

Summary
Subterranean clover is an important annual forage legume, whose diploidy and inbreeding nature make it an ideal model for genomic analysis in Trifolium. We reported a draft genome assembly of the subterranean clover TSUd_r1.1. Here we evaluate genome mapping on nanochannel arrays and generation of a transcriptome atlas across tissues to advance the assembly and gene annotation. Using a BioNano‐based assembly spanning 512 Mb (93% genome coverage), we validated the draft assembly, anchored unplaced contigs and resolved misassemblies. Multiple contigs (264) from the draft assembly coalesced into 97 super‐scaffolds (43% of genome). Sequences longer than >1 Mb increased from 40 to 189 Mb giving 1.4‐fold increase in N50 with total genome in pseudomolecules improved from 73 to 80%. The advanced assembly was re‐annotated using transcriptome atlas data to contain 31 272 protein‐coding genes capturing >96% of the gene content. Functional characterization and GO enrichment confirmed gene expression for response to water deprivation, flavonoid biosynthesis and embryo development ending in seed dormancy, reflecting adaptation to the harsh Mediterranean environment. Comparative analyses across Papilionoideae identified 24 893 Trifolium‐specific and 6325 subterranean‐clover‐specific genes that could be mined further for traits such as geocarpy and grazing tolerance. Eight key traits, including persistence, improved livestock health by isoflavonoid production in addition to important agro‐morphological traits, were fine‐mapped on the high‐density SNP linkage map anchored to the assembly. This new genomic information is crucial to identify loci governing traits allowing marker‐assisted breeding, comparative mapping and identification of tissue‐specific gene promoters for biotechnological improvement of forage legumes.
Keywords: forage legumes, advanced reference assembly, BioNano, transcriptome, gene expression, Legume comparative genomics
Introduction
Forage legumes are highly valued feed for extensive livestock production. There is an increasing interest worldwide in using annual forage legumes as cover crops to supply soil nitrogen (Sulas, 2005; Piano et al., 2010). Symbiotic nitrogen fixation in legumes leads to high protein fodder content and rejuvenated soils for a sustainable feed system. The clovers in particular are among the most effective to break the ‘infernal circle of the fallow’ a technique known to the Germans as ‘Besömmerung’ (Blanning, 2008). Subterranean clover (Trifolium subterraneum L.) makes the greatest contribution to livestock feed production and soil improvement in terms of total worldwide usage among annual clovers (McGuire, 1985), particularly in Australia, where it is sown over 29 mill. ha. The self‐reseeding ability and grazing tolerance of subterranean clover, even under suboptimal and variable environmental conditions (Nichols et al., 2013), contribute to its widespread distribution.
Subterranean clover is a diploid (2n = 2x = 16), predominantly inbreeding, annual species with a relatively small genome size of 540 Mbp (1C = 0.55 pg DNA; Vižintin et al., 2006) that can be readily hybridized, and exhibits wide diversity for both qualitative and quantitative agronomic and morphological characters (Ghamkhar et al., 2011). Within the genus Trifolium, it is established as a reference species for genetic and genomic studies. We reported a de novo draft genome assembly of the subterranean clover TSUd_r1.1, generated using a combination of long‐ and short‐read sequencing platforms (Figure S1) (Hirakawa et al., 2016). Genetic and genomic analyses of the internationally commercially important perennial legumes white clover and red clover are difficult, as they are outcrossing and have self‐incompatible fertilization, with white clover also being an allotetraploid (2n = 4x = 32) (Abberton and Marshall, 2005; Ghamkhar et al., 2011). As molecular markers developed for white and red clovers were readily transferable to subterranean clover (Ghamkhar et al., 2011), it is likely that subterranean clover QTLs and genes are applicable to these other species. Close synteny with the model legume, Medicago truncatula, also provides opportunities for genomic comparisons and the identification of candidate genes.
A variety of approaches are required for the de novo assemblies to improve draft genomes, and include a method developed by Dovetail Genomics for genome scaffolding using long‐range genomic information obtained by Chicago method (Putnam et al., 2016). In this study, we evaluated genome mapping on nanochannel arrays employing BioNano Irys® system (www.bionanogenomics.com) to validate the initial draft assembly, anchor additional unplaced contigs and to resolve misassemblies, with the objective of improving the overall assembly coverage. BioNano genome mapping is a technique of optical mapping in which specific sequence motifs in single DNA molecules are fluorescently labelled. The labelled DNA molecules are loaded onto the IrysChip where they are electrophoretically linearized in thousands of silicon channels. Fluorescence imaging allows the construction of maps of the physical distances between occurrences of the sequence motifs (Lam et al., 2012). The aim was to advance the pseudomolecule assembly of the eight chromosomes of subterranean clover, based on the direct visualizations of sequence motifs on long single DNA molecules. Gene annotations for the de novo draft genome assembly of the subterranean clover TSUd_r1.1 were conducted using in silico automated Augustus and Maker pipelines only. This was compared with direct transcriptome analysis using whole genome RNA sequencing technology across five different tissues of subterranean clover to generate a valuable resource for the identification and characterization of genes and pathways underlying plant growth and development. This also provided a basis to investigate specific processes, biological functions and gene interactions for key agronomic traits.
Results and discussion
The advanced assembly
The first draft genome assembly of the Australian subterranean clover variety, cv. Daliak, covers 85.4% of the estimated genome in eight pseudomolecules of 401.1 Mb length and was constructed using a linkage map consisting of 35 341 SNPs (Hirakawa et al., 2016). In this study, we evaluated genome mapping via scaffolding using de novo physical maps generated using the BioNano Genomics (BNG) Irys platform to improve the genome assembly. A total of 221.7 Gb (401× genome coverage) of filtered data (molecules >150 kb) was generated on the Irys instrument. After filtering out low‐quality single molecules, a total of 188.5 Gb (341× genome coverage) of data was included in the final BioNano‐based de novo physical map assembly. This physical map assembly consisted of 309 075 individual molecules and 468 consensus maps that spanned 512 Mb (93% genome coverage) with N50 of 1.4 Mb. Multiple contigs (264) from the first draft assembly coalesced into 97 super‐scaffolds (containing 43% of the total genome captured) (Figure 1). In the advanced assembly (Tsub_Refv2.0), the size of sequences longer than >1 Mb increased dramatically from 40 to 189 Mb. This resulted in a 1.4‐fold increase in the N50 with the total percentage of genome captured in pseudomolecules improving from 73 to 80% with a substantial reduction of sequence gaps (Table 1).
Figure 1.

(a) In silico map of super‐scaffold 9 aligned to the BioNano optical maps (401× coverage). XMAP alignments for in silico map of sequence scaffolds 103, 143, 1029, 292 and 166 are shown. Consensus genome maps (blue with molecule map coverage shown in dark blue) align to the in silico maps of scaffolds (green with contigs overlaid as translucent coloured squares). An illustration of using BioNano optical maps to assist contig placement, scaffolding and inversion correction. Sequence scaffolds 103, 143, 1029, 292 and 166 were placed within super‐scaffold 9 by the optical maps and among these scaffolds; scaffolds 103, 143 and 166 were reversed in the super‐scaffold. (b) Illustration of misassemblies in the genome examined using BioNano optical maps. The super‐scaffold (colourful bar) contains scaffold Tsud_sc00127.10.1, scaffold Tsud_sc00415.00.1 and a gap between them. Compared with the optical map (blue bar), there are more Bsp QI restriction cut sites (displayed as straight lines) in the super‐scaffold at positions 2; there are some Bsp QI restriction cut sites missing at positions 1. At positions 3, 4 and 5, the super‐scaffold does not consist of BioNano consensus map. All those discordances can be examined in detail in the corresponding contigs or gaps (indicated above the super‐scaffold).
Table 1.
BioNano genome mapping statistics (cv. Daliak)
| N Genome Maps | 468 |
| Total Genome Map Len (Mb) | 512.439 |
| Avg. Genome Map Len (Mb) | 1.095 |
| Genome Map N50 (Mb) | 1.408 |
| Molecule Stats | |
| Contig Coverage (x) | 367.81 |
| Molecules Aligned to the first draft genome assembly TSUd_r1.1 | |
| N mol align | 30 9075 |
| Mol fraction align | 0.37 |
| Tot align len (Mb) | 59930.8 |
| Avg align len (kb) | 193.9 |
In this study, the BNG Irys platform provided affordable, high‐throughput physical maps of improved contiguity to validate the draft assembly (Hirakawa et al., 2016) generated across a combination of long‐read and short‐read platforms and extended scaffolds. In contrast to alternative, sequencing‐based approaches (e.g. Chicago method of Dovetail Genomics, GemCode Technology of 10× Genomics, or Hi‐C), it enables a highly accurate sizing of gaps in sequence assemblies and provides a real picture of genomic regions intractable to current sequencing technologies, such as long arrays of tandem repeats (Staňková et al., 2016). Thus, combinations of platforms are recommended and no one platform is a perfect technology to use in answering every research question (Chaney et al., 2016).
The transcriptome ATLAS and gene features
The identification and annotation of expressed genes within the T. subterraneum genome assembly used high‐throughput whole genome RNA sequencing analyses to predict a total of 32 333 transcripts for 31 272 protein‐coding genes, with evidence for their expression across five different tissue types. The process‐involved annotations that combined evidence from transcriptome alignments obtained from protein homology and in silico gene prediction derived from different tissue types (roots, stem and peduncles, leaf and petioles, flowers and developing seeds) of cv. Daliak (Table S1). Phytozome and TrEMBL were the most informative databases for assigning functional annotations to subterranean clover proteins, with 29 157 (90.2%) and 29 278 (89.9%) proteins annotated, respectively (Table S2). In the draft TSUd_r1.1, the presence of 42 706 genes was predicted from in silico evidence using homology studies, domain searches and ab initio gene predictions. The reduction in genes annotated in the present study was achieved because the latter computational methods are unable to provide definitive evidence about which genes are actually expressed (Guigó et al., 2000; Wheelan and Boguski, 1998).
Repetitive element analysis predicted that 64% of the genome comprises repeat sequences, of which 15.9% and 8.4% are within introns and exons, respectively. About a quarter (23.9%) of repetitive elements were classified in the unknown zone. Like most eukaryotes, transposable elements (TEs) were most commonly long terminal repeats (LTR) retrotransposons (9.3%), with Gypsy‐like elements as the most frequently classified retrotransposons. Other TEs such as DNA, rolling circle (RC) and non‐LTR long retrotransposons such as long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) comprised a relatively small proportion (2.2% and 1.9%, respectively) (Figure 2; Table S3). Non‐coding RNA was estimated to comprise 0.12% of the genome, the majority being ribosomal RNA (0.02%) and transfer RNA (0.01%), with predicted snRNA and miRNA representing 0.01% and 0.07%, respectively (Table S4).
Figure 2.
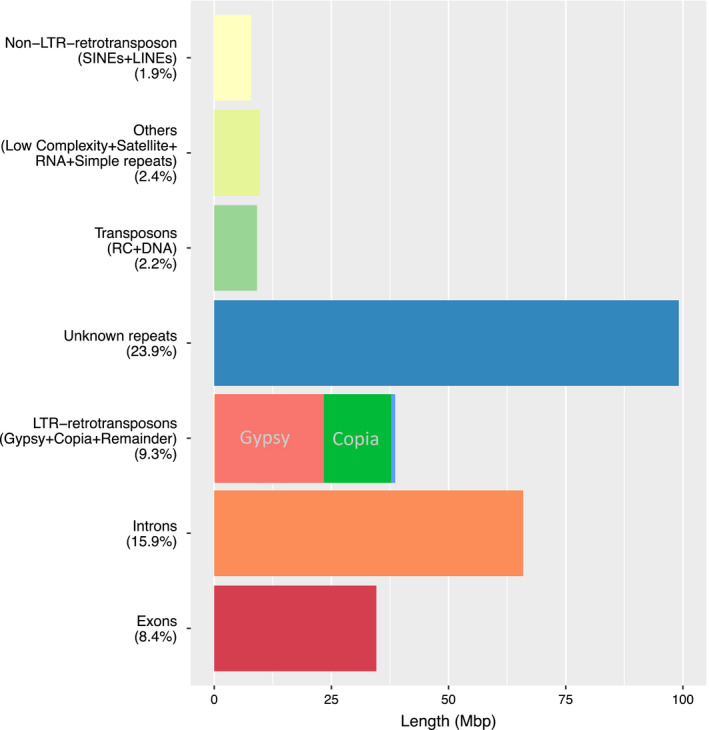
Functional composition of the assembled reference sequence Trifolium subterraneum L. genome.
To test the quality of the advanced genome assembly and re‐annotation, we conducted a CEGMA analysis (Parra et al., 2007) to identify the presence of core eukaryotic genes. From the core set of 248 eukaryotic genes, there were 240 complete and 247 partial genes present in the advanced assembly, representing 96.8% and 99.6% [in comparison with the 95.6% and 98.0% for the draft assembly TSUd_r1.1] genes of the core set, respectively (Table S5).
Functional characterization and GO enrichment analyses of the advanced genome
The advanced assembly was functionally characterized by assigning gene ontology (GO) terms to proteins by manually transferring the GO terms for Swiss‐Prot IDs using the UniProt‐GOA database (Huntley et al., 2015). All these protein‐coding genes/transcripts were grouped into the three main GO categories: biological processes, molecular function and cellular components. A total of 21 210 (65.6%) of the subterranean clover protein‐coding genes/transcripts were assigned GO terms (Table S1). Of the 131 324 GO terms identified, 5,648 appear only once. There are 1013 GO terms appearing between 50 and 200 times with a total sum of 35 383 (Figure S2).
Among the 5648 GO terms appearing once, the top five most highly represented groups in the biological processes category were genes/transcripts associated with signal transduction, response to abscisic acid, cellular response to DNA damage stimulus, methylation and response to water deprivation (Table S6, for enriched cellular components and molecular functions see Tables S7 and S8, respectively). More detailed classification of the biological process GO category also showed enrichment in comparison with whole UniProt for response to cold and oxidative stress, flower development, flavonoid biosynthesis and embryo development ending in seed dormancy as highly represented groups. Overall, this functional characterization and GO enrichment analysis confirmed gene expression for response to water deprivation, cold and oxidative stress, flavonoid biosynthetic process and embryo development ending in seed dormancy reflecting the adaptation of subterranean clover to the harsh Mediterranean environment.
Global gene expression trends
To investigate the representation of genes among the five tissue types, hierarchical clustering of tissues, based on global gene expression and GO terms, was performed on 100 genes with the highest sum of all transcripts per million (TPM). The biological identity of the tissues was clearly reflected in the analysis by hierarchical clustering of the RNA‐Seq‐based transcriptome (Figure 3; Table S9). Predicted genes showed substantial variation in their expression over tissues, reflecting tissue‐specific biological activities. For instance, in leaf and petiole tissue GO‐based clusters G1 and G2 were enriched and genes‐encoding proteins involved in photosynthesis, binding and chloroplast thylakoid membranes were over‐represented consistent with the specialized biological function of these tissues. Likewise, the root transcriptome profile showed enriched expression for stress and defence responses with genes encoding for cysteine‐type endopeptidase inhibitor activity. However, the G3 cluster indicates commonalities in the transcriptome of all above ground vegetative tissue types and showed enrichments for translational and structural ribosomal pathways.
Figure 3.
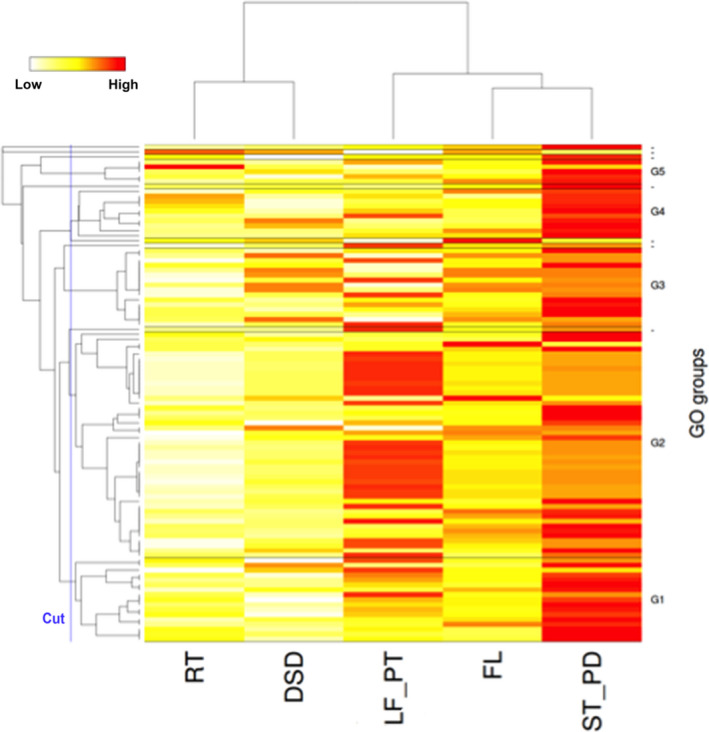
Heat map showing hierarchical clustering of tissues based on global gene expression and GO terms. Clustering was based on 100 genes with highest sum of all TPM (transcripts per million) of the five tissue types. The dendrogram of the selected genes [vertical axis] is visualized along with the expression patterns that are used to cluster the five tissue samples, i.e. root (RT), developing seed (DSD), leaf and petioles (LF_PT), stem and peduncles (ST_PD) and flower (FL) [horizontal axis]. There are five GO‐based clusters named G1…G5 that contain more than one gene. The GO clusters are separated by a horizontal bar in the heat map. Genes without annotations are omitted from the heat map. Highly expressed genes are shown with red colour and lower expressed genes with white or yellow colour, respectively.
For tissues with predominance of a specific biological activity, a large proportion of reads may have represented abundantly expressed genes and, therefore, deeper sequencing may be needed to detect genes with relatively low expression levels. However, the high correlation between biological replicates and the clustering of tissues based on their biology indicates that the sampling depth in this study is sufficient to draw inferences about the transcriptome.
Dynamic spatial gene expression analysis
The tissues sampled in the RNA library preparations allow the determination of dynamic spatial gene expression and characterization. A comparative gene expression analysis between five tissue types (Table S1), using the advanced genome assembly (Tsub_Refv2.0) as the reference, revealed tissue‐specific functional diversification of paralogous genes. Specific gene expression in each of the five different tissues was indicated by the comparison of global gene expression across tissues. To explore gene expression by tissue‐type further, differentially expressed genes were identified in all pairwise tissue‐type comparisons. These differentially expressed genes were then used to identify clusters of co‐expressed genes, which represent spatially enriched expression patterns. Such co‐expression clusters were identified by the Mfuzz package (Futschik and Carlisle, 2005) using the soft clustering approach with fuzzy c‐means algorithm. This analysis generated five clusters, which revealed dynamic gene expression patterns across the five tissue types (Figure 4a; Table S10).
Figure 4.
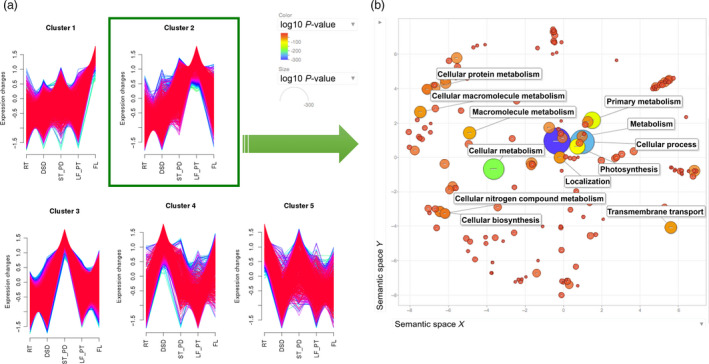
Dynamic tissue‐specific gene co‐expression clusters. (a) Five clusters were generated using Fuzzy C‐means soft clustering algorithm implemented in Mfuzz. Data points on the X‐axis represent root (RT), developing seed (DSD), stem and peduncles (ST_PD), leaf and petioles (LF_PT), flowers (FL) tissues, respectively. The Y‐axis represents gene expression values, where gene expression values were standardized to have zero mean and one standard deviation. (b) The scatterplot generated using REVIGO web browser shows the cluster representatives (i.e. terms remaining after the redundancy reduction) for leaf and petiole tissue‐enriched Cluster 2 biological processes in a two‐dimensional space derived by applying multidimensional scaling to a matrix of the GO terms’ semantic similarities. Bubble colour indicates the P‐value (legend in upper left‐hand corner); size indicates the frequency of the GO term in the wholeUniProt GOA database (bubbles of more general terms are larger).
Cluster 1 included genes with the highest expression in floral tissue (Figure 4b). Among genes in this cluster several are of interest. For example, this cluster includes many genes defining floral organ identity such as floral homeotic protein AGAMOUS, APETALA 2, PMADS 2 and DEFICIENS. In addition, Cluster 1 also included chromatin structure‐remodelling complex protein SYD, MADS‐box protein CMB1 and many chloroplast and carotenoid genes. Leaf‐enriched Cluster 2 included transcripts‐encoding enzymes involved in photosynthesis, Rubisco and oxygenase activity in addition to many chloroplast and plastid‐regulating genes. Cluster 3 contained genes with high expression in stem and peduncle tissue, and included genes/transcripts‐encoding enzymes involved in nutrient transport, structural growth with a high activity of various receptor kinases, expansins and transferases. Developing seed‐enriched Cluster 4 showed genes genes/transcripts involved with embryogenesis and growth. Cluster 5 enriched with root tissue gene expression profiles included different nodulins, binding proteins, MYB‐transcription factors and both stress‐ and defence‐related genes.
Genes within different clusters are potential sources of tissue‐specific promoters. For example, to improve forage nutritional quality by isoflavonoid biosynthesis or digestibility through processes such as cell wall loosening and lignin biosynthesis, promoters specific to leaf tissue are required. Options for transformation have recently broadened to include genome‐editing tools such as CRISPR‐cas9 (Rani et al., 2016).
Functional classification of the tissue‐enriched gene expression clusters
Genes representing tissue‐enriched clusters were subjected to GO enrichment analysis to further characterize and identify over‐represented functional groups in subterranean clover using REVIGO web server (Supek et al., 2011) to graphically represent the results. The scatterplot depicting leaf‐enriched cluster (Cluster 2) showed enrichment for genes associated with photosynthesis, transport and response to stimulus (Figure 4b; Table S11). The flower‐enriched cluster (1) showed a concentration of genes associated with the processes of nitrogen compound cellular biosynthesis, multicellular organismal organization and development (Table S12). The stem‐ and peduncle‐enriched cluster (3) displayed a high representation of genes associated with response to stimulus, stress, oxidation‐reduction process, transport and signal transduction. The root‐enriched cluster (5) had a high representation of genes associated with response to stress, stimulus, nutrient levels, transport and nitrate metabolism. Clearly, the GO enrichment analyses of tissue‐enriched clusters showed over‐representation of predicted classes of genes in different tissue types and thereby validated our tissue‐enriched gene co‐expression clusters. Additionally, putative genes‐encoding enzymes in subterranean clover were assigned to various pathways in the KEGG database using BLASTKOALA (Kanehisa et al., 2016). This analysis distributed 4,094 genes‐encoding enzymes (12.7%) into 133 different KEGG pathways (Figure S3; Table S13).
Papilionoideae whole genome phylogenetic analyses
Comparative analyses across the Papilionoideae using the published whole genome sequences for 13 legumes and the Tsub_Refv2.0 (Figure 5a and b; Table S14) revealed an overlap of 12 170 orthologous gene clusters between Papilionoideae and Arabidopsis thaliana as the outgroup species. Within the Papilionoideae, the number of orthologous gene clusters specific to galegoid (Lotus japonicus, Trifolium pratense, T. subterraneum, M. truncatula and Cicer arietinum), millettioid (Glycine max, Cajanus cajan, Vigna radiata, V. angularis and Phaseolus vulgaris) and dalbergoid (Arachis ipaensis and A. duranensis) was 10 873, 7976 and 4004, respectively. There were 3171 orthologous gene clusters in common among galegoid, millettioid and dalbergoid species.
Figure 5.
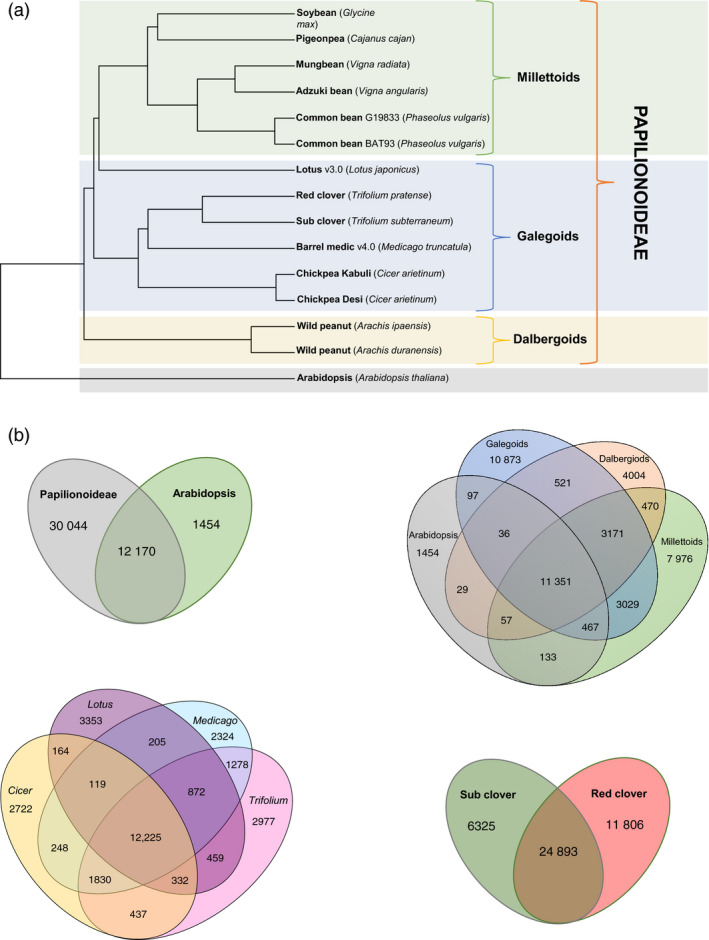
(a) Phylogenetic tree using the published predicted protein sequences for 13 legume genomes including the newly annotated Trifolium subterraneum L. and Arabidopsis thaliana. Mash v1.1 (preprint: http://biorxiv.org/content/early/2016/04/19/029827) was used to calculate a distance matrix and pairwise mutation distance estimation using the published whole genome sequences for all the legumes except the transcriptome assembly for Cajanus cajan as a whole genome assembly was not available. The phylogeny was constructed using UPGMA as implemented in the R‐package ‘phangorn’ v2.0.3. (b) Shared and unique gene clusters in A) Papilionoideae species and A. thaliana; B) Millettioid, Galegoid and Dalbergioid clade or and A. thaliana; C) Lotus, Trifoliums, Medicago and Cicers; D) Shared and unique genes in sub‐clover and red clover.
To find Trifolium‐specific genes among the galegoids, we identified 18 131 genes representing 24.3% of the total 74 556 genes using BLAST searches (Table S15). This proportion is higher than reported in chickpea (10%; Garg et al., 2011; Jain et al., 2013) Arabidopsis (4.9%; Lin et al., 2010) and rice (17.4%; Campbell et al., 2007). Trifolium‐specific gene clusters are a source of unique genes controlling important traits as drought tolerance, and disease resistance. Within the genus Trifolium, similar BLAST searches identified 6,325 (19.6%) and 11 806 (28.0%) genes as subterranean‐clover‐specific and red‐clover‐specific genes, respectively (Tables S16 and S17). These candidate subterranean‐clover‐specific genes could be mined further for such key traits as geocarpy and other factors related to the grazing tolerance of subterranean clover. Examining orthologous gene clusters provides an important foundation for comparative biology and functional inference in subterranean clover, because genes with simple orthologous relationships often have conserved functions, whereas genes duplicated more recently relative to speciation often underlie functional diversification. Comparative genomic analysis of the advanced subterranean assembly with the model legume, Medicago truncatula, showed close synteny and extensive collinearity of large sequence blocks (Figure 6). This provides opportunities for genomic comparisons and translation to identify candidate genes for traits of interest in the two species.
Figure 6.
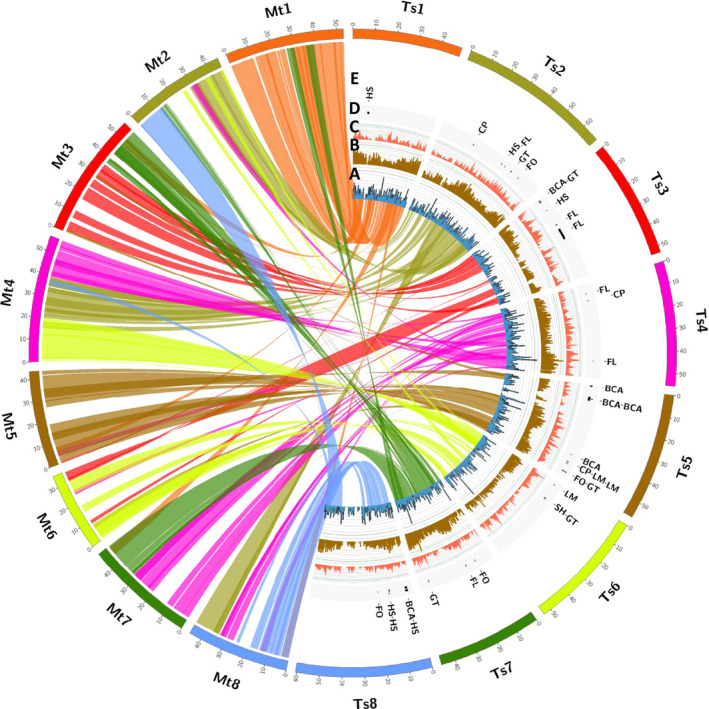
Graphical view of the genome structure of Trifolium subterraneum L. A) Syntenic relationship between Medicago truncatula (left) and T. subterraneum (right) with synteny links and synteny density histograms; B) T. subterraneum gene densities C) T. subterraneum SNP densities; D) T. subterraneum QTLs mapped for important traits using the high‐density SNP linkage map E) Names for the QTLs mapped for important traits using the high‐density SNP linkage map [FL Flowering time, LM Leaf marks, CP Calyx pigmentation, SH Stem Hairiness, FO Formononetin, GT Genistein, BCA Biochanin A and HS Hardseededness].
Marker‐trait association studies
Phenotypic information for eight important traits described by Ghamkhar et al. (2011) (flowering time, hardseededness, leaf marks, calyx tube pigmentation, stem hairiness and the isoflavonoids, formononetin, genistein, and biochanin A) was associated with specific regions in the revised high‐density SNP linkage map (Tables S18 and S19) previously described by Hirakawa et al., 2016;. Significant associations were then mapped onto the advanced assembly (Tsub_Refv2.0) to identify possible candidate genes (Figure 6; Table 2; Table 3). The effective anchoring of sequence scaffolds onto the high‐density SNP linkage map in a high‐quality chromosome‐level genome assembly was a major factor in the identification of loci governing key traits. For example, QTLs for leaf marks (LM), calyx pigmentation (CP) and the isoflavonoids [formononetin (FO), genistein (GT) and biochanin A (BCA)] were found to map adjacently to a region on Chr 5. The linkage between these traits was demonstrated by Francis and Millington (1965), who used mutagenesis on cv. Geraldton, which has a high formononetin content, a prominent leaf mark and anthocyanin pigmentation of leaves, calyx tubes, stipules and stems to produce the low formononetin cv. Uniwager, which concomitantly lost its leaf mark and anthocyanin pigmentation. Co‐localization of the QTLs identified for the traits in the present study explains the observation of breeders that isoflavone content is linked to pigmentation traits (leaf and calyx) (Figure 6; Table 3) (Francis and Millington, 1965). This information illustrates links between the new assembly, the high‐density linkage map and key quantitative traits that can assist future marker‐assisted selection and comparative mapping with other species to improve forage legumes and increase livestock productivity.
Table 2.
Assembly and predicted gene statistics for advanced Tsub_Refv2.0 assembly using RNAseq supported data in comparison to the first draft genome assembly TSUd_r1.1
| TSUd_r1.1 contigs | TSUd_r1.1_pseudomolecules | Tsub_Refv2.0 scaffolds | Tsub_Refv2.0_pseudomolecules | |
|---|---|---|---|---|
| Number of Sequences | 27 424 | 1702 | 27 257 | 1545 |
| Maximum length (bases) | 2 878 652 | 63 731 624 | 7 089 608 | 67 952 282 |
| Minimum length (bases) | 300 | 42 658 284 | 300 | 46 363 611 |
| Average length (bases) | 17 205 | 50 143 517 | 19 481 | 55 397 738 |
| Total % of genome captured in pseudomolecules (bp)a | – | 72.62 | – | 80.23 |
| N50 length (bases) | 287 605 | – | 410 493 | – |
| Number sequences >100 Kbp | 1 302 | – | 1 135 | – |
| Size of sequences >100 Kbp | 386 665 122 | – | 445 838 546 | – |
| Number sequences >1 Mbp | 31 | – | 86 | – |
| Size of sequences >1 Mbp | 40 489 272 | – | 189 281 499 | – |
| Number of sequences contained in super–scaffolds | – | – | 97b | – |
| Length of sequences contained in super–scaffolds | – | – | 193 110 501 | – |
| GC% | 33.3 | – | 33.3 | – |
| Number of predicted genes | 42 706 | – | 32 333 | – |
| Total length of predicted genes (bp) | 47 965 017 | – | 34 758 167 | – |
| Average length of predicted genes (bp) | 1123 | – | 1075 | – |
| Max length of predicted genes (bp) | 15 417 | – | 15 309 | – |
| Min length of predicted genes (bp) | 150 | – | 201 | – |
| N50 length of predicted genes (bp) | 1548 | – | 1437 | – |
Estimated genome size 552.4 Mb.
Consists of 264 contigs from TSUd_r1.1.
Table 3.
Major QTLs and candidate genes mapped for the important traits using the high‐density SNP linkage map anchoring to the advanced Tsub_Refv2.0 assembly
| Trait Name | Trait ID | Chr | Left Marker | Right Marker | LOD | PVE (%) | Candidate genes |
|---|---|---|---|---|---|---|---|
| Flowering time | FL | 3 | Tsud_sc00634.00_113902 | Tsud_sc01407.00_29664 | 36.36 | 61.94 | 87 |
| 3 | Tsud_sc00557.00_23548 | Tsud_sc01682.00_2842 | 4.78 | 4.93 | 6 | ||
| 2 | Tsud_sc01286.00_17178 | Tsud_sc01608.00_15349 | 3.25 | 3.14 | 5 | ||
| 4 | Tsud_sc00001.20_668469 | Tsud_sc00001.20_681817 | 3.10 | 2.99 | 2 | ||
| 4 | Tsud_sc00277.00_152530 | Tsud_sc00277.00_243760 | 2.76 | 2.43 | 8 | ||
| 7 | Tsud_sc00124.00_567396 | Tsud_sc00124.00_311320 | 2.98 | 2.85 | 28 | ||
| Leaf marks | LM | 5 | Tsud_sc01769.00_33298 | Tsud_sc01806.00_29808 | 91.74 | 52.58 | 3 |
| 5 | Tsud_sc00041.00_165499 | Tsud_sc00041.00_347862 | 25.67 | 8.90 | 16 | ||
| 6 | Tsud_sc00011.20_420838 | Tsud_sc00011.20_536648 | 2.76 | 0.61 | 15 | ||
| Calyx pigmentation | CP | 5 | Tsud_sc00695.00_95603 | Tsud_sc00094.10_42823 | 111.79 | 96.88 | 13 |
| 4 | Tsud_sc00164.00_12669 | Tsud_sc00164.00_104886 | 2.96 | 0.50 | 6 | ||
| 2 | Tsud_sc00213.10_37103 | Tsud_sc00274.00_337396 | 2.70 | 0.48 | 3 | ||
| Stem hairiness | SH | 6 | Tsud_sc00057.00_9596 | Tsud_sc00057.00_401981 | 136.43 | 98.71 | 30 |
| Formononetin | FO | 5 | Tsud_sc00724.00_157584 | Tsud_sc00955.00_89974 | 17.25 | 25.53 | 30 |
| 2 | Tsud_sc00651.20_25763 | Tsud_sc01310.00_44071 | 12.43 | 15.53 | 5 | ||
| 7 | Tsud_sc00459.00_51828 | Tsud_sc00459.00_195702 | 7.42 | 9.03 | 13 | ||
| 8 | Tsud_sc00629.00_42778 | Tsud_sc00629.00_69362 | 4.12 | 4.74 | 3 | ||
| Genistein | GT | 2 | Tsud_sc01915.00_18283 | Tsud_sc00906.00_202766 | 5.80 | 12.07 | 19 |
| 3 | Tsud_sc00026.00_688983 | Tsud_sc00178.00_122069 | 5.57 | 11.50 | 34 | ||
| 7 | Tsud_sc00024.00_176871 | Tsud_sc00024.00_366227 | 5.05 | 10.00 | 25 | ||
| 6 | Tsud_sc01326.00_38538 | Tsud_sc01503.00_12748 | 3.12 | 6.17 | 0 | ||
| 5 | Tsud_sc00724.00_157584 | Tsud_sc00955.00_89974 | 2.75 | 5.38 | 30 | ||
| Biochanin_A | BCA | 3 | Tsud_sc00211.20_34823 | Tsud_sc00026.00_63478 | 9.60 | 23.45 | 19 |
| 8 | Tsud_sc00654.20_18111 | Tsud_sc01006.00_41002 | 4.53 | 10.40 | 30 | ||
| 5 | Tsud_sc00222.00_346664 | Tsud_sc00242.00_222297 | 2.92 | 6.22 | 37 | ||
| 5 | Tsud_sc00285.00_86451 | Tsud_sc00285.00_325955 | 2.76 | 5.84 | 10 | ||
| 5 | Tsud_sc02041.00_8358 | Tsud_sc00674.20_21693 | 2.70 | 5.76 | 1 | ||
| 5 | Tsud_sc00073.20_492966 | Tsud_sc00076.10_146128 | 2.64 | 5.60 | 87 | ||
| Hardseededness | HS | 1 | Tsud_sc00068.20_1257208 | Tsud_sc00855.00_29720 | 9.29 | 36.24 | 80 |
| 3 | Tsud_sc00009.30_108575 | Tsud_sc00009.30_115638 | 2.76 | 6.62 | 2 | ||
| 8 | Tsud_sc00654.20_18111 | Tsud_sc01006.00_41002 | 2.70 | 6.50 | 30 | ||
| 8 | Tsud_sc00309.00_532001 | Tsud_sc00204.10_27738 | 2.58 | 6.46 | 10 | ||
| 8 | Tsud_sc00204.10_27738 | Tsud_sc00431.00_91208 | 2.61 | 6.29 | 12 | ||
| 2 | Tsud_sc00682.00_120236 | Tsud_sc01560.00_10224 | 2.55 | 6.16 | 6 |
Conclusion
To improve the draft assembly of subterranean clover, the BNG Irys® system provided affordable, high‐throughput physical maps of high contiguity to validate the draft and extend existing scaffolds. Effective anchoring of sequence scaffolds to genetic linkage groups, coupled with the use of the BioNano system, resulted in a high‐confidence chromosome‐level genome assembly. Tissue‐specific transcript profiling with RNA‐Seq technology delivered gene expression data of high value for gene annotation of the assembly (Tsub_Refv2.0) and transcriptional dynamics to understand tissue‐specific pathways. This new genomic information is the key to identifying loci governing traits that allow marker‐assisted breeding in subterranean clover for comparative mapping with other species and the identification of tissue‐specific gene promoters for biotechnological improvement of forage legumes.
Experimental procedures
Plant materials
Suspensions of intact cell nuclei were prepared according to Vrána et al. (2016). Briefly, mature dry seeds of subterranean clover (Trifolium subterraneum L.) cv. Daliak (approx. 20 g) were germinated on moistened paper towels in the dark at 25° ± 0.5 °C until roots achieved 2–3 cm in length. Roots were cut to 1 cm from the apex and transferred into 25 mL formaldehyde fixative (2% v/v) for 20 min at 5 °C, followed by three 5‐min washes in Tris buffer. Finally, the root tips (approx. 40) were cut, transferred into 1 mL IB buffer (Šimková et al., 2003) and homogenized using a mechanical homogenizer (blender) at 13 000 RPM for 18 s. The crude homogenate was filtered through 50‐μm (pore size) nylon mesh and stained with DAPI (2 μg/mL final concentration). A total of fifteen samples were prepared.
For transcriptome work, subterranean clover cv. Daliak plants were sown on 1 May 2015 in the field at Shenton Park, Western Australia (31o57′S, 115o50′E). Five different tissue types (roots, stem and peduncles, leaf and petioles, flowers and developing seeds) were harvested from a single Daliak plant on 24 September, when it was flowering and setting seeds. All samples were taken between 10.00 am and noon to eliminate any diurnal variations.
Preparation of high molecular weight (HMW) DNA for BioNano mapping
Cell nuclei were purified by a FACSAria II SORP flow cytometer and sorter (BD Biosciences, San Jose, CA) equipped with UV laser for DAPI excitation. Populations of G1 nuclei were sorted in batches of 700 000 into 1.5‐mL polystyrene tubes containing 660 μL IB buffer. In total, four batches of nuclei were sorted and each batch was used for preparation of one 20‐μL agarose miniplug. DNA embedded in miniplugs was purified by proteinase K (Roche, Basel, Switzerland) treatment according to Šimková et al. (2003). The miniplugs were washed four times in wash buffer (10 mm Tris, 50 mm EDTA, pH 8.0) and five times in TE buffer (10 mm Tris, 1 mm EDTA, pH 8.0) and then melted for 5 min at 70 °C and solubilized with GELase (Epicentre, Madison, WI) for 45 min. A drop dialysis against TE buffer (Merck Millipore, Billerica, MA) was performed for 90 min to purify DNA from any residues and subsequently quantified using a Quant‐iTTM PicoGreen® dsDNA assay (Thermo Fisher Scientific, Waltham, MA).
RNA extractions and library preparation
Total RNA from all the tissue samples (Figure S4) was extracted using the Spectrum™ Plant Total RNA Kit (Sigma‐Aldrich, USA) following the manufacturer's instructions. Aliquots of purified RNA were stored at −80 °C. The concentration of RNA was confirmed using Qubit fluorometer with Qubit RNA assay kit (Life Technologies, Australia). The integrity of total RNA was determined by electrophoretic separation on 1.2% (w/v) denaturing agarose gels. Sequencing libraries were constructed using 500 ng of total RNA with a TruSeq® Stranded Total RNA Sample Prep Kits with Ribo‐Zero (Illumina, San‐Diego, USA) following the manufacturer's instructions. Library concentrations were measured using a Qubit fluorometer with Qubit dsDNA BR assay kit (Life Technologies, USA) and Agilent high‐sensitivity DNA chips (Agilent Technologies, USA). The amplified libraries were pooled in equimolar amounts, and quality was assessed with Agilent high‐sensitivity DNA chips (Agilent Technologies, USA). Paired‐end 100‐bp x 2 sequencing was performed with HiSeq2000 (Illumina, San‐Diego, USA).
BioNano mapping
The genome sequence of subterranean clover (TSUd_r1.1) (Hirakawa et al., 2016) was analysed with Nickers software to assess the frequency of recognition sites for four different nicking enzymes (Nt.BbvC1, Nt.BsPQ1, Nb.bsm1 and Nb.BsrD1). The optimal labelling frequency was calculated for endonuclease Nt.BsPQ1 (7.6 sites/100 kb). DNA was then processed using NLRS protocol using the IrysPrep® Reagent Kit (BioNano Genomics, San Diego, CA) following manufacturer's instructions. DNA was nicked using 8U of Nt.BspQ1 (New England BioLabs, Beverly, MA) for two hours at 37 °C in NEBuffer 3. The nicked DNA was labelled with a fluorescent‐dUTP nucleotide analogue using Taq polymerase (New England BioLabs) for one hour at 72 °C. After labelling, the nicks were ligated with Taq ligase (New England BioLabs) in the presence of dNTPs for 30 min at 37 °C. The backbone of the labelled DNA was stained with IrysPrep® DNA Stain (BioNano Genomics). The NLRS DNA concentration was measured again with the Quant‐iTTM PicoGreen® dsDNA assay.
Labelled and stained DNA was loaded on the Irys chip, and four consecutive runs were performed (each run consisting of 30 cycles). A total of 489.8 Gb data were generated, of which 221.7 Gb exceeded 150 kb, the threshold for map assembly. These filtered data (>150 kb), corresponding to 401× coverage of the subterranean clover genome, were compiled from 994 895 molecules with N50 of 215.4 kb. De novo assembly of the BioNano map was performed by a pairwise comparison of all single molecules and graph building (Cao et al., 2014). A P‐value threshold of 1e−9 was used during the pairwise assembly, 1e−10 for extension and refinement steps and 1e−15 for a final refinement.
Stitch: super scaffolding and correcting potential misassemblies
The complete pipeline of the Stitch algorithm by Shelton et al. (2015) (https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/assemble_XeonPhi_LAB.md) was run on the online cluster zythos provided by the Pawsey Supercomputing Center, Western Australia. The first draft genome assembly of T. subterraneum (TSUd_r1.1) (Hirakawa et al., 2016) was used as the reference genome. Super‐scaffolds were generated, and BioNano IrysView was used to examine the new assemblies and the alignments between BioNano consensus maps and the in silico scaffolds. Based on these alignments, misassemblies were identified in TSUd_r1.1 (Figure 1b).
Reference genome‐guided transcriptome sequence assembly
All RNASeq libraries were Illumina, San‐Diego, USA TruSeq adapter trimmed and quality trimmed (sliding window, minimum quality score: 20) using Trimmomatic v 0.36 (Bolger et al., 2014). Trimmed libraries were aligned to the advanced reference sequence using HISAT2 v 2.0.1 (insert size 0 to 1000) (Kim et al., 2015). The resulting SAM files were converted to BAM format using samtools v1.3 (Li et al., 2009).
BRAKER1 v 1.9 (Hoff et al., 2016) was used to predict genes using GeneMark‐ET v 4.32 (Lomsadze et al., 2014) and AUGUSTUS v 3.2.1 (Stanke et al., 2008) based on the RNASeq alignments.
Functional annotation and classification of the transcriptome
The resulting predicted proteins were aligned to several databases using blastp v 2.2.31+ (Altschul et al., 1990) (minimum e‐value 1e−10). The database used were Swiss‐Prot and TrEMBL downloaded on March 13 2016 (Boeckmann et al., 2003) and all 2 542 385 predicted proteins downloaded from Phytozome v11 on 14 March 2016 (Goodstein et al., 2012). For each predicted protein, the hit with the highest score and lowest e‐value was chosen as annotation.
GO terms were assigned to proteins by manually transferring the GO terms for Swiss‐Prot IDs using the UniProt‐GOA database downloaded on 13 March 2016 (Huntley et al., 2015). KEGG K numbers were assigned to all predicted proteins using BLASTKOALA (taxonomy group: Plants, KEGG GENES database: family_eukaroytes) (Kanehisa et al., 2016).
Tissue‐specific expression analysis
Tissue expression was estimated using kallisto v 0.42.4 (Bray et al., 2015). All genes with total transcripts per million (TPM) count below 1 or where more than two tissue types had a TPM of 0 were removed. To make log‐normalization possible, 0.25 was added to the remaining TPM values. Expression was log‐normalized and clustered into 12 clusters using Mfuzz v. 2.30.0 (Futschik and Carlisle, 2005) (m = 1.25).
Hierarchical clustering of tissues based on global gene expression
Clustering was based on 100 genes with highest sum of all TPM (transcripts per million) of the five tissue types. The dendrogram of the selected genes [vertical axis] is visualized along with the expression patterns used to cluster the five tissue samples, i.e. root (RT), developing seed (DSD), leaf and petioles (LF_PT), stem and peduncles (ST_PD) and flower (FL) [horizontal axis]. There are five GO‐based clusters named G1, G2, G3, G4 and G5 that contain more than one gene. The GO clusters are separated by a horizontal bar in the heat map. Genes without annotations are omitted from the heat map. Over‐expressed genes are shown with red colour and under expressed genes with white or yellow colour, respectively (Figure 3).
Genome structure and synteny analysis
Syntenic relationships between M. truncatula and T. subterraneum were calculated using SyMAP v4.2 (Soderlund et al., 2006, 2011). The T. subterraneum genome was mined for repeats using RepeatModeler and RepeatMasker (http://www.repeatmasker.org).
Linkage map construction and quantitative trait locus (QTL) analyses
The linkage map was constructed using MultiPoint 3.3 (http://www.multiqtl.com/) as described in (Hirakawa et al., 2016). The complete set of 188 F2 lines of a biparental population 92S80 (cv. Woogenellup x cv. Daliak) phenotypic data as reported by Ghamkhar et al., 2011 was used for QTL screens for the following morphological and agronomic traits: levels of the oestrogenic isoflavones, formononetin (FO), genistein (GT) and biochanin A (BCA); days to first flowering (FT); leaf marks (LM); pigmentation of calyces (CP); hairiness of stems (SH); and hardseededness (HS). Levels of the isoflavones FO, GT and BCA were measured using the technique of Francis and Millington (1965). FT was measured as the number of days from sowing to appearance of the first flower. The morphological traits, LM, CP and SH, were scored 120 days after sowing, using the rating systems given in Nichols et al., (1996). Hardseededness was measured in the laboratory using the method of Quinlivan (1961).
QTL screens for the traits reported (Table 3) were conducted using an inclusive composite interval mapping (ICIM) approach implemented in QTL IciMapping v4.0 (Wang et al., 2014). Missing phenotypes were deleted using the ‘Deletion’ command in the software. The walking speed was set at 1 cm. A suitable probability for entering marker variables in stepwise regression was chosen so that the variation explained by the model approximated the trait heritability. The regression model was then used for background genetic variation control in the ICIM QTL mapping. The LOD was calculated using 1000 permutations, with a Type 1 error being 0.05, and significant QTLs were defined accordingly.
Papilionoideae whole genome phylogenetic analyses
A phylogenetic tree was constructed, based on published predicted protein sequences for 13 legume genomes, including those of the newly annotated Trifolium subterraneum L. and A. thaliana. Mash v1.1 (Ondov et al., 2016) was used to calculate a distance matrix using k‐mer counting and pairwise mutation distance estimation, using the predicted amino acids from the published whole genome sequences for all the legumes, except Cajanus cajan, for which only the transcriptome assembly was used (as the whole genome assembly is not available). The tree was then constructed using UPGMA as implemented in the R‐package ‘phangorn’ v2.0.3. Shared and unique gene families were called using BLAST and OrthoMCL (Li et al., 2003). OrthomclToVenn (https://github.com/philippbayer/orthomcltovenn) was used to calculate the number of unique and shared genes between: (i) the Papilionoideae species and A. thaliana; (ii) the Millettioid, Galegoid and Dalbergioid clades; (iii) A. thaliana and the Lotus, Trifolium, Medicago and Cicer genera; and (iv) T. subterranean and T. pratense.
Accession code
Advanced genome sequence assembly and annotation data have been made available (under embargoed) at https://zenodo.org/record/161479, DOI: http://doi.org/10.5281/zenodo.161479
Author contributions
P.K. conceived and performed the research and wrote the manuscript with contributions from P.E.B, Z.M, J.V, Y.Y, R.A, D.E, J.B, P.N, J.D and W.E. The BioNano Irys®;System genome mapping experiments were led and designed by J.D and performed and analysed by Z.M, J.V and Y.Y. P.E.B and Y.Y did the bioinformatics analysis and helped with the figure preparations. All authors read the manuscript and approved the content.
Supporting information
Figure S1 The strategy and status of sequencing and assembly.
Figure S2 Functional GO characterization of Trifolium subterraneum L. genes.
Figure S3 Distribution of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the Trifolium subterranean annotation.
Figure S4 Five tissue types of Trifolium subterraneum L. used for the transcriptome ATLAS.
Table S1 Functional annotation of the predicted protein‐coding genes and transcripts.
Table S2 Functional annotation of the predicted protein‐coding genes.
Table S3 Functional composition of the assembled reference sequence Trifolium subterraneum L. genome assembly TSub_v2.0.
Table S4 Non‐coding RNA genes in the advanced draft genome assembly.
Table S5 Statistics of the completeness of the advanced genome Tsub_Refv2.0 assembly based using CEGMA pipeline.
Table S6 List of 562 enriched GO terms among the unique 5648 GO terms, in the biological processes category using REVIGO web server.
Table S7 List of 134 enriched GO terms among the unique 5648 GO terms, in the cellular processes category using REVIGO web server.
Table S8 List of 316 enriched GO terms among the unique 5648 GO terms, in the molecular function category using REVIGO web server.
Table S9 Hierarchical clustering of tissues based on global gene expression and GO terms.
Table S10 Clustering of co‐expressed genes based on spatially enriched expression patterns.
Table S11 List of GO terms for the leaf‐enriched Cluster 2, using REVIGO web server.
Table S12 List of GO terms for the flower‐enriched Cluster 1, using REVIGO web server.
Table S13 Number of putative genes‐encoding enzymes those mapped on KEGG pathways.
Table S14 Distance matrix and pairwise mutation distance estimation calculated by Mash v1.1 (preprint: http://biorxiv.org/content/early/2016/04/19/029827) using the published whole genome sequences for all the legumes except the transcriptome assembly for Cajanus cajan as a whole genome assembly not available.
Table S15 List of Trifolium‐specific genes among the galegoids identified using BLAST searches.
Table S16 List of subterranean‐clover‐specific genes among the Trifoliums identified using BLAST searches.
Table S17 List of red‐clover‐specific genes among the Trifoliums identified using BLAST searches.
Table S18 Statistics of a SNP linkage map and numbers of anchored scaffolds.
Table S19 High‐density SNP linkage map and anchoring information for Trifolium subterraneum L. genome assembly TSub_v2.0.
Acknowledgements
Meat & Livestock Australia provided financial support to the study. Z.M., J.V. and J.D. were supported by grant award LO1204 from the National Program of Sustainability I and by the Czech Science Foundation (award no. P501/12/G090). We thank Zdeňka Dubská for assistance with nuclei flow sorting, Helena Staňková for help with BioNano mapping and Hana Šimková for advice on BioNano mapping. We acknowledge the supercomputing resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. The authors declare no conflicts of interest.
References
- Abberton, M.T. and Marshall, A.H. (2005) Progress in breeding perennial clovers for temperate agriculture. J. Agric. Sci. 143, 117–135. [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Blanning, T. (2008) The Pursuit of Glory: The Five Revolutions that Made Modern Europe: 1648–1815. New York: Penguin Books. [Google Scholar]
- Boeckmann, B. , Bairoch, A. , Apweiler, R. , Blatter, M.‐C. , Estreicher, A. , Gasteiger, E. , Martin, M.J. et al. (2003) The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N. , Pimentel, H. , Melsted, P. and Pachter, L. (2015) Near‐optimal RNA‐Seq quantification. arXiv:1505.02710 [cs, q‐bio].
- Campbell, M.A. , Zhu, W. , Jiang, N. , Lin, H. , Ouyang, S. , Childs, K.L. , Haas, B.J. et al. (2007) Identification and characterization of lineage‐specific genes within the Poaceae. Plant Physiol. 145, 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H. , Hastie, A.R. , Cao, D. , Lam, E.T. , Sun, Y. , Huang, H. , Liu, X. et al. (2014) Rapid detection of structural variation in a human genome using nanochannel‐based genome mapping technology. GigaScience 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney, L. , Sharp, A.R. , Evans, C.R. and Udall, J.A. (2016) Genome Mapping in Plant Comparative Genomics. Trends Plant Sci. 21, 770–780. [DOI] [PubMed] [Google Scholar]
- Francis, C.M. and Millington, A.J. (1965) Isoflavone mutations in subterranean clover. 1. Their production, characterisation and inheritance. Aust. J. Agric. Res. 16, 567–573. [Google Scholar]
- Futschik, M.E. and Carlisle, B. (2005) Noise‐robust soft clustering of gene expression time‐course data. J. Bioinform. Comput. Biol. 03, 965–988. [DOI] [PubMed] [Google Scholar]
- Garg, R. , Patel, R.K. , Tyagi, A.K. and Jain, M. (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamkhar, K. , Isobe, S. , Nichols, P.G.H. , Faithfull, T. , Ryan, M.H. , Snowball, R. , Sato, S. et al. (2011) The first genetic maps for subterranean clover (Trifolium subterraneum L.) and comparative genomics with T. pratense L. and Medicago truncatula Gaertn. to identify new molecular markers for breeding. Mol. Breed. 30, 213–226. [Google Scholar]
- Goodstein, D.M. , Shu, S. , Howson, R. , Neupane, R. , Hayes, R.D. , Fazo, J. , Mitros, T. et al. (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigó, R. , Agarwal, P. , Abril, J.F. , Burset, M. and Fickett, J.W. (2000) An assessment of gene prediction accuracy in large DNA Sequences. Genome Res. 10, 1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa, H. , Kaur, P. , Shirasawa, K. , Nichols, P. , Nagano, S. , Appels, R. , Erskine, W. et al. (2016) Draft genome sequence of subterranean clover, a reference for genus Trifolium . Sci. Rep. 6, 30358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K.J. , Lange, S. , Lomsadze, A. , Borodovsky, M. and Stanke, M. (2016) BRAKER1: unsupervised RNA‐Seq‐based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32, 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley, R.P. , Sawford, T. , Mutowo‐Meullenet, P. , Shypitsyna, A. , Bonilla, C. , Martin, M.J. and O'Donovan, C. (2015) The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 43, D1057–D1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, M. , Misra, G. , Patel, R.K. , Priya, P. , Jhanwar, S. , Khan, A.W. , Shah, N. et al. (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 74, 715–729. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. , Sato, Y. and Morishima, K. (2016) BlastKOALA and GhostKOALA: KEGG Tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Meth. 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, E.T. , Hastie, A. , Lin, C. , Ehrlich, D. , Das, S.K. , Austin, M.D. , Deshpande, P. et al. (2012) Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H. , Moghe, G. , Ouyang, S. , Iezzoni, A. , Shiu, S.‐H. , Gu, X. and Buell, C.R. (2010) Comparative analyses reveal distinct sets of lineage‐specific genes within Arabidopsis thaliana . BMC Evol. Biol. 10, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze, A. , Burns, P.D. and Borodovsky, M. (2014) Integration of mapped RNA‐Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119. 10.1093/nar/gku557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire, W.S. (1985) Subterranean clover: Clover science and technology. American Society of Agronomy, Crop Science Society of America and Soil Science Society of America 1985, 515–534.
- Nichols, P.G.H. , Collins, W.J. and Barbetti, M.J. (1996) Registered cultivars of subterranean clover ‐ their characteristics, origin and identification. Agriculture Western Australia Bulletin No. 4327, 61. [Google Scholar]
- Nichols, P.G.H. , Foster, K.J. , Piano, E. , Pecetti, L. , Kaur, P. , Ghamkhar, K. and Collins, W.J. (2013) Genetic improvement of subterranean clover (Trifolium subterraneum L.). 1. Germplasm, traits and future prospects. Crop Pasture Sci. 64, 312–346. [Google Scholar]
- Ondov, B.D. , Treangen, T.J. , Melsted, P. , Mallonee, A.B. , Bergman, N.H. , Koren, S. and Phillippy, A.M. (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biology 17, 132. doi 10.1186/s13059-016-0997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Piano, E. , Pecetti, L. , Boller, B. , Posselt, U.K. and Veronesi, F. (2010) Minor Legume Species. In Fodder Crops and Amenity Grasses ( Boller, F. , Posselt, U.K. , and Veronesi, F. , eds), pp. 477–500. New York: Springer; . [Google Scholar]
- Putnam, N.H. , O'Connell, B.L. , Stites, J.C. , Rice, B.J. , Blanchette, M. , Calef, R. et al. (2016) Chromosome‐scale shotgun assembly using an in vitro method for long‐range linkage. Genome Res. 26, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlivan, B.J. (1961) The effect of constant and fluctuating temperatures on the permeability of the hard seeds of some legume species. Aust. J. Agric. Res. 12, 1009–1022. [Google Scholar]
- Rani, R. , Yadav, P. , Barbadikar, K.M. , Baliyan, N. , Malhotra, E.V. , Singh, B.K. , Kumar, A. et al. (2016) CRISPR/Cas9: a promising way to exploit genetic variation in plants. Biotechnol. Lett. 38, 1991–2006. [DOI] [PubMed] [Google Scholar]
- Shelton, J.M. , Coleman, M.C. , Herndon, N. , Lu, N. , Lam, E.T. , Anantharaman, T. , Sheth, P. et al. (2015) Tools and pipelines for BioNano data: molecule assembly pipeline and FASTA super scaffolding tool. BMC Genom. 16, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimková, H. , Číhalíková, J. , Vrána, J. , Lysák, M.A. and Doležel, J. (2003) Preparation of HMW DNA from plant nuclei and chromosomes isolated from root tips. Biol. Plant. 46, 369–373. [Google Scholar]
- Soderlund, C. , Nelson, W. , Shoemaker, A. and Paterson, A. (2006) SyMAP: a system for discovering and viewing syntenic regions of FPC maps. Genome Res. 16, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund, C. , Bomhoff, M. and Nelson, W.M. (2011) SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Diekhans, M. , Baertsch, R. and Haussler, D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Staňková, H. , Hastie, A.R. , Chan, S. , Vrána, J. , Tulpová, Z. , Kubaláková, M. et al. (2016) BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotech. J. 14, 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulas, L. , (2005) The future role of forage legumes in Mediterranean‐climate areas. In Grasslands: developments opportunities perspectives. ( Reynolds, S. and Frame, J. , eds), pp. 29–54. Rome: FAO and Plymouth UK: Science Publishers, Inc. [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. and Šmuc, T. (2011) REVIGO Summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vižintin, L. , Javornik, B. and Bohanec, B. (2006) Genetic characterization of selected Trifolium species as revealed by nuclear DNA content and ITS rDNA region analysis. Plant Sci. 170, 859–866. [Google Scholar]
- Vrána, J. , Cápal, P. , Číhalíková, J. , Kubaláková, M. and Doležel, J. (2016) Flow sorting plant chromosomes. Methods Mol. Biol. 1429, 119–134. [DOI] [PubMed] [Google Scholar]
- Wang, J.K. , Li, H.H. , Zhang, Y. and Meng, L. (2014) Users’ Manual of QTL IciMapping v4.0. Beijing: Institute of Crop Science, CAAS. [Google Scholar]
- Wheelan, S.J. and Boguski, M.S. (1998) Late‐night thoughts on the sequence annotation problem. Genome Res. 8, 168–169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The strategy and status of sequencing and assembly.
Figure S2 Functional GO characterization of Trifolium subterraneum L. genes.
Figure S3 Distribution of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the Trifolium subterranean annotation.
Figure S4 Five tissue types of Trifolium subterraneum L. used for the transcriptome ATLAS.
Table S1 Functional annotation of the predicted protein‐coding genes and transcripts.
Table S2 Functional annotation of the predicted protein‐coding genes.
Table S3 Functional composition of the assembled reference sequence Trifolium subterraneum L. genome assembly TSub_v2.0.
Table S4 Non‐coding RNA genes in the advanced draft genome assembly.
Table S5 Statistics of the completeness of the advanced genome Tsub_Refv2.0 assembly based using CEGMA pipeline.
Table S6 List of 562 enriched GO terms among the unique 5648 GO terms, in the biological processes category using REVIGO web server.
Table S7 List of 134 enriched GO terms among the unique 5648 GO terms, in the cellular processes category using REVIGO web server.
Table S8 List of 316 enriched GO terms among the unique 5648 GO terms, in the molecular function category using REVIGO web server.
Table S9 Hierarchical clustering of tissues based on global gene expression and GO terms.
Table S10 Clustering of co‐expressed genes based on spatially enriched expression patterns.
Table S11 List of GO terms for the leaf‐enriched Cluster 2, using REVIGO web server.
Table S12 List of GO terms for the flower‐enriched Cluster 1, using REVIGO web server.
Table S13 Number of putative genes‐encoding enzymes those mapped on KEGG pathways.
Table S14 Distance matrix and pairwise mutation distance estimation calculated by Mash v1.1 (preprint: http://biorxiv.org/content/early/2016/04/19/029827) using the published whole genome sequences for all the legumes except the transcriptome assembly for Cajanus cajan as a whole genome assembly not available.
Table S15 List of Trifolium‐specific genes among the galegoids identified using BLAST searches.
Table S16 List of subterranean‐clover‐specific genes among the Trifoliums identified using BLAST searches.
Table S17 List of red‐clover‐specific genes among the Trifoliums identified using BLAST searches.
Table S18 Statistics of a SNP linkage map and numbers of anchored scaffolds.
Table S19 High‐density SNP linkage map and anchoring information for Trifolium subterraneum L. genome assembly TSub_v2.0.