

Abstract
Identification of dynamic protein–protein interactions at the peptide level on a proteomic scale is a challenging approach that is still in its infancy. We have developed a system to cross-link cells directly in culture with the special lysine cross-linker bis(succinimidyl)-3-azidomethyl-glutarate (BAMG). We used the Gram-positive model bacterium Bacillus subtilis as an exemplar system. Within 5 min extensive intracellular cross-linking was detected, while intracellular cross-linking in a Gram-negative species, Escherichia coli, was still undetectable after 30 min, in agreement with the low permeability in this organism for lipophilic compounds like BAMG. We were able to identify 82 unique interprotein cross-linked peptides with <1% false discovery rate by mass spectrometry and genome-wide database searching. Nearly 60% of the interprotein cross-links occur in assemblies involved in transcription and translation. Several of these interactions are new, and we identified a binding site between the δ and β′ subunit of RNA polymerase close to the downstream DNA channel, providing a clue into how δ might regulate promoter selectivity and promote RNA polymerase recycling. Our methodology opens new avenues to investigate the functional dynamic organization of complex protein assemblies involved in bacterial growth. Data are available via ProteomeXchange with identifier PXD006287.
Keywords: Bacillus subtilis, in vivo cross-linking, bis(succinimidyl)-3-azidomethyl-glutarate (BAMG), diagonal strong cation exchange chromatography, RNA polymerase, delta subunit, mass spectrometry, NusA, glutamate dehydrogenase, ribosome biogenesis
Introduction
Understanding how biological assemblies function at the molecular level requires knowledge of the spatial arrangement of their composite proteins. Chemical protein cross-linking coupled to identification of proteolytic cross-linked peptides by mass spectrometry (CX–MS) has been successfully used to obtain information about the 3D topology of isolated protein complexes.1 In this approach, the amino acid sequences of a cross-linked peptide pair reveal the interacting protein domains. However, because in vitro reconstitution of labile protein complexes can be difficult and might not properly reflect the natural situation, in vivo cross-linking methods can be crucial to gain insight into the configuration of dynamic multiprotein complexes. The continued increase in peptide identification sensitivity by improved MS techniques and equipment combined with affinity purification of low abundant target proteins has opened the door to proteome-wide protein interaction studies by cross-linking living cells. Such a systems-level view on dynamic protein interactions would be a tremendously powerful tool to study cell biology.
The only large-scale in vivo CX–MS studies with bacteria thus far have been performed with four Gram-negative species after repeated washings by pelleting and resuspension in phosphate-buffered saline of the cells before the addition of the cross-linker.2−5 This approach has provided valuable data about the 3D topology of outer membrane and periplasmic protein complexes. However, there is uncertainty whether all protein–protein interactions that exist under a defined physiological condition survive the pretreatment of the cells before the addition of the cross-linker used in this approach. Here we have developed conditions that enable rapid cross-linking of intracellular proteins by adding the cross-linking agent directly in the growth medium of bacteria. Avoiding harvesting and washing the cells before cross-linking offers the best possible surety that labile and dynamic interactions occurring under a defined condition can be experimentally trapped in a way that the in vivo spatial arrangement of proteins in the cell can be studied in great detail.
We used the Gram-positive model Bacillus subtilis, widely studied for processes guided by dynamic protein–protein interactions involved in gene expression, cell division, sporulation, and germination.6 Cross-linking was accomplished by a previously designed reagent, bis(succinimidyl)-3-azidomethyl-glutarate (BAMG) (Figure S1).7 This bifunctional N-hydroxysuccinimidyl ester covalently links proximal lysine residues on protein surfaces via two amide bonds bridged by a spacer of five carbon atoms. The relatively short spacer results in high-resolution cross-link maps. A cross-linker with the same spacer length and similar hydrophobicity, disuccinimidyl glutarate (DSG) (Figure S1), is membrane permeable and has been used before for cross-linking in living human cells.8 Importantly, to prevent possible dissociation of transient intracellular protein interactions by washing and medium change, we added the cross-linker directly in the culture medium containing a low concentration of primary amines to minimize reaction with and quenching of the cross-linker.
A main limitation of cross-linking studies is the identification of proteolytic cross-linked peptides obtained from complex cell extracts. This is facilitated both by separation of cross-linked peptides from the bulk of unmodified species and by determination of the masses of the two linked peptides. To this end, BAMG provides the cross-linked peptides with additional chemical properties that greatly facilitate cross-link identification by virtue of the presence of a 3-azidomethylene group in the spacer domain. The azido group can be reduced to an amine group, enabling isolation of the low abundant cross-linked peptides by 2D strong cation exchange chromatography.9 In addition, chemical reduction renders the two cross-link amide bonds of BAMG-cross-linked peptides scissile in the gas phase by collision-induced dissociation in a way that the masses of the two composite peptides can be determined from an MS/MS spectrum, thereby facilitating peptide identification by searching an entire genomic database.10 Identification of cross-linked peptides from a single MS/MS spectrum provides BAMG with a large advantage over the two other cleavable reagents used up to now that require multistage tandem mass spectrometry to map cross-links formed in vivo.3,11 To obtain sufficient cross-linked material by labeling directly in culture, adequate amounts of cross-linker are necessary, and in this report we also include a scalable new synthesis route for BAMG.
Using our novel in vivo cross-linking procedure, we were able to detect several transient protein–protein interactions at the peptide level in B. subtilis cells. Many of the interprotein cross-links could be corroborated by structural data from previous studies, but other cross-links represent new interactions. This approach can be readily modified to allow the identification of less abundant protein complexes or to investigate in depth the dynamic assembly of specific protein complexes.
Experimental Procedures
Synthesis of BAMG
Tetra-Ester 1
Dimethyl malonate (3.42 mL, 30 mmol) was added dropwise to a stirred suspension of sodium hydride (60% dispersion in oil, 2.46 g, 66.0 mmol) in THF (120 mL) at RT (Scheme 1). After stirring for 45 min tert-butyl bromoacetate (9.45 mL, 64 mmol) was added dropwise. The reaction was stirred for 16 h and cooled in ice, and the excess sodium hydride was carefully neutralized with acetic acid (ca. 6 mmol). Extractive workup with sat. aqueous NH4Cl and ether, drying over MgSO4, and evaporation gave tetra-ester 1 as an oil (quantitative), which was immediately used for the next step. 1H NMR (400 MHz, CDCl3): δ 3.77 (s, 6H); 3.06 (s, 4H); 1.45 (s, 18H).
Scheme 1. Formation of BAMG.

Carboxylic Acid 2
A solution of tetra-ester 1 (30 mmol) in THF (150 mL) and methanol (40 mL) was diluted with a solution of lithium hydroxide (2.94 g, 70 mmol) in water (150 mL) and refluxed for 2 h. After removal of the organic solvents in vacuo the aqueous layer was extracted with a 1:1 mixture of diethyl ether and PE 40/60. Acidification of the water layer (pH ca. 1), extraction with diethyl ether, drying with MgSO4, and evaporation gave a mixture of mono- and dicarboxylic acids. This mixture was refluxed in toluene (150 mL) for 2 h. Evaporation of the toluene gave carboxylic acid 2 (5.5 g, 19.1 mmol, 64% from dimethyl malonate) as a slowly solidifying oil. 1H NMR (400 MHz, CDCl3): δ 3.23 (m, 1H); 2.68 (dd, 1H, J = 7.2, J = 16.6 Hz); 2.54 (dd, 1H, J = 6.2, J = 16.6 Hz); 1.46 (s, 18H). 13C NMR (100 MHz, CDCl3): δ 179.7, 170.4, 81.1, 37.5, 36.2, 27.8. IR (film, cm–1): 3200, 1728, 1711 cm–1.
Alcohol 3
Borane dimethylsulfide (1.45 mL, 15 mmol) was added dropwise to a solution of carboxylic acid 2 (1.3 g, 4.5 mmol) in anhydrous THF (30 mL) at 0 °C. The reaction was stirred at RT for 16 h and carefully quenched with saturated aqueous NH4Cl and diethyl ether. Extractive workup and flash chromatography with a mixture of PE 40/60 and ethyl acetate (3:1 an 1:1) gave alcohol 3 (0.78 g, 2.85 mmol, 63%) as an oil. 1H NMR (400 MHz, CDCl3): δ 3.65 (d, 2H, J = 5.2 Hz); 2.45 (m, 1H); 2.3–2.4 (m, 4H); 1.47 (s, 18JH). 13C NMR (100 MHz, CDCl3): δ 172.1, 80.6, 80.55, 65.0, 64.9, 37.2, 37.1, 34.8, 27.9. IR: 3500, 1726 cm–1. IR (film, cm–1): 3500, 1726.
Mesylate 4
Methanesulfonyl chloride (0.255 mL, 3.3 mmol) was added dropwise to a solution of alcohol 3 (0.78 g, 2.85 mmol) and triethylamine (0.526 mL, 4.0 mL) in anhydrous dichloromethane (20 mL) at 0 °C. After stirring for 1 h at 0 °C the reaction was diluted with diethyl ether (ca. 50 mL) and quenched with water. Extractive workup gave mesylate 4 (1.0 g, 2.84 mmol, quantitative). 1H NMR (400 MHz, CDCl3): δ 4.31 (d, 2H, J = 5.1 Hz); 2.67 (m, 1H); 2.35–2.47 (m, 4H); 1.47 (m, 18H). 13C NMR (100 MHz, CDCl3): δ 170.6, 80.9, 80.85, 71.1, 36.95, 35.9, 31.9, 27.9. IR (film, cm–1): 1725.
Azide 5
A mixture of mesylate 4 (1.0 g, 2.84 mmol) and sodium azide (0.554 g, 8.5 mmol) in anhydrous DMF (10 mL) was stirred at 80 °C for 3 h. Extractive workup with water and diethyl ether, followed by chromatography (PE 40/60/ethyl acetate 6:1). gave pure azide 5 (0.84 g, 2.8 mmol, 98% from 3). 1H NMR (400 MHz, CDCl3): δ 3.44 (d, 2H, J = 5.7 Hz); 2.50 (m, 1H); 2.3–0.45 (m, 4H); 1.48 (s, 18H). 13C NMR (100 MHz, CDCl3): δ 170.8, 80.6, 54.1, 32.4, 27.9. IR (film, cm–1): 2102, 1728.
3-(Azidomethyl)-glutaric Acid 6
Azide 5 (0.638 g, 2.13 mmol) was stirred in a mixture of dichloromethane (16 mL) and trifluoroacetic acid (4 mL) for 6 h at RT. Toluene (30 mL) was added and the solvents were removed in vacuo. Drying of the resulting glass (0.02 mbar, 50 °C) gave pure diacid 6 in quantitative yield. 1H NMR (400 MHz, CDCl3 + 10% CD3OD): δ 3.44 (d, 2H, J = 5.7 Hz). 13C NMR (100 MHz, CDCl3 + 10% CD3OD): δ 176.2, 54.0, 35.7, 31.8. IR (film, cm–1): 3100 (broad), 2103, 1708.
BAMG: Bis(succinimidyl) 3-Azidomethyl-glutarate
This step was carried out according to a described procedure.12 Trifluoroacetic anhydride (1.4 mL) was added to a solution of diacid 6 (0.415 g, 2.1 mmol) and N-hydroxysuccinimide (1.15 g, 10.0 mmol) in a mixture of dichloromethane (8 mL) and anhydrous pyridine (4 mL) at 0 °C. The cooling bath was removed, and stirring was continued for 1.5 h. The reaction mixture was diluted with dichloromethane and extracted with three 50 mL portions of 1 M HCl and finally with NaHCO3 (2 × 50 mL). Drying over MgSO4, evaporation of the solvent and drying (0.02 mbar, 40 °C) gave BAMG (0.76 g, 2.0 mmol, 95%) as a slightly yellow syrup. BAMG was stored at −80 °C. Before storage, BAMG was dissolved in acetonitrile, divided in aliquots, and dried by vacuum centrifugation. 1H NMR (400 MHz, CDCl3): δ 3.65 (d, 2H, J = 5.5 Hz); 2.83–2.90 (m, 12H), 2.75 (m, 1H). 13C NMR (100 MHz,CDCl3): δ 169.0, 166.6, 52.5, 32.3, 32.2, 25.4. IR (film, cm–1): 2108, 1814, 1783, 1735.
Growth of Bacteria
B. subtilis strain 168 (trp–) was grown in a MOPS minimal medium13 modified as described for B. subtilis(14) and supplemented with 0.2% glucose, 1.2 mM glutamine, and 0.2 mM tryptophan. To obtain an exponentially growing culture for cross-linking, streaks from a glycerol stock of cells grown on liquid LB medium were first put on an LB agar plate. Following overnight growth at 37 °C a single colony was suspended in 10 mL of minimal medium in 100 mL culture flasks. From the suspension, dilutions were made in 10 mL of minimal medium for overnight growth in 100 mL flasks placed at 37 °C in a water bath shaking at 240 rpm. An overnight culture in midexponential growth as determined by an OD600nm = 0.3 to 0.5 was used for dilution to OD600nm = 0.01 in prewarmed minimal medium in Erlenmeyer flasks to obtain exponentially growing cultures for cross-linking. Escherichia coli strain MC4100 was cultured in MOPS medium supplemented with 0.16% (w/v) glucosamine and 0.1 mM NH4Cl. To obtain an exponentially growing culture for cross-linking, an overnight culture in this growth medium was 40 times diluted in fresh medium to an OD600nm = 0.08.
Cross-Linking in Vivo
In exponentially growing B. subtilis cultures at OD600nm = 0.45 to 0.50, cross-linking was started by the addition of 2.0 mM BAMG from a freshly prepared stock solution of 1 M in DMSO. A magnetic stirrer was used for rapid mixing with the culture. Cross-linking was for 5 min in the shaking water bath at 37 °C. The cross-linking reaction was quenched by the addition of 1 M Tris-Cl (pH 8.0) to a final concentration of 50 mM. Cross-linked cells were harvested by centrifugation for 5 min at 4000g, and cell pellets were stored frozen at −20 °C.
Exponentially growing Escherichia coli cells were cross-linked at OD600nm = 0.7, corresponding to 0.21 μg dry weight per ml,15 with 2 mM BAMG for 10, 30, and 50 min. The cross-linking reaction was quenched by the addition of 50 mM Tris-Cl (pH 8.0). We assess that 2.76 mM glucose, formed from an equivalent amount of N-acetylglucosamine,16 has been consumed for energy and biomass production at the time of cross-linking.13 Deamination of glucosamine, after deacetylation of N-acetylglucosamine, results in the formation of an equivalent amount of NH4+, of which 1.88 mM NH4+ has been consumed for biomass production.13 This implies that the culture contains 0.88 mM free NH4+ that can react with BAMG, leaving 1.12 mM BAMG for protein cross-linking. Cross-linked cells were harvested by centrifugation for 5 min at 4000g, and cell pellets were stored frozen at −20 °C.
Protein Extraction
Frozen B. subtilis cell pellets from 2 to 40 mL culture medium were resuspended in 1 mL of a solution containing 10 mM Tris-HCl and 0.1 mM EDTA (pH 7.5). Cell suspensions of 1 mL in 2 mL polypropylene Eppendorf vials placed in ice water were lysed by sonication with a micro tip mounted in an MSE ultrasonic integrator operated at 21 kHz and amplitude setting 3 in six periods of 15 s with 15 s intervals in between. Lysates were centrifuged for 15 min at 16 000g. Supernatants were used for further analysis. Cell extracts from Escherichia coli were prepared by suspending cell pellets from 1 mL cultures in SDS-PAGE sample buffer17 without β-mercaptoethanol. Suspensions were incubated for 1 h at 60 °C and then centrifuged for 2 min at 13 000g. Proteins were concentrated with 0.5 mL of Amicon Ultra 10 kDa cutoff centrifugal filters (Millipore) before SDS-PAGE analysis.
Gel Filtration
A cross-linked protein fraction with a size distribution of approximately 400 kDa to 1 to 2 MDa was obtained by gel filtration on a Superose 6 10/300 GL column (GE Healthcare) operated on an Akta FPLC system (GE Healthcare) in a buffer containing 20 mM HEPES pH 7.9, 300 mM KCl, 0.2 mM EDTA, 0.1 mM DTT, and 20% glycerol (gel filtration buffer) at a flow rate of 0.5 mL min–1. Fractions of 1 mL were collected and snap-frozen in liquid nitrogen for storage at −20 °C.
Protein Determination and Polyacrylamide Gel Electrophoresis in the Presence of Sodium Dodecyl Sulfate (SDS-PAGE)
Protein was measured with the bicinchoninic acid method18 using a protein assay kit (Pierce). SDS-PAGE17 was carried out using 10 or 12% precast Novex gels (Themo Fisher Scientific).
Protein Digestion
Pooled gel filtration fractions of extracted cross-linked proteins in the 400 kDa to 1 to 2 MDa range were concentrated to ∼10 mg protein/mL with 0.5 mL of Amicon Ultra 10 kDa cut off centrifugal filters (Millipore). Prior to digestion, cysteines were alkylated by the addition of a solution of 0.8 M iodoacetamide (Sigma–Aldrich), followed by the addition of solutions of 1 M Tris-HCl pH 8.0 and 9.6 M urea (Bioreagent grade, Sigma–Aldrich) to obtain final concentrations of 40 mM iodoacetamide, 0.1 M Tris HCl and 6 M urea, respectively. Incubation was for 30 min at room temperature in the dark. The solution was diluted six times by the addition of 0.1 M Tris–HCl pH 8.0 and digested with trypsin (Trypsin Gold, Promega, Madison, WI) overnight at 30 °C at a 1:50 (w/w) ratio of enzyme and substrate. Peptides were desalted on C18 reversed-phase TT3 top tips (Glygen, Columbia, MD), eluted with 0.1% TFA in 50% acetonitrile, and dried in a vacuum centrifuge.
Diagonal SCX Chromatography
A protocol previously described9 was used with several modifications. The main difference was the use of a solution of ammonium formate instead of KCl for salt gradient elution. The use of the volatile ammonium formate avoids time-consuming desalting steps and prevents loss of material. Dry desalted peptides (240 μg) were reconstituted with 10 μL of a solution containing 0.1% TFA and 25% acetonitrile, followed by the addition of 0.2 mL of 10 mM ammonium formate and 25% acetonitrile pH 3.0 (buffer A), and 0.2 mL of the mixture was loaded on a polysulfoethyl aspartamide column (2.1 mm ID, 10 cm length) (Poly LC, Columbia, MD) operated on an Ultimate HPLC system (LC Packings, Amsterdam, The Netherlands). For elution, at a flow rate of 0.4 mL min–1, increasing amounts of buffer B (500 mM ammonium formate pH 3.0) were mixed with buffer A, according to the following scheme. At t = 5 min, 1% buffer B was added; at t = 10 min a linear gradient from 1 to 50% buffer B was started over 10 min, followed by a gradient from 50 to 100% buffer B over 3 min. Elution with 100% B lasted 2 min, after which the column was washed with buffer A for 19 min. A UV detector was used to measure absorbance at 280 nm of the eluent. Peptides started to elute at t = 14 min and were manually collected in 0.2 mL fractions and lyophilized. For secondary SCX runs, dried cross-linked enriched peptides (fractions 7–16) were dissolved in 20 μL of 40 mM TCEP (BioVectra) in 20% acetonitrile and incubated under argon for 2 h at 60 °C. The peptide solution was then diluted with 0.19 mL buffer A just before loading for the secondary SCX runs. Elution occurred under the same conditions as in the primary SCX run. Material was collected when the absorbance at 280 nm started to rise again (∼30 s after the end of the elution time window of the primary fraction, until it came back to base level (high salt shifted fraction)). Collected eluent was lyophilized.
LC–MS/MS for Protein Identification
Identification of proteins by LC–MS/MS analysis of peptides in SCX fractions was performed with an amaZon Speed Iontrap with a CaptiveSpray ion source (Bruker) coupled to an EASY-nLC II (Proxeon, Thermo Scientific) chromatographic system. About 200 ng peptides from the first 12 fractions obtained by SCX chromatography were injected and separated at flow rate of 300 nL/min on an EASY-Column 10 cm (SC 200 Thermo Scientific coupled with an SC001 2 cm precolumn) using a 30 min gradient of 0–50% acetonitrile and 0.1% formic acid. MS1 scans were recorded in enhanced mode (resolution about 3000) at an MS1 mass range m/z 400–1500; CID was performed in SmartFrag mode with variable energy. MS/MS scan were recorded in extreme mode in a mass window starting from m/z 100. Five precursor ions per scan cycle were selected at a mass window of m/z 4 using data-dependent acquisition with active exclusion for 30 s. LC–MS/MS data were processed with the Bruker Daltonics DataAnalysis software version 4.2. The processed data from the 12 SCX fractions combined into one mgf file were searched with the MASCOT server program 2.3.02 against B. subtilis strain 168 protein database from the UniProt consortium with both forward and reversed (decoy) sequences (March 2015 release; 8436 entries in total). Trypsin was used as the enzyme, with two missed cleavage allowed. Carbamidomethylation of cysteine was used as a fixed modification. Both precursor and MS/MS fragment mass tolerance was set at 0.3 Da. A threshold peptide ions score of 20 and a protein score of 20 were applied as criteria for peptide assignment and protein identification at a false discovery rate (FDR) of 4.4%.
LC–MS/MS for Identification of Cross-Linked Peptides
Identification of cross-linked peptides enriched by diagonal SCX chromatography by LC–MS/MS analysis was performed with an Eksigent Expert nanoLC 425 system connected to the Nano spray source of a TripleTOF 5600+ mass spectrometer. Peptides were loaded onto an Eksigent trap column (Nano LC trap set, ChromXP C18, 120 Å, 350 μm × 0.5 mm) in a solution containing 0.1% TFA and 2% acetonitrile and desalted with 3% TFA and 0.1% formic acid at 2 μL/min. After loading, peptides were separated on an in-house-packed 7 cm long, 75 μm inner diameter analytical column (Magic C18 resin, 100 Å pore size, 5 μm) at 300 nL/min. Mobile phase A consisted of 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in acetonitrile. The gradient consisted of 5% B for 5 min, then 5–10% B over 10 min, followed by 10–35% B over 60 min, and then the gradient was constant at 80% B for 10 min. After each run the column was equilibrated for 20 min under starting conditions. The TripleTOF 5600+ mass spectrometer was operated with nebulizer gas of 6 PSI, curtain gas of 30 PSI, an ion spray voltage of 2.4 kV, and an interface temperature of 150 °C. The instrument was operated in high-sensitivity mode. For information-dependent acquisition, survey scans were acquired in 50 ms in the m/z range 400–1250 Da. In each cycle, 20 product ion scans were collected for 50 ms in the m/z range 100–1800 Da, if exceeding 100 counts per seconds and if the charge state was 3+ to 5+. Dynamic exclusion was used for half of the peak width (15 s), and rolling collision energy was used.
Before acquisition of two samples the mass spectrometer was calibrated using the built-in autocalibration function of Analyst 1.7. For MS calibration, 25 fmol of β-galactosidase digest (Sciex) was injected. For TOF MS calibration, ions with the following m/z values were selected: 433.88, 450.70, 528.93, 550.28, 607.86, 671.34, 714.85, and 729.40 Da. The ion at m/z 729.4 Da was selected for fragmentation, and product ions were used for TOF MS/MS calibration.
For 27 out of 29 LC–MS/MS runs, average mass deviations from calculated values of identified components varied from −4.0 ± 2.4 to 15.3 ± 4.1 ppm. For data processing of MS/MS (MS1MS2) data by Reang (described below) and database searching of MS/MS (MS1MS2) data by Mascot, 25 ppm mass tolerance was allowed in these cases for both M1 and MS2. In the two remaining runs average mass deviations of identified components were 31.9 ± 12.0 and 62.5 ± 7.8 ppm, respectively. In these cases, a mass tolerances of 50 and 75 ppm, respectively, was allowed for both MS1 and MS2
Data Processing
Raw LC–MS1MS2 data were processed with Mascot Distiller version 2.2.6.0 and MS2 data were deconvoluted to MH+ values at the QStar default settings using the option to calculate masses for 3+ to 6+ charged precursor ions in case the charge state could not be assessed unambiguously.
Identification of Candidate Cross-Linked Peptides
For cross-link identification using the entire B. subtilis sequence database, a software tool named Reang10 was used for further MS1MS2 data processing. The rationale of the processing by Reang described below is based on the notion that an MS1MS2 spectrum of BAMG-cross-linked peptides provides both the information for the masses of the candidate composing peptides as well as the fragment ions for identification of the composing peptides. In brief, Reang identifies precursor ions with mass >1500 Da, potentially corresponding to a BAMG-cross-linked peptide pair A and B with the azide reduced to an amine, showing evidence of cleavage of the cross-linked amide bonds in the presumed cross-link. Such cleavage events result in product ions of the unmodified peptides A and B and in modified peptides Am and Bm fulfilling the following mass relationships
| 1 |
| 2 |
where MAm and MBm, respectively, are the masses of peptides A and B modified with the remnant m of the cross-linker in the form of a γ-lactam with elemental composition C6H7NO2, corresponding to a mass of 125.0477 Da, MA and MB are the masses of peptide A and peptide B, respectively, and MP is the mass of the precursor P.
Reang identifies among the 30 product ions of highest signal intensity within a mass error of 25, 50, or 75 ppm depending on the LC–MS/MS run (see LC–MS/MS for Identification of Cross-Linked Peptides) (i) pairs of mass values of fragment ions >500 Da differing 125.0477 Da, that is, a candidate A and Am pair or B and Bm pair, (ii) pairs of mass values for A and B fulfilling the equation
| 3 |
and (iii) pairs of mass values for Am and Bm fulfilling the equation
| 4 |
The mass values of the other pairs in the cases (i), (ii), and (iii) are calculated from eqs 1 and 2.
MS1 values of entries in the MS1MS2 data files with MS2 data fulfilling at least one of the eqs 1, 3, or 4 are replaced by MS1 values corresponding to MA, MAm, MB, and MBm. Furthermore, fragment ions corresponding to MA, MAm, MB, and MBm are removed from the new MS1MS2 entries as well as fragments ions larger than the new MS1 values.
The new MS1MS2 files in pkl format are input for Mascot to nominate candidate peptides for A, Am, B, and Bm by interrogating the B. subtilis strain 168 database containing both forward and reversed sequences. Reang combines the nominated peptides with a Mascot score ≥1 into candidate cross-linked peptides and assigns these candidates with a mass tolerance of 25, 50, or 75 ppm (depending on the LC–MS/MS run; see LC–MS/MS for Identification of Cross-Linked Peptides) to precursor ions in the original MS1MS2 data file. Candidates are validated based on the original MS1MS2 data files. The principle of our approach is that an MS1MS2 spectrum of cross-linked peptides provides both the information for the masses of the candidate composing peptides as well as the fragment ions for identification of the composition of the peptides.
Cross-Link Mapping and Validation
Validation and FDR determination is facilitated by a software tool called Yeun Yan.10 Only one candidate cross-linked peptide or cross-linked decoy peptide is assigned for each precursor ion, at least if the candidate fulfills certain criteria with respect to a minimum number of y ions that should be assigned with a mass tolerance of 25, 50, or 75 ppm (depending on the LC–MS/MS run; see LC–MS/MS for Identification of Cross-Linked Peptides) to each of the composing peptides in a cross-linked peptide pair. Only assigned y ions among the 100 fragments of highest signal intensity are taken into account. The number of required assigned y ions differs for intraprotein and interprotein cross-linked peptides, with the latter type of cross-links requiring more stringent criteria for assignment than the former type. This difference is based on the notion10,19−21 that the probability of identifying cross-links as the result of a random event from a sequence database of many proteins is higher for cross-linked peptides from different protein sequences (interprotein cross-links) than for cross-linked peptides comprising different peptide sequences from the same protein sequence (intraprotein cross-links). Intraprotein cross-links comprise peptides from the same protein sequence, whereas interprotein cross-links comprise peptides from different protein sequences, unless the peptides have identical sequences and therefore must have originated from two identical protein molecules in a complex, assuming that a given protein sequence does not yield two or more identical tryptic peptides. In the case of an intraprotein cross-link, at least one unambiguous y ion should be assigned for each composing peptide and both the number of assigned y ions for each composing peptide, and the score, called the Yeun Yan score, defined below, should be the same as or more than the number of assigned y ions and the score for other possible candidates with forward sequences or one or more decoy sequences for the same precursor. No intraprotein cross-link decoy sequences consisting of reversed sequences from the same protein or hybrid forward and reversed sequences from the same protein were observed. For an interprotein cross-linked peptide pair between different proteins or decoy cross-links, the number of assigned y ions should be at least 3 for each peptide built up from up to 10 amino acid residues and at least 4 for peptides consisting of 11 amino acids or more. The number of assigned y ions for each peptide should be the same or more than the number of assigned y ions for each peptide of other possible candidates for the same precursor ion. Both the total number of y ions and the Yeun Yan score for a candidate cross-linked peptide should exceed the total number of y ions and the score for other possible candidates for the same precursor. These criteria are also used for the assignment of interprotein cross-links comprising two identical sequences. For both intra- and interprotein cross-links, a Yeun Yan score of more than 40 is required. We do not take into account the number of b ions as a requirement for assignment because b ions in our data set occur more than four times less than y ions, and taking them into account would require application of different statistical weights for assignment of b and y ions, which would complicate the calculations. Some spectra with a precursor mass difference of +1 Da compared with an identified cross-linked peptide were manually inspected to verify whether the precursor represents a cross-linked peptide in which the azide group was converted by TCEP to a hydroxyl group instead of an amine group.7 This appeared to be the case on a single occasion.
For proposed candidate cross-linked peptides, Yeun Yan calculates the masses of possible b and y fragments, b and y fragments resulting from water loss (b0, y0) and ammonia loss (b*, y*), fragment ions resulting from cleavage of the amide bonds of the cross-link, and b, b0, b*, y, y0, and y* fragments resulting from secondary fragmentations of cleavage products. A prerequisite for nomination by Yeun Yan as a candidate and calculation of the corresponding score is the presence in the MS2 spectrum of at least ten fragment ions and assignment of one unambiguous y ion per peptide. A y ion is considered ambiguous if it can also be assigned to one or more other fragments. A y ion resulting from primary and secondary cleavage at the same position is counted only once for the requirement with respect to the minimal number of unambiguous y ions for validation and assignment.
The YY score is calculated according to the equation
| 5 |
in which fassigned is the total number of matching fragment ions, including primary b and y fragments, b and y fragments resulting from water loss (b0, y0) and ammonia loss (b*, y*), fragment ions resulting from cleavage of the amide bonds of the cross-link, and b, b0, b*, y, y0, and y* fragments resulting from secondary fragmentation of products resulting from cross-link amide bond cleavages, and ftotal is the total number of fragments ions in the spectrum with a maximum of 40, starting from the fragment ion of highest intensity.
Cross-Linking of Isolated RNAP
RNAP was purified from a pellet from 2 L of culture of B. subtilis BS200 (trpC2 spo0A3 rpoC-6his spc) as follows. Following lysis in 20 mM KH2PO4 pH8.0, 500 mM NaCl, 0.1 mM DTT and clarification, RNAP was initially purified by Ni2+ affinity chromatography. Pooled RNAP containing fractions were dialyzed in 20 mM Tris-HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 0.1 mM DTT and loaded onto a MonoQ column(GE Healthcare) in dialysis buffer without EDTA. RNAP was eluted using a gradient over 10 column volumes in dialysis buffer supplemented with 2 M NaCl. RNAP-containing fractions were pooled and dialyzed in 20 mM Tris-HCl pH 7.8, 150 mM NaCl, 10 mM MgCl2, 30% glycerol, 0.1 mM DTT prior to flash freezing and storage at −80 °C. Before cross-linking RNAP was dialyzed in 20 mM HEPES, 150 mM NaCl, 10% glycerol, pH 7.4 (cross-linking buffer). RNAP was cross-linked at a protein concentration of 0.5 mg/mL for 30 min at room temperature. The cross-link reaction was started by the addition of a solution containing 80 mM BAMG in acetonitrile to obtain a final concentration of 0.4 mM BAMG and 0.5% acetonitrile. The reaction was quenched by adding 1 M Tris–HCl pH 8.0 to a final concentration of 50 mM. Digestion of the cross-linked protein and isolation and identification of cross-linked peptides were carried out as previously described.22
Determination of Spatial Distances between Cross-Linked Residues
PDB files of structural models were downloaded from the protein data bank (http://www.rcsb.org/pdb/home/home.do). Only PDB files of B. subtilis proteins or proteins with at least 40% sequence identity were used. Sequences were aligned using the BLAST algorithm to identify corresponding residues (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). For RNAP, a homology model of B. subtilis elongation complex (EC)23 was used. It gives similar results as with PDB file 2O5I.24 Structures were inspected with DeepView - Swiss-PdbViewer (http://spdbv.vital-it.ch/refs.html) for distance measurements.
In Silico Docking
A homology model of B. subtilis EC23 was used along with other published structures identified by their protein data bank IDs (PDB ID) detailed below. The N-terminal domain of δ (PDB ID 2M4K) was used along with the EC model and in vitro and in vivo cross-linking data to produce a model using the HADDOCK2.2 web server Easy interface:25 40 models in 10 clusters (4 models per cluster) were obtained and analyzed for compliance to the maximum Cα–Cα cross-link distance permitted by BAMG (29.7 Å) in PyMol v1.8.2.0. The total cumulative distance of β′K208-δ48, β′K1104-δ48, and β′K1152-δ48 Cα measurements was used to identify models that were most compliant (lowest cumulative distance) with cross-link criteria. To colocalize δ and σA region 1.1, E. coli RNAP holoenzyme in which σ70 region 1.1 was present (PDB ID 4LK1)26 was superimposed over the B. subtilis EC model, and all but σ70 region 1.1 were deleted.
Results
Defined Growth Medium for in Vivo Cross-Linking
The addition of the cross-linker directly to a growth medium enables trapping of transient protein interactions in living cells that may otherwise dissociate upon washing and medium exchange. This requires a low concentration of primary amines to minimize quenching of the cross-linker in the medium. We found that the growth rate of B. subtilis in minimal medium containing only 1.2 mM glutamine as the nitrogen source was almost identical to the growth rate using the standard 5 mM glutamine, with doubling times of 45 and 43 min, respectively (Figure 1a). The addition of 2 mM of the cross-linker BAMG resulted in an immediate end to the increase in OD600 nm, indicating that biomass production ceased instantaneously (Figure S2). As shown by SDS-PAGE analysis (Figure 1b), most extracted proteins become cross-linked upon treatment of the cells with 2 mM BAMG for 5 min. The same results were obtained with DSG (Figure 1b). This indicates that the azidomethylene group in BAMG does not affect membrane permeability, with DSG and BAMG having about the same protein cross-linking efficiency.7 SDS-PAGE analysis (Figure S3) shows that the cross-linked proteins could be digested efficiently, establishing a set of experimental conditions suitable for the identification of in vivo cross-linked peptides.
Figure 1.
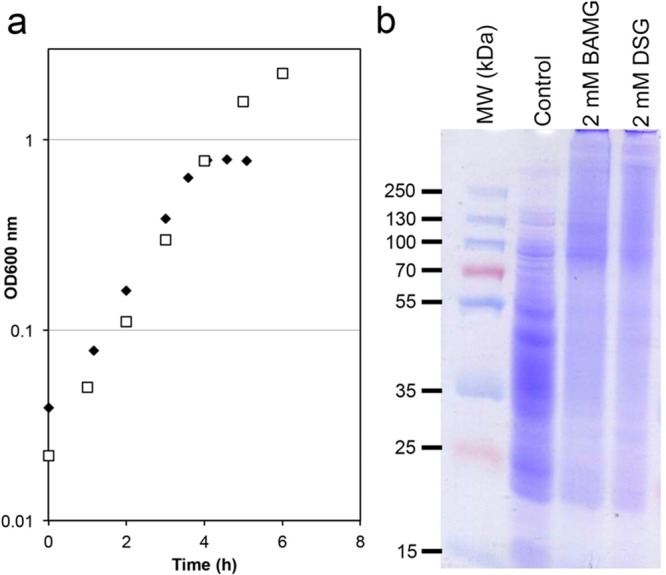
In vivo cross-linking in of B. subtilis in culture. (a) Growth curves of B. subtilis in minimal medium with 1.2 mM glutamine (filled diamonds) and 5 mM glutamine (open squares). (b) SDS-PAGE analysis of in vivo cross-linking with BAMG and DSG of exponentially growing B. subtilis directly in the growth medium. Control, soluble proteins from untreated exponentially growing B. subtilis. 2 mM BAMG or DSG, soluble proteins from exponentially growing B. subtilis treated with 2 mM BAMG or DSG, respectively. Molecular weights (kDa) are shown on the left-hand side adjacent to prestained molecular weight markers (MW markers).
In Vivo Cross-Linking of Gram-Negative Species
It is well known that the outer membrane of Gram-negative bacteria forms a barrier for the diffusion of lipophilic compounds due to the relatively low fluidity of the bilayer imposed by the lipopolysaccharide outer leaflet.27 To test whether this property prevents the use of BAMG as an effective cross-linker for soluble proteins in Gram-negative species, we used Escherichia coli as an example. Cells were grown in a MOPS medium with 0.16% N-acetylglucosamine as the only source for energy, carbon, and nitrogen. The culture medium was also supplied with 0.1 mM NH4Cl to provide the cells with a small amount of a nitrogen source to enable a rapid start of growth. When the NH4+ in the medium has been consumed, cells have to rely on the ammonia that is formed intracellularly during catabolism of N-acetylglucosamine. On the basis of published data13 it can be calculated that the amount of NH4+ thus formed will be enough for fast growth and will not accumulate to concentrations that will decrease the concentration of BAMG by >1 mM by reaction with the cross-linker (Experimental Procedures). The doubling time under these conditions is 56 min during midexponential growth. This compares favorably with a similar, but slightly faster, doubling time of 50–55 min reported by others using a medium containing N-acetylglucosamine supplemented with 9.5 mM NH4Cl as a nitrogen source.16 Upon the addition of 2 mM BAMG to the exponentially growing cells in this medium, inhibition of growth occurred (Figure S4). However, in striking contrast with the Bacillus subtilis results (Figure 1b), SDS-PAGE analysis provided no evidence of large-scale cross-linking of extracted proteins from BAMG-treated E. coli cells because the Coomassie-blue-stained patterns in the lanes from control cells and cross-linked cells were indistinguishable (Figure S5). These results are in agreement with the known slow diffusion rate of hydrophobic compounds through the outer membrane and put limitations on the use of Gram-negative organisms for rapid in vivo cross-linking by N-hydroxysuccinimidyl esters.
Mass Spectrometric Analysis Reveals a Large Number of Cross-Linked Peptides at a Low False Discovery Rate
The workflow for sample preparation of cross-linked peptides from in vivo cross-linked B. subtilis cells for LC–MS/MS analysis is shown in Figure 2b. After cross-linking and protein extraction, cross-linked proteins were fractionated by size exclusion chromatography to obtain a sample expected to be enriched in cross-links formed during transient interaction. To this end, samples with a size distribution of roughly 400 to 2000 kDa were used for further analysis. This fraction was enriched in RNAP and also contained ribosomes, that is, protein complexes involved in processes guided by many transient protein–protein interactions. A list of proteins present in this fraction, identified by peptide fragment fingerprinting and sorted according to their abundance index,28 is presented in Table S1. Besides subunits from ribosomes and RNAP, we also detected many proteins of high abundance with a known molecular weight far below 400 kDa that included all glycolytic and TCA cycle enzymes and many enzymes involved in amino acid synthesis, indicating that these proteins have cross-linked in vivo with other proteins.
Figure 2.

Workflow for peptide level identification of protein cross-links introduced by BAMG in exponentially growing B. subtilis. (a) Overview. (b) Left part: Reaction products formed (1) in the cross-link reaction with BAMG, (2) by TCEP-induced reduction, and (3) by cross-link amide bond cleavages and peptide bond cleavages by collision with gas molecules during LC–MS/MS, leading to the formation of unmodified peptide ions and peptide ions modified by the cross-linker remnant in the form of a γ-lactam, along with b and y ions. A, peptide A; B, peptide B. Depicted peptide charge states after (1) and (2) are calculated for pH 3, assuming full protonation of basic amino acids and carboxylic acids. Depicted charge states in the gas phase after (3) are arbitrary, assuming a net charge state of +4 of the intact precursor ion. Right part: Principles of isolation of cross-linked peptides by diagonal strong cation exchange (DSCX) chromatography. After digestion, the peptide mixture from a protein extract is fractionated by SCX chromatography using a mobile phase of pH 3 and a salt gradient of ammonium formate to elute bound peptides (first run). Cyan, cross-linked peptides; gray, unmodified peptides. Subsequently, fractions containing cross-linked peptides are treated with TCEP to reduce the azido group to an amine group, which becomes protonated at pH 3, adding one positive charge to cross-linked peptides. TCEP-treated fractions are then separately subjected to a second run of diagonal chromatography. The change in chromatographic behavior caused by the charge increase in cross-linked peptides leads to their separation from the bulk of unmodified peptides present in the same primary SCX fraction.
After trypsin digestion of the extracted proteins in the high-molecular-weight fraction, cross-linked peptides were enriched by diagonal strong cation exchange (SCX) chromatography.9 The principle of the enrichment is schematically depicted in Figure 2b. Peptides in the cross-link-enriched SCX fractions were subjected to LC–MS/MS, data processing, and database searching according to the workflow schematically depicted in Figure 3.10 For efficient identification of cross-linked peptides from the entire B. subtilis sequence database with MS/MS data, it is necessary to know the masses of the two peptides in a cross-link. This is possible due to abundant signals in MS/MS spectra arising from cleavage of the two cross-linked amide bonds,10 shown as an example in the mass spectrum in Figure 4. Following this protocol, we identified 82 unique interprotein cross-links (Table S2) and 369 unique intraprotein cross-links (Table S3) in 299 and 1920 precursor ions, respectively, that fulfilled all criteria mentioned in Figure 3. Importantly, no decoy peptides fulfilled these criteria, indicating a low FDR.
Figure 3.

Overview of identification and validation of cross-linked peptides by mass spectrometry and database searching. A, B, Am, and Bm: free peptides A and B and peptides A and B modified by the cross-linker in the form of a γ-lactam. MA, MB, MAm, and MBm: Masses of peptides A and B and their γ-lactam modifications. MP: precursor mass. fassigned: total number of assigned fragment ions. ftotal: total number of fragment ions of highest intensity taken into account with a minimum of 10 and a maximum of 40 fragments. totaldecoy: total number of assigned decoy peptides. totaltarget: total number of assigned target peptides.
Figure 4.

Mass spectrum of product ions generated by collision-induced dissociation of a precursor ion of a BAMG-cross-linked peptide pair. The spectrum shows characteristic features of the fragmentation pattern of a cross-linked peptide in which the azido group in the spacer of the cross-linker has been reduced to an amine group. These features are (i) signals of high intensity resulting from cleavage of the cross-linked amide bonds leading to unmodified peptide A (CA) and peptide A modified by the remnant of the cross-linker in the form of a γ-lactam (CAm), adding 125.0477 Da to the mass of peptide, and (ii) secondary fragments resulting from cleavage of a cross-linked amide bond along with peptide bond cleavages of an unmodified peptide (blue, subscript An, Bn) or a peptide lactam (red, subscript Am, Bm). These secondary cleavages occur along with primary cleavages of the peptide bonds (black, subscript A, B). The presence of both primary fragments (resulting from cleavages of the cross-link amide bonds and peptide bonds) and secondary fragments tremendously facilitates the identification of cross-linked peptides according to the work flow schematically depicted in Figure 3b. *, fragment with NH3 loss.
About 39% of the 82 unique interprotein cross-linked peptides are from enzymes involved in intermediary metabolism, protein and RNA folding, and protein and RNA degradation. Most of these cross-links comprise peptides with identical sequences, showing that the parent proteins occurred in symmetric homodimers, possibly organized in higher order assemblies. About 40% of all interprotein cross-links are from translation complexes, that is, ribosomes and auxiliary proteins involved in translation, and ∼18% are from transcription complexes, that is, RNAP and initiation and elongation factors (Table S1).
It is interesting to note that the ratio of identified inter- and intraprotein cross-links is about 1:1 and about 1:2, respectively, in the group of proteins associated with transcription and translation, while this ratio is only about 1:9 in the group of other proteins that end up in the >400 kDa gel filtration fraction (Tables S2 and S3). The paucity of identified interprotein cross-links in this group can be explained by assuming that a certain degree of random distribution of many low-molecular-weight proteins and protein complexes in the crowded cytoplasm has resulted in heterogeneous cross-linking, leading to the formation of covalent complexes >400 kDa. Identification of such heterogeneous cross-links with respect to protein identity and linked residues is likely below the detection limit, in contrast with cross-links formed between proteins engaged in a more defined spatial distribution, as required for transcription and translation.
Use of Different Assignment Criteria for Interprotein and Intraprotein Cross-Linked Peptides
To obtain a low FDR of both intraprotein and interprotein cross-links we used different assignment criteria for these two types of cross-links (Figure 3). Because the probability of identifying false-positive cross-linked peptide pairs from a complete sequence database is much higher for interprotein cross-links than for intraprotein cross-links, false discoveries are practically all confined to interprotein cross-links if the same assignment criteria are employed for both cross-link types.10,20,21 In Table S4 it is shown how variations in assignment criteria affect the number of identified cross-links and the FDR. Applying the more stringent criteria for assignment of interprotein cross-links to intraprotein cross-links only leads to a decrease of ∼20% assigned unique cross-linked peptides. Consequently, the number of assigned interprotein cross-links slightly increases upon relaxing the stringency of the criteria for assignment. However, this increase is accompanied by a relatively large increase in FDR. So, the stringent criteria that we apply here for interprotein cross-links result in efficient identification and a low FDR.
Biological Consistency of Identified Cross-Linked Peptides
To corroborate identified cross-linked peptides by comparison with published data, we determined spatial distances between Cα atoms of linked residues. In models of crystal structures, the maximal distance that can be spanned by BAMG varies between 25.7 and 29.7 Å, assuming a spacer length of BAMG of 7.7 Å, a lysine side chain length of 6.5 Å, and a coordinate error of 2.5–4.5 Å. The distances between 95.6% (n = 135) of Cα atom pairs of linked residues in cross-links with nonoverlapping sequences from one protein (denoted intraprotein cross-links) are <25.7 Å, including 14 interprotein cross-links between identical proteins that fit better than intraprotein species (Table S3 and Figure S6). The distances between Cα atoms of linked residues of only 2 cross-links out of the 135 exceed 29.7 Å. These results underscore the high biological consistency and thereby reliability of identified cross-linked peptides.
Table 1 lists the interprotein cross-linked peptides from transcription and translation complexes. The distances between the Cα atoms of interlinked lysine residues of all nine cross-links comprising peptides from proteins involved in transcription are in agreement with models based on crystal structures. Also, five small ribosomal interprotein cross-linked peptides nicely fit in the available structural model of a stalled ribosome.29 However, five small ribosomal interprotein cross-links that exceed 29.7 Å by >45 Å were notable exceptions. Because the FDR is low and the large majority of our data set is biologically consistent, it is reasonable to assume that the formation of these cross-links actually took place. Most likely these distance measurements represent the detection of ribosomal assembly intermediates or covalent links between proximal ribosomes.
Table 1. Interprotein Cross-Linked Peptides from Proteins Involved in Transcription and Translation.
| mass (Da) | sequence peptide A | Uniprot entry name | position XL residue | sequence peptide B | Uniprot entry name | position XL residue | (template) PDB file model | distance (Å) in structure model |
|---|---|---|---|---|---|---|---|---|
| 1788.0 | XSLEEVK | RPOA | 294 | XSLEEVK | RPOA | 294 | n.a. | |
| 2194.2 | IXELGPR | RSPB | 112 | DTXLGPEEITR | RPOB | 803 | n.a. | |
| 2298.2 | MYLXEIGR | SIGA | 107 | QLLSEXEYR | RPOC | 153 | 4IGC | 16.1 |
| 2527.4 | LLTVXIPVR | GUDB | 52 | XDVVDEVYDQR | NUSA | 62 | n.a. | |
| 2629.4 | IGAEVXDGDLLVGK | RPOB | 837 | GYTPADANXR | RPOA | 155 | 2O5I$ | 12.3 |
| 2672.5 | XLALK | RPOA | 84 | VAVAANSLXNVTFTEEQR | RPOC | 545 | 2O5I$ | 20.0 |
| 2762.5 | AQEXVFPMTAEGK | GREA | 5 | XGFTATVIPNR | RPOB | 156 | 4WQT | n.a. |
| 2974.6 | DTXLGPEEITR | RPOB | 803 | TLXPEKDGLFCER | RPOC | 40 | 2O5I$ | 12.7 |
| 3026.6 | XGFTATVIPNR | RPOB | 156 | DXQQEIVVQGAVETR | RPOC | 987 | 2O5I$ | 21.1 |
| 3066.7 | IAAQTAXQVVTQR | NUSA | 111 | VTPXGVTELTAEER | RPOB | 849 | n.a. | |
| 3387.6 | VTPXGVTELTAEER | RPOB | 849 | IFGPTXDWECHCGK | RPOC | 56 | 2O5I$ | 14.4 |
| 3510.8 | LVPAGTGMMXYR | RPOC | 1179 | ALEEIDAGLLSFEXEDRE | RPOZ | 63 | 2O5I$ | 13.8 |
| 3780.0 | XEELGDR | RPOE | 48 | VIDAGDTDVLPGTLLDIHQFTEANXK | RPOC | 1104 | n.a. | |
| 4572.3 | GILAKPLXEGTETIER | RPOC | 830 | SFGDLSENSEYDSAXEEQAFVEGR | GREA | 55 | 4WQT | 28.0 |
| 4695.7 | IGAEVXDGDLLVGK | RPOB | 837 | GYTPADANXRDDQPIGVIPIDSIYTPVSR | RPOA | 155 | 2O5I$ | 12.3 |
| 6461.1 | DTXLGPEEITR | RPOB | 803 | EILXIAQEPVSLETPIGEEDDSHLGDFIEDQEATSPSDHAAYELLK | SIGA | 258 | 4IGC | 11.6 |
| 1694.0 | VIXVVR | RL7 | 73 | GPXGELTR | RL6 | 31 | n.a. | |
| 1838.1 | XEVVQLK | RS2 | 132 | GEVLPTXK | RS3 | 210 | 3J9W | 22.3 |
| 1947.0 | DIIDXLK | RS13 | 62 | QXFASADGR | RL31 | 48 | n.a. | |
| 2083.2 | GXILPR | RS18 | 43 | SVSXTGTLQEAR | RS21 | 25 | n.a. | |
| 2217.2 | XFVSER | RS18 | 36 | IDPSXLELEER | RS5 | 8 | 3J9W | 86.2 |
| 2383.4 | XAVIER | RS6 | 20 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | 149.7 |
| 2392.3 | AEDVAXLR | RS5 | 155 | VFLXYGQNNER | RS8 | 65 | 3J9W | 12.4 |
| 2429.3 | XNEEGGK | RS3 | 212 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | <20.9* |
| 2433.3 | XFVSER | RS18 | 36 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | 116.1 |
| 2438.3 | GEVLPTXK | RS3 | 210 | VXVLDVNENEER | RS1H | 326 | n.a. | |
| 2457.3 | ILDQSAEXIVETAKR | RS10 | 24 | NEEGGX | RS3 | 218 | n.a. | |
| 2498.5 | XALNSLTGK | RS3 | 88 | VXVLDVNENEER | RS1H | 326 | n.a. | |
| 2500.3 | GIVTXVEDK | RS1H | 25 | QAQDSVXEEAQR | RS2 | 81 | n.a. | |
| 2539.4 | GEVLPTXK | RS3 | 210 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | 18.5 |
| 2557.4 | XKNEEGGK | RS3 | 211 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | 16.9 |
| 2571.4 | XALNSLTGK | RS3 | 88 | XQAQDSVKEEAQR | RS2 | 74 | 3J9W | 87.4 |
| 2654.4 | EITGLGLXEAK | RL7 | 84 | XAAGIESGSGEPNR | RL11 | 81 | n.a. | |
| 2701.6 | VVXVVK | RL11 | 7 | MLVITPYDXTAIGDIEK | RRF | 72 | n.a. | |
| 2725.4 | GPQAANVTXEA | CSPB | 65 | XQAQDSVKEEAQR | RS2 | 74 | n.a. | |
| 2728.5 | SVSXTGTLQEAR | RS21 | 25 | IDPSXLELEER | RS5 | 8 | n.a. | |
| 2733.4 | XNESLEDALR | RS21 | 8 | XLSEYGLQLQEK | RS4 | 43 | n.a. | |
| 2739.4 | YEVGEGIEXR | EFTS | 278 | AEVYVLSXEEGGR | EFTU | 316 | 1EFU | 18.6 |
| 2749.4 | ELVDNTPXPLK | RL7 | 95 | XAAGIESGSGEPNR | RL11 | 81 | n.a. | |
| 2845.5 | AXLSGTAERPR | RL18 | 21 | GGDDTLFAXIDGTVK | RL28 | 70 | n.a. | |
| 2861.5 | XNESLEDALR | RS21 | 8 | XKLSEYGLQLQEK | RS4 | 42 | n.a. | |
| 2881.5 | IDPSXLELEER | RS5 | 8 | VXVLSVDRDNER | RS1H | 240 | n.a. | |
| 2882.6 | IAXIEVVR | RL19 | 83 | XLLDYAEAGDNIGALLR | EFTU | 266 | n.a. | |
| 3080.6 | VHINILEIXR | RS3 | 106 | ELEETPXADQEDYR | RS1H | 349 | n.a. | |
| 3153.8 | EAXELVDNTPKPLK | RL7 | 87 | LALETGTAFIEXR | MTNK | 377 | n.a. | |
| 3184.7 | XQAQDSVKEEAQR | RS2 | 74 | ILDQSAEXIVETAK | RS10 | 24 | 3J9W | 75.4 |
| 3427.8 | EAXELVDNTPKPLK | RL7 | 87 | SLLGNMVEGVSXGFER | RL6 | 82 | n.a. | |
| 3524.8 | VNITIHTAXPGMVIGK | RS1 | 71 | ELEETPXADQEDYR | RS1H | 349 | n.a. | |
| 3789.9 | EAXELVDNTPKPLK | RL7 | 87 | AXEAEAAGADFVGDTDYINK | RL1 | 85 | n.a. |
X, cross-linked K residue; *, linked residue K212 (RS3) is not in structure model; distance is assumed based on a maximal distance of 4 Å between Cα atoms of K211 and K212; $, the structure of the RNAP elongation complex was modeled as described in the Experimental Procedures; n.a., model not available or linked residue not in structure. Uniprot entry names RPOA, RPOB, RPOC, RPOE, and RPOZ correspond to subunits α, β, β′, δ, and ω of RNAP.
Many Cross-Links Reveal Transient Protein–Protein Interactions
The power of our approach was demonstrated by the detection of several transient interactions between translation factors and ribosomes and between transcription factors and core RNAP (Table 1). Ribosome-recycling factor RRF forms a cross-link with ribosomal protein RL11, in agreement with cryo-EM data showing an interaction between these two proteins in the post-termination complex.30 A cross-link between RL19 and EF-Tu is in agreement with the presence of RL19 near the EF-Tu binding site on the ribosome.31 Cross-linked peptides were found between K4 and K55 of the transcription elongation factor GreA and residues β-K156 and β′-K830, respectively, in the RNAP secondary channel. This position fits with the known function of GreA and with a crystal structure of a chimeric Gfh1–GreA in complex with RNAP.32 Likewise, the binding of NusA close to the RNA exit channel of RNAP, as revealed by a cross-link between NusA-K111 and β-K849, is in agreement with results previously obtained that indicate the N-terminal domain of NusA binds to the β-flap tip of RNAP.23,33 Two cross-linked peptides between the sigma A factor (σA) and RNAP were identified. The distances between Cα atoms of corresponding residues in the structure of the E. coli RNAP holoenzyme34 are 16.1 and 11.6 Å. Thus the spatial arrangements of the proteins involved in these transient interactions are in agreement with previously published in vitro data, underscoring the reliability of our in vivo cross-link approach.
Of great interest was the identification of novel transient interactions. A binding site of the RNA chaperone CspB on ribosomes, as revealed by a cross-link between CspB and RS2, has not been observed before to our knowledge. This interaction makes sense because cold shock proteins colocalize with ribosomes in live cells and are involved in coupling transcription and translation.35,36 The biological significance of the interaction between glutamate dehydrogenase GudB and transcription elongation factor NusA is not known, but recent work may suggest a functional link between the two proteins. The gudB gene encodes a cryptic glutamate dehydrogenase (GDH), which is highly expressed but not active. If the main GDH (RocG) is inactivated, then a frame-shift mutation activates GudB. This mutation depends on transcription of gudB and requires the transcription–repair coupling factor Mfd.37 Interestingly, NusA is also involved in transcription-coupled repair.38 Whether the interaction of GudB with NusA is relevant for the regulation of this gene decryption remains to be established.
Another noteworthy interaction is revealed by a cross-link between the β′ subunit of RNAP and a protein originally found to be associated with isolated RNAP named δ.39 Importantly, δ has a complex effect on transcription. It inhibits initiation from weak promoters mediated by σA,40,41 stimulates or inhibits transcription from certain other promotors,42,43 and increases RNAP recycling speed41,42 in synergy with the DNA helicase HelD,44 probably by dissociation of stalled RNAP-DNA or RNAP–RNA complexes. Transcriptomics experiments indicate that δ reduces nonspecific initiation of transcription, which is relatively prevalent in Gram-positive bacteria.45,46 Up to now it has remained elusive how these different effects on transcription are brought about.
The 20.4 kDa δ occurs exclusively in Gram-positive bacteria. It consists of an amino-terminal globular domain and a nucleic acid-mimicking highly acidic unstructured C-terminal half.47,48 The δ protein forms a complex with the RNAP core enzyme in a 1:1 stoichiometry.42,47,49 A truncated form of δ lacking the C-terminal half is sufficient for binding to RNAP. Intact δ, as well as the acidic unstructured C-terminal domain but not the truncated N-terminal domain, inhibits the binding of nucleic acids to RNAP. It has been proposed that the amino-terminal RNAP-binding domain may act both to orient and increase the local concentration of the flexible negatively charged carboxyl terminal domain to effectively shield nearby positively charged nucleic acid binding sites on RNAP.47 The effect of δ on promoter selectivity by σA-mediated initiation suggests an interaction of δ with the preinitiation complex. Indeed, it has been reported that δ and σA can bind simultaneously to core RNAP with negative cooperativity.50 However, other experiments indicated mutual exclusion of the binding of δ and σA to core RNAP.42,51
We identified a cross-link between K48 of the δ subunit (RpoE) and K1104 of the RNAP β′ subunit (RpoC) (Figure 4). K1104 is located in the so-called downstream clamp region. This suggests a binding site for the δ subunit on RNAP close to the downstream DNA binding cleft. To confirm this finding, we performed in vitro cross-linking with purified δ-containing RNAP. This resulted in two additional cross-links, one between K48 of δ and residue β′-K208 and one between δ-K48 and β′-K1152, both in close proximity to β′-K1104, thereby corroborating our in vivo findings. Because only one residue in the N-terminal domain of δ was involved in cross-linking, further evidence is required to assign a preferential orientation of delta with respect to the clamp region. To this end we used in an in silico docking analysis using a B. subtilis RNAP EC model and the known N-terminal domain structure of δ.23,52 The best 10 output models were analyzed to establish which complied with the maximum Cα–Cα cross-link distance achievable with BAMG (Table S5). In all but one model at least one cross-link exceeded the 29.7 Å maximum distance; however, in published structures of RNAP, crystallographic B factors are relatively high around positions β′-K1104 and β′-K1152, implying some conformational flexibility in those regions. The model that gave the lowest cumulative Cα-Cα cross-link distance, with all predicted cross-links <29.7 Å, places δ in the downstream side of the DNA binding cleft of RNAP (Figure 5a). A position of δ inside the DNA binding cleft, as shown in Figure 5b, suggests that the N-terminal domain of δ could sterically inhibit the binding of downstream DNA. In this position, δ could also sterically inhibit the binding of the 1.1 region of σA because this region is expected to interact with an overlapping binding site based on crystal structures of E. coli RNAP holoenzyme with the homologous σ70 factor.26,34 The model with the next lowest aggregate score also placed δ in this region, but the remaining eight placed it on top of the β′ subunit outside of the DNA binding cleft (Figure 5c). This position of the N-terminal domain of δ outside the downstream DNA channel but close to its entrance implies that interference with DNA binding and binding of the 1.1 region of the σ factor requires penetration of the C-terminal unstructured acid domain into the channel to interact with positive charges of the polymerase involved in DNA binding and in binding of the σ 1.1 region. Because δ is known to displace RNA more efficiently than it can DNA47 and is important in RNAP recycling following the termination of transcription,44 we expect that it must be oriented so that the acidic C-terminal domain is able to influence RNA binding through contact with the transcript close to or even within the RNA exit channel. However, the data prevent discrimination between models placing δ inside or outside the downstream DNA channel because both the aggregate distance scores (Table S5) and the HADDOCK scores of the best models show only small differences.
Figure 5.

Model of B. subtilis RNAP in complex with δ. (a) Zoomed region of δ (blue) located within the DNA binding cleft of RNAP (gray). The cross-linked amino acids are shown in red, and the distances in angstroms between the Cα carbon of δ K48 and RNAP β′ K208, K1104, and K1152 are indicated. (b) Model of RNAP (gray) in complex with δ (blue) with σ region 1.1 (purple) and DNA (green, template strand; orange, nontemplate strand) shown as semitransparent cartoons. The active site Mg2+ is shown as a cyan sphere and RNA is shown as a red cartoon. Part of the unstructered C-terminal domain, attached at the C-terminal end of the structured N-terminal domain, is depicted as a yellow squiggle, pointing in the direction of the RNA export channel. (c) Compilation of all 10 docked models (all cyan) with the C-terminal 5 amino acids of the structured N-terminal domain colored red.
Discussion
We have developed a new method for large-scale identification of cross-links introduced in vivo by N-hydroxysuccinimidyl esters directly in a bacterial cell culture. Within as little as 5 min extensive cross-linking was observed using 2 mM BAMG. This implies that cross-link analysis on a time scale of seconds could be a future development, enabling, for instance, monitoring at the peptide level transient protein–protein interactions involved in rapid cellular adaptation. Rapid in vivo cross-linking requires a Gram-positive organism because the outer membrane of Gram-negative species forms a barrier for diffusion.
With our method we identified many interprotein cross-links confirming several known stable and transient protein interactions, underscoring the reliability of the approach. In addition, we found intriguing new interactions that deserve further investigation to understand their functional significance, like the interaction between GudB and NusA. Another intriguing cross-link was found between the δ and β′ subunits of RNAP. This cross-link revealed that δ binds to the clamp region of RNAP close to the entrance of the downstream DNA binding cleft. However, the cross-link data combined with in silico docking experiments did not provide enough evidence to assign an unambiguous orientation of δ with respect to the channel. This implies that different scenarios are possible for the molecular mechanism by which δ regulates promoter selectivity and promotes RNAP recycling. However, the knowledge that δ binds to the clamp region of β′ will help us to design experiments aimed at developing a better understanding of the mechanism of action of δ.
Besides membrane permeability, the unique chemical properties of BAMG, combined with our statistical analysis, are at the heart of the large number of identified cross-linked peptides with high biological consistency and low FDR. This is also the first study in which a large number of cross-links from intracellular protein complexes generated in undisturbed growing cells have been identified by mass spectrometry and database searching using a complete species specific database. Our data set consists of about 18% interprotein and 82% intraprotein cross-links. This percentage of interprotein cross-links is relatively low in comparison with other data sets obtained either by in vivo cross-linking of bacteria2−5 or by cross-linking bacterial cellular protein extracts of high complexity.53,54 Also, the reported fractions of interprotein cross-links identified in human cells, 23–25% of the total number of cross-links,55,56 is relatively high compared with the 10% that we obtained in a previous study.10 We consider it important for the future development of the technology to understand the causes of such differences. In one of these studies56 we noticed that many sequences of the peptides from identified cross-links, classified as intermolecular species, are not unique and therefore that many cross-linked peptide pairs could be either from the same protein or from different proteins. Furthermore, in protein extracts nonspecific interactions may be formed dependent on extraction conditions. However, a key difference between these reports2−5,53−56 and our approach concerns the statistical analysis, in which we make a distinction between inter- and intramolecular cross-links. If this distinction is not made, then an overestimation of intermolecular cross-links occurs at the expense of a relatively large number of false-positives.
In this study we provide an efficient layout for large-scale cross-link identification using in-culture cross-linking. Several modifications to the method can be made to tailor specific experimental requirements. For example, in complex mixtures, low-abundance cross-links can escape detection by LC–MS/MS. The use of affinity-tagged target proteins will enable enrichment of these complexes for subsequent interpeptide cross-link identification of transient interactions. Furthermore, in this study, we have focused on the soluble fraction of cross-linked cells. Further extraction and digestion of the insoluble fraction, enriched in membrane and cell wall proteins, is likely to reveal a rich source of interesting protein cross-links. Relative quantification of cross-linked peptides can also be employed with commercially available isotope-labeled starting materials for the synthesis route of BAMG presented here. Finally, our analytical strategy may also benefit from the option of mass spectrometry to combine collision-induced dissociation with electron-transfer dissociation56,57 to increase efficiency of identification of cross-linked peptides. The high average precursor charge state of slightly more than +4 of all identified BAMG-cross-linked peptides in our data set is favorable for the latter fragmentation method.58 Overall, we believe that the in vivo cross-linking and data analysis methods developed here will pave the way to a systems level view on dynamic protein interactions. Such a view will lead to a deeper understanding of the molecular mechanisms of biological processes guided by dynamic protein–protein interactions in the cell.
Conclusions
A system has been developed for rapid in vivo protein cross-linking by an amine-specific bifunctional reagent added directly to a culture of Bacillus subtilis. We identified several stable and dynamic interactions in intracellular protein complexes with a size range of about 400 to 2000 kDa by mass spectrometric analysis of isolated cross-linked peptides and database searching using the entire species-specific sequence database. In-culture cross-linking of cytoplasmic protein in Gram-positive bacteria is much faster than in Gram-negative species. In combination with affinity purification of target proteins, our in vivo cross-link technology will be useful to obtain insight into the molecular mechanisms of processes guided by dynamic protein–protein interactions in large assemblies.
Acknowledgments
Work in P.J.L.’s laboratory was supported by a grant from the Australian Research Council (ARC, DP110100190). Work in L.W.H.’s laboratory was funded by The Netherlands Organization for Scientific Research (NWO, STW-Vici 12128).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00068.
Table S4. Effect of variations of assignment criteria on the number of identified cross-linked peptides and the false discovery rate. Table S5. Distance measurements for HADDOCK output data on the combined invitro and in vivo cross-linking data for the interaction of δ with the β′ subunit of RNAP. Figure S1. Structures of cross-linkers used in this study. Figure S2. Growth arrest of Bacillus subtilis after the addition of various concentrations of BAMG. Figure S3. Extracted proteins after in vivo cross-linking with different concentrations BAMG of exponentially growing B. subtilis can be digested efficiently. Figure S4. Growth curve of E. coli in MOPS medium supplemented with 0.16% N-acetylglucosamine and 0.1 mM NH4Cl. Figure S5. Coomassie Briliant Blue stained SDS-PAGE of extracted proteins from exponentially growing E. coli before and after the addition of 2 mM of the cross-linker BAMG directly in the growth medium. Figure S6. Distribution of 135 cross-link distances derived from 31 crystal structures comprising 43 different proteins with nonoverlapping cross-linked peptide sequences from the same protein sequence, denoted intraprotein cross-links. (PDF)
Table S1. Proteins identified in SCX fractions 1–12. (XLSX)
Table S2. Interprotein cross-linked peptides. (XLSX)
Table S3. Intraprotein cross-linked peptides (cross-links with different sequences from the same protein). (XLSX)
The authors declare no competing financial interest.
Notes
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE59 partner repository with the data set identifier PXD006287. The programs REANG and YEUN YAN, written in Visual Basics for Applications will be sent on request by the authors W.R. (w.roseboom@uva.nl) or H.B. (h.buncherd@gmail.com).
Supplementary Material
References
- Leitner A.; Faini M.; Stengel F.; Aebersold R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci. 2016, 41, 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Navare A. T.; Chavez J. D.; Zheng C.; Weisbrod C. R.; Eng J. K.; Siehnel R.; Singh P. K.; Manoil C.; Bruce J. E. Probing the protein interaction network of Pseudomonas aeruginosa cells by chemical cross-linking mass spectrometry. Structure 2015, 23, 762–773. 10.1016/j.str.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Chavez J. D.; Schweppe D. K.; Zheng C.; Weisbrod C. R.; Eng J. K.; Murali A.; Lee S. A.; Ramage E.; Gallagher L. A.; Kulasekara H. D.; Edrozo M. E.; Kamischke C. N.; Brittnacher M. J.; Miller S. I.; Singh P. K.; Manoil C.; Bruce J. E. In vivo protein interaction network analysis reveals porin-localized antibiotic inactivation in Acinetobacter baumannii strain AB5075. Nat. Commun. 2016, 7, 13414. 10.1038/ncomms13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Tang X.; Munske G. R.; Zakharova N.; Yang L.; Zheng C.; Wolff M. A.; Tolic N.; Anderson G. A.; Shi L.; Marshall M. J.; Fredrickson J. K.; Bruce J. E. In vivo identification of the outer membrane protein OmcA-MtrC interaction network in Shewanella oneidensis MR-1 cells using novel hydrophobic chemical cross-linkers. J. Proteome Res. 2008, 7, 1712–1720. 10.1021/pr7007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; Yang L.; Hoopmann M. R.; Eng J. K.; Tang X.; Weisbrod C. R.; Bruce J. E. Cross-linking measurements of in vivo protein complex topologies. Mol. Cell. Proteomics 2011, 10, M110 006841. 10.1074/mcp.M110.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graumann P.Bacillus: Cellular and Molecular Biology, 2nd ed.; Caister Academic Press: Norfold, U.K., 2012. [Google Scholar]
- Kasper P. T.; Back J. W.; Vitale M.; Hartog A. F.; Roseboom W.; de Koning L. J.; van Maarseveen J. H.; Muijsers A. O.; de Koster C. G.; de Jong L. An aptly positioned azido group in the spacer of a protein cross-linker for facile mapping of lysines in close proximity. ChemBioChem 2007, 8, 1281–1292. 10.1002/cbic.200700150. [DOI] [PubMed] [Google Scholar]
- Nowak D. E.; Tian B.; Brasier A. R. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. BioTechniques 2005, 39, 715–725. 10.2144/000112014. [DOI] [PubMed] [Google Scholar]
- Buncherd H.; Roseboom W.; Ghavim B.; Du W.; de Koning L. J.; de Koster C. G.; de Jong L. Isolation of cross-linked peptides by diagonal strong cation exchange chromatography for protein complex topology studies by peptide fragment fingerprinting from large sequence databases. J. Chromatogr A 2014, 1348, 34–46. 10.1016/j.chroma.2014.04.083. [DOI] [PubMed] [Google Scholar]
- Buncherd H.; Roseboom W.; de Koning L. J.; de Koster C. G.; de Jong L. A gas phase cleavage reaction of cross-linked peptides for protein complex topology studies by peptide fragment fingerprinting from large sequence database. J. Proteomics 2014, 108, 65–77. 10.1016/j.jprot.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Kaake R. M.; Wang X.; Burke A.; Yu C.; Kandur W.; Yang Y.; Novtisky E. J.; Second T.; Duan J.; Kao A.; Guan S.; Vellucci D.; Rychnovsky S. D.; Huang L. A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol. Cell. Proteomics 2014, 13, 3533–3543. 10.1074/mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard N. M.; Brunckova J. In situ formation of N-trifluoroacetoxy succinimide (TFA-NHS): one-pot formation of succinimidyl esters, N-trifluoroacetyl amino acid succinimidyl esters, and N-maleoyl amino acid succinimidyl esters. J. Org. Chem. 2011, 76, 9169–9174. 10.1021/jo201686e. [DOI] [PubMed] [Google Scholar]
- Neidhardt F. C.; Bloch P. L.; Smith D. F. Culture medium for enterobacteria. J. Bacteriol. 1974, 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P.; Leighton T.; Ishkhanova G.; Kustu S. Sensing of nitrogen limitation by Bacillus subtilis: comparison to enteric bacteria. J. Bacteriol. 1999, 181, 5042–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soini J.; Ukkonen K.; Neubauer P. High cell density media for Escherichia coli are generally designed for aerobic cultivations - consequences for large-scale bioprocesses and shake flask cultures. Microb. Cell Fact. 2008, 7, 26. 10.1186/1475-2859-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Anorve L. I.; Calcagno M. L.; Plumbridge J. Why does Escherichia coli grow more slowly on glucosamine than on N-acetylglucosamine? Effects of enzyme levels and allosteric activation of GlcN6P deaminase (NagB) on growth rates. J. Bacteriol. 2005, 187, 2974–2982. 10.1128/JB.187.9.2974-2982.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Rinner O.; Seebacher J.; Walzthoeni T.; Mueller L.; Beck M.; Schmidt A.; Mueller M.; Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat. Methods 2008, 5, 315–318. 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka M. J.; Baker P. R.; Robinson P. J.; Burlingame A. L.; Chalkley R. J. Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 2014, 13, 420–434. 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzthoeni T.; Claassen M.; Leitner A.; Herzog F.; Bohn S.; Forster F.; Beck M.; Aebersold R. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 2012, 9, 901–903. 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- Glas M.; van den Berg van Saparoea H. B.; McLaughlin S. H.; Roseboom W.; Liu F.; Koningstein G. M.; Fish A.; den Blaauwen T.; Heck A. J.; de Jong L.; Bitter W.; de Esch I. J.; Luirink J. The soluble periplasmic domains of Escherichia coli cell division proteins FtsQ/FtsB/FtsL form a trimeric complex with submicromolar affinity. J. Biol. Chem. 2015, 290, 21498–21509. 10.1074/jbc.M115.654756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Mobli M.; Yang X.; Keller A. N.; King G. F.; Lewis P. J. RNA polymerase-induced remodelling of NusA produces a pause enhancement complex. Nucleic Acids Res. 2015, 43, 2829–2840. 10.1093/nar/gkv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev D. G.; Vassylyeva M. N.; Perederina A.; Tahirov T. H.; Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature 2007, 448, 157–162. 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- van Zundert G. C.; Rodrigues J. P.; Trellet M.; Schmitz C.; Kastritis P. L.; Karaca E.; Melquiond A. S.; van Dijk M.; de Vries S. J.; Bonvin A. M. The HADDOCK2.2 Web Server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Bae B.; Davis E.; Brown D.; Campbell E. A.; Wigneshweraraj S.; Darst S. A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma70 domain 1.1. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19772–19777. 10.1073/pnas.1314576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y.; Oda Y.; Tabata T.; Sato T.; Nagasu T.; Rappsilber J.; Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 2005, 4, 1265–1272. 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Sohmen D.; Chiba S.; Shimokawa-Chiba N.; Innis C. A.; Berninghausen O.; Beckmann R.; Ito K.; Wilson D. N. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 2015, 6, 6941. 10.1038/ncomms7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T.; Shaikh T. R.; Iwakura N.; Kaji H.; Kaji A.; Agrawal R. K. Structural insights into initial and intermediate steps of the ribosome-recycling process. EMBO J. 2012, 31, 1836–1846. 10.1038/emboj.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan C. E.; Dunkle J. A.; Maehigashi T.; Dang M. N.; Devaraj A.; Miles S. J.; Qin D.; Fredrick K.; Dunham C. M. Reorganization of an intersubunit bridge induced by disparate 16S ribosomal ambiguity mutations mimics an EF-Tu-bound state. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9716–9721. 10.1073/pnas.1301585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S.; Murayama Y.; Svetlov V.; Nudler E.; Yokoyama S. The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Mol. Cell 2015, 57, 408–421. 10.1016/j.molcel.2014.12.014. [DOI] [PubMed] [Google Scholar]
- Ha K. S.; Toulokhonov I.; Vassylyev D. G.; Landick R. The NusA N-terminal domain is necessary and sufficient for enhancement of transcriptional pausing via interaction with the RNA exit channel of RNA polymerase. J. Mol. Biol. 2010, 401, 708–725. 10.1016/j.jmb.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K. S. X-ray crystal structure of Escherichia coli RNA polymerase sigma70 holoenzyme. J. Biol. Chem. 2013, 288, 9126–9134. 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sharoud W. M.; Graumann P. L. Cold shock proteins aid coupling of transcription and translation in bacteria. Sci. Prog. 2007, 90 (Pt 1), 15–27. 10.3184/003685007780440549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M. H.; Volkov A. V.; Fricke I.; Marahiel M. A.; Graumann P. L. Localization of cold shock proteins to cytosolic spaces surrounding nucleoids in Bacillus subtilis depends on active transcription. J. Bacteriol. 2001, 183, 6435–6443. 10.1128/JB.183.21.6435-6443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunka K.; Tholen S.; Gerwig J.; Herzberg C.; Stulke J.; Commichau F. M. A high-frequency mutation in Bacillus subtilis: requirements for the decryptification of the gudB glutamate dehydrogenase gene. J. Bacteriol. 2012, 194, 1036–1044. 10.1128/JB.06470-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. E.; Lewis C. A.; Mooney R. A.; Kohanski M. A.; Collins J. J.; Landick R.; Walker G. C. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15517–15522. 10.1073/pnas.1005203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pero J.; Nelson J.; Fox T. D. Highly asymmetric transcription by RNA polymerase containing phage-SP01-induced polypeptides and a new host protein. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 1589–1593. 10.1073/pnas.72.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobinson K. F.; Spiegelman G. B. Effect of the delta subunit of Bacillus subtilis RNA polymerase on initiation of RNA synthesis at two bacteriophage phi 29 promoters. Biochemistry 1987, 26, 8206–8213. 10.1021/bi00399a028. [DOI] [PubMed] [Google Scholar]
- Juang Y. L.; Helmann J. D. The delta subunit of Bacillus subtilis RNA polymerase. An allosteric effector of the initiation and core-recycling phases of transcription. J. Mol. Biol. 1994, 239, 1–14. 10.1006/jmbi.1994.1346. [DOI] [PubMed] [Google Scholar]
- Prajapati R. K.; Sengupta S.; Rudra P.; Mukhopadhyay J. Bacillus subtilis delta factor functions as a transcriptional regulator by facilitating the open complex formation. J. Biol. Chem. 2016, 291, 1064–1075. 10.1074/jbc.M115.686170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapati R. K.; Sur R.; Mukhopadhyay J. A novel function of delta factor from Bacillus subtilis as a transcriptional tepressor. J. Biol. Chem. 2016, 291, 24029–24035. 10.1074/jbc.M116.746065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedermannova J.; Sudzinova P.; Koval T.; Rabatinova A.; Sanderova H.; Ramaniuk O.; Rittich S.; Dohnalek J.; Fu Z.; Halada P.; Lewis P.; Krasny L. Characterization of HelD, an interacting partner of RNA polymerase from Bacillus subtilis. Nucleic Acids Res. 2014, 42, 5151–5163. 10.1093/nar/gku113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas P.; Mader U.; Dervyn E.; Rochat T.; Leduc A.; Pigeonneau N.; Bidnenko E.; Marchadier E.; Hoebeke M.; Aymerich S.; Becher D.; Bisicchia P.; Botella E.; Delumeau O.; Doherty G.; Denham E. L.; Fogg M. J.; Fromion V.; Goelzer A.; Hansen A.; Hartig E.; Harwood C. R.; Homuth G.; Jarmer H.; Jules M.; Klipp E.; Le Chat L.; Lecointe F.; Lewis P.; Liebermeister W.; March A.; Mars R. A.; Nannapaneni P.; Noone D.; Pohl S.; Rinn B.; Rugheimer F.; Sappa P. K.; Samson F.; Schaffer M.; Schwikowski B.; Steil L.; Stulke J.; Wiegert T.; Devine K. M.; Wilkinson A. J.; van Dijl J. M.; Hecker M.; Volker U.; Bessieres P.; Noirot P. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 2012, 335, 1103–1106. 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Weiss A.; Shaw L. N. Small things considered: the small accessory subunits of RNA polymerase in Gram-positive bacteria. FEMS Microbiol. Rev. 2015, 39, 541–554. 10.1093/femsre/fuv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Saro F. J.; Moon Woody A.-Y.; Helmann J. D. Structural analysis of the Bacillus subtilis delta factor: a protein polyanion which displaces RNA from RNA polymerase. J. Mol. Biol. 1995, 252, 189–202. 10.1006/jmbi.1995.0487. [DOI] [PubMed] [Google Scholar]
- Papouskova V.; Kaderavek P.; Otrusinova O.; Rabatinova A.; Sanderova H.; Novacek J.; Krasny L.; Sklenar V.; Zidek L. Structural study of the partially disordered full-length delta subunit of RNA polymerase from Bacillus subtilis. ChemBioChem 2013, 14, 1772–1779. 10.1002/cbic.201300226. [DOI] [PubMed] [Google Scholar]
- Doherty G. P.; Fogg M. J.; Wilkinson A. J.; Lewis P. J. Small subunits of RNA polymerase: localization, levels and implications for core enzyme composition. Microbiology 2010, 156 (Pt 12), 3532–3543. 10.1099/mic.0.041566-0. [DOI] [PubMed] [Google Scholar]
- Hyde E. I.; Hilton M. D.; Whiteley H. R. Interactions of Bacillus subtilis RNA polymerase with subunits determining the specificity of initiation. Sigma and delta peptides can bind simultaneously to core. J. Biol. Chem. 1986, 261, 16565–16570. [PubMed] [Google Scholar]
- Lopez de Saro F. J.; Yoshikawa N.; Helmann J. D. Expression, abundance, and RNA polymerase binding properties of the delta factor of Bacillus subtilis. J. Biol. Chem. 1999, 274, 15953–15958. 10.1074/jbc.274.22.15953. [DOI] [PubMed] [Google Scholar]
- Motackova V.; Sanderova H.; Zidek L.; Novacek J.; Padrta P.; Svenkova A.; Korelusova J.; Jonak J.; Krasny L.; Sklenar V. Solution structure of the N-terminal domain of Bacillus subtilis delta subunit of RNA polymerase and its classification based on structural homologs. Proteins: Struct., Funct., Genet. 2010, 78, 1807–1810. 10.1002/prot.22708. [DOI] [PubMed] [Google Scholar]
- Yang B.; Wu Y. J.; Zhu M.; Fan S. B.; Lin J.; Zhang K.; Li S.; Chi H.; Li Y. X.; Chen H. F.; Luo S. K.; Ding Y. H.; Wang L. H.; Hao Z.; Xiu L. Y.; Chen S.; Ye K.; He S. M.; Dong M. Q. Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 9, 904–906. 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- Tan D.; Li Q.; Zhang M. J.; Liu C.; Ma C.; Zhang P.; Ding Y. H.; Fan S. B.; Tao L.; Yang B.; Li X.; Ma S.; Liu J.; Feng B.; Liu X.; Wang H. W.; He S. M.; Gao N.; Ye K.; Dong M. Q.; Lei X. Trifunctional cross-linker for mapping protein-protein interaction networks and comparing protein conformational states. eLife 2016, 5, e12509. 10.7554/eLife.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J. D.; Weisbrod C. R.; Zheng C.; Eng J. K.; Bruce J. E. Protein interactions, post-translational modifications and topologies in human cells. Mol. Cell. Proteomics 2013, 12, 1451–1467. 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rijkers D. T.; Post H.; Heck A. J. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 2015, 12, 1179–1184. 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Arlt C.; Gotze M.; Ihling C. H.; Hage C.; Schafer M.; Sinz A. Integrated workflow for structural proteomics studies based on cross-linking/mass spectrometry with an MS/MS cleavable cross-Linker. Anal. Chem. 2016, 88, 7930–7937. 10.1021/acs.analchem.5b04853. [DOI] [PubMed] [Google Scholar]
- Syka J. E.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q. W.; Wang R.; Hermjakob H. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44 (D1), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.