

Summary
Human longevity is a complex phenotype influenced by genetic and environmental components. Unraveling the contribution of genetic vs. nongenetic factors to longevity is a challenging task. Here, we conducted a large‐scale RNA‐sequencing‐based expression quantitative trait loci study (eQTL) with subsequent heritability analysis. The investigation was performed on blood samples from 244 individuals from Germany and Denmark, representing various age groups including long‐lived subjects up to the age of 104 years. Our eQTL‐based approach revealed for the first time that human longevity is associated with a depletion of metabolic pathways in a genotype‐dependent and independent manner. Further analyses indicated that 20% of the differentially expressed genes are influenced by genetic variants in cis. The subsequent study of twins showed that the transcriptional activity of a third of the differentially regulated genes is heritable. These findings suggest that longevity‐associated biological processes such as altered metabolism are, to a certain extent, also the driving force of longevity rather than just a consequence of old age.
Keywords: functional genomics, human, longevity, RNA‐ sequencing, transcriptome
Introduction
Human longevity is likely to be influenced by multiple genetic and environmental factors as well as by chance (Martin et al., 2007). In contrast to aging, which is a continuous process occurring in all individuals, longevity is achieved only by a very small proportion of a birth cohort. The genetic component to this rare phenotype has been estimated at ~40% in long‐lived individuals (LLI) who survive beyond 85 years (Murabito et al., 2012). Interestingly, many of the elderly who attain such an extreme age also tend to be healthier for a longer period of their overall lifetime than their peers who died decades earlier. They achieve this extension of ‘healthspan’ by postponing the onset of major age‐related diseases and the beginning of functional decline (Andersen et al., 2012). In addition, LLI often show beneficial profiles for some metabolic parameters such as lipid and lipoprotein particle profiles (Barzilai et al., 2003). On the genetic level, nonagenarians and centenarians have been hypothesized to harbor specific alleles with protective effects, so‐called longevity variants, that may buffer or counteract the numerous disease variants they carry (Bergman et al., 2007; Beekman et al., 2010; Sebastiani et al., 2012). However, so far, variation in only three loci, the APOE gene (Schächter et al., 1994; Rea et al., 2015), the FOXO3A gene in the insulin‐IGF1 pathway (Willcox et al., 2008; Flachsbart et al., 2009; Soerensen et al., 2010), and a region of unknown function on chromosome 5q33.3 (Deelen et al., 2014), has been reported to influence survival beyond 90 years of age in various populations. Many more genes are assumed to play a role in human longevity, but they have remained undetected as yet despite large‐scale genome‐wide efforts (Deelen et al., 2011, 2014; Nebel et al., 2011; Beekman et al., 2013). As the genetic approaches have thus far provided only limited information about the determinants and molecular mechanisms underlying longevity, several studies have focused on the molecular functional aspects of the phenotype by analyzing the miRNA, epigenome, and transcriptome profiles of LLI (Rodwell et al., 2004; Bollati et al., 2009; Harries et al., 2011; ElSharawy et al., 2012; Passtoors et al., 2012). Yet many other studies focused primarily on aging mechanisms rather than on longevity (Van den Akker et al., 2014; Peters et al., 2015). The transcriptome of an individual reflects the influence of both genetic variation and the environment. A majority of transcriptome studies in longevity research use a cross‐sectional design (Rodwell et al., 2004; Harries et al., 2011; Passtoors et al., 2012), in which data of LLI are compared with that of younger participants who were born generations later. However, such a setup is not suitable to distinguish between the changes in gene expression that predispose to longevity and those that result from it (Deelen et al., 2013). To partly overcome this limitation, we performed in this study RNA‐sequencing‐based transcriptome profiling in blood samples of 244 LLI and younger controls and subsequently employed an expression quantitative trait loci (eQTL) analysis to investigate the association between mRNA expression levels and single‐nucleotide polymorphisms (SNPs). As the SNPs involved in the eQTLs are constitutional, they cannot be a consequence of longevity but represent one of its underlying factors. Taken together, integrating genotype information with high‐resolution transcriptome data from this sample does not only help elucidate functional mechanisms associated with longevity, but also provides for the first time the opportunity to assess the genetic contribution to it.
Results
Differential gene expression between LLI and CI
The transcriptome analysis of 55 LLI and 73 CI (CI: control individuals, German samples) identified 19 483 of 24 934 genes analyzed as expressed in blood. Of these, 6214 were significantly differentially expressed (Benjamini‐Hochberg corrected P‐value ≤ 0.001; false discovery rate FDR ≤ 0.1%) between LLI and CI including 3525 downregulated and 2689 upregulated genes in LLI (Fig. 1, Table S1, Supporting information, Appendix S1: Supplemental raw data, Supporting information). Of those 6214 differentially expressed genes, 139 exhibited a high correlation (r ≥ 0.8) with individual blood cell types (Palmer et al., 2006) and were excluded from further analysis to control for altered cell compositions due to age. Subjecting the remaining 6075 differentially expressed genes to a gene ontology analysis resulted in 107 significant biological processes. We found that eight of the ten most significantly enriched or depleted biological processes were functionally linked to metabolism, which is very unlikely to have occurred by chance (P = 3.51 × 10−5; Fischer's exact test). In addition, a larger number of downregulated genes than expected by chance were associated with these eight metabolic processes, which represents the most prominent finding of the study. Most gene ontology categories were relatively broad, for example, such as cellular macromolecule metabolic process, nucleobase‐containing compound metabolic process, heterocycle metabolic process, or cellular aromatic compound metabolic process (Table S2, Supporting information); however, upon closer inspection, many of them seemed to point to nucleotide metabolism. Modulation was also found in processes functionally linked to defense as well as to cell and tissue regeneration, yet to a much lesser extent than in metabolism (Fig. 2).
Figure 1.
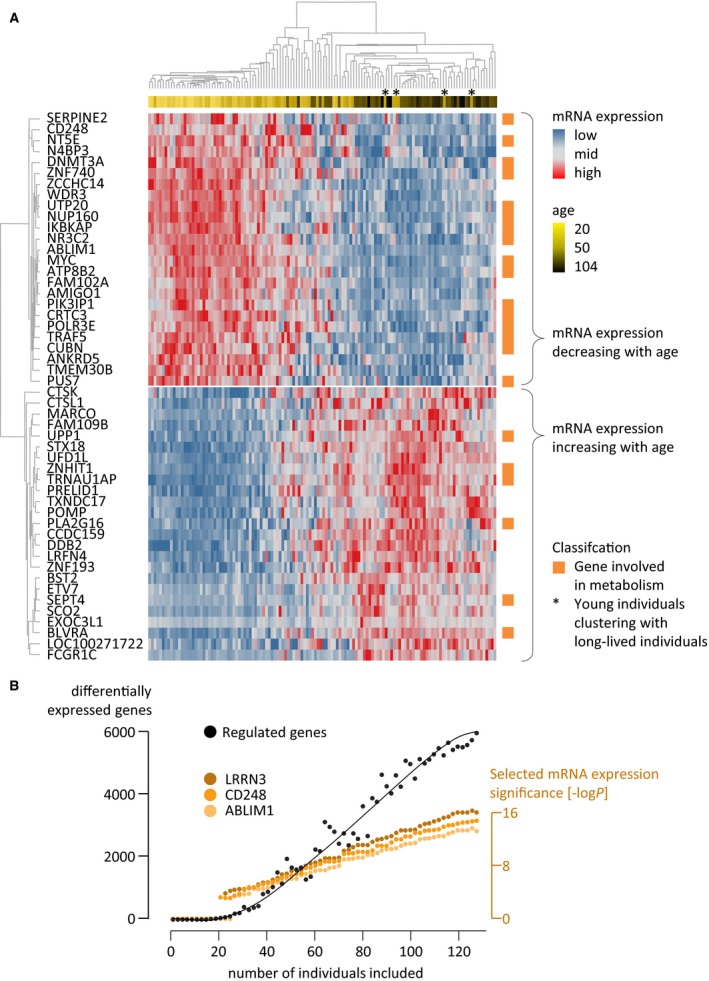
Genes differentially expressed with chronological age. (A) Hierarchical clustering of the top 50 genes, exhibiting the strongest correlation with chronological age. Each column represents an individual, while each row represents a transcript that is labeled with the corresponding gene symbol. Column dendrograms display similarities between samples and row dendrograms display transcript similarities. The orange column on the far right, which classifies genes by their involvement in metabolism, illustrates the downregulation of metabolism‐associated transcripts with age. For the top ten mRNA transcripts with positive or negative correlation with chronological age, please refer to Fig. S4 (Supporting information). (B) Differentially expressed genes identified vs. individuals included in the study setup, exemplified for the German cohort (max. n = 128 individuals). Randomly selected individuals were added to the analysis to estimate the resulting number of significantly differentially expressed genes (adjusted P‐value ≤ 0.001). For better visualization, a curve fit was added. In addition, three previously published genes (LRRN4,CD248, and ABLIM1) and the strongest pathway signal (GO‐term) are displayed with their significances (−log10 P‐value).
Figure 2.
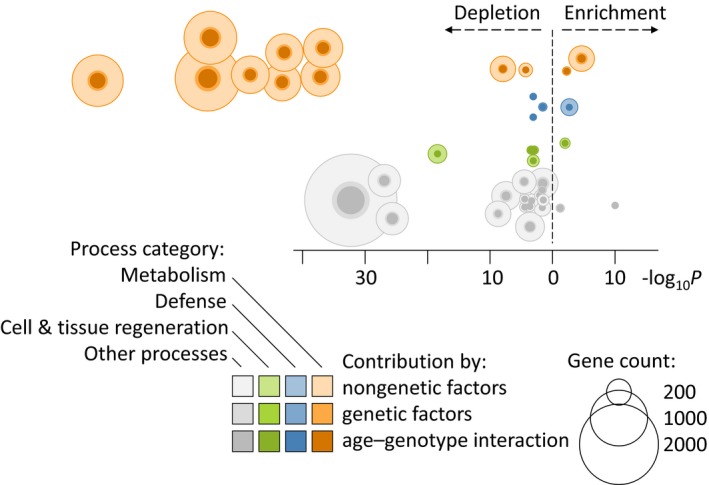
Biological processes in longevity are controlled by genetic and nongenetic factors. For better readability, only processes with at least 150 genes are displayed. A strong depletion of processes associated with metabolism (orange) is the most prominent finding, while the effects on defense (blue) and cell and tissue regeneration (green) are less dominant. Processes that are not part of these three categories are displayed in grey. The contribution of age–genotype interaction (dark shading, inner circle) represents a part of the genetic contribution ( cis ‐ eQTL , middle circle), while the remaining effects (environment, epigenetics etc.) are labeled nongenetic contribution (light shading, outer circle). The y ‐axis is arranged by process category, while the x ‐axis illustrates the degree of enrichment or depletion of the individual processes (−log 10 P). The ten depleted metabolism‐associated processes are (orange, top left section, from left to right): cellular macromolecule metabolic process, regulation of metabolic process, macromolecule metabolic process, nucleobase‐containing compound metabolic process, cellular aromatic compound metabolic process, heterocycle metabolic process, organic cyclic compound metabolic process, cellular nitrogen compound metabolic process, protein metabolic process, and phosphorus metabolic process.
Comparing the P‐values of differentially expressed genes with the findings from five previous aging transcriptomics studies yielded Spearman rank correlation coefficients from 0.44 to 0.72. The overlap in the direction of the fold change of each gene ranged from 65% to 100% per gene (Table 2, Fig. S1, Supporting information). One specific gene, namely LRRN3 (Leucine Rich Repeat Neuronal 3), was identified to be the most significantly regulated gene by all studies including the present one. LRRN3 was consistently found to be downregulated in older and/or LLI.
Table 2.
Overlap with previous studies in aging/longevity transcriptomics
| Hong | Harries | Passtoors | Akker | Peters | |
|---|---|---|---|---|---|
| Study year | 2008 | 2011 | 2012 | 2014 | 2015 |
| Overlap | 100% | 100% | 98% | 65% | 73% |
| Overlap P‐value | 3.2 × 10−14 | 3.8 × 10−51 | 6.9 × 10−154 | <1.0 × 10−300 a | <1.0 × 10−300 a |
| SRCC | 0.73 | 0.62 | 0.60 | 0.44 | 0.61 |
| Shared genes in top 10 |
FCGBP LRRN3b NRCAM PDGFRB |
ABLIM1 CCR7 CD248 FAM102A LRRN3b NELL2 |
ABLIM1 CAMK4 CD248 LRRN3b NOG |
LRRN3b
NELL2 |
ABLIM1 CD248 FAM102A LRRN3b NELL2 |
The following publications were used to generate this table: Hong (Hong et al., 2008), Harries (Harries et al., 2011), Passtoors (Passtoors et al., 2012), Akker (Van den Akker et al., 2014), and Peters (Peters et al., 2015). The overlap was calculated based on how many identified genes were significantly regulated and concordant in their regulation direction. Spearman rank correlation coefficient (SRCC) was calculated using all genes identified in the corresponding study. Shared genes in top 10 describes which genes were found in both studies to be among the top 10 most significantly regulated transcripts, ranked by false discovery rate (Van den Akker) or by P‐value (all others).
Fisher's exact test P‐value could not be calculated (P < 1.0 × 10−300).
LRRN3 (Leucine Rich Repeat Neuronal 3) was identified as the most significantly regulated gene by all studies including the present one.
eQTL and genotype–age (G×A) interaction analysis of the differentially expressed genes
After quality control, 636 904 SNPs were used for the eQTL analysis in the German sample. The cis‐eQTL analysis identified 8757 statistically significant cis‐eQTLs associated with 1200 differentially expressed genes (Fig. 3, Table S3, Supporting information). A subsequent G×A analysis showed that 599 of these 1200 genes displayed an additional G×A interaction (Table S4, Supporting information). An exemplary gene exhibiting G×A interaction is shown in Fig. 3A and an example for a gene with no GxA and no significant eQTL finding is shown in Fig. 3B. The analysis of the frequency of the variants revealed that the majority (25th to 50th percentile) of significant eQTL signals are found in 20–50% of all individuals (Fig. 3C).
Figure 3.
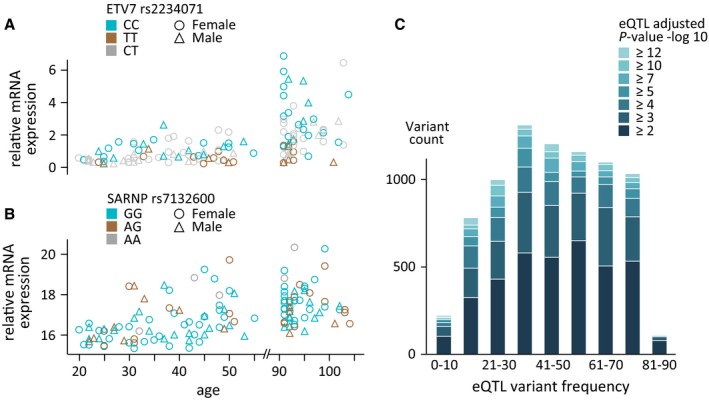
Age‐associated transcripts and influence of genetic variation. One under genetic control (A, ETV 7) and one independent of genetic control (B, SARNP ), while ETV 7 shows an additional age–genotype interaction. Frequencies of eQTL variants (C) are shown color‐coded with the corresponding P ‐value (‐log 10 P).
Integrating the identified biological processes with the transcriptome and genotype data results in a complex hierarchical organization. To illustrate this architecture, the complexity was reduced by including only biological processes that contain 150 or more eQTL genes. We found that 5119 variants influence 722 genes (eQTLs), further contributing to 40 biological processes that can be grouped into four major biological categories, all of them affecting the longevity phenotype (Fig. 4).
Figure 4.
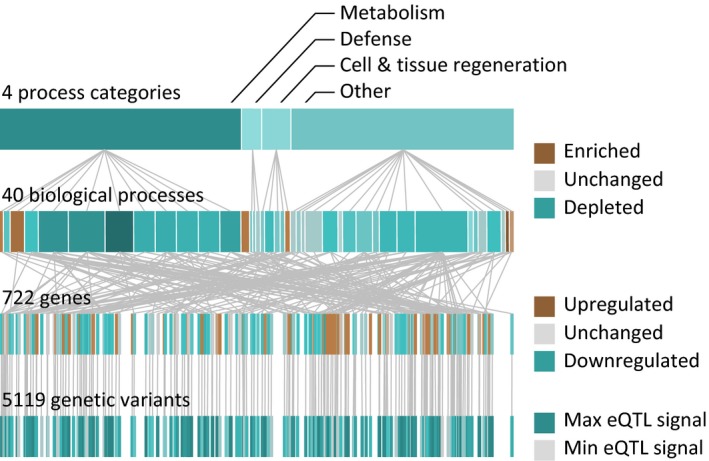
Hierarchical organization of eQTL effects on longevity. Four different layers are represented with their hierarchical connections: 5119 variants influencing the longevity‐associated mRNA expression of 722 genes, contributing to 40 biological processes, which are grouped in four categories. The graph is based on biological processes that contain at least 150 genes significantly associated with longevity (corresponding to Fig. 2). For better readability, the number of displayed connections between two hierarchical levels was limited to a maximum of 200. The y ‐axis corresponds to the hierarchical level, while the x ‐axis represents the genomic location of genes and variants.
Finally, comparing our eQTL findings with previously published genetic variants associated with age‐related diseases (GWAS catalog), we did not observe an enrichment of these variants in our set of identified eQTLs, keeping in mind that our experimental setup may not be suitable to capture the enormous genetic variability of this trait.
Technical validation of selected differentially expressed genes
We replicated our results from the differential expression analysis by measuring the expression levels of 80 candidates (selected based on fold change, significance of differential expression, and eQTL P‐value, Table S6, Supporting information) employing RT‐qPCR in the German samples (53 LLI and 70 CI; five samples were excluded due to lack of RNA). Of the 80 genes, 74 were detectable, of which 50 were significantly differentially expressed between LLI and CI. Moreover, these 50 genes were regulated in the same direction as in the transcriptome sequencing data. A permutation analysis revealed that by chance, one would have expected 0.042 of the 80 genes to be differentially expressed; therefore, finding 50 differentially expressed genes corresponds to a high validation rate, as reflected by a low probability of detecting 50 genes by chance (Fisher's exact test P‐value= 7.57 × 10−17).
Biological validation of differentially expressed genes and cis‐eQTLs
The 6075 differentially expressed genes in Germans were subjected to further validation in the Danish samples (34 older individuals [83–92 years], 34 younger individuals [58–60 years]). Of these genes, 5183 were regulated in the same direction as in the German dataset, with 1661 of those showed a Benjamini‐Hochberg corrected P‐value ≤ 0.05 and a FDR ≤ 5%. This concordance is significantly higher than expected by chance (Fisher's exact test, expected overlap vs. observed overlap; P‐value = 2.4 × 10−13). A gene ontology analysis of these 1661 genes found that nine of the 25 most significant biological processes were functionally linked to metabolism, which is in concordance with the findings in the German samples.
We further correlated the transcriptome and genotype data from the 68 Danish samples to validate the cis‐eQTLs found in the German dataset. Of the 1200 genes, which exhibited a significant association with cis‐variants in the German sample, 374 genes showed significant cis association in the Danish dataset as well. In total, we found 3332 statistically significant cis‐eQTLs associated with these 374 genes, of which 2017 were unique to the Danish dataset.
Heritability estimation of gene expression
Of the 24 934 genes described in the annotation file (hg19), 15 640 were expressed in more than 95% of the blood samples of monozygotic (MZ) and dizygotic (DZ) twins from Denmark (Appendix S2, Supporting information, raw FPKM‐values for all detected genes: Supplemental raw data, Danish samples). Of these, 3029 genes are controlled by a heritable component, which ranges from approximately 30% to 99% (Table S5, Supporting information). Combining this with the above‐mentioned eQTL results (German dataset), an overlap of 334 genes was found: These genes were differentially expressed and associated with a genetic variant in cis, while at the same time also exhibited heritable transcriptional activity. By chance, one would have expected 182 genes to overlap. The observed 1.8‐fold enrichment was highly significant (Fisher's exact test, P = 1.90 × 10−36).
Discussion
Understanding the nature of human longevity is a challenging task, as this heterogeneous phenotype is influenced by both genetic and environmental components. Conservative genetic approaches in longevity research have resulted so far in only very few associations that were consistently replicated in various samples (Schächter et al., 1994; Willcox et al., 2008; Flachsbart et al., 2009; Nebel et al., 2011; Deelen et al., 2014). The majority of genetic findings could not be verified independently (Christensen et al., 2006), suggesting that a large number of different variants may lead to the same phenotypic outcome and rendering it difficult to identify susceptibility factors shared by several populations. To overcome the limitation of a purely genetic study design, some investigations focused on the transcriptome of LLI that reflects both environmental and genetic influences. In the present study, this approach was expanded by integrating, for the first time in longevity research, RNA‐sequencing data from blood samples of 244 LLI and younger controls with genotype information from the same individuals, followed by a heritability analysis in twins. Employing this setup, the study's objective was to identify biological processes associated with longevity and to assess the genetic and nongenetic contribution to those.
In contrast to previous studies, a substantial higher number of genes were observed to be differentially expressed. This observation may be mostly attributed to the high dynamic range of the applied RNA‐sequencing technology (Zhao et al., 2014). The top candidates identified here (LRRN3, P = 5.40 × 10−20; ABLIM1, P = 2.50 × 10−11; CD248, P = 1.50 × 10−23, see Table S1, Supporting information) were in agreement with previously published findings (Hong et al., 2008; Harries et al., 2011; Passtoors et al., 2012; Van den Akker et al., 2014; Peters et al., 2015), which confirms the validity of our setup (Table 2, Fig. S1, Supporting information). The overlap between previous findings with the results presented here is illustrated in Fig. S2 (Supporting information). The top three genes, prominently identified by all previous studies, are shown with their cross‐sectional mRNA expression in Fig. S3 (Supporting information, ABLIM1, CD248, LRRN3). An earlier blood‐based eQTL meta‐study (Westra et al., 2013), despite its large sample size, is not suitable to display longevity effects, as the samples were composed of continuous age‐ranges, excluding extremely long‐lived individuals. Consequently, the previously observed effects reflect mostly aging, while the results presented here are likely to be a combination of both aging and longevity. To critically assess the power of our analysis, we conducted a permutation‐based power estimation, illustrating that the study setup selected is appropriate to deliver significant results and therefore appropriate to support our conclusions (Fig. 1B). Similarly, the successful technical validation of selected genes by quantitative real‐time PCR documents that RNA‐sequencing is a precise method for identifying differentially expressed genes as indicated by the low probability of our results being validated by chance (P‐value = 7.57 × 10−17). In the same context, the biological validation of findings from the German study population in Danes showed a high concordance (P‐value of this overlap not occurring by chance: 2.4 × 10−13).
Quantitative data from whole blood are known to be influenced by altered cell compositions, yet focusing on only one selected cell population in blood might not be sufficient to create a broad picture of longevity‐associated transcriptome patterns, as it is unclear which cell type contributes dominantly to these patterns. Moreover, whole blood has been shown to reflect the age‐related molecular changes seen in other tissues quite well (Passtoors et al., 2008). The presented approach, that is, omitting all genes that show a high correlation with specific blood cell types (Palmer et al., 2006), introduces a degree of robustness, because it removes all signals which are likely to fluctuate as a result of age‐related cell composition changes.
Further analysis revealed that about 20% of the identified differentially expressed genes were influenced by cis‐variants (Table S3, Supporting information), while about half of those exhibited an additional genotype–age interaction: The same genotype exerts a different effect at different ages (Table S4, Supporting information, exemplified in Fig. 3A). Interestingly, we could not find an enrichment of genetic variants associated with age‐related phenotypes such as Parkinson's disease, type II diabetes, or others in our group of cis‐variants.
Monitoring rare or private variants in longevity is not possible with the setup presented here. Consequently, our approach can focus only on shared effects: The majority of eQTL signals occur in 20 to 50% of all individuals (Fig. 3C). This is in concordance with a previous large‐scale eQTL study by the GEUVADIS consortium, employing 462 transcriptomes with corresponding genomes, where the majority of eQTLs identified were shared as well (Lappalainen et al., 2013).
As each gene can potentially interact with all the detected variants (730 525 SNPs) in trans, we categorized all non‐cis interactions as nongenetic: In any given experimental setup, trans‐interactions are almost undetectable, despite their presence and potential relevance. We are aware that categorizing the remaining 80% as nongenetic is likely to be an overestimation due to the contribution of trans‐regulatory effects. Keeping this in mind, the conclusion that 80% of the genes were not under cis‐regulation still supports the notion that longevity is influenced to a large extent by nongenetic factors (Talens et al., 2012).
The genetic influence observed here was further emphasized by the subsequent heritability analysis revealing that only 19% of all genes are influenced by detectable heritability (Table S5, Supporting information). Moreover, the variation in those heritable genes was explained by heritability in a range from 30% to 99%. This highlights that most genes with a heritable component are under additional control of nongenetic factors, while the majority of genes show no heritable influence. Consequently, it appears there is no biological process that is independent of a nongenetic contribution.
Interestingly, about one‐third of the genes that were influenced by cis‐variants also displayed a significant heritable component in an independent set of samples, which is substantially more than expected by chance (enrichment P‐value P = 1.90 × 10−34). In contrast to that, several genetic variants might require additional environmental or genetic effects to display a functional impact on mRNA expression, explaining why a large number of genes that were influenced by cis‐variants did not show a heritable component. Finally, our observations on the heritability on the molecular level are in concordance with previous findings on the phenotype level of familial aggregation of longevity as reviewed in Murabito et al., 2012.
As a primary result of the subsequent gene ontology analysis, we found 107 biological processes associated with longevity, most of which were assigned to one of the three categories: metabolism, tissue and cell regeneration as well as immune system and defense. Within this, the most prominent finding is the significant depletion of processes functionally linked with metabolism (Fig. 2), which dominates by number of genes per process (median: 1103) as well as by significance (min. P = 3.5 × 10−24). Keeping the limitations of pathway‐analysis approaches in mind, the unusually large number of genes associated with the individual metabolic process (1085–1555) further increases our confidence in this finding. Here, the large number of metabolism‐associated processes identified might indicate a key role of this process category in longevity. Modulations in cell and tissue regeneration and in defense processes were significant as well, yet less dominant as the number of genes contributing to these observations was substantially lower. The depletion of metabolic processes resulted mostly from a downregulation of metabolism‐associated genes in both the German and Danish LLI. The downregulation may point to an existing reduced (resting) metabolic rate that in turn has been linked with increased lifespan in humans (Ruggiero et al., 2008; Rozing et al., 2010). Unfortunately, no metabolic rate data were available for our study participants.
Our results support the hypothesis that a subgroup of individuals, exhibiting patterns of reduced metabolism (whether due to genetic and/or environmental factors), is more likely to reach old age in a healthy state. An interesting question that arises in this context is if these patterns in LLI are a consistent feature throughout their lives or are they restricted only to advanced age? The approach applied here using cross‐sectional data does not allow us to address this issue. If the former was the case, the younger individuals that cluster with the LLI based on downregulation of metabolism‐associated genes (highlighted in Fig. 1A) might represent good candidates for people living up to their nineties and beyond.
It is not unlikely that the downregulation of metabolism‐associated genes illustrates the lifestyle of the LLI when compared to younger controls, attributed to moderate food intake and reduced physical activity (Von Wurmb‐Schwark et al., 2010). In this context, it is important to mention the hypothesis of longevity being linked to caloric restriction, which is supported by several prominent observations: For example in Okinawans, caloric restriction and traditional functional food were suggested to play a role in extended lifespan (Willcox & Willcox, 2014). Similarly, caloric restriction has been proposed to have beneficial effects for age‐related outcomes in the CALERIE cohort (Ravussin et al., 2015). While our findings seem to further support this hypothesis, our data do not allow creating a direct link to caloric restriction, as we have demonstrated a depletion of metabolic processes without measuring metabolic rates or caloric intake.
Considering that the majority (~80%) of the observed depletion of metabolic processes is independent of genetics, one could assume that this is indeed the result of longevity. In contrast to this, the genetic contribution (~20%) to this depletion allows a second implication: Individuals with a given genetic setup will have an altered metabolic pattern, resulting in a higher probability of reaching old age. Therefore, our eQTL findings support the concept that longevity is not only a consequence of environmental factors, but also driven by genetics. Moreover, the complexity of the genetic contribution is further increased by the fact that some variants have different impacts at different ages, as demonstrated by our findings on genotype–age interaction (Fig. 3).
It is important to note that the observed depletion of metabolic processes has limited predictive value for other samples: The cross‐sectional study design employed here may overestimate this depletion by reflecting differences between birth cohorts, yet the correlation between altered expression and increasing chronological age (Fig. 3A, Fig. S4, Supporting information) supports a noncohort‐specific effect. This is facilitated by the large age windows covered by our cohorts (see also Fig. S4, Supporting information); our study setup neither allows nor aims at monitoring aging effects but instead is designed to identify characteristics of long‐lived individuals. In contrast to that, an alternative longitudinal setup suffers similar limitations as findings in longitudinal studies potentially have validity only for the birth cohort investigated. Keeping the limitations of the cross‐sectional study design in mind, the integration of stable genomic data with dynamic transcriptome data from different age windows allows us to illustrate the functional consequences of genetic variations. As the SNPs observed in the eQTLs cannot be the result of longevity, this approach offers the unique opportunity to identify potentially causative factors contributing to the phenotype.
Finally, our results might also partially explain why replication of genetic longevity associations is a challenging task. We observed a hierarchical structure, where a large number of heterogeneous heritable and nonheritable, genetic and nongenetic components potentially lead to altered pathway patterns, which finally may result in longevity. The heterogeneity of factors contributing to the same phenotype, as illustrated in Fig. 4, might explain why some genetic associations appear to be valid only in a specific population. Moreover, the independent cohort from Denmark we employed to biologically validate our findings in German individuals displayed the same modulation in metabolic pathways, yet the genes and variants of origin did only partially overlap between Danes and Germans. This provides further support for the concept of a hierarchical structure, where different genetic variations lead to the same phenotype.
Taken together, our systems biology‐based analysis on unique longevity samples illustrates, for the first time, how human longevity is associated with a depletion of metabolic pathways in a genotype‐dependent and independent manner. At the same time, our eQTL findings indicate that those processes are, to some extent, also a driving force of longevity rather than only a consequence of old age.
Experimental procedures
Study populations
This study included a total of 244 whole blood samples from LLI and control individuals (CI) (Table 1). The samples of the German subjects were collected with the support of the biobank PopGen, as previously described (Nebel et al., 2005). The Danish individuals were recruited from two population‐based and nationwide twin surveys conducted at the Danish Twin Registry: the Longitudinal Study of Middle‐aged Danish Twins and the Longitudinal Study of Aging Danish Twins (Skytthe et al., 2013) (Table 1). All participants signed a written informed consent. Approval for the study was received from the Ethics Committees of Kiel University and the Regional Scientific Ethical Committees for Southern Denmark.
Table 1.
Overview of study participants
| Number of individuals | Age range (years) | Gender (f/m) | Country | |
|---|---|---|---|---|
| Long‐lived individuals (LLI) | 55 | 90–104 | 40/15 | GER |
| Control individuals (CI) | 73 | 20–55 | 45/28 | GER |
| Long‐lived twins (LLT) | 48 (28 DZ, 20 MZ) | 83–92 | 32/16 | DK |
| Control twins (CT) | 48 (24 DZ, 24 MZ) | 58–60 | 32/16 | DK |
| Unrelated LLI | 10 | 83–92 | 8/2 | DK |
| Unrelated CI | 10 | 58–60 | 4/6 | DK |
Country: Germany (GER), Denmark (DK).
DZ: dizygotic, MZ: monozygotic.
Sample processing
Total RNA was extracted for all 244 individuals from frozen blood samples using the PAXgene Blood miRNA kit (Qiagen) according to the manufacturer's protocol. Paired‐end libraries were prepared with the Illumina TruSeq RNA Sample Preparation Kit, multiplexed with four samples per lane and sequenced on an Illumina HiSeq 2000.
For the German study population, DNA was extracted from EDTA whole blood using the Invisorb Blood Giga Kit following the manufacturer's instructions. DNA samples from all German individuals (55 LLI and 73 CI) were subjected to genotyping employing the HumanOmniExpress BeadChip (Illumina) that monitors 730 525 SNPs. For the Danish population, DNA was extracted by a salting‐out procedure either performed manually or with the AutoPure LS instrument (Qiagen). One individual from each of the 22 MZ pairs (LLT = 10 pairs and CT = 12 pairs) as well as 52 DZ twin individuals (LLT = 14 pairs and CT = 12 pairs) and 20 unrelated individuals (10 LLI and 10 CI) were selected for genotyping.
Data analysis
The sequencing reads that failed the Illumina chastity filter (chastity threshold = 0.6) were removed with the Illumina CASAVA‐1.8 FASTQ filter v0.1. Subsequent alignment to the UCSC Homo sapiens reference genome (build hg19) was performed with TopHat v2.0.4 (Trapnell et al., 2009). The aligned reads were assembled into transcripts and gene‐level abundance in terms of fragments per kilobase of exon per million fragments mapped (FPKM) was estimated by Cufflinks v2.0.2 (Trapnell et al., 2010). After quantile sample‐to‐sample normalization, the FPKM estimates from the German samples were used to identify the differentially expressed genes. Only genes with a log2 FPKM > 0 in more than 5% of the samples were subjected to further analysis, while differential expression was determined using a nonparametric Wilcoxon rank sum test. Genes with a corrected P‐value ≤ 0.001 (employing Benjamini and Hochberg's correction) and a false discovery rate (FDR) ≤ 0.1% based on a Westfall and Young permutation of the fold changes were considered to be significantly differentially expressed. Differentially expressed genes that highly correlated (r ≥ 0.8) with blood cell counts (Palmer et al., 2006) were omitted from further analysis.
Gene expression data from the Danish twins were used to estimate the heritability of transcriptional activity. Only genes that were expressed (i.e., log2 FPKM > 0) in more than 95% of the samples were subjected to heritability estimation that assessed additive genetic effects. An ACE model (A: additive genetics, C: common environment, E: unique environment) was fitted using biometric modeling (R package mets v0.1‐13) and was allowed to compete with the parsimonious nested models, namely AE, CE, and E. The best fitting model was chosen based on the Akaike information criterion (AIC) (Akaike, 1974) for non‐nested models and the likelihood ratio test for nested models. The final heritability estimate was obtained from the best fitting model.
Genes that were differentially expressed between LLI and CI (German dataset) were subjected to cis‐eQTL analysis. Only variants located within a 1‐Mb up‐ and downstream of the starting and end points of the gene were included in the analysis. SNPs with minor allele frequencies below 1%, call rate below 95%, and Hardy–Weinberg equilibrium testing P‐value ≤ 0.0001 were filtered out. The eQTL analysis for the German samples was performed with PLINK v1.07 (Purcell et al., 2007). The obtained cis‐eQTLs were adjusted using a Benjamini and Hochberg correction for multiple testing at an α level of 5%. Furthermore, the SNPs that exhibited significant association with gene expression (cis‐eQTLs) were compared with genetic variants from GWAS catalog (https://www.ebi.ac.uk/gwas/) to examine whether there is an enrichment of variants associated with age‐related diseases in our identified eQTLs. To assess the frequency of variances per gene, all significant eQTL findings were binned according to their occurrence in the German study population.
To assess the genotype–age interaction (G×A), only differentially expressed genes with at least one significant cis‐eQTL were considered. The G×A interaction for each gene and the most significant cis‐eQTL were investigated by employing a linear mixed model as described previously (Glass et al., 2013).
The overlap with previous studies (Hong et al., 2008; Harries et al., 2011) was assessed by transforming the observed P‐values to −log(P,100), multiplied with the sign of the fold change, to properly reflect the direction of regulation. For one study (Harries et al., 2011), only the false discovery rate data were available which was used equivalently. Subsequently, the transformed P‐values of the genes that were found in both our study and the previous one were subjected to a correlation analysis using the Spearman rank correlation. The expected overlap between studies was based on the number of identified genes in each study, assuming a genomewide approach. The overlap P‐value was calculated employing a Fisher's exact test comparing observed vs. expected overlap.
Functional analysis: gene ontology
Gene ontology analysis was performed by associating genes of interest to gene ontology terms (retrieved from http://www.geneontology.org), followed by a two‐sided Fisher's exact test to determine significance of enrichment or depletion. Finally, resulting P‐values were adjusted employing a Benjamini and Hochberg's correction for multiple testing.
Technical validation of differential gene expression by real‐time PCR
Eighty candidate genes were selected based on the ranks of their fold changes, P‐values, and regulation of their expression by cis‐eQTLs and were subjected to validation in the German samples. Real‐time PCR (TaqMan) was performed according to the manufacturer's instructions (Applied Biosystems) on a 7900HT real‐time PCR system. Gene expression levels were quantified relative to the median of three genes (RER1, E2F4, BFAR) that showed minimum variation in the expression levels across the samples. Differences between LLI and CI were determined with the Wilcoxon rank sum test and the P‐values were corrected using Benjamini and Hochberg's method.
Biological validation of differential gene expression and cis‐eQTLs using Danish samples
In total, 68 individuals were included in the validation set (10 unrelated LLI, 10 unrelated controls, and 48 individuals selected from 48 twin pairs: 22 MZ and 26 DZ – Table 1). The RNA‐sequencing‐based transcriptome data from these 68 samples were analyzed as described above to identify and validate the differentially expressed genes detected in the German dataset. As the validation dataset was smaller than the German sample, the genes with a corrected P‐value ≤ 0.05 and a FDR ≤ 5% were considered to be significantly differentially expressed. The genotype and gene expression data were analyzed using PLINK.
Funding
The Danish Twin Registry is supported by grants from The National Program for Research Infrastructure 2007 (09‐063256) from the Danish Agency for Science Technology and Innovation, the Velux Foundation, and the US National Institute of Health (P01 AG08761). This study was funded by INTERREG 4A Syddanmark‐Schleswig‐K.E.R.N., the Excellence Cluster ‘Inflammation at Interfaces’ (DFG), the PopGen 2.0 network (BMBF, 01EY1103), and by the German National Genome Research Network (NGFN, BMBF).
Author contributions
RH participated in designing the study, supervising the analysis, and writing the manuscript. GV carried out the analysis and contributed to writing the manuscript. QT participated in the analysis and supervised the analysis. FF participated in designing and coordinating the study. PR participated in the sequencing analysis, coordinating the study, and drafting the manuscript. WL contributed to the study design and the analysis. SS, KC, LC, and AN participated in study design, coordination of the study, and drafting and finalizing the manuscript.
Conflict of interest
All authors read and approved the final manuscript and declare no conflict of interest.
Supporting information
Fig. S1 Comparison of findings to previous transcriptomic studies.
Fig. S2 Unique and overlapping features compared to previous studies.
Fig. S3 Cross sectional mRNA regulation of three selected genes.
Fig. S4 mRNA expression of genes correlating with age.
Table S1 The top 25 up‐ and downregulated genes in LLI.
Table S2 All the significantly represented biological processes with respect to the genes upregulated and downregulated in LLI (in the German samples).
Table S3 The top 50 cis‐eQTLs associated with differentially expressed genes.
Table S4 The top 50 G×A interaction effects exhibited by differentially expressed genes.
Table S5 The top 50 genes with heritable transcriptional activity; the best fit model for these genes was AE (A=additive genetic effect, E=unique environmental effect).
Table S6 List of 80 candidate genes and the TaqMan assays used for validation.
Appendix S1 This file contains FPKM (fragments per kilobase of exon per million fragments mapped) values for all German samples.
Appendix S2 This file contains FPKM (fragments per kilobase of exon per million fragments mapped) values for all Danish samples.
Acknowledgments
First, we would like to thank all study participants in Germany and Denmark for enabling us to conduct this study. In addition, we would like to thank Ina Clefsen, Melanie Friskovec, Catharina von der Lancken, Dorina Oelsner, Markus Schilhabel, Melanie Schlapkohl, Ilona Urbach, and Tanja Wesse at the Institute of Clinical Molecular Biology for the excellent technical assistance as well as Gunnar Jacobs for the recruitment via PopGen.
References
- Akaike H (1974) A new look at the statistical model identification. IEEE Trans. Autom. Control 19, 716–723. [Google Scholar]
- Andersen SL, Sebastiani P, Dworkis DA, Feldman L, Perls TT (2012) Health span approximates life span among many supercentenarians: compression of morbidity at the approximate limit of life span. J. Gerontol. A. Biol. Sci. Med. Sci. 67, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Atzmon G, Schechter C, Schaefer EJ, Cupples AL, Lipton R, Cheng S, Shuldiner AR (2003) Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA. 290, 2030–2040. [DOI] [PubMed] [Google Scholar]
- Beekman M, Nederstigt C, Suchiman HED, Kremer D, van der Breggen R, Lakenberg N, Alemayehu WG, de Craen AJM, Westendorp RGJ, Boomsma DI, de Geus EJC, Houwing‐Duistermaat JJ, Heijmans BT, Slagboom PE (2010) Genome‐wide association study (GWAS)‐identified disease risk alleles do not compromise human longevity. Proc. Natl. Acad. Sci. USA 107, 18046–18049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman M, Blanché H, Perola M, Hervonen A, Bezrukov V, Sikora E, Flachsbart F, Christiansen L, De Craen AJM, Kirkwood TBL, Rea IM, Poulain M, Robine JM, Valensin S, Stazi MA, Passarino G, Deiana L, Gonos ES, Paternoster L, Sørensen TIA, Tan Q, Helmer Q, Van Den Akker EB, Deelen J, Martella F, Cordell HJ, Ayers KL, Vaupel JW, Törnwall O, Johnson TE, Schreiber S, Lathrop M, Skytthe A, Westendorp RGJ, Christensen K, Gampe J, Nebel A, Houwing‐Duistermaat JJ, Slagboom PE, Franceschi C (2013) Genome‐wide linkage analysis for human longevity: genetics of healthy aging study. Aging Cell 12, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman A, Atzmon G, Ye K, MacCarthy T, Barzilai N (2007) Buffering mechanisms in aging: a systems approach toward uncovering the genetic component of aging. PLoS Comput. Biol. 3, 1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A (2009) Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 130, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K, Johnson TE, Vaupel JW (2006) The quest for genetic determinants of human longevity: challenges and insights. Nat. Rev. Genet. 7, 436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Uh HW, Helmer Q, Kuningas M, Christiansen L, Kremer D, van der Breggen R, Suchiman HED, Lakenberg N, van den Akker EB, Passtoors WM, Tiemeier H, van Heemst D, de Craen AJ, Rivadeneira F, de Geus EJ, Perola M, van der Ouderaa FJ, Gunn DA, Boomsma DI, Uitterlinden AG, Christensen K, van Duijn CM, Heijmans BT, Houwing‐Duistermaat JJ, Westendorp RGJ, Slagboom PE (2011) Genome‐wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell 10, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Capri M, Franceschi C, Slagboom PE (2013) Identifying the genomic determinants of aging and longevity in human population studies: progress and challenges. BioEssays 35, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Uh H‐W, Broer L, Ayers KL, Tan Q, Kamatani Y, Bennet AM, Tamm R, Trompet S, Guðbjartsson DF, Flachsbart F, Rose G, Viktorin A, Fischer K, Nygaard M, Cordell HJ, Crocco P, van den Akker EB, Böhringer S, Helmer Q, Nelson CP, Saunders GI, Alver M, Andersen‐Ranberg K, Breen ME, van der Breggen R, Caliebe A, Capri M, Cevenini E, Collerton JC, Dato S, Davies K, Ford I, Gampe J, Garagnani P, de Geus EJC, Harrow J, van Heemst D, Heijmans BT, Heinsen F‐A, Hottenga J‐J, Hofman A, Jeune B, Jonsson PV, Lathrop M, Lechner D, Martin‐Ruiz C, McNerlan SE, Mihailov E, Montesanto A, Mooijaart SP, Murphy A, Nohr EA, Paternoster L, Postmus I, Rivadeneira F, Ross OA, Salvioli S, Sattar N, Schreiber S, Stefánsson H, Stott DJ, Tiemeier H, Uitterlinden AG, Westendorp RGJ, Willemsen G, Samani NJ, Galan P, Sørensen TIA, Boomsma DI, Jukema JW, Rea IM, Passarino G, de Craen AJM, Christensen K, Nebel A, Stefánsson K, Metspalu A, Magnusson P, Blanché H, Christiansen L, Kirkwood TBL, van Duijn CM, Franceschi C, Houwing‐Duistermaat JJ, Slagboom PE (2014) Genome‐wide association meta‐analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum. Mol. Genet. 23, 4420–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSharawy A, Keller A, Flachsbart F, Wendschlag A, Jacobs G, Kefer N, Brefort T, Leidinger P, Backes C, Meese E, Schreiber S, Rosenstiel P, Franke A, Nebel A (2012) Genome‐wide miRNA signatures of human longevity. Aging Cell 11, 607–616. [DOI] [PubMed] [Google Scholar]
- Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller‐Eberstein H, Nikolaus S, Schreiber S, Nebel A (2009) Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA 106, 2700–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D, Viñuela A, Davies MN, Ramasamy A, Parts L, Knowles D, Brown AA, Hedman AK, Small KS, Buil A, Grundberg E, Nica AC, Meglio P, Nestle FO, Ryten M, Durbin R, McCarthy MI, Deloukas P, Dermitzakis ET, Weale ME, Bataille V, Spector TD (2013) Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 14, R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harries LW, Hernandez D, Henley W, Wood AR, Holly AC, Bradley‐Smith RM, Yaghootkar H, Dutta A, Murray A, Frayling TM, Guralnik JM, Bandinelli S, Singleton A, Ferrucci L, Melzer D (2011) Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10, 868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M‐G, Myers AJ, Magnusson PKE, Prince JA (2008) Transcriptome‐wide assessment of human brain and lymphocyte senescence. PLoS ONE 3, e3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PAC, Monlong J, Rivas MA, Gonzàlez‐Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlöf J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HPJ, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Pirinen M, Montgomery SB, Donnelly P, McCarthy MI, Flicek P, Strom TM, Geuvadis Consortium , Lehrach H, Schreiber S, Sudbrak R, Carracedo A, Antonarakis SE, Häsler R, Syvänen A‐C, van Ommen G‐J, Brazma A, Meitinger T, Rosenstiel P, Guigó R, Gut IG, Estivill X, Dermitzakis ET (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Bergman A, Barzilai N (2007) Genetic determinants of human health span and life span: progress and new opportunities. PLoS Genet. 3, e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murabito JM, Yuan R, Lunetta KL (2012) The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long‐lived individuals. J. Gerontol. A Biol. Sci. Med. Sci. 67, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel A, Croucher PJP, Stiegeler R, Nikolaus S, Krawczak M, Schreiber S (2005) No association between microsomal triglyceride transfer protein (MTP) haplotype and longevity in humans. Proc. Natl. Acad. Sci. USA 102, 7906–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel A, Kleindorp R, Caliebe A, Nothnagel M, Blanch H, Junge O, Wittig M, Ellinghaus D, Flachsbart F, Wichmann HE, Meitinger T, Nikolaus S, Franke A, Krawczak M, Lathrop M, Schreiber S (2011) A genome‐wide association study confirms APOE as the major gene influencing survival in long‐lived individuals. Mech. Ageing Dev. 132, 324–330. [DOI] [PubMed] [Google Scholar]
- Palmer C, Diehn M, Alizadeh A, Brown P (2006) Cell‐type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom. 7, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passtoors WM, Beekman M, Gunn D, Boer JM, Heijmans BT, Westendorp RGJ, Zwaan BJ, Slagboom PE (2008) Genomic studies in ageing research: the need to integrate genetic and gene expression approaches. J. Intern. Med. 263, 153–166. [DOI] [PubMed] [Google Scholar]
- Passtoors WM, Boer JM, Goeman JJ, van den Akker EB, Deelen J, Zwaan BJ, Scarborough A, van der Breggen R, Vossen RHAM, Houwing‐Duistermaat JJ, van Ommen GJB, Westendorp RGJ, van Heemst D, de Craen AJM, White AJ, Gunn DA, Beekman M, Slagboom PE (2012) Transcriptional profiling of human familial longevity indicates a role for ASF1A and IL7R. PLoS ONE 7, e27759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, Reinmaa E, Sutphin GL, Zhernakova A, Schramm K, Wilson YA, Kobes S, Tukiainen T, NABEC/UKBEC Consortium , Ramos YF, Göring HHH, Fornage M, Liu Y, Gharib SA, Stranger BE, De Jager PL, Aviv A, Levy D, Murabito JM, Munson PJ, Huan T, Hofman A, Uitterlinden AG, Rivadeneira F, van Rooij J, Stolk L, Broer L, Verbiest MMPJ, Jhamai M, Arp P, Metspalu A, Tserel L, Milani L, Samani NJ, Peterson P, Kasela S, Codd V, Peters A, Ward‐Caviness CK, Herder C, Waldenberger M, Roden M, Singmann P, Zeilinger S, Illig T, Homuth G, Grabe H‐J, Völzke H, Steil L, Kocher T, Murray A, Melzer D, Yaghootkar H, Bandinelli S, Moses EK, Kent JW, Curran JE, Johnson MP, Williams‐Blangero S, Westra H‐J, McRae AF, Smith JA, Kardia SLR, Hovatta I, Perola M, Ripatti S, Salomaa V, Henders AK, Martin NG, Smith AK, Mehta D, Binder EB, Nylocks KM, Kennedy EM, Klengel T, Ding J, Suchy‐Dicey AM, Enquobahrie DA, Brody J, Rotter JI, Chen Y‐DI, Houwing‐Duistermaat J, Kloppenburg M, Slagboom PE, Helmer Q, den Hollander W, Bean S, Raj T, Bakhshi N, Wang QP, Oyston LJ, Psaty BM, Tracy RP, Montgomery GW, Turner ST, Blangero J, Meulenbelt I, Ressler KJ, Yang J, Franke L, Kettunen J, Visscher PM, Neely GG, Korstanje R, Hanson RL, Prokisch H, Ferrucci L, Esko T, Teumer A, van Meurs JBJ, Johnson AD (2015) The transcriptional landscape of age in human peripheral blood. Nat. Commun. 6, 8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravussin E, Redman LM, Rochon J, Das SK, Fontana L, Kraus WE, Romashkan S, Williamson DA, Meydani SN, Villareal DT, Smith SR, Stein RI, Scott TM, Stewart TM, Saltzman E, Klein S, Bhapkar M, Martin CK, Gilhooly CH, Holloszy JO, Hadley EC, Roberts SB, CALERIE Study Group (2015) A 2‐year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea JNM, Carvalho A, McNerlan SE, Alexander HD, Rea IM (2015) Genes and life‐style factors in BELFAST nonagenarians: nature, nurture and narrative. Biogerontology 16, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodwell GEJ, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, Kim SK (2004) A transcriptional profile of aging in the human kidney. PLoS Biol. 2, e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozing MP, Houwing‐Duistermaat JJ, Slagboom PE, Beekman M, Frölich M, De Craen AJM, Westendorp RGJ, Van Heemst D (2010) Familial longevity is associated with decreased thyroid function. J. Clin. Endocrinol. Metab. 95, 4979–4984. [DOI] [PubMed] [Google Scholar]
- Ruggiero C, Metter EJ, Melenovsky V, Cherubini A, Najjar SS, Ble A, Senin U, Longo DL, Ferrucci L (2008) High basal metabolic rate is a risk factor for mortality: the Baltimore Longitudinal Study of Aging. J. Gerontol. A Biol. Sci. Med. Sci. 63, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schächter F, Faure‐Delanef L, Guénot F, Rouger H, Froguel P, Lesueur‐Ginot L, Cohen D (1994) Genetic associations with human longevity at the APOE and ACE loci. Nat. Genet. 6, 29–32. [DOI] [PubMed] [Google Scholar]
- Sebastiani P, Solovieff N, Dewan AT, Walsh KM, Puca A, Hartley SW, Melista E, Andersen S, Dworkis DA, Wilk JB, Myers RH, Steinberg MH, Montano M, Baldwin CT, Hoh J, Perls TT (2012) Genetic signatures of exceptional longevity in humans. PLoS ONE 7, e29848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skytthe A, Christiansen L, Kyvik KO, Bødker FL, Hvidberg L, Petersen I, Nielsen MMF, Bingley P, Hjelmborg J, Tan Q, Holm NV, Vaupel JW, McGue M, Christensen K (2013) The Danish Twin Registry: linking surveys, national registers, and biological information. Twin Res. Hum. Genet. 16, 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soerensen M, Dato S, Christensen K, McGue M, Stevnsner T, Bohr VA, Christiansen L (2010) Replication of an association of variation in the FOXO3A gene with human longevity using both case‐control and longitudinal data. Aging Cell 9, 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, Suchiman HED, Slagboom PE, Boomsma DI, Heijmans BT (2012) Epigenetic variation during the adult lifespan: cross‐sectional and longitudinal data on monozygotic twin pairs. Aging Cell 11, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Akker EB, Passtoors WM, Jansen R, van Zwet EW, Goeman JJ, Hulsman M, Emilsson V, Perola M, Willemsen G, Penninx BWJH, Heijmans BT, Maier AB, Boomsma DI, Kok JN, Slagboom PE, Reinders MJT, Beekman M (2014) Meta‐analysis on blood transcriptomic studies identifies consistently coexpressed protein‐protein interaction modules as robust markers of human aging. Aging Cell 13, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Wurmb‐Schwark N, Schwark T, Caliebe A, Drenske C, Nikolaus S, Schreiber S, Nebel A (2010) Low level of the mtDNA(4977) deletion in blood of exceptionally old individuals. Mech. Ageing Dev. 131, 179–184. [DOI] [PubMed] [Google Scholar]
- Westra H‐J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, Zhernakova A, Zhernakova DV, Veldink JH, Van den Berg LH, Karjalainen J, Withoff S, Uitterlinden AG, Hofman A, Rivadeneira F, ‘t Hoen PAC, Reinmaa E, Fischer K, Nelis M, Milani L, Melzer D, Ferrucci L, Singleton AB, Hernandez DG, Nalls MA, Homuth G, Nauck M, Radke D, Völker U, Perola M, Salomaa V, Brody J, Suchy‐Dicey A, Gharib SA, Enquobahrie DA, Lumley T, Montgomery GW, Makino S, Prokisch H, Herder C, Roden M, Grallert H, Meitinger T, Strauch K, Li Y, Jansen RC, Visscher PM, Knight JC, Psaty BM, Ripatti S, Teumer A, Frayling TM, Metspalu A, van Meurs JBJ, Franke L (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Willcox DC (2014) Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa: controversies and clinical implications. Curr. Opin. Clin. Nutr. Metab. Care 17, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD (2008) FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 105, 13987–13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Fung‐Leung WP, Bittner A, Ngo K, Liu X (2014) Comparison of RNA‐Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 9, e78644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Comparison of findings to previous transcriptomic studies.
Fig. S2 Unique and overlapping features compared to previous studies.
Fig. S3 Cross sectional mRNA regulation of three selected genes.
Fig. S4 mRNA expression of genes correlating with age.
Table S1 The top 25 up‐ and downregulated genes in LLI.
Table S2 All the significantly represented biological processes with respect to the genes upregulated and downregulated in LLI (in the German samples).
Table S3 The top 50 cis‐eQTLs associated with differentially expressed genes.
Table S4 The top 50 G×A interaction effects exhibited by differentially expressed genes.
Table S5 The top 50 genes with heritable transcriptional activity; the best fit model for these genes was AE (A=additive genetic effect, E=unique environmental effect).
Table S6 List of 80 candidate genes and the TaqMan assays used for validation.
Appendix S1 This file contains FPKM (fragments per kilobase of exon per million fragments mapped) values for all German samples.
Appendix S2 This file contains FPKM (fragments per kilobase of exon per million fragments mapped) values for all Danish samples.