulmonary hypertension model, rapamycin was found to protect the pulmonary vasculature, effects that were reversed by inhibition of heme oxygenase activity (645). Notably, simvastatin was able to reverse established pulmonary hypertension changes. These effects were lost with concomitant inhibition of heme oxygenase activity by tin protoporphyrin, highlighting a key role of heme oxygenase activity in this process (187). Also, more recently, erythropoietin was found to ameliorate the vascular changes of pulmonary hypertension, findings that were dependent on Hmox1 activation. Notably, inhibition of heme oxygenase activity reversed and accentuated the progression of vascular remodeling despite the erythropoietin treatment (554).
G. HMOX1 Promoter Polymorphisms and CVD
The substantial evidence gained in preclinical studies in support of a beneficial role of Hmox1 in various injury settings has led to multiple investigations addressing its significance in human diseases. To this end, several studies have been conducted evaluating the relationship between Hmox1 promoter polymorphisms and development of various clinical conditions particularly CVD (Figure 10). This is based on unique polymorphisms in the 5'-flanking region of the Hmox1 gene among individuals that are associated with varying Hmox1 expression/activity. These polymorphisms include a (GT)n dinucleotide length polymorphism and two single nucleotide polymorphisms (SNP), G(-1135)A and T(-413)A (reviewed in Ref. 129). The T(-413)A SNP has been reported to modulate the Hmox1 promoter activity, with the “A” allele promoter having higher activity than the “T” allele (402). However, current clinical findings on the role of T(-413)A SNP is limited and inconsistent (402, 403). The functional significance of the G(-1135)A SNP remains to be elucidated. In contrast, the polymorphism of (GT)n repeats in the human Hmox1 proximal promoter region has been studied extensively and reported to have functional significance in different clinical settings (reviewed in Refs. 22, 103, 129, 432).
FIGURE 10.
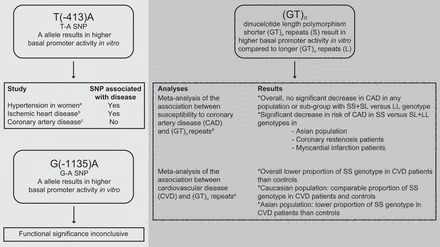
Promoter polymorphisms in HMOX1 and their clinical significance. Two single nucleotide polymorphisms (SNP) have been identified in the HMOX1 promoter, G(-1135)A, and T(-413)A. Their clinical significance remains inconclusive, as a result of limited and inconsistent data. The more extensively studied association of the dinucleotide length polymorphism (GT)n with various cardiovascular diseases has been reviewed in two recent meta-analyses. The results suggest that this polymorphism may have clinical implications in some populations. Superscripts refer to the following references: a403, b402, c323, d432, and e103.
There are numerous regions of purine-pyrimidine repeat sequences that are scattered throughout the human genome and that frequently exhibit length polymorphism. Such purine-pyrimidine repeat regions result in a Z-DNA confirmation that can negatively affect transcription (389). The number of GT repeats varies between 12 and 40. While there is no general consensus regarding the precise cut-off, it is evident that the lower GT repeats are associated with more robust Hmox1 promoter activity and expression. This notion has been confirmed by utilizing transient transfection assays. For instance, promoter/luciferase fusion constructs containing <25 GT repeats in transfected rat aortic smooth muscle cells revealed higher Hmox1 expression under basal conditions (72). In transiently transfected cells, H2O2 exposure increased the transcriptional activity of the Hmox1 promoter/luciferase fusion genes with <21 GT but not >28 GT repeats (606). Similarly, lymphoblastoid cell lines from subjects possessing short (<27) GT repeats showed higher Hmox1 mRNA and enzyme activity in response to oxidative stress compared with cell lines from subjects with long alleles (≥33) GT repeats (180). These results have been corroborated in another study where human umbilical vein endothelial cells with short (=23) GT repeats had higher basal Hmox1 expression as well as a stronger induction in response to several stimuli (523).
The first study to investigate a functional significance of (GT)n repeats in the human Hmox1 gene promoter region in a clinical condition was published in 1997 (248). Kimpara and colleagues investigated the Hmox1 (GT)n promoter polymorphism and its potential association with Alzheimer's disease and Parkinson's disease. While this study did not observe these neurological diseases to associate with any particular alleles, it provided a momentous area of investigation for other clinical conditions (248). In fact, the association of Hmox1 promoter polymorphism has now been studied in various clinical settings. These include emphysema (606), abdominal aortic aneurysm (468), ischemic stroke in patients with CVD risk factors especially low HDL cholesterol (16), rheumatoid arthritis (448), arteriovenous fistula patency in hemodialysis patients (307), type 2 diabetes mellitus (reviewed in Ref. 22), risk of recurrent venous thromboembolism (377), kidney allograft function (14, 128), and CVD (reviewed in Refs. 103, 432), as well as others with variable outcomes.
Among the clinical entities, CVD has been studied most extensively, with overall conflicting results. This has led to multiple reanalyses of the literature to elucidate potential causes of discrepant results. A recent Human Genome Epidemiology (HuGE) review was performed to closely examine the association of Hmox1 promoter polymorphism and susceptibility to coronary artery disease (CAD) (432). This meta-analysis included 11 studies between 2002 and 2013 that involved 10,170 patients with CAD and 6,868 controls. Defining short (S) and long (L) allele as <25 and >25 GT repeats, respectively, the authors observed no difference in CAD between the SS and LL + SL genotype of the Hmox1 (GT)n repeat length polymorphism. However, significant heterogeneity among the studies examined was noted with regards to age and sex matching, and the extent of coronary stenosis. As a result, subgroup analyses were carried out, and these revealed significantly reduced risks of CAD in Asians, in those with coronary-artery-narrowing = 50%, myocardial infarction, the age- and sex-matched subgroup, and the good-quality-reports subgroup (432). These findings highlight the paramount importance of vigilant randomization and adjustment for relevant factors so that accurate conclusions can be drawn from such diverse studies.
A more recent meta-analysis on the role of Hmox1 promoter polymorphism in CVD analyzed 41 publications between 1997 and 2013 (103). Subjects were divided into four groups to minimize confounding effects based on heterogeneity of the studies: 1) patients with established CVD, 2) patients without CVD, 3) controls (unknown cardiovascular status, and 4) children <20 yr of age (unselected). Overall, the proportion of short (SS, <25) GT repeat genotype was lower in CVD compared with non-CVD and control subjects in whom the cardiovascular status was not fully evaluated (103). These results overall support the presumed protective effects of Hmox1 against CVD. The study also revealed racial disparities in the distribution of the Hmox1 (GT)n repeat length, with Asian individuals having a significantly higher proportion of the SS genotype than Caucasians that might influence the associations of the genotype with CVD status (103).
The above comprehensive reviews overall corroborate the beneficial role of Hmox1 in CVD. However, several limitations remain and need to be considered in future studies to minimize confounding factors and to clarify the role of Hmox1 promoter polymorphism in clinical conditions, including CVD. Specifically, such studies would benefit from careful analysis and account for factors such as age, sex, race, uniform definition of CAD/CVD, and background comorbidities and risk factors. The latter is particularly important, as multiple studies reported a protective role of Hmox1 against CVD only in the presence of risk factors and increased oxidative stress (reviewed in Ref. 103). Given the current emphasis on personalized medicine, screening for Hmox1 promoter polymorphisms in CVD is worth considering.
H. Neoangiogenesis and Neovascularization
Angiogenesis is a physiological process that involves sprouting of EC from preexisting blood vessels that migrate and proliferate forming tubelike structures. Neovascularization is defined as a process involving the formation of new functional microvascular networks through which blood cells are able to circulate. Neovascularization differs from angiogenesis, which is characterized by endothelial outgrowths from preexisting blood vessels. Vascularization of the otherwise avascular cornea of the eye is a classic example for neovascularization. Angiogenesis could be either physiological (as would occur in the placenta during pregnancy) or pathological (e.g., following wound healing or in tumors). Hmox1 plays a key role in both physiological and pathological angiogenesis (reviewed in Ref. 119).
1. Physiological angiogenesis
During pregnancy, angiogenesis in the placenta is critical for fetal development. Hmox1 expression facilitates angiogenesis, and its importance in maintaining a normal pregnancy is corroborated by several observations (reviewed in Ref. 635). 1) Inhibition of heme oxygenase enzyme activity results in fetal loss, while Hmox1 overexpression improves pregnancy outcomes in animal models (633). 2) Several factors implicated in placental angiogenesis such as VEGF, SDF-1, and NO induce Hmox1 expression. 3) Hmox1−/− mice do not breed well (426), exhibit smaller placentas, and are associated with higher rates of intrauterine death, an effect that is reversed by administration of exogenous CO (634). 4) Increased length polymorphisms in a GT repeat sequence in the proximal Hmox1 promoter are associated with preeclampsia, albeit of a late-onset and less severe form (222). 5) Finally, maternal cigarette smoke (an exogenous source of CO) and higher ambient CO exposure during pregnancy have been reported to reduce incidence of preeclampsia in women (358, 591, 612).
The effects of Hmox1 on key cellular processes such as cellular proliferation, cytoprotection, angiogenesis, and immunomodulation have highlighted similarities between pregnancy and cancer biology (reviewed in Ref. 641). Recent studies showed that Hmox1 expression along with progesterone was reduced in placentas in a mouse model of fetal-growth restriction triggered by stress challenge during midgestation (491). Using this stress model, the authors provided evidence that progesterone increased placental Hmox1 expression. A dichotomous CD8+ T-cell response was identified in stress-challenged dams, as shown by an increase in the proportion of cytotoxic CD8+ T cells along with decreased frequencies of CD8+ T-cells coexpressing CD122. The authors suggested that Hmox1 promotes the generation of CD8+CD122+ T-cells and that adoptive transfer of CD8+CD122+ T-cells ameliorates impaired fetal growth and placental vascularization in Hmox1−/− mice (491).
2. Wound healing
Following injury, the healing process entails replacement of damaged capillaries with new vessels, and this is accomplished by neovascularization. The multiple phases involved in wound healing including the coagulation and inflammatory phases are integrally dependent on neovascularization. The involvement of Hmox1 in impaired wound healing was demonstrated by observations in Hmox1−/− mice. Compared with wild-type littermate mice, Hmox1−/− mice exhibit impaired wound healing, and this is associated with decreased recruitment of endothelial progenitor cells and capillary formation at the site of injury (110). Hmox1−/− mice also exhibit impaired regeneration and revascularization in hindlimb ischemia models in vivo compared with wild-type mice, possibly due to reduced elaboration of pro-angiogenic factors such as VEGF and SDF-1 (161). Another study by Grochot-Przeczek et al. (162) further corroborates the essential role of Hmox1 in wound healing. The authors reported chemical inhibition of Hmox to be associated with impaired neovascularization, ineffective wound healing and closure, similar to the situation in Hmox1−/− mice. That study also showed improved wound healing by overexpression of Hmox1 in keratinocytes. Furthermore, while diabetic mice exhibit impaired wound healing, local delivery of Hmox1 via adenoviral vector increased vascularization and accelerated wound healing (162).
Supporting these observations are recent studies from Kozakowska et al. (262), who demonstrated improved blood flow recovery and increased angiogenesis by injecting conditioned media from Hmox1 overexpressing myoblasts into diabetic mice after hindlimb ischemia. Analysis of the conditioned media from Hmox1 overexpressing cells revealed increased concentrations of SDF-1 and other proteins such as peptidyl-prolyl-cis–trans isomerase A, macrophage inhibitory factor, haptoglobin, and galectin-1 (262). On the other hand, conditioned media from Hmox1-deficient mesenchymal stem cells exposed to hypoxia shows reduced concentrations of proangiogenic factors such as VEGF and SDF-1 (Figure 11) (631). Under permissible conditions, Hmox1-deficient mesenchymal stem cells are capable of differentiating into osteogenic and adipocytic lineages, but display limited potential to differentiate into capillary-like structures (631). Other groups further substantiated these proangiogenic and pro-wound healing properties of Hmox1 (563, 564)
FIGURE 11.
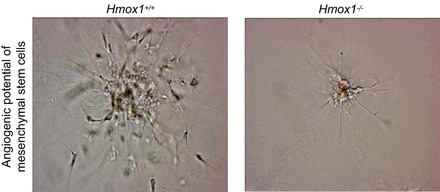
Multipotent mesenchymal stem cells (MSC) derived from Hmox1−/− mice have lower angiogenic potential than MSC from wild-type (Hmox1+/+) animals. An equal number of cells were placed on growth factor-reduced Matrigel and cultured in endothelial basal medium-2 for 9 days. The figure shows representative images of 3 separate experiments. Bar = 100 μm. [From Zarjou et al. (631).]
While the effects of Hmox2 in corneal wound healing have been reported (166), more recent studies have also evaluated the role of Hmox2 in cutaneous wound healing (324). Hmox2−/− mice show delayed cutaneous wound closure that was associated with decreased collagen deposition and vessel density along with an inflammatory response. The authors discovered delayed expression profiles of the chemokine (C-X-C) ligand-11 (CXCL-11), a protein linked to wound healing and angiogenesis, in wounds of Hmox2−/− mice (324). Furthermore, both Hmox1 and Hmox2 are highly expressed in the skin of patients suffering from psoriasis (169), although the precise role of heme oxygenases in this inflammatory condition remains to be established.
3. Cancer and tumor angiogenesis
Studies by Fang et al. (132) were the first to demonstrate the role of Hmox1 expression in tumor growth. Using pegylated zinc protoporphyrin, they showed that inhibition of Hmox1 enzyme activity facilitated shrinkage of tumors in an animal model (132). The expression of Hmox1 is increased in human cancers involving the prostate, brain, pancreas, liver, skin, and blood as well as others (reviewed in Ref. 578). Such expression is localized mainly to tumor cells but also observed in surrounding macrophages, particularly in tumors associated with significant neovascularization and hemorrhage, wherein heme serves as one potential inducer of Hmox1 expression. Hmox1 expression promotes cell proliferation and cell survival, while inhibition of heme oxygenase enzyme activity limits tumor growth in animal models. Hmox1 expression has also been observed in tissue samples from patients with Kaposi sarcoma, as well as in EC infected with herpes virus (349). Importantly, inhibition of Hmox has been shown to mitigate tumor growth (342, 344).
Proangiogenic factors such as VEGF and SDF-1 also promote tumor angiogenesis. VEGF regulates specific cell survival pathways and also participates in cell-matrix interactions through integrins. Proangiogenic bone marrow cells and endothelial progenitor cells differentiate into vascular cells and contribute to tumor angiogenesis. SDF-1 plays a major role in the recruitment and retention of CXCR4+ bone marrow-derived cells to areas of tumor growth (266). In addition to its proangiogenic effects, SDF-1 also promotes tumor cell growth, migration, and invasion. The importance of SDF-1 in tumor angiogenesis is highlighted by findings that blockade of the SDF-1/CXCR-4 axis decreases angiogenesis and tumor growth (65, 266).
Recent studies have demonstrated that tumor growth and angiogenesis is accelerated in mice with sickle cell disease in a melanoma model (570). Given this link between sickle cell disease and Hmox1, the authors demonstrated increased expression of Hmox1 in the tumors and, more importantly, showed that inhibition of Hmox1 activity with zinc protoporphyrin blocked angiogenesis and tumor growth in this animal model (570).
While blockade of Hmox1 is mostly beneficial in multiple tumor models, the exact mechanisms underlying these effects are not entirely known. One or more of the molecules involved in Hmox1-catalyzed heme degradation have been implicated, including heme, CO iron, and biliverdin (reviewed in Refs. 119, 578). The effects include pro-proliferative actions through cell cycle proteins such as p21; effects of CO on caveolin-1 and others (119, 578); tumor differentiation, anti-apoptotic effects of Hmox1, and tumor-related angiogenesis. The anti-inflammatory effects of Hmox1 can also enhance tumor progression by promoting an immunosuppressed environment. While previous studies have suggested that CO exhibits pro-angiogenic effects (321), recent work has suggested the contrary (7). Thus Wegiel et al. (581) observed that CO (250 ppm, 1 h/day) actually decreased tumor angiogenesis in prostate and lung cancer models, by targeting mitochondria and inducing a transient “anti-Warburg effect,” metabolic exhaustion, and cancer cell death. CO (using CORM-2) was shown to have pro-angiogenic effects in human and murine EC in vitro as well as in aortic rings from Hmox1−/− mice (110). In contrast, however, other studies have reported comparatively higher concentrations of CO (50 μM) to inhibit VEGF-induced EC proliferation and formation of capillary-like tubes (7). Furthermore, mice exposed to 250 ppm CO (1 h/day for 14 days) exhibited a marked decrease in angiogenesis in implanted Matrigel plugs (7). Using newer CO-releasing molecules such as CORM-401 (capable of releasing up to 3 CO/mol of the compound and also being sensitive to oxidants and increasing CO release), a recent study reported CO to accelerate EC migration in vitro and to increase VEGF and IL-8, supporting a proangiogenic effect (134). Taken together, the angiogenic potential of Hmox1 in tumors is a complex process and has yielded contrasting results and likely involves independent/parallel pathways. Therefore, its effect in the context of tumor-related angiogenesis requires more elaborate studies (for a review of the current understanding of the field and its future direction, see Ref. 322).
4. Dual role of Hmox1 in angiogenesis
Bussolati et al. (55) have demonstrated that the role of Hmox1 in angiogenesis is context dependent and may vary depending on the underlying condition, particularly focusing on the presence or absence of inflammation. For example, VEGF-induced angiogenesis requires Hmox1 activity, while inflammation-induced blood vessel formation is attenuated by overexpression of Hmox1 (55, 56). Leukocyte infiltration is a key process involved in LPS-induced angiogenesis and is attenuated by prior induction of Hmox1. Conversely, in VEGF-induced noninflammatory angiogenesis, pharmacological inhibition of Hmox1 induces leukocyte infiltration and enhances VEGF-induced angiogenesis (55, 56). However, blocking Hmox1 with interruption of leukocyte infiltration inhibits VEGF-induced angiogenesis. Therefore, it is thought that during chronic inflammation Hmox1 inhibits leukocyte infiltration and facilitates tissue repair by promoting VEGF-driven noninflammatory angiogenesis. This complex interaction indicates that when Hmox1 expression occurs in the environment free of the inflammatory reactions, the products of Hmox1 activity can be proangiogenic. In contrast, when angiogenesis is driven by LPS-induced inflammation, the expression of Hmox1 may inhibit the formation of new vessel (56).
VI. THERAPEUTIC POTENTIAL OF HMOX AND ITS PRODUCTS IN THE PREVENTION OF VASCULAR DISEASES
As demonstrated in the previous sections, substantive data indicate that heme oxygenases, in particular Hmox1, can modulate the outcome of several vascular diseases, and hence are a potential therapeutic target for these conditions (505). Indeed, several clinical trials have been undertaken, or are currently being recruited for, to examine whether the modulation of Hmox1 expression or the application of CO gas provides health benefits (Tables 6 AND 7). Principally, the therapeutic potential of the heme oxygenase pathway can be targeted at different levels. These include Hmox gene transcription, Hmox protein and activity, as well as its products CO and biliverdin, and the by-product bilirubin (Figure 12).
Table 6.
Hmox1 inducers in human studies of vascular disease
| Agent | Indication | Status | Outcome | NCT Nos. |
|---|---|---|---|---|
| Hemin | Plasma Hmox1 protein concentration | Completed | Increase in Hmox1 protein and activity at 24 and 48 h | NCT00882804 (36) |
| Resveratrol | Hmox1 mRNA expression | Completed | NCT01768507 | |
| Aspirin and simvastatin | Plasma Hmox1 protein concentration and venous monocyte Hmox1 protein activity | Completed | No increase in Hmox1 protein concentration or activity at 2 or 7 days | (35) |
| Nizatidine | Monocyte Hmox1 activity and plasma Hmox1 protein concentration | Completed | NCT02232308 | |
| Lisinopril | Monocyte Hmox1 activity and plasma Hmox1 protein concentration | Completed | NCT02232308 | |
| Curcumin | Peripheral blood monocyte Hmox1 protein concentration and mRNA expression | Completed | NCT00895167 | |
| Heme arginate | Peripheral blood monocyte Hmox1 protein concentration and mRNA expression | Completed | Increase in Hmox1 mRNA and protein content over 48 h | NCT00682370 (113) |
| Acute kidney injury after cardiac surgery | Not yet recruiting | NCT02142699 | ||
| Cardiac injury after myocardial infarction | Completed | NCT00483587 | ||
| Ischemia reperfusion injury following deceased donor renal transplants | Completed | NCT01430156 | ||
| Oxidative stress in the human heart | Not yet recruiting | NCT02314780 | ||
| Adenosine-induced vasodilation | Terminated | NCT00856817 | ||
| Reperfusion patterns after ischemia | Completed | Improvement in reperfusion patterns during ischemia-reperfusion injury measured via blood oxygen level dependent (BOLD) functional magnetic resonance imaging | NCT01461512 (12) | |
| Probucol | Xanthoma regression in patients with hypercholesterolemia | Completed | Decrease in Achilles tendon thickness, serum total cholesterol, and HDL | (607) |
| Probucol Quantitative Regression Swedish Study (PQRST) trial | Completed | Decrease in serum total cholesterol, LDL, HDL, and HDL2. No change in lumen volume, arterial edge roughness and amount of aorto-femoral atherosclerosis | 567 | |
| SECURE study | Completed | Decrease in serum total cholesterol, LDL, HDL, and ApoAI; no significant change in plaque volume or composition between cilostazol plus probucol combination therapy versus cilostazol monotherapy | (255) | |
| Fukuoka Atherosclerosis Trial (FAST) | Completed | Decrease in serum LDL, HDL, and total cholesterol; intima-to-media ratio in common carotid artery; and incidence of cardiac events | (465) | |
| Efficacy of rapamycin and probucol co-eluting stent | Completed | Comparable differential safety profile of single or co-eluting stents in terms of death/MI, definite stent thrombosis, and target lesion revascularization at 2 yr | NCT00598533 (57) | |
| Succinobucol | Canadian Antioxidant Restenosis Trial (CART)-1 | Completed | Improved lumen dimension at site of percutaneous coronary intervention; decrease in restenosis | (532) |
| Canadian Antioxidant Restenosis Trial (CART)-2 | Completed | No significant improvement in atherosclerosis regression | (531) | |
| Aggressive Reduction of inflammation stop events (ARISE) | Completed | No benefit on time to first occurrence of cardiovascular event (primary end point); fewer incidences of composite secondary end point. Succinobucol increased occurrence of atrial fibrillation and bleeding episodes, and decreased LDL and HDL, systolic blood pressure, and glycated hemoglobin | NCT00066898 (533) | |
| Nrf2 induction | Bardoxolone methyl in pulmonary hypertension (LARIAT) | Currently recruiting | NCT02036970 |
NCT numbers refer to the unique identification code allocated to each clinical trial registered on http://clinicaltrials.gov. Reference numbers are given in parentheses. ApoAI, apolipoprotein A-I; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
FIGURE 12.
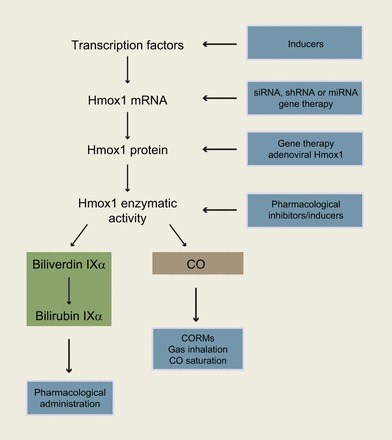
Potential therapeutic modulation of Hmox1 and its products. Upstream of its enzymatic activity, Hmox1 may be modulated via transcription factors and gene therapy to increase or decrease expression of Hmox1 at the mRNA and protein level employing microRNA (miRNA), short interfering RNA (siRNA), and/or short hairpin RNA (shRNA). Pharmacological inhibitors or inducers may be used to modulate Hmox1 enzymatic activity. CO (via inhalation or via CORMs), biliverdin, and bilirubin may also be used therapeutically.
A. Modulating Transcription Factor Activity
The transcription factor Nrf2 is a key modulator of the expression of Hmox1 in response to a wide variety of stresses, including oxidative and nucleophile stress (reviewed in Ref. 329). Numerous natural and synthetic compounds can activate Nrf2, leading to increased expression of Hmox1 in experimental systems (140), and activation/inhibition of Nrf2 is a therapeutic target in vascular diseases. Many of the Nrf2-activating compounds interfere with the protein-protein interaction between Keap1 and Nrf2 by modifying cysteine residue(s) in Keap1 that results in increased Nrf2 activity (reviewed in Ref. 586). Dimethyl fumarate is an Nrf2-activator that has been approved for use in multiple sclerosis, while another bardoxolone methyl is currently being trialed in the context of pulmonary hypertension. It is important to note that a clinical trial using bardoxolone methyl in patients with diabetic kidney disease had to be terminated due to adverse cardiovascular events and negative results with respect to risk of end-stage kidney disease (106), despite encouraging results from earlier studies (420). A major potential drawback with targeting Nrf2 activation is the lack of specificity for Hmox1, because activation of Nrf2 leads to the differential expression of a multitude of genes, and potentially deleterious off-target effects may occur. Furthermore, inhibition of Bach1 has been suggested as a potential therapeutic target to increase Hmox1 expression (196), as Bach1 is a nuclear heme sensor and repressor of Hmox1 transcription (517). When the cellular concentration of heme is increased, heme binds Bach1, and this decreases its ability to bind to DNA, leading to the activation of Hmox1 by transcriptional activators such as Nrf2. Heme-containing Bach1 may also be responsive to CO, as has been reported for another heme-binding transcription factor, neuronal PAS domain protein 2 (Npas2). Npas2 forms a heterodimer with BMAL1, an active transcriptional complex that regulates components of the circadian rhythm (438, 449). Npas2 contains two PAS domains that are responsible for heme binding. The transcriptional activity of the Npas2-BMAL1 heterodimer is modulated by the redox state of cellular pyridine nucleotides, with NADH and NADPH increasing, and NAD+ and NADP+ decreasing, binding (449). CO disrupts Npas2-heme binding, thereby inhibiting the transcriptional activation induced by reduced pyridine nucleotides (112). It is possible that CO may similarly modulate the transcriptional activity of heme-containing Bach1.
Several small molecule inhibitors of AP-1 have been developed (reviewed in Ref. 613), as this transcription factor can modulate a variety of pro-inflammatory cytokines in addition to Hmox1. In terms of Hmox1, inhibition of AP-1 may be appropriate in the setting of cancer, where increased Hmox1 is likely detrimental and exacerbates chemoresistance. Similarly, inhibition of NF-κB is also studied widely in terms of potentially decreasing the proliferation of cancer cells. Of the NF-κB inhibitors being developed, bortezomib has been approved for the treatment of multiple myeloma (313).
A major issue with targeting of transcription factor-activating compounds is their lack of specificity for Hmox1, with increased transcription factor activation potentially leading to the differential expression of a large set of genes and off-target effects. In this regard, a study in a human keratinocyte cell line reported that knockdown of Bach1 induced Hmox1 expression 135-fold but only induced other Bach1-repressed-activated genes, including Nrf2, by a maximum of 3-fold (331). While only a selected set of genes was studied in response to Bach1 knockdown, this work suggests that in some conditions inhibition of Bach1 may predominantly affect Hmox1 expression. Modulation of transcription factors to selectively affect gene expression may increase their utility as therapeutic targets. Differences in the molecular recognition of the human versus rodent Hmox1 genes will be an important factor to take into consideration for further drug discovery programs targeting the Hmox1 gene.
B. mRNA and Protein Modulation
Hmox1 gene transfer via adenoviral or lentiviral-based vectors has been utilized extensively as an experimental tool in a range of disease models, as outlined in section V. Interestingly, hypoxia/HIF-driven HMOX1 adenoviral therapeutic gene delivery has been reported to be effective as a preemptive treatment strategy in models of I/R injury (210, 411). While this strategy is potentially relevant to the human situation, it has not been translated to the clinical situation, and its applicability is limited due to its preemptive nature. In addition, RNAi-based drugs have been developed as a method to transcriptionally silence gene expression. However, no Hmox1 gene therapy trial has been undertaken to date, although inhibition of Hmox1 in conjunction with chemotherapeutics has been suggested as a strategy to minimize chemoresistance in the treatment of various cancers. In this situation, RNAi-based inhibition of Hmox1 could be utilized as a potential therapy.
In experimental work, hemin and heme arginate are used widely to induce Hmox1, and both forms of heme have been demonstrated to stimulate Hmox1 expression in healthy subjects (36, 113). Heme arginate has been tested in or is currently undergoing testing in 13 separate clinical trials, including several in vascular diseases. As shown in Table 6, heme arginate has been trialed as a therapy for MI, I/R injury, AKI after cardiac surgery, and vasodilation. In addition, a pilot-study (NCT01768507) was undertaken to analyze the effect of resveratrol on Hmox1 expression in peripheral blood mononuclear cells, although the results have not been published to date.
C. Activity
Inhibition of Hmox activity is a widely used strategy in experimental studies, with synthetic metalloporphyrins such as proto, meso, and deuteron porphyrins representing the first generation and best-known Hmox inhibitors. Some of these porphyrins have been tested in preclinical models of hyperbilirubinemia. Since then, modified derivatives have been developed, such as zinc protoporphyrin conjugated with polyethylene glycol (456) that has increased solubility in water and has been demonstrated to effectively inhibit heme oxygenase activity in murine solid tumors in vivo (456). As indicated earlier, however, the major limitations with metalloporphyrins, particularly when used at concentrations above 0.5 μM, are lack of selectivity/specificity and light sensitivity (163) that limit their therapeutic applicability. Given these limitations, nonporphyrin inhibitors of heme oxygenase are being developed, with some of them showing Hmox isoform specificity (249, 560, 561), although to date none of these has become mainstream tools in experimental studies.
D. Products of Hmox-Mediated Heme Catabolism
In experimental disease models where increased Hmox1 expression is considered beneficial, administration of CO, biliverdin, or bilirubin often (though not always) yields similar beneficial effects. Of these products, CO has received most interest as a potential therapeutic agent. Indeed, there is considerable evidence for CO administration, as a gas or via CORMs, representing a promising strategy to treat a variety of vascular diseases based on rodent studies (reviewed in Ref. 374). In humans, two clinical trials (NCT00122694 and NCT00094406) have been completed to date using inhaled CO (Table 7), with the first of these studies published. In that pilot study (25), the anti-inflammatory effects of CO (100–125 ppm for 2 h on 4 consecutive days) were examined in stable patients with chronic obstructive pulmonary disease. CO inhalation was safe with peak carboxyhemoglobin (COHb) values ≤4.5%, but CO had no material impact on the primary end point, i.e., sputum neutrophil percentage (25). This observation is consistent with an earlier human study that reported no effect on systemic cytokine concentrations with CO inhalation (500 ppm for 1 h, COHb <10%) after LPS administration (347). These findings in humans contrast the situation in non-human primates, where CO inhalation (500 ppm for 6 h after LPS injection) protects against LPS-induced lung neutrophil influx (362). While these comparatively higher exposures of CO provided anti-inflammatory benefit in non-human primates, they resulted in unacceptably high COHb values of >30%, and lower concentrations of CO (250 ppm, that are associated with anti-inflammatory protection in rodents) were ineffective in non-human primates (362). Together, these findings highlight the interspecies variation in both dose-response relationship between CO and COHb, and anti-inflammatory activities of CO. Appropriately, Mitchell et al. (362) concluded that the translation of CO therapy to human disease will require additional extensive rigorous proof-of-concept studies in humans in the future.
Table 7.
Carbon monoxide inhalation in human studies of vascular disease
| Indication | Status | Outcome | NCT Nos. |
|---|---|---|---|
| CO to prevent lung inflammation | Completed | NCT00094406 | |
| CO therapy for severe pulmonary arterial hypertension | Active, not recruiting | NCT01523548 | |
| Inhaled CO to treat idiopathic pulmonary fibrosis | Active, not recruiting | NCT01214187 | |
| Modification of chronic inflammation by inhaled CO in patients with stable COPD | Completed | Inhalation of 100–125 ppm CO feasible in patients with COPD in a stable phase. No significant decrease in sputum eosinophils and responsiveness to methacholine | NCT00122694 (25) |
| CO inhalation on cytokine production during experimental human endotoxemia | Completed | No effect of inhalation of 500 ppm CO on systemic inflammation markers (TNF-α, IL-6, IL-8, IL-1α, and IL-1β) | (347) |
NCT numbers refer to the unique identification code allocated to each clinical trial registered on http://clinicaltrials.gov. Reference numbers are given in parentheses. CO, carbon monoxide; COPD, chronic obstructive pulmonary disease; IL, interleukin; ppm, parts per million; TNF-α, tumor necrosis factor-α.
In addition to CO gas, there is also support for the use of CORMs as a potential strategy to treat vascular diseases (reviewed in Ref. 374). An advantage of CORMs is that they deliver CO to tissues in a more controlled manner (374) and without substantial increases in blood COHb (104, 264). Different types of CORMs have been developed, including water-soluble CORM-3 that has been shown to restore vascular function in experimental pathological conditions such as vasoconstriction, systemic hypertension, vascular thrombosis, and pulmonary hypertension (1, 77, 85, 142, 264). At the present time, however, there have been no human studies using CORMs.
Similar to the situation with CORMs, we are unaware of studies or clinical trials examining the effects of biliverdin or bilirubin in humans. There are several critical aspects surrounding the potential use of bile pigments as therapeutics, including the supply of biliverdin/bilirubin of sufficient quality and quantity, their formulation, and the general lack of sufficient knowledge in relation to pharmacokinetic and route of administration. Bulmer et al. (51) recently assessed the pharmacokinetics of biliverdin and bilirubin administered intravenously, intraperitoneally, and intraduodenally to rats at 2.7 mg/kg (51). Among the different routes of administration tested, intraperitoneal injection showed the highest bioavailability, with administered biliverdin being converted rapidly to bilirubin.
E. Atherosclerosis
Atherosclerosis can result in serious clinical manifestations such as thrombus formation, vessel occlusion, and ischemia, underscoring the importance of effective treatments to minimize plaque formation and rupture. Current treatment regimes are centered on lifestyle changes and the treatment of risk factors such as hypertension and hypercholesterolemia. Given that atherosclerosis is the leading cause of morbidity and mortality in most developed countries, the development of effective anti-atherosclerotic therapies is critical, particularly as several recent strategies have failed.
A large body of data indicates that increased expression of Hmox1 is beneficial against several pathological aspects associated with atherosclerosis, as outlined in section VA. Thus increasing Hmox1 content may provide a useful therapeutic target to attenuate endothelial dysfunction and arterial inflammation, decrease atherosclerotic plaque size, and increase plaque stability (505). In terms of therapeutic modulation of Hmox1 in atherosclerosis and related CVD, several clinical trials have been undertaken (Table 6). The Hmox1 inducers trialed include the old cholesterol-lowering drug probucol and its derivatives that inhibit experimental atherosclerosis and related vascular disease in an Hmox1-dependent manner (597). Overall, the outcome of clinical trials with probucol are controversial (reviewed in Ref. 501), with both positive (57, 464, 465, 607) and neutral results observed (255, 567). Probucol is no longer available in most countries, primarily due to concern of undesirable side effects, including lowering of high-density lipoprotein cholesterol and prolongation of cardiac depolarization (501). Succinobucol (a monosuccinate derivative of probucol also referred to as AGI-1067) has also been subjected to clinical trials for the treatment of restenosis and atherosclerosis-related CVD. Succinobucol decreased restenosis following angioplasty (532), but did not improve the coronary atherosclerosis (531) or acute coronary syndrome outcome (533) in patients managed with conventional treatments. In addition to these studies, a clinical trial examining the effect of heme arginate on adenosine-induced vasodilation (NCT00856817) was terminated due to recruitment failure before initiation of the treatment phase.
Several preclinical studies suggest that statin therapy may lead to increases in Hmox1 expression (see, e.g., Refs. 188, 375). Heeba et al. (176) treated atherosclerotic New Zealand white rabbits with atorvastatin or simvastatin and reported cotreatment with tin-protoporphyrin to attenuate the extent of disease inhibition observed with statin treatment alone. These results suggest that statins may inhibit experimental atherosclerosis in part via their ability to increase Hmox1 expression. However, the relevance of these and related studies to the human situation is questionable, as the dose of statins used routinely in experimental systems are unachievable in human patients (for a discussion, see Ref. 41). Indeed, two recent human studies failed to observe induction of Hmox1 in subjects treated with clinically relevant doses of simvastatin (35) or atorvastatin (360).
While preclinical studies suggest a potential beneficial role for bilirubin in modulating atherosclerosis, to date no clinical trials using Hmox1-derived reaction products have been carried out in an atherosclerosis setting. As pointed out in earlier sections, however, there are several aspects surrounding their efficacy and safety (e.g., formulation, route of administration, pharmacokinetics) that need to be addressed first. Similarly, gene therapy is also a potential manner in which Hmox1 could be modulated in atherosclerosis, although currently unresolved issues related to delivery, safety, and efficacy of gene vectors limit its clinical application. Once overcome, this form of Hmox1 modulation may provide a novel method to treat atherosclerotic lesion progression.
F. Pulmonary Hypertension
The progressive nature of pulmonary hypertension has created major awareness and fueled vigorous research to identify and utilize novel modalities and/or pharmacological agents that could mitigate the advancement of this disease. There is also great interest to potentially reverse the pulmonary vascular lesions that are the fundamental culprits of the initiation and propagation of this disease. Current treatment options are based on the classification provided by the World Health Organization and include calcium channel blockers, prostanoids, endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and inducers of the NO pathway, such as sGC inducers (149). Despite available treatment options, mortality from severe pulmonary hypertension remains high with lung transplantation as the last effective treatment option. Hence, it is evident that introduction of an effective approach to slow or reverse pulmonary hypertension is of paramount importance.
As discussed above, there is strong evidence to support the protective role of Hmox1 induction and the products of Hmox enzymatic activity in slowing the progression as well as reversal of established pulmonary hypertension. Based on in vitro and animal models, strategies that could enable targeted overexpression of Hmox1 in pulmonary vasculature and/or lung parenchyma may provide novel avenues that could accelerate research and could potentially authenticate such strategy as a novel approach to treat pulmonary hypertension. If successful, such approaches would translate to potential inhibition of VSMC proliferation and reduced inflammation with subsequent mitigation of pulmonary hypertension and its related complications.
Another approach would be to use biliverdin, bilirubin, or CO (either in inhaled form or CORMs). As mentioned above, these compounds have potent protective effects against pulmonary hypertension-induced arteriolar remodeling, right ventricular failure, and fibrosis. Also, there is unexplored potential in pharmacological agents that provide beneficial effects in this disease based on their ability to induce Hmox1. These agents include rapamycin, simvastatin, and erythropoietin, all of which have already been approved for clinical applications for other settings. A recent ongoing phase II clinical trial involves the use of bardoxolone methyl, a synthetic triterpenoid and potent inducer of Nrf2 and Hmox1 (304), for patients with pulmonary hypertension (NCT02036970).
The potential application of small molecules to induce Hmox1 is timely, germane, and merits more in-depth investigation that could introduce novel agents with application in multiple disease settings. One major obstacle in identification of such agents has been the significant differences in Hmox1 gene regulation observed between humans and rodents. However, generation of “humanized” Hmox1 mice that solely harbor the human Hmox1 with all of its regulatory regions will enable a closer examination of small molecules that induce human Hmox1 in vivo (241). There are other major challenges that need to be addressed prior to being able to implement Hmox1 induction or its metabolites to treat pulmonary hypertension. For instance, there is still considerable debate regarding suitable animal models of pulmonary hypertension and their resemblance of the human disease (498). Furthermore, identification of patients at risk who could develop pulmonary hypertension (either through novel biomarkers, imaging techniques or other clinical criteria) would certainly provide valuable information that could be used to implement some of the above-mentioned approaches. Additionally, clinical application of products of the enzymatic reaction (such as the use of CO or CORMs) of heme oxygenase would require meticulous and systematic analysis that would allow identification of appropriate doses to provide the anticipated beneficial effects without major unwanted side effects.
G. Neoangiogenesis
Given the involvement of angiogenesis in a number of physiological and pathological conditions, its modulation by Hmox1 is an exciting and promising therapeutic approach. However, careful design and strategies are warranted to achieve the desirable effects. For example, it would be desirable to enhance neoangiogenesis in conditions not amenable to revascularization, such as wound healing, peripheral artery disease, and MI. On the other hand, the approach for tumor angiogenesis would be to institute interventions to block Hmox1.
Modulation of Hmox1 to induce local angiogenesis may be achieved via several mechanisms. Hmox1 gene delivery is an approach that has significant therapeutic potential. However, there are at least two major obstacles that need to be overcome for successful gene delivery. First, evidence is needed from ongoing clinical trials that vectors such as adeno-associated viral vectors or other systems are safe and efficient. Second, Hmox1 gene delivery must be specific to the targeted cell/tissue. Targeted and tissue-specific modulation of Hmox1 to induce neoangiogenesis could also be achieved by delivery of molecules such as SDF-1 and VEGF, both of which are known to induce Hmox-1 in addition to their proangiogenic properties (119).
Another approach that is gaining popularity and may be applied to a number of clinical settings is utilization of mesenchymal stem cells. Several studies have shown that Hmox1 expression enhances the protective and proangiogenic effects of mesenchymal stem cells (228, 631, 636, 637). Furthermore, several ongoing clinical trials are investigating the safety and efficacy of mesenchymal stem cells in various clinical conditions. Once approved, induction of Hmox1 prior to administration of mesenchymal stem cells may provide additional beneficial effects that could also be utilized to enhance angiogenesis. It should be noted however that such approaches will also require vigilant design to ensure delivery of the mesenchymal stem cells to the targeted region/tissue to prevent neoangiogenesis at distant sites.
Pharmacological inducers of Hmox1 may also be utilized to enhance angiogenesis at the desired site. An agent with such applicability would be resveratrol, a polyphenolic compound present in red wine. Resveratrol increases neovascularization in the infarcted rat myocardium via induction of VEGF in a thioredoxin-1 and Hmox1-dependent manner (223). Moreover, identification of potential small molecules that are able to stimulate Hmox1 expression could also facilitate the implementation of using Hmox1 as a proangiogenic target. Similarly, the products of Hmox1 reaction could also be considered to improve neovascularization by means of local delivery. The evidence to support their role in the process of neoangiogenesis has been discussed above. However, and as pointed out, these molecules/compounds need to be closely examined to ensure their safety, and careful pharmacokinetic type studies are required to elucidate the optimal dose that would exert the desired effects within the local environment.
While neoangiogenesis is highly beneficial in various settings, inhibition of this process would be highly desirable for cancer therapy. In addition to blocking Hmox1 as a therapeutic strategy for tumors, its effects can also be exploited as a sensitizing approach to enhance the efficacy of chemotherapeutic drugs, radiation, or photodynamic therapy (398). The very same principles that were highlighted in promotion of angiogenesis apply to inhibition of this process. These include safety and efficacy of potential vectors that would carry genes to silence Hmox1 in tumors, delivery of potential agents and/or molecules that would inhibit Hmox1 expression or activity, and investigation of strategies that would enable precise and local delivery to tumors. If successful, such approaches would be beneficial for rapidly growing tumors as well as tumors that are rich in blood vessels such as renal cell cancer.
VII. CONCLUSIONS
Since the identification of heme oxygenases, there has been much focus on their biological functions and roles, and it is currently well accepted that Hmox1 in particular is a key mediator of cytoprotection in response to a wide range of stimuli and stress. Heme oxygenases are highly conserved enzymes evolutionary with homologs found in organisms such as plants, bacteria, and yeast, underscoring their importance in a multitude of biological systems. The discovery of bilirubin having important biological activities rather than simply being a waste product of heme catabolism marked a turning point in the understanding of, and research centered around, heme oxygenases that since has been extended to CO and biliverdin. As described in section V, the role of Hmox1 and Hmox2 in vascular disease modulation is firmly cemented in a wide range of disease settings. The mechanisms that mediate this modulation are beginning to be unraveled, although much more is to be learned. For example, the role of Hmox1 and posttranslationally modified form(s) of this isozyme in various subcellular localizations and the potential nonenzymatic functions of Hmox1 are exciting new concepts for how Hmox1 could mediate cell protection beyond just heme degradation. In addition, the discoveries of noncanonical Hmox in bacteria and insects highlight that there are still many issues on a basic science level that need to be uncovered to fully understand the functions of Hmox.
The translation of experimental findings to human disease is another exciting, albeit challenging, area of research. Many challenges still need to be overcome in this regard. These challenges include issues related to safety and optimal delivery methods of CO and bile pigments, as well as their optimal dosage required to elicit the desired benefits in humans. Improvements in technologies such as gene-delivery and CO-releasing molecules, and the development of specific Hmox1 inducing agents will likely be key to the safe and effective modulation of Hmox1 in various cardiovascular disease settings. Undoubtedly, the future will see advances made in these areas and the development of novel methods to harness the heme oxygenase systems in the context of cardiovascular health and disease in humans.
GRANTS
We acknowledge support from the Ben J. Lipps fellowship grant from the American Society of Nephrology (to A. Zarjou), National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK59600 and P30 DK079337 (to A. Agarwal), and the National Health & Medical Research Council of Australia Program Grant 1052616 and Project Grant 1080604 (to R. Stocker). R. Stocker is supported by Senior Principal Research Fellowship 1111632 from the National Health & Medical Research Council of Australia.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGEMENTS
We thank Drs. Louise Dunn and Chris Stanley for useful comments on part of the manuscript and Dr. Stephanie Kong for providing Figure 3.
A. Ayer and A. Zarjou contributed equally.
Address for reprint requests and other correspondence: R. Stocker, Vascular Biology Division, Victor Chang Cardiac Research Institute, Lowy Packer Building, 405 Liverpool St., Darlinghurst NSW 2010, Australia (e-mail: r.stocker@victorchang.edu.au).
REFERENCES
- 1.Abid S, Houssaini A, Mouraret N, Marcos E, Amsellem V, Wan F, Dubois-Rande JL, Derumeaux G, Boczkowski J, Motterlini R, Adnot S. P21-dependent protective effects of a carbon monoxide-releasing molecule-3 in pulmonary hypertension. Arterioscler Thromb Vasc Biol 34: 304–312, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Abraham NG, Lavrovsky Y, Schwartzman ML, Stoltz RA, Levere RD, Gerritsen ME, Shibahara S, Kappas A. Transfection of the human heme oxygenase gene into rabbit coronary microvessel endothelial cells: protective effect against heme and hemoglobin toxicity. Proc Natl Acad Sci USA 92: 6798–6802, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abraham NG, Scapagnini G, Kappas A. Human heme oxygenase: cell cycle-dependent expression and DNA microarray identification of multiple gene responses after transduction of endothelial cells. J Cell Biochem 90: 1098–1111, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Abran D, Dumont I, Hardy P, Peri K, Li DY, Molotchnikoff S, Varma DR, Chemtob S. Characterization and regulation of prostaglandin E2 receptor and receptor-coupled functions in the choroidal vasculature of the pig during development. Circ Res 80: 463–472, 1997. [PubMed] [Google Scholar]
- 5.Agarwal A, Balla J, Balla G, Croatt AJ, Vercellotti GM, Nath KA. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am J Physiol Renal Fluid Electrolyte Physiol 271: F814–F823, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Ahluwalia N, Genoux A, Ferrieres J, Perret B, Carayol M, Drouet L, Ruidavets JB. Iron status is associated with carotid atherosclerotic plaques in middle-aged adults. J Nutr 140: 812–816, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmad S, Hewett PW, Fujisawa T, Sissaoui S, Cai M, Gueron G, Al-Ani B, Cudmore M, Ahmed SF, Wong MK, Wegiel B, Otterbein LE, Vitek L, Ramma W, Wang K, Ahmed A. Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation. Thromb Haemost 113: 329–337, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed A, Rahman M, Zhang X, Acevedo CH, Nijjar S, Rushton I, Bussolati B, St John J. Induction of placental heme oxygenase-1 is protective against TNFα-induced cytotoxicity and promotes vessel relaxation. Mol Med 6: 391–409, 2000. [PMC free article] [PubMed] [Google Scholar]
- 9.Aizawa T, Ishizaka N, Taguchi J, Kimura S, Kurokawa K, Ohno M. Balloon injury does not induce heme oxygenase-1 expression, but administration of hemin inhibits neointimal formation in balloon-injured rat carotid artery. Biochem Biophys Res Commun 261: 302–307, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Akboga MK, Canpolat U, Sahinarslan A, Alsancak Y, Nurkoc S, Aras D, Aydogdu S, Abaci A. Association of serum total bilirubin level with severity of coronary atherosclerosis is linked to systemic inflammation. Atherosclerosis 240: 110–114, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Almeida AS, Queiroga CS, Sousa MF, Alves PM, Vieira HL. Carbon monoxide modulates apoptosis by reinforcing oxidative metabolism in astrocytes: role of Bcl-2. J Biol Chem 287: 10761–10770, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreas M, Schmid AI, Doberer D, Schewzow K, Weisshaar S, Heinze G, Bilban M, Moser E, Wolzt M. Heme arginate improves reperfusion patterns after ischemia: a randomized, placebo-controlled trial in healthy male subjects. J Cardiovasc Magn Reson 14: 55, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andria B, Bracco A, Attanasio C, Castaldo S, Cerrito MG, Cozzolino S, Di Napoli D, Giovannoni R, Mancini A, Musumeci A, Mezza E, Nasti M, Scuderi V, Staibano S, Lavitrano M, Otterbein LE, Calise F. Biliverdin protects against liver ischemia reperfusion injury in swine. PloS One 8: e69972, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F, Ijzermans J, Roodnat J, Weimar W. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant 4: 811–818, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Bach FH, Ferran C, Soares M, Wrighton CJ, Anrather J, Winkler H, Robson SC, Hancock WW. Modification of vascular responses in xenotransplantation: inflammation and apoptosis. Nat Med 3: 944–948, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Bai CH, Chen JR, Chiu HC, Chou CC, Chau LY, Pan WH. Shorter GT repeat polymorphism in the heme oxygenase-1 gene promoter has protective effect on ischemic stroke in dyslipidemia patients. J Biomed Sci 17: 12, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bak I, Papp G, Turoczi T, Varga E, Szendrei L, Vecsernyes M, Joo F, Tosaki A. The role of heme oxygenase-related carbon monoxide and ventricular fibrillation in ischemic/reperfused hearts. Free Radic Biol Med 33: 639–648, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Bak I, Szendrei L, Turoczi T, Papp G, Joo F, Das DK, de Leiris J, Der P, Juhasz B, Varga E, Bacskay I, Balla J, Kovacs P, Tosaki A. Heme oxygenase-1-related carbon monoxide production and ventricular fibrillation in isolated ischemic/reperfused mouse myocardium. FASEB J 17: 2133–2135, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Bakrania B, Du Toit EF, Wagner KH, Headrick JP, Bulmer AC. Pre- or post-ischemic bilirubin ditaurate treatment reduces oxidative tissue damage and improves cardiac function. Int J Cardiol 202: 27–33, 2016. [DOI] [PubMed] [Google Scholar]
- 20.Balla G, Jacob HS, Eaton JW, Belcher JD, Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler Thromb 11: 1700–1711, 1991. [DOI] [PubMed] [Google Scholar]
- 21.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA 90: 9285–9289, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao W, Song F, Li X, Rong S, Yang W, Wang D, Xu J, Fu J, Zhao Y, Liu L. Association between heme oxygenase-1 gene promoter polymorphisms and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am J Epidemiol 172: 631–636, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Barañano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA 99: 16093–16098, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartz RR, Suliman HB, Piantadosi CA. Redox mechanisms of cardiomyocyte mitochondrial protection. Front Physiol 6: 291, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, van der Toorn M, Boezen HM, Kerstjens HA. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J 30: 1131–1137, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Baum M, Schiff E, Kreiser D, Dennery PA, Stevenson DK, Rosenthal T, Seidman DS. End-tidal carbon monoxide measurements in women with pregnancy-induced hypertension and preeclampsia. Am J Obstet Gynecol 183: 900–903, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Bayer SB, Maghzal G, Stocker R, Hampton MB, Winterbourn CC. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. FASEB J 27: 3315–3322, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Beck K, Wu BJ, Ni J, Santiago FS, Malabanan KP, Li C, Wang Y, Khachigian LM, Stocker R. Interplay between heme oxygenase-1 and the multifunctional transcription factor Yin Yang 1 in the inhibition of intimal hyperplasia. Circ Res 107: 1490–1497, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Beckman JD, Chen C, Nguyen J, Thayanithy V, Subramanian S, Steer CJ, Vercellotti GM. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J Biol Chem 286: 3194–3202, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellner L, Marrazzo G, van Rooijen N, Dunn MW, Abraham NG, Schwartzman ML. Heme oxygenase-2 deletion impairs macrophage function: implication in wound healing. FASEB J 29: 105–115, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellner L, Martinelli L, Halilovic A, Patil KA, Puri N, Dunn MW, Regan RF, Schwartzman ML. Heme oxygenase-2 deletion causes endothelial cell activation marked by oxidative stress, inflammation, and angiogenesis. J Pharmacol Exp Ther 331: 925–932, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellner L, Vitto M, Patil KA, Dunn MW, Regan R, Laniado-Schwartzman M. Exacerbated corneal inflammation and neovascularization in the HO-2 null mice is ameliorated by biliverdin. Exp Eye Res 87: 268–278, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellner L, Wolstein J, Patil KA, Dunn MW, Laniado-Schwartzman M. Biliverdin rescues the HO-2 null mouse phenotype of unresolved chronic inflammation following corneal epithelial injury. Invest Ophthalmol Vis Sci 52: 3246–3253, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Amotz R, Bonagura J, Velayutham M, Hamlin R, Burns P, Adin C. Intraperitoneal bilirubin administration decreases infarct area in a rat coronary ischemia/reperfusion model. Front Physiol 5: 53, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bharucha AE, Choi KM, Saw JJ, Gibbons SJ, Farrugia GF, Carlson DA, Zinsmeister AR. Effects of aspirin & simvastatin and aspirin, simvastatin, & lipoic acid on heme oxygenase-1 in healthy human subjects. Neurogastroenterol Motil 26: 1437–1442, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bharucha AE, Kulkarni A, Choi KM, Camilleri M, Lempke M, Brunn GJ, Gibbons SJ, Zinsmeister AR, Farrugia G. First-in-human study demonstrating pharmacological activation of heme oxygenase-1 in humans. Clin Pharmacol Ther 87: 187–190, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilbija D, Gravning JA, Haugen F, Attramadal H, Valen G. Protecting the heart through delivering DNA encoding for heme oxygenase-1 into skeletal muscle. Life Sci 91: 828–836, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, Bandyopadhyay U. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem 286: 39387–39402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bisht K, Tampe J, Shing C, Bakrania B, Winearls J, Fraser J, Wagner KH, Bulmer AC. Endogenous tetrapyrroles influence leukocyte responses to lipopolysaccharide in human blood: pre-clinical evidence demonstrating the anti-inflammatory potential of biliverdin. J Clin Cell Immunol 5: 1000218, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas C, Shah N, Muthu M, La P, Fernando AP, Sengupta S, Yang G, Dennery PA. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J Biol Chem 289: 26882–26894, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Björkhem-Bergman L, Lindh JD, Bergman P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br J Clin Pharmacol 72: 164–165, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blann AD, Balakrishnan B, Ryan P, Lip GY. Increased levels of plasma haemoxygenase-1 in prostate cancer. Prostate Cancer Prostatic Dis 14: 114–117, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Bolisetty S, Zarjou A, Hull TD, Traylor AM, Perianayagam A, Joseph R, Kamal AI, Arosio P, Soares MP, Jeney V, Balla J, George JF, Agarwal A. Macrophage and epithelial cell H-ferritin expression regulates renal inflammation. Kidney Int 88: 95–108, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 368: 850–853, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Boon AC, Hawkins CL, Coombes JS, Wagner KH, Bulmer AC. Bilirubin scavenges chloramines and inhibits myeloperoxidase-induced protein/lipid oxidation in physiologically relevant hyperbilirubinemic serum. Free Radic Biol Med 86: 259–268, 2015. [DOI] [PubMed] [Google Scholar]
- 46.Botros FT, Dobrowolski L, Navar LG. Renal heme oxygenase-1 induction with hemin augments renal hemodynamics, renal autoregulation, and excretory function. Int J Hypertens 2012: 189512, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Botros FT, Schwartzman ML, Stier CT Jr, Goodman AI, Abraham NG. Increase in heme oxygenase-1 levels ameliorates renovascular hypertension. Kidney Int 68: 2745–2755, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Bouche D, Chauveau C, Roussel JC, Mathieu P, Braudeau C, Tesson L, Soulillou JP, Iyer S, Buelow R, Anegon I. Inhibition of graft arteriosclerosis development in rat aortas following heme oxygenase-1 gene transfer. Transplant Immunol 9: 235–238, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J, Kohler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119: 2323–2332, 2009. [DOI] [PubMed] [Google Scholar]
- 50.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192: 1015–1026, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bulmer AC, Coombes JS, Blanchfield JT, Toth I, Fassett RG, Taylor SM. Bile pigment pharmacokinetics and absorption in the rat: therapeutic potential for enteral administration. Br J Pharmacol 164: 1857–1870, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunn HF, Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem 243: 465–475, 1968. [PubMed] [Google Scholar]
- 53.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science 317: 1393–1397, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Burton MJ, Kapetanaki SM, Chernova T, Jamieson AG, Dorlet P, Santolini J, Moody PC, Mitcheson JS, Davies NW, Schmid R, Raven EL, Storey NM. A heme-binding domain controls regulation of ATP-dependent potassium channels. Proc Natl Acad Sci USA 113: 3785–3790, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, Mason JC. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood 103: 761–766, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Bussolati B, Mason JC. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid Redox Signal 8: 1153–1163, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Byrne RA, Kastrati A, Tiroch K, Schulz S, Pache J, Pinieck S, Massberg S, Seyfarth M, Laugwitz KL, Birkmeier KA, Schömig A, Mehilli J. 2-year clinical and angiographic outcomes from a randomized trial of polymer-free dual drug-eluting stents versus polymer-based Cypher and Endeavor drug-eluting stents. J Am Coll Cardiol 55: 2536–2543, 2010. [DOI] [PubMed] [Google Scholar]
- 58.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78: 415–423, 1996. [DOI] [PubMed] [Google Scholar]
- 59.Cao L, Zhang Z, Cai B, Bai W, Zhang Y, Sun W, Xie X, Sun W, Cai Q, Li Z, Liu D, Xiong Y, Ma M, Liu X, Xu G. Association of heme oxygenase-1 gene rs2071746 polymorphism with vascular outcomes in patients with atherosclerotic stroke. J Neurol Sci 344: 154–157, 2014. [DOI] [PubMed] [Google Scholar]
- 60.Cao YA, Kusy S, Luong R, Wong RJ, Stevenson DK, Contag CH. Heme oxygenase-1 deletion affects stress erythropoiesis. PloS One 6: e20634, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao YA, Wagers AJ, Karsunky H, Zhao H, Reeves R, Wong RJ, Stevenson DK, Weissman IL, Contag CH. Heme oxygenase-1 deficiency leads to disrupted response to acute stress in stem cells and progenitors. Blood 112: 4494–4502, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cary SP, Marletta MA. The case of CO signaling: why the jury is still out. J Clin Invest 107: 1071–1073, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan KH, Ng MK, Stocker R. Haem oxygenase-1 and cardiovascular disease: mechanisms and therapeutic potential. Clin Sci 120: 493–504, 2011. [DOI] [PubMed] [Google Scholar]
- 64.Chang T, Wu L, Wang R. Inhibition of vascular smooth muscle cell proliferation by chronic hemin treatment. Am J Physiol Heart Circ Physiol 295: H999–H1007, 2008. [DOI] [PubMed] [Google Scholar]
- 65.Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res 124: 31–82, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chauveau C, Bouchet D, Roussel JC, Mathieu P, Braudeau C, Renaudin K, Tesson L, Soulillou JP, Iyer S, Buelow R, Anegon I. Gene transfer of heme oxygenase-1 and carbon monoxide delivery inhibit chronic rejection. Am J Transplant 2: 581–592, 2002. [DOI] [PubMed] [Google Scholar]
- 67.Chen KD, Hsu LW, Goto S, Huang KT, Nakano T, Weng WT, Lai CY, Kuo YR, Chiu KW, Wang CC, Cheng YF, Lin CC, Ma YY, Chen CL. Regulation of heme oxygenase 1 expression by miR-27b with stem cell therapy for liver regeneration in rats. Transplant Proc 46: 1198–2200, 2014. [DOI] [PubMed] [Google Scholar]
- 68.Chen RJ, Yuan HH, Zhang TY, Wang ZZ, Hu AK, Wu LL, Yang ZP, Mao YJ, Ji DJ, Zhu XR. Heme oxygenase-2 suppress TNF-α and IL6 expression via TLR4/MyD88-dependent signaling pathway in mouse cerebral vascular endothelial cells. Mol Neurobiol 50: 971–978, 2014. [DOI] [PubMed] [Google Scholar]
- 69.Chen S, Ding Y, Tao W, Zhang W, Liang T, Liu C. Naringenin inhibits TNF-α induced VSMC proliferation and migration via induction of HO-1. Food Chem Toxicol 50: 3025–3031, 2012. [DOI] [PubMed] [Google Scholar]
- 70.Chen S, Kapturczak MH, Wasserfall C, Glushakova OY, Campbell-Thompson M, Deshane JS, Joseph R, Cruz PE, Hauswirth WW, Madsen KM, Croker BP, Berns KI, Atkinson MA, Flotte TR, Tisher CC, Agarwal A. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc Natl Acad Sci USA 102: 7251–7256, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen YC, Gines P, Yang J, Summer SN, Falk S, Russell NS, Schrier RW. Increased vascular heme oxygenase-1 expression contributes to arterial vasodilation in experimental cirrhosis in rats. Hepatology 39: 1075–1087, 2004. [DOI] [PubMed] [Google Scholar]
- 72.Chen YH, Lin SJ, Lin MW, Tsai HL, Kuo SS, Chen JW, Charng MJ, Wu TC, Chen LC, Ding YA, Pan WH, Jou YS, Chau L. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet 111: 1–8, 2002. [DOI] [PubMed] [Google Scholar]
- 73.Chen-Roetling J, Song W, Schipper HM, Regan CS, Regan RF. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 46: 1093–1098, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng C, Noordeloos AM, Jeney V, Soares MP, Moll F, Pasterkamp G, Serruys PW, Duckers HJ. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 119: 3017–3027, 2009. [DOI] [PubMed] [Google Scholar]
- 75.Cheng C, Noorderloos M, van Deel ED, Tempel D, den Dekker W, Wagtmans K, Duncker DJ, Soares MP, Laman JD, Duckers HJ. Dendritic cell function in transplantation arteriosclerosis is regulated by heme oxygenase 1. Circ Res 106: 1656–1666, 2010. [DOI] [PubMed] [Google Scholar]
- 76.Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, Gallo D, Cszimadia E, Bach FH, Lee PJ, Otterbein LE. Hypoxia-inducible factor 1α stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci USA 104: 5109–5114, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res 71: 393–401, 2006. [DOI] [PubMed] [Google Scholar]
- 78.Choi BM, Kim YM, Jeong YR, Pae HO, Song CE, Park JE, Ahn YK, Chung HT. Induction of heme oxygenase-1 is involved in anti-proliferative effects of paclitaxel on rat vascular smooth muscle cells. Biochem Biophys Res Commun 321: 132–137, 2004. [DOI] [PubMed] [Google Scholar]
- 79.Choi HC, Kim HS, Lee KY, Chang KC, Kang YJ. NS-398, a selective COX-2 inhibitor, inhibits proliferation of IL-1β-stimulated vascular smooth muscle cells by induction of HO-1. Biochem Biophys Res Commun 376: 753–757, 2008. [DOI] [PubMed] [Google Scholar]
- 80.Choi HC, Lee KY, Lee DH, Kang YJ. Heme oxygenase-1 induced by aprotinin inhibits vascular smooth muscle cell proliferation through cell cycle arrest in hypertensive rats. Korean J Physiol Pharmacol 13: 309–313, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res 86: 1224–1229, 2000. [DOI] [PubMed] [Google Scholar]
- 82.Cisowski J, Loboda A, Jozkowicz A, Chen S, Agarwal A, Dulak J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem Biophys Res Commun 326: 670–676, 2005. [DOI] [PubMed] [Google Scholar]
- 83.Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J 348: 615–619, 2000. [PMC free article] [PubMed] [Google Scholar]
- 84.Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol 278: H643–H651, 2000. [DOI] [PubMed] [Google Scholar]
- 85.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 93: e2–e8, 2003. [DOI] [PubMed] [Google Scholar]
- 86.Clarke HM, Shrivastava S, Motterlini R, Sawle P, Chen D, Dorling A. Donor HO-1 expression inhibits intimal hyperplasia in unmanipulated graft recipients: a potential role for CD8+ T-cell modulation by carbon monoxide. Transplantation 88: 653–661, 2009. [DOI] [PubMed] [Google Scholar]
- 87.Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM. Mechanism of nitric oxide-induced vasodilatation: refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ Res 84: 210–219, 1999. [DOI] [PubMed] [Google Scholar]
- 88.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46: 205–229, 1994. [PubMed] [Google Scholar]
- 89.Collinson EJ, Wimmer-Kleikamp S, Gerega SK, Yang YH, Parish CR, Dawes IW, Stocker R. The yeast homolog of heme oxygenase-1 affords cellular antioxidant protection via the transcriptional regulation of known antioxidant genes. J Biol Chem 286: 2205–2214, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Conde de la Rosa L, Vrenken TE, Hannivoort RA, Buist-Homan M, Havinga R, Slebos DJ, Kauffman HF, Faber KN, Jansen PL, Moshage H. Carbon monoxide blocks oxidative stress-induced hepatocyte apoptosis via inhibition of the p54 JNK isoform. Free Radic Biol Med 44: 1323–1333, 2008. [DOI] [PubMed] [Google Scholar]
- 91.Cooper R, Cutler J, Desvigne-Nickens P, Fortmann SP, Friedman L, Havlik R, Hogelin G, Marler J, McGovern P, Morosco G, Mosca L, Pearson T, Stamler J, Stryer D, Thom T. Trends and disparities in coronary heart disease, stroke, and other cardiovascular diseases in the United States: findings of the national conference on cardiovascular disease prevention. Circulation 102: 3137–3147, 2000. [DOI] [PubMed] [Google Scholar]
- 92.Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, Campanella A, Brunelli S, Manfredi AA, Apostoli P, Silvestri L, Camaschella C, Rovere-Querini P. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 95: 1814–1822, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Corriu C, Feletou M, Canet E, Vanhoutte PM. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br J Pharmacol 119: 959–964, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Corriu C, Feletou M, Edwards G, Weston AH, Vanhoutte PM. Differential effects of prostacyclin and iloprost in the isolated carotid artery of the guinea-pig. Eur J Pharmacol 426: 89–94, 2001. [DOI] [PubMed] [Google Scholar]
- 95.Cortese-Krott MM, Fernandez BO, Santos JL, Mergia E, Grman M, Nagy P, Kelm M, Butler A, Feelisch M. Nitrosopersulfide (SSNO−) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biol 2: 234–244, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cortese-Krott MM, Kuhnle GG, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, Nagy P, King SB, Saavedra JE, Keefer LK, Singer M, Kelm M, Butler AR, Feelisch M. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc Natl Acad Sci USA 112: E4651–E4660, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol 553: 183–189, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cruse I, Maines MD. Evidence suggesting that the two forms of heme oxygenase are products of different genes. J Biol Chem 263: 3348–5333, 1988. [PubMed] [Google Scholar]
- 99.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, Ahmed A. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 115: 1789–1797, 2007. [DOI] [PubMed] [Google Scholar]
- 100.Cudmore MJ, Ahmad S, Sissaoui S, Ramma W, Ma B, Fujisawa T, Al-Ani B, Wang K, Cai M, Crispi F, Hewett PW, Gratacos E, Egginton S, Ahmed A. Loss of Akt activity increases circulating soluble endoglin release in preeclampsia: identification of inter-dependency between Akt-1 and heme oxygenase-1. Eur Heart J 33: 1150–1158, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Czibik G, Martinov V, Ruusalepp A, Sagave J, Skare O, Valen G. In vivo remote delivery of DNA encoding for hypoxia-inducible factor 1 alpha reduces myocardial infarct size. Clin Transl Sci 2: 33–40, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Da Silva JL, Tiefenthaler M, Park E, Escalante B, Schwartzman ML, Levere RD, Abraham NG. Tin-mediated heme oxygenase gene activation and cytochrome P450 arachidonate hydroxylase inhibition in spontaneously hypertensive rats. Am J Med Sci 307: 173–181, 1994. [DOI] [PubMed] [Google Scholar]
- 103.Daenen KE, Martens P, Bammens B. Association of HO-1 (GT)n promoter polymorphism and cardiovascular disease: a reanalysis of the literature. Can J Cardiol 32: 160–168, 2016. [DOI] [PubMed] [Google Scholar]
- 104.De Backer O, Elinck E, Blanckaert B, Leybaert L, Motterlini R, Lefebvre RA. Water-soluble CO-releasing molecules reduce the development of postoperative ileus via modulation of MAPK/HO-1 signalling and reduction of oxidative stress. Gut 58: 347–356, 2009. [DOI] [PubMed] [Google Scholar]
- 105.De Mey JG, Vanhoutte PM. End o' the line revisited: moving on from nitric oxide to CGRP. Life Sci 118: 120–128, 2014. [DOI] [PubMed] [Google Scholar]
- 106.De Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM, BEACON . Trial Investigators. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369: 2492–2503, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng YM, Wu BJ, Witting PK, Stocker R. Probucol protects against smooth muscle cell proliferation by upregulating heme oxygenase-1. Circulation 110: 1855–1860, 2004. [DOI] [PubMed] [Google Scholar]
- 108.Dennery PA, Spitz DR, Yang G, Tatarov A, Lee CS, Shegog ML, Poss KD. Oxygen toxicity and iron accumulation in the lungs of mice lacking heme oxygenase-2. J Clin Invest 101: 1001–1011, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Deramaudt BM, Braunstein S, Remy P, Abraham NG. Gene transfer of human heme oxygenase into coronary endothelial cells potentially promotes angiogenesis. J Cell Biochem 68: 121–127, 1998. [DOI] [PubMed] [Google Scholar]
- 110.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med 204: 605–618, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 49: 717–727, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dioum EM, Rutter J, Tuckerman JR, Gonzalez G, Gilles-Gonzalez MA, McKnight SL. NPAS2: a gas-responsive transcription factor. Science 298: 2385–2387, 2002. [DOI] [PubMed] [Google Scholar]
- 113.Doberer D, Haschemi A, Andreas M, Zapf TC, Clive B, Jeitler M, Heinzl H, Wagner O, Wolzt M, Bilban M. Haem arginate infusion stimulates haem oxygenase-1 expression in healthy subjects. Br J Pharmacol 161: 1751–1762, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doré S, Goto S, Sampei K, Blackshaw S, Hester LD, Ingi T, Sawa A, Traystman RJ, Koehler RC, Snyder SH. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 99: 587–592, 2000. [DOI] [PubMed] [Google Scholar]
- 115.Doré S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci USA 96: 2445–2450, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Du D, Chang S, Chen B, Zhou H, Chen ZK. Adenovirus-mediated heme oxygenase transfer inhibits graft arteriosclerosis in rat aortic transplants. Transplant Proc 39: 3446–3448, 2007. [DOI] [PubMed] [Google Scholar]
- 117.Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, Clinton Webb R, Lee ME, Nabel GJ, Nabel EG. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat Med 7: 693–698, 2001. [DOI] [PubMed] [Google Scholar]
- 118.Duckles H, Boycott HE, Al-Owais MM, Elies J, Johnson E, Dallas ML, Porter KE, Giuntini F, Boyle JP, Scragg JL, Peers C. Heme oxygenase-1 regulates cell proliferation via carbon monoxide-mediated inhibition of T-type Ca2+ channels. Pflügers Arch 467: 415–427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117: 231–241, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, Green CJ, Pachinger O, Weidinger F, Motterlini R. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid Redox Signal 4: 229–240, 2002. [DOI] [PubMed] [Google Scholar]
- 121.Dunn LL, Midwinter RG, Ni J, Hamid HA, Parish CR, Stocker R. New insights into intracellular locations and functions of heme oxygenase-1. Antioxid Redox Signal 20: 1723–1742, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Durante W, Christodoulides N, Cheng K, Peyton KJ, Sunahara RK, Schafer AI. cAMP induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle. Am J Physiol Heart Circ Physiol 273: H317–H323, 1997. [DOI] [PubMed] [Google Scholar]
- 123.Durante W, Kroll MH, Christodoulides N, Peyton KJ, Schafer AI. Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ Res 80: 557–564, 1997. [DOI] [PubMed] [Google Scholar]
- 124.Durante W, Peyton KJ, Schafer AI. Platelet-derived growth factor stimulates heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 19: 2666–2672, 1999. [DOI] [PubMed] [Google Scholar]
- 125.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396: 269–272, 1998. [DOI] [PubMed] [Google Scholar]
- 126.Ekelund U, Adner M, Edvinsson L, Mellander S. Effects of selective ETB-receptor stimulation on arterial, venous and capillary functions in cat skeletal muscle. Br J Pharmacol 112: 887–894, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87: 474–479, 2000. [DOI] [PubMed] [Google Scholar]
- 128.Exner M, Böhmig GA, Schillinger M, Regele H, Watschinger B, Hörl WH, Raith M, Mannhalter C, Wagner OF. Donor heme oxygenase-1 genotype is associated with renal allograft function. Transplantation 77: 538–542, 2004. [DOI] [PubMed] [Google Scholar]
- 129.Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic Biol Med 37: 1097–1104, 2004. [DOI] [PubMed] [Google Scholar]
- 130.Exner M, Schillinger M, Minar E, Mlekusch W, Schlerka G, Haumer M, Mannhalter C, Wagner O. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with restenosis after percutaneous transluminal angioplasty. J Endovasc Ther 8: 433–440, 2001. [DOI] [PubMed] [Google Scholar]
- 131.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol 47: C7–12, 2006. [DOI] [PubMed] [Google Scholar]
- 132.Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G, Hamada A, Maeda H. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res 63: 3567–3574, 2003. [PubMed] [Google Scholar]
- 133.Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int J Cancer 109: 1–8, 2004. [DOI] [PubMed] [Google Scholar]
- 134.Fayad-Kobeissi S, Ratovonantenaina J, Dabiré H, Wilson JL, Rodriguez AM, Berdeaux A, Dubois-Randé JL, Mann BE, Motterlini R, Foresti R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem Pharmacol 102: 64–77, 2016. [DOI] [PubMed] [Google Scholar]
- 135.Fernandez M, Bonkovsky HL. Vascular endothelial growth factor increases heme oxygenase-1 protein expression in the chick embryo chorioallantoic membrane. Br J Pharmacol 139: 634–640, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, Tysoe SA, Wolosker H, Barañano DE, Doré S, Poss KD, Snyder SH. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol 1: 152–157, 1997. [DOI] [PubMed] [Google Scholar]
- 137.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401: 493–497, 1999. [DOI] [PubMed] [Google Scholar]
- 138.Fleischhacker AS, Sharma A, Choi M, Spencer AM, Bagai I, Hoffman BM, Ragsdale SW. The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 54: 2709–2718, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Flynn CR, Albaugh VL, Cai S, Cheung-Flynn J, Williams PE, Brucker RM, Bordenstein SR, Guo Y, Wasserman DH, Abumrad NN. Bile diversion to the distal small intestine has comparable metabolic benefits to bariatric surgery. Nat Commun 6: 7715, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Foresti R, Bains SK, Pitchumony TS, de Castro Bras LE, Drago F, Dubois-Rande JL, Bucolo C, Motterlini R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76: 132–148, 2013. [DOI] [PubMed] [Google Scholar]
- 141.Foresti R, Goatly H, Green CJ, Motterlini R. Role of heme oxygenase-1 in hypoxia-reoxygenation: requirement of substrate heme to promote cardioprotection. Am J Physiol Heart Circ Physiol 281: H1976–H1984, 2001. [DOI] [PubMed] [Google Scholar]
- 142.Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe A, Green CJ, Motterlini R. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol 142: 453–460, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fraser ST, Midwinter RG, Berger BS, Stocker R. Heme Oxygenase-1: a critical link between iron metabolism, erythropoiesis, and development. Adv Hematol 2011: 473709, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fraser ST, Midwinter RG, Coupland LA, Kong S, Berger BS, Yeo JH, Andrade OC, Cromer D, Suarna C, Lam M, Maghzal GJ, Chong BH, Parish CR, Stocker R. Heme oxygenase-1 deficiency alters erythroblastic island formation, steady-state erythropoiesis and red blood cell lifespan in mice. Haematologica 100: 601–610, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Frey GH, Krider DW. Serum ferritin and myocardial infarct. W V Med J 90: 13–15, 1994. [PubMed] [Google Scholar]
- 146.Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R. Carbon monoxide protects against cardiac ischemia–reperfusion injury in vivo via MAPK and Akt–eNOS pathways. Arterioscler Thromb Vasc Biol 24: 1848–1853, 2004. [DOI] [PubMed] [Google Scholar]
- 147.Furchgott RF, Jothianandan D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels 28: 52–61, 1991. [DOI] [PubMed] [Google Scholar]
- 148.Gaetano C, Massimo L, Alberto M. Control of iron homeostasis as a key component of macrophage polarization. Haematologica 95: 1801–1803, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, Klepetko W, McGoon MD, McLaughlin VV, Preston IR, Rubin LJ, Sandoval J, Seeger W, Keogh A. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 62: D60–72, 2013. [DOI] [PubMed] [Google Scholar]
- 150.Gao C, Peng FH, Peng LK. MiR-200c sensitizes clear-cell renal cell carcinoma cells to sorafenib and imatinib by targeting heme oxygenase-1. Neoplasma 61: 680–689, 2014. [DOI] [PubMed] [Google Scholar]
- 151.Gauthier KM, Chawengsub Y, Goldman DH, Conrow RE, Anjaiah S, Falck JR, Campbell WB. 11(R),12(S),15(S)-trihydroxyeicosa-5(Z),8(Z),13(E)-trienoic acid: an endothelium-derived 15-lipoxygenase metabolite that relaxes rabbit aorta. Am J Physiol Heart Circ Physiol 294: H1467–H1472, 2008. [DOI] [PubMed] [Google Scholar]
- 152.Gauthier KM, Goldman DH, Aggarwal NT, Chawengsub Y, Falck JR, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in acetylcholine-induced relaxations of mouse arteries. Am J Physiol Heart Circ Physiol 300: H725–H735, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gauthier KM, Spitzbarth N, Edwards EM, Campbell WB. Apamin-sensitive K+ currents mediate arachidonic acid-induced relaxations of rabbit aorta. Hypertension 43: 413–419, 2004. [DOI] [PubMed] [Google Scholar]
- 154.Gauthier KM, Zhang DX, Cui L, Nithipatikom K, Campbell WB. Angiotensin II relaxations of bovine adrenal cortical arteries: role of angiotensin II metabolites and endothelial nitric oxide. Hypertension 52: 150–155, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gauthier KM, Zhang DX, Edwards EM, Holmes B, Campbell WB. Angiotensin II dilates bovine adrenal cortical arterioles: role of endothelial nitric oxide. Endocrinology 146: 3319–3324, 2005. [DOI] [PubMed] [Google Scholar]
- 156.Gemsa D, Woo CH, Fudenberg HH, Schmid R. Stimulation of heme oxygenase in macrophages and liver by endotoxin. J Clin Invest 53: 647–651, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goda N, Suzuki K, Naito M, Takeoka S, Tsuchida E, Ishimura Y, Tamatani T, Suematsu M. Distribution of heme oxygenase isoforms in rat liver. Topographic basis for carbon monoxide-mediated microvascular relaxation. J Clin Invest 101: 604–612, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goel SA, Guo LW, Liu B, Kent KC. Mechanisms of post-intervention arterial remodelling. Cardiovasc Res 96: 363–371, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc Soc Exp Biol Med 214: 54–61, 1997. [DOI] [PubMed] [Google Scholar]
- 160.Govindaraju V, Teoh H, Hamid Q, Cernacek P, Ward ME. Interaction between endothelial heme oxygenase-2 and endothelin-1 in altered aortic reactivity after hypoxia in rats. Am J Physiol Heart Circ Physiol 288: H962–H970, 2005. [DOI] [PubMed] [Google Scholar]
- 161.Grochot-Przeczek A, Kotlinowski J, Kozakowska M, Starowicz K, Jagodzinska J, Stachurska A, Volger OL, Bukowska-Strakova K, Florczyk U, Tertil M, Jazwa A, Szade K, Stepniewski J, Loboda A, Horrevoets AJ, Dulak J, Jozkowicz A. Heme oxygenase-1 is required for angiogenic function of bone marrow-derived progenitor cells: role in therapeutic revascularization. Antioxid Redox Signal 20: 1677–1692, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grochot-Przeczek A, Lach R, Mis J, Skrzypek K, Gozdecka M, Sroczynska P, Dubiel M, Rutkowski A, Kozakowska M, Zagorska A, Walczynski J, Was H, Kotlinowski J, Drukala J, Kurowski K, Kieda C, Herault Y, Dulak J, Jozkowicz A. Heme oxygenase-1 accelerates cutaneous wound healing in mice. PloS One 4: e5803, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grundemar L, Ny L. Pitfalls using metalloporphyrins in carbon monoxide research. Trends Pharmacol Sci 18: 193–195, 1997. [DOI] [PubMed] [Google Scholar]
- 164.Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, Dawn B, Motterlini R, Bolli R. Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol 286: H1649–H1653, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Haider A, Olszanecki R, Gryglewski R, Schwartzman ML, Lianos E, Kappas A, Nasjletti A, Abraham NG. Regulation of cyclooxygenase by the heme-heme oxygenase system in microvessel endothelial cells. J Pharmacol Exp Ther 300: 188–194, 2002. [DOI] [PubMed] [Google Scholar]
- 166.Halilovic A, Patil KA, Bellner L, Marrazzo G, Castellano K, Cullaro G, Dunn MW, Schwartzman ML. Knockdown of heme oxygenase-2 impairs corneal epithelial cell wound healing. J Cell Physiol 226: 1732–1740, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219: 1–14, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. J Pharmacol Exp Ther 329: 94–101, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hanselmann C, Mauch C, Werner S. Haem oxygenase-1: a novel player in cutaneous wound repair and psoriasis? Biochem J 353: 459–466, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hara E, Takahashi K, Tominaga T, Kumabe T, Kayama T, Suzuki H, Fujita H, Yoshimoto T, Shirato K, Shibahara S. Expression of heme oxygenase and inducible nitric oxide synthase mRNA in human brain tumors. Biochem Biophys Res Commun 224: 153–158, 1996. [DOI] [PubMed] [Google Scholar]
- 171.Hartsfield CL, Alam J, Cook JL, Choi AM. Regulation of heme oxygenase-1 gene expression in vascular smooth muscle cells by nitric oxide. Am J Physiol Lung Cell Mol Physiol 273: L980–L988, 1997. [DOI] [PubMed] [Google Scholar]
- 172.Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, Higashimura Y, Katada K, Handa O, Ishikawa T, Yagi N, Kokura S, Ichikawa H, Muto A, Igarashi K, Yoshikawa T. BTB and CNC homolog 1 (Bach1) deficiency ameliorates TNBS colitis in mice: role of M2 macrophages and heme oxygenase-1. Inflamm Bowel Dis 19: 740–753, 2013. [DOI] [PubMed] [Google Scholar]
- 173.Haudenschild CC, Schwartz SM. Endothelial regeneration. II. Restitution of endothelial continuity. Lab Invest 41: 407–418, 1979. [PubMed] [Google Scholar]
- 174.He JZ, Ho JJ, Gingerich S, Courtman DW, Marsden PA, Ward ME. Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J Biol Chem 285: 9452–9461, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hedlund BE, Hallaway PE. High-dose systemic iron chelation attenuates reperfusion injury. Biochem Soc Trans 21: 340–343, 1993. [DOI] [PubMed] [Google Scholar]
- 176.Heeba G, Moselhy ME, Hassan M, Khalifa M, Gryglewski R, Malinski T. Anti-atherogenic effect of statins: role of nitric oxide, peroxynitrite and haem oxygenase-1. Br J Pharmacol 156: 1256–1266, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297, 2004. [DOI] [PubMed] [Google Scholar]
- 178.Hill MA, Falcone JC, Meininger GA. Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am J Physiol Heart Circ Physiol 259: H1586–H1594, 1990. [DOI] [PubMed] [Google Scholar]
- 179.Hinkel R, Lange P, Petersen B, Gottlieb E, Ng JK, Finger S, Horstkotte J, Lee S, Thormann M, Knorr M, El-Aouni C, Boekstegers P, Reichart B, Wenzel P, Niemann H, Kupatt C. Heme oxygenase-1 gene therapy provides cardioprotection via control of post-ischemic inflammation: an experimental study in a pre-clinical pig model. J Am Coll Cardiol 66: 154–165, 2015. [DOI] [PubMed] [Google Scholar]
- 180.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, Suzuki S, Shibahara S, Sasaki H. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood 102: 1619–1621, 2003. [DOI] [PubMed] [Google Scholar]
- 181.Hirai K, Sasahira T, Ohmori H, Fujii K, Kuniyasu H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int J Cancer 120: 500–505, 2007. [DOI] [PubMed] [Google Scholar]
- 182.Hiwasa T, Sakiyama S. Increase in the synthesis of a Mr 32,000 protein in BALB/c 3T3 cells after treatment with tumor promoters, chemical carcinogens, metal salts, and heat shock. Cancer Res 46: 2474–2481, 1986. [PubMed] [Google Scholar]
- 183.Hock TD, Liby K, Wright MM, McConnell S, Schorpp-Kistner M, Ryan TM, Agarwal A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J Biol Chem 282: 6875–6886, 2007. [DOI] [PubMed] [Google Scholar]
- 184.Holay MP, Choudhary AA, Suryawanshi SD. Serum ferritin-a novel risk factor in acute myocardial infarction. Indian Heart J 64: 173–177, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hori R, Kashiba M, Toma T, Yachie A, Goda N, Makino N, Soejima A, Nagasawa T, Nakabayashi K, Suematsu M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide-induced cytotoxicity. J Biol Chem 277: 10712–10718, 2002. [DOI] [PubMed] [Google Scholar]
- 186.Hou W, Tian Q, Steuerwald NM, Schrum LW, Bonkovsky HL. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim Biophys Acta 1819: 1113–1122, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hsu HH, Ko WJ, Hsu JY, Chen JS, Lee YC, Lai IR, Chen CF. Simvastatin ameliorates established pulmonary hypertension through a heme oxygenase-1 dependent pathway in rats. Respir Res 10: 32, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hsu M, Muchova L, Morioka I, Wong RJ, Schröder H, Stevenson DK. Tissue-specific effects of statins on the expression of heme oxygenase-1 in vivo. Biochem Biophys Res Commun 343: 738–744, 2006. [DOI] [PubMed] [Google Scholar]
- 189.Hu C, Dandapat A, Chen J, Liu Y, Hermonat PL, Carey RM, Mehta JL. Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis 199: 288–294, 2008. [DOI] [PubMed] [Google Scholar]
- 190.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res 96: 376–383, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Huang JS, Ramamurthy SK, Lin X, Le Breton GC. Cell signalling through thromboxane A2 receptors. Cell Signal 16: 521–533, 2004. [DOI] [PubMed] [Google Scholar]
- 192.Huber WJ 3rd, Backes WL. Expression and characterization of full-length human heme oxygenase-1: the presence of intact membrane-binding region leads to increased binding affinity for NADPH cytochrome P450 reductase. Biochemistry 46: 12212–12219, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 1: e85817, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 280: 605–613, 1998. [DOI] [PubMed] [Google Scholar]
- 195.Hyvelin JM, Maurel B, Uzbekov R, Motterlini R, Lermusiaux P. Hemin prevents in-stent stenosis in rat and rabbit models by inducing heme-oxygenase-1. J Vasc Surg 51: 417–428, 2010. [DOI] [PubMed] [Google Scholar]
- 196.Igarashi K, Watanabe-Matsui M. Wearing red for signaling: the heme-bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J Exp Med 232: 229–253, 2014. [DOI] [PubMed] [Google Scholar]
- 197.Ignarro LJ, Harbison RG, Wood KS, Wolin MS, McNamara DB, Hyman AL, Kadowitz PJ. Differences in responsiveness of intrapulmonary artery and vein to arachidonic acid: mechanism of arterial relaxation involves cyclic guanosine 3':5'-monophosphate and cyclic adenosine 3':5'-monophosphate. J Pharmacol Exp Ther 233: 560–569, 1985. [PubMed] [Google Scholar]
- 198.Ishikawa K, Matera KM, Zhou H, Fujii H, Sato M, Yoshimura T, Ikeda-Saito M, Yoshida T. Identification of histidine 45 as the axial heme iron ligand of heme oxygenase-2. J Biol Chem 273: 4317–4322, 1998. [DOI] [PubMed] [Google Scholar]
- 199.Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest 100: 1209–1216, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ishikawa K, Sato M, Yoshida T. Expression of rat heme oxygenase in Escherichia coli as a catalytically active, full-length form that binds to bacterial membranes. Eur J Biochem 202: 161–165, 1991. [DOI] [PubMed] [Google Scholar]
- 201.Ishikawa K, Sugawara D, Goto J, Watanabe Y, Kawamura K, Shiomi M, Itabe H, Maruyama Y. Heme oxygenase-1 inhibits atherogenesis in Watanabe heritable hyperlipidemic rabbits. Circulation 104: 1831–1836, 2001. [DOI] [PubMed] [Google Scholar]
- 202.Ishikawa K, Sugawara D, Wang XP, Suzuki K, Itabe H, Maruyama Y, Lusis AJ. Heme oxygenase-1 inhibits atherosclerotic lesion formation in LDL-receptor knockout mice. Circ Res 88: 506–512, 2001. [DOI] [PubMed] [Google Scholar]
- 203.Ishikawa M, Kajimura M, Adachi T, Maruyama K, Makino N, Goda N, Yamaguchi T, Sekizuka E, Suematsu M. Carbon monoxide from heme oxygenase-2 is a tonic regulator against NO-dependent vasodilatation in the adult rat cerebral microcirculation. Circ Res 97: e104–114, 2005. [DOI] [PubMed] [Google Scholar]
- 204.Ishizaka N, Griendling KK. Heme oxygenase-1 is regulated by angiotensin II in rat vascular smooth muscle cells. Hypertension 29: 790–795, 1997. [DOI] [PubMed] [Google Scholar]
- 205.Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, Maeda Y, Urabe M, Mizukami H, Kume A, Takahashi M, Ikeda U, Shimada K, Ozawa K. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res 101: 734–741, 2007. [DOI] [PubMed] [Google Scholar]
- 206.Iyer AK, Greish K, Fang J, Murakami R, Maeda H. High-loading nanosized micelles of copoly(styrene-maleic acid)-zinc protoporphyrin for targeted delivery of a potent heme oxygenase inhibitor. Biomaterials 28: 1871–1881, 2007. [DOI] [PubMed] [Google Scholar]
- 207.Jadhav A, Tiwari S, Lee P, Ndisang JF. The heme oxygenase system selectively enhances the anti-inflammatory macrophage-M2 phenotype, reduces pericardial adiposity, and ameliorated cardiac injury in diabetic cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther 345: 239–249, 2013. [DOI] [PubMed] [Google Scholar]
- 208.Jamal Uddin M, Joe Y, Kim SK, Oh Jeong S, Ryter SW, Pae HO, Chung HT. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell Mol Immunol 13: 170–179, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jansen T, Hortmann M, Oelze M, Opitz B, Steven S, Schell R, Knorr M, Karbach S, Schuhmacher S, Wenzel P, Münzel T, Daiber A. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J Mol Cell Cardiol 49: 186–195, 2010. [DOI] [PubMed] [Google Scholar]
- 210.Jazwa A, Stepniewski J, Zamykal M, Jagodzinska J, Meloni M, Emanueli C, Jozkowicz A, Dulak J. Pre-emptive hypoxia-regulated HO-1 gene therapy improves post-ischaemic limb perfusion and tissue regeneration in mice. Cardiovasc Res 97: 115–124, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis 45: 173–202, 2002. [DOI] [PubMed] [Google Scholar]
- 212.Johnson FK, Johnson RA. Carbon monoxide promotes endothelium-dependent constriction of isolated gracilis muscle arterioles. Am J Physiol Regul Integr Comp Physiol 285: R536–R541, 2003. [DOI] [PubMed] [Google Scholar]
- 213.Johnson FK, Johnson RA, Durante W, Jackson KE, Stevenson BK, Peyton KJ. Metabolic syndrome increases endogenous carbon monoxide production to promote hypertension and endothelial dysfunction in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 290: R601–R608, 2006. [DOI] [PubMed] [Google Scholar]
- 214.Johnson RA, Colombari E, Colombari DS, Lavesa M, Talman WT, Nasjletti A. Role of endogenous carbon monoxide in central regulation of arterial pressure. Hypertension 30: 962–967, 1997. [DOI] [PubMed] [Google Scholar]
- 215.Johnson RA, Lavesa M, Askari B, Abraham NG, Nasjletti A. A heme oxygenase product, presumably carbon monoxide, mediates a vasodepressor function in rats. Hypertension 25: 166–169, 1995. [DOI] [PubMed] [Google Scholar]
- 216.Jones AW, Durante W, Korthuis RJ. Heme oxygenase-1 deficiency leads to alteration of soluble guanylate cyclase redox regulation. J Pharmacol Exp Ther 335: 85–91, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jones DP, Sies H. The redox code. Antioxid Redox Signal 23: 734–746, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Józkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid Redox Signal 5: 155–162, 2003. [DOI] [PubMed] [Google Scholar]
- 219.Józkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Effect of prostaglandin-J2 on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid Redox Signal 4: 577–585, 2002. [DOI] [PubMed] [Google Scholar]
- 220.Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation 104: 1519–1525, 2001. [DOI] [PubMed] [Google Scholar]
- 221.Jung SS, Moon JS, Xu JF, Ifedigbo E, Ryter SW, Choi AM, Nakahira K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am J Physiol Lung Cell Mol Physiol 308: L1058–L1067, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kaartokallio T, Klemetti MM, Timonen A, Uotila J, Heinonen S, Kajantie E, Kere J, Kivinen K, Pouta A, Lakkisto P, Laivuori H. Microsatellite polymorphism in the heme oxygenase-1 promoter is associated with nonsevere and late-onset preeclampsia. Hypertension 64: 172–177, 2014. [DOI] [PubMed] [Google Scholar]
- 223.Kaga S, Zhan L, Matsumoto M, Maulik N. Resveratrol enhances neovascularization in the infarcted rat myocardium through the induction of thioredoxin-1, heme oxygenase-1 and vascular endothelial growth factor. J Mol Cell Cardiol 39: 813–822, 2005. [DOI] [PubMed] [Google Scholar]
- 224.Kang L, Grande JP, Farrugia G, Croatt AJ, Katusic ZS, Nath KA. Functioning of an arteriovenous fistula requires heme oxygenase-2. Am J Physiol Renal Physiol 305: F545–F552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kaur H, Hughes MN, Green CJ, Naughton P, Foresti R, Motterlini R. Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett 543: 113–119, 2003. [DOI] [PubMed] [Google Scholar]
- 226.Kawamura K, Ishikawa K, Wada Y, Kimura S, Matsumoto H, Kohro T, Itabe H, Kodama T, Maruyama Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler Thromb Vasc Biol 25: 155–160, 2005. [DOI] [PubMed] [Google Scholar]
- 227.Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I. Heme oxygenase-1 deficiency: the first autopsy case. Hum Pathol 33: 125–130, 2002. [DOI] [PubMed] [Google Scholar]
- 228.Kawashiri MA, Nakanishi C, Tsubokawa T, Shimojima M, Yoshida S, Yoshimuta T, Konno T, Yamagishi M, Hayashi K. Impact of enhanced production of endogenous heme oxygenase-1 by pitavastatin on survival and functional activities of bone marrow-derived mesenchymal stem cells. J Cardiovasc Pharmacol 65: 601–606, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kayser H, Wray V, Nimtz M. Structure of a novel farnesylated bilin from an insect–formation by α-cleavage of heme A of mitochondrial cytochrome c oxidases? FEBS J 281: 2366–2376, 2014. [DOI] [PubMed] [Google Scholar]
- 230.Ke K, Sul OJ, Rajasekaran M, Choi HS. MicroRNA-183 increases osteoclastogenesis by repressing heme oxygenase-1. Bone 81: 237–246, 2015. [DOI] [PubMed] [Google Scholar]
- 231.Kelsen S, Patel BJ, Parker LB, Vera T, Rimoldi JM, Gadepalli RS, Drummond HA, Stec DE. Heme oxygenase attenuates angiotensin II-mediated superoxide production in cultured mouse thick ascending loop of Henle cells. Am J Physiol Renal Physiol 295: F1158–F1165, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Keyse SM, Applegate LA, Tromvoukis Y, Tyrrell RM. Oxidant stress leads to transcriptional activation of the human heme oxygenase gene in cultured skin fibroblasts. Mol Cell Biol 10: 4967–4969, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Keyse SM, Tyrrell RM. Both near ultraviolet radiation and the oxidizing agent hydrogen peroxide induce a 32-kDa stress protein in normal human skin fibroblasts. J Biol Chem 262: 14821–14825, 1987. [PubMed] [Google Scholar]
- 234.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA 86: 99–103, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kharitonov VG, Sharma VS, Pilz RB, Magde D, Koesling D. Basis of guanylate cyclase activation by carbon monoxide. Proc Natl Acad Sci USA 92: 2568–2571, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kikuchi G, Yoshida T. Function and induction of the microsomal heme oxygenase. Mol Cell Biochem 53/54: 163–183, 1983. [DOI] [PubMed] [Google Scholar]
- 237.Kim D, Yukl ET, Moenne-Loccoz P, Montellano PR. Fungal heme oxygenases: Functional expression and characterization of Hmx1 from Saccharomyces cerevisiae and CaHmx1 from Candida albicans. Biochemistry 45: 14772–14780, 2006. [DOI] [PubMed] [Google Scholar]
- 238.Kim HP, Wang X, Galbiati F, Ryter SW, Choi AM. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J 18: 1080–1089, 2004. [DOI] [PubMed] [Google Scholar]
- 239.Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, Ryter SW, Choi AM. Caveolin-1 expression by means of p38β mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci USA 102: 11319–11324, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, Choi AM. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38b MAPK and heat shock factor-1. J Immunol 175: 2622–2629, 2005. [DOI] [PubMed] [Google Scholar]
- 241.Kim J, Zarjou A, Traylor AM, Bolisetty S, Jaimes EA, Hull TD, George JF, Mikhail FM, Agarwal A. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int 82: 278–291, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kim JE, Kang YJ, Lee KY, Choi HC. Isoproterenol inhibits angiotensin II-stimulated proliferation and reactive oxygen species production in vascular smooth muscle cells through heme oxygenase-1. Biol Pharm Bull 32: 1047–1052, 2009. [DOI] [PubMed] [Google Scholar]
- 243.Kim JW, Lim SC, Lee MY, Lee JW, Oh WK, Kim SK, Kang KW. Inhibition of neointimal formation by trans-resveratrol: role of phosphatidyl inositol 3-kinase-dependent Nrf2 activation in heme oxygenase-1 induction. Mol Nutr Food Res 54: 1497–1505, 2010. [DOI] [PubMed] [Google Scholar]
- 244.Kim JY, Cho HJ, Sir JJ, Kim BK, Hur J, Youn SW, Yang HM, Jun SI, Park KW, Hwang SJ, Kwon YW, Lee HY, Kang HJ, Oh BH, Park YB, Kim HS. Sulfasalazine induces haem oxygenase-1 via ROS-dependent Nrf2 signalling, leading to control of neointimal hyperplasia. Cardiovasc Res 82: 550–560, 2009. [DOI] [PubMed] [Google Scholar]
- 245.Kim N, Hwangbo C, Lee S, Lee JH. Eupatolide inhibits PDGF-induced proliferation and migration of aortic smooth muscle cells through ROS-dependent heme oxygenase-1 induction. Phytother Res 27: 1700–1707, 2013. [DOI] [PubMed] [Google Scholar]
- 246.Kim SE, Lee MY, Lim SC, Hien TT, Kim JW, Ahn SG, Yoon JH, Kim SK, Choi HS, Kang KW. Role of Pin1 in neointima formation: down-regulation of Nrf2-dependent heme oxygenase-1 expression by Pin1. Free Radic Biol Med 48: 1644–1653, 2010. [DOI] [PubMed] [Google Scholar]
- 247.Kim SJ, Lee SM. NLRP3 inflammasome activation in d-galactosamine and lipopolysaccharide-induced acute liver failure: role of heme oxygenase-1. Free Radic Biol Med 65: 997–1004, 2013. [DOI] [PubMed] [Google Scholar]
- 248.Kimpara T, Takeda A, Watanabe K, Itoyama Y, Ikawa S, Watanabe M, Arai H, Sasaki H, Higuchi S, Okita N, Takase S, Saito H, Takahashi K, Shibahara S. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum Genet 100: 145–147, 1997. [DOI] [PubMed] [Google Scholar]
- 249.Kinobe RT, Dercho RA, Nakatsu K. Inhibitors of the heme oxygenase: carbon monoxide system: on the doorstep of the clinic? Can J Physiol Pharmacol 86: 577–599, 2008. [DOI] [PubMed] [Google Scholar]
- 250.Kirk EA, Heinecke JW, LeBoeuf RC. Iron overload diminishes atherosclerosis in apoE-deficient mice. J Clin Invest 107: 1545–1553, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, Kedzierski RM, Hammer RE, Yanagisawa H, Williams SC, Richardson JA, Suzuki T, Yanagisawa M. Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension 56: 121–128, 2010. [DOI] [PubMed] [Google Scholar]
- 252.Knauert M, Vangala S, Haslip M, Lee PJ. Therapeutic applications of carbon monoxide. Oxid Med Cell Longev 2013: 360815, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Knot HJ, Zimmermann PA, Nelson MT. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J Physiol 492: 419–430, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ko YG, Choi SH, Chol Kang W, Kwon Lee B, Wook Kim S, Shim WH. Effects of combination therapy with cilostazol and probucol versus monotherapy with cilostazol on coronary plaque, lipid and biomarkers: SECURE study, a double-blind randomized controlled clinical trial. J Atheroscler Thromb 21: 816–830, 2014. [DOI] [PubMed] [Google Scholar]
- 256.Koller A, Sun D, Kaley G. Role of shear stress and endothelial prostaglandins in flow- and viscosity-induced dilation of arterioles in vitro. Circ Res 72: 1276–1284, 1993. [DOI] [PubMed] [Google Scholar]
- 257.Kong D, Melo LG, Mangi AA, Zhang L, Lopez-Ilasaca M, Perrella MA, Liew CC, Pratt RE, Dzau VJ. Enhanced inhibition of neointimal hyperplasia by genetically engineered endothelial progenitor cells. Circulation 109: 1769–1775, 2004. [DOI] [PubMed] [Google Scholar]
- 258.Kostoglou-Athanassiou I, Forsling ML, Navarra P, Grossman AB. Oxytocin release is inhibited by the generation of carbon monoxide from the rat hypothalamus–further evidence for carbon monoxide as a neuromodulator. Brain Res 42: 301–306, 1996. [DOI] [PubMed] [Google Scholar]
- 259.Kovarnik T, Kral A, Skalicka H, Mintz GS, Kralik L, Chval M, Horak J, Skalicka L, Sonka M, Wahle A, Downe RW, Uhrova J, Benakova H, Cernohousova L, Martasek P, Belohlavek J, Aschermann M, Linhart A. The prediction of coronary artery disease based on non-invasive examinations and heme oxygenase 1 polymorphism versus virtual histology. J Invasive Cardiol 25: 32–37, 2013. [PubMed] [Google Scholar]
- 260.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116: 6054–6062, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kozakowska M, Ciesla M, Stefanska A, Skrzypek K, Was H, Jazwa A, Grochot-Przeczek A, Kotlinowski J, Szymula A, Bartelik A, Mazan M, Yagensky O, Florczyk U, Lemke K, Zebzda A, Dyduch G, Nowak W, Szade K, Stepniewski J, Majka M, Derlacz R, Loboda A, Dulak J, Jozkowicz A. Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid Redox Signal 16: 113–127, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kozakowska M, Kotlinowski J, Grochot-Przeczek A, Ciesla M, Pilecki B, Derlacz R, Dulak J, Jozkowicz A. Myoblast-conditioned media improve regeneration and revascularization of ischemic muscles in diabetic mice. Stem Cell Res Ther 6: 61, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kral A, Kovarnik T, Kralik L, Skalicka H, Horak J, Mintz GS, Uhrova J, Sonka M, Wahle A, Downe R, Aschermann M, Martasek P, Linhart A. Genetic variants in haem oxygenase-1 and endothelial nitric oxide synthase influence the extent and evolution of coronary artery atherosclerosis. Folia Biol 57: 182–190, 2011. [DOI] [PubMed] [Google Scholar]
- 264.Kramkowski K, Leszczynska A, Mogielnicki A, Chlopicki S, Fedorowicz A, Grochal E, Mann B, Brzoska T, Urano T, Motterlini R, Buczko W. Antithrombotic properties of water-soluble carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol 32: 2149–2157, 2012. [DOI] [PubMed] [Google Scholar]
- 265.Krönke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol 27: 1276–1282, 2007. [DOI] [PubMed] [Google Scholar]
- 266.Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol 292: C987–C995, 2007. [DOI] [PubMed] [Google Scholar]
- 267.Kurinna S, Werner S. NRF2 and microRNAs: new but awaited relations. Biochem Soc Trans 43: 595–601, 2015. [DOI] [PubMed] [Google Scholar]
- 268.Kutty RK, Maines MD. Oxidation of heme c derivatives by purified heme oxygenase. Evidence for the presence of one molecular species of heme oxygenase in the rat liver. J Biol Chem 257: 9944–9952, 1982. [PubMed] [Google Scholar]
- 269.Kwak JY, Takeshige K, Cheung BS, Minakami S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim Biophys Acta 1076: 369–373, 1991. [DOI] [PubMed] [Google Scholar]
- 270.Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem 272: 27345–27352, 1997. [DOI] [PubMed] [Google Scholar]
- 271.Laslett LJ, Alagona P Jr, Clark BA 3rd Drozda JP Jr, Saldivar F, Wilson SR, Poe C, Hart M. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol 60: S1–49, 2012. [DOI] [PubMed] [Google Scholar]
- 272.Lau AK, Leichtweis SB, Hume P, Mashima R, Hou JY, Chaufour X, Wilkinson B, Hunt NH, Celermajer DS, Stocker R. Probucol promotes functional reendothelialization in balloon-injured rabbit aortas. Circulation 107: 2031–2036, 2003. [DOI] [PubMed] [Google Scholar]
- 273.Lauffer RB. Iron depletion and coronary disease. Am Heart J 119: 1448–1449, 1990. [DOI] [PubMed] [Google Scholar]
- 274.Ledoux J, Bonev AD, Nelson MT. Ca2+-activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol 131: 125–135, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lee CY, Yen MH. Nitric oxide and carbon monoxide, collaborative and competitive regulators of hypertension. Chang Gung Med J 32: 12–21, 2009. [PubMed] [Google Scholar]
- 276.Lee S, Seo J, Ryoo S, Cuong TD, Min BS, Lee JH. Malabaricone C inhibits PDGF-induced proliferation and migration of aortic smooth muscle cells through induction of heme oxygenase-1. J Cell Biochem 113: 2866–2876, 2012. [DOI] [PubMed] [Google Scholar]
- 277.Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: a novel mechanism of vessel protection. Circulation 110: 1296–1302, 2004. [DOI] [PubMed] [Google Scholar]
- 278.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 8: 240–246, 2002. [DOI] [PubMed] [Google Scholar]
- 279.Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation 99: 1222–1229, 1999. [DOI] [PubMed] [Google Scholar]
- 280.Leo MD, Bulley S, Bannister JP, Kuruvilla KP, Narayanan D, Jaggar JH. Angiotensin II stimulates internalization and degradation of arterial myocyte plasma membrane BK channels to induce vasoconstriction. Am J Physiol Cell Physiol 309: C392–C402, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Levere RD, Martasek P, Escalante B, Schwartzman ML, Abraham NG. Effect of heme arginate administration on blood pressure in spontaneously hypertensive rats. J Clin Invest 86: 213–219, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li Calzi S, Purich DL, Chang KH, Afzal A, Nakagawa T, Busik JV, Agarwal A, Segal MS, Grant MB. Carbon monoxide and nitric oxide mediate cytoskeletal reorganization in microvascular cells via vasodilator-stimulated phosphoprotein phosphorylation: evidence for blunted responsiveness in diabetes. Diabetes 57: 2488–2494, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Li H, Zhou X, Zhang J. Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced inflammasome activation in human gingival epithelial cells. Int J Mol Med 34: 1039–1044, 2014. [DOI] [PubMed] [Google Scholar]
- 284.Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation 107: 1053–1058, 2003. [DOI] [PubMed] [Google Scholar]
- 285.Li M, Gallo D, Csizmadia E, Otterbein LE, Wegiel B. Carbon monoxide induces chromatin remodelling to facilitate endothelial cell migration. Thromb Haemost 111: 951–959, 2014. [DOI] [PubMed] [Google Scholar]
- 286.Li M, Liu Y, Shi H, Zhang Y, Wang G, Xu J, Lu J, Zhang D, Xie X, Han D, Wu Y, Li S. Statins inhibit pulmonary artery smooth muscle cell proliferation by upregulation of HO-1 and p21WAF1. Naunyn-Schmiedebergs Arch Pharmacol 385: 961–968, 2012. [DOI] [PubMed] [Google Scholar]
- 287.Li M, Tanaka Y, Alioua A, Wu Y, Lu R, Kundu P, Sanchez-Pastor E, Marijic J, Stefani E, Toro L. Thromboxane A2 receptor and MaxiK-channel intimate interaction supports channel trans-inhibition independent of G-protein activation. Proc Natl Acad Sci USA 107: 19096–19101, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Li N, Yi F, dos Santos EA, Donley DK, Li PL. Role of renal medullary heme oxygenase in the regulation of pressure natriuresis and arterial blood pressure. Hypertension 49: 148–154, 2007. [DOI] [PubMed] [Google Scholar]
- 289.Li Q, Guo Y, Ou Q, Wu WJ, Chen N, Zhu X, Tan W, Yuan F, Dawn B, Luo L, Hunt GN, Bolli R. Gene transfer as a strategy to achieve permanent cardioprotection II: rAAV-mediated gene therapy with heme oxygenase-1 limits infarct size 1 year later without adverse functional consequences. Basic Res Cardiol 106: 1367–1377, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Li T, Tian H, Zhao Y, An F, Zhang L, Zhang J, Peng J, Zhang Y, Guo Y. Heme oxygenase-1 inhibits progression and destabilization of vulnerable plaques in a rabbit model of atherosclerosis. Eur J Pharmacol 672: 143–152, 2011. [DOI] [PubMed] [Google Scholar]
- 291.Li Volti G, Wang J, Traganos F, Kappas A, Abraham NG. Differential effect of heme oxygenase-1 in endothelial and smooth muscle cell cycle progression. Biochem Biophys Res Commun 296: 1077–1082, 2002. [DOI] [PubMed] [Google Scholar]
- 292.Li W, Iyer S, Lu L, Buelow R, Fung JJ, Rao AS, Woo J, Qian S. Attenuation of aortic graft arteriosclerosis by systemic administration of Allotrap peptide RDP58. Transpl Int 16: 849–856, 2003. [DOI] [PubMed] [Google Scholar]
- 293.Li W, Tanaka K, Morioka K, Uesaka T, Yamada N, Takamori A, Handa M, Tanabe S, Ihaya A. Thymidine phosphorylase gene transfer inhibits vascular smooth muscle cell proliferation by upregulating heme oxygenase-1 and p27KIP1. Arterioscler Thromb Vasc Biol 25: 1370–1375, 2005. [DOI] [PubMed] [Google Scholar]
- 294.Li XC, Tong GX, Zhang Y, Liu SX, Jin QH, Chen HH, Chen P. Neferine inhibits angiotensin II-stimulated proliferation in vascular smooth muscle cells through heme oxygenase-1. Acta Pharmacol Sin 31: 679–686, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Li Y, Gao C, Shi Y, Tang Y, Liu L, Xiong T, Du M, Xing M, Liu L, Yao P. Carbon monoxide alleviates ethanol-induced oxidative damage and inflammatory stress through activating p38 MAPK pathway. Toxicol Appl Pharmacol 273: 53–58, 2013. [DOI] [PubMed] [Google Scholar]
- 296.Li Y, Wang Q, Xu Q, Cai S, Zhou J, Ren B, Sun T, Liu X, Yu H. Valsartan decreases neointimal hyperplasia in balloon-injured rat aortic arteries by upregulating HO-1 and inhibiting angiotensin II type 1 receptor. Life Sci 110: 70–76, 2014. [DOI] [PubMed] [Google Scholar]
- 297.Li YG, Wang DM, Chen SM, Tan XR, Fang XY, Wu JW, Zhang GH, Mai RQ. Haem oxygenase-1 expression and coronary heart disease–association between levels of haem oxygenase-1 expression and angiographic morphology as well as the quantity of coronary lesions. Acta Cardiol 61: 295–300, 2006. [DOI] [PubMed] [Google Scholar]
- 298.Li Z, Wang Y, Man RY, Vanhoutte PM. Upregulation of heme oxygenase-1 potentiates EDH-type relaxations in the mesenteric artery of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 305: H1471–H1483, 2013. [DOI] [PubMed] [Google Scholar]
- 299.Li Z, Wang Y, Vanhoutte PM. Upregulation of heme oxygenase 1 by hemin impairs endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Hypertension 58: 926–934, 2011. [DOI] [PubMed] [Google Scholar]
- 300.Liang GH, Xi Q, Leffler CW, Jaggar JH. Hydrogen sulfide activates Ca2+ sparks to induce cerebral arteriole dilatation. J Physiol 590: 2709–2720, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Liang KW, Sheu WH, Lee WL, Lee IT, Lin SY, Ting CT, Lee WJ. Shorter GT repeats in the heme oxygenase-1 gene promoter are associated with a lower severity score in coronary artery disease. J Chin Med Assoc 76: 312–318, 2013. [DOI] [PubMed] [Google Scholar]
- 302.Libby P. Inflammation in atherosclerosis. Nature 420: 868–874, 2002. [DOI] [PubMed] [Google Scholar]
- 303.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 473: 317–325, 2011. [DOI] [PubMed] [Google Scholar]
- 304.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 65: 4789–4798, 2005. [DOI] [PubMed] [Google Scholar]
- 305.Lieu PT, Heiskala M, Peterson PA, Yang Y. The roles of iron in health and disease. Mol Aspects Med 22: 1–87, 2001. [DOI] [PubMed] [Google Scholar]
- 306.Lim HJ, Lee KS, Lee S, Park JH, Choi HE, Go SH, Kwak HJ, Park HY. 15d-PGJ2 stimulates HO-1 expression through p38 MAP kinase and Nrf-2 pathway in rat vascular smooth muscle cells. Toxicol Appl Pharmacol 223: 20–27, 2007. [DOI] [PubMed] [Google Scholar]
- 307.Lin CC, Yang WC, Lin SJ, Chen TW, Lee WS, Chang CF, Lee PC, Lee SD, Su TS, Fann CS, Chung MY. Length polymorphism in heme oxygenase-1 is associated with arteriovenous fistula patency in hemodialysis patients. Kidney Int 69: 165–172, 2006. [DOI] [PubMed] [Google Scholar]
- 308.Lin CY, Peng CY, Huang TT, Wu ML, Lai YL, Peng DH, Chen PF, Chen HF, Yen BL, Wu KK, Yet SF. Exacerbation of oxidative stress-induced cell death and differentiation in induced pluripotent stem cells lacking heme oxygenase-1. Stem Cells Dev 21: 1675–1687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Lin HH, Chen YH, Yet SF, Chau LY. After vascular injury, heme oxygenase-1/carbon monoxide enhances re-endothelialization via promoting mobilization of circulating endothelial progenitor cells. J Thromb Haemost 7: 1401–1408, 2009. [DOI] [PubMed] [Google Scholar]
- 310.Lin JP, Vitek L, Schwertner HA. Serum bilirubin and genes controlling bilirubin concentrations as biomarkers for cardiovascular disease. Clin Chem 56: 1535–1543, 2010. [DOI] [PubMed] [Google Scholar]
- 311.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 282: 20621–20633, 2007. [DOI] [PubMed] [Google Scholar]
- 312.Lin SH, Song W, Cressatti M, Zukor H, Wang E, Schipper HM. Heme oxygenase-1 modulates microRNA expression in cultured astroglia: implications for chronic brain disorders. Glia 63: 1270–1284, 2015. [DOI] [PubMed] [Google Scholar]
- 313.Lin Y, Bai L, Chen W, Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets 14: 45–55, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Linnenbaum M, Busker M, Kraehling JR, Behrends S. Heme oxygenase isoforms differ in their subcellular trafficking during hypoxia and are differentially modulated by cytochrome P450 reductase. PloS One 7: e35483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Linzke N, Schumacher A, Woidacki K, Croy BA, Zenclussen AC. Carbon monoxide promotes proliferation of uterine natural killer cells and remodeling of spiral arteries in pregnant hypertensive heme oxygenase-1 mutant mice. Hypertension 63: 580–588, 2014. [DOI] [PubMed] [Google Scholar]
- 316.Liu PL, Liu JT, Kuo HF, Chong IW, Hsieh CC. Epigallocatechin gallate attenuates proliferation and oxidative stress in human vascular smooth muscle cells induced by interleukin-1β via heme oxygenase-1. Mediators Inflamm 2014: 523684, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Liu XM, Azam MA, Peyton KJ, Ensenat D, Keswani AN, Wang H, Durante W. Butylated hydroxyanisole stimulates heme oxygenase-1 gene expression and inhibits neointima formation in rat arteries. Cardiovasc Res 74: 169–179, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation 105: 79–84, 2002. [DOI] [PubMed] [Google Scholar]
- 319.Liu XM, Peyton KJ, Ensenat D, Wang H, Hannink M, Alam J, Durante W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res 75: 381–389, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Lo WC, Lu PJ, Ho WY, Hsiao M, Tseng CJ. Induction of heme oxygenase-1 is involved in carbon monoxide-mediated central cardiovascular regulation. J Pharmacol Exp Ther 318: 8–16, 2006. [DOI] [PubMed] [Google Scholar]
- 321.Loboda A, Jozkowicz A, Dulak J. Carbon monoxide: pro- or anti-angiogenic agent? Comment on Ahmad et al. Thromb Haemost 114: 432–433, 2015. [DOI] [PubMed] [Google Scholar]
- 322.Loboda A, Jozkowicz A, Dulak J. HO-1/CO system in tumor growth, angiogenesis and metabolism–Targeting HO-1 as an anti-tumor therapy. Vascul Pharmacol 74: 11–22, 2015. [DOI] [PubMed] [Google Scholar]
- 323.Lublinghoff N, Winkler K, Winkelmann BR, Seelhorst U, Wellnitz B, Boehm BO, März W, Hoffmann MM. Genetic variants of the promoter of the heme oxygenase-1 gene and their influence on cardiovascular disease (the Ludwigshafen Risk and Cardiovascular Health study). BMC Med Genet 10: 36, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lundvig DM, Scharstuhl A, Cremers NA, Pennings SW, te Paske J, van Rheden R, van Run-van Breda C, Regan RF, Russel FG, Carels CE, Maltha JC, Wagener FA. Delayed cutaneous wound closure in HO-2 deficient mice despite normal HO-1 expression. J Cell Mol Med 18: 2488–2498, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Luo D, Vincent SR. Metalloporphyrins inhibit nitric oxide-dependent cGMP formation in vivo. Eur J Pharmacol 267: 263–267, 1994. [DOI] [PubMed] [Google Scholar]
- 326.Luo YP, Jiang L, Kang K, Fei DS, Meng XL, Nan CC, Pan SH, Zhao MR, Zhao MY. Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol 20: 24–32, 2014. [DOI] [PubMed] [Google Scholar]
- 327.Lyall F, Barber A, Myatt L, Bulmer JN, Robson SC. Heme oxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J 14: 208–219, 2000. [DOI] [PubMed] [Google Scholar]
- 328.Ma N, Xiang Y, Zhang Y, Zhao X, Zhou L, Gao X. The balance mediated by miRNAs and the heme oxygenase 1 feedback loop contributes to biological effects. J Cell Biochem 114: 2637–2642, 2013. [DOI] [PubMed] [Google Scholar]
- 329.Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Ma YH, Gebremedhin D, Schwartzman ML, Falck JR, Clark JE, Masters BS, Harder DR, Roman RJ. 20-Hydroxyeicosatetraenoic acid is an endogenous vasoconstrictor of canine renal arcuate arteries. Circ Res 72: 126–136, 1993. [DOI] [PubMed] [Google Scholar]
- 331.MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 30: 1571–1580, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Maghzal GJ, Krause KH, Stocker R, Jaquet V. Detection of reactive oxygen species derived from the family of NOX NADPH oxidases. Free Radic Biol Med 53: 1903–1918, 2012. [DOI] [PubMed] [Google Scholar]
- 333.Maghzal GJ, Leck MC, Collinson E, Li C, Stocker R. Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J Biol Chem 284: 29251–29259, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet 383: 999–1008, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology 47: 727–733, 1996. [DOI] [PubMed] [Google Scholar]
- 336.Maines MD, Eke BC, Zhao X. Corticosterone promotes increased heme oxygenase-2 protein and transcript expression in the newborn rat brain. Brain Res 722: 83–94, 1996. [DOI] [PubMed] [Google Scholar]
- 337.Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. J Biol Chem 261: 411–419, 1986. [PubMed] [Google Scholar]
- 338.Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 261: 411–419, 1986. [PubMed] [Google Scholar]
- 339.Mancuso C, Bonsignore A, Di Stasio E, Mordente A, Motterlini R. Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem Pharmacol 66: 2355–2363, 2003. [DOI] [PubMed] [Google Scholar]
- 340.Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem 286: 13460–13469, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Manttari M, Manninen V, Huttunen JK, Palosuo T, Ehnholm C, Heinonen OP, Frick MH. Serum ferritin and ceruloplasmin as coronary risk factors. Eur Heart J 15: 1599–1603, 1994. [DOI] [PubMed] [Google Scholar]
- 342.Marinissen MJ, Tanos T, Bolos M, de Sagarra MR, Coso OA, Cuadrado A. Inhibition of heme oxygenase-1 interferes with the transforming activity of the Kaposi sarcoma herpesvirus-encoded G protein-coupled receptor. J Biol Chem 281: 11332–11346, 2006. [DOI] [PubMed] [Google Scholar]
- 343.Martasek P, Schwartzman ML, Goodman AI, Solangi KB, Levere RD, Abraham NG. Hemin and l-arginine regulation of blood pressure in spontaneous hypertensive rats. J Am Soc Nephrol 2: 1078–1084, 1991. [DOI] [PubMed] [Google Scholar]
- 344.Martín MJ, Tanos T, García AB, Martin D, Gutkind JS, Coso OA, Marinissen MJ. The Ga12/13 family of heterotrimeric G proteins and the small GTPase RhoA link the Kaposi sarcoma-associated herpes virus G protein-coupled receptor to heme oxygenase-1 expression and tumorigenesis. J Biol Chem 282: 34510–34524, 2007. [DOI] [PubMed] [Google Scholar]
- 345.Mather S, Dora KA, Sandow SL, Winter P, Garland CJ. Rapid endothelial cell-selective loading of connexin 40 antibody blocks endothelium-derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 97: 399–407, 2005. [DOI] [PubMed] [Google Scholar]
- 346.Matz P, Turner C, Weinstein PR, Massa SM, Panter SS, Sharp FR. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res 713: 211–222, 1996. [DOI] [PubMed] [Google Scholar]
- 347.Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med 171: 354–360, 2005. [DOI] [PubMed] [Google Scholar]
- 348.Mazzone GL, Rigato I, Ostrow JD, Bossi F, Bortoluzzi A, Sukowati CH, Tedesco F, Tiribelli C. Bilirubin inhibits the TNFα-related induction of three endothelial adhesion molecules. Biochem Biophys Res Commun 386: 338–344, 2009. [DOI] [PubMed] [Google Scholar]
- 349.McAllister SC, Hansen SG, Ruhl RA, Raggo CM, DeFilippis VR, Greenspan D, Fruh K, Moses AV. Kaposi sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1 expression and activity in KSHV-infected endothelial cells. Blood 103: 3465–3473, 2004. [DOI] [PubMed] [Google Scholar]
- 350.McCoubrey WK Jr, Huang TJ, Maines MD. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J Biol Chem 272: 12568–12574, 1997. [DOI] [PubMed] [Google Scholar]
- 351.McCoubrey WK Jr, Maines MD. Domains of rat heme oxygenase-2: the amino terminus and histidine 151 are required for heme oxidation. Arch Biochem Biophys 302: 402–408, 1993. [DOI] [PubMed] [Google Scholar]
- 352.McCoubrey WK Jr, Maines MD. The structure, organization and differential expression of the gene encoding rat heme oxygenase-2. Gene 139: 155–161, 1994. [DOI] [PubMed] [Google Scholar]
- 353.McDonagh AF. The biliverdin-bilirubin antioxidant cycle of cellular protection: missing a wheel? Free Radic Biol Med 49: 814–820, 2010. [DOI] [PubMed] [Google Scholar]
- 354.McLaughlin BE, Hutchinson JM, Graham CH, Smith GN, Marks GS, Nakatsu K, Brien JF. Heme oxygenase activity in term human placenta. Placenta 21: 870–873, 2000. [DOI] [PubMed] [Google Scholar]
- 355.McNeish AJ, Garland CJ. Thromboxane A2 inhibition of SKCa after NO synthase block in rat middle cerebral artery. Br J Pharmacol 151: 441–449, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007. [DOI] [PubMed] [Google Scholar]
- 357.Midwinter RG, Maghzal GJ, Dennis JM, Wu BJ, Cai H, Kapralov AA, Belikova NA, Tyurina YY, Dong LF, Khachigian L, Neuzil J, Kagan VE, Stocker R. Succinobucol induces apoptosis in vascular smooth muscle cells. Free Radic Biol Med 52: 871–879, 2012. [DOI] [PubMed] [Google Scholar]
- 358.Miller EC, Cao H, Wen SW, Yang Q, Lafleche J, Walker M. The risk of adverse pregnancy outcomes is increased in preeclamptic women who smoke compared with nonpreeclamptic women who do not smoke. Am J Obstet Gynecol 203: 334 e331–338, 2010. [DOI] [PubMed] [Google Scholar]
- 359.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA 98: 8798–8803, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Mirjanic-Azaric B, Rizzo M, Jürgens G, Hallstroem S, Srdic S, Marc J, Cerne D. Atorvastatin treatment increases plasma bilirubin but not HMOX1 expression in stable angina patients. Scand J Clin Lab Invest 75: 382–389, 2015. [DOI] [PubMed] [Google Scholar]
- 361.Mirza A, Eder V, Rochefort GY, Hyvelin JM, Machet MC, Fauchier L, Bonnet P. CO inhalation at dose corresponding to tobacco smoke worsens cardiac remodeling after experimental myocardial infarction in rats. Toxicol Sci 85: 976–982, 2005. [DOI] [PubMed] [Google Scholar]
- 362.Mitchell LA, Channell MM, Royer CM, Ryter SW, Choi AM, McDonald JD. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am J Physiol Lung Cell Mol Physiol 299: L891–L897, 2010. [DOI] [PubMed] [Google Scholar]
- 363.Mitra AK, Gangahar DM, Agrawal DK. Cellular, molecular and immunological mechanisms in the pathophysiology of vein graft intimal hyperplasia. Immunol Cell Biol 84: 115–124, 2006. [DOI] [PubMed] [Google Scholar]
- 364.Momotani K, Artamonov MV, Utepbergenov D, Derewenda U, Derewenda ZS, Somlyo AV. p63RhoGEF couples Gaq/11-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ Res 109: 993–1002, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13: 709–721, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Moreno-Dominguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, Cole WC. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol 591: 1235–1250, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori K, Takenouchi T, Takahashi T, Ishii I, Matsubara K, Kabe Y, Uchiyama S, Nagata E, Gadalla MM, Snyder SH, Suematsu M. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci USA 109: 1293–1298, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem 272: 32804–32809, 1997. [DOI] [PubMed] [Google Scholar]
- 369.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci USA 92: 1475–1479, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, Otterbein SL, Otterbein LE, Choi AM. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem 278: 36993–36998, 2003. [DOI] [PubMed] [Google Scholar]
- 371.Morsi WG, Shaker OG, Ismail EF, Ahmed HH, El-Serafi TI, Maklady FA, Abdel-Aziz MT, El-Asmar MF, Atta HM. HO-1 and VGEF gene expression in human arteries with advanced atherosclerosis. Clin Biochem 39: 1057–1062, 2006. [DOI] [PubMed] [Google Scholar]
- 372.Motterlini R, Foresti R, Intaglietta M, Winslow RM. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am J Physiol Heart Circ Physiol 270: H107–H114, 1996. [DOI] [PubMed] [Google Scholar]
- 373.Motterlini R, Green CJ, Foresti R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid Redox Signal 4: 615–624, 2002. [DOI] [PubMed] [Google Scholar]
- 374.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9: 728–743, 2010. [DOI] [PubMed] [Google Scholar]
- 375.Muchova L, Wong RJ, Hsu M, Morioka I, Vitek L, Zelenka J, Schröder H, Stevenson DK. Statin treatment increases formation of carbon monoxide and bilirubin in mice: a novel mechanism of in vivo antioxidant protection. Can J Physiol Pharmacol 85: 800–810, 2007. [DOI] [PubMed] [Google Scholar]
- 376.Muller PM, Gnugge R, Dhayade S, Thunemann M, Krippeit-Drews P, Drews G, Feil R. H2O2 lowers the cytosolic Ca2+ concentration via activation of cGMP-dependent protein kinase Iα. Free Radic Biol Med 53: 1574–1583, 2012. [DOI] [PubMed] [Google Scholar]
- 377.Mustafa S, Weltermann A, Fritsche R, Marsik C, Wagner O, Kyrle PA, Eichinger S. Genetic variation in heme oxygenase 1 (HMOX1) and the risk of recurrent venous thromboembolism. J Vasc Surg 47: 566–570, 2008. [DOI] [PubMed] [Google Scholar]
- 378.Muzaffar S, Shukla N, Lobo C, Angelini GD, Jeremy JY. Iloprost inhibits superoxide formation and gp91phox expression induced by the thromboxane A2 analogue U46619, 8-isoprostane F2a, prostaglandin F2a, cytokines and endotoxin in the pig pulmonary artery. Br J Pharmacol 141: 488–496, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Naik JS, Walker BR. Heme oxygenase-mediated vasodilation involves vascular smooth muscle cell hyperpolarization. Am J Physiol Heart Circ Physiol 285: H220–H228, 2003. [DOI] [PubMed] [Google Scholar]
- 380.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Nakao A, Murase N, Ho C, Toyokawa H, Billiar TR, Kanno S. Biliverdin administration prevents the formation of intimal hyperplasia induced by vascular injury. Circulation 112: 587–591, 2005. [DOI] [PubMed] [Google Scholar]
- 382.Nakao A, Neto JS, Kanno S, Stolz DB, Kimizuka K, Liu F, Bach FH, Billiar TR, Choi AM, Otterbein LE, Murase N. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant 5: 282–291, 2005. [DOI] [PubMed] [Google Scholar]
- 383.Nakayama M, Takahashi K, Komaru T, Fukuchi M, Shioiri H, Sato K, Kitamuro T, Shirato K, Yamaguchi T, Suematsu M, Shibahara S. Increased expression of heme oxygenase-1 and bilirubin accumulation in foam cells of rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol 21: 1373–1377, 2001. [DOI] [PubMed] [Google Scholar]
- 384.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 90: 267–270, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Nath KA, d'Uscio LV, Juncos JP, Croatt AJ, Manriquez MC, Pittock ST, Katusic ZS. An analysis of the DOCA-salt model of hypertension in HO-1−/− mice and the Gunn rat. Am J Physiol Heart Circ Physiol 293: H333–H342, 2007. [DOI] [PubMed] [Google Scholar]
- 386.Nath KA, Hernandez MC, Croatt AJ, Katusic ZS, Juncos L. Heme oxygenase activity as a determinant of the renal hemodynamic response to low-dose ANG II. Am J Physiol Regul Integr Comp Physiol 299: R1183–R1191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Nausch LW, Bonev AD, Heppner TJ, Tallini Y, Kotlikoff MI, Nelson MT. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. Am J Physiol Heart Circ Physiol 302: H594–H602, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Navarra P, Pozzoli G, Costa A, Grossman A. Endotoxin, prostanoids and corticotrophin-releasing hormone: an integrated view. Funct Neurol 16: 217–225, 2001. [PubMed] [Google Scholar]
- 389.Naylor LH, Clark EM. d(TG)n d(CA)n sequences upstream of the rat prolactin gene form Z-DNA and inhibit gene transcription. Nucleic Acids Res 18: 1595–1601, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Ndisang JF, Mishra M. The heme oxygenase system selectively suppresses the proinflammatory macrophage M1 phenotype and potentiates insulin signaling in spontaneously hypertensive rats. Am J Hypertens 26: 1123–1131, 2013. [DOI] [PubMed] [Google Scholar]
- 391.Neuzil J, Gebicki JM, Stocker R. Radical-induced chain oxidation of proteins and its inhibition by chain-breaking antioxidants. Biochem J 293: 601–606, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Neuzil J, Stocker R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett 331: 281–284, 1993. [DOI] [PubMed] [Google Scholar]
- 393.Neuzil J, Stocker R. Free and albumin-bound bilirubin are efficient co-antioxidants for a-tocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J Biol Chem 269: 16712–16719, 1994. [PubMed] [Google Scholar]
- 394.Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 313: 1256–1262, 1985. [DOI] [PubMed] [Google Scholar]
- 395.Nimura T, Weinstein PR, Massa SM, Panter S, Sharp FR. Heme oxygenase-1 (HO-1) protein induction in rat brain following focal ischemia. Brain Res 37: 201–208, 1996. [DOI] [PubMed] [Google Scholar]
- 396.Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, Ito Y, Inamura T, Ikezaki K, Fukui M, Iwaki T, Kuwano M. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res 5: 1107–1113, 1999. [PubMed] [Google Scholar]
- 397.Noguchi M, Yoshida T, Kikuchi G. Specific requirement of NADPH-cytochrome c reductase for the microsomal heme oxygenase reaction yielding biliverdin IXα. FEBS Lett 98: 281–284, 1979. [DOI] [PubMed] [Google Scholar]
- 398.Nowis D, Legat M, Grzela T, Niderla J, Wilczek E, Wilczynski GM, Glodkowska E, Mrowka P, Issat T, Dulak J, Jozkowicz A, Was H, Adamek M, Wrzosek A, Nazarewski S, Makowski M, Stoklosa T, Jakobisiak M, Golab J. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 25: 3365–3374, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Ogawa T, Hanggi D, Wu Y, Michiue H, Tomizawa K, Ono S, Matsui H, Date I, Steiger HJ. Protein therapy using heme-oxygenase-1 fused to a polyarginine transduction domain attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 31: 2231–2242, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Oh CJ, Park S, Kim JY, Kim HJ, Jeoung NH, Choi YK, Go Y, Park KG, Lee IK. Dimethylfumarate attenuates restenosis after acute vascular injury by cell-specific and Nrf2-dependent mechanisms. Redox Biol 2: 855–864, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Ollinger R, Bilban M, Erat A, Froio A, McDaid J, Tyagi S, Csizmadia E, Graca-Souza AV, Liloia A, Soares MP, Otterbein LE, Usheva A, Yamashita K, Bach FH. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation 112: 1030–1039, 2005. [DOI] [PubMed] [Google Scholar]
- 402.Ono K, Goto Y, Takagi S, Baba S, Tago N, Nonogi H, Iwai N. A promoter variant of the heme oxygenase-1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis 173: 315–319, 2004. [DOI] [PubMed] [Google Scholar]
- 403.Ono K, Mannami T, Iwai N. Association of a promoter variant of the haeme oxygenase-1 gene with hypertension in women. J Hypertens 21: 1497–1503, 2003. [DOI] [PubMed] [Google Scholar]
- 404.Ono S, Komuro T, Macdonald RL. Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. J Neurosurg 96: 1094–1102, 2002. [DOI] [PubMed] [Google Scholar]
- 405.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, Deshane J, Bolisetty S, Shaposhnik Z, Shih DM, Agarwal A, Lusis AJ, Araujo JA. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res 100: 1703–1711, 2007. [DOI] [PubMed] [Google Scholar]
- 406.Ortiz de Montellano PR. The mechanism of heme oxygenase. Curr Opin Chem Biol 4: 221–227, 2000. [DOI] [PubMed] [Google Scholar]
- 407.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000. [DOI] [PubMed] [Google Scholar]
- 408.Otterbein LE, Mantell LL, Choi AMK. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 276: L688–L694, 1999. [DOI] [PubMed] [Google Scholar]
- 409.Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, Stachulak C, Bodyak N, Smith RN, Csizmadia E, Tyagi S, Akamatsu Y, Flavell RJ, Billiar TR, Tzeng E, Bach FH, Choi AM, Soares MP. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 9: 183–190, 2003. [DOI] [PubMed] [Google Scholar]
- 410.Overhaus M, Moore BA, Barbato JE, Behrendt FF, Doering JG, Bauer AJ. Biliverdin protects against polymicrobial sepsis by modulating inflammatory mediators. Am J Physiol Gastrointest Liver Physiol 290: G695–G703, 2006. [DOI] [PubMed] [Google Scholar]
- 411.Pachori AS, Melo LG, Hart ML, Noiseux N, Zhang L, Morello F, Solomon SD, Stahl GL, Pratt RE, Dzau VJ. Hypoxia-regulated therapeutic gene as a preemptive treatment strategy against ischemia/reperfusion tissue injury. Proc Natl Acad Sci USA 101: 12282–12287, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Pae HO, Jeong GS, Jeong SO, Kim HS, Kim SA, Kim YC, Yoo SJ, Kim HD, Chung HT. Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp Mol Med 39: 267–277, 2007. [DOI] [PubMed] [Google Scholar]
- 413.Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 72: 1187–1203, 1999. [DOI] [PubMed] [Google Scholar]
- 414.Pang JH, Jiang MJ, Chen YL, Wang FW, Wang DL, Chu SH, Chau LY. Increased ferritin gene expression in atherosclerotic lesions. J Clin Invest 97: 2204–2212, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Parfenova H, Leffler CW. Cerebroprotective functions of HO-2. Curr Pharm Des 14: 443–453, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Park SJ, Kang SJ, Virmani R, Nakano M, Ueda Y. In-stent neoatherosclerosis: a final common pathway of late stent failure. J Am Coll Cardiol 59: 2051–2057, 2012. [DOI] [PubMed] [Google Scholar]
- 417.Pechlaner R, Willeit P, Summerer M, Santer P, Egger G, Kronenberg F, Demetz E, Weiss G, Tsimikas S, Witztum JL, Willeit K, Iglseder B, Paulweber B, Kedenko L, Haun M, Meisinger C, Gieger C, Muller-Nurasyid M, Peters A, Willeit J, Kiechl S. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with progressive atherosclerosis and incident cardiovascular disease. Arterioscler Thromb Vasc Biol 35: 229–236, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Pendrak ML, Chao MP, Yan SS, Roberts DD. Heme oxygenase in Candida albicans is regulated by hemoglobin and is necessary for metabolism of exogenous heme and hemoglobin to α-biliverdin. J Biol Chem 279: 3426–3433, 2004. [DOI] [PubMed] [Google Scholar]
- 419.Peng YJ, Makarenko VV, Nanduri J, Vasavda C, Raghuraman G, Yuan G, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Inherent variations in CO-H2S-mediated carotid body O2 sensing mediate hypertension and pulmonary edema. Proc Natl Acad Sci USA 111: 1174–1179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG, BEAM Study Investigators . Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365: 327–336, 2011. [DOI] [PubMed] [Google Scholar]
- 421.Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H, Schafer AI, Durante W. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 99: 4443–4448, 2002. [DOI] [PubMed] [Google Scholar]
- 422.Peyton KJ, Shebib AR, Azam MA, Liu XM, Tulis DA, Durante W. Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front Pharmacol 3: 48, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Pflueger A, Croatt AJ, Peterson TE, Smith LA, d'Uscio LV, Katusic ZS, Nath KA. The hyperbilirubinemic Gunn rat is resistant to the pressor effects of angiotensin II. Am J Physiol Renal Physiol 288: F552–F558, 2005. [DOI] [PubMed] [Google Scholar]
- 424.Pietrangelo A. Mechanism of iron toxicity. Adv Exp Med Biol 509: 19–43, 2002. [DOI] [PubMed] [Google Scholar]
- 425.Poss KD, Thomas MJ, Ebralidze AK, O'Dell TJ, Tonegawa S. Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron 15: 867–873, 1995. [DOI] [PubMed] [Google Scholar]
- 426.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA 94: 10919–10924, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci USA 94: 10925–10930, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Protchenko O, Philpott CC. Regulation of intracellular heme levels by HMX1, a homologue of heme oxygenase, in Saccharomyces cerevisiae. J Biol Chem 278: 36582–36587, 2003. [DOI] [PubMed] [Google Scholar]
- 429.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18: 286–290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Pulkkinen KH, Ylä-Herttuala S, Levonen AL. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med 51: 2124–2131, 2011. [DOI] [PubMed] [Google Scholar]
- 431.Purdy KE, Arendshorst WJ. EP1 and EP4 receptors mediate prostaglandin E2 actions in the microcirculation of rat kidney. Am J Physiol Renal Physiol 279: F755–F764, 2000. [DOI] [PubMed] [Google Scholar]
- 432.Qiao H, Sai X, Gai L, Huang G, Chen X, Tu X, Ding Z. Association between heme oxygenase 1 gene promoter polymorphisms and susceptibility to coronary artery disease: a HuGE review and meta-analysis. Am J Epidemiol 179: 1039–1048, 2014. [DOI] [PubMed] [Google Scholar]
- 433.Quan S, Yang L, Shnouda S, Schwartzman ML, Nasjletti A, Goodman AI, Abraham NG. Expression of human heme oxygenase-1 in the thick ascending limb attenuates angiotensin II-mediated increase in oxidative injury. Kidney Int 65: 1628–1639, 2004. [DOI] [PubMed] [Google Scholar]
- 434.Radhakrishnan N, Yadav SP, Sachdeva A, Pruthi PK, Sawhney S, Piplani T, Wada T, Yachie A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol 33: 74–78, 2011. [DOI] [PubMed] [Google Scholar]
- 435.Ragsdale SW, Yi L. Thiol/Disulfide redox switches in the regulation of heme binding to proteins. Antioxid Redox Signal 14: 1039–1047, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Rajapurkar MM, Shah SV, Lele SS, Hegde UN, Lensing SY, Gohel K, Mukhopadhyay B, Gang S, Eigenbrodt ML. Association of catalytic iron with cardiovascular disease. Am J Cardiol 109: 438–442, 2012. [DOI] [PubMed] [Google Scholar]
- 437.Raju VS, McCoubrey WK Jr, Maines MD. Regulation of heme oxygenase-2 by glucocorticoids in neonatal rat brain: characterization of a functional glucocorticoid response element. Biochim Biophys Acta 1351: 89–104, 1997. [DOI] [PubMed] [Google Scholar]
- 438.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science 293: 506–509, 2001. [DOI] [PubMed] [Google Scholar]
- 439.Ren Y, D'Ambrosio MA, Wang H, Liu R, Garvin JL, Carretero OA. Heme oxygenase metabolites inhibit tubuloglomerular feedback (TGF). Am J Physiol Renal Physiol 295: F1207–F1212, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, Cherry AD, Wester TE, Natoli MJ, Massey EW, Moon RE, Suliman HB. Carbon monoxide, skeletal muscle oxidative stress, and mitochondrial biogenesis in humans. Am J Physiol Heart Circ Physiol 297: H392–H399, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Ricchetti GA, Williams LM, Foxwell BM. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J Leukoc Biol 76: 719–726, 2004. [DOI] [PubMed] [Google Scholar]
- 442.Rivera M, Zeng Y. Heme oxygenase, steering dioxygen activation toward heme hydroxylation. J Inorg Biochem 99: 37–354, 2005. [DOI] [PubMed] [Google Scholar]
- 443.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 265: C299–C303, 1993. [DOI] [PubMed] [Google Scholar]
- 444.Rodriguez AI, Gangopadhyay A, Kelley EE, Pagano PJ, Zuckerbraun BS, Bauer PM. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler Thromb Vasc Biol 30: 98–104, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Rodriguez F, Kemp R, Balazy M, Nasjletti A. Effects of exogenous heme on renal function: role of heme oxygenase and cyclooxygenase. Hypertension 42: 680–684, 2003. [DOI] [PubMed] [Google Scholar]
- 446.Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, Pan C, Lee S, Vakili L, Yang WP, Kayne P, Mungrue IN, Araujo JA, Berliner JA, Lusis AJ. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res 109: e27–e41, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Roos TU, Heiss EH, Schwaiberger AV, Schachner D, Sroka IM, Oberan T, Vollmar AM, Dirsch VM. Caffeic acid phenethyl ester inhibits PDGF-induced proliferation of vascular smooth muscle cells via activation of p38 MAPK, HIF-1α, and heme oxygenase-1. J Nat Prod 74: 352–356, 2011. [DOI] [PubMed] [Google Scholar]
- 448.Rueda B, Oliver J, Robledo G, López-Nevot MA, Balsa A, Pascual-Salcedo D, González-Gay MA, González-Escribano MF, Martin J. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum 56: 3953–3958, 2007. [DOI] [PubMed] [Google Scholar]
- 449.Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293: 510–514, 2001. [DOI] [PubMed] [Google Scholar]
- 450.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650, 2006. [DOI] [PubMed] [Google Scholar]
- 451.Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res 167: 7–34, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Sabaawy HE, Zhang F, Nguyen X, ElHosseiny A, Nasjletti A, Schwartzman M, Dennery P, Kappas A, Abraham NG. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension 38: 210–215, 2001. [DOI] [PubMed] [Google Scholar]
- 453.Sacerdoti D, Bolognesi M, Di Pascoli M, Gatta A, McGiff JC, Schwartzman ML, Abraham NG. Rat mesenteric arterial dilator response to 11,12-epoxyeicosatrienoic acid is mediated by activating heme oxygenase. Am J Physiol Heart Circ Physiol 291: H1999–H2002, 2006. [DOI] [PubMed] [Google Scholar]
- 454.Sacerdoti D, Colombrita C, Di Pascoli M, Schwartzman ML, Bolognesi M, Falck JR, Gatta A, Abraham NG. 11,12-Epoxyeicosatrienoic acid stimulates heme-oxygenase-1 in endothelial cells. Prostaglandins Other Lipid Mediat 82: 155–161, 2007. [DOI] [PubMed] [Google Scholar]
- 455.Sacerdoti D, Escalante B, Abraham NG, McGiff JC, Levere RD, Schwartzman ML. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science 243: 388–390, 1989. [DOI] [PubMed] [Google Scholar]
- 456.Sahoo SK, Sawa T, Fang J, Tanaka S, Miyamoto Y, Akaike T, Maeda H. Pegylated zinc protoporphyrin: a water-soluble heme oxygenase inhibitor with tumor-targeting capacity. Bioconjug Chem 13: 1031–1038, 2002. [DOI] [PubMed] [Google Scholar]
- 457.Sakamoto H, Omata Y, Palmer G, Noguchi M. Ferric α-hydroxyheme bound to heme oxygenase can be converted to verdoheme by dioxygen in the absence of added reducing equivalents. J Biol Chem 274 18196–18200, 1999. [DOI] [PubMed] [Google Scholar]
- 458.Salonen JT, Nyyssonen K, Korpela H, Tuomilehto J, Seppanen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation 86: 803–811, 1992. [DOI] [PubMed] [Google Scholar]
- 459.Sanchorawala H, Keaney JF Jr. Physiology of vascular homeostasis. In: Textbook of Coronary Thrombosis and Thrombolysis. New York: Springer, 1997, p. 139–153. [Google Scholar]
- 460.Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels KCa and connexins: possible relationship to vasodilator function? J Anat 209: 689–698, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Santiago FS, Ishii H, Shafi S, Khurana R, Kanellakis P, Bhindi R, Ramirez MJ, Bobik A, Martin JF, Chesterman CN, Zachary IC, Khachigian LM. Yin Yang-1 inhibits vascular smooth muscle cell growth and intimal thickening by repressing p21WAF1/Cip1 transcription and p21WAF1/Cip1-Cdk4-cyclin D1 assembly. Circ Res 101: 146–155, 2007. [DOI] [PubMed] [Google Scholar]
- 462.Sarady-Andrews JK, Liu F, Gallo D, Nakao A, Overhaus M, Ollinger R, Choi AM, Otterbein LE. Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 289: L1131–L1137, 2005. [DOI] [PubMed] [Google Scholar]
- 463.Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 275: 21722–21729, 2000. [DOI] [PubMed] [Google Scholar]
- 464.Sawayama Y, Maeda S, Ohnishi H, Okada K, Hayashi J. Effect of probucol on elderly hypercholesterolemic patients in the FAST study. Fukuoka Igaku Zasshi 97: 15–24, 2006. [PubMed] [Google Scholar]
- 465.Sawayama Y, Shimizu C, Maeda N, Tatsukawa M, Kinukawa N, Koyanagi S, Kashiwagi S, Hayashi J. Effects of probucol and pravastatin on common carotid atherosclerosis in patients with asymptomatic hypercholesterolemia. Fukuoka Atherosclerosis Trial (FAST). J Am Coll Cardiol 39: 610–616, 2002. [DOI] [PubMed] [Google Scholar]
- 466.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121: 1276–1284, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, Gallo D, Otterbein LE, Hanafy KA. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest 125: 2609–2625, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Schillinger M, Exner M, Mlekusch W, Domanovits H, Huber K, Mannhalter C, Wagner O, Minar E. Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aortic aneurysm. Thromb Res 106: 131–136, 2002. [DOI] [PubMed] [Google Scholar]
- 469.Schweda F, Klar J, Narumiya S, Nusing RM, Kurtz A. Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am J Physiol Renal Physiol 287: F427–F433, 2004. [DOI] [PubMed] [Google Scholar]
- 470.Sedlak TW, Snyder SH. Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 113: 1776–1782, 2004. [DOI] [PubMed] [Google Scholar]
- 471.Sessa WC, Abraham NG, Escalante B, Schwartzman ML. Manipulation of cytochrome P-450 dependent renal thromboxane synthase activity in spontaneously hypertensive rats. J Hypertens 7: 37–42, 1989. [DOI] [PubMed] [Google Scholar]
- 472.Seta F, Bellner L, Rezzani R, Regan RF, Dunn MW, Abraham NG, Gronert K, Laniado-Schwartzman M. Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am J Pathol 169: 1612–1623, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Shah ZA, Nada SE, Dore S. Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience 180: 248–255, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Shan Y, Lambrecht RW, Ghaziani T, Donohue SE, Bonkovsky HL. Role of Bach-1 in regulation of heme oxygenase-1 in human liver cells: insights from studies with small interfering RNAS. J Biol Chem 279: 51769–51774, 2004. [DOI] [PubMed] [Google Scholar]
- 475.Sharma HS, Das DK, Verdouw PD. Enhanced expression and localization of heme oxygenase-1 during recovery phase of porcine stunned myocardium. Mol Cell Biochem 196: 133–139, 1999. [DOI] [PubMed] [Google Scholar]
- 476.Sharma HS, Maulik N, Gho BC, Das DK, Verdouw PD. Coordinated expression of heme oxygenase-1 and ubiquitin in the porcine heart subjected to ischemia and reperfusion. Mol Cell Biochem 157: 111–116, 1996. [DOI] [PubMed] [Google Scholar]
- 477.Sheng-Long C, Yan-Xin W, Yi-Yi H, Ming F, Jian-Gui H, Yi-Li C, Wen-Jing X, Hong M. AVE0991, a nonpeptide compound, attenuates Angiotensin II-induced vascular smooth muscle cell proliferation via induction of heme oxygenase-1 and downregulation of p-38 MAPK phosphorylation. Int J Hypertens 2012: 958298, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Shimada Y, Tsunoda H, Zang L, Hirano M, Oka T, Tanaka T. Synergistic induction of heme oxygenase-1 by nicaraven after subarachnoid hemorrhage to prevent delayed cerebral vasospasm. Eur J Pharmacol 620: 16–20, 2009. [DOI] [PubMed] [Google Scholar]
- 479.Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J, Kohler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res 99: 537–544, 2006. [DOI] [PubMed] [Google Scholar]
- 480.Sies H. Biological redox systems and oxidative stress. Cell Mol Life Sci 64: 2181–2188, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Sies H. Oxidative Stress: Oxidants and Antioxidants. London: Academic, 1991, p. 1–650. [Google Scholar]
- 482.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol 4: 180–183, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Sies H. Oxidative stress: introductory remarks. In: Oxidative Stress, edited by Sies H. New York: Academic, 1985, p. 1–8. [Google Scholar]
- 484.Sies H. What is oxidative stress? In: Oxidative Stress and Vascular Disease, edited by Keaney JFJr. Boston: Kluwer Academic, 2000, p. 1–8. [Google Scholar]
- 485.Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: molecular regulation of the heme oxygenase-1 gene in renal injury. Am J Physiol Renal Physiol 286: F425–F441, 2004. [DOI] [PubMed] [Google Scholar]
- 486.Silva G, Cunha A, Grégoire IP, Seldon MP, Soares MP. The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38α MAPK isoform. J Immunol 177: 1894–1903, 2006. [DOI] [PubMed] [Google Scholar]
- 487.Skaar EP, Gaspar AH, Schneewind OI. sdG and IsdI, heme degrading enzymes in the cytoplasm of Staphylococcus aureus. J Biol Chem 279 436–443, 2004. [DOI] [PubMed] [Google Scholar]
- 488.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, Bach FH. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med 4: 1073–1077, 1998. [DOI] [PubMed] [Google Scholar]
- 489.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol 172: 3553–3563, 2004. [DOI] [PubMed] [Google Scholar]
- 490.Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, Bach FH. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antioxid Redox Signal 4: 321–329, 2002. [DOI] [PubMed] [Google Scholar]
- 491.Solano ME, Kowal MK, O'Rourke GE, Horst AK, Modest K, Plosch T, Barikbin R, Remus CC, Berger RG, Jago C, Ho H, Sass G, Parker VJ, Lydon JP, DeMayo FJ, Hecher K, Karimi K, Arck PC. Progesterone and HMOX-1 promote fetal growth by CD8+ T cell modulation. J Clin Invest 125: 1726–1738, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Solymoss BC, Marcil M, Gilfix BM, Gelinas F, Poitras AM, Campeau L. The place of ferritin among risk factors associated with coronary artery disease. Coron Artery Dis 5: 231–235, 1994. [DOI] [PubMed] [Google Scholar]
- 493.Song J, Sumiyoshi S, Nakashima Y, Doi Y, Iida M, Kiyohara Y, Sueishi K. Overexpression of heme oxygenase-1 in coronary atherosclerosis of Japanese autopsies with diabetes mellitus: Hisayama study. Atherosclerosis 202: 573–581, 2009. [DOI] [PubMed] [Google Scholar]
- 494.Srivastava P, Pandey VC. Mitochondrial heme oxygenase of Mastomys coucha. Int J Biochem Cell Biol 28: 1071–1077, 1996. [DOI] [PubMed] [Google Scholar]
- 495.Stanford SJ, Walters MJ, Hislop AA, Haworth SG, Evans TW, Mann BE, Motterlini R, Mitchell JA. Heme oxygenase is expressed in human pulmonary artery smooth muscle where carbon monoxide has an anti-proliferative role. Eur J Pharmacol 473: 135–141, 2003. [DOI] [PubMed] [Google Scholar]
- 496.Stec DE, Drummond HA, Vera T. Role of carbon monoxide in blood pressure regulation. Hypertension 51: 597–604, 2008. [DOI] [PubMed] [Google Scholar]
- 497.Stein AB, Guo Y, Tan W, Wu WJ, Zhu X, Li Q, Luo C, Dawn B, Johnson TR, Motterlini R, Bolli R. Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction. J Mol Cell Cardiol 38: 127–134, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 499.Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal 6: 841–849, 2004. [DOI] [PubMed] [Google Scholar]
- 500.Stocker R. Induction of haem oxygenase as a defence against oxidative stress. Free Radical Res Commun 9: 101–112, 1990. [DOI] [PubMed] [Google Scholar]
- 501.Stocker R. Molecular mechanisms underlying the antiatherosclerotic and antidiabetic effects of probucol, succinobucol, and other probucol analogues. Curr Opin Lipidol 20: 227–235, 2009. [DOI] [PubMed] [Google Scholar]
- 502.Stocker R, Ames BN. Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc Natl Acad Sci USA 84: 8130–8134, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci USA 84: 5918–5922, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Stocker R, Keaney JF Jr.. Role of oxidative modifications in atherosclerosis. Physiol Rev 84: 1381–1478, 2004. [DOI] [PubMed] [Google Scholar]
- 505.Stocker R, Perrella MA. Heme oxygenase-1. A novel drug target for atherosclerotic diseases? Circulation 114: 2178–2189, 2006. [DOI] [PubMed] [Google Scholar]
- 506.Stocker R, Peterhans E. Antioxidant properties of conjugated bilirubin and biliverdin: biologically relevant scavenging of hypochlorous acid. Free Radical Res Commun 6: 57–66, 1989. [DOI] [PubMed] [Google Scholar]
- 507.Stocker R, Peterhans E. Synergistic interaction between vitamin E and the bile pigments bilirubin and biliverdin. Biochim Biophys Acta 1002: 238–244, 1989. [DOI] [PubMed] [Google Scholar]
- 508.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043–1046, 1987. [DOI] [PubMed] [Google Scholar]
- 509.Stoeckius M, Erat A, Fujikawa T, Hiromura M, Koulova A, Otterbein L, Bianchi C, Tobiasch E, Dagon Y, Sellke FW, Usheva A. Essential roles of Raf/extracellular signal-regulated kinase/mitogen-activated protein kinase pathway, YY1, and Ca2+ influx in growth arrest of human vascular smooth muscle cells by bilirubin. J Biol Chem 287: 15418–15426, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry 33: 5636–5640, 1994. [DOI] [PubMed] [Google Scholar]
- 511.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res 100: 884–893, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Stubbert D, Prysyazhna O, Rudyk O, Scotcher J, Burgoyne JR, Eaton P. Protein kinase G Iα oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 64: 1344–1351, 2014. [DOI] [PubMed] [Google Scholar]
- 513.Suematsu M, Goda N, Sano T, Kashiwagi S, Egawa T, Shinoda Y, Ishimura Y. Carbon monoxide: an endogenous modulator of sinusoidal tone in the perfused rat liver. J Clin Invest 96: 2431–2437, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest 117: 3730–3741, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Sullivan JL. Iron and the sex difference in heart disease risk. Lancet 1: 1293–1294, 1981. [DOI] [PubMed] [Google Scholar]
- 516.Sullivan JL. Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochim Biophys Acta 1790: 718–723, 2009. [DOI] [PubMed] [Google Scholar]
- 517.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J 21: 5216–5224, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Sun JJ, Kim HJ, Seo HG, Lee JH, Yun-Choi HS, Chang KC. YS 49, 1-(a-naphtylmethyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, regulates angiotensin II-stimulated ROS production, JNK phosphorylation and vascular smooth muscle cell proliferation via the induction of heme oxygenase-1. Life Sci 82: 600–607, 2008. [DOI] [PubMed] [Google Scholar]
- 519.Sun Y, Rotenberg MO, Maines MD. Developmental expression of heme oxygenase isozymes in rat brain. Two HO-2 mRNAs are detected. J Biol Chem 265: 8212–8217, 1990. [PubMed] [Google Scholar]
- 520.Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, Matsuno S, Shibahara S, Abraham NG. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 6: 15–24, 2003. [DOI] [PubMed] [Google Scholar]
- 521.Sylvester JT, McGowan C. The effects of agents that bind to cytochrome P-450 on hypoxic pulmonary vasoconstriction. Circ Res 43: 429–437, 1978. [DOI] [PubMed] [Google Scholar]
- 522.Tada T, Reidy MA. Endothelial regeneration. IX. Arterial injury followed by rapid endothelial repair induces smooth-muscle-cell proliferation but not intimal thickening. Am J Pathol 129: 429–433, 1987. [PMC free article] [PubMed] [Google Scholar]
- 523.Taha H, Skrzypek K, Guevara I, Nigisch A, Mustafa S, Grochot-Przeczek A, Ferdek P, Was H, Kotlinowski J, Kozakowska M, Balcerczyk A, Muchova L, Vitek L, Weigel G, Dulak J, Jozkowicz A. Role of heme oxygenase-1 in human endothelial cells: lesson from the promoter allelic variants. Arterioscler Thromb Vasc Biol 30: 1634–1641, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Taillé C, El-Benna J, Lanone S, Dang MC, Ogier-Denis E, Aubier M, Boczkowski J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J Biol Chem 279: 28681–28688, 2004. [DOI] [PubMed] [Google Scholar]
- 525.Tanaka S, Akaike T, Fang J, Beppu T, Ogawa M, Tamura F, Miyamoto Y, Maeda H. Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer 88: 902–909, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendelsohn ME. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med 9: 1506–1512, 2003. [DOI] [PubMed] [Google Scholar]
- 527.Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin E2 on the renal afferent arteriole: role of EP3 and EP4 receptors. Circ Res 86: 663–670, 2000. [DOI] [PubMed] [Google Scholar]
- 528.Tang YL, Tang Y, Zhang YC, Agarwal A, Kasahara H, Qian K, Shen L, Phillips MI. A hypoxia-inducible vigilant vector system for activating therapeutic genes in ischemia. Gene Ther 12: 1163–1170, 2005. [DOI] [PubMed] [Google Scholar]
- 529.Tang YL, Tang Y, Zhang YC, Qian K, Shen L, Phillips MI. Protection from ischemic heart injury by a vigilant heme oxygenase-1 plasmid system. Hypertension 43: 746–751, 2004. [DOI] [PubMed] [Google Scholar]
- 530.Tanous D, Bräsen JH, Choy K, Wu BJ, Kathir K, Lau A, Celermajer DS, Stocker R. Probucol inhibits in-stent thrombosis and neointimal hyperplasia by promoting re-endothelialization. Atherosclerosis 189: 342–349, 2006. [DOI] [PubMed] [Google Scholar]
- 531.Tardif JC, Gregoire J, L'Allier PL, Ibrahim R, Anderson TJ, Reeves F, Title LM, Schampaert E, LeMay M, Lesperance J, Scott R, Guertin MC, Brennan ML, Hazen SL, Bertrand OF. Effects of the antioxidant succinobucol (AGI-1067) on human atherosclerosis in a randomized clinical trial. Atherosclerosis 197: 480–486, 2008. [DOI] [PubMed] [Google Scholar]
- 532.Tardif JC, Grégoire J, Lavoie MA, L'Allier PL. Pharmacologic prevention of both restenosis and atherosclerosis progression: AGI-1067, probucol, statins, folic acid and other therapies. Curr Opin Lipidol 14: 615–620, 2003. [DOI] [PubMed] [Google Scholar]
- 533.Tardif JC, McMurray JJ, Klug E, Small R, Schumi J, Choi J, Cooper J, Scott R, Lewis EF, L'Allier PL, Pfeffer MA. Effects of succinobucol (AGI-1067) after an acute coronary syndrome: a randomised, double-blind, placebo-controlled trial. Lancet 371: 1761–1768, 2008. [DOI] [PubMed] [Google Scholar]
- 534.Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res 89: 224–236, 2001. [DOI] [PubMed] [Google Scholar]
- 535.Tenhunen R, Marver H, Schmid R. Microsomal heme oxygenase, characterization of the enzyme. J Biol Chem 44: 6388–6394, 1969. [PubMed] [Google Scholar]
- 536.Tenhunen R, Marver HS, Schmid R. The enzymatic catabolism of hemoglobin: stimulation of microsomal heme oxygenase by hemin. J Lab Clin Med 75: 410–421, 1970. [PubMed] [Google Scholar]
- 537.Tenhunen R, Ross ME, Marver S, Schmid R. Reduced nicotinamide-adenine dinucleotide phosphate dependent biliverdin reductase: partial purification and characterization. Biochemistry 20: 298–303, 1970. [DOI] [PubMed] [Google Scholar]
- 538.Teran FJ, Johnson RA, Stevenson BK, Peyton KJ, Jackson KE, Appleton SD, Durante W, Johnson FK. Heme oxygenase-derived carbon monoxide promotes arteriolar endothelial dysfunction and contributes to salt-induced hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 288: R615–R622, 2005. [DOI] [PubMed] [Google Scholar]
- 539.Thorup C, Jones CL, Gross SS, Moore LC, Goligorsky MS. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am J Physiol Renal Physiol 277: F882–F889, 1999. [DOI] [PubMed] [Google Scholar]
- 540.Togane Y, Morita T, Suematsu M, Ishimura Y, Yamazaki JI, Katayama S. Protective roles of endogenous carbon monoxide in neointimal development elicited by arterial injury. Am J Physiol Heart Circ Physiol 278: H623–H632, 2000. [DOI] [PubMed] [Google Scholar]
- 541.Tong S, Kaitu'u-Lino TJ, Onda K, Beard S, Hastie R, Binder NK, Cluver C, Tuohey L, Whitehead C, Brownfoot F, De Silva M, Hannan NJ. Heme oxygenase-1 is not decreased in preeclamptic placenta and does not negatively regulate placental soluble fms-like tyrosine kinase-1 or soluble endoglin secretion. Hypertension 66: 1073–1081, 2015. [DOI] [PubMed] [Google Scholar]
- 542.Torisu-Itakura H, Furue M, Kuwano M, Ono M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res 91: 906–910, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, Kane GC, Terzic A, Griffin MD, Nath KA. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int 72: 1073–1080, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem 261: 11131–11137, 1986. [PubMed] [Google Scholar]
- 545.Tran CH, Taylor MS, Plane F, Nagaraja S, Tsoukias NM, Solodushko V, Vigmond EJ, Furstenhaupt T, Brigdan M, Welsh DG. Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am J Physiol Cell Physiol 302: C1226–C1242, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, Gladwin MT, Nabel EG. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res 101: 893–901, 2007. [DOI] [PubMed] [Google Scholar]
- 547.Tsoyi K, Lee TY, Lee YS, Kim HJ, Seo HG, Lee JH, Chang KC. Heme-oxygenase-1 induction and carbon monoxide-releasing molecule inhibit lipopolysaccharide (LPS)-induced high-mobility group box 1 release in vitro and improve survival of mice in LPS- and cecal ligation and puncture-induced sepsis model in vivo. Mol Pharmacol 76: 173–182, 2009. [DOI] [PubMed] [Google Scholar]
- 548.Tulis DA, Durante W, Liu X, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 104: 2710–2715, 2001. [DOI] [PubMed] [Google Scholar]
- 549.Tulis DA, Durante W, Peyton KJ, Chapman GB, Evans AJ, Schafer AI. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem Biophys Res Commun 279: 646–652, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Tulis DA, Durante W, Peyton KJ, Evans AJ, Schafer AI. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis 155: 113–122, 2001. [DOI] [PubMed] [Google Scholar]
- 551.Tulis DA, Keswani AN, Peyton KJ, Wang H, Schafer AI, Durante W. Local administration of carbon monoxide inhibits neointima formation in balloon injured rat carotid arteries. Cell Mol Biol 51: 441–446, 2005. [PMC free article] [PubMed] [Google Scholar]
- 552.Tuomainen TP, Punnonen K, Nyyssonen K, Salonen JT. Association between body iron stores and the risk of acute myocardial infarction in men. Circulation 97: 1461–1466, 1998. [DOI] [PubMed] [Google Scholar]
- 553.Turner CP, Bergeron M, Matz P, Zegna A, Noble LJ, Panter SS, Sharp FR. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab 18: 257–273, 1998. [DOI] [PubMed] [Google Scholar]
- 554.Van Loon RL, Bartelds B, Wagener FA, Affara N, Mohaupt S, Wijnberg H, Pennings SW, Takens J, Berger RM. Erythropoietin attenuates pulmonary vascular remodeling in experimental pulmonary arterial hypertension through interplay between endothelial progenitor cells and heme oxygenase. Front Pediatr 3: 71, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Varadi J, Lekli I, Juhasz B, Bacskay I, Szabo G, Gesztelyi R, Szendrei L, Varga E, Bak I, Foresti R, Motterlini R, Tosaki A. Beneficial effects of carbon monoxide-releasing molecules on post-ischemic myocardial recovery. Life Sci 80: 1619–1626, 2007. [DOI] [PubMed] [Google Scholar]
- 556.Vera T, Granger JP, Stec DE. Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am J Physiol Regul Integr Comp Physiol 297: R738–R743, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol 5: 94, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Visner GA, Lu F, Zhou H, Liu J, Kazemfar K, Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation 107: 911–916, 2003. [DOI] [PubMed] [Google Scholar]
- 559.Vitali SH, Mitsialis SA, Liang OD, Liu X, Fernandez-Gonzalez A, Christou H, Wu X, McGowan FX, Kourembanas S. Divergent cardiopulmonary actions of heme oxygenase enzymatic products in chronic hypoxia. PloS One 4: e5978, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Vlahakis JZ, Kinobe RT, Bowers RJ, Brien JF, Nakatsu K, Szarek WA. Imidazole-dioxolane compounds as isozyme-selective heme oxygenase inhibitors. J Med Chem 49: 4437–4441, 2006. [DOI] [PubMed] [Google Scholar]
- 561.Vlahakis JZ, Vukomanovic D, Nakatsu K, Szarek WA. Selective inhibition of heme oxygenase-2 activity by analogs of 1-(4-chlorobenzyl)-2-(pyrrolidin-1-ylmethyl)-1H-benzimidazole (clemizole): exploration of the effects of substituents at the N-1 position. Bioorg Med Chem 21: 6788–6795, 2013. [DOI] [PubMed] [Google Scholar]
- 562.Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res 97: 908–915, 2005. [DOI] [PubMed] [Google Scholar]
- 563.Wagener FA, Scharstuhl A, Tyrrell RM, Von den Hoff JW, Jozkowicz A, Dulak J, Russel FG, Kuijpers-Jagtman AM. The heme-heme oxygenase system in wound healing; implications for scar formation. Curr Drug Targets 11: 1571–1585, 2010. [DOI] [PubMed] [Google Scholar]
- 564.Wagener FA, van Beurden HE, von den Hoff JW, Adema GJ, Figdor CG. The heme-heme oxygenase system: a molecular switch in wound healing. Blood 102: 521–528, 2003. [DOI] [PubMed] [Google Scholar]
- 565.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev 55: 551–571, 2003. [DOI] [PubMed] [Google Scholar]
- 566.Wagner KH, Wallner M, Molzer C, Gazzin S, Bulmer AC, Tiribelli C, Vitek L. Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin Sci 129: 1–25, 2015. [DOI] [PubMed] [Google Scholar]
- 567.Walldius G, Erikson U, Olsson AG, Bergstrand L, Hadell K, Johansson J, Kaijser L, Lassvik C, Molgaard J, Nilsson S, et al. The effect of probucol on femoral atherosclerosis: the Probucol Quantitative Regression Swedish Trial (PQRST). Am J Cardiol 74: 875–883, 1994. [DOI] [PubMed] [Google Scholar]
- 568.Wang G, Hamid T, Keith RJ, Zhou G, Partridge CR, Xiang X, Kingery JR, Lewis RK, Li Q, Rokosh DG, Ford R, Spinale FG, Riggs DW, Srivastava S, Bhatnagar A, Bolli R, Prabhu SD. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 121: 1912–1925, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 130: 1643–1652, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Wang J, Tran J, Wang H, Luo W, Guo C, Harro D, Campbell AD, Eitzman DT. Melanoma tumor growth is accelerated in a mouse model of sickle cell disease. Exp Hematol Oncol 4: 19, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Wang L, Bautista LE. Serum bilirubin and the risk of hypertension. Int J Epidemiol 44: 142–152, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Wang LJ, Lee TS, Lee FY, Pai RC, Chau LY. Expression of heme oxygenase-1 in atherosclerotic lesions. Am J Pathol 152: 711–720, 1998. [PMC free article] [PubMed] [Google Scholar]
- 573.Wang R, Shamloul R, Wang X, Meng Q, Wu L. Sustained normalization of high blood pressure in spontaneously hypertensive rats by implanted hemin pump. Hypertension 48: 685–692, 2006. [DOI] [PubMed] [Google Scholar]
- 574.Wang R, Wang Z, Wu L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol 121: 927–934, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Wang R, Wu L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J Biol Chem 272: 8222–8226, 1997. [DOI] [PubMed] [Google Scholar]
- 576.Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163: 1585–1595, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med 43: 995–1022, 2007. [DOI] [PubMed] [Google Scholar]
- 578.Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets 11: 1551–1570, 2010. [DOI] [PubMed] [Google Scholar]
- 579.Weber CM, Eke BC, Maines MD. Corticosterone regulates heme oxygenase-2 and NO synthase transcription and protein expression in rat brain. J Neurochem 63: 953–962, 1994. [DOI] [PubMed] [Google Scholar]
- 580.Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, Chin BY, Kaczmarek E, Alam J, Bach FH, Zuckerbraun BS, Otterbein LE. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem 284: 21369–21378, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, Penacho N, Seth P, Sukhatme V, Ahmed A, Pandolfi PP, Helczynski L, Bjartell A, Persson JL, Otterbein LE. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res 73: 7009–7021, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Wegiel B, Gallo D, Csizmadia E, Roger T, Kaczmarek E, Harris C, Zuckerbraun BS, Otterbein LE. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci USA 108: 18849–18854, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Wegiel B, Hedblom A, Li M, Gallo D, Csizmadia E, Harris C, Nemeth Z, Zuckerbraun BS, Soares M, Persson JL, Otterbein LE. Heme oxygenase-1 derived carbon monoxide permits maturation of myeloid cells. Cell Death Dis 5: e1139, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP, Otterbein LE. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest 124: 4926–4940, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Weinberg ED. The hazards of iron loading. Metallomics 2: 732–740, 2010. [DOI] [PubMed] [Google Scholar]
- 586.Wells G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem Soc Trans 43: 674–679, 2015. [DOI] [PubMed] [Google Scholar]
- 587.Wenzel P, Rossmann H, Müller C, Kossmann S, Oelze M, Schulz A, Arnold N, Simsek C, Lagrange J, Klemz R, Schönfelder T, Brandt M, Karbach SH, Knorr M, Finger S, Neukirch C, Hauser F, Beutel ME, Kröller-Schön S, Schulz E, Schnabel RB, Lackner K, Wild PS, Zeller T, Daiber A, Blankenberg S, Münzel T. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J 36: 3437–3446, 2015. [DOI] [PubMed] [Google Scholar]
- 588.Westcott EB, Goodwin EL, Segal SS, Jackson WF. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J Physiol 590: 1849–1869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Weston AH, Feletou M, Vanhoutte PM, Falck JR, Campbell WB, Edwards G. Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol 145: 775–784, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Wiesel P, Patel AP, Carvajal IM, Wang ZY, Pellacani A, Maemura K, DiFonzo N, Rennke HG, Layne MD, Yet SF, Lee ME, Perrella MA. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ Res 88: 1088–1094, 2001. [DOI] [PubMed] [Google Scholar]
- 591.Wikström AK, Stephansson O, Cnattingius S. Tobacco use during pregnancy and preeclampsia risk: effects of cigarette smoking and snuff. Hypertension 55: 1254–1259, 2010. [DOI] [PubMed] [Google Scholar]
- 592.Wilks A, Ortiz de Montellano PR. Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species J Biol Chem 268: 22357–22362, 1993. [PubMed] [Google Scholar]
- 593.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Heme oxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 306: 2093–2097, 2004. [DOI] [PubMed] [Google Scholar]
- 594.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med 2: 87–90, 1996. [DOI] [PubMed] [Google Scholar]
- 595.Winterbourn CC. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim Biophys Acta 1840: 730–738, 2014. [DOI] [PubMed] [Google Scholar]
- 596.Wong RJ, Zhao H, Stevenson DK. A deficiency in haem oxygenase-1 induces foetal growth restriction by placental vasculature defects. Acta Paediatr 101: 827–834, 2012. [DOI] [PubMed] [Google Scholar]
- 597.Wu BJ, Kathir K, Witting PK, Beck K, Choy K, Li C, Croft KD, Mori TA, Tanous D, Adams MR, Lau AK, Stocker R. Antioxidants protect from atherosclerosis by a heme oxygenase-1 pathway that is independent of free radical scavenging. J Exp Med 203: 1117–1127, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Wu BJ, Midwinter RG, Cassano C, Beck K, Wang Y, Changsiri D, Gamble JR, Stocker R. Heme oxygenase-1 increases endothelial progenitor cells. Arterioscler Thromb Vasc Biol 29: 1537–1542, 2009. [DOI] [PubMed] [Google Scholar]
- 599.Wu J, Ma J, Fan ST, Schlitt HJ, Tsui TY. Bilirubin derived from heme degradation suppresses MHC class II expression in endothelial cells. Biochem Biophys Res Commun 338: 890–896, 2005. [DOI] [PubMed] [Google Scholar]
- 600.Wu TW, Wu J, Li RK, Mickle D, Carey D. Albumin-bound bilirubins protect human ventricular myocytes against oxyradical damage. Biochem Cell Biol 69: 683–688, 1991. [DOI] [PubMed] [Google Scholar]
- 601.Wysoczynski M, Ratajczak J, Pedziwiatr D, Rokosh G, Bolli R, Ratajczak MZ. Identification of heme oxygenase 1 (HO-1) as a novel negative regulator of mobilization of hematopoietic stem/progenitor cells. Stem Cell Rev Rep 11: 110–118, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res 102: 1118–1126, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of a-subunits. Am J Physiol Heart Circ Physiol 286: H610–H618, 2004. [DOI] [PubMed] [Google Scholar]
- 604.Xiao S, Wang X, Ni H, Li N, Zhang A, Liu H, Pu F, Xu L, Gao J, Zhao Q, Mu Y, Wang C, Sun Y, Du T, Xu X, Zhang G, Hiscox JA, Goodfellow IG, Zhou EM. MicroRNA miR-24-3p promotes porcine reproductive and respiratory syndrome virus replication through suppression of heme oxygenase-1 expression. J Virol 89: 4494–4503, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 103: 129–135, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, Sasaki H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet 66: 187–195, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Yamamoto A, Matsuzawa Y, Yokoyama S, Funahashi T, Yamamura T, Kishino B. Effects of probucol on xanthomata regression in familial hypercholesterolemia. Am J Cardiol 57: 29H–35H, 1986. [DOI] [PubMed] [Google Scholar]
- 608.Yamamoto Y, Klemm MF, Edwards FR, Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. J Physiol 535: 181–195, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Yang H, Wang Q, Li S. MicroRNA-218 promotes high glucose-induced apoptosis in podocytes by targeting heme oxygenase-1. Biochem Biophys Res Commun 471: 582–588, 2016. [DOI] [PubMed] [Google Scholar]
- 610.Yang L, Quan S, Abraham NG. Retrovirus-mediated HO gene transfer into endothelial cells protects against oxidant-induced injury. Am J Physiol Lung Cell Mol Physiol 277: L127–L133, 1999. [DOI] [PubMed] [Google Scholar]
- 611.Yang L, Quan S, Nasjletti A, Laniado-Schwartzman M, Abraham NG. Heme oxygenase-1 gene expression modulates angiotensin II-induced increase in blood pressure. Hypertension 43: 1221–1226, 2004. [DOI] [PubMed] [Google Scholar]
- 612.Yang Q, Wen SW, Smith GN, Chen Y, Krewski D, Chen XK, Walker MC. Maternal cigarette smoking and the risk of pregnancy-induced hypertension and eclampsia. Int J Epidemiol 35: 288–293, 2006. [DOI] [PubMed] [Google Scholar]
- 613.Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem 57: 6930–6948, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Yet SF, Layne MD, Liu X, Chen YH, Ith B, Sibinga NE, Perrella MA. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J 17: 1759–1761, 2003. [DOI] [PubMed] [Google Scholar]
- 615.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest 103: R23–R29, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, Dzau VJ, Lee ME, Perrella MA. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ Res 89: 168–173, 2001. [DOI] [PubMed] [Google Scholar]
- 617.Yi L, Morgan JT, Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem 285: 20117–20127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Yi L, Ragsdale SW. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J Biol Chem 282: 21056–21067, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Yi N, Chen SY, Ma A, Chen PS, Yao B, Liang TM, Liu C. Tunicamycin inhibits PDGF-BB-induced proliferation and migration of vascular smooth muscle cells through induction of HO-1. Anat Rec 295: 1462–1472, 2012. [DOI] [PubMed] [Google Scholar]
- 620.Yoshida T, Kikuchi G. Features of the reaction of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem 253: 4230–4236, 1978. [PubMed] [Google Scholar]
- 621.Yoshida T, Kikuchi G. Sequence of the reaction of heme catabolism catalyzed by the microsomal heme oxygenase system. FEBS Lett 48: 256–261, 1974. [DOI] [PubMed] [Google Scholar]
- 622.Yoshida T, Maulik N, Ho YS, Alam J, Das DK. Hmox-1 constitutes an adaptive response to effect antioxidant cardioprotection: A study with transgenic mice heterozygous for targeted disruption of the Heme oxygenase-1 gene. Circulation 103: 1695–1701, 2001. [DOI] [PubMed] [Google Scholar]
- 623.Yoshida T, Migita CT. Mechanism of heme degradation by heme oxygenase. J Inorg Biochem 82: 33–41, 2000. [DOI] [PubMed] [Google Scholar]
- 624.Yoshida T, Noguchi M, Kikuchi G.. Oxygenated form of heme·heme oxygenase complex and requirement for second electron to initiate heme degradation from the oxygenated complex. J Biol Chem 255 4418–4420, 1980. [PubMed] [Google Scholar]
- 625.Yoshida T, Noguchi M, Kikuchi G. The step of carbon monoxide liberation in the sequence of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem 257: 9345–9348, 1982. [PubMed] [Google Scholar]
- 626.Yoshinaga T, Sassa S, Kappas A. A comparative study of heme degradation by NADPH-cytochrome c reductase alone and by the complete heme oxygenase system. Distinctive aspects of heme degradation by NADPH-cytochrome c reductase. J Biol Chem 257: 7794–7802, 1982. [PubMed] [Google Scholar]
- 627.Yu X, Tao W, Jiang F, Li C, Lin J, Liu C. Celastrol attenuates hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via induction of heme oxygenase-1. Am J Hypertens 23: 895–903, 2010. [DOI] [PubMed] [Google Scholar]
- 628.Yuan G, Vasavda C, Peng YJ, Makarenko VV, Raghuraman G, Nanduri J, Gadalla MM, Semenza GL, Kumar GK, Snyder SH, Prabhakar NR. Protein kinase G-regulated production of H2S governs oxygen sensing. Science Signaling 8: ra37, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Zager RA, Johnson AC, Becker K. Plasma and urinary heme oxygenase-1 in AKI. J Am Soc Nephrol 23: 1048–1057, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Zakhary R, Poss KD, Jaffrey SR, Ferris CD, Tonegawa S, Snyder SH. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci USA 94: 14848–14853, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Zarjou A, Kim J, Traylor AM, Sanders PW, Balla J, Agarwal A, Curtis LM. Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am J Physiol Renal Physiol 300: F254–F262, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Zellers TM, McCormick J, Wu Y. Interaction among ET-1, endothelium-derived nitric oxide, and prostacyclin in pulmonary arteries and veins. Am J Physiol Heart Circ Physiol 267: H139–H147, 1994. [DOI] [PubMed] [Google Scholar]
- 633.Zenclussen ML, Anegon I, Bertoja AZ, Chauveau C, Vogt K, Gerlof K, Sollwedel A, Volk HD, Ritter T, Zenclussen AC. Over-expression of heme oxygenase-1 by adenoviral gene transfer improves pregnancy outcome in a murine model of abortion. J Reprod Immunol 69: 35–52, 2006. [DOI] [PubMed] [Google Scholar]
- 634.Zenclussen ML, Casalis PA, El-Mousleh T, Rebelo S, Langwisch S, Linzke N, Volk HD, Fest S, Soares MP, Zenclussen AC. Haem oxygenase-1 dictates intrauterine fetal survival in mice via carbon monoxide. J Pathol 225: 293–304, 2011. [DOI] [PubMed] [Google Scholar]
- 635.Zenclussen ML, Linzke N, Schumacher A, Fest S, Meyer N, Casalis PA, Zenclussen AC. Heme oxygenase-1 is critically involved in placentation, spiral artery remodeling, and blood pressure regulation during murine pregnancy. Front Pharmacol 5: 291, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Zeng B, Chen H, Zhu C, Ren X, Lin G, Cao F. Effects of combined mesenchymal stem cells and heme oxygenase-1 therapy on cardiac performance. Eur J Cardiothorac Surg 34: 850–856, 2008. [DOI] [PubMed] [Google Scholar]
- 637.Zeng B, Lin G, Ren X, Zhang Y, Chen H. Over-expression of HO-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. J Biomed Sci 17: 80, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Zhang DX, Gauthier KM, Chawengsub Y, Campbell WB. ACh-induced relaxations of rabbit small mesenteric arteries: role of arachidonic acid metabolites and K+. Am J Physiol Heart Circ Physiol 293: H152–H159, 2007. [DOI] [PubMed] [Google Scholar]
- 639.Zhang J, Vandevenne P, Hamdi H, Van Puyvelde M, Zucchi A, Bettonville M, Weatherly K, Braun MY. Micro-RNA-155-mediated control of heme oxygenase 1 (HO-1) is required for restoring adaptively tolerant CD4+ T-cell function in rodents. Eur J Immunol 45: 829-442, 2015. [DOI] [PubMed] [Google Scholar]
- 640.Zhang M, Zhang BH, Chen L, An W. Overexpression of heme oxygenase-1 protects smooth muscle cells against oxidative injury and inhibits cell proliferation. Cell Res 12: 123–132, 2002. [DOI] [PubMed] [Google Scholar]
- 641.Zhao H, Ozen M, Wong RJ, Stevenson DK. Heme oxygenase-1 in pregnancy and cancer: similarities in cellular invasion, cytoprotection, angiogenesis, and immunomodulation. Front Pharmacol 5: 295, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Zhao H, Wong RJ, Kalish FS, Nayak NR, Stevenson DK. Effect of heme oxygenase-1 deficiency on placental development. Placenta 30: 861–868, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Zhen G, Zhang Z, Xu Y. The role of endogenous carbon monoxide in the hypoxic vascular remodeling of rat model of hypoxic pulmonary hypertension. J Huazhong Univ Sci Technol Med Sci 23: 356–358, 2003. [DOI] [PubMed] [Google Scholar]
- 644.Zheng Q, Liu S, Song Z. Mechanism of arterial remodeling in chronic allograft vasculopathy. Front Med 5: 248–253, 2011. [DOI] [PubMed] [Google Scholar]
- 645.Zhou H, Liu H, Porvasnik SL, Terada N, Agarwal A, Cheng Y, Visner GA. Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension. Lab Invest 86: 62–71, 2006. [DOI] [PubMed] [Google Scholar]
- 646.Zhu W, Hunt DJ, Richardson AR, Stojiljkovic I. Use of heme compounds as iron sources by pathogenic Neisseriae requires the product of the hemO gene. J Bacteriol 182: 439–447, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Zhu W, Wilks A, Stojiljkovic I. Degradation of heme gram-negative bacteria: the product of the hemO Gene of Neisseriae is a heme oxygenase. J Bacteriol 182: 6783–6790, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S, Otterbein LE. Carbon monoxide reverses established pulmonary hypertension. J Exp Med 203: 2109–2119, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]