

Abstract
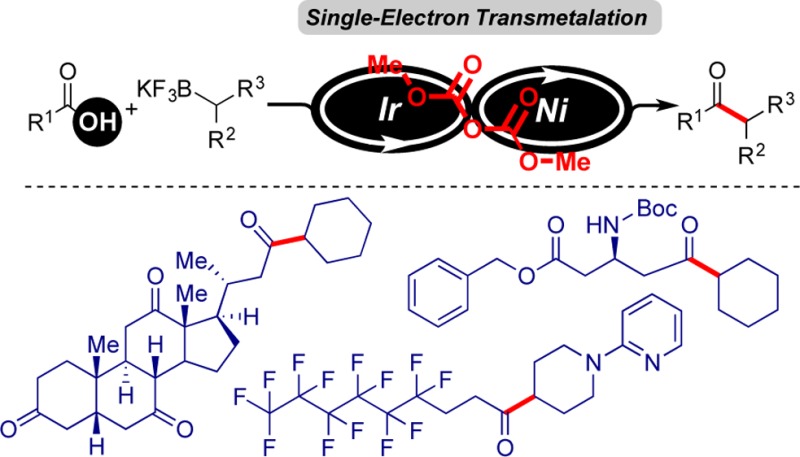
An efficient and mild method for acyl–Csp3 bond formation based on the direct conversion of carboxylic acids has been established. This protocol is enabled by the synergistic, Ir-photoredox/nickel catalytic cross-coupling of in situ activated carboxylic acids and alkyltrifluoroborates. This versatile method is amenable to the cross-coupling of structurally diverse carboxylic acids with various potassium alkyltrifluoroborates, affording the corresponding ketones with high yields. In this operationally simple cross-coupling protocol, aliphatic ketones are obtained in one step from bench stable, readily available carboxylic acids.
The direct conversion of carboxylic acids to alkyl ketones represents a fundamental transformation of importance, but one that has yet to reach its enormous potential. Carboxylic acids can be converted directly to alkyl ketones by treatment with excess organolithium compounds and Grignard reagents,1 but such protocols severely limit functional group incorporation in both partners. Greater toleration of sensitive functional groups can derive from reaction of stoichiometric alkylmetallics (e.g., R-[M], M = Cu,2 Mn,3 Zn,4 Zr,5 B6) with activated carboxylic acid derivatives such as acyl halides, anhydrides, thioesters, or amides. Further enhancements can stem from transition metal-catalyzed protocols, which overcome many limitations associated with traditional approaches to aliphatic ketones.7 In particular, palladium- and nickel-catalyzed cross-couplings of carboxylic acid derivatives such as acyl chlorides, anhydrides, activated esters, thioesters, and amides have been developed for the synthesis of alkyl ketones.8 However, only a few protocols have been reported for the highly desirable, direct conversion of carboxylic acids in transition-metal-catalyzed synthesis of ketones. Thus, although a number of elegant protocols, including in situ activation of carboxylic acids, have been disclosed by Gooßen,9 Yamamoto,10 and Zou,11 they are largely limited to the synthesis of aromatic ketones. Reductive coupling of in situ activated carboxylic acids provides another means to generate aliphatic ketones, but the employment of superstoichiometric amounts of Zn or Mn as the reductant and, in some cases, the use of three to four equivalent of carboxylic acid are distinct disadvantages of these methods.12
Recent efforts have demonstrated that photoredox/transition metal dual catalysis is a valuable mechanistic protocol that ameliorates chemical transformations that suffer from limitations in traditional cross-coupling paradigms.13 In these approaches, a novel concept for transmetalation based on a single-electron transfer (SET) pathway is applied that lowers the high activation energy barrier associated with the transmetalation step in the coupling of Csp3-hybridized nucleophiles.14 In this context and to broaden the range of electrophiles to which the metallaphotoredox cross-coupling can be applied, our laboratory has recently developed a visible light-enabled acylation of alkyltrifluoroborates with both carboxylic acid chlorides15 and amides16 to access aliphatic ketones. The MacMillan17 and Doyle18 groups previously employed related, but mechanistically and operationally distinct, strategies for the synthesis of ketones using anhydrides. Those protocols employed mixed anhydrides, formed in situ from carboxylic acids and acyl chlorides, and symmetrical anhydrides, respectively, with both methods being focused on the synthesis of α-amino ketones.
A general, robust method for the direct synthesis of alkyl ketones from carboxylic acids via Csp3–acyl bond construction under mild conditions and without the limitations of the more traditional two-electron protocols would be highly valued. For realization of this idea and to expand the scope of the photoredox/Ni dual catalysis cross-coupling strategy, we investigated the potential of the coupling of carboxylic acids with alkyltrifluoroborates via in situ activation of the carboxylic acids (Figure 1).
Figure 1.

Photoredox/Ni dual catalysis cross-coupling of carboxylic acids with alkyltrifluoroborates.
Initial investigations to explore the feasibility of the proposed transformation began with the coupling of hydrocinnamic acid with cyclohexyltrifluoroborate as a model reaction in the presence of a variety of activating reagents (see Supporting Information, Figure S1). The investigations revealed the feasibility of the cross-coupling reaction by using dimethyl dicarbonate (DMDC) as the activator. It was satisfying to see that DMDC, which is a commodity chemical used as a beverage preservative, would serve as a nearly ideal activating agent that generates an activated carbonic anhydride in situ. Upon reaction, the byproducts of the subsequent acylation include gaseous CO2 and two equivalents of methanol, neither of which interferes with the separation of the produced ketones.9dFigure 2 shows the proposed mechanistic cycle for this acyl-sp3 coupling protocol adapted from our studies on cross-coupling of potassium alkyltrifluoroborates with aryl halides.19
Figure 2.
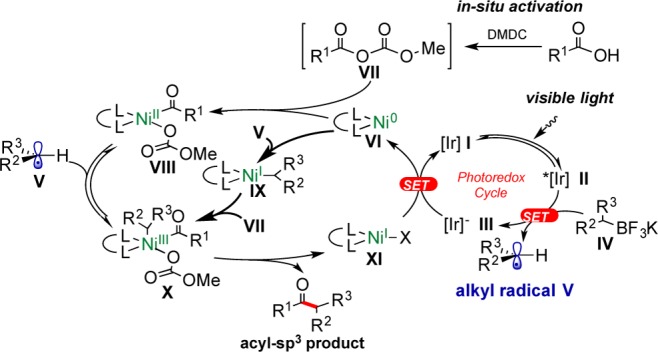
Proposed mechanism for the acyl-sp3 cross-coupling.
Further investigations showed that the presence of any type of base decreased the yield of the reaction (Figures S1 and S2). As reported by the MacMillan group,20 the main reason for the lowering of the yield of the reaction under basic conditions is likely the single electron oxidation of the carboxylate anion derived from the deprotonation of the acid to form carboxyl radicals. The resulting carboxyl radicals undergo CO2 extrusion, producing the corresponding alkyl radicals that generate side products. To avoid this phenomenon in the protocol developed herein, weak proton donor additives were examined (Figure S5). Interestingly, the use of NH4Cl as an additive afforded higher yields.
Screening of various reaction parameters (e.g., photocatalyst, solvent, Ni catalyst, and ligand) via microscale high-throughput experimentation21 to enhance the yields and improve the robustness of this transformation revealed that the best conditions involved treatment of the carboxylic acid with potassium alkyltrifluoroborate in the presence of 3 mol % of photocatalyst 1 and 6 mol % of [Ni(dtbbpy)(H2O)4]Cl2, 3.0 equiv of DMDC, and 1.0 equiv of NH4Cl in i-PrOAc/THF (2:1) as a solvent mixture at room temperature. As expected, control experiments showed that all parameters were essential for the reaction to proceed, and the Ir complex 1 was the best photocatalyst for this cross-coupling reaction (Table S1).
Subsequently, the generality of the cross-coupling reaction with respect to the alkyltrifluoroborate component was examined. As illustrated in Scheme 1, a variety of alkyltrifluoroborates were acylated under mild conditions with good to excellent yields. The aliphatic, heterocyclic trifluoroborates containing Boc- and Cbz-protected nitrogen atoms (2i–k) and nitrogenous substrates bearing Lewis basic moieties (2h) reacted efficiently under the reaction conditions to provide the desired aliphatic ketones with good yields (65%–89%). Also, N-methylsulfonamides such as N-methylthiophene-2-sulfonamide (2m) were well tolerated under the reaction conditions (77% yield). Notably, α-aminoalkyltrifluoroborates can be incorporated with high efficiency in the protocol to generate α-amino substituted ketones, widespread in pharmaceuticals and nature (2j, 2l, and 2m).22 α-Alkoxymethyltrifluoroborates underwent the coupling in excellent yields (2f and 2g, 86%–90%). Therefore, this new protocol provides a strategy for direct access to α-alkoxy ketones as valuable and versatile building blocks in construction of various natural compounds by the direct use of carboxylic acids.23 Furthermore, the acylation of 2-methylcyclopentyltrifluoroborate generated the trans-diastereomer as the only product (2b, 69%). We found that β-trifluoroboratoketones and -esters were successfully coupled as well to generate 1,4-dicarbonyl adducts (2c and 2d), which are useful precursors to generate substituted pyrroles, furans, and thiophenes by the Paal–Knorr synthesis.24 The scalable nature of the coupling was evaluated by performing the reaction on a 7.0 mmol scale with 1.5 mol % of photocatalyst 1 and 3.0 mol % of [Ni(dtbbpy)(H2O)4]Cl2, affording product 2a in 70% yield (Table S1).
Scheme 1. Scope of Potassium Alkyltrifluoroborates in Cross-Coupling with Carboxylic Acids.

See Supporting Information for experimental details. All cited yields are isolated.
Reaction performed on 7.0 mmol scale with 1.5 mol % of Ir photocatalyst 1 and 3.0 mol % of [Ni(dtbbpy)(H2O)4]Cl2.
Next, we focused our attention on an examination of the scope of the carboxylic acid partner. As illustrated in Scheme 1, a broad range of carboxylic acids readily participated in this acylative coupling reaction with great efficiency. Many different functional groups could be accommodated under the reaction conditions. N-Boc-protected amino acids exhibited excellent reactivity in this coupling protocol to afford the corresponding ketones in high yields (3b–c, 3n, and 3r, 81%–87%). Carboxylic acids containing reactive functional groups such as esters (3g and 3r, 78% and 87% yield, respectively) and nitriles (3m, 70% yield), which are susceptible to side reactions in the presence of strong, nucleophilic organometallic reagents, participated in the reaction as well. Somewhat surprisingly, a carboxylic acid possessing a hydroxyl group such as 6-hydroxycaproic acid proved to be a useful coupling partner (3j, 86% yield). Moreover, this protocol could be applied to carboxylic acids containing aliphatic halides to produce adducts that could be used for further elaboration. The acylation of 3-bromoadamantane-1-acetic acid generated the corresponding aliphatic ketone with 88% yield (3i). The reaction was insensitive to steric hindrance. The carboxylic acids bearing bulky groups, 1-adamantanecarboxylic acid (3h) and 3-oxotricyclo[2.2.1.02,6]heptane-7-carboxylic acid (3s), thus provided the corresponding ketones with 70% and 83% yield, respectively. The cross-coupling reaction of oleic acid with 2-(4-(trifluoroborato)piperidin-1-yl)pyridine generated ketone 3t with 57% yield. The power of this method is further demonstrated by the preparation of ketone 3p from the acylation of a synthetic bile acid, dehydrocholic acid, with cyclohexyltrifluoroborate in 60% isolated yield.
In conclusion, we have established a robust strategy for the formation of C(O)–Csp3 bonds from carboxylic acids and potassium alkyltrifluoroborates. This new platform for acyl–sp3 bond construction is enabled by the synergistic Ir-photoredox/nickel catalytic cross-coupling of in situ activated carboxylic acids using alkyltrifluoroborates as the coupling partners through low-barrier, open-shell processes. This new protocol is amenable to the efficient coupling of a wide array of both carboxylic acids and alkyltrifluoroborates under mild conditions at room temperature.
Acknowledgments
This research was supported by the NIGMS (R01 GM-113878). Frontier Scientific is thanked for the donation of boron compounds used in this investigation. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
Supporting Information Available
This material is available free of charge on the ACS Publications Web site at DOI: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b01588.
Experimental procedures, HTE data, compound characterization data, and NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Huston R. C.; Bailey D. L. J. Am. Chem. Soc. 1946, 68, 1382. 10.1021/ja01211a504. [DOI] [Google Scholar]; b House H. O.; Bare T. M. J. Org. Chem. 1968, 33, 943. 10.1021/jo01267a003. [DOI] [Google Scholar]; c Suga K.; Watanabe S.; Fujita T.; Takahashi Y. Aust. J. Chem. 1973, 26, 2123. 10.1071/CH9732123. [DOI] [Google Scholar]; d Levine R.; Karten M. J.; Kadunce W. M. J. Org. Chem. 1975, 40, 1770. 10.1021/jo00900a020. [DOI] [Google Scholar]
- a Dubois J. E.; Boussu M.; Lion C. Tetrahedron Lett. 1971, 12, 829. 10.1016/S0040-4039(01)96567-0. [DOI] [Google Scholar]; b Dieter R. K.; Sharma R. R.; Yu H.; Gore V. K. Tetrahedron 2003, 59, 1083. 10.1016/S0040-4020(02)01526-0. [DOI] [Google Scholar]
- a Cahiez G.; Laboue B. Tetrahedron Lett. 1989, 30, 7369. 10.1016/S0040-4039(00)70699-X. [DOI] [Google Scholar]; b Cahiez G.; Laboue B. Tetrahedron Lett. 1992, 33, 4439. 10.1016/S0040-4039(00)60104-1. [DOI] [Google Scholar]; c Cahiez G.; Razafintsalama L.; Laboue B.; Chau F. Tetrahedron Lett. 1998, 39, 849. 10.1016/S0040-4039(97)10747-X. [DOI] [Google Scholar]
- a Blaise E. E.; Maire M. Compt. Rend. 1907, 145, 73. [Google Scholar]; b Blaise E. E.; Koehler A. Bull. Soc. Chim. Paris 1910, 7, 215. [Google Scholar]; c Blaise E. E. Bull. Soc. Chim. 1911, 9, 1. [Google Scholar]; d Reddy C. K.; Knochel P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1700. 10.1002/anie.199617001. [DOI] [Google Scholar]
- Hart D. W.; Schwartz J. J. Am. Chem. Soc. 1974, 96, 8115. 10.1021/ja00833a048. [DOI] [Google Scholar]
- a Negishi E.; Chiu K.-W.; Yosida T. J. Org. Chem. 1975, 40, 1676. 10.1021/jo00899a046. [DOI] [Google Scholar]; b Negishi E.; Abramovitch A.; Merrill R. E. J. Chem. Soc., Chem. Commun. 1975, 138. 10.1039/c39750000138. [DOI] [Google Scholar]
- a Larock R. C.Comprehensive Organic Transformations: A Guide to Functional Group Preparations; VCH: New York, 1989; pp 685–702. [Google Scholar]; b Dieter R. K. Tetrahedron 1999, 55, 4177. 10.1016/S0040-4020(99)00184-2. [DOI] [Google Scholar]; c Rubottom G. M.; Kim C. J. Org. Chem. 1983, 48, 1550. 10.1021/jo00157a038. [DOI] [Google Scholar]; d Ahn Y.; Cohen T. Tetrahedron Lett. 1994, 35, 203. 10.1016/S0040-4039(00)76511-7. [DOI] [Google Scholar]
- a Wu F.; Lu W.; Qian Q.; Ren Q.; Gong H. Org. Lett. 2012, 14, 3044. 10.1021/ol3011198. [DOI] [PubMed] [Google Scholar]; b Wotal A. C.; Weix D. J. Org. Lett. 2012, 14, 1476. 10.1021/ol300217x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Urawa Y.; Ogura K. Tetrahedron Lett. 2003, 44, 271. 10.1016/S0040-4039(02)02501-7. [DOI] [Google Scholar]; d Haddach M.; McCarthy J. R. Tetrahedron Lett. 1999, 40, 3109. 10.1016/S0040-4039(99)00476-1. [DOI] [Google Scholar]; e Kakino R.; Yasumi S.; Shimizu I.; Yamamoto A. Bull. Chem. Soc. Jpn. 2002, 75, 137. 10.1246/bcsj.75.137. [DOI] [Google Scholar]; f Kakino R.; Shimizu I.; Yamamoto A. Bull. Chem. Soc. Jpn. 2001, 74, 371. 10.1246/bcsj.74.371. [DOI] [Google Scholar]; g Tatamidani H.; Kakiuchi F.; Chatani N. Org. Lett. 2004, 6, 3597. 10.1021/ol048513o. [DOI] [PubMed] [Google Scholar]; h Liebeskind L. S.; Srogl J. J. Am. Chem. Soc. 2000, 122, 11260. 10.1021/ja005613q. [DOI] [Google Scholar]; i Weires N. A.; Baker E. L.; Garg N. K. Nat. Chem. 2016, 8, 75. 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]; j Meng G.; Szostak M. Org. Lett. 2015, 17, 4364. 10.1021/acs.orglett.5b02209. [DOI] [PubMed] [Google Scholar]; k Lei P.; Meng G.; Szostak M. ACS Catal. 2017, 7, 1960. 10.1021/acscatal.6b03616. [DOI] [Google Scholar]; l Meng G.; Szostak M. Org. Biomol. Chem. 2016, 14, 5690. 10.1039/C6OB00084C. [DOI] [PubMed] [Google Scholar]; m Dander J. E.; Garg N. K. ACS Catal. 2017, 7, 1413. 10.1021/acscatal.6b03277. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Liu C.; Liu Y.; Liu R.; Lalancette R.; Szostak M. Org. Lett. 2017, 19, 1434. 10.1021/acs.orglett.7b00373. [DOI] [PubMed] [Google Scholar]; o Meng G.; Shi S.; Szostak M. ACS Catal. 2016, 6, 7335. 10.1021/acscatal.6b02323. [DOI] [Google Scholar]; p Onaka M.; Matsuoka Y.; Mukaiyama T. Chem. Lett. 1981, 10, 531. 10.1246/cl.1981.531. [DOI] [Google Scholar]
- a Gooßen L. J.; Ghosh K. Angew. Chem., Int. Ed. 2001, 40, 3458.. [DOI] [PubMed] [Google Scholar]; b Gooßen L. J.; Ghosh K. Eur. J. Org. Chem. 2002, 2002, 3254.. [DOI] [Google Scholar]; c Gooßen L. J.; Ghosh K. Chem. Commun. 2001, 2084. 10.1039/b106779f. [DOI] [PubMed] [Google Scholar]; d Gooßen L. J.; Winkel L.; Doehring A.; Ghosh K.; Paetzold J. Synlett 2002, 1237. [Google Scholar]
- a Kakino R.; Narahashi H.; Shimizu I.; Yamamoto A. Chem. Lett. 2001, 30, 1242. 10.1246/cl.2001.1242. [DOI] [Google Scholar]; b Kakino R.; Narahashi H.; Shimizu I.; Yamamoto A. Bull. Chem. Soc. Jpn. 2002, 75, 1333. 10.1246/bcsj.75.1333. [DOI] [Google Scholar]
- Si S.; Wang C.; Zhang N.; Zou G. J. Org. Chem. 2016, 81, 4364. 10.1021/acs.joc.6b00421. [DOI] [PubMed] [Google Scholar]
- a Yin H.; Zhao C.; You H.; Lin Q.; Gong H. Chem. Commun. 2012, 48, 7034. 10.1039/c2cc33232a. [DOI] [PubMed] [Google Scholar]; b Zhao C.; Jia X.; Wang X.; Gong H. J. Am. Chem. Soc. 2014, 136, 17645. 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shu X.-Z.; Zhang M.; He Y.; Frei H.; Toste F. D. J. Am. Chem. Soc. 2014, 136, 5844. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tellis J. C.; Amani J.; Molander G. A. Org. Lett. 2016, 18, 2994. 10.1021/acs.orglett.6b01357. [DOI] [PubMed] [Google Scholar]
- a Amani J.; Sodagar E.; Molander G. A. Org. Lett. 2016, 18, 732. 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Amani J.; Molander G. A. J. Org. Chem. 2017, 82, 1856. 10.1021/acs.joc.6b02897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amani J.; Alam R.; Badir S.; Molander G. A. Org. Lett. 2017, 19, 2426. 10.1021/acs.orglett.7b00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le C. C.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 11938. 10.1021/jacs.5b08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe C. L.; Doyle A. G. Angew. Chem., Int. Ed. 2016, 55, 4040. 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble A.; McCarver S. J.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 624. 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For more details about High Throughput Experimentations, see Supporting Information; b Schmink J. R.; Bellomo A.; Berritt S. Aldrichimica Acta 2013, 46, 71. [Google Scholar]
- a Meltzer P. C.; Butler D.; Deschamps J. R.; Madras B. K. J. Med. Chem. 2006, 49, 1420. 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bouteiller C.; Becerril-Ortega J.; Marchand P.; Nicole O.; Barre L.; Buisson A.; Perrio C. Org. Biomol. Chem. 2010, 8, 1111. 10.1039/b923255a. [DOI] [PubMed] [Google Scholar]
- a Mori M.; Inouye Y.; Kakisawa H. Chem. Lett. 1989, 18, 1021. 10.1246/cl.1989.1021. [DOI] [Google Scholar]; b Edwards R. L.; Whalley A. J. S. J. Chem. Soc., Perkin Trans. 1 1979, 803. 10.1039/P19790000803. [DOI] [Google Scholar]
- a Paal C. Ber. Dtsch. Chem. Ges. 1884, 17, 2756. 10.1002/cber.188401702228. [DOI] [Google Scholar]; b Knorr L. Ber. Dtsch. Chem. Ges. 1884, 17, 2863. 10.1002/cber.188401702254. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.