

Summary
A decline in mitochondrial electron transport chain (ETC) function has long been implicated in aging and various diseases. Recently, moderate mitochondrial ETC dysfunction has been found to prolong lifespan in diverse organisms, suggesting a conserved and complex role of mitochondria in longevity determination. Several nuclear transcription factors have been demonstrated to mediate the lifespan extension effect associated with partial impairment of the ETC, suggesting that compensatory transcriptional response to be crucial. In this study, we showed that the transcription factors CEP‐1/p53 and CEH‐23 act through a similar mechanism to modulate longevity in response to defective ETC in Caenorhabditis elegans. Genomewide gene expression profiling comparison revealed a new link between these two transcription factors and AAK‐2/AMP kinase (AMPK) signaling. Further functional analyses suggested that CEP‐1/p53 and CEH‐23 act downstream of AAK‐2/AMPK signaling and CRTC‐1 transcriptional coactivator to promote stress resistance and lifespan. As AAK‐2, CEP‐1, and CEH‐23 are all highly conserved, our findings likely provide important insights for understanding the organismal adaptive response to mitochondrial dysfunction in diverse organisms and will be relevant to aging and pathologies with a mitochondrial etiology in human.
Keywords: aging, AMPK, C. elegans, mitochondrial ETC, p53, transcription
Introduction
The mitochondrial electron transport chain (ETC) produces the majority of ATP in cells and broadly influences diverse biological processes. Accordingly, perturbations in mitochondrial ETC function are usually detrimental. However, in some cases, moderate reduction of mitochondrial ETC function prolongs organismal longevity in organisms ranging from yeast to mice (Dillin et al., 2002; Kirchman et al., 1999; Liu et al., 2005; Lee et al., 2003; Copeland et al., 2009). In Caenorhabditis elegans, several mutants that harbor point mutations in distinct mitochondrial ETC subunits have been identified. These mutants show altered lifespan, with some living longer, while others living shorter, than wild‐type worms (Wong et al.,1994; Feng et al., 2001; Yang & Hekimi, 2010; Ishii et al., 1998; Kayser et al., 1999; Hartman et al., 2001). Moreover, RNAi knockdown of many different ETC subunits also impact lifespan (Dillin et al., 2002; Lee et al., 2003), and RNAi dilution experiments clearly demonstrating that the degree of knockdown of a single ETC subunit can determine whether the lifespan becomes extended or shortened (Rea et al., 2007). Together, these observations suggest that mitochondria play a crucial and complex role in determining the lifespan of an organism.
Proper communication between mitochondria and the nucleus is essential for organismal survival. In yeast, a retrograde signaling pathway relays signals from mitochondria to the nucleus and is key to the aging phenotypes associated with yeast cells with compromised mitochondrial ETC function (Parikh et al., 1987; Jazwinski et al., 2013). In C. elegans, mitochondrial ETC dysfunction is accompanied by specific changes in nuclear gene expression (Falk et al., 2008; Cristina et al., 2009; Yee et al., 2014), and particular transcription factors have been shown to mediate the altered longevity associated with the various ETC mutants (Lee et al., 2010; Khan et al., 2013; Walter et al., 2011; Ventura et al., 2010).
The C. elegans homeodomain protein CEH‐23 plays an important role in mediating lifespan extension associated with ETC dysfunction (Walter et al., 2011). Inactivation of ceh‐23 partially suppresses the prolonged lifespan of the isp‐1(qm150) mutant without affecting its development and reproduction phenotypes, suggesting that CEH‐23 specifically mediates the longevity of the isp‐1 mutant (Walter et al., 2011). isp‐1 encodes the Rieske iron sulfur protein, a key subunit of the mitochondrial ETC complex III, and the isp‐1 mutant harbors a point mutation that reduces the electron transport efficiency of complex III and extends lifespan by up to 50% (Feng et al., 2001). Interestingly, loss of ceh‐23 has no impact on the lifespan of wild‐type animals or long‐lived mutants in the insulin‐like signaling and the eat‐2 caloric restriction pathways, suggesting that CEH‐23 modulates longevity specifically in response to mitochondrial ETC dysfunction (Walter et al., 2011). How CEH‐23 performs this function is unknown. Besides longevity modulation, earlier studies suggested a role for CEH‐23 in neuronal differentiation (Altun‐Gultekin et al., 2001), although the ceh‐23(ms23) null mutant has no obvious neuronal or behavioral defects (Walter et al., 2011).
In addition to CEH‐23, CEP‐1 is another transcription factor that is required for the longevity of the isp‐1 mutant (Baruah et al., 2014; Ventura et al., 2010). cep‐1 encodes the sole C. elegans ortholog of the mammalian p53 family, which acts as a transcription factor in response to various stresses (Derry et al., 2001; Baruah et al., 2014). Mammalian p53 is well known to participate in DNA repair, cell cycle regulation, and apoptosis, processes that have also been implicated in aging (Levine, 1997). C. elegans CEP‐1, similar to its mammalian ortholog, participates in apoptosis and cell cycle regulation (Derry et al., 2001; Greiss et al., 2008). Recent studies in C. elegans revealed intriguing opposing roles of CEP‐1 in the longevity of different ETC mutants (Baruah et al., 2014; Ventura et al., 2010).
In this study, we investigated the possible collaboration of the transcription factors CEH‐23 and CEP‐1 in modulating longevity in response to ETC dysfunction. Our data suggested that ceh‐23 and cep‐1 act in the same pathway to mediate the longevity of the complex III isp‐1 mutant. We also demonstrated that CEH‐23 and CEP‐1 regulate a common set of target genes, which are overrepresented by kinases and phosphatases of specific families, with likely roles in signal transduction. Intriguingly, the majority of the CEH‐23 and CEP‐1 co‐regulated transcriptional targets are also regulated by activated AAK‐2/AMPK signaling, suggesting a link between these two transcription factors and AAK‐2/AMPK signaling. Further analyses indicated that CEH‐23 and CEP‐1 play key roles downstream of AAK‐2 to modulate stress response and lifespan. As AMPK, CEP‐1, and CEH‐23 are all highly conserved, the findings reported here suggested that the mammalian counterparts of these proteins likely have similar roles in mediating the adaptive response to mitochondrial dysfunction.
Results
ceh‐23 and cep‐1 act in the same genetic pathway to modulate longevity of the ETC mutants
Both CEH‐23 and CEP‐1 have been shown to be critical for the extended lifespan associated with the isp‐1 ETC mutant (Baruah et al., 2014; Ventura et al., 2010; Walter et al., 2011). We performed epistasis analysis to assess how these two transcription factors might interact to modulate the lifespan of mitochondrial ETC mutants. While ceh‐23 and cep‐1 contribute to part of the extended lifespan of the isp‐1 mutant, we found that the triple mutant cep‐1; ceh‐23; isp‐1 had a lifespan similar to the cep‐1; isp‐1 and ceh‐23; isp‐1 double mutants (Fig. 1A). Similar to that reported for each single mutation (Baruah et al., 2014; Walter et al., 2011), combining the cep‐1 and ceh‐23 mutations had no effect on wild‐type lifespan (Fig. S2A), suggesting that these two transcription factors are not required to maintain normal lifespan. Because combining the ceh‐23 and cep‐1 mutations did not result in an additive suppression of isp‐1 mutant lifespan, we concluded that ceh‐23 and cep‐1 act in the same genetic pathway to modulate isp‐1 mutant longevity.
Figure 1.
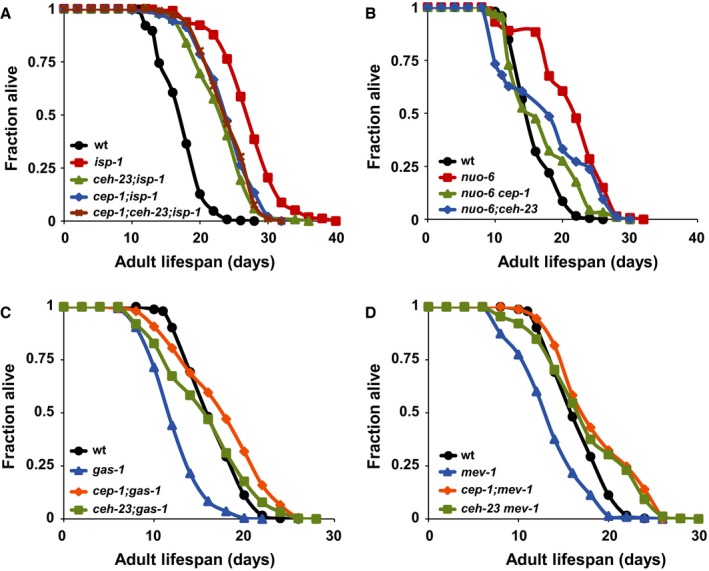
ceh‐23 and cep‐1 act in the same genetic pathway to modulate longevity when mitochondrial electron transport chain (ETC) function is impaired. (A) Both ceh‐23 and cep‐1 mutations partially suppressed the extended lifespan of the isp‐1(qm150) mutant (P < 0.0005), and inactivation of ceh‐23 and cep‐1 did not additively suppress isp‐1 mutant lifespan, as the triple mutant lived as long as the double mutants (P = 0.529 compared to cep‐1;isp‐1 and P = 0.003 compared to ceh‐23;isp‐1). Inactivation of ceh‐23 and cep‐1 partially suppressed the long lifespan of the nuo‐6(qm200) mutant (P < 0.0005 and 0.001, respectively) (B) and restored lifespan in the short‐lived gas‐1(fc21) (C) and mev‐1(kn1) (D) mutants (all with P < 0.0005). Survival curves represent data pooled from multiple biological replicates. Quantitative data for the individual and pooled experiments are shown in Table S1 (Supporting information).
cep‐1 has been shown to have opposing effects on longevity in different mitochondrial ETC mutants. It promotes longevity in the nuo‐6 and isp‐1 mutants, as cep‐1 inactivation suppresses the long life of these mutants. In contrast, cep‐1 limits the lifespan of the short‐lived gas‐1 and mev‐1 mutants, as cep‐1 mutation restores normal lifespan in these mutants (Baruah et al., 2014; Ventura et al., 2010). Because our data suggested that ceh‐23 and cep‐1 act in the same genetic pathway to modulate longevity of the isp‐1 mutant, we expected that ceh‐23 would have similar opposing roles in the various ETC mutants. Consistent with our hypothesis, the ceh‐23 mutation suppressed the extended lifespan of the long‐lived nuo‐6 mutant (Fig. 1B) but restored lifespan in the short‐lived gas‐1 and mev‐1 mutants (Fig. 1C,D). Thus, ceh‐23 and cep‐1 are required for the altered lifespans of the various ETC mutants, suggesting that they play a key role in modulating lifespan in responding to ETC dysfunction.
CEH‐23 and CEP‐1 share a large group of transcriptional targets in response to mild ETC dysfunction
To elucidate the possible molecular basis by which CEH‐23 and CEP‐1 modulate lifespan in the long‐lived ETC complex III isp‐1 mutant, we compared the transcriptional outputs of these transcription factors in the isp‐1 mutants. We previously identified the transcriptional responses regulated by CEP‐1 in the isp‐1 mutant and found that CEP‐1 regulates the expression of a broad array of genes in response to ETC stress, including those involved in phosphate metabolism, lipid modification, and neuropeptide signaling (Baruah et al., 2014). We performed similar microarray analyses to profile the CEH‐23 dependent transcriptomic response in the long‐lived isp‐1 mutant by comparing the global gene expression profiles between isp‐1 and ceh‐23; isp‐1 mutant worms synchronized at larval stage 4 (L4), which is a clearly identifiable developmental stage and a temporal window reported to be important for some ETC dysfunctions to influence adult lifespan (Dillin et al., 2002). Using the statistical tool SAM (Statistical Analysis of Microarray) (Tusher et al., 2001) with stringent parameters (false discovery rate (FDR) = 0.59%, fold change > 1.5‐fold), we identified 1878 genes whose expression change was ceh‐23 dependent. Of these genes, 1244 were upregulated, and 634 were downregulated, in the isp‐1 mutant with respect to the ceh‐23; isp‐1 double mutant (Fig. 2). Gene Ontology (GO) analyses revealed that the 1244 genes (upregulated in the isp‐1 mutant where CEH‐23 is functional) were enriched for genes involved in many fundamental biological processes, in particular cell cycle, reproduction, lipid transport, and phosphate metabolism (Fig. 2). The 634 genes (downregulated in the isp‐1 mutant) were especially enriched for the biological function neuropeptide signaling (Fig. 2). These data suggested that CEH‐23 promotes the expression of development, reproduction, and metabolism genes and represses neuronal signaling genes in response to mild complex III dysfunction. Our previous works showed that ceh‐23 deficiency in the isp‐1 mutant did not affect the slow development or reduced reproduction phenotypes (Walter et al., 2011); however, cep‐1 mutation partially restored normal development in the isp‐1 mutant (Baruah et al., 2014). Interestingly, we found that functional ceh‐23 was required for cep‐1 to modulate development in the isp‐1 mutant (Fig. 3C). Exactly how these two factors interact in this context awaits further investigation.
Figure 2.
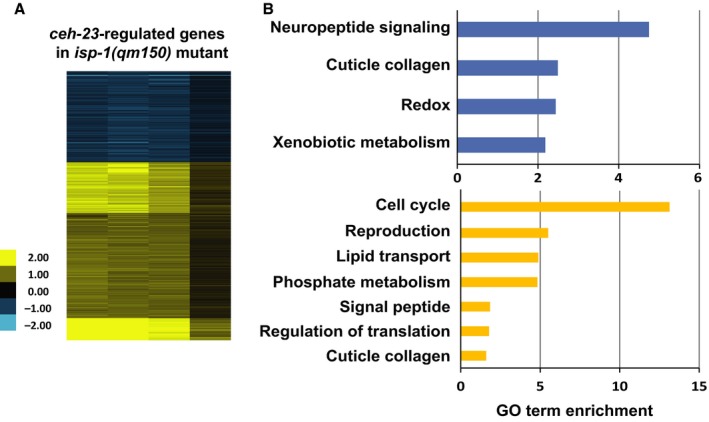
Transcriptomic targets of CEH‐23. (A) The heat map shows the genes whose expression changes significantly between the isp‐1(qm150) mutant compared to the ceh‐23(ms23); isp‐1(qm150) double mutant. Gene changes were identified by Statistical Analysis of Microarray (SAM) one‐class analysis false discovery rate (FDR) = 0, fold change > 1.5). Genes downregulated in isp‐1 relative to ceh‐23; isp‐1 are shown in blue and genes upregulated are shown in yellow. The intensity of the heat map represents the log2 ratio of the expression comparison. The significant gene lists are presented in Table S2 (Supporting information). (B) The most enriched Gene Ontology (GO) terms among the CEH‐23‐upregulated genes (yellow bars) and CEH‐23‐downregulated genes (blue bars). X‐axis represents GO term enrichment scores.
Figure 3.
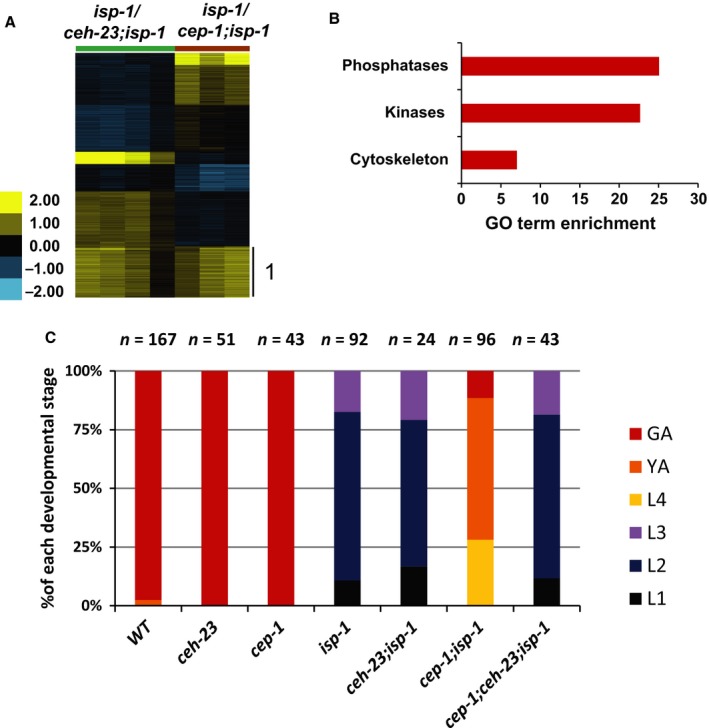
Transcriptomic analyses revealed that CEH‐23 and CEP‐1 co‐regulate a large set of genes in response to electron transport chain (ETC) dysfunction. (A) Genomewide comparison of expression changes between isp‐1(qm150) and ceh‐23(ms23); isp‐1(qm150) or cep‐1(gk138); isp‐1(qm150). The common targets of CEH‐23 and CEP‐1 in the isp‐1 mutant false discovery rate (FDR = 0%, fold change > 1.5 fold) are indicated as Cluster 1. Gene clustering was performed using K‐mean clustering with K = 7. The intensity of the heat map represents the log2 ratio of the expression comparison. The isp‐1(qm150) vs. cep‐1(gk138);isp‐1(qm150) microarray were performed in the Baruah et al. study. (B) The most enriched Gene Ontology (GO) terms among the CEH‐23 and CEP‐1 common targets. X‐axis represents GO term enrichment scores. Gene lists used for the GO terms analysis are presented in Table S3 (Supporting information). (C) Postembryonic developmental phenotypes of the indicated strains (wild‐type, ceh‐23(ms23), cep‐1(gk138), isp‐1(qm150), ceh‐23(ms23);isp‐1(qm150), cep‐1(gk138);isp‐1(qm150), cep‐1(gk138);ceh‐23(ms23);isp‐1(qm150)). Bar graph shows the fraction of each developmental stage 60 h after egg lay for each genotype. The sample size (n numbers) for each genotype is denoted above the bar graph. (L1‐L4: larval stage 1–4, YA: young adults, GA: gravid adults.)
Given that earlier epistasis analyses revealed that ceh‐23 and cep‐1 likely act in the same genetic pathway to modulate longevity in ETC mutants (Fig. 1A), we hypothesized that these two factors would share a transcriptional outcome that is important for longevity determination during ETC dysfunction. We compared the CEH‐23‐dependent transcriptional response to isp‐1 mutation with the previously published CEP‐1‐dependent response (Baruah et al., 2014) to identify any possible shared transcriptional outputs between these transcription factors. Consistent with our hypothesis, statistical analyses with stringent criteria (FDR = 0%, fold change > 1.5‐fold) revealed a substantial number of genes that were upregulated in the isp‐1 mutant in a CEH‐23‐ and CEP‐1‐dependent manner (897 genes) (Fig. 3A Cluster 1, Table S3). Interestingly, this analysis revealed very few genes that were downregulated in the isp‐1 mutant compared to the ceh‐23; isp‐1 and cep‐1; isp‐1 mutants (19 genes) (Table S3). We herein term the group of genes whose expression changed similarly when comparing isp‐1 vs. ceh‐23; isp‐1 or isp‐1 vs. cep‐1; isp‐1 the ‘CEH‐23 and CEP‐1 common target genes’ (916 genes). GO term analyses revealed that the CEH‐23 and CEP‐1 common target genes were enriched with kinases and phosphatases (Fig. 3B), suggesting that CEH‐23 and CEP‐1 regulate signaling networks to prolong isp‐1 mutant lifespan.
CEH‐23 and CEP‐1/p53 share many transcriptional targets with active AAK‐2/AMPK signaling
Although the CEH‐23 and CEP‐1 common transcriptional targets were enriched with kinases and phosphatases, these proteins had not been directly implicated in known longevity pathways. To further assess the roles of CEH‐23 and CEP‐1, we performed an unbiased search for factors that also regulate the expression of the CEH‐23 and CEP‐1 common target genes using the WormMine tool at www.wormbase.org. Interestingly, this analysis revealed a highly significant overlap between CEH‐23 and CEP‐1 common target genes and the genes that are upregulated by constitutively active AAK‐2, the AMP kinase (AMPK) catalytic subunit homolog in C. elegans (424 Genes) (Fig. 4A,B, Cluster 1) (Mair et al., 2011).
Figure 4.
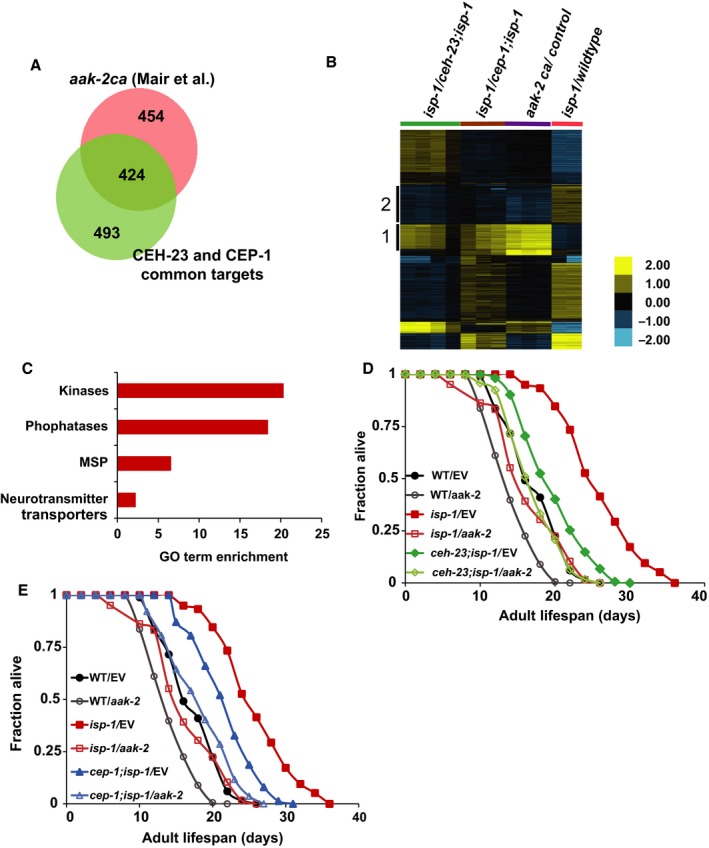
CEH‐23, CEP‐1, and AAK‐2 act in the same pathway to mediate isp‐1 mutant lifespan. (A) The Venn diagram shows a substantial overlap between the genes that are commonly regulated by CEH‐23 and CEP‐1 in the isp‐1 mutant and those regulated by constitutively active AAK‐2 (aak‐2 ca) (Representation factor: 7.0; P < 0.000e+00). The representation factor and P‐value (determined by hypergeometric probability test) were calculated using the web‐based tool (http://nemates.org/MA/progs/overlap_stats.html). The aak‐2ca microarray data are obtained from Mair et al., 2011 ; (B) The heat map represents a genomewide comparison of expression patterns between isp‐1(qm150) vs. ceh‐23(ms23); isp‐1(qm150) or cep‐1(gk138); isp‐1(qm150), aak‐2ca vs. transgenic control (data from Mair et al., 2011), and isp‐1(qm150) vs. wild‐type. Genes without significant differences in any of the pairwise comparisons have been filtered out. Genes are grouped using K‐mean clustering with K = 7. Cluster 1 highlights genes that are commonly regulated by CEH‐23, CEP‐1, and AAK‐2, and Cluster 2 highlights genes that shared targets between CEH‐23 and AAK‐2. (C) The most enriched Gene Ontology (GO) terms among the CEH‐23, CEP‐1, and AAK‐2 common targets. X‐axis represents GO term enrichment scores. Gene lists for the GO terms are presented in Table S5 (Supporting information). ceh‐23; isp‐1 (D) and cep‐1; isp‐1 (E) mutants treated with aak‐2 RNAi had similar lifespan as the isp‐1 single mutant treated with aak‐2 RNAi, suggesting that ceh‐23 and cep‐1 and aak‐2 act in the same genetic pathway to modulate isp‐1 mutant lifespan.
AMPK is a well‐conserved kinase that serves as a cellular energy sensor. It regulates diverse biological processes in response to different environmental stresses and cellular energy levels, in particular a change in AMP:ATP ratio. AMPK has been implicated in several pathways that are key to organismal lifespan (Apfeld et al., 2004; Greer et al., 2007a,b). Furthermore, overexpression of a constitutively active AAK‐2 is sufficient to extend C. elegans lifespan (Mair et al., 2011). Given that mitochondria are important for cellular fuel production, defects in the mitochondrial ETC are likely to trigger a cellular energy imbalance and thus activate AMPK. Indeed, AAK‐2/AMPK activation appears to be enhanced in the isp‐1 mutant (Hwang et al., 2014) and is required for the extended lifespan of the isp‐1 mutant (Curtis et al., 2006). Together, our findings suggest that AAK‐2 signaling, CEH‐23, and CEP‐1 converge to modulate longevity in the isp‐1 mutant. GO term analyses of the genes that are commonly regulated by AAK‐2, CEH‐23, and CEP‐1 revealed a highly significant enrichment for kinases, phosphatases, major sperm proteins (MSPs), and neurotransmitter transport proteins (Fig. 4C). Therefore, AAK‐2, CEP‐1, CEH‐23 may regulate downstream signal transduction to promote longevity in mutant worms with mild ETC dysfunction. Although AAK‐2/AMPK is known to modulate lifespan under a broad range of conditions (Burkewitz et al., 2014), the mechanism by which AAK‐2 mediates the longevity of mitochondrial ETC mutants remains unclear. Our results revealed a new connection between AAK‐2 and the transcription factors CEH‐23 and CEP‐1.
We next compared the transcriptional profiles of the isp‐1 mutants with or without ceh‐23 or cep‐1 with that of wild‐type worms (Fig. 4B). This comparison revealed a somewhat surprising pattern where the genes that were commonly upregulated in the isp‐1 mutant in a CEH‐23‐ and CEP‐1‐dependent manner appeared to be downregulated in the isp‐1 mutant relative to wild‐type worms (Fig. 4B, Cluster 1), suggesting that functional CEH‐23 and CEP‐1 are required for maintaining the expression of these genes in the isp‐1 mutant, albeit at levels lower than wild‐type. Inactivation of ceh‐23 and cep‐1 in the isp‐1 mutant results in further repression of these genes. As the isp‐1 mutant is long‐lived compared to the ceh‐23; isp‐1 or cep‐1; isp‐1 mutants, we proposed that activation of AAK‐2, CEH‐23, and CEP‐1 in the isp‐1 mutant acts to buffer the downregulation of these genes, allowing the worms to live long, and the absence of AAK‐2, CEH‐23, or CEP‐1 results in further repression of these genes and a detrimental effect on lifespan.
CEH‐23 and CEP‐1 are downstream effectors of AAK‐2 signaling
To further delineate the relationship of AAK‐2, CEH‐23, and CEP‐1 in response to ETC stress, we assessed the effect of aak‐2 depletion in isp‐1 mutants lacking functional ceh‐23 or cep‐1. First, we sought to confirm the role of aak‐2 in lifespan modulation. Consistent with previous findings (Curtis et al., 2006), we found that RNAi knockdown of aak‐2 slightly shortened the lifespan of wild‐type animals and substantially suppressed the extended lifespan of the isp‐1 mutant (Fig. 4D,E, Table S1C), indicating that AAK‐2 signaling is essential for normal lifespan and required for the full lifespan extension of isp‐1 mutant. If AAK‐2, CEH‐23, and CEP‐1 act together to modulate lifespan in the isp‐1 mutant, we would predict that loss of aak‐2 and ceh‐23 or cep‐1 would not additively suppress isp‐1 mutant lifespan. Indeed, we observed that isp‐1 mutants devoid of both ceh‐23 and aak‐2 (ceh‐23; isp‐1; aak‐2(RNAi or mutation)) or cep‐1 and aak‐2 (cep‐1; isp‐1; aak‐2(RNAi or mutation)) lived as long as isp‐1 mutants with only aak‐2 depleted (Fig. 4D,E, Table S1C). Taken together, our data showed that inactivation of ceh‐23 or cep‐1 and depletion of aak‐2 do not additively suppress isp‐1 mutant lifespan, thus supporting the model that AAK‐2/AMPK signaling acts with CEH‐23 and CEP‐1/p53 to prolong the lifespan of the isp‐1 mutant. It is important to note that aak‐2 RNAi had a greater effect in suppressing the extended lifespan of the isp‐1 mutant compared to ceh‐23 or cep‐1 inactivation, suggesting that AAK‐2 likely works with additional effectors, beyond CEH‐23 and CEP‐1, to modulate longevity in the isp‐1 mutant.
As the microarray and epistasis data suggested that AAK‐2, CEH‐23, and CEP‐1 act together to modulate the lifespan of the isp‐1 mutant, we tested whether CEH‐23 and CEP‐1 could influence AAK‐2/AMPK activity by measuring the levels of phosphorylated form of AAK‐2 in isp‐1 mutants with or without ceh‐23 or cep‐1. We found that upregulation of active AAK‐2/AMPK in the isp‐1 mutant did not depend on functional ceh‐23 or cep‐1, as neither ceh‐23 nor cep‐1 mutation reduced phospho‐AAK‐2 levels in the isp‐1 mutants (Fig. 5A). Moreover, inactivation of CEH‐23 or CEP‐1 also had no detectable effect on the mRNA levels of aak‐2 in the isp‐1 mutants (Fig. S1C). Taken together, our data suggested that CEP‐1 and CEH‐23 likely do not regulate AAK‐2 activity in the isp‐1 mutant.
Figure 5.
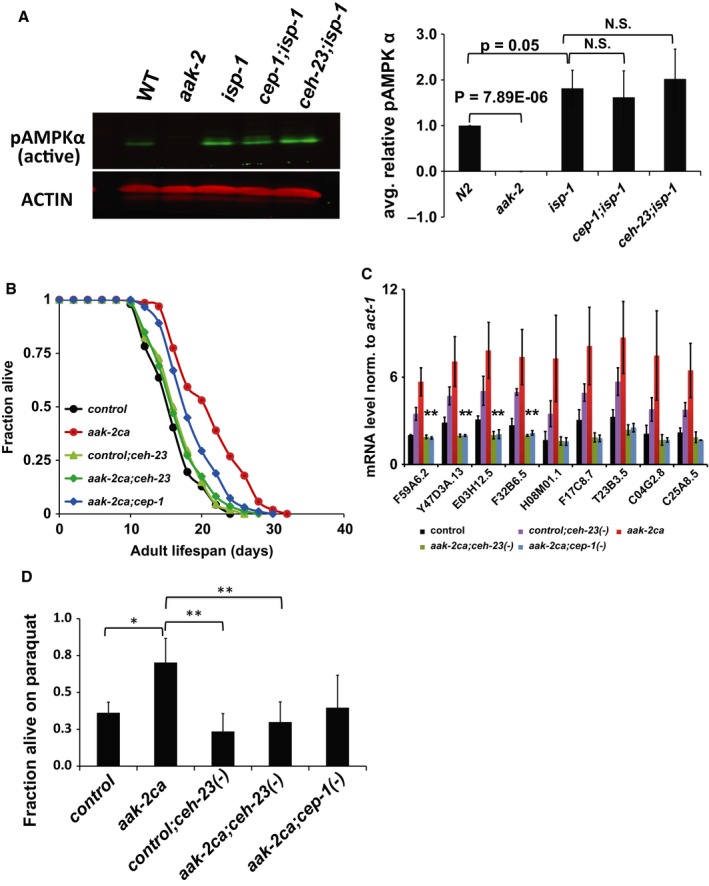
CEH‐23, CEP‐1 act downstream of AAK‐2/AMPK signaling. (A) Western blot analysis showed that activated AAK‐2 levels are similarly elevated in the isp‐1 single mutant and in the ceh‐23; isp‐1 and cep‐1; isp‐1 double mutants. Quantified pAAK‐2/AMPKα levels from three independent experiments are presented in the bar graph. Both ceh‐23 and cep‐1 mutations suppressed the extended lifespan (B), transcriptional response (C), and oxidative stress resistance (D) of aak‐2ca worms. In (D), P‐value is represented as following: *< 0.05, **< 0.01. Although the difference in oxidative stress resistance between aak‐2ca; cep‐1(‐) and aak‐2ca is not significant, we consistently observed this difference in all our trials. aak‐2ca refers to WBM60 and control refers to WBM59. Our attempt to introduce the cep‐1 mutation into the control strain was not successful, so only control strain carrying the ceh‐23 mutation was included in this experiment.
We next tested whether aak‐2 RNAi, like that of ceh‐23 or cep‐1 deletion, would restore lifespan in the short‐lived gas‐1 and mev‐1 mutants. To our surprise, knocking down aak‐2 has no effect on the lifespan of the short‐lived ETC mev‐1 and gas‐1 mutants (Fig.S2B). Therefore, whereas aak‐2, ceh‐23, cep‐1 appear to act in the same pathway to promote lifespan in the isp‐1 mutant, they do not act the same way in response to the complex I gas‐1 and complex II mev‐1 mutations. As discussed above, CEH‐23 and CEP‐1 likely represent only a subset of the effectors of AAK‐2/AMPK signaling, and it is therefore possible that aak‐2 RNAi has a different effect on the lifespan of the gas‐1 and mev‐1 mutants due to additional outputs beyond CEH‐23 and CEP‐1.
We next investigated whether CEH‐23 and CEP‐1 could mediate the biological outputs of AAK‐2 beyond the context of mitochondrial dysfunction by testing whether functional CEH‐23 and CEP‐1 are required for the extended lifespan and the increased resistance to oxidative stress of worms overexpressing constitutively active AAK‐2 (aak‐2ca) in an otherwise wild‐type background (Mair et al., 2011; Hwang et al., 2014). The data showed that both ceh‐23 and cep‐1 were required for the extended lifespan of the aak‐2ca animals (Fig. 5B), suggesting that CEH‐23 and CEP‐1 act downstream of AMPK signaling for longevity determination. Consistent with this finding, functional CEH‐23 and CEP‐1 were also required for the increased tolerance to oxidative stress of the aak‐2ca animals (Fig. 5D). Interestingly, ceh‐23 inactivation appeared to have a greater effect on suppressing the prolonged lifespan of the aak‐2ca animals compared to cep‐1 inactivation. This observation suggested that CEH‐23 mediates a greater portion of the longevity effect of AMPK than CEP‐1, which might be reflected by the gene expression comparison where CEH‐23, but not CEP‐1, repressed the expression of a group of genes in the isp‐1 mutants that are also downregulated in aak‐2ca worms (Fig. 4B, Cluster 2). To further test whether CEH‐23 and CEP‐1 act downstream of AAK‐2, we asked whether CEH‐23 and CEP‐1 could mediate some of the transcriptional responses caused by constitutive active AAK‐2. Microarray analyses revealed a group of genes that were regulated by CEH‐23 and CEP‐1 in the isp‐1 mutant and were upregulated when AAK‐2 is overactive (Fig. 4B, Cluster 1). Quantitative PCR analyses demonstrated that the induction of these genes in the aak‐2ca animals required ceh‐23 and cep‐1 (Fig. 5C). Together, our data supported the model that CEH‐23 and CEP‐1 act downstream of AAK‐2 to modulate some of its biological outputs, including longevity and oxidative stress response.
CEH‐23 and CEP‐1 likely act downstream of CRTC‐1 to modulate longevity
AAK‐2 is well established to modulate longevity by acting through the transcription factor DAF‐16/FOXO and the transcriptional coactivator CRTC‐1/CRTCs. Both DAF‐16 and CRTC‐1 can be phosphorylated by AMPK and both play crucial roles in mediating the longevity effect of AAK‐2, albeit in an opposing manner (Greer et al., 2007a,b; Mair et al., 2011). daf‐16 encodes the sole C. elegans ortholog of the FOXO transcription factors and is a key effector of the insulin/IGF‐1 signaling (IIS) pathway (Kenyon et al., 1993; Ogg et al., 1997; Lin et al., 1997). However, IIS and daf‐16 have been shown to modulate longevity through a mechanism distinct from that associated with mild ETC dysfunction (Feng et al., 2001), and we have previously demonstrated that ceh‐23 and daf‐16 likely act separately to mediate the lifespan of the isp‐1 mutant worms (Walter et al., 2011). crtc‐1 encodes the sole C. elegans ortholog of the CREB‐regulated transcription coactivator, and AAK‐2 has been shown to promote longevity by sequestering CRTC‐1 outside of the nucleus through phosphorylation (Mair et al., 2011). The subcellular localization and the longevity effect of CRTC‐1 in worms with a mild ETC dysfunction have not been explored. We used a tdTomato‐fused crtc‐1 transgene to monitor the subcellular localization of CRTC‐1 in the isp‐1(qm150) mutant. Similar to that seen in the aak‐2ca animals (Mair et al., 2011), we observed greater nuclear exclusion of CRTC‐1 in the isp‐1 mutants compared to wild‐type (Fig. 6A), which is consistent with the idea that AAK‐2 is activated in the isp‐1 mutant. We next tested whether CEH‐23 or CEP‐1 might influence the subcellular location of CRTC‐1 in the isp‐1 mutant. We found that the nuclear exclusion of CRTC‐1::tdTomato in the isp‐1 mutant was not reduced by either cep‐1 or ceh‐23 mutations, suggesting that cep‐1 and ceh‐23 are not required for active AAK‐2 signaling to expel CRTC‐1 from the nucleus. In fact, the data suggested that more CRTC‐1 was excluded from the nucleus when cep‐1 and ceh‐23 were mutated, which might point to a possible feedback regulation.
Figure 6.
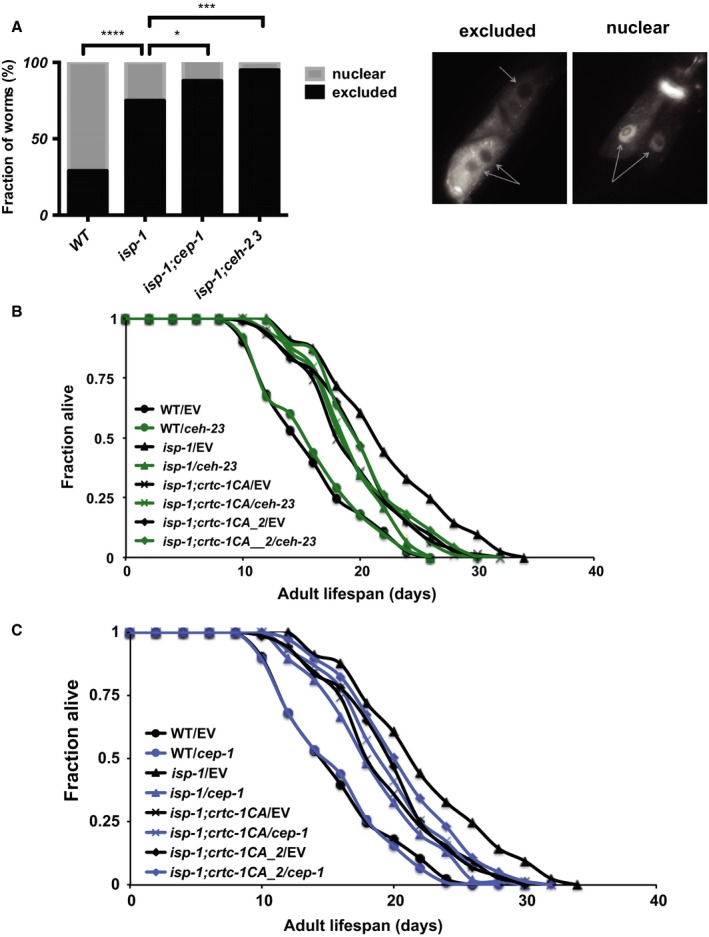
CEH‐23, CEP‐1 likely act downstream of CRTC‐1. (A) More CRTC‐1 was excluded from the nucleus in the isp‐1 mutant compared to wild‐type worms, and the nuclear exclusion was slightly enhanced when ceh‐23 and cep‐1 were mutated. CRTC‐1 localization experiments were performed using 40 young adults (YA) worms for each genotypes and the localization of CRTC‐1 in intestinal cells was scored. Microscopic images show representative nuclear‐excluded and nuclear‐localized CRTC‐1 pattern. Gray arrows denote intestinal nuclei (P‐value: ****< 0.0001, ***< 0.0005, *< 0.05). (B, C) Forced expression of constitutively nuclear CRTC‐1 partially suppressed the prolonged lifespan of the isp‐1 mutant and ceh‐23 (B) or cep‐1 (C) RNAi knockdown did not further impact the suppressed lifespan.
It is known that constitutively nuclear‐localized CRTC‐1 (CRTC‐1CA) has a negative impact on the extended lifespan of the aak‐2ca mutant (Mair et al., 2011); we therefore tested whether CRTC‐1CA expression also suppressed the extended lifespan of the isp‐1 mutants. We found that constitutive nuclear expression of CRTC‐1 (CRTC‐1CA) partially suppressed the extended lifespan of the isp‐1 mutant, consistent with the expectation that AAK‐2 activation and CRTC‐1 nuclear exclusion are important for mediating the prolonged lifespan of the isp‐1 mutant (Fig. 6B,C). We next tested whether crtc‐1 and ceh‐23 and cep‐1 might act in parallel or interdependently to mediate the lifespan of the isp‐1 mutant. We found that inactivating either ceh‐23 or cep‐1 with either RNAi or mutations did not further reduce the shortened lifespan of the isp‐1; crtc‐1ca worms (Fig. 6B,C, Fig. S3B,C), indicating that ceh‐23, cep‐1, and crtc‐1 likely act in the same genetic pathway to mediate the extended longevity of the isp‐1 mutant. As it is the forced expression of CRTC‐1 in the nucleus and the loss of CEH‐23 and CEP‐1 that suppressed the extended lifespan of the isp‐1(qm150) mutant, we interpreted the results to suggest that nuclear CRTC‐1 can act as a negative regulator of CEH‐23 and CEP‐1 to limit longevity in the isp‐1 mutants.
Discussion
Highly regulated communications between mitochondria and the nucleus are required for maintaining biological functions in response to compromised mitochondrial function. The transcription factors CEH‐23 and CEP‐1 have previously been identified as key mediators of the longevity extension phenotype of the mitochondrial ETC complex III mutant isp‐1(qm150) (Baruah et al., 2014; Ventura et al., 2010; Walter et al., 2011). In this study, we investigated how these transcription factors mediate longevity specifically focusing on the possible collaboration between them. Epistasis analyses demonstrated that ceh‐23 and cep‐1 act through the same genetic pathway to mediate the longevity of several different ETC mutants. Consistent with the genetic results, microarray analyses revealed that CEH‐23 and CEP‐1 share a large set of transcriptional targets in response to mitochondrial complex III dysfunction. Intriguingly, the majority of the CEH‐23 and CEP‐1 co‐regulated genes are also transcriptional targets of constitutively active AAK‐2 (Fig. 4A,B), thus pointing to a new link between CEH‐23, CEP‐1, and the AAK‐2 signaling pathway. We note that only one allele of cep‐1 and ceh‐23 mutants were used, so we cannot rule out allele‐specific interactions. Nevertheless, further functional analyses suggested that CEH‐23 and CEP‐1 act downstream of AAK‐2 to modulate lifespan, stress response, and gene expression, either with or without mitochondrial dysfunction.
The new link between ceh‐23, cep‐1, and aak‐2 is bolstered by their genetic interactions with the AAK‐2/AMPK target, CRTC‐1 (Fig. 6B,C). CRTC‐1 is known to be regulated by AMPK through phosphorylation, and the nuclear exclusion of CRTC‐1 is important for the lifespan extension associated with constitutively active AAK‐2/AMPK (Mair et al., 2011; Burkewitz et al., 2015). Our data indicated that nuclear‐excluded CRTC‐1 is also required for the extended lifespan of the isp‐1 mutant (Fig. 6B,C), suggesting that CRTC‐1 plays a broad role in modulating longevity in response to various stresses that activate AMPK activity. Additional functional analyses suggested a model that CRTC‐1 acts as a negative regulator of CEH‐23 and CEP‐1 in the nucleus and when AAK‐2/AMPK is activated, like in the isp‐1 mutant, phosphorylated CRTC‐1 is expelled from the nucleus, enabling CEH‐23 and CEP‐1 to regulate gene expression that prolong lifespan. We noted that the expression of many of the AAK‐2, CEH‐23, and CEP‐1 common targets were repressed in the isp‐1 mutant relative to wild‐type worms, but their expression were further reduced in the ceh‐23; isp‐1 and cep‐1; isp‐1 double mutants relatively to the isp‐1 single mutant. We therefore propose that the AAK‐2, CRTC‐1, CEH‐23, CEP‐1 signaling nexus act to maintain the expression levels of a subset of target genes that is important for the prolongevity effect associated with the isp‐1 mutation.
In further support of the notion that CEH‐23 and CEP‐1 might act downstream of AAK‐2/AMPK, sequence alignment analyses revealed that both CEH‐23 and CEP‐1 contain putative AMPK phosphorylation motifs that appear to be highly conserved (http://scansite3.mit.edu/#home). Mammalian p53 is a known substrate of AMPK (Imamura et al., 2001), and multiple sequence alignment analysis revealed that C. elegans CEP‐1 also contains a highly conserved AMPK motif (Fig. S1A). Moreover, the predicted AMPK site on CEH‐23 is conserved across various nematode species (Fig. S1B), and EMX2, the putative mammalian homolog of CEH‐23, also contains two predicted AMPK motifs. The conserved nature of the predicted AMPK motifs in both CEP‐1 and CEH‐23 suggests that these factors could be AMPK substrates in diverse organisms. It is important to note that whereas we discovered a connection between AAK‐2 and CEH‐23 & CEP‐1 by studying the mitochondrial mutant isp‐1, our results with constitute active aak‐2 animals suggested that CEH‐23 and CEP‐1 are likely downstream mediators of AAK‐2 in a broad contexts beyond mitochondrial dysfunction. Further elaboration of the molecular nature of the regulatory relationship among CEP‐1, CEH‐23, and AAK‐2 will provide important mechanistic insights that will likely be broadly relevant.
Bioinformatic analyses of the AAK‐2, CEH‐23, and CEP‐1 common targets revealed a possible interesting connection to neuronal function. Among the kinases that are commonly regulated by CEH‐23 and CEP‐1 in the isp‐1 mutant and by activated AAK‐2, most belong to the TTBKL (Tau‐tubulin kinase‐like) serine/threonine kinase family. Human TTBK kinases have been associated with neurodegeneration, where TTBK1 is thought to phosphorylate Tau protein and promote the progression of Alzheimer's disease (Sato et al., 2006) and TTBK2 has been linked to another Tau‐related disease, spinocerebellar ataxia 11 (SCA11) (Ikezu & Ikezu, 2014). C. elegans has a single TTBK ortholog and 31 kinases that display sequence similarity to TTBK (Manning, 2005). One of these TTBK‐like kinases has recently been implicated in the pathogenesis of a C. elegans model of the neurodegenerative disease amyotrophic lateral sclerosis (ALS) (Ikezu & Ikezu, 2014). Another interesting group of genes revealed as CEH‐23, CEP‐1, and AAK‐2 common targets encode MSPs. The MSP domain is highly conserved, and a substitution mutation in the human MSP domain protein VAPB has been associated with ALS (Nishimura et al., 2004), suggesting a possible role of MSP domain proteins in neuronal function. Lastly, several sodium:neurotransmitter symporters are overrepresented among the common transcriptional targets of CEH‐23, CEP‐1, and AAK‐2. Taken together, it appears that neuronal signaling might represent a key downstream output of active AAK‐2 and CEH‐23 and CEP‐1 in the isp‐1 mutant. Consistent to our bioinformatic observation, recent study has demonstrated that CRTC1‐dependent transcription is impaired in the early stage of Alzheimer's disease (Parra‐Damas et al., 2014). Moreover, it is interesting to note that ETC dysfunction in the neurons are thought to have a particularly key role in modulating longevity (Dillin et al., 2002). Furthermore, CEH‐23, CRTC‐1, and AAK‐2 are all expressed in neurons (Mair et al., 2011; Spencer et al., 2011; Walter et al., 2011), and the action of AAK‐2 and CRTC‐1 in neurons has also been demonstrated to play a particularly important role in modulating longevity (Burkewitz et al., 2015). An intriguing possibility is that the ETC dysfunction in the isp‐1 mutant induces the activation of AMPK, CEH‐23, and CEP‐1 in the neurons, which elicits a protective effect on lifespan.
AMPK is a critical metabolic sensor and regulator and its deregulation has been linked to major diseases, such as cancer and neurodegenerative disease (Burkewitz et al., 2014; Cai et al., 2012). Likewise, mitochondrial ETC dysfunction is tightly linked to aging and many diseases (García‐Escudero et al., 2013). AMPK has been implicated as a key responder to mitochondrial dysfunction (Curtis et al., 2006; Hwang et al., 2014; Burkewitz et al., 2014), especially because of its ability to respond directly to altered AMP/ATP ratio and redox imbalance (Burkewitz et al., 2014). Our study reveals a new link between the transcription factors CEP‐1/p53 and CEH‐23 to AAK‐2/AMPK and implicates additional downstream signaling as an adaptive response that leads to prolonged lifespan in mutants with reduced ETC function. Importantly, our findings suggest that CEH‐23 and CEP‐1/p53 are key effectors of AAK‐2/AMPK in contexts beyond mitochondrial ETC dysfunction. Further investigation of the molecular relationship of AAK‐2/AMPK, CEH‐23, and CEP‐1/p53 will likely provide important insights that are broadly relevant to aging and age‐related diseases in diverse organisms.
Experimental procedures
C. elegans strains
All strain stocks were maintained on OP50 Escherichia coli at 20 °C and were handled under standard growth conditions. We used the following strains: Wild‐type N2 Bristol, IU279.1 ceh‐23(ms23), IU445 cep‐1(gk138), IU291.4 isp‐1(qm150), IU398.1 nuo‐6(qm200), IU231.2 mev‐1(kn1), IU338.1 gas‐1(fc21), IU293.2 ceh‐23(ms23); isp‐1(qm150), IU448 cep‐1(gk138); isp‐1(qm150), IU418.1 cep‐1(gk138); ceh‐23(ms23); isp‐1(qm150), IU450.1 mev‐1(kn1); cep‐1(gk138), IU451.1 cep‐1(gk138); gas‐1(fc21). Standard genetic methods were used to construct the following strains: IU399.2 nuo‐6(qm200); ceh‐23(ms23), IU481.3 ceh‐23(ms23) mev‐1(kn1), IU477.2 ceh‐23(ms23); gas‐1(fc21), IU507.1 uthIs248; ceh‐23(ms23), IU508.1 uthIs248; cep‐1(gk138), and IU506.1 uthIs272; ceh‐23(ms23). We failed to generate uthIs272; cep‐1(gk138) using the same cross‐strategy possibly because the transgene and cep‐1(gk138) are linked. nuo‐6(qm200) cep‐1(gk138) was generously provided by Dr. Siegfried Hekimi at McGill University. WBM60 uthIs248[Paak‐2::aak‐2 genomic (aa1‐321)::GFP::unc54 3′UTR, Pmyo‐2::tdTomato] is the transgenic strain with overexpressed aak‐2ca and WBM59 uthIs272[Pmyo‐2::tdTomato, unc‐54 3′UTR] is the transgenic control. Both WBM59 and WBM60 were generous gifts from Dr. Willian B. Mair at Harvard University.
Lifespan analysis
All lifespan experiments were performed at 20 °C on Nematode Growth Media (NGM) plates seeded with E. coli OP50 or HT115 for RNAi experiments. For experiments with OP50, bacteria were cultured overnight at 37 °C, the OD600 was measured, and the overnight culture was concentrated to OD600 = 4.0. For RNAi experiments, HT115 bacteria containing vectors expressing dsRNA were concentrated to OD600 = 4. IPTG (4 mm) was added to RNAi bacteria‐seeded plates at room temperature overnight prior to use to induce dsRNA expression. Well‐fed gravid adult worms were allowed to lay eggs at 20 °C, and the progeny were grown at 20 °C. Survival was scored every other day, and the survival curves of each population were estimated using the Kaplan–Meier method, and a log‐rank test was performed for statistical analysis. A P ≤ 0.01 was considered significantly different from the control population. The independent trials were analyzed both separately and pooled between independent trials, and the pooled experiments are presented in the figures. Data from individual trials and pooled trials are presented in Table S1 (Supporting information).
Oxidative stress assay
L4 worms were transferred to NGM plates with 10 mm paraquat or vehicle control (M9). Survival of worms was scored 96 h after exposure to paraquat. The experiment was performed at 20 °C.
Microarray
isp‐1(qm150) and ceh‐23(ms23); isp‐1(qm150) mutant worms were grown on NGM plates with live OP50 bacteria. Worms were harvested when the majority of the population reached mid‐L4 stage. Total RNA was isolated using Tri‐reagent (Molecular Research Center, Inc., Cincinnati, OH USA) and purified with the RNeasy kit (Qiagen). cRNA synthesis/amplification, Cy3/Cy5 dye labeling, and hybridization onto Agilent (Santa Clara, CA USA) 4X44K C. elegans oligonucleotide microarrays (v2) were performed as previously described (Shaw et al., 2007). Hybridized microarray slides were washed according to Agilent instructions and scanned immediately on the Agilent DNA microarray scanner (G2505B) using one color scan setting for 4x44k array slides (scan area 61 × 21.6 mm, scan resolution 5um, dye channel is set to red & green and both the Red and Green PMT is set to 100%). The scanned images were analyzed with Feature Extraction Software 9.1 (Agilent) using parameters (protocol GE1‐105_DEC8 and Grid: 012391_D_20060331) to obtain background subtracted and spatially detrended processed signal intensities. Features flagged in Feature Extraction as Feature Nonuniform outliers were excluded. Two of the four arrays were dye‐flip replicates, and three independent biological experiments were performed. The microarray result was validated using RT‐qPCR. Detailed description of microarray analyses is listed in the Data S1 (supporting information).
Western blot
Worms were grown on 10‐cm NGM plates seeded with OP50 E. coli, synchronized at the mid‐L4 stage, and harvested in M9 buffer. Worms were lysed by boiling. The protein concentrations were quantified using Quick Start Bradford Protein Assay (Bio‐Rad, Hercules, CA USA); 100 ng of total protein was loaded on 8% SDS‐PAGE, electrophoresed, and transferred to a nitrocellulose membrane. The membranes were blocked in TBST with 5% BSA and subsequently incubated with primary antibody, anti‐actin (mouse, Chemicon, Temecula, CA USA), and anti‐phospho‐AMPKα (rabbit, #4188 Cell Signaling Beverly, MA, USA). Anti‐phopho‐AMPKα incubation was performed at 4 °C for 6–9 h, and anti‐actin incubation was performed at room temperature for 1 h. Anti‐mouse or anti‐rabbit secondary antibodies conjugated to fluorescent dyes were used for anti‐actin and anti‐phopho‐AMPKα, respectively. The Western blot images were obtained with the Odyssey infrared imaging system and quantified using Image Studio ver. 2.0. Phospho‐AMPKα levels were normalized to actin as an internal control and normalized to WT.
Development assay
Gravid adult worms were placed onto 60‐mm NGM plates seeded with OP50 E. coli and allowed to lay eggs for 3–5 h. Subsequently, the adults were removed, and the embryos were allowed to develop at 20 °C. The developmental stages were scored 60 h after egg lay. The L1‐L3 larval stages were scored based on gonad structure using a DIC microscope (Leica DM 5000B microscope, Mannheim, Germany). The L4 larval stage and adult worms were identified based on vulval morphology.
Accession number
The GEO accession number for the isp‐1(qm150) vs. ceh‐23(ms23); isp‐1(qm150) microarray dataset in this paper is GSE67754.
Conflict of interest
None declared.
Funding
This work was supported by NIH R01 grant AG024425 to S.S.L. [Correction added on 15 June 2017, after first online publication: The funding information has been added in this current version.]
Author contribution
H‐W. C. and S.S.L. conceived all the experiments, interpreted the results, and wrote the paper. H‐W. C designed and performed most of the experiments. S.B. performed the CRTC‐1 subcellular localization assay. A.C. performed some of the pAAK‐2 Western blots. J.C. performed the oxidative stress assay, and S.G. performed the developmental assay. A.B. performed the microarray experiment comparing transcription profile between isp‐1 mutant and cep‐1; isp‐1 mutant.
Supporting information
Fig. S1 CEH‐23 and CEP‐1 harbor putative AMPK phosphorylation sites.
Fig. S2 (A) Wild‐type C. elegans lifespan was not altered by the combined loss of ceh‐23 and cep‐1. The ceh‐23(ms23) and cep‐1(gk138) single and double mutants were used in the lifespan analysis. (B) aak‐2 RNAi had little effect on the lifespan of the short‐lived gas‐1(fc21) and mev‐1(kn1) mutants. EV: empty vector RNAi control.
Fig. S3 CRTC‐1 antagonizes CEH‐23 and CEP‐1 to modulate the lifespan of isp‐1(qm150) mutant.
Table S1 Quantitative data of individual lifespan experiments.
Table S2 Genes that are differentially expressed between isp‐1(qm150) and ceh‐23(ms23); isp‐1(qm150) (identified by Statistical Analysis of Microarray (SAM) 1 class analysis with false discovery rate (FDR) = 0.59%, 1.5 fold change cutoff).
Table S3 Genes that are commonly regulated by CEH‐23 and CEP‐1 in the isp‐1 mutant (identified by Statistical Analysis of Microarray (SAM) 1 class analysis with false discovery rate (FDR) = 0, 1.5 fold change cutoff).
Table S4 Genes that are differentially expressed between aak‐2ca strain and transgenic control strain. (Data from Mair et al. and reanalyzed using SAM analysis. Gene list was identified by SAM 1 class analysis with FDR = 0, 1.5 fold change cutoff.)
Table S5 Genes that are commonly regulated by CEH‐23, CEP‐1 and AAK‐2 (identified by overlap between gene lists in Table S3 and S4).
Data S1 Supplemental experimental procedures.
Acknowledgments
We thank the Caenorhabditis Genetics Center for providing some of the strains used in this study, Dr. Willian B. Mair at Harvard University for the aak‐2ca strain, and Dr. Siegfried Hekimi at McGill University for the nuo‐6 cep‐1 strain. We thank Dr. Erich Schwarz and Lee laboratory members at Cornell University for helpful discussion and suggestions. We also thank Dr. Kenneth Kemphues and Dr. Thomas D. Fox at Cornell University for helpful discussion and critical reading of the manuscript.
References
- Altun‐Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O (2001) A regulatory cascade of three homeobox genes, ceh‐10, ttx‐3 and ceh‐23, controls cell fate specification of a defined interneuron class in C. elegans . Development 128, 1951–1969. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R (2004) The AMP‐activated protein kinase AAK‐2 links energy levels and insulin‐like signals to lifespan in C. elegans . Genes Dev. 18, 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruah A, Chang H‐W, Hall M, Yuan J, Gordon S, Johnson E, Shtessel LL, Yee C, Hekimi S, Derry WB, Lee SS (2014) CEP‐1, the Caenorhabditis elegans p53 Homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet. 10, e1004097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Zhang Y, Mair WB (2014) AMPK at the nexus of energetics and aging. Cell Metab. 20, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Morantte I, Weir HJM, Yeo R, Zhang Y, Huynh FK, Ilkayeva OR, Hirschey MD, Grant AR, Mair WB (2015) Neuronal CRTC‐1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 160, 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Yan L‐J, Li K, Quazi SH, Zhao B (2012) Roles of AMP‐activated protein kinase in Alzheimer's Disease. Neuromolecular Med. 14, 1–14. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. [DOI] [PubMed] [Google Scholar]
- Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C (2009) A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans . PLoS Genet. 5, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis R, O'Connor G, DiStefano PS (2006) Aging networks in Caenorhabditis elegans: AMP‐activated protein kinase (aak‐2) links multiple aging and metabolism pathways. Aging Cell 5, 119–126. [DOI] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu A‐L, Arantes‐Oliveira N, Lehrer‐Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. [DOI] [PubMed] [Google Scholar]
- Falk MJ, Zhang Z, Rosenjack JR, Nissim I, Daikhin E, Sedensky MM, Yudkoff M, Morgan PG (2008) Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans . Mol. Genet. Metab. 93, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussière F, Hekimi S (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans . Dev. Cell 1, 633–644. [DOI] [PubMed] [Google Scholar]
- García‐Escudero V, Martín‐Maestro P, Perry G, Avila J (2013) Deconstructing mitochondrial dysfunction in alzheimer disease. Oxid. Med. Cell. Longev. 201, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A (2007a) An AMPK‐FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans . Curr. Biol. 17, 1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A (2007b) The energy sensor AMP‐activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107–30119. [DOI] [PubMed] [Google Scholar]
- Greiss S, Schumacher B, Grandien K, Rothblatt J, Gartner A (2008) Transcriptional profiling in C. elegans suggests DNA damage dependent apoptosis as an ancient function of the p53 family. BMC Genom. 9, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman PS, Ishii N, Kayser EB, Morgan PG, Sedensky MM (2001) Mitochondrial mutations differentially affect aging, mutability and anesthetic sensitivity in Caenorhabditis elegans . Mech. Ageing Dev. 122, 1187–1201. [DOI] [PubMed] [Google Scholar]
- Hwang AB, Ryu E‐AE‐A, Artan M, Chang H‐WH‐W, Kabir MH, Nam H‐JH‐J, Lee D, Yang J‐SJ‐S, Kim S, Mair WB, Lee C, Lee SSS‐JS‐J, Lee SSS‐JS‐J (2014) Feedback regulation via AMPK and HIF‐1 mediates ROS‐dependent longevity in Caenorhabditis elegans . Proc. Natl Acad. Sci. 111, 1411199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezu S, Ikezu T (2014) Tau‐tubulin kinase. Front. Mol. Neurosci. 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H (2001) Cell cycle regulation via p53 phosphorylation by a 5′‐AMP activated protein kinase activator, 5‐aminoimidazole‐ 4‐carboxamide‐1‐beta‐D‐ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287, 562–567. [DOI] [PubMed] [Google Scholar]
- Ishii N, Tsuda M, Yasuda K, Yanase S, Suzuki K (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697. [DOI] [PubMed] [Google Scholar]
- Jazwinski SM (2013) The retrograde response: when mitochondrial quality control is not enough. Biochim. Biophys. Acta 1833, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser EB, Morgan PG, Sedensky MM (1999) GAS‐1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans . Anesthesiology 90, 545–554. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. [DOI] [PubMed] [Google Scholar]
- Khan MH, Ligon M, Hussey LR, Hufnal B, Farber R, Munkácsy E, Rodriguez A, Dillow A, Kahlig E, Rea SL (2013) TAF‐4 is required for the life extension of isp‐1, clk‐1 and tpk‐1 Mit mutants. Aging (Albany. NY). 5, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM (1999) Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 152, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Lee S‐J, Hwang AB, Kenyon C (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF‐1 activity. Curr. Biol. 20, 2131–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf‐16: an HNF‐3/forkhead family member that can function to double the life‐span of Caenorhabditis elegans . Science 278, 1319–1322. [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S (2005) Evolutionary conservation of the clk‐1‐dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues APC, Manning G, Montminy M, Shaw RJ, Dillin A (2011) Lifespan extension induced by AMPK and calcineurin is mediated by CRTC‐1 and CREB. Nature 470, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G (2005) Genomic overview of protein kinases. WormBook 13, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Mitne‐Neto M, Silva HC, Richieri‐Costa A, Middleton S, Cascio D, Kok F, Oliveira JRM, Gillingwater T, Webb J, Skehel P, Zatz M (2004) A mutation in the vesicle‐trafficking protein VAPB causes late‐onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF‐16 transduces insulin‐like metabolic and longevity signals in C. elegans . Nature 389, 994–999. [DOI] [PubMed] [Google Scholar]
- Parikh VS, Morgan MM, Scott R, Clements LS, Butow RA (1987) The mitochondrial genotype can influence nuclear gene expression in yeast. Science 235, 576–580. [DOI] [PubMed] [Google Scholar]
- Parra‐Damas A, Valero J, Chen M, Espana J, Martin E, Ferrer I, Rodriguez‐Alvarez J, Saura CA (2014) Crtc1 activates a transcriptional program deregulated at early Alzheimer's disease‐related stages. J. Neurosci. 34, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE (2007) Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans . PLoS Biol. 5, e259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Cerny RL, Buescher JL, Ikezu T (2006) Tau‐tubulin kinase 1 (TTBK1), a neuron‐specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J. Neurochem. 98, 1573–1584. [DOI] [PubMed] [Google Scholar]
- Shaw WM, Luo S, Landis J, Ashraf J, Murphy CT (2007) The C. elegans TGF‐beta Dauer pathway regulates longevity via insulin signaling. Curr. Biol. 17, 1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, Reinke V, Petrella L, Strome S, Von Stetina SE, Katz M, Shaham S, Rätsch G, Miller DM (2011) A spatial and temporal map of C. elegans gene expression. Genome Res. 21, 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura N, Rea SL, Schiavi A, Torgovnick A, Testi R, Johnson TE (2010) p53/CEP‐1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell 8, 380–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Baruah A, Chang H‐W, Pace HM, Lee SS (2011) The homeobox protein CEH‐23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans . PLoS Biol. 9, e1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Boutis P, Hekimi S (1994) Mutations in the clk‐1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139, 1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S (2010) Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans . Aging Cell 9, 433–447. [DOI] [PubMed] [Google Scholar]
- Yee C, Yang W, Hekimi S (2014) The intrinsic apoptosis pathway mediates the pro‐longevity response to mitochondrial ROS in C. elegans . Cell 157, 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 CEH‐23 and CEP‐1 harbor putative AMPK phosphorylation sites.
Fig. S2 (A) Wild‐type C. elegans lifespan was not altered by the combined loss of ceh‐23 and cep‐1. The ceh‐23(ms23) and cep‐1(gk138) single and double mutants were used in the lifespan analysis. (B) aak‐2 RNAi had little effect on the lifespan of the short‐lived gas‐1(fc21) and mev‐1(kn1) mutants. EV: empty vector RNAi control.
Fig. S3 CRTC‐1 antagonizes CEH‐23 and CEP‐1 to modulate the lifespan of isp‐1(qm150) mutant.
Table S1 Quantitative data of individual lifespan experiments.
Table S2 Genes that are differentially expressed between isp‐1(qm150) and ceh‐23(ms23); isp‐1(qm150) (identified by Statistical Analysis of Microarray (SAM) 1 class analysis with false discovery rate (FDR) = 0.59%, 1.5 fold change cutoff).
Table S3 Genes that are commonly regulated by CEH‐23 and CEP‐1 in the isp‐1 mutant (identified by Statistical Analysis of Microarray (SAM) 1 class analysis with false discovery rate (FDR) = 0, 1.5 fold change cutoff).
Table S4 Genes that are differentially expressed between aak‐2ca strain and transgenic control strain. (Data from Mair et al. and reanalyzed using SAM analysis. Gene list was identified by SAM 1 class analysis with FDR = 0, 1.5 fold change cutoff.)
Table S5 Genes that are commonly regulated by CEH‐23, CEP‐1 and AAK‐2 (identified by overlap between gene lists in Table S3 and S4).
Data S1 Supplemental experimental procedures.