

Abstract
Background
Toxoplasma gondii is a pathogen implicated in psychiatric disorders. As elevated antibodies to T. gondii are also present in non-symptomatic individuals, we hypothesized that the age during first exposure to the pathogen may affect symptom manifestation. We tested this hypothesis by evaluating neurobehavioral abnormalities and the immune response in mice following adolescent or adult T. gondii infection.
Methods
Mice were infected with T. gondii at postnatal day 33 (adolescent/juvenile) or61 (adult). At 8 weeks post-infection (wpi), pre-pulse inhibition of the acoustic startle (PPI) in mice administered MK-801 (0.1 and 0.3 mg/kg) and amphetamine (5 and 10 mg/kg) was assessed. Peripheral (anti-T. gondii, C1q-associated IgG and anti-GLUN2 antibodies) and central (C1q and Iba1) markers of the immune response were also evaluated. In addition, regional brain expression of N-methyl-D-aspartate receptor (NMDAR) subunits (GLUN1 and GLUN2A), glutamatergic (vGLUT1, PSD95) and GABAergic (GAD67) markers, and monoamines (DA, NE, 5-HT) and their metabolites were measured.
Results
Juvenile and adult infected mice exhibited opposite effects of MK-801 on PPI, with decreased PPI in juveniles and increased PPI in adults. There was a significantly greater elevation of GLUN2 autoantibodies in juvenile- compared to adult-infected mice. In addition, age-dependent differences were found in regional expression of NMDAR subunits and markers of glutamatergic, GABAergic, and monoaminergic systems. Activated microglia and C1q elevations were found in both juvenile- and adult-T. gondii infected mice.
Conclusions
Our study demonstrates that the age at first exposure to T. gondii is an important factor in shaping distinct behavioral and neurobiological abnormalities. Elevation in GLUN2 autoantibodies or complement protein C1q may be a potential underlying mechanism. A better understanding of these age-related differences may lead to more efficient treatments of behavioral disorders associated with T. gondii infection.
Keywords: NMDAR, Toxoplasma, C1q, schizophrenia, autoantibody, neurodevelopment
Introduction
Psychiatric disorders such as schizophrenia are etiologically complex and likely arise from a combination of genetic and environmental contributions throughout life. One environmental factor that has been implicated in psychiatric disorders is pathogen exposure (Yolken and Torrey, 2008). Infection with the protozoan parasite Toxoplasma gondii (T. gondii) is suggested to increase the risk of developing schizophrenia, bipolar disorder, or other alterations in human behavior (Flegr, 2007; Hamidinej at et al., 2010; Hodkova et al., 2007; Torrey et al., 2012; Torrey et al., 2007; Wang et al., 2006). The association of T. gondii infection and abnormal human behavior hascome out of serological studies looking at T. gondii-specific antibodies. Elevated anti-T. gondii antibodies are detected in a greater percentage of psychiatric patients than control subjects, indicating there are individuals that have antibodies to T. gondii but do not exhibit clinical symptoms (Torrey et al., 2007). Abnormal behavior in only a subset of infected individuals may be partly explained by the age at which an individual is first exposed to T. gondii. Indeed, environmental stressors during critical periods of brain development have been suggested to lead to altered behavior in adulthood (Jaaro-Peled et al., 2009).
The neuro developmental hypothesis of schizophrenia and related disorders postulates that abnormal brain maturation leads to the onset of psychiatric disease during early adulthood. Adolescence is one of the most critical developmental periods and is characterized by a variety of processes including maturation of GABA interneurons and dopaminergic projections, and pruning of glutamatergic synapses (Bale et al., 2010; Casey et al., 2008a; Casey et al., 2008b; Jaaro-Peled et al., 2009). Synapse maturation and plasticity can be modulated by complement protein C1q and the N-methyl-D-aspartate receptor (NMDAR) (Stephen et al., 2012; Stephenson et al., 2008). C1q is a molecule suggested to be a player in neurodegenerative diseases and is associated with risk of developing schizophrenia (Stevens et al., 2007; Severance et al., 2012). Classically, however, C1q and other molecules in the complement system have been studied in the context of the immune system and pathogen clearance (Stephen et al., 2012). As with C1q, NMDAR dysfunction has been suggested to play a role in the development of schizophrenia (Kantrowitz and Javitt, 2010). The underlying cause of receptor dysfunction is unclear, however, NMDAR autoantibodies have been postulated to play a role in the pathogenesis of mental illness (Kayser and Dalmau, 2014). In addition, pathogens have been associated with elevated NMDAR antibodies (Pruss et al., 2012; Hammer et al., 2014). As infections can elevate C1q and NMDAR antibodies, and both are involved in modulating synapses, it is possible that chronic infection with T. gondii may affect postnatal brain development by altering synaptic maturation. Indeed, environmental stress during adolescence can affect normal maturation processes, and subsequently behavior during adulthood (Adriani et al., 2004; Kameda et al., 2011; Niwa et al., 2013). We therefore hypothesized that juvenile exposure to T. gondii would lead to different behavioral and neurobiological consequences as compared to adult exposure.
Our data demonstrate that the age of the host during T. gondii infection may shape distinct behavioral and neurobiological abnormalities. Juvenile infection intensified MK-801-induced impairment in PPI, while adult infection enhanced MK-801-induced PPI facilitation. In addition, we observed age-dependent differences in regional expression of N-methyl-D-aspartate receptor (NMDAR) subunits, C1q, and markers of glutamatergic, GABAergic, and monoaminergic systems. Also, GLUN2 autoantibodies were more elevated in juvenile than adult infected mice. Our findings suggest that the age of exposure to T. gondii could play a critical role in differential behavioral outcomes in humans.
Materials and Methods
Animals
Male BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) were used in this study. Mice were sacrificed either pre-behavioral testing (8 weeks post-infection) or post-behavioral testing (20 weeks post-infection). Animal protocols were reviewed and approved by the Animal Care and Use Committee of Johns Hopkins University (JHU). Mice were housed 5 per cage, unless separated due to fighting, with 14.5/ 9.5 hours of light/dark cycle with free access to food and water.
Toxoplasma tachyzoite purification
Prugniaud (PRU) tachyzoites were maintained in human foreskin fibroblast monolayers (HFF) and purified as previously described (Kannan et al., 2010). Mice were either intraperitoneally (ip)mock-infected with sterile Dulbecco's phosphate buffered saline (DPBS) or administered 400 tachyzoites (2 parasites/μL) at post-natal day (PND) 33 (termed adolescent/juvenile) or PND 61 (termed adult).
Sensorimotor Gating
Sensorimotor gating was assessed using pre-pulse inhibition of the acoustic startle (PPI). Behavioral testing was performed 2-4 months post-infection (mpi) using a within-group design in the following order: pre-pulse inhibition of the acoustic startle (PPI) with saline administration, PPI with MK-801 (0.1 then 0.3 mg/kg) administration (Juvenile, n=18; Adult, n=18), PPI with saline administration, and PPI with amphetamine (5 mg/kg then 10 mg/kg) administration (Juvenile, n=17; Adult, n=18). Mice were given 1 week of rest between each test. All handling and testing was performed by the same female experimenter.
Mice were intraperitoneally injected with 200 μL bacteriostatic 0.9% sodium chloride (Saline) (Hospira, INC; Lake Forest, IL), 0.1 mg/kg or 0.3 mg/kg (+)-MK-801 hydrogen maleate (Sigma-Aldrich), and D-amphetamine hemisulfate salt C-II (Sigma-Aldrich) 5 min prior to placement in PPI chambers (SR Labs, San Diego Instruments, San Diego, CA). All drugs were diluted with saline and prepared fresh in the morning of use for each individual mouse. In the testing apparatus, mice were acclimatized to a 76dB background noise for 5 min. They were then given 10 presentations of a 120dB pulse and 10 presentations of no pulse. This was followed by 5 presentations in randomized order of a 120 dB pulse, no pulse, or pre-pulses with a pulse (77, 78, 80, 84, 88 dB- 120 dB). The intervals between each presentation varied from 10-19 seconds. Mean PPI % was calculated by averaging all PPI% values for presentations of all pre-pulses for each experimental group. The mean PPI% for each infection group was normalized to that of age-matched infection controls. These values were then further normalized to the juvenile-saline group, and the ratio multiplied by 100 to convert to percent, which was used for graphing and statistical analysis.
Histology and Biochemistry
Brain Processing
For histology, brains (5 mice per group) were fixed using standard procedures. Briefly, mice were anesthetized and blood cleared with 1× phosphate-buffered saline (PBS). Brains were perfused with 4% paraformaldehyde (PFA) and fixed at 4°C in 4% PFA, 10% sucrose, and then 30% sucrose dissolved in PBS (3×). Brains were frozen and 40 μM sections cut for staining. Brains used for western blotting (5-6 mice per group) were cleared of blood. Brains used for HPLC (9-10 mice per group) were not cleared of blood.
Hematoxylin and Eosin
Sections were stained with hematoxylin and eosin (H&E) following standard procedures (5 mice per group; 4-5 sections per mouse). Parasite cysts were visualized and counted under bright-field illumination in a blinded manner.
Iba1 Immunofluorescence
Sections were stained following standard procedures (5 mice per group; 3 sections per mouse). Briefly, sections were placed in blocking solution [2.5% Triton X and 5% Horse Serum in 1× PBS] for 30 min, followed by primary antibody incubation with anti-Iba1 (1:250, WAKO) overnight. Sections were incubated in secondary antibody goat α-Rb AlexaFluor594 (1:400; Life Technologies) for 1 hr, and DAPI (1:3000; Invitrogen) for 10 min. Images were visualized using a Nikon eclipse E400 microscope and images taken with the program Meta Vue version 6.2r6.
Western Blot Analysis
Hippocampal and frontal cortex protein expression was measured via western blotting following standard protocols (5-6 mice per group). Lysis buffer was composed of RIPA buffer (Sigma), 1 × protease inhibitor (Sigma), 1× phosphatase inhibitor 3 (Sigma) and 1× phosphatase inhibitor II (Sigma). For all blots, 34-40 μg of protein per lane was loaded into NuPage 4-12% Bis-Tris, 1.0mm gels (Invitrogen), and transferred onto nitrocellulose 0.45 um (BioRad) membranes using the Invitrogen xcell surelock and blot modules. All primary and secondary antibodies used are listed in Supplementary Table 1. Gel images were quantified using Image J (Schneider et al., 2012). Eight wpi samples were run in quadruplicate and 20 wpi samples and C1q were run in duplicate. Data were first normalized to loading control β-tubulin and then averaged between gels. The data from all mice were then normalized to juvenile DBPS infection controls and this ratio then used for graphing and statistical analysis.
Monoamine Analysis
Monoamine measurements were made using high-performance liquid chromatography (HPLC) analysis. Brain regions obtained from each animal (9-10 mice per group) were weighed, ultrasonicated in 0.01 M perchloric acid, and then centrifuged at 20,000 ×g for 15 min. Concentrations of norepinephrine (NE), dopamine (DA), 3, 4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), serotonin (5-HT)and 5-hydroxyindoleacetic acid (5-HIAA) were measured in brain tissue extracts by HPLC with electrochemical detection as described earlier (Krasnova et al., 2000; Krasnova et al., 2001). In brief, the analytical column was Sunfire C-18 (5 μm, 4.6×150.0 mm; Waters); the mobile phase was 0.01 M sodium dihydrogenphosphate, 0.01 M citric acid,2 mM sodium EDTA, 1 mM sodium octylsulfate, 10% methanol, pH 3.5 at flow rate 1.0 ml/min and temperature 25°C. The installation consisted of 717 Plus automated injection system, 1525 binary pump, a thermostat (all from Waters, USA) and a Coulochem III electrochemical detector (ESA). The electrode was set at +0.75 V. Peak areas and monoamine concentrations were calculated with Breeze 3.3 software (Waters). Contents of NE, DA, DOPAC, HVA, 5-HT and 5-HIAA were calculated as ng/mg of tissue weight. Data was normalized to DPBS controls and this ratio used for statistical analysis.
Serum Antibody Measurements
Blood was taken during sacrifice and centrifuged at 10,000×g to collect serum. Antibodies to T. gondii, GLUN2, and C1q were measured in serum via enzyme-linked immunosorbent assay (ELISA) (10 mice per group). Total anti-Toxoplasma IgG was measured as described previously (Xiao et al., 2009). Serum antibodies to GLUN2 were measured using the Gold Dot GLUN2 Antibody Test Kit (CIS Biotech, Inc). Serum C1q activity was measured as previously described (Severance et al., 2012). This assay measures antibody immune complexes associated with C1q as well as antibodies directed against the C1q molecule. Absorbance values of infected mice were normalized to DPBS controls and this value used for statistical analysis.
Statistical Analysis
Age-infection interaction and age-drug interaction was tested using multiple linear regression. For multiple-comparison testing data were then checked for normality and equal variance using SPSS (v.21). If data passed normality and equal variance tests, t-test was performed. If normality and equal variance tests failed, non-parametric Mann-Whitney U test was used to compare all groups. A p ≤ 0.05 was considered significant.
Results
Differences in psychostimulant-induced PPI changes in juvenile and adult T. gondii infected mice
Sensorimotor gating deficits, as measured by pre-pulse inhibition (PPI) of the acoustic startle, are observed in many psychiatric disorders (Kohl et al., 2013). We explored whether juvenile and adult T. gondii infection would differentially affect PPI in mice and found that the infection did not alter PPI of the acoustic startle in either age group (data not shown).
To further probe for possible latent impairment in PPI, we administered MK-801 and amphetamine, psychostimulants that are widely used in rodent models to unmask potential alterations to the brain (van den Buuse et al., 2005). Potential alterations in the glutamate system were explored with MK-801, an N-methyl-D-aspartate Receptor (NMDAR) antagonist, while possible changes in the dopaminergic system were assessed with amphetamine, a dopamine transporter inhibitor. We found age-dependent PPI disruptions with MK-801 and amphetamine administration in the juvenile and adult T. gondii-infected groups (Figure 1). Juvenile-infected mice showed a dose-dependent decrease in MK-801-induced PPI, while adult-infected mice showed a dose-dependent increase in MK-801- induced PPI. Linear regression analysis revealed a drug effect (b= 31.644, t=3.975, p<0.001) and age × drug interaction (b=-41.179, t= -3.167, p<0.01) (Fig 1A). In contrast, adult-infected mice showed significant PPI increase with 5mg/kg amphetamine administration as compared to juvenile-infected mice. However, linear regression analysis did not reveal an age × drug interaction (Fig 1B)
Figure 1. Age of host when infected with T. gondii affects pre-pulse inhibition (PPI) susceptibility to MK-801 and Amphetamine.
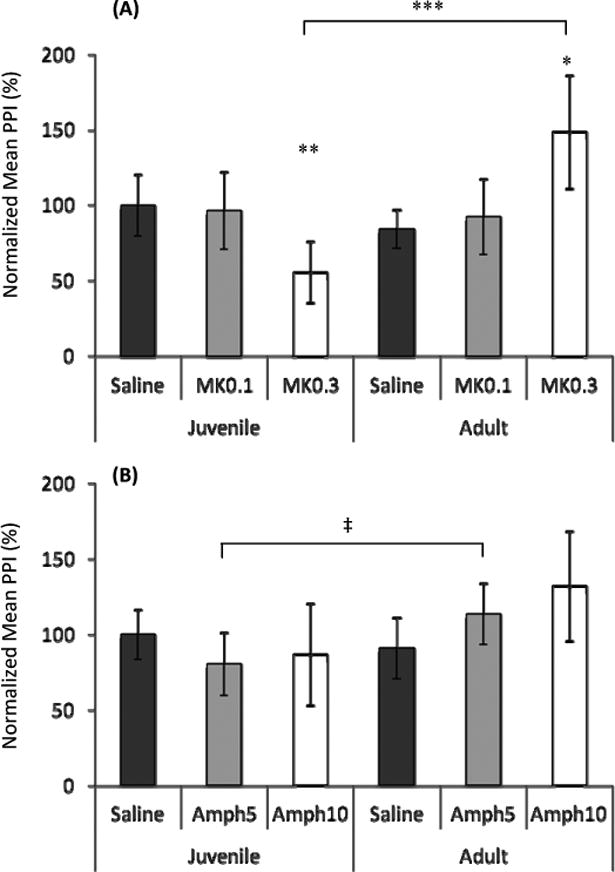
A) MK-801 (0.3mg/kg) administration revealed decreased PPI in juvenile-infected mice and increased PPI in adult-infected mice. Linear regression analysis showed an age x drug interaction (b= -41.179, t= -3.167, p<0.01). B) Amphetamine (5mg/kg) administration showed increased PPI in adult-infected mice. Liner regression analysis determined no age x drug interaction.
Juvenile saline and MK-801 (0.1 and 0.3 mg/kg), n=18; Adult saline and MK-801 (0.1 and 0.3 mg/kg), n=18; Juvenile saline and amphetamine (5 and 10 mg/kg), n=17; Adult saline and amphetamine (5 and 10 mg/kg), n=18
Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons.
* denotes p ≤ 0.05 compared to saline and 0.1 mg/kg MK-801;
** denotes p≤ 0.01 compared to saline and 0.1 mg/kg MK-801;
*** denotes p≤ 0.001
‡ denotes p ≤ 0.05
As differences in startle between groups could confound PPI results, we also looked at the startle response and found no group differences in basal or drug-induced startle amplitude (data not shown). Taken together, our data indicate that juvenile and adult infection with T. gondii differentially modulates the effects of MK-801 and amphetamine on PPI.
Brain changes associated with glutamatergicsynaptic markers in juvenile and adult T. gondii infected mice
Because the MK-801 effects were most strikingly different between juvenile and adult infected mice, we sought to explore the impact of T. gondii infection on the glutamate (GLU) system. Cortical and hippocampal expression of NMDAR subunits (GLUN1 and GLUN2A)and markers of GLU synapses (vGLUT1, PSD95) were measured at 8 wpi, a time point post-infection and prior to behavior testing with psychostimulants to avoid confounding effects of drug administration and testing (Kinney et al., 2006; Mostany et al., 2013).
We found that juvenile and adult infection with T. gondii led to age-dependent alterations in GLUN2A and GLUN1 expression (Figure 2). Frontal cortex GLUN2A expression was decreased and GLUN1 was increased in adult-infected mice as compared to both age-matched controls and juvenile-infected mice. In the hippocampus, GLUN2A expression was decreased in both juvenile- and adult-infected groups, while GLUN1 levels were unchanged. Linear regression analysis revealed that both cortical and hippocampal GLUN2A showed a significant age × infection interaction (b= -0.251, t=-5.124, p= 0.000; b= 0.241, t= 3.331, p= 0.004, respectively). In addition, both cortical GLUN2A and GLUN1 showed an age effect (b=-0.189, t=-3.859, p< 0.01; b= -1.465, t=-2.353, p<0.05, respectively), while hippocampal GLUN2A and GLUN1 showed an infection effect (b=-0.281, t=-3.888, p<0.01; b=-0.243, t=-3.076, p<0.01, respectively). Age-dependent alterations in NMDAR subunit expression differed but were also present in mice after behavioral testing (20 wpi)(Supp Fig 1). Linear regression analysis showed an effect of age, infection, and an age × infection interaction in only cortical GLUN1 expression (b=16.024, t=5.107, p<0.001; b=4.443, t=3.206, p<0.01; and b=-3.986, t=-4.548, p<0.01, respectively). Thus, while only adult-infection affected cortical GLUN1 and GLUN2A expression, both juvenile- and adult-infection decreased levels of GLUN2A in the hippocampus.
Figure 2. N-methyl-D- aspartate receptor (NMDAR) subunit proteins are altered in juvenile- and adult-infected mice at 8 wpi.
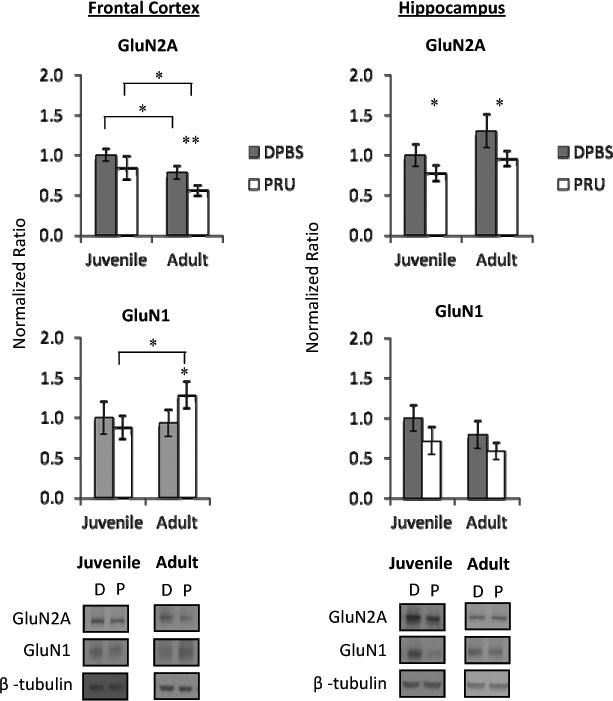
Frontal cortex GluN2A expression is decreased and GluN1 expression is increased in adult-infected mice. Linear regression revealed an age × infection interaction in cortical Glun2A expression (b= -0.251, t=-5.124, p=0.000), but not GluN1. Hippocampal GluN2A expression is decreased in juvenile- and adult-infected mice, while GluN1 was not significantly changed. Linear regression showed an age × infection interaction of Glun2A (b=0.241, t=3.331, p=0.0004).
Data represent an average of 5 mice per group and 4 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. Horizontal lines with symbols denote groups compared and * denotes p≤ 0.05. For symbols alone,
* denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group and
** denotes p≤ 0.01 compared to age-matched mock-infected (DPBS) group.
Because NMDAR functioning is involved in synaptic plasticity and T. gondii infection differentially altered the expression of NMDAR subunits in the brain, we analyzed the effects of infection on glutamatergic synapses by measuring expression of vesicular glutamate 1 transporter (VGLUT1) as a pre-synaptic marker and post-synaptic density 95 (PSD-95) as a post-synaptic marker (Figure 3). No changes in these markers were found in the frontal cortex. In the hippocampus, juvenile infection significantly decreased both glutamate markers, while adult infection significantly decreased VGLUT1 only. There was a significant age × infection interaction and infection effect on hippocampal PSD95 expression (b= -0.261, t=-2.435, p=0.026; b=-0.416, t=-3.885, p<0.01, respectively), and a significant infection effect on hippocampal VGLUT1 expression (b=-0.42, t=-3.909, p<0.01). In addition, age-related alterations in VGLUT1 and PSD-95 levels were more pronounced after behavior testing (20 wpi) (Suppl Fig 2). Linear regression analysis revealed that after behavior testing, cortical VGLUT1 showed an age × infection interaction and infection effect (b= 0.433, t=3.695, p<0.01; b=-0.613, t=-5.228, p<0.001, respectively), while cortical PSD95 expression showed an age effect (b=1.167, t=2.272, p<0.05). Our data shows that compared to adult infection, juvenile infection has a greater impact on expression of pre- and post- glutamatergic synapse markers, with the hippocampus being more affected than the cortex by infection at either age.
Figure 3. Glutamate pre- and post-synaptic marker expression is altered in T. gondii- infected mice at 8 wpi.
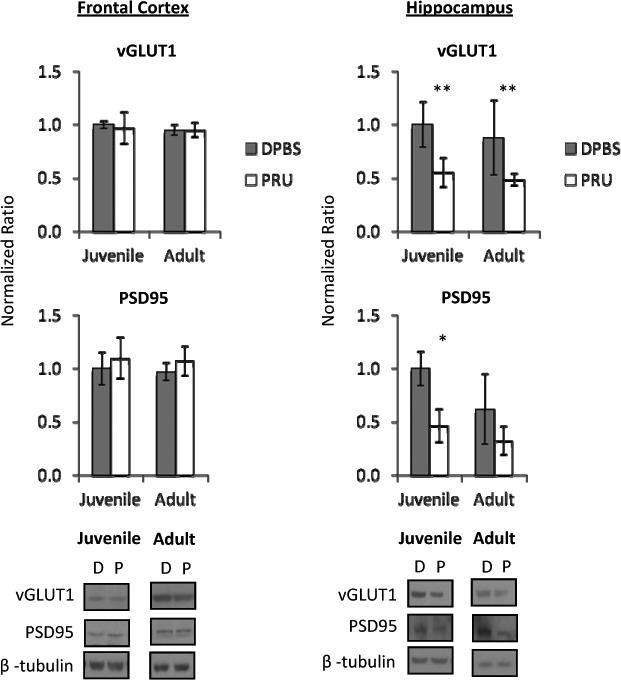
Frontal cortex pre-synaptic vesicular glutamate transporter 1 (vGLUT1) and post-synaptic density 95 (PSD95) expression were unchanged by infection. Hippocampal vGLUT1 was decreased in juvenile- and adult- infected mice. Linear regression analysis revealed no age × infection interaction. Hippocampal PSD95 was decreased in juvenile-infected mice and an age × infection interaction was determined (b=-0.261, t=-2.435, p=0.026). Data represent an average of 5 mice per group and 4 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons.
* denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group
** denotes p≤ 0.01 compared to age-matched mock-infected (DPBS) group
To determine whether T. gondii infection may be selectively targeting the GLU system, we evaluated markers of other brain systems at 20 wpi. As found with the GLU markers, differential regional alterations in juvenile and adult infected mice were observed in levels of dopamine (DA) and metabolites (DOPAC, HVA), norepinephrine (NE), and serotonin (5-HT) and metabolite (5H1AA) (Supp Table 2). In addition, expression of frontal cortex glutamate decarboxylase 67 (GAD67) was decreased in juvenile- and adult-infected mice, but showed an age × infection interaction and age effect (b= 0.435, t=3.695, p=0.002; b=-0.513, t=-2.306, p<0.05) (Supp Fig 3). Thus, our data indicate that T. gondii infection produced alterations in multiple neurotransmitter systems in the brain.
Immune changes involved in synaptic plasticity injuvenile and adult T. gondii infected mice
We found T. gondii infection differentially affected behavior and the expression of synaptic markers and monoamines in juvenile and adult infected mice. As a possible mechanism for these changes, we explored whether the age at infection would also differently impact immune factors that could affect maturation and/or functioning of synapses. We focused on autoantibodies to GLUN2 and complement protein C1q due to their targeting of synapses and association with psychiatric disorders (Lancaster et al., 2011; Levite, 2014; Severance et al., 2012; Stephan et al., 2012; Tsutsui et al., 2012).
We detected elevated serum GLUN2 antibodies in infected mice, with a greater increase in juveniles than adults (Figure 4). We also found elevated Toxoplasma-induced serum and brain C1q levels in infected mice (Figure 5). Serum C1q antibody levels were significantly elevated due to juvenile- and adult- infection, however more antibodies were detected in adult-compared to juvenile-controls. In addition to a peripheral elevation of C1q-associated IgG, C1q expression was increased in blood-cleared cortical and hippocampal samples of juvenile- and adult-infected mice. While frontal cortex C1q was more highly expressed in adult- infected mice compared to juveniles, hippocampus C1q was more highly expressed in juvenile-infected and control mice compared to infection-matched adults. Linear regression revealed no age × infection interaction in GLUN2 antibodies or C1q levels. Our data shows that while infection increases GLUN2 antibodies and C1q levels, juvenile infection has a greater impact on GLUN2 antibody elevation, while adult infection has a greater impact on frontal cortex C1q expression. This indicates potentially separate mechanisms for altering synaptic functioning in juvenile- and adult-infection.
Figure 4. Serum antibodies to GluN2 are elevated in T. gondii infected mice at 8 wpi.
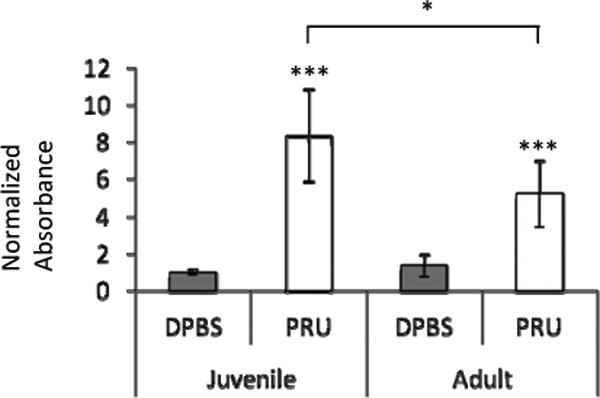
Antibodies to GluN2 were elevated in the serum of juvenile- and adult-infected mice, with a higher elevation in juveniles. Linear regression did not reveal an age × infection interaction. Data represent an average of 10 mice per group. DPBS denotes mock-infected controls and PRU denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons.
The horizontal line with symbol denotes groups compared and * denotes p≤ 0.05. For the symbol alone, *** denotes p≤ 0.001 compared to age-matched mock-infected (DPBS) group
Figure 5. C1q is elevated in the serum and brain of T. gondii infected mice at 8 wpi.
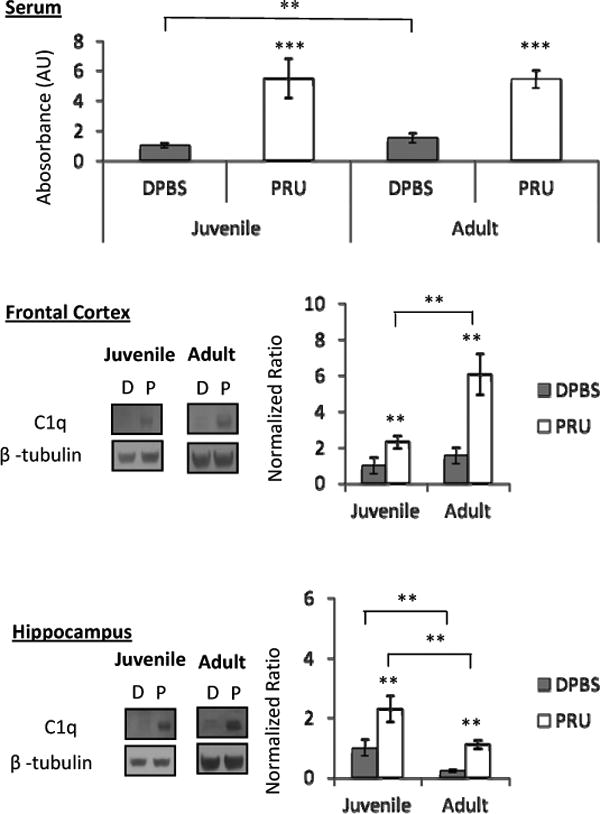
Serum, frontal cortex, and hippocampus C1q is increased in juvenile- and adult-infected mice. Frontal cortex C1q expression is higher in adult than juvenile-infected mice. Linear regression determined no age × infection interaction for serum, frontal cortex, or hippocampus C1q. Data represent an average of 10 mice per group for serum measurements, and 5 mice per group for frontal cortex and hippocampus western blots. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. Horizontal lines with symbols denote groups compared and ** denotes p≤ 0.01 . For symbols alone, ** denotes p≤ 0.01 compared to age-matched mock-infected (DPBS) group and *** denotes p≤ 0.001 compared to age-matched mock-infected (DPBS) group.
As activated microglia can secrete C1q (Stephen et al., 2012), we stained for microglia in the brain and found activated microglia in the brains of infected mice (Supp Fig 4). In addition, because of age-related differences in GLUN2 autoantibody levels, we measured anti-T.gondii IgG in serum and parasite cysts in the brain as markers of infectivity (Supp Table 3) and found these also differed between the two age groups at 8 wpi.
Taken together our data show that T. gondii infection leads to elevated GLUN2 autoantibodies in an age-dependent manner, and similarly elevates C1q levels in the periphery and brain in both juvenile- and adult- infected mice.
Discussion
Psychiatric disorders and other behavioral alterations have been associated with pathogen infection, such as T. gondii (Flegr, 2007; Lamberton et al., 2008; Torrey et al., 2012; Webster, 2007). Yet individuals who do not demonstrate behavioral symptoms also have antibodies to the parasite, as demonstrated by antibodies to the parasite in non-psychiatric controls (Torrey et al., 2007). It has been suggested that the parasite strain, sex of the host, host strain, and genetic predisposition of the host can contribute to differential behavioral outcomes of chronic T. gondii infection (Eells et al., 2015; Kannan et al., 2010; Kannan and Pletnikov, 2012; Xiao et al., 2012). An important contributing factor that has not previously been explored in the context of T. gondii infection is the age at first exposure to the pathogen. One critical window in neurodevelopment is the period of adolescence. While animal models of mental disorders have long focused on insults during the pre-natal period, some deficits during adulthood may not manifest unless presented with a second stress during adolescence (Giovanoli et al., 2013). Even one time exposure to environmental adversities during adolescence can produce significant long-term pathological changes in adulthood (Adriani et al., 2004; Kameda et al., 2011; Niwa et al., 2013). Therefore, we hypothesized that juvenile infection would have greater consequences on behavior and brain alterations than adult infection.
Our data clearly demonstrate that juvenile and adult infection with T. gondii can lead to different psychostimulant-induced alterations in pre-pulse inhibition (PPI) of the acoustic startle. Consistent with prior work, we show T. gondii infection per se does not alter PPI (Kannan et al., 2010; Xiao et al., 2012). However, when the NMDAR antagonist MK-801 was administered to unmask potential impairment, we found a robust PPI decrease in juvenile-infected mice and a significant PPI increase in adult-infected mice. Both PPI inhibition and facilitation have been observed in patients with schizophrenia and other neuropsychiatric disorders (Wynn et al., 2004). It is therefore conceivable that infection or other insults during different ages may lead to these opposite PPI alterations in humans. Since psychostimulant administration was required to observe PPI changes in infected mice, it potentially indicates that a one time exposure to T. gondii may only modestly affect the brain. It is tempting to speculate that because of this, a second “hit” may be needed to produce more robust neurobiological consequences, consistent with the concepts of gene-environment (GxE) and environment-environment (ExE) interactions (Kannan et al., 2013; Abazyan et al., 2010; Eells et al., 2015; Giovanoli et al., 2013). Nevertheless, our PPI data suggests T. gondii infection differentially impairs rodents infected as juveniles and adults, which may model different aspects of schizophrenia in humans. Future studies will provide a more comprehensive evaluation of other age-dependent behavioral alterations in infected mice.
Consistent with age-dependent differences in relation to the PPI effects of an NMDAR antagonist, we found decreased expression of GLUN2A in the hippocampus of juvenile- and adult-infected mice, and a decrease in GLUN2A but increase in GLUN1 in the cortex of adult-infected mice at 8 wpi. Because hippocampus GLUN2A is decreased in both age groups, it suggests this reduction may be a general consequence of T. gondii infection. In addition, markers of excitatory synapses (vGLUT1 and PSD95) were predominantly decreased in the hippocampus of juvenile-infected mice. We see a different effect after behavior testing, which could be due to long-term effects of the psychostimulants used during testing (Li et al., 2015; Thomases et al., 2013). Our data differs from previous studies on T. gondii-infected rodents that revealed no changes in vGLUT1 and a decrease in PSD95 in the cortex (Parlog et al, 2014; Brooks et al., 2015), likely due to differences in the parasite strain, rodent strain, and rodent sex used. In addition, such differences may partly explain phenotypic differences in a lack of seizures observed in our model compared with others (Brooks et al., 2015). We speculate these changes reflect a loss of GLUN2A-GLUN1 heterodimers at synapses in the hippocampus and an increase in GLUN2B-GLUN1 heterodimers at the synapses in the cortex of adult-infected mice. Future work should provide a more detailed analysis of synaptic abnormalities induced by T. gondii infection.
T. gondii infected mice also exhibited differential abnormalities in the expression of other GABAergic and monoaminergic system markers in juvenile- and adult-infected mice. These results support the hypothesis that distinct behavioral abnormalities displayed by juvenile-and adult-infected mice could be partly explained by age-dependent abnormalities in expression of NMDAR subunits and markers of other brain systems.
In order to elucidate a potential mechanism for age-dependent brain and behavior changes, we assessed peripheral and central immune factors that have been found to impact synaptic maturation and plasticity. As GLUN2 autoantibodies have been shown to affect NMDAR functioning (Faust et al., 2010), we measured GLUN2 autoantibodies in chronically T. gondii-infected mice. We found that T. gondii elevated serum levels of GLUN2A antibodies, with a significantly greater increase being observed in juvenile- compared to adult-infected mice. It is tempting to speculate that the age differences in GLUN2 antibodies might explain differences in PPI observed. In addition to autoantibodies, we found T. gondii infection led to increased serum levels of C1q-associated IgG, increased immune reactivity of a microglia marker, Iba1, and up-regulation of regional brain C1q expression. Interestingly, we found a significantly greater increase in frontal cortex C1q in adult- compared to juvenile-infected mice. We conjecture increased cortical C1q expression may contribute to behavioral differences in adult- infected mice compared to juveniles. As microglia activation and C1q are associated with synaptic pruning during development, an increase in these immune factors may contribute to behavioral alterations (Miyamoto et al., 2004; Zabel and Kirsch, 2013). Future studies will address the potential role of GLUN2 autoantibodies and C1q in mediating age-dependent behavior alterations in chronic T. gondii infection.
In conclusion, our findings show for the first time that age of initial exposure to T. gondii can determine differential neurobehavioral abnormalities in infected mice, possibly as a result of age-dependent up-regulation of GLUN2A autoantibodies and associated down-regulation of NMDAR subunits. In light of the association of T. gondii exposure and psychiatric disorders, one could hypothesize that an increase in NMDAR autoantibodies and/or C1q due to T. gondii exposure might be the underlying mechanism for behavioral alterations in symptomatic individuals. Future research will address this possibility in detail.
Supplementary Material
Supplementary Figure 1. N-methyl-D-aspartate receptor (NMDAR) subunit proteins are altered in juvenile- and adult-infected mice at 20 wpi. Frontal cortex GluN2A is decreased in both juvenile-and adult-infected mice. Frontal cortex GluN1 is increased in juvenile-infected mice and decreased in adult-infected mice. Linear regression revealed an age × infection interaction of frontal cortex GluN1 only (b=-3.986, t=-4.548, p<0.01). Hippocampus GluN2A was unchanged, while GluN1 was increased in adult-infected mice. No age × infection interaction was determined for the hippocampus.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. The horizontal line with symbol denote groups compared and ** denotes p≤ 0.01. For symbols alone,
* denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group and
** denotes p≤ 0.01 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 2. Glutamate pre- and post-synaptic marker expression is altered in T. gondii- infected mice at 20 wpi. Frontal cortex pre-synaptic vesicular glutamate transporter 1 (vGLUT1) is decreased in juvenile- and adult-infected mice, with a greater decrease in juveniles. Linear regression revealed an age × infection interaction (b=0.433, t=3.695, p<0.01). Frontal cortex post-synaptic density 95 (PSD95) was increased in adult controls and infected groups, with no age × infection interaction. Hippocampus vGLUT1 was decreased in adult-infected mice and hippocampus PSD95 was unchanged. Linear regression analysis revealed no age × infection interaction.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. Horizontal lines with symbols denote groups compared and * denotes p≤ 0.05 while ** denotes p≤ 0.01. For symbols alone, * denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group and ** denotes p ≤ 0.01 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 3. GABA marker expression is altered in T. gondii- infected mice at 20 wpi. Frontal cortex glutamate decarboxylase 67 (GAD67) is decreased in juvenile- and adult-infected mice. Linear regression analysis revealed an age × infection interaction (b=0.0435, t=3.695, p=0.002). Hippocampus GAD67 was unchanged and no age × infection interaction was found.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons with * denoting p≤ 0.05 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 4. Microglia are activated in the brains of T. gondii- infected mice at 8 wpi. Ionized calcium binding adaptor molecule 1 (IBA1) positive microglia show an activated morphology juvenile- and adult- infected brain.
Red denotes microglia marker Iba1. Blue denotes nuclear staining with DAPI.
Scale bar represents 150 μM.
Inset show representative Iba1 positive cells.
Supplementary Table 1. Western blot antibodies. Primary and secondary antibodies, dilutions, and company information.
Supplementary Table 2. Monoamine and metabolite changes in T. gondii infected mice at 20 wpi. Dopamine and metabolites were changed in the hippocampus and striatum of juvenile- and adult-infected mice. Serotonin and metabolites were altered in the hippocampus of infected mice. A ratio < 1.0 signifies a decrease in the monoamine/metabolite compared to DPBS while a ratio > 1.0 signifies an increase in the monoamine/metabolite compared to DPBS.
Data shown is the average of 9-10 mice per group plus/minus standard error. Symbols represent statistical significance from Mann-Whitney U-test comparisons with * denoting p≤ 0.05 compared to age-matched mock-infected (DPBS) group.
Monoamines and metabolites measured include: dopamine (DA), 3,4-Dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), norepinephrine (NE)
Serotonin (5HT), 5-hycroxyindoleacetic acid (5H1AA).
Supplementary Table 3. T. gondii-infected mice have serum T. gondii antibodies and parasite cysts in brain at 8 wpi. T. gondii IgG was elevated in juvenile- and adult-infected mice, with higher levels in adult-infected mice. Parasites cysts were found in juvenile- and adult-infected mice, with fewer cysts/ section in adult-infected mice. T. gondii IgG data shown is the average of 9-10 mice per group. Parasite cyst data shown is the average of 5 mice and 5 sections per mouse. Symbols represent statistical significance from independent sample T-test comparisons with * denoting p<0.05 compared to age-matched mock-infected control (DPBS) and ^ denoting p<0.05 compared to juvenile-infected (PRU) mice.
Acknowledgments
The authors would like to thank Dr. Lorraine Jones-Brando for critical reading of the manuscript. Funding for this work was provided by the Stanley Medical Research Institute (SMRI), Chevy Chase, MD, USA and the Silvio O. Conte Grant MH-094268.
References
- Abazyan B, et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psych. 2010;68:1172–81. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriani W, et al. Behavioral and neurochemical vulnerability during adolescence in mice: studies with nicotine. Neuropsychopharmacology. 2004;29:869–78. doi: 10.1038/sj.npp.1300366. [DOI] [PubMed] [Google Scholar]
- Bale TL, et al. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–9. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JM, et al. Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous sytem. mBio. 2015;6:e01428–15. doi: 10.1128/mBio.01428-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BJ, et al. The adolescent brain. Dev Rev. 2008a;28:62–77. doi: 10.1016/j.dr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BJ, et al. The adolescent brain. Ann N Y Acad Sci. 2008b;1124:111–26. doi: 10.1196/annals.1440.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eells JB, et al. Chronic Toxoplasma gondii in Nurr1-Null Heterozygous Mice Exacerbates Elevated Open Field Activity. PLoS One. 2015;10:e0119280. doi: 10.1371/journal.pone.0119280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegr J. Effects of toxoplasma on human behavior. Schizophr Bull. 2007;33:757–60. doi: 10.1093/schbul/sbl074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust TW. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci. 2010;107:18569–74. doi: 10.1073/pnas.1006980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanoli S, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. 2013;339:1095–1099. doi: 10.1126/science.1228261. [DOI] [PubMed] [Google Scholar]
- Hamidinejat H, et al. Toxoplasma gondii infection in first-episode and inpatient individuals with schizophrenia. Int J Infect Dis. 2010;14:e978–81. doi: 10.1016/j.ijid.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Hammer C, et al. Neuropsychiatric disease relevance of circulating anti-nmda receptor autoantibodies depends on blood brain barrier integrity. Mol Psychiatry. 2014;19:1143–9. doi: 10.1038/mp.2013.110. [DOI] [PubMed] [Google Scholar]
- Hodkova H, et al. Higher perceived dominance in Toxoplasma infected men--a new evidence for role of increased level of testosterone in toxoplasmosis-associated changes in human behavior. Neuro Endocrinol Lett. 2007;28:110–4. [PubMed] [Google Scholar]
- Infurna RN, Spear LP. Developmental changes in amphetamine-induced taste aversions. Pharmacol Biochem Behav. 1979;11:31–5. doi: 10.1016/0091-3057(79)90293-4. [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 2009;32:485–95. doi: 10.1016/j.tins.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pahtway on the road to schizophrenia? Brain Res Bull. 2010;83:108–21. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda SR, et al. Adolescent mice are more vulnerable than adults to single injection-induced behavioral sensitization to amphetamine. Pharmacol Biochem Behav. 2011;98:320–4. doi: 10.1016/j.pbb.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Kannan G, et al. Toxoplasma gondii strain-dependent effects on mouse behaviour. Folia Parasitol (Praha) 2010;57:151–5. doi: 10.14411/fp.2010.019. [DOI] [PubMed] [Google Scholar]
- Kannan G, Pletnikov MV. Toxoplasma Gondii and Cognitive Deficits in Schizophrenia: An Animal Model Perspective. Schizophr Bull. 2012 doi: 10.1093/schbul/sbs079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan G, et al. Mouse models of gene-environment interactions in schizophrenia. Neurobiol Dis. 2013;57:5–11. doi: 10.1016/j.nbd.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser MS, Dalmau J. Anti-nmda receptor encephalitics, autoimmunity, and psychosis. Schizophr Res. 2014 doi: 10.1016/j.schres.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, et al. A specific role for GLUN2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–15. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, et al. Prepulse inhibition in psychiatric disorders--apart from schizophrenia. J Psychiatr Res. 2013;47:445–52. doi: 10.1016/j.jpsychires.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Krasnova IN, et al. Intracerebroventricular administration of substance P increases dopamine content in the brain of 6-hydroxydopamine-lesioned rats. Neuroscience. 2000;95:113–7. doi: 10.1016/s0306-4522(99)00400-5. [DOI] [PubMed] [Google Scholar]
- Krasnova IN, et al. Amphetamine-induced toxicity in dopamine terminals in CD-1 and C57BL/6J mice: complex roles for oxygen-based species and temperature regulation. Neuroscience. 2001;107:265–74. doi: 10.1016/s0306-4522(01)00351-7. [DOI] [PubMed] [Google Scholar]
- Lamberton PH, et al. Specificity of the Toxoplasma gondii-altered behaviour to definitive versus non-definitive host predation risk. Parasitology. 2008;135:1143–50. doi: 10.1017/S0031182008004666. [DOI] [PubMed] [Google Scholar]
- Lancaster E, et al. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77:179–89. doi: 10.1212/WNL.0b013e318224afde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levite M. Glutamate receptor antibodies in neurological diseases: anti-AMPA-GluR3 antibodies, anti-NMDA-GLUN1 antibodies, anti-NMDA-GLUN2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren's syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor's expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J Neural Transm. 2014;121:1029–75. doi: 10.1007/s00702-014-1193-3. [DOI] [PubMed] [Google Scholar]
- Li JT, et al. Long-term effects of neonatal exposure to MK-801 on recognition memory and excitatory-inhibitory balance in rat hippocampus. Neuroscience. 2015;308:134–43. doi: 10.1016/j.neuroscience.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, et al. Behavioural adaptations to addictive drugs in mice lacking the NMDA receptor epsilon1 subunit. Eur J Neurosci. 2004;19:151–8. doi: 10.1111/j.1460-9568.2004.03086.x. [DOI] [PubMed] [Google Scholar]
- Mostany R, et al. Altered synaptic dynamics during normal brain aging. J Neurosci. 2013;33:4094–104. doi: 10.1523/JNEUROSCI.4825-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, et al. Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science. 2013;339:335–9. doi: 10.1126/science.1226931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlog A, et al. Chronic murine toxoplasmosis is defined by subtle changes in neuronal connectivity. Dis Mod and Mech. 2014;7:459–69. doi: 10.1242/dmm.014183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss H, et al. N-methyl-d-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol. 2012;72:902–11. doi: 10.1002/ana.23689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, et al. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance EG, et al. Complement C1q formation of immune complexes with milk caseins and wheat glutens in schizophrenia. Neurobiol Dis. 2012;48:447–53. doi: 10.1016/j.nbd.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk J, et al. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neuro. 2015;9:82. doi: 10.3389/fncel.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, et al. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- Stephenson, et al. Assembly and forward trafficking of nmda receptors (review) Mol memb biol. 2008;25:311–20. doi: 10.1080/09687680801971367. [DOI] [PubMed] [Google Scholar]
- Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Thomases DR, et al. Periadolescent exposure to the NMDA antagonist MK-801 impairs the functional maturation of local GABAergic circuits in the adult prefrontal cortex. J Neurosci. 2013;33:26–34. doi: 10.1523/JNEUROSCI.4147-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, et al. Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr Bull. 2012;38:642–7. doi: 10.1093/schbul/sbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, et al. Antibodies to Toxoplasma gondii in patients with schiozphrenia: a meta-analysis. Schizophr Bull. 2007;33:729–36. doi: 10.1093/schbul/sbl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui K, et al. Anti-NMDA-receptor antibody detected in encephalitis, schizophrenia, and narcolepsy with psychotic features. BMC Psychiatry. 2012;12:37. doi: 10.1186/1471-244X-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Buuse M, et al. Reduced effects of amphetamine on prepulse inhibition of startle in gastrin-deficient mice. Neurosci Lett. 2005;373:237–42. doi: 10.1016/j.neulet.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Wang HL, et al. Prevalence of Toxoplasma infection in first-episode schizophrenia and comparison between Toxoplasma-seropositive and Toxoplasma-seronegative schizophrenia. Acta Psychiatr Scand. 2006;114:40–8. doi: 10.1111/j.1600-0447.2006.00780.x. [DOI] [PubMed] [Google Scholar]
- Webster JP. The effect of Toxoplasma gondii on animal behavior: playing cat and mouse. Schizophr Bull. 2007;33:752–6. doi: 10.1093/schbul/sbl073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn JK, et al. Prepulse facilitation and prepulse inhibition in schizophrenia patients and their unaffected siblings. Biol Psychiatry. 2004;55:518–23. doi: 10.1016/j.biopsych.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Xiao J, et al. Serological pattern consistent with infection with type I Toxoplasma gondii in mothers and risk of psychosis among adult offspring. Microbes Infect. 2009;11:1011–8. doi: 10.1016/j.micinf.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Xiao J, et al. Sex-specific changes in gene expression and behavior induced by chronic Toxoplasma infection in mice. Neuroscience. 2012 doi: 10.1016/j.neuroscience.2011.12.051. [DOI] [PubMed] [Google Scholar]
- Yolken RH, Torrey EF. Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol Psychiatry. 2008;13:470–9. doi: 10.1038/mp.2008.5. [DOI] [PubMed] [Google Scholar]
- Zabel MK, Kirsch WM. From development to dysfunction: microglia and the complement cascade in CNS homeostasis. Ageing Res Rev. 2013;12:749–56. doi: 10.1016/j.arr.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. N-methyl-D-aspartate receptor (NMDAR) subunit proteins are altered in juvenile- and adult-infected mice at 20 wpi. Frontal cortex GluN2A is decreased in both juvenile-and adult-infected mice. Frontal cortex GluN1 is increased in juvenile-infected mice and decreased in adult-infected mice. Linear regression revealed an age × infection interaction of frontal cortex GluN1 only (b=-3.986, t=-4.548, p<0.01). Hippocampus GluN2A was unchanged, while GluN1 was increased in adult-infected mice. No age × infection interaction was determined for the hippocampus.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. The horizontal line with symbol denote groups compared and ** denotes p≤ 0.01. For symbols alone,
* denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group and
** denotes p≤ 0.01 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 2. Glutamate pre- and post-synaptic marker expression is altered in T. gondii- infected mice at 20 wpi. Frontal cortex pre-synaptic vesicular glutamate transporter 1 (vGLUT1) is decreased in juvenile- and adult-infected mice, with a greater decrease in juveniles. Linear regression revealed an age × infection interaction (b=0.433, t=3.695, p<0.01). Frontal cortex post-synaptic density 95 (PSD95) was increased in adult controls and infected groups, with no age × infection interaction. Hippocampus vGLUT1 was decreased in adult-infected mice and hippocampus PSD95 was unchanged. Linear regression analysis revealed no age × infection interaction.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons. Horizontal lines with symbols denote groups compared and * denotes p≤ 0.05 while ** denotes p≤ 0.01. For symbols alone, * denotes p≤ 0.05 compared to age-matched mock-infected (DPBS) group and ** denotes p ≤ 0.01 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 3. GABA marker expression is altered in T. gondii- infected mice at 20 wpi. Frontal cortex glutamate decarboxylase 67 (GAD67) is decreased in juvenile- and adult-infected mice. Linear regression analysis revealed an age × infection interaction (b=0.0435, t=3.695, p=0.002). Hippocampus GAD67 was unchanged and no age × infection interaction was found.
Data represent an average of 5-6 mice per group and 2 western blots per sample, with representative western blot images shown. DPBS (D) denotes mock-infected controls and PRU (P) denotes T. gondii infected mice. Bars represent 95% confidence interval and symbols represent statistical significance from Mann-Whitney U-test comparisons with * denoting p≤ 0.05 compared to age-matched mock-infected (DPBS) group.
Supplementary Figure 4. Microglia are activated in the brains of T. gondii- infected mice at 8 wpi. Ionized calcium binding adaptor molecule 1 (IBA1) positive microglia show an activated morphology juvenile- and adult- infected brain.
Red denotes microglia marker Iba1. Blue denotes nuclear staining with DAPI.
Scale bar represents 150 μM.
Inset show representative Iba1 positive cells.
Supplementary Table 1. Western blot antibodies. Primary and secondary antibodies, dilutions, and company information.
Supplementary Table 2. Monoamine and metabolite changes in T. gondii infected mice at 20 wpi. Dopamine and metabolites were changed in the hippocampus and striatum of juvenile- and adult-infected mice. Serotonin and metabolites were altered in the hippocampus of infected mice. A ratio < 1.0 signifies a decrease in the monoamine/metabolite compared to DPBS while a ratio > 1.0 signifies an increase in the monoamine/metabolite compared to DPBS.
Data shown is the average of 9-10 mice per group plus/minus standard error. Symbols represent statistical significance from Mann-Whitney U-test comparisons with * denoting p≤ 0.05 compared to age-matched mock-infected (DPBS) group.
Monoamines and metabolites measured include: dopamine (DA), 3,4-Dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), norepinephrine (NE)
Serotonin (5HT), 5-hycroxyindoleacetic acid (5H1AA).
Supplementary Table 3. T. gondii-infected mice have serum T. gondii antibodies and parasite cysts in brain at 8 wpi. T. gondii IgG was elevated in juvenile- and adult-infected mice, with higher levels in adult-infected mice. Parasites cysts were found in juvenile- and adult-infected mice, with fewer cysts/ section in adult-infected mice. T. gondii IgG data shown is the average of 9-10 mice per group. Parasite cyst data shown is the average of 5 mice and 5 sections per mouse. Symbols represent statistical significance from independent sample T-test comparisons with * denoting p<0.05 compared to age-matched mock-infected control (DPBS) and ^ denoting p<0.05 compared to juvenile-infected (PRU) mice.