

Abstract
Here we use a systems biology approach to comprehensively assess the conservation of gene networks in naive pluripotent stem cells (PSCs) with preimplantation embryos. While gene networks in murine naive and primed pluripotent states are reproducible across data sets, different sources of human stem cells display high degrees of variation, partly reflecting disparities in culture conditions. Finally, naive gene networks between human and mouse PSCs are not well conserved and better resemble their respective blastocysts.
Pluripotent stem cells (PSCs) can exist in various metastable states such as the naive or primed state. These two phases of pluripotency are distinguished by prominent molecular and cellular features (Hackett and Surani, 2014). In mouse, naive embryonic stem cells (ESCs), which are derived from the ICM of the E3.5 blastocyst embryo, display two active X chromosomes in female cells and frequently give rise to chimeric embryos. By contrast, primed PSCs represent a relatively later stage in mouse development and are poised for lineage commitment. For example, mouse epiblast stem cells (mEpiSCs) are derived from the E4.5– E5.5 embryo, exhibit X chromosome inactivation, and rarely contribute to blastocyst chimeras (Brons et al., 2007; Tesar et al., 2007).
Naive mouse ESCs (mESCs) can also be subdivided into two subclasses based on culture conditions. Culture in the presence of two pharmacological agents (GSK3b and MAP2K inhibitors, simply termed “2i/LIF”) stabilizes a so called ground state of pluripotency. By undergoing simultaneous inhibition of GSK3b of the Wnt pathway and MAP2K of the ERK signaling cascade, mESCs conventionally cultured in serum/LIF-containing medium (abbreviated as serum/LIF) are induced into a more homogeneous and pluripotent state that selfrenews in serum-free medium (Ying et al., 2008). The 2i/LIF pluripotent state is also characterized by dramatic changes in the transcriptome and epigenome, including global DNA hypomethylation and genome-wide redistribution of H3K27me3 (reviewed in Hackett and Surani, 2014).
Until recently, it was unclear whether human PSCs (hPSCs) including hESCs and induced pluripotent stem cells (iPSCs) could adopt a naive-like pluripotent state. Conventional human ESCs have been suggested to resemble a primed pluripotent state because they share many characteristics with mEpiSCs such as the completion of X chromosome inactivation and a more flattened morphology in culture (Brons et al., 2007; Hanna et al., 2010; Tesar et al., 2007). More recently, several studies have reported the generation of naive hPSCs using different combinations of pharmacological agents and cytokines (Chan et al., 2013; Gafni et al., 2013; Hanna et al., 2010; Takashima et al., 2014; Theunissen et al., 2014; Ware et al., 2014). These naive hPSCs share several morphological and molecular similarities with naive mESCs, suggesting a conserved naive pluripotent state in vitro. However, whether the transcriptomes of mouse or human naive states resemble those of early embryogenesis has not been studied extensively.
We previously used a systems biology approach to identify conserved changes in gene networks during early human and mouse preimplantation development (Xue et al., 2013). This conserved genetic program was characterized by stepwise changes in functional gene networks including cell cycle, transcription regulation, RNA processing, translation, and bioenergetic processes. Here, we performed weighted gene coexpression network analysis (WGCNA) (Zhang and Horvath, 2005) to comprehensively identify gene networks in naive and primed PSCs, and we asked whether the transcriptional organization of naive or primed PSCs resembles transitional stages of preimplantation development.
WGCNA uses an unsupervised and unbiased approach to identify coexpression modules representing clusters of correlated genes. Gene coexpression modules can be cross-analyzed in different data sets and stringently tested for gene network topology preservation across multiple data sets and different species. High preservation scores indicate similar transcriptional organization between two modules including the identity of intra-modular hub genes, which are genes that have high module membership (or gene connectivity). Hub genes are centrally located in their respective module, are representative of the module’s overall function, and have a high likelihood to be critical components within the network. WGCNA has been applied in different biological contexts to effectively uncover functional modules that are representative of the underlying biology. Together, this systems-level approach provides a powerful method to assess the relevance of gene networks between various states of pluripotency and the developing preimplantation embryo.
The Transcriptional Organization of Ground State mESCs Is Most Relevant to the ICM
It has been suggested that different states of mouse PSCs (mPSCs) exhibit transcriptional similarities to different stages of the developing embryos; however, those analyses were limited in sample sizes and number of assayed genes. Inorder to comprehensively analyze data sets in different laboratories, we first curated 125 mouse samples comprising data from naive mESCs in 2i/LIF (n = 45), conventional mESCs in serum/LIF (n = 51), and primed EpiSCs (n = 29) (samples listed in Table S1A available online). These data were divided fairly evenly between two separate microarray platforms and were used for independent cross-validation. In the first data set, WGCNA identified a total of 10 modules, but only 3 modules were specifically correlated with 2i/LIF, serum/LIF, and primed states (Figure S1A available online). The second validation data set likewise identified distinct modules that specifically correlated with 2i/LIF, serum/LIF, and primed states (Figure S1B). Direct module comparisons between the two data sets revealed that 2i/LIF, serum/LIF, and primed modules shared significant gene overlap (p < 1 × 10−30, hypergeometric test) (Figure S1C). Module preservation analysis, a suite of rigorous statistical tests that determine network density and topology consistency between two data sets (see Supplemental Experimental Procedures), showed that all the state-specific modules share concordant transcriptional organization (Figure S1D). Together, these results demonstrated that mPSCs under 2i/LIF, serum/LIF, and primed states have a unique, robust, and reproducible transcriptional organization that could be represented through a small number of gene modules.
To determine whether the transcriptional organization of the naive or primed states resembled early embryos, we cross-referenced our identified modules with mouse preimplantation expression data sets spanning the developmental spectrum from one-cell to blastocyst embryos using either single-cell RNA-sequencing (Tang et al., 2011) or whole-embryo microarray (Xie et al., 2010) platforms. By overlaying mouse 2i/LIF, serum/LIF, and primed modules, we found that mESCs cultured in 2i/LIF most resembled late-stage preimplantation embryos (p < 1 × 10−15 and Figure 1A). By contrast, conventiona mESCs cultured in serum/LIF were not enriched for any particular stage of preimplantation development. Instead, the serum/LIF module overlapped significantly (p < 1 × 10−16) with day3 and day5 ESCs derived from ICM outgrowths (Tang et al., 2011). These data suggest that mESCs cultured in serum/LIF exist in a unique pluripotent state outside of preimplantation embryos, likely as a consequence of adapting to conventiona ESC culture conditions. Our results are consistent with previous findings by Tang et al. (2010) and Boroviak et al (2014), who showed that ESCs cultured in serum/LIF are distinctly different from the ICM of blastocysts while 2i/LIF ESC colonies share resemblance to E4.5 preimplantation embryos based on expression of lineage markers and pathway genes (Boroviak et al., 2014; Tang et al. 2010). Furthermore, our results identified a significant overlap between the primed EpiSC module and blastocyst/epiblast modules (p < 9 × 10−5). Interestingly, the primed module also appeared to show some significant overlap with the one-cell embryo. However, using more stringent module preservation analyses, we found that neither the one-cell nor the epiblast module is preserved in mESCs. By contrast, blastocyst and ESC outgrowth modules are significantly preserved in 2i/LIF and serum/LIF mESCs, respectively (Figure 1B). Since module preservation analysis takes into consideration correlations between individual genes, this result indicates that while individual genes may significantly overlap between primed and one-cel embryo, the organization of these genes (i.e., coexpression relationships with other genes) is dissimilar. Overlapping genes that do not share topologica structures probably represent different biological subtexts in each cell type and likely do not share any biologically meaningful overlap. Together, these results indicate that the transcriptional architecture of the murine ground state stem cells most closely resembles the blastocyst stage.
Figure 1. Transcription Networks in Preimplantation Embryos and Naive/Primed Pluripotent States.
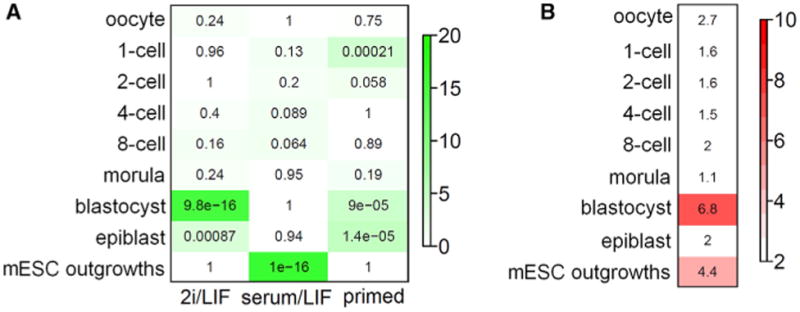
(A) Heatmap showing the significance of gene overlaps between mouse 2i/LIF, serum/LIF, and primed modules (x axis) and stage-specific mouse preimplantation embryo modules (y axis). Mouse stem cell modules were defined by identifying clusters of correlated transcripts to 2i/LIF, serum/LIF, and primed state asshown in Figure S1. Each cell contains the average p value (geometric mean) from gene intersects of 2i/LIF, serum, and primed modules with two separate mouse preimplantation data sets (Tang et al., 2011; Xie et al., 2010). Color legend represents –log10 p value based on the hypergeometric test. (B) Heatmap of module preservation scores between mouse preimplantation stages and mPSCs. Module preservation scores are represented by the median Zsummary statistic from three independent mouse preimplantation data sets to ensure sufficient coverage of all preimplantation stages (Tang et al., 2011; Xie et al., 2010; Xue et al., 2013). Shown is the preservation level of stage-specific preimplantation reference modules (y axis) within the mPSC data sets. Generally, a Zsummary score of less than 2 indicates that the reference module is not preserved in the test data set, Zsummary of 5 is moderately preserved, and a Zsummary score above 10 is strongly preserved. Note: epiblast cells (y axis) are single-cell transcriptome data from E4.5 embryos (Tang et al., 2011), and EpiSCs (x axis) are stabilized culture derived from E5.75 embryos (Table S1A).
Naive hPSC Transcriptomes Vary across Different Methods but Share a Consensus Gene Network in RNA Processing, Ribosome Biogenesis, and Mitochondrial Metabolism
Recent studies have reported the use of various molecules targeting signaling pathways or epigenetic regulators to generate naive hPSCs with different molecular and cellular characteristics (Chan et al., 2013; Gafni et al., 2013; Hanna et al., 2010; Takashima et al., 2014; Theunissen et al., 2014; Ware et al., 2014). To evaluate whether these different naive hPSCs correspond to similar or different states of pluripotency, we used WGCNA to define individual gene networks in six data sets for pairwise comparison. It should be noted that some data sets have relatively small sample sizes, which makes the network analysis more prone to noise and less robust. Nevertheless, we found that the gene networks of various naive hPSCs generally shared significant overlap, suggesting some core component that is shared across all existing lines of naive hPSCs (Figure S2A). Notably, the two data sets from Takashima et al. (2014) and Theunissen et al. (2014) have the best pairwise overlap of naive modules, suggesting that these two naive pluripotency gene networks are closely related. Surprisingly, we found that the primed modules among different data sets showed limited overlap (Figure S2A). A closer look at experimental approaches showed biological differences due to variable culture conditions and technical noise in transcriptome data (Table S1B and data not shown). Thus, it would be useful to standardize culture conditions as well as increase transcriptome sample numbers for more effective systems biology analysis.
Nevertheless, we applied WGCNA to construct consensus modules across all six data sets to stringently scrutinize overlapping gene networks. Consensus modules group together highly coexpressed genes across multiple data sets and by definition are present in multiple data sets. Thus, consensus modules signify common and robust coexpression relationships that are more resistant to technical noise and are therefore more representative of the underlying biology. In our analysis, we identified a single consensus module of317 genes that was significantly correlated to the naive state in three data sets (p < 0.05; Figure S2B). Gene ontology (GO) analysis of the consensus naive module revealed enrichment in RNA processing, ribosome biogenesis, and mitochondrial genes (p < 1 × 10−4; Figure S2C). Importantly, increased mitochondrial activity has been previously reported in naive hPSCs as well as in naive mESCs (Takashima et al., 2014; Ware et al., 2014; Zhou et al., 2012). The increased mitochondrial activity and increased expression of housekeeping genes may reflect overall higher bioenergetic requirements in naive PSCs. However, our data showed that the conserved naive hPSC module is relatively small (approximately 10%–15%) compared to the large transcriptome changes observed for each independently established line of naive hPSCs. This indicated that while all established naive hPSCs share a conserved component, the vast majority of transcriptional changes represent a unique pluripotent state that is unlike others. Nonetheless, these data suggest that naive hPSCs unanimously exhibit fundamentally different molecular and metabolic activities from those of primed hESCs.
Cross-Comparisons of Human-Mouse Naive and Primed PSCs
Conventionally, the gold standard to determine whether primed hPSCs have successfully converted to a naive state has been to benchmark against naive mESCs. To determine whether naive gene networks between human and mouse are preserved, we cross-referenced human and mouse naive and primed PSCs. Our analysis indicated that most existing lines of naive hESCs do not share meaningful overlap with naive mESCs, with exception of the data sets by Takashima et al. (2014) and Theunissen et al. (2014), which showed significant overlap with the 2i/LIF mESC module (p < 0.01) (Figure 2A). On the other hand, primed hPSCs generally had significant overlap with the mouse EpiSC primed module, in agreement with previous observations (Brons et al., 2007; Tesar et al., 2007). The overlapped genes between human primed modules and mouse EpiSCs were enriched for genes in cell-adhesion categories. However, in general the transcriptional networks between hPSCs and mPSCs were either weakly or not preserved (data not shown). Overall, our data suggests that the transcriptional organization of naive hESCs has variable resemblance to naive mESCs.
Figure 2. Transcription Networks in Preimplantation Embryos and Naive/Primed Pluripotent States.
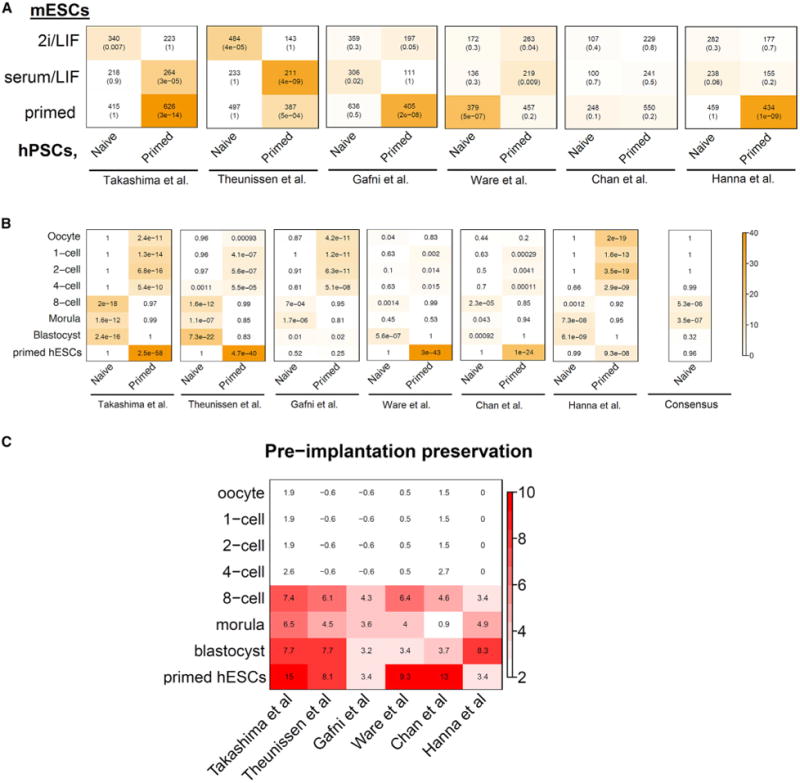
(A) Heatmap containing the significance levels of gene intersects between 2i/LIF, serum/LIF, and primed modules (y axis) with naive and primed hESCs from six different methods (x axis). Each cell contains the number of intersecting genes and the p value of the intersection. (B) Heatmap showing the significance of gene overlaps between human preimplantation embryos and six different methods for producing naive hESCs. The x axis shows naive and primed hESC modules from different groups and the y axis shows stage-specific modules of human preimplantation development. Comparison with consensus naive and primed modules (as defined in Figure S2B) is also shown. Each cell contains the average p value (geometric mean) from gene intersects of naive and primed modules with three separate human preimplantation data sets (Vassena et al., 2011; Xie et al., 2010; Yan et al., 2013). Color legend represents –log10 p value based on the hypergeometric test. (C) Heatmap of human preimplantation module preservation across multiple naive hESC data sets. Module preservation scores are represented by the median Zsummary statistic from three independent human preimplantation data sets (Vassena et al., 2011; Xie et al., 2010; Yan et al., 2013). Preservation levels of pre-implantation stage-specific modules (y axis) across multiple naive/primed hESCs data sets are shown.
Human Naive PSCs More Closely Resemble the Human Preimplantation Blastocyst
Because mouse naive modules did not return consistent results between different naive hESCs, we reasoned that the human preimplantation transcriptome may present a more reliable reference for the human naive state. By comparing naive and primed hESCs with the gene networks of early human embryos from three data sets that span one-cell to blastocyst stages (Vassena et al., 2011; Xie et al., 2010; Yan et al., 2013), we found that each data set showed improved but variable similarity to human preimplantation embryos compared to that found from cross-referencing with mouse naive ESCs (Figure 2B). Again, naive hPSCs generated by the Takashima et al. (2014) and Theunissen et al. (2014) protocols most closely resembled the expression profile of the human blastocyst. Interestingly, the consensus naive hPSC module had significant overlaps with the eight-cell and morula preimplantation stages (Figure 2B), consistent with the conserved activation of gene networks in RNA processing, translation, and mitochondrial genes after the first major wave of embryonic genome activation (EGA) (Xue et al., 2013). This suggests that naive PSCs may share some cellular and metabolic features with post-EGA blastomeres. By contrast, the primed pluripotent states exhibited significant overlap with early passage (p0 and p10) hESCs derived from ICM outgrowths (p < 5 × 10−4). This similarity is consistent with previous observations that primed hESCs transcriptionally adapt to culture conditions from the very beginning (Yan et al., 2013). In addition, gene networks of primed hESCs from different labs also had significant overlap with modules of pre-EGA cleavage embryos including the one-cell to four-cell stages (Figure 2B), similar to observations in the mouse primed EpiSC module that showed similarity with prezygotic genome activation one-cell embryos (Figure 1A). Although overlap in primed hPSCs with pre-EGA cleavage embryos is enriched for genes involved in cell cycle and mitosis, module preservation analysis showed that pre-EGA networks are absent in hPSCs. This discrepancy indicated that gene network topologies between pre-EGA embryos and primed hPSCs are rather different (Figure 2C), and therefore should not share meaningful biological properties.
Analysis of Intramodular Hub Genes in Human Naive Stem Cells and post-EGA Embryos
WGCNA provides a measure of intramodular gene membership (kME; see Supplemental Experimental Procedures), which is closely related to the measure of gene connectivity. Genes with high module membership are regarded as intramodular hub genes, which are centrally located in their respective module and have a high likelihood to be critical components within the network. To identify conserved hub genes (kME > 0.8 and p < 10−22) between preimplantation embryos and naive hPSCs, we focused on consensus hub genes derived from the consensus naive hPSC module (Figure S2B). As expected, RNA processing genes CLP1 and PRPF38A were conserved hub genes between eight-cell embryos and naive hPSCs. RRP12, a ribosomal RNA processing gene, is also a common intramodular hub. In general, these hub genes reflect the overall function of eight-cell/morula and consensus naive hPSC modules. Unexpectedly, we identified the cell cycle regulator, CCNE1, as a shared hub gene between naive hESCs and eight-cell embryos. CCNE1 encodes cyclin E1, a regulatory subunit of CDK2. Importantly, these hub genes do not appear to be conserved in mPSCs. Thus, our analysis lends further support to the idea that intramodular hub genes are more conserved between naive hPSCs and human blastocyst embryos than that of naive hPSCs and mPSCs.
Concluding Remarks
Gene network analyses revealed that the mouse ground state PSCs share a robust and highly conserved genetic program with blastocyst embryos in vivo, whereas conventional mESCs in serum/LIF appear to represent a pluripotent state unlike that of preimplantation development. Therefore, our analysis of gene networks in stem cells with preimplantation embryos allowed us to form a robust standard to define naive versus primed pluripotency in murine stem cells. In contrast, different sets of established naive hPSCs exhibit large variations in their transcriptomes when compared to either naive mESCs or human blastocysts. These variations likely represent unique pluripotent states defined by their distinctive culture conditions (Table S1B). Nonetheless, regardless of variations, we found that all established lines of naive hPSCs show clear resemblance to human late preimplantation embryos when compared with their primed counterparts. This relationship is partially explained by a convergent network of increased cellular metabolic activity reminiscent of genes activated during the first major wave of embryonic genome activation. Taken together, our transcriptome analysis of stem cells and early embryos in both murine and human has consistently suggested a high conservation of gene networks underlying naive and primed states of pluripotency with different stages of preimplantation and postimplantation embryos.
In this issue of Cell Stem Cell, Fang et al. (2014) report the generation of naive iPSCs using the rhesus monkey model. In contrast to human naive iPSCs, this study suggests that FGF signaling is indispensable to naive nonhuman primate iPSCs (Fang et al., 2014). Although the limited naive monkey iPSC transcriptome data set prevented us from conducting a meaningful systems biology comparison with naive mPSCs and hPSCs, this study further illustrates the complexity of species-specific signaling pathways underlying mammalian naive pluripotency networks. A unique aspect of this study is their extensive description of interspecies mouse-monkey embryo chimera formation as a way to test naive pluripotency of nonhuman primate PSCs. Of course, analogous to the mouse model system, a rigorous test of the naive pluripotency or totipotency of nonhuman primate PSCs should be conducted within nonhuman primate animal models in the future.
Our analyses suggest that a systems-level comparison of transcriptome data from mammalian preimplantation embryos with naive PSCs is a useful metric to benchmark ground state pluripotency. However, systems-level analysis requires a relatively large sample size to safeguard against technical noise and to form statistically confident conclusions. For example, our current analysis indicates that some naive hESC data sets have substantial intra-data-set variations (data not shown) which potentially limit some of our conclusions. Nevertheless, we partially overcame this challenge by constructing consensus modules that scan for shared modules across multiple data sets. This method also identified consensus hub genes that may be useful markers for quickly assessing transitions between primed and naive hPSCs. Although our study identifies commonalities in gene networks between various defined states of naive hPSCs, the epigenetic landscape, such as DNA methylation and X chromosome inactivation, also represents a crucial feature of naive pluripotency (Takashima et al., 2014). The epigenetic states of many established “naive” state hPSCs remain to be characterized and perhaps to be compared with human preimplantation embryos as well. Therefore, future work shall continue to investigate and identify the “authentic” naive state of hPSCs at both transcriptome and epigenome levels using the profile of human preimplantation embryos as one of the gold standards.
Supplementary Material
Acknowledgments
We would like to thank Rudolf Jaenisch and Thorold W. Theunissen for their insightful comments on our paper. This study is supported by NIH grant DE 022928. K.H. is supported by the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA.
Footnotes
Supplemental Information: Supplemental Information for this article includes Supplemental Experimental Procedures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2014.09.014.
Author Contributions: K.H. and G.F. conceived the study and designed the analyses. K.H. and T.M. performed WGCNA and statistical analyses. K.H., T.M., and G.F. wrote the manuscript.
References
- Boroviak T, Loos R, Bertone P, Smith A, Nichols J. Nat Cell Biol. 2014;16:516–528. doi: 10.1038/ncb2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, Vallier L. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Chan YS, G(x00F6);ke J, Ng JH, Lu X, Gonzales KA, Tan CP, Tng WQ, Hong ZZ, Lim YS, Ng HH. Cell Stem Cell. 2013;13:663–675. doi: 10.1016/j.stem.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Fang R, Liu K, Zhao Y, Li H, Zhu D, Du Y, Xiang C, Li X, Liu H, Miao Z, et al. Cell Stem Cell. 2014;15(this issue):488–496. doi: 10.1016/j.stem.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viu-kov S, Maza I, Zviran A, et al. Nature. 2013;504:282–286. doi: 10.1038/nature12745. [DOI] [PubMed] [Google Scholar]
- Hackett JA, Surani MA. Cell Stem Cell. 2014;15(this issue):416–429. doi: 10.1016/j.stem.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, Jaenisch R. Proc Natl Acad Sci USA. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, et al. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Bao S, Lee C, Nord-man E, Wang X, Lao K, Surani MA. Cell Stem Cell. 2010;6:468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Nordman E, Bao S, Lee C, Wang X, Tuch BB, Heard E, Lao K, Surani MA. PLoS ONE. 2011;6:e21208. doi: 10.1371/journal.pone.0021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RD. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Theunissen TW, Powell BE, Wang H, Mitali-pova M, Faddah DA, Reddy J, Fan ZP, Maet-zel D, Ganz K, Shi L, et al. Cell Stem Cell. 2014;15(this issue):471–487. [Google Scholar]
- Vassena R, Boué S, Gonza´lez-Roca E, Aran B, Auer H, Veiga A, Izpisua Belmonte JC. Development. 2011;138:3699–3709. doi: 10.1242/dev.064741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Nelson AM, Mecham B, Hesson J, Zhou W, Jonlin EC, Jimenez-Caliani AJ, Deng X, Cavanaugh C, Cook S, et al. Proc Natl Acad Sci USA. 2014;111:4484–4489. doi: 10.1073/pnas.1319738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie D, Chen CC, Ptaszek LM, Xiao S, Cao X, Fang F, Ng HH, Lewin HA, Cowan C, Zhong S. Genome Res. 2010;20:804–815. doi: 10.1101/gr.100594.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Huang K, Cai C, Cai L, Jiang CY, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, et al. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. Nat Struct Mol Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. Statistical Applications in Genetics and Molecular Biology. 2005;4:17. doi: 10.2202/1544-6115.1128. Article. [DOI] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.