

Abstract

We present a mild way of converting secondary methyl ethers into ketones using calcium hypochlorite in aqueous acetonitrile with acetic acid as activator. The reaction is compatible with various oxygen- and nitrogen-containing functional groups and afforded the corresponding ketones in up to 98% yield. The use of this methodology could expand the application of the methyl group as a useful protecting group.
Introduction
Well-established methodologies have been developed for the selective deprotection of aryl, allyl, and benzyl ethers, which are commonly used by organic chemists. In contrast, selective cleavage of aliphatic ethers is much less explored. These ethers, which in fact are often used as solvents, are extraordinarily unreactive toward a variety of reagents,1 including most well-known oxidants. Historically, the methyl group has been used as a protecting group for phenols and carboxylic acids, but rarely for aliphatic alcohols. For instance, the methyl group of an aryl methyl ether can be selectively removed by boron tribromide,2 sodium ethanethiolate in refluxing DMF,3 or lithium iodide in refluxing collidine,4 whereas aliphatic methyl ethers remain unaffected. Due to its inertness, it is difficult to selectively remove the methyl group from an aliphatic methyl ether while keeping other functional groups intact. Rigorous conditions are required to successfully cleave off the methyl group of methyl ethers, such as aqueous sulfuric, hydroiodic, hydrobromic, or hydrochloric acid.5 Other methods to transform methyl ethers into more reactive functional groups are based on oxidation. Olah et al.6 utilized uranium hexafluoride as the oxidant to transform secondary methyl ethers into the corresponding ketones. Other research groups observed the same result by using HOF,7 a manganese complex and m-CPBA as the stoichiometric oxidant,8 hydrogen peroxide over titanosilicates,9 and Bobbitt’s salt (2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate).10,11 Mayhoub et al.12 also showed the selective oxidation of benzyl methyl ethers using NBS and UV light to afford aldehydes or esters. It may be clear that oxidation of ethers requires either the ether to be prone to oxidation (such as benzyl and allyl ethers) or expensive transition-metal catalysts. Herein, we describe an oxidation method to selectively transform secondary methyl ethers into ketones with the versatile and cheap oxidant calcium hypochlorite under mild acidic conditions, without the aid of any transition-metal catalyst. Various oxygen- and nitrogen-bearing functional groups are compatible with the reaction. Other aliphatic ethers can undergo the same conversion, but we focused on the methyl ether, because of the observed regioselectivity, the unreactive nature, and the possible use as a versatile protecting group. It was previously reported by Nwaukwa et al.13 that ethers can be oxidized with sodium and calcium hypochlorite. However, the reactions were only carried out on symmetrical ethers, such as tetrahydrofuran (THF) and diethyl ether. Remarkably, the examined ethers were oxidized to esters. We herewith report that the oxidation of methyl-protected secondary alcohols reproducibly afforded the corresponding ketones in up to 98% yield.
Results and Discussion
We envisioned that multifunctionalized molecules would selectively form ketones from methyl or benzyl ethers leaving other functional groups unchanged. First, we looked for mild reaction conditions to oxidize ethers. We observed that THF (1) was oxidized to γ-butyrolactone (9) using both sodium and calcium hypochlorite (entries 1–2, Table 1). Calcium hypochlorite was the preferred oxidant, because it was easier to use stoichiometrically compared to the alkaline sodium hypochlorite solution. The oxidation of 2-methyl-tetrahydrofuran (2) was completely regioselective and overoxidation of primary alcohol 10 was not observed within 1 h (entry 3). Dioxane, 2-(chloromethyl)tetrahydro-2H-pyran, and isosorbide were completely unaffected under these reaction conditions. Several benzyl ethers (3–7) were synthesized and submitted to the conditions used by Nwaukwa et al.13 for the oxidation of symmetrical ethers (6 equiv of oxidant, 9 equiv of acetic acid in a 1:3 mixture of acetonitrile/water). Benzyl ethers 3 and 4 (entries 4–5) were oxidized to ketones 11 and 12, respectively. The benzyl group of the ether was oxidized to chlorinated benzaldehydes and benzoic acids giving rise to an inseparable mixture. Primary alkyl benzyl ether 5 (entry 6) was partially oxidatively deprotected to give 13, but multiple unidentified side products were formed. Linear secondary alkyl benzyl ethers 6 and 7 (entries 7–9) were oxidized to the corresponding methyl ketones 14 and 15. Chlorination of the aromatic ring could be reduced (entry 9) by lowering the reaction temperature and by adding less oxidant portion wise. Then, we moved on to secondary methyl ether 8 for which similar conditions were used (entries 10–11) as for the oxidation of the aforementioned benzyl ethers. Reducing the amounts of oxidant (to 1.6 equiv) and acid (to 3.5 equiv) and lowering the reaction temperature to 0 °C (entries 12–13) eventually led to a clean reaction in which ketone 15 was obtained in 89% yield without the need for further purification.
Table 1. Optimization of Ether Oxidation.

Equivalents of oxidant; Ca(OCl)2 contains two equivalents of oxidant.
Conversions are based on 1H NMR analysis.
Isolation of products was only achieved for entries 11–13.
NaOCl was used as oxidant in a pH 6 phosphate buffer.
Optimal results were obtained when the methyl ether (1.0 equiv) was stirred for 20–24 h in a 1:1 mixture of acetonitrile/water (0.25 M) and acetic acid (3.5 equiv), while calcium hypochlorite (1.6 equiv) was added portion wise at 0 °C. To explore the scope and limitations of this oxidative demethylation various methyl ethers carrying other functional groups were synthesized from readily available starting materials (see Supporting Information). Oxidation reactions were conducted under the optimal reaction conditions, typically on a 1 mmol scale, unless otherwise stated (Table 2). If necessary, additional oxidant was added after 1 day. The reaction was tested in three series of compounds: acyclic compounds, cyclohexane derivatives, and functionalized piperidines. Benzoyl protected primary alcohol 16 (entry 1) gave the desired ketone 29 in excellent yield (98%). TBDMS-protected primary alcohol 17 (entry 2) was partially hydrolyzed to the hydroxy ketone 10, but still the corresponding ketone 30 was obtained in good yield (68%).
Table 2. Scope of Methyl Ethers as Substrates.
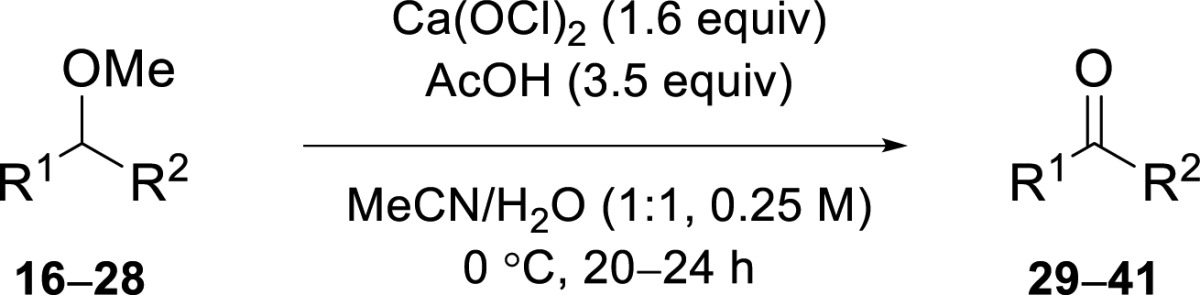
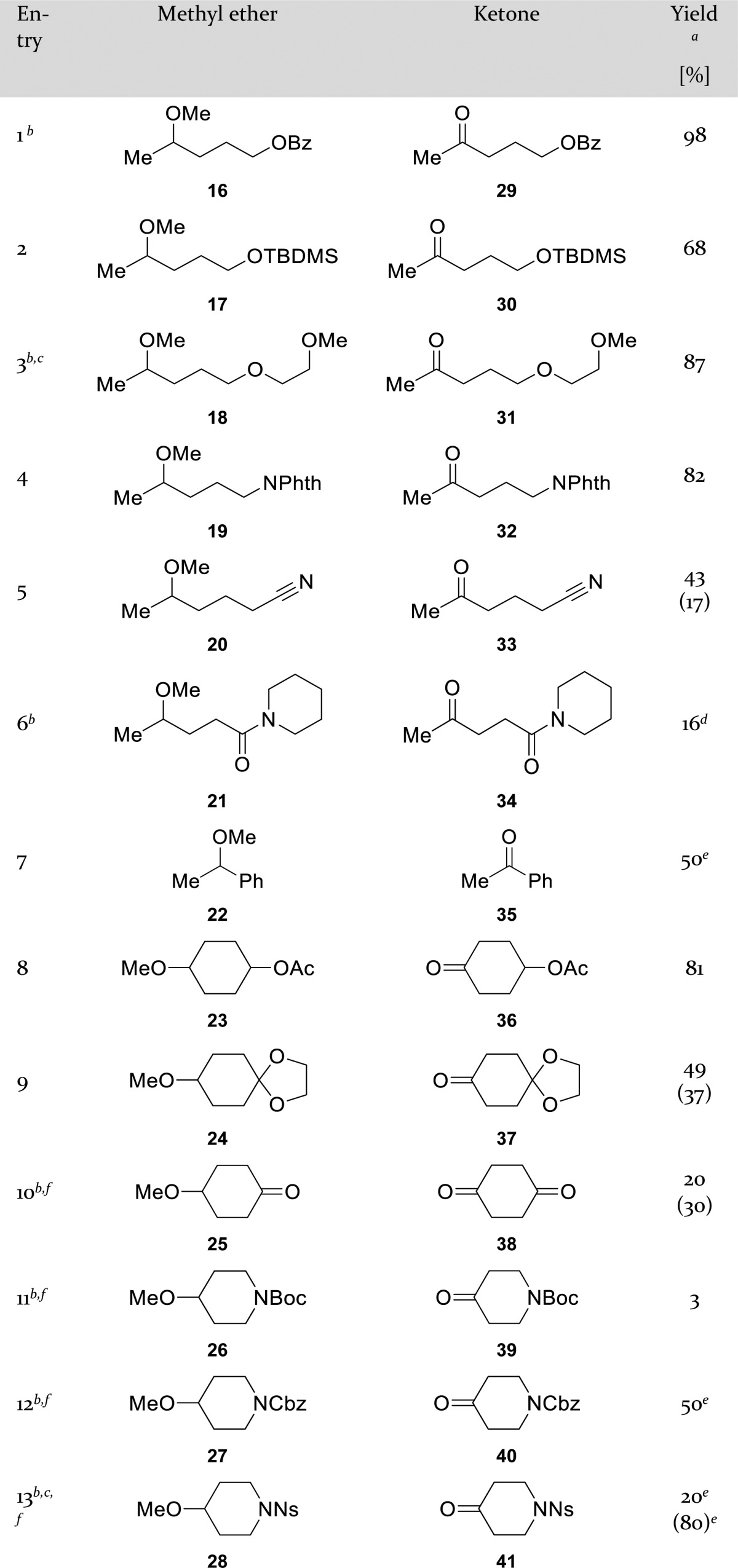
The recovered starting material [%] is given between brackets.
Additional oxidant was added after 1 day and stirring was continued for another day.
Reaction was performed on smaller scale (see Experimental Section).
Isolated yield of the corresponding 2,4-dinitrophenyl hydrazone 34a.
Conversion based on 1H NMR analysis of the crude mixture.
Total reaction time exceeded 48 h (see Experimental Section).
Remarkably, compound 31 (entry 3) was the only product (87%) from the oxidation reaction of poly ether 18, leaving the ether tail completely intact. The explanation for this regioselectivity was supported by failed attempts to oxidize dioxane and isosorbide under similar conditions. All those substrates have their heteroatoms in an ethylene glycol-like connectivity. We hypothesize that the inductive effect of one of the oxygen atoms lowers the nucleophilicity of the other oxygen. We observed similar unreactivity of the 1,3-dioxolane toward oxidation while forming ketone 37 (entry 9). Finally, we tried the reaction with the corresponding unprotected primary alcohol (4-methoxypentan-1-ol), although without success. Primary amine 19 (entry 4) protected as phthalimide afforded the corresponding amino ketone 32 in good yield (82%). The reaction of nitrile 20 (entry 5) was stopped after 27.5 h, purified, and product 33 was obtained in 43% yield, alongside 17% of 20. As expected, the reaction with a tertiary amine as substrate failed. The reaction of secondary amide 21 (entry 6) worked to a certain extent, but eventually we discovered that the amide functionality itself was prone to oxidation, leading to several water-soluble unidentified side products (detected by NMR). Therefore, the crude mixture after aqueous extraction only contained methyl ether 21 and ketone 34. Due to negligible difference in polarity between these two compounds, we decided to isolate the ketone as the corresponding 2,4-dinitrophenyl hydrazone 34a (16%) using Brady’s reagent (2,4-dinitrophenyl-hydrazine).14 Benzylic methyl ether 22 (entry 7) heavily suffered from chlorination of the phenyl ring as a side reaction. Two successive rounds of silica gel column chromatography were insufficient to separate 35 from the complex mixture.
Besides linear substrates, cyclic substrates derived from cyclohexane were considered to be suitable for this reaction. The acetoxy group of methyl ether 23 (entry 8) was completely unreactive under the mild reaction conditions and ketone 36 was formed in 81% yield. As expected, a TMS-protected secondary alcohol (1-methoxy-4-[(trimethylsilyl)oxy]cyclohexane) was hydrolyzed before any observable oxidation took place. The 1,3-dioxolane protecting group of compound 24 (entry 9) was not completely unreactive. The reaction was stopped after 20 h because of the formation of an additional product according to TLC. After purification, the monoprotected diketone 37 was isolated in 49% yield alongside 37% of the starting material. 4-Methoxycyclohexanone 25 (entry 10) reacted extremely slowly and after 5 days of stirring with additional oxidant, diketone 38 was isolated in only 20% yield, alongside 30% of the starting material. Then, we moved on to piperidine derivatives. Boc-protected 4-methoxypiperidine 26 (entry 11) was not unreactive under the acidic reaction conditions, giving rise to a number of unidentified side products. The corresponding 4-piperidone 39 was just isolated as a minor component (3% yield). Cbz-protection of compound 27 (entry 12) indeed made the carbamate functional group unreactive, but the aromatic ring was prone to chlorination. Within the crude mixture, product 40 was the predominant one, but isolation was not achieved. Sulfonamide 28 (entry 13) was cleanly converted into the corresponding ketone 41. However, the oxidation was extremely slow and after two successive rounds of oxidation (three and 6 days, respectively) the conversion was only 20%. Most of the material was recovered, but due to the negligible difference in polarity, attempts to isolate 41 were not successful, hence conversions are mentioned.
We propose a reaction sequence for the oxidative transformation of secondary methyl ethers into ketones (Figure 1). First, protonation of hypochlorite anion by acetic acid is required to generate hypochlorous acid, which is the active species. The chlorinating species hypochlorous acid is in equilibrium with acetyl hypochlorite and molecular chlorine, which are other chlorinating species.
Figure 1.

Proposed reaction sequence for the oxidation of methyl ethers to ketones.
Then, chlorination occurs at the nucleophilic ether oxygen of I to form oxonium ion II. Subsequent selective HCl elimination via an E2 mechanism at the most substituted carbon forms the most stable oxocarbenium intermediate III. The stabilized cation III is trapped by a water molecule and the formed hemiacetal IV collapses to form the corresponding ketone V upon release of one molecule of methanol. The regioselectivity is supported by the observation that during the oxidation of secondary methyl ethers, the corresponding secondary alcohol was never detected with TLC analysis. In contrast, this secondary alcohol was always detected as an intermediate in the oxidation of secondary benzyl ethers.
Conclusions
A novel and versatile method to transform secondary methyl ethers into ketones has been developed. From our perspective, the secondary methyl ether can now be considered as a masked ketone, and hence, this reaction should find use in organic synthesis where it might reduce the number of protection and oxidation steps. The reaction is rather slow, but highly regioselective. The scope and limitations have been determined and we can safely state that a variety of oxygen- and nitrogen-containing functional groups are tolerated. Under the mild acidic reaction conditions used, in particular some acid labile groups are tolerated. However, nondeactivated aromatic systems were chlorinated, making this reaction not suitable for aromatic compounds of this particular kind.
Experimental Section
General Information
Reagents were obtained from commercial suppliers and were used without purification. Reactions were followed using thin-layer chromatography (TLC) on silica gel-coated plates (Merck 60 F254). Detection was performed with UV light and/or by charring at 150 °C after dipping in a solution of Brady’s reagent (2,4-dinitophenylhydrazine) or a solution of KMnO4. Column chromatography was performed manually using Acros silica gel, 0.035–0.070 mm, 60 A. Low-resolution ESI mass spectra were recorded on a Thermo Finnigan LCQ Advantage Max Ion Trap mass spectrometer. Low-resolution EI mass spectra were recorded on a JEOL AccuTOF-GCv JMS-100GCv mass spectrometer. High-resolution ESI mass spectra were recorded on a JEOL AccuTOF CS JMS-T100CS mass spectrometer. High-resolution FD and FI mass spectra were recorded on a JEOL AccuTOF GC v 4g, JMS-T100GCV mass spectrometer. NMR spectra were recorded at 298 K on a Varian Inova 4003 spectrometer (400 MHz) and on a Bruker Avance III 400 spectrometer (400 MHz) in CDCl3. Chemical shifts (δ) are given in parts per million (ppm) with respect to tetramethylsilane (0.00 ppm) as internal standard for 1H NMR; and CDCl3 (77.16 ppm) as internal standard for 13C NMR. Coupling constants are reported as J values in Hertz (Hz). Data for 1H and 13C NMR spectra are reported as follows: chemical shift (multiplicity, coupling constant, integration).
General Procedure for the Oxidation of Methyl Ethers to Ketones
The methyl ether (1.0 mmol, 1.0 equiv) was dissolved in acetonitrile/water (1:1 v/v, 0.25 M, 4 mL). Acetic acid (0.2 mL, 3.5 mmol, 3.5 equiv) was added and the solution was cooled to 0 °C. Calcium hypochlorite (4 × 44 mg, 4 × 0.2 mmol, 4 × 0.4 equiv, 65%) was added in four portions over 3 h (one portion every hour). The solution was stirred at 0 °C until TLC indicated full conversion of the starting material (typically 20–48 h). Additional calcium hypochlorite was added after 20–30 h if necessary. Upon completion of the reaction, the ice bath was removed and the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL) and the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL); then dried over magnesium sulfate, filtered, and the solvent was evaporated in vacuo to obtain the crude mixture. The crude mixture was purified with silica gel column chromatography (different eluent systems) to afford the ketone when necessary.
2-Methoxyundecane (8)15
Sodium hydride (865 mg, 21.6 mmol, 1.5 equiv, 60% dispersion in mineral oil) was added at 0 °C to a solution of undecan-2-ol 42 (2.48 g, 14.4 mmol, 1.0 equiv) in THF (20 mL). The mixture was stirred for 30 min; then methyl iodide (1.35 mL, 21.6 mmol, 1.5 equiv) was added. The mixture was allowed to warm to room temperature and it was stirred for 60 h. Then, the reaction was quenched with water (25 mL) and the product was extracted with ethyl acetate (3 × 40 mL), washed with brine (40 mL), dried over magnesium sulfate, and the solvent was evaporated in vacuo to afford a brownish oil. The crude mixture was dissolved in ethyl acetate (20 mL) and washed with aqueous sodium thiosulfate (10% in water, 20 mL), dried over magnesium sulfate and concentrated. Silica gel column chromatography (pentane/dichloro-methane, 4:1) furnished 8 (2.60 g, 97%) as a transparent yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.31 (s, 3 H), 3.33–3.23 (m, 1 H), 1.59–1.46 (m, 1 H), 1.43–1.20 (m, 15 H), 1.12 (d, J = 6.1 Hz, 3 H), 0.92–0.85 (m, 3 H); 13C NMR (101 MHz, CDCl3) δ 76.9, 55.9, 36.3, 31.9, 29.8, 29.7, 29.6, 29.3, 25.5, 22.7, 19.0, 14.1.
Undecan-2-one (15)16
According to the general procedure, substrate 8 (186 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. The reaction mixture was stirred for 21.5 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), and the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL); then dried over magnesium sulfate and the solvent was removed in vacuo yielding ketone 15 (151 mg, 89%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 2.43 (t, J = 7.5 Hz, 3 H), 2.15 (s, 3 H), 1.58 (p, J = 7.2 Hz, 2 H), 1.38–1.20 (m, 12 H), 0.95–0.85 (m, 3 H).
4-Methoxypentyl Benzoate (16)17
Benzoyl chloride (209 μL, 1.8 mmol, 1.2 equiv) was added at 0 °C to a solution of alcohol 43 (177 mg, 1.5 mmol, 1.0 equiv), DMAP (37 mg, 0.3 mmol, 20 mol%), and triethylamine (314 μL, 2.25 mmol, 1.5 equiv) in dichloromethane (8 mL). The reaction was allowed to warm to 20 °C and it was stirred for 15 h. The reaction was quenched with saturated aqueous sodium bicarbonate (5 mL), stirred for another 30 min and then extracted with dichloromethane (3 × 5 mL). The combined organic extracts were washed with 1 M HCl (2 × 3 mL), saturated aqueous sodium bicarbonate (3 mL), and brine (3 mL); then dried over magnesium sulfate and the solvent was removed in vacuo to obtain ester 16 (317 mg, 95%) as a pale yellow liquid. 1H NMR (400 MHz, CDCl3) δ 8.07–8.02 (m, 2 H), 7.58–7.53 (m, 1 H), 7.47–7.41 (m, 2 H), 4.39–4.28 (m, 2 H), 3.42–3.32 (m, 1 H), 3.33 (s, 3 H), 1.95–1.74 (m, 2 H), 1.71–1.52 (m, 2 H), 1.17 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 166.6, 132.8, 130.4, 129.5, 128.3, 76.3, 65.1, 56.0, 32.8, 24.8, 19.0; MS (EI+) calcd for (C13H18O3 – CH3)+ 207.102, found 207.126; HRMS (FD+) calcd for (C13H18O3)+ 222.1256, found 222.1261.
tert-Butyl[(4-methoxypentyl)oxy]dimethylsilane (17)18
Triethylamine (20.5 mL, 147 mmol, 3.75 equiv) was added at 0 °C to a stirring solution of TBDMSCl (5.9 g, 39.1 mmol, 1.0 equiv) in dichloromethane (60 mL) followed by addition of DMAP (0.6 g, 4.91 mmol, 12.5 mol%). Then, a solution of 5-hydroxypentan-2-one 10 (5.0 g, 49 mmol, 1.25 equiv) in dichloromethane (15 mL) was added to the stirring mixture at the same temperature. The reaction mixture was stirred for 30 min at 0 °C and then brought to 20 °C with continued stirring for another 3 h. The reaction mixture was then quenched with saturated aqueous ammonium chloride (40 mL) and the product was extracted with ethyl acetate (3 × 65 mL). The combined organic extracts were washed with water (15 mL) and brine (15 mL); then dried over sodium sulfate and the solvent was removed in vacuo to give the crude product (9.6 g), which was purified by flash chromatography (ethyl acetate/heptane 1:9) to recover starting material 10 (2.33 g) and to afford the silyl ether (3.74 g, 65% brsm) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.1 Hz, 2 H), 2.51 (t, J = 7.3 Hz, 2 H), 2.15 (s, 3 H), 1.78 (tt, J = 7.2, 6.1 Hz, 2 H), 0.89 (s, 9 H), 0.04 (s, 6 H). Sodium borohydride (612 mg, 16.1 mmol, 1.0 equiv) was added portion wise over 5 min to a solution of the aforementioned silyl ether (3.5 g, 16.1 mmol, 1.0 equiv) in methanol (75 mL). The reaction was stirred at 20 °C for 15 min and then most methanol was evaporated under reduced pressure. Water (50 mL) was added to the residue, followed by 1 N HCl to make the solution acidic. The product was extracted with diethyl ether (3 × 20 mL). The combined ethereal extracts were washed with water (10 mL) and brine (10 mL); then dried over magnesium sulfate and the solvent was evaporated in vacuo to afford the secondary alcohol (3.52 g, 99%) as a yellow transparent oil. 1H NMR (400 MHz, CDCl3) δ 3.87–3.76 (m, 1 H), 3.73–3.61 (m, 2 H), 2.64 (bs, 1 H), 1.73–1.55 (m, 3 H), 1.55–1.41 (m, 1 H), 1.19 (d, J = 6.2 Hz, 3 H), 0.90 (s, 9 H), 0.07 (s, 6 H). Sodium hydride (660 mg, 16.5 mmol, 1.2 equiv, 60% dispersion in mineral oil) was added at 0 °C to a solution of the aforementioned secondary alcohol (3.00 g, 13.7 mmol, 1.0 equiv) in THF (20 mL). The mixture was stirred for 15 min and then methyl iodide (1.03 mL, 16.5 mmol, 1.2 equiv) was added. The mixture was allowed to warm to 20 °C and it was stirred for 18 h. Then, the reaction was quenched with aqueous sodium thiosulfate (5% in water, 30 mL) and the product was extracted with ethyl acetate (3 × 30 mL), washed with brine (15 mL), then dried over magnesium sulfate and concentrated. Silica gel column chromatography (pentane → ethyl acetate) yielded substrate 17 (3.05 g, 95%) as a transparent yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.67–3.57 (m, 2 H), 3.31 (s, 3 H), 3.35–3.27 (m, 1 H), 1.65–1.39 (m, 4 H), 1.13 (d, J = 6.1 Hz, 3 H), 0.89 (s, 9 H), 0.05 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 76.6, 63.2, 55.9, 32.5, 28.7, 26.0, 19.1, 18.4, – 5.3; MS (ESI+) calcd for (C12H28O2Si + H)+ 233, found 233. HRMS (FI+) calcd for (C12H28O2Si – C4H9)+ 175.1154, found 175.1160.
4-Methoxy-1-(2-methoxyethoxy)pentane (18)
Sodium hydride (72 mg, 1.8 mmol, 1.2 equiv, 60% dispersion in mineral oil) was added at 0 °C to a solution of compound 43 (177 mg, 1.5 mmol, 1.0 equiv) in THF (4 mL). The mixture was stirred for 30 min and then 2-chloroethyl methyl ether (164 μL, 1.8 mmol, 1.2 equiv) was added. Stirring was continued for 2 h, while no conversion was observed. Sodium iodide (22 mg, 0.15 mmol, 10 mol%) was added to catalyze the reaction. After stirring for another 16 h, conversion was barely observed so additional THF was added (5 mL), together with sodium hydride (120 mg, 3.0 mmol, 2.0 equiv, 60% dispersion in mineral oil) and 2-chloroethyl methyl ether (275 μL, 3.0 mmol, 2.0 equiv). The temperature was elevated and the mixture was refluxed for 2 days. Then, the reaction was carefully quenched with water (6 mL) and 1 M HCl (3 mL) and the product was extracted with ethyl acetate (3 × 6 mL). The combined organic extracts were washed with brine (3 mL), dried over magnesium sulfate, and concentrated. Consecutive silica gel column chromatography (ethyl acetate/heptane 1:3) afforded ether 18 (124 mg, 47%) as a yellow oil which was slightly contaminated with unknown impurities originating from the eluent (bottle ethyl acetate). 1H NMR (400 MHz, CDCl3) δ 3.61–3.52 (m, 4 H), 3.51–3.44 (m, 2 H), 3.39 (s, 3 H), 3.31 (s, 3 H), 3.35–3.26 (m, 1 H), 1.75–1.41 (m, 4 H), 1.13 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 76.6, 72.0, 71.5, 70.0, 59.1, 56.0, 32.7, 25.6, 19.0; HRMS (ESI+) calcd for (C9H20O3 + Na)+ 199.1310, found 199.1296.
2-(4-Methoxypentyl)isoindoline-1,3-dione (19)19
Sulfonate 44 (294 mg, 1.5 mmol, 1.0 equiv) was added to a suspension of potassium phthalimide (556 mg, 3.0 mmol, 2.0 equiv) in DMF (25 mL) and the mixture was stirred at 80 °C for 2 h until TLC indicated full conversion of the sulfonate. Upon completion, the mixture was cooled to 20 °C and subsequently diluted with dichloromethane (45 mL), washed with water (2 × 45 mL) and brine (2 × 45 mL); then dried over magnesium sulfate and the solvent was removed in vacuo yielding a crude mixture that still contained DMF. The crude mixture was redissolved in dichloromethane (30 mL) and washed with water (2 × 15 mL) and brine (15 mL); then it was dried over magnesium sulfate and concentrated. The residue was purified with silica gel column chromatography (ethyl acetate/heptane 1:4) to afford compound 19 (271 mg, 73%) as a transparent sticky oil. 1H NMR (400 MHz, CDCl3) δ 7.87–7.81 (m, 2 H), 7.74–7.68 (m, 2 H), 3.70 (t, J = 7.3 Hz, 2 H), 3.37–3.28 (m, 1 H), 3.30 (s, 3 H), 1.86–1.66 (m, 2 H), 1.61–1.40 (m, 2 H), 1.12 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 168.4, 133.9, 132.1, 123.2, 76.2, 56.1, 38.0, 33.5, 24.7, 19.0; HRMS (ESI+) calcd for (C14H17NO3 + Na)+ 270.1106, found 270.1080.
5-Methoxyhexanenitrile (20)20,21
Sodium cyanide (221 mg, 4.5 mmol, 3.0 equiv) was added to a solution of sulfonate 44 (294 mg, 1.5 mmol, 1.0 equiv) in DMSO (12 mL) and the mixture was stirred at 80 °C for 2.5 h, and subsequently cooled to 20 °C. Then, the reaction was carefully quenched with water (60 mL), and the reaction mixture was extracted with ethyl acetate (3 × 40 mL). The combined organic extracts were washed with brine (2 × 20 mL), dried over sodium sulfate and concentrated. Purification by silica gel column chromatography (dichloromethane) afforded nitrile 20 (130 mg, 68%) as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 3.39–3.30 (m, 1 H), 3.32 (s, 3 H), 2.46–2.28 (m, 2 H), 1.86–1.64 (m, 2 H), 1.62–1.56 (m, 2 H), 1.15 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 119.7, 75.8, 56.0, 35.3, 21.5, 18.9, 17.2; MS (EI+) calcd for (C7H13NO – CH3)+ 112.076, found 112.089.
4-Methoxy-1-(piperidin-1-yl)pentan-1-one (21)22
Trimethylaluminum (2 M in toluene, 2.5 mL, 5 mmol, 2 equiv) was added at −78 °C to a 1 M solution of freshly distilled piperidine (1.25 mL, 12.5 mmol, 5 equiv) in THF (12.5 mL) and the resulting mixture was stirred at −78 °C for 30 min. Then, a solution of γ-valerolactone 45 (0.24 mL, 2.5 mmol, 1 equiv) in THF (6 mL) was added dropwise to the stirring piperidine solution after which the reaction mixture was allowed to warm to 20 °C. Stirring was continued for another 1.5 h before the reaction was quenched by careful addition of dichloromethane (25 mL) and 0.1 M HCl (50 mL). The product was extracted with dichloromethane (5 × 10 mL) and the combined organic extracts were washed with 1 M HCl (25 mL), dried over magnesium sulfate and the solvent was removed in vacuo to afford alcohol intermediate as a sticky yellow liquid. Then, the alcohol was redissolved in THF (5 mL) and to this solution sodium hydride (120 mg, 3.0 mmol, 1.2 equiv, 60% dispersion in mineral oil) was added at 0 °C. The mixture was stirred for 30 min and then methyl iodide (188 μL, 3.0 mmol, 1.2 equiv) was added. The mixture was allowed to warm to 20 °C and it was stirred for 2 h. Then, the reaction was quenched with aqueous sodium thiosulfate (5% in water, 8 mL) and the product was extracted with ethyl acetate (3 × 10 mL), washed with brine (5 mL), dried over magnesium sulfate, and concentrated. Silica gel column chromatography (ethyl acetate) yielded ether 21 (408 mg, 82% over 2 steps) as a transparent yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.59–3.51 (m, 2 H), 3.44–3.40 (m, 2 H), 3.40–3.33 (m, 1 H), 3.32 (s, 3 H), 2.43 (ddd, J = 15.3, 9.2, 6.0 Hz, 1 H), 2.36 (ddd, J = 15.4, 9.0, 6.5 Hz, 1 H), 1.85 (dddd, J = 13.7, 9.2, 6.5, 4.4 Hz, 1 H), 1.74 (dddd, J = 14.1, 9.1, 7.4, 6.0 Hz, 1 H), 1.68–1.60 (m, 2 H), 1.60–1.49 (m, 4 H), 1.15 (d, J = 6.2 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 171.2, 76.1, 56.0, 46.6, 42.7, 31.8, 28.9, 26.5, 25.6, 24.6, 19.0; HRMS (ESI+) calcd for (C11H21NO2 + Na)+ 222.1470, found 222.1456.
(1-Methoxyethyl)benzene (22)23
Sodium hydride (180 mg, 4.5 mmol, 1.5 equiv, 60% dispersion in mineral oil) was added at 0 °C to a stirred solution of 1-phenylethanol (46, 363 μL, 3.0 mmol, 1.0 equiv) in THF (12 mL) and DMF (3 mL). After stirring for 40 min, methyl iodide (375 μL, 6.0 mmol, 2.0 equiv) was added and the reaction mixture was allowed to warm to 20 °C. After stirring for 21 h the reaction was carefully quenched with water (20 mL) and the product was extracted with ethyl acetate (3 × 40 mL). The combined organic extracts were successively washed with aqueous sodium thiosulfate (5% in water, 20 mL) and brine (2 × 20 mL); then dried over sodium sulfate and concentrated. The residue was purified with silica gel column chromatography (ethyl acetate/heptane 1:9) to give compound 22 (276 mg, 68%) as a transparent liquid, which was not dried at the oil pump to avoid evaporation of the product. 1H NMR (400 MHz, CDCl3) δ 7.38–7.25 (m, 5 H), 4.29 (q, J = 6.5 Hz, 1 H), 3.23 (s, 3 H), 1.44 (d, J = 6.5 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 143.5, 128.4, 127.4, 126.2, 79.6, 56.4, 23.9.
4-Methoxycyclohexyl Acetate (23)24
Acetic anhydride (350 μL, 3.7 mmol, 2.0 equiv) was added to a solution of compound 48 (237 mg, 1.8 mmol, 1.0 equiv, mixture of cis and trans) in pyridine (3 mL). The mixture was stirred at 20 °C for 20.5 h and subsequently diluted with ethyl acetate (50 mL); washed with 1 M HCl (2 × 20 mL), saturated aqueous sodium bicarbonate (2 × 20 mL), and brine (20 mL); and then dried over magnesium sulfate and the solvent was removed in vacuo to afford ester 23 (240 mg, 77%, mixture of cis and trans) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 4.82 (tt, J = 7.1, 3.5 Hz, 0.7 H), 4.78–4.70 (m, 0.3 H), 3.34 (s, 0.9 H), 3.33 (s, 2.1 H), 3.29 (tt, J = 6.6, 3.3 Hz, 0.7 H), 3.25–3.18 (m, 0.3 H), 2.04 (s, 2.1 H), 2.03 (s, 0.9 H), 2.02–1.93 (m, 1.2 H), 1.86–1.70 (m, 2.8 H), 1.70–1.56 (m, 2.8 H), 1.46–1.37 (m, 1.2 H); 13C NMR (101 MHz, CDCl3) δ 170.7, 170.6, 77.3, 75.8, 71.8, 70.6, 55.9, 55.6, 28.3, 28.3, 27.1, 27.1, 21.4, 21.4.
8-Methoxy-1,4-dioxaspiro[4.5]decane (24)25,26
Magnesium sulfate (361 mg, 3.0 mmol, 2.0 equiv) was added to a solution of ketone 25 (192 mg, 1.5 mmol, 1.0 equiv), ethylene glycol (0.3 mL, 5.3 mmol, 3.5 equiv) and a catalytic amount of p-toluenesulfonic acid (26 mg, 0.15 mmol, 10 mol%) in toluene (5 mL). The resulting mixture was refluxed for 4 h. The reaction was cooled to 20 °C and quenched with saturated aqueous sodium bicarbonate (10 mL). The product was extracted with diethyl ether (3 × 10 mL) and the combined ethereal extracts were successively washed with brine (10 mL), dried over sodium sulfate, and the solvent was removed in vacuo to obtain ketal 24 (219 mg, 85%) as a colorless transparent oil. NMR (400 MHz, CDCl3) δ 3.98–3.90 (m, 4 H), 3.33 (s, 3 H), 3.32–3.27 (m, 1 H), 1.88–1.65 (m, 6 H), 1.59–1.50 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 108.5, 76.2, 64.3, 64.3, 55.8, 31.3, 28.2.
4-Methoxycyclohexan-1-one (25)27
A solution of alcohol 48 (520 mg, 4.0 mmol, 1.0 equiv) in dichloromethane (5 mL) was added slowly poured into a solution of PCC (1.7 g, 8.0 mmol, 2.0 equiv) in dichloromethane (10 mL). The resulting orange mixture was stirred for 3.5 h at 20 °C while the color changed from orange to brown. Upon completion of the reaction, the mixture was filtered through Celite, concentrated, and purified with silica gel column chromatography (ethyl acetate/heptane 1:1) to afford ketone 25 (416 mg, 81%) as a yellow transparent oil. 1H NMR (400 MHz, CDCl3) δ 3.62 (tt, J = 5.7, 3.0 Hz, 1 H), 3.41 (s, 3 H), 2.62–2.51 (m, 3 H), 2.31–2.22 (m, 2 H), 2.15–2.05 (m, 2 H), 1.99–1.89 (m, 1 H); 13C NMR (101 MHz, CDCl3) δ 211.2, 74.2, 56.1, 37.1, 30.1.
tert-Butyl 4-methoxypiperidine-1-carboxylate (26)28
Boc anhydride (982 mg, 4.5 mmol, 1.5 equiv) and palladium on carbon (750 mg, 10 wt% palladium) were added to a solution of compound 27 (748 mg, 3.0 mmol, 1.0 equiv) in ethanol (20 mL). The flask was placed under a hydrogen atmosphere and the mixture was stirred at 20 °C for 2.5 h. Upon completion, the mixture was filtered through Celite and the filtrate was concentrated in vacuo. Purification of the residue by silica gel column chromatography (methanol/dichloromethane 1:49 → 1:9) gave compound 26 (537 mg, 83%) as a yellow transparent liquid. 1H NMR (400 MHz, CDCl3) δ 3.83–3.69 (m, 2 H), 3.41–3.30 (m, 1 H), 3.35 (s, 3 H), 3.22 (ddd, J = 13.2, 9.2, 3.5 Hz, 2 H), 1.92–1.76 (m, 2 H), 1.56–1.40 (m, 2 H), 1.46 (s, 9 H); 13C NMR (101 MHz, CDCl3) δ 154.8, 79.4, 76.0, 55.6, 41.2, 30.6, 28.4; HRMS (ESI+) calcd for (C11H21NO3 + Na)+ 238.1419, found 238.1413.
Benzyl 4-Methoxypiperidine-1-carboxylate (27)
Sodium borohydride (189 mg, 5.0 mmol, 1.0 equiv) was added to a solution of ketone 40 (1.17 g, 5.0 mmol, 1.0 equiv) in methanol (20 mL) and the reaction mixture was stirred for 40 min. Upon completion, most methanol was evaporated under reduced pressure and the residue was diluted with water (15 mL) and 0.1 M HCl was added to adjust the pH to 7. The product was extracted with ethyl acetate (3 × 10 mL) and the combined organic extracts were washed with brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford the secondary alcohol (1.19 g, 99%) as a sticky colorless oil, slightly contaminated with ethyl acetate. 1H NMR (400 MHz, CDCl3) δ 7.41–7.27 (m, 5 H), 5.13 (s, 2 H), 4.01–3.81 (m, 3 H), 3.15 (ddd, J = 13.3, 9.5, 3.5 Hz, 2 H), 1.95–1.76 (m, 2 H), 1.57–1.40 (m, 2 H). Sodium hydride (240 mg, 6.0 mmol, 1.2 equiv, 60% dispersion in mineral oil) was added at 0 °C to a solution of the aforementioned secondary alcohol (1.19 g, 5.0 mmol, 1.0 equiv) in THF (10 mL). After 25 min of stirring, methyl iodide (375 μL, 6.0 mmol, 1.2 equiv) was added. Then, the mixture was allowed to warm to 20 °C and it was stirred for another 18.5 h. The reaction was quenched with aqueous sodium thiosulfate (5% in water, 15 mL) and the product was extracted with ethyl acetate (3 × 15 mL). The combined organic extract were washed with brine (10 mL), dried over magnesium sulfate, and concentrated. Silica gel column chromatography (ethyl acetate/heptane 1:2) of the residue gave ether 27 (1.15 g, 89%) as yellow transparent liquid. 1H NMR (400 MHz, CDCl3) δ 7.41–7.28 (m, 5 H), 5.13 (s, 2 H), 3.88–3.73 (m, 2 H), 3.42–3.34 (m, 1 H), 3.35 (s, 3 H), 3.22 (ddd, J = 13.3, 8.8, 3.6 Hz, 2 H), 1.93–1.75 (m, 2 H), 1.60–1.45 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 155.3, 136.9, 128.5, 127.9, 127.8, 75.6, 67.1, 55.7, 41.2. HRMS (ESI+) calcd for (C14H19NO3 + Na)+ 272.1263, found 272.1250.
4-Methoxy-1-(4-nitrobenzenesulfonyl)piperidine (28)29
A solution of compound 26 (249 mg, 1.16 mmol, 1.05 equiv) in TFA/dichloromethane (1:1, 25 mL) was stirred at 20 °C for 40 min. The solvent was evaporated and the residue was dissolved in dichloromethane (12 mL) and at 0 °C treated with triethylamine (383 μL, 2.75 mmol, 2.5 equiv) and p-nosyl chloride (244 mg, 1.1 mmol, 1.0 equiv). After stirring at 20 °C for 21.5 h, the reaction was quenched with saturated aqueous sodium bicarbonate (12 mL). The product was extracted with dichloromethane (3 × 25 mL) and the combined organic extracts were washed with brine (25 mL), dried over magnesium sulfate and the solvent was removed in vacuo to obtain an orange solid. The crude mixture was dissolved in dichloromethane (20 mL) and successively washed with 0.1 M HCl (20 mL), saturated aqueous sodium bicarbonate (20 mL) and brine (20 mL); then dried over magnesium sulfate and the solvent was removed in vacuo to give sulfonamide 28 (238 mg, 72%) as a pale orange solid. 1H NMR (400 MHz, CDCl3) δ 8.41–8.35 (m, 2 H), 7.97–7.92 (m, 2 H), 3.34 (tt, J = 6.3, 3.2 Hz, 1 H), 3.25 (s, 3 H), 3.20–3.06 (m, 4 H), 1.93–1.83 (m, 2 H), 1.83–1.72 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 150.1, 142.6, 128.7, 124.3, 73.2, 55.8, 42.7, 29.5. MS (EI+) calcd for (C12H16N2O5S – CH3OH)+ 268.052, found 268.079; HRMS (FD+) calcd for (C12H16N2O5S)+ 300.0780, found 300.0783.
4-Oxopentyl Benzoate (29)30
According to the general procedure, compound 16 (222 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. After stirring for 26 h, additional calcium hypochlorite (44 mg, 0.2 mmol, 0.4 equiv) was added at 0 °C and stirring was continued for another 20 h before the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford ketone 29 (202 mg, 98%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.06–8.00 (m, 2 H), 7.60–7.53 (m, 1 H), 7.48–7.41 (m, 2 H), 4.34 (t, J = 6.4 Hz, 2 H), 2.61 (t, J = 7.2 Hz, 2 H), 2.18 (s, 3 H), 2.07 (tt, J = 7.2, 6.4 Hz, 2 H); 13C NMR (101 MHz, CDCl3) δ 207.7, 166.5, 133.0, 130.2, 129.5, 128.4, 64.1, 40.0, 30.0, 22.9.
5-{[tert-Butyl(dimethyl)silyl]oxy}pentan-2-one (30)18
According to the general procedure, 17 (232 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. The reaction mixture was stirred for 23 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo. The residue was purified with silica gel column chromatography (ethyl acetate/pentane 1:5) to afford 30 (148 mg, 68%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.1 Hz, 2 H), 2.51 (t, J = 7.3 Hz, 2 H), 2.15 (s, 3 H), 1.78 (tt, J = 7.2, 6.1 Hz, 2 H), 0.89 (s, 9 H), 0.04 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 208.9, 62.1, 40.1, 30.0, 26.9, 25.9, 18.3, – 5.4.
5-(2-Methoxyethoxy)pentan-2-one (31)
According to the general procedure, methyl ether 18 (88 mg, 0.5 mmol, 1.0 equiv) was dissolved in acetonitrile (1 mL), water (1 mL) and acetic acid (0.1 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (88 mg, 0.4 mmol, 1.6 equiv) was added portion wise over 3 h. After stirring for 23 h, additional calcium hypochlorite (22 mg, 0.1 mmol, 0.4 equiv) was added at 0 °C and stirring was continued for another 24 h before the reaction was quenched with aqueous sodium thiosulfate (10% in water, 2.5 mL). The product was extracted with dichloromethane (3 × 5 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (5 mL) and brine (5 mL),dried over magnesium sulfate, and the solvent was removed in vacuo to afford ketone 31 (70 mg, 87%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.58–3.50 (m, 4 H), 3.48 (t, J = 6.2 Hz, 2 H), 3.38 (s, 3 H), 2.54 (t, J = 7.2 Hz, 2 H), 2.15 (s, 3 H), 1.87 (tt, J = 7.2, 6.2 Hz, 2 H); 13C NMR (101 MHz, CDCl3) δ 208.7, 71.9, 70.3, 70.0, 59.1, 40.3, 30.0, 23.7; HRMS (ESI+) calcd for (C8H16O3 + Na)+ 183.0997, found 183.0980.
2-(4-Oxopentyl)isoindoline-1,3-dione (32)31
According to the general procedure, phthalimide 19 (247 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL) and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. The reaction mixture was stirred for 22.5 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated. Purification by silica gel column chromatography (ethyl acetate/heptane 1:3) yielded ketone 32 (190 mg, 82%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.87–7.82 (m, 2 H), 7.75–7.69 (m, 2 H), 3.71 (t, J = 6.7 Hz, 2 H), 2.50 (t, J = 7.2 Hz, 2 H), 2.14 (s, 3 H), 1.96 (p, J = 7.0 Hz, 2 H); 13C NMR (101 MHz, CDCl3) δ 207.4, 168.5, 134.0, 132.1, 123.2, 40.6, 37.2, 29.9, 22.7.
5-Oxohexanenitrile (33)32
According to the general procedure, nitrile 20 (127 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 0.8 equiv) was added portion wise over 3 h. The reaction mixture was stirred for 27.5 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated. Purification by silica gel column chromatography (ethyl acetate/heptane 1:3) separately gave starting material 20 (22 mg, 17%) and ketone 33 (48 mg, 43%) as an off-white liquid. 1H NMR (400 MHz, CDCl3) δ 2.65 (t, J = 6.8 Hz, 2 H), 2.44 (t, J = 7.0 Hz, 2 H), 2.19 (s, 3 H), 1.92 (p, J = 6.9 Hz, 2 H).
4-[2-(2,4-Dinitrophenyl)hydrazinylidene]-1-(piperidin-1-yl)pentan-1-one (34a)12
According to the general procedure, compound 21 (199 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. Stirring was continued for 46 h, while additional calcium hypochlorite (44 mg, 0.2 mmol, 0.4 equiv) was added after 24 h. Finally, the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (5 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated to give a transparent yellow oil (116 mg) that contained both starting material 21 and ketone 34. Separation of these two compounds by means of silica gel column chromatography was not achieved so another separation method was used. Treatment of the crude mixture with Brady’s reagent (2,4-dinitrophenyl hydrazine in ethanol and sulfuric acid) transformed the ketone into the corresponding 2,4-dinitrophenyl hydrazone whereas the methyl ether remained intact. The solution was poured into diethyl ether (75 mL). The layers were separated and the ethereal extract was washed with water (25 mL) and concentrated to give a red precipitate. The red precipitate was redissolved in diethyl ether (10 mL) and successively washed with 0.1 M HCl (3 × 10 mL), dried over magnesium sulfate, and concentrated. The residue was purified with silica gel column chromatography (ethyl acetate/heptane 1:2 → 1:0) to give hydrazone 34a (59 mg, 16%, 8:1 mixture of E and Z, ratio based on the 1H NMR signals between 12 and 11 ppm) as an orange solid. E-isomer: 1H NMR (400 MHz, CDCl3) δ 11.06 (s, 1 H), 9.13 (d, J = 2.5 Hz, 1 H), 8.27 (ddd, J = 9.6, 2.6, 0.7 Hz, 1 H), 7.89 (d, J = 9.6 Hz, 1 H), 3.61–3.53 (m, 2 H), 3.53–3.47 (m, 2 H), 2.81 (t, J = 6.4 Hz, 2 H), 2.72 (t, J = 6.4 Hz, 2 H), 2.13 (s, 3 H), 1.74–1.60 (m, 4 H), 1.60–1.50 (m, 2 H); 13C NMR (400 MHz, CDCl3) δ 169.7, 157.4, 145.2, 137.6, 129.8, 129.0, 123.6, 116.2, 46.5, 42.9, 33.8, 28.8, 26.6, 25.6, 24.6, 16.9. Z-isomer: Signals could not be identified due to overlap with the signals of the E-isomer. HRMS (ESI+) calcd for (C16H21N5O5 + Na)+ 386.1440, found 386.1437.
Acetophenone (35)
According to the general procedure, compound 22 (136 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. The reaction was stirred for 26 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The products were extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford a yellow oil (103 mg), that contained the product. The crude NMR spectrum revealed that a mixture of six compounds was obtained: (1-methoxyethyl)benzene (22), 1-chloro-2-(1-methoxyethyl)benzene, 1-chloro-4-(1-methoxyethyl)benzene, acetophenone (35), 1-(2-chloro-phenyl)ethan-1-one and 1-(4-chlorophenyl)ethan-1-one. The ratio 35:side products was roughly 1:1. Attempts to separate the product by silica gel column chromatography (pentane → dichloromethane) failed.
4-Oxocyclohexyl Acetate (36)33
According to the general procedure, ester 23 (172 mg, 1.0 mmol, 1.0 equiv, mixture of cis and trans) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. The reaction mixture was stirred for 47 h before it was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated. Purification by silica gel column chromatography (pentane → methanol/dichloromethane 1:19) gave ketone 36 (126 mg, 81%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 5.17 (p, J = 4.9 Hz, 1 H), 2.61–2.49 (m, 2 H), 2.42–2.32 (m, 2 H), 2.11 (s, 3 H), 2.13–2.03 (m, 4 H); 13C NMR (101 MHz, CDCl3) δ 209.8, 170.4, 68.6, 37.3, 30.4, 21.3.
1,4-Dioxaspiro[4.5]decan-8-one (37)34
According to the general procedure, ketal 24 (172 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 0.8 equiv) was added portion wise over 3 h. After 20 h of stirring TLC analysis indicated that, besides the starting material and the desired product, a third compound was being produced. Therefore, the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The products were extracted with dichloromethane (5 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated. Purification by silica gel column chromatography (ethyl acetate/heptane 1:3) separately gave starting material 24 (64 mg, 37%) and ketone 37 (76 mg, 48%) as a white solid. The third compound, cyclohexane-1,4-dione (trace amounts), was detected in the crude mixture, but not isolated. 1H NMR (400 MHz, CDCl3) δ 4.03 (s, 4 H), 2.55–2.48 (m, 4 H), 2.06–1.97 (m, 4 H); 13C NMR (101 MHz, CDCl3) δ 210.4, 107.1, 64.7, 38.2, 33.9.
Cyclohexane-1,4-dione (38)35
According to the general procedure, compound 25 (128 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 0.8 equiv) was added portion wise over 3 h. Stirring was continued for 116 h, while additional calcium hypochlorite (308 mg, 1.4 mmol, 1.4 equiv) was added in portions after 19, 27, and 51 h. Finally, the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The product was extracted with dichloromethane (5 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and concentrated. Purification by silica gel column chromatography (ethyl acetate/heptane 1:1) separately gave starting material 25 (38 mg, 30%) and diketone 38 (22 mg, 20%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 2.72 (s, 8 H).
tert-Butyl 4-oxopiperidine-1-carboxylate (39)36
According to the general procedure, compound 26 (215 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. Stirring was continued for 119 h, while additional calcium hypochlorite (2 × 44 mg, 2 × 0.2 mmol, 0.8 equiv) was added after 23 and 29 h. When TLC indicated that side products became abundant the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The products were extracted with dichloromethane (5 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford a yellow mixture (32 mg), that predominantly contained the product. Gray crystals formed on the adapter of the rotary evaporator were compound 39 (6 mg, 3%). Most material lost the Boc group and therefore remained in the aqueous phases. 1H NMR (400 MHz, CDCl3) δ 3.75–3.68 (m, 4 H), 2.48–2.41 (m, 4 H), 1.50 (s, 9 H).
Benzyl 4-Oxopiperidine-1-carboxylate (40)
According to the general procedure, compound 27 (249 mg, 1.0 mmol, 1.0 equiv) was dissolved in acetonitrile (2 mL), water (2 mL), and acetic acid (0.2 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (176 mg, 0.8 mmol, 1.6 equiv) was added portion wise over 3 h. Stirring was continued for 70 h, while additional calcium hypochlorite (308 mg, 1.4 mmol, 2.8 equiv) was added after 23 (0.4 equiv), 47 (0.4 equiv) and 51 h (2.0 equiv). NMR analysis of an aliquot elucidated that besides the starting material and the product multiple side products had been formed so the reaction was quenched with aqueous sodium thiosulfate (10% in water, 5 mL). The products were extracted with dichloromethane (3 × 10 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (10 mL) and brine (10 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford a yellow mixture (250 mg) that contained the product. The crude NMR spectrum revealed that a mixture of six compounds was obtained: benzyl 4-methoxypiperidine-1-carboxylate (27), 2-chlorobenzyl 4-methoxy-piperidine-1-carboxylate, 4-chlorobenzyl 4-methoxypiperidine-1-carboxylate, the desired product (40), 2-chlorobenzyl 4-oxopiperidine-1-carboxylate and 4-chlorobenzyl 4-oxopiperidine-1-carboxylate. The ratio 40:side products was roughly 1:1. Separation was not achieved.
1-(4-Nitrobenzenesulfonyl)piperidin-4-one (41)
According to the general procedure, sulfonamide 28 (150 mg, 0.5 mmol, 1.0 equiv) was dissolved in acetonitrile (1 mL), water (1 mL), and acetic acid (0.1 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (88 mg, 0.4 mmol, 1.6 equiv) was added portion wise over 3 h. Stirring was continued for 72 h, while additional calcium hypochlorite (22 mg, 0.1 mmol, 0.4 equiv) was added after 22 h. According to TLC, the reaction was clean and proceeded well, whereas NMR analysis elucidated that the actual conversion after 72 h was only 10–15%. The reaction was quenched with aqueous sodium thiosulfate (10% in water, 2.5 mL). The products were extracted with dichloromethane (3 × 5 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (5 mL) and brine (5 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford a white-orange solid (144 mg), that exclusively contained starting material and the product. The crude mixture was submitted to the same reaction conditions: it was dissolved in acetonitrile (2 mL), water (1 mL), and acetic acid (0.1 mL, 3.5 equiv). The solution was cooled to 0 °C and calcium hypochlorite (88 mg, 0.4 mmol, 1.6 equiv) was added portion wise over 3 h. Stirring was continued for 7 days, while additional calcium hypochlorite (242 mg, 1.1 mmol, 4.4 equiv) was added after 22 h (0.2 equiv), 2 days (1.0 equiv), and 3 days (1.0 equiv). Apparently, the reaction did not proceed any further than 20% conversion. Eventually, the reaction was quenched with aqueous sodium thiosulfate (10% in water, 2.5 mL). The products were extracted with dichloromethane (3 × 5 mL), the combined organic extracts were subsequently washed with saturated aqueous sodium bicarbonate (5 mL) and brine (5 mL), dried over magnesium sulfate, and the solvent was removed in vacuo to afford a white-orange solid (139 mg). Attempts to separate product 41 via silica gel column chromatography failed. Analytical data could be acquired from the crude 1H NMR spectrum. 1H NMR (400 MHz, CDCl3) δ 8.43–8.39 (m, 2 H), 8.02–7.99 (m, 2 H), 3.48 (t, J = 6.3 Hz, 4 H), 2.59 (t, J = 6.3 Hz, 4 H).
4-Methoxypentan-1-ol (43)37
Compound 17 (2.32 g, 10 mmol, 1.0 equiv) was dissolved in THF (20 mL) and triethylamine trihydrofluoride (1.63 mL, 10 mmol, 1.0 equiv) was added. The mixture was stirred for 27.5 h at 20 °C before it was concentrated at the rotary evaporator. The residue was purified with silica gel column chromatography (ethyl acetate/heptane 1:1) to afford 43 (1.063 g, 90%) as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.0 Hz, 2 H), 3.41–3.32 (m, 1 H), 3.34 (s, 3 H), 2.09 (bs, 1 H), 1.74–1.54 (m, 4 H), 1.16 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 76.8, 63.0, 56.0, 33.2, 28.8, 18.8; MS (EI+) calcd for (C6H14O2 – CH3)+ 103.076, found 103.089.
4-Methoxypentyl Methanesulfonate (44)
Methanesulfonyl chloride (0.5 mL, 6.4 mmol, 1.3 equiv) and triethylamine (1.0 mL, 7.2 mmol, 1.4 equiv) were successively added at 0 °C to a solution of alcohol 43 (591 mg, 5.0 mmol, 1.0 equiv) in dichloromethane (50 mL). The reaction mixture was stirred for 1 h and then allowed to warm to 20 °C. The mixture was poured into water (50 mL) and the product was extracted with dichloromethane (3 × 25 mL). The combined organic extracts were dried over magnesium sulfate and the solvent was removed in vacuo to afford sulfonate 44 (987 mg, 99%) as a yellow oil. Compound 44 was immediately used in the next steps without further purification. 1H NMR (400 MHz, CDCl3) δ 4.31–4.20 (m, 2 H), 3.38–3.30 (m, 1 H), 3.31 (s, 3 H), 3.01 (s, 3 H), 1.94–1.74 (m, 2 H), 1.61–1.53 (m, 2 H), 1.15 (d, J = 6.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 76.0, 70.2, 56.0, 37.4, 32.2, 25.3, 18.9.
4-Methoxycyclohexan-1-ol (48)38
Sodium hydride (1.0 g, 25 mmol, 1.0 equiv, 60% dispersion in mineral oil) was added at 0 °C to a solution of cyclohexane-1,4-diol (47, 2.9 g, 25 mmol, 1.0 equiv, mixture of cis and trans) in THF (50 mL). The mixture was stirred for 30 min and then methyl iodide (1.9 mL, 30 mmol, 1.2 equiv) was added. NMR analysis showed an actual conversion of only 10% after stirring for 18 h. Therefore, extra sodium hydride (0.5 g, 12.5 mmol, 0.5 equiv, 60% dispersion in mineral oil) and methyl iodide (1.55 mL, 25 mmol, 1.0 equiv) were added. Stirring was continued for 60 h and the reaction was quenched by the careful addition of water (40 mL). The product was extracted with ethyl acetate (3 × 50 mL), and the combined organic extracts were washed with aqueous sodium thiosulfate (10%, 50 mL) and brine (50 mL), dried over magnesium sulfate, and concentrated. Purification with silica gel column chromatography (ethyl acetate/heptane 1:1) separately gave 1,4-dimethoxycyclohexane (375 mg, 10%, mixture of cis and trans) and 48 (580 mg, 18%, mixture of cis and trans) as a clear transparent liquid. 1H NMR (400 MHz, CDCl3) δ 3.74 (p, J = 5.8 Hz, 0.7 H), 3.71–3.64 (m, 0.3 H), 3.34 (s, 0.9 H), 3.32 (s, 2.1 H), 3.28 (tt, J = 6.1, 3.1 Hz, 0.7 H), 3.21–3.15 (m, 0.3 H), 2.07–1.94 (m, 1.2 H), 1.89–1.78 (m, 1.4 H), 1.69–1.62 (m, 2.8 H), 1.59–1.50 (m, 1.4 H), 1.36–1.28 (m, 1.2 H).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b00632.
Substrate synthesis schemes and copies of 1H and 13C NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Reactivities, Reagents, and Reactivity Charts. In Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wuts P. G. M., Greene T. W., Eds.; John Wiley & Sons, Inc., 2006; pp 986–1051. [Google Scholar]
- McOmie J. F. W.; Watts M. L.; West D. E. Tetrahedron 1968, 24, 2289–2292. 10.1016/0040-4020(68)88130-X. [DOI] [Google Scholar]
- Kende A. S.; Rizzi J. P. Tetrahedron Lett. 1981, 22, 1779–1782. 10.1016/S0040-4039(01)90437-X. [DOI] [Google Scholar]
- Harrison I. T. J. Chem. Soc. D, Chem. Commun. 1969, 616. 10.1039/C2969000616A. [DOI] [Google Scholar]
- Bhatt M. V.; Kulkarni S. U. Synthesis 1983, 1983, 249–282. 10.1055/s-1983-30301. [DOI] [Google Scholar]
- Olah G. A.; Welch J. J. Am. Chem. Soc. 1978, 100, 5396–5402. 10.1021/ja00485a024. [DOI] [Google Scholar]
- Rozen S.; Dayan S.; Bareket Y. J. Org. Chem. 1995, 60, 8267–8269. 10.1021/jo00130a029. [DOI] [Google Scholar]
- Kamijo S.; Matsumura S.; Inoue M. Org. Lett. 2010, 12, 4195–4197. 10.1021/ol1018079. [DOI] [PubMed] [Google Scholar]
- Sasidharan M.; Bhaumik A. J. Mol. Catal. A: Chem. 2011, 338, 105–110. 10.1016/j.molcata.2011.02.003. [DOI] [Google Scholar]
- Pradhan P. P.; Bobbitt J. M.; Bailey W. F.; et al. J. Org. Chem. 2009, 74, 9524–9527. 10.1021/jo902144b. [DOI] [PubMed] [Google Scholar]
- Kelly C. B.; Ovian J. M.; Cywar R. M.; Gosselin T. R.; Wiles R. J.; Leadbeater N. E. Org. Biomol. Chem. 2015, 13, 4255–4259. 10.1039/C5OB00270B. [DOI] [PubMed] [Google Scholar]
- Mayhoub A. S.; Talukdar A.; Cushman M. J. Org. Chem. 2010, 75, 3507–3510. 10.1021/jo1004313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaukwa S. O.; Keehn P. M. Tetrahedron Lett. 1982, 23, 35–38. 10.1016/S0040-4039(00)97525-7. [DOI] [Google Scholar]
- Behforouz M.; Bolan J. L.; Flynt M. S. J. Org. Chem. 1985, 50, 1186–1189. 10.1021/jo00208a009. [DOI] [Google Scholar]
- Fujioka H.; Yahata K.; Hamada T.; Kubo O.; Okitsu T.; Sawama Y.; Ohnaka T.; Maegawa T.; Kita Y. Chem. - Asian J. 2012, 7, 367–373. 10.1002/asia.201100812. [DOI] [PubMed] [Google Scholar]
- Liu C.; Achtenhagen M.; Szostak M. Org. Lett. 2016, 18, 2375–2378. 10.1021/acs.orglett.6b00842. [DOI] [PubMed] [Google Scholar]
- Guo S.; Zhang X.; Tang P. Angew. Chem., Int. Ed. 2015, 54, 4065–4069. 10.1002/anie.201411807. [DOI] [PubMed] [Google Scholar]
- Singh N.; Pulukuri K. K.; Chakraborty T. K. Tetrahedron 2015, 71, 4608–4615. 10.1016/j.tet.2015.05.026. [DOI] [Google Scholar]
- Schering Cooperation. US2008/45568A1, 2008.
- Nimbus Iris. WO2012/97013A1, 2012.
- Fujioka H.; Yamanaka T.; Takuma K.; Miyazaki M.; Kita Y. J. Chem. Soc., Chem. Commun. 1991, 533–534. 10.1039/C39910000533. [DOI] [Google Scholar]
- Sugimura H.; Sato S.; Tokudome K.; Yamada T. Org. Lett. 2014, 16, 3384–3387. 10.1021/ol501446w. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Kim I. S.; Li Q. R.; Dong G. R.; Jeong L. S.; Jung Y. H. J. Org. Chem. 2011, 76, 10011–10019. 10.1021/jo201794k. [DOI] [PubMed] [Google Scholar]
- Zefirov N. S.; Samoshin V. V.; Kurbanova V. A.; Lutsenko A. I.; Yartseva I. V.; Mursakulov I. G. Russ. J. Org. Chem. 1991, 27, 2456–2457. [Google Scholar]
- Srikrishna A.; Viswajanani R. Tetrahedron 1995, 51, 3339–3344. 10.1016/0040-4020(95)00055-D. [DOI] [Google Scholar]
- Dibble D. J.; Ziller J. W.; Woerpel K. A. J. Org. Chem. 2011, 76, 7706–7719. 10.1021/jo200950s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M. M.; Clouthier C. M. J. Org. Chem. 2006, 71, 8424–8430. 10.1021/jo061349t. [DOI] [PubMed] [Google Scholar]
- Bajwa J. S. Tetrahedron Lett. 1992, 33, 2955–2956. 10.1016/S0040-4039(00)79570-0. [DOI] [Google Scholar]
- Blanco-Ania D.; Gawade S. A.; Zwinkels L. J. L.; Maartense L.; Bolster M. G.; Benningshof J. C. J.; Rutjes F. P. J. T. Org. Process Res. Dev. 2016, 20, 409–413. 10.1021/acs.oprd.5b00354. [DOI] [Google Scholar]
- Linclau B.; Wang Z.; Compain G.; Paumelle V.; Fontenelle C. Q.; Wells N.; Weymouth-Wilson A. Angew. Chem., Int. Ed. 2016, 55, 674–678. 10.1002/anie.201509460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantet A.-C.; Carreyre H.; Gesson J.-P.; Jouannetaud M.-P.; Renoux B. J. Org. Chem. 2008, 73, 2875–2878. 10.1021/jo702441p. [DOI] [PubMed] [Google Scholar]
- Fischer D. F.; Sarpong R. J. Am. Chem. Soc. 2010, 132, 5926–5927. 10.1021/ja101893b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinpeter E.; Heydenreich M.; Koch A.; Linker T. Tetrahedron 2012, 68, 2363–2373. 10.1016/j.tet.2012.01.022. [DOI] [Google Scholar]
- Ren K.; Zhao M.; Hu B.; Lu B.; Xie X.; Ratovelomanana-Vidal V.; Zhang Z. J. Org. Chem. 2015, 80, 12572–12579. 10.1021/acs.joc.5b02519. [DOI] [PubMed] [Google Scholar]
- Chambers R. D.; Hutchinson J.; Sandford G.; Shah A.; Vaughan J. F. S. Tetrahedron 1997, 53, 15833–15842. 10.1016/S0040-4020(97)10043-6. [DOI] [Google Scholar]
- Moreno-Mañas M.; Pérez M.; Pleixats R. Tetrahedron 1994, 50, 515–528. 10.1016/S0040-4020(01)80773-0. [DOI] [Google Scholar]
- Biernacki W.; Gdula A. Polym. J. Chem. 1981, 55, 1063–1067. [Google Scholar]
- Bhatia S.; Spahlinger G.; Boukhumseen N.; Boll Q.; Li Z.; Jackson J. E. Eur. J. Org. Chem. 2016, 2016, 4230–4235. 10.1002/ejoc.201600719. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.