

Abstract
The intestinal epithelium forms a key protective barrier that separates internal organs from the harmful environment of the gut lumen. Increased permeability of the gut barrier is a common manifestation of different inflammatory disorders contributing to the severity of disease. Barrier permeability is controlled by epithelial adherens junctions and tight junctions. Junctional assembly and integrity depend on fundamental homeostatic processes such as cell differentiation, rearrangements of the cytoskeleton, and vesicle trafficking. Alterations of intestinal epithelial homeostasis during mucosal inflammation may impair structure and remodeling of apical junctions, resulting in increased permeability of the gut barrier. In this review, we summarize recent advances in our understanding of how altered epithelial homeostasis affects the structure and function of adherens junctions and tight junctions in the inflamed gut. Specifically, we focus on the transcription reprogramming of the cell, alterations in the actin cytoskeleton, and junctional endocytosis and exocytosis. We pay special attention to knockout mouse model studies and discuss the relevance of these mechanisms to human gastrointestinal disorders.
Keywords: adherens junctions, actin cytoskeleton, endocytosis, exocytosis, non-muscle myosin II, tight junctions
1. Introduction
The epithelial lining of the gut represents the largest interface that separates the internal organs from the environment. It plays several vital roles including absorption of nutrients and water, and creation of the environment for symbiotic luminal microflora. One of the most important functions of the intestinal epithelium is formation of the protective barrier between the internal organs and microbial pathogens and antigens of the gut lumen [1, 2]. This barrier includes many different components such as secreted mucus and antimicrobial peptides as well as a powerful immune system populating the gut wall. However, one of the most important manifestations of the intestinal barrier is a single layer of tightly-packed columnar-shaped epithelial cells with extended lateral surfaces that interacts with adjacent cells via multiple adhesive contacts.
Structure and permeability of the intestinal epithelial barrier is known to be modulated by a variety of physiological and environmental factors. For example, metabolites derived by luminal microbes increase barrier tightness, which is important for limiting exposure of gut microbiota to intestinal immune cells [3]. Conversely, some nutrients such as glucose increase permeability of the intestinal barrier, thereby accelerating fluid and nutrient absorption [3, 4]. Such physiological plasticity of the gut barrier is essential for normal gastrointestinal functions, epithelial rejuvenation, and tissue morphogenesis. In disease conditions, however, the prolonged increase in permeability of the gut barrier enhances body exposure to luminal bacteria and their products, leading to overactivation of intestinal immune cells and perpetuation of mucosal inflammation [4, 5].
Mounting evidence highlights increased intestinal epithelial permeability as a common pathophysiologic mechanism for a large group of diseases. This mechanism is primarily characterized in different gastrointestinal disorders such as inflammatory bowel disease (IBD; composed of Crohn's disease [CD] and ulcerative colitis [UC]), celiac disease (CeD), irritable bowel syndrome (IBS), necrotizing enterocolitis, and enteric infections [2, 5, 6]. Furthermore, a leaky gut appears to contribute to the development of extraintestinal and systemic immune diseases including obesity, type I diabetes, and liver steatosis [7, 8]. Dysfunctions of the gut barrier have been under extensive investigation during the recent decade and these investigations have produced an exponential growth in the number of related publications. Several excellent recent reviews summarized the progress achieved in this field [2, 3, 5, 9, 10]. Many of these reviews discuss barrier disruption from the immunological perspective by focusing on the effects of immune cells and mediators, or by describing the signaling events triggered by activation of epithelial pathogen-recognizing proteins and cytokine receptors. Less attention was dedicated to the fact that mucosal inflammation causes profound changes in the intestinal epithelial homeostasis, which may directly or indirectly impair intercellular adhesions leading to increased barrier permeability. The present review focuses on recent developments in the understanding of fundamental cellular mechanisms that regulate the integrity of epithelial barrier in normal gut and drive barrier disruption during mucosal inflammation. Specifically, we focus on the molecular composition of epithelial junctions, vesicle trafficking events, and the organization and remodeling of the cortical actomyosin cytoskeleton. Due to the space limitation, we opted not to include several important barrier-protective and barrier-disruptive mechanisms such as mucin synthesisand secretion as well as epithelial cell death during intestinal inflammation. These mechanisms have been recently discussed in several excellent reviews [2, 11, 12].
2. Molecular composition of intestinal epithelial apical junctions
Permeability of the intestinal epithelial barrier is controlled by several multiprotein adhesive complexes (junctions) located along the lateral surface of contacting epithelial cells. These structures include the most apical tight junctions (TJs), subjacent adherens junctions (AJs), and desmosomes [13]. While all three types of epithelial junctions cooperate in regulating the passage of ions and other molecules across the gut barrier, this review is focused on AJs and TJs, which have been the most extensively characterized.
2.1. Adherens Junctions
AJs, the most ancient and abundant adhesion structures, exist in virtually all mammalian tissues. They have many crucial functions ranging from mediating tissue integrity to controlling cell proliferation and motility. A general model of the AJ structure postulates the existence of its transmembrane adhesive core interacting with several cytoplasmic scaffolds that physically links AJs to the underlying actomyosin cytoskeleton [14, 15]. The transmembrane adhesive core of epithelial AJs is composed of two major classes of integral membrane proteins, cadherins and nectins, which regulate calcium-dependent and calcium-independent intercellular adhesions, respectively [14, 15]. Mammalian AJs contain so called ‘classical cadherins’: single-spanned membrane proteins composed of an extended extracellular amino-terminal region or ectodomain, a transmembrane segment, and a short cytoplasmic domain [16]. Intestinal epithelial cells predominantly express epithelial (E) cadherin, and to a lesser extent they express placental (P) cadherin [17]. As a result of E-cadherin molecules engaging in homotypic trans-dimerization with partners on the opposing plasma membrane and engaging in cis-dimerization with partners on the same membrane, large cadherin clusters form that mediate epithelial adhesions. Assembly and stability of these adhesive clusters is regulated by scaffolding proteins that bind to the cytoplasmic tail of E-cadherin. Two armadillo-repeat proteins, β-catenin and p120-catenin, directly interact with the E-cadherin cytoplasmic tail and control cadherin trafficking and retention at the plasma membrane [14, 16]. Furthermore, β-catenin interacts with an actin binding protein (ABP), α-catenin, which mediates AJ linkage to the underlying actomyosin bundles.
In contrast to the well-studied E-cadherin-catenin complexes, little is known about the functional roles and regulation of nectins in the intestinal epithelium. In mammals, the nectin protein family is composed of four different immunoglobulin-like proteins: nectins-1 through -4 [18]. Nectin-2and -3 were shown to be abundantly expressed in the colon of mice, whereas the two other isoforms were barely detectable [19]. Unlike classical cadherins, different nectins are readily engaged in both homotypic and heterotypic trans-dimerization. A conserved four amino-acid motif on the end of the cytoplasmic domain of nectins interacts with a cytoplsmic scaffolding protein, afadin. Afadin is a known ABP and it may provide an additional link between AJ proteins and the actin cytoskeleton [18].
2.2. Tight Junctions
TJs represent an elaborate network of protein-based fibrils embedded in the plasma membrane [20-22]. The transmembrane proteins of TJs are engaged in variety of cis- and trans- interactions at sites of intercellular contacts, thereby creating the paracellular barrier. The peripheral membrane proteins interacting with these transmembrane molecules form a cytosolic plaque that is crucial for TJ stability and dynamics (assembly and disassembly) [20-22]. Three major classes of transmembrane proteins cooperate in the establishment of the TJ barrier. They include the claudin protein family, the tight junction-associated MARVEL proteins (TAMP), as well as several immunoglobulin-like proteins such as junctional adhesion molecule (JAM)-A and coxsackievirus and adenovirus receptor (CAR). A number of scaffolding and signaling molecules have been associated with the cytosolic plaque of TJs. They include zonula occludens (ZO) proteins, MAGI proteins, a PAR3/PAR6 polarity complex, and cingulin [20-22].
Barrier properties in the intestinal epithelium critically depend on claudins that self-assemble into the intramembranous fibrils and form size-restrictive and charge-selective paracellular pores. Claudins are four-transmembrane domain (tetraspan) proteins. Their two extracellular loops are enriched in hydrophobic amino acids, which favors different adhesive interactions with other claudin molecules at the same and the the opposing plasma membranes. This large protein family consists of 27 members in mammalian epithelia [5, 21, 23, 24]. Expression of almost all mammalian claudins (except for claudin 6, 16 and 19) was detected in the human and murine intestinal mucosa [5, 23, 24]. Functionally, claudin proteins have been divided into two groups: ‘sealing’ or ‘tight’ claudins, and ‘leaky’ claudins. Overexpression of ‘sealing’ claudins is known to increase transepithelial electrical resistance, which indicates tightening of the paracellular barrier. By contrast, expression of ‘leaky’ claudins was shown to increase permeability of epithelial barriers. The majority of mammalian claudins (claudin-1, 4, 5, 7, 8, 11, 14, 16, 18, and 19) have been assigned to the sealing group, and only claudins 2, 10, and 15 were found to be leaky [5, 20, 21, 25].
The TAMP family of TJ proteins has thee homologous members: occludin, tricellulin, and MARVEL (MAL and related proteins for vesicle trafficking and membrane link) D3 [20, 26]. Similar to claudins, these are tetraspan proteins that possess two hydrophobic extracellular loops engaging into adhesive homotypical trans-interactions [27]. Occludin and MARVELD3 were shown to be uniformly distributed in all TJs in model epithelial cell monolayers and epithelial tissues in vivo. By contrast, tricellulin was found to be selectively accumulated at tricellular junctions [28]. Despite an extensive research focus on TAMPs during the last decade, the physiologic roles of these proteins remain poorly defined. Conflicting reports exist on whether different TAMPs are important for the establishment and regulation of TJs in model epithelial cellmonolayers [22, 26]. Some studies suggested dispensability of occludin and MARVELD3 for the maintenance of the intestinal epithelial barrier. This most likely reflects the functional redundancy between these adhesive proteins. Alternatively, TAMPs may have intercellular adhesion-independent functions such as regulation of epithelial cell death and proliferation [29].
JAM-A and CAR are structurally different from claudins and TAMPs. They are single span proteins with one transmembrane domain, two extracellular immunoglobulin-like domains, and a cytoplasmic tail [26, 30]. The extracellular domains of these proteins can be engaged into homotypical or heterotypical cis- and trans-interactions. Different experimental approaches using inhibitory antibodies, soluble ectodomains, or siRNA-mediated knockdown of these proteins provide strong evidence implicating JAM-A and CAR in the regulation of TJ structure and functions in the model intestinal epithelium [26, 30].
The cytosolic TJ plaque represents a large multiprotein complex that contains various scaffolding and signaling molecules and associates with underlying cortical actin cytoskeleton. This complex is important for the assembly, stability and remodeling of TJs. Members of ZO protein (ZO-1, 2, and 3) are the best-characterized molecular components of the cytosolic TJ plaque. These scaffolds have several PDZ and SH3 domains, which are essential for binding of all major transmembrane TJ proteins [21, 26, 31]. In vitro studies involving knockdown of different ZO isoforms in epithelial cells revealed their unique and redundant functions in regulating structure and permeability of apical junctions. While downregulation of neither ZO-2 nor ZO-3 affected the formation of TJs, knockdown of ZO-1 attenuated TJ assembly and establishment of the epithelial barrier [32]. Interestingly, only a simultaneous loss of all ZO proteins caused a dramatic disruption of the TJ architecture and opening of the paracellular barrier in model epithelial cell monolayers [33].
3. Regulation of AJ and TJ protein expression in inflamed intestinal mucosa
Increased permeability of inflamed intestinal mucosa is well-documented in both clinical settings and different experimental models of colitis and gastrointestinal infection [2, 3, 5, 25, 34, 35]. Since AJs and TJs are critical regulators of paracellular permeability, structural abnormalities of these junctional complexes are thought to underlie the compromised barrier properties of the gut during mucosal inflammation. Indeed, ultrastructural studies have demonstrated a dilation of apical junctions and increased intercellular space in mucosal biopsies of CD, UC, and CeD patients [36-38]. Furthermore, freeze-fracture electron microscopy revealed abnormal fragmentation of TJ strands in active CD, which correlates with the increased epithelial permeability [39]. A significant body of literature documents multiple mechanisms that may mediate the impaired structure and functions of epithelial apical junctions in the inflamed intestinal mucosa. These mechanisms can be classified into three large categories: decreased expression of AJ and TJ proteins; altered trafficking of junctional components; and remodeling of the perijunctional actomyosin cytoskeleton (Fig. 1).
Figure 1. Different mechanisms that mediate disassembly of adherens junctions and tight junctions in the inflamed intestinal mucosa.
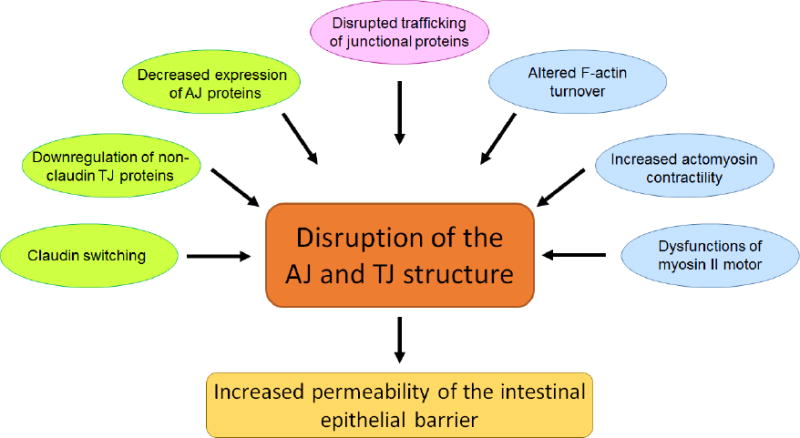
This schematic diagram summarizes the major mechanisms that contribute to intestinal AJ and TJ disassembly during mucosal inflammation. Different cellular responses such as altered expression of junctional proteins, cytoskeletal abnormalities, and altered vesicle trafficking are marked by different colors.
3.1. Decreased expression of AJ proteins during intestinal inflammation
E-cadherin based AJs are crucial regulators of epithelial differentiation and homeostasis in different tissues. Surprisingly, the role of this junctional complex in the disruption of the gut barrier during inflammation remains poorly understood, and research on this subject is somewhat overshadowed by the focus on the dysfunctions of TJs. Clinical and experimental studies demonstrate disruption of AJ architecture during intestinal inflammation, which may significantly contribute to functional defects of the gut barrier. The available data are limited to the E-cadherin-catenin module of AJs and they document either downregulation or mislocalization of these proteins during mucosal inflammation. Indeed, decreased expression of E-cadherin has been reported in tissue biopsies of CD, UC, CeD, and IBS patients [40-44]. Both total level of E-cadherin and its junctional-associated fraction is decreased during gut inflammation. Importantly, genome-wide association studies revealed significant association of E-cadherin polymorphism with susceptibility to UC [45] and CD [46]. One IBD-associated haplotype of E-cadherin is characterized by a 72-amino acid deletion in the N-terminal region of the precursor protein [46]. This truncated molecule was not properly targeted to AJs and showed diffuse cytoplasmic localization that resembles E-cadherin labeling in tissue biopsies of CD patients.
Interestingly, inflammation and injury of the intestinal mucosa does not simply downregulate E-cadherin expression, but rather they lead to the ‘cadherin switching’, which manifests itself by the increased expression of P-cadherin [17, 40]. Although E-cadherin and P-cadherin are highly homologous proteins, they are functionally different. In addition to regulating cell-cell adhesions, P-cadherin controls stem cell proliferation and differentiation as well as epithelial cell migration [47]. While the roles of P-cadherin in inflamed intestinal mucosa have not been investigated, the described functions of this protein in cancer cells suggest that P-cadherin overexpression could be essential for mucosal restitution during the remission stage of gut inflammation.
E-cadherin is not the only AJ protein that has its expression downregulated during mucosal inflammation as several studies showed decreased levels of its cytosolic binding partners, p120-catenin, and β-catenin [41, 42, 44, 48]. Since catenins are essential for the delivery and stabilization of E-cadherin at AJs, loss of these scaffolding proteins should result in depletion of the AJ-associated pool of E-cadherin in inflamed intestinal mucosa, further weakening epithelial cell-cell adhesions. Disruption of E-cadherin-catenin complexes at AJs may have other profound homeostatic consequences, such as nuclear translocation of β-catenin. Nuclear β-catenin acts as transcriptional co-activator that drives intestinal epithelial cell proliferation [49]. There are two different functional outcomes of nuclear translocation of β-catenin in inflamed intestinal mucosa: acceleration of wound healing during mucosal restitution; and promotion of colitis-associated colon cancer.
Studies that used knockout animal models support a strong causal connection between the disruption of AJs and the development of mucosal inflammation. Interestingly, the first report that demonstrated the essential anti-inflammatory role of the intact gut barrier involved a targeted disruption (by overexpressing a dominant-negative mutant of N-cadherin) of intestinal E-cadherin-based cell-cell adhesions in mice [50]. This disruption resulted in the development of spontaneous colitis resembling human CD. A subsequent study characterized an inducible knockout of E-cadherin in small intestinal and colonic epithelium [51]. Elimination of intestinal epithelial E-cadherin caused a bloody diarrhea associated with enhanced cell death and loss of the mucosal architecture. Similarly, dramatic disruption of epithelial cell-cell adhesions and induction of mucosal inflammation has been reported in mice with intestinal epithelial-specific knockout of p120-catenin [52]. By contrast, inducible ablation of intestinal epithelial β-catenin, while blocking cell proliferation, did not impair the integrity of epithelial junctions [53]. Consistently, downregulation of E-cadherin and p120-catenin expression was shown to increase paracellular permeability and to disrupt TJ integrity in model epithelial cell monolayers [52, 54]. Together, these data strongly suggest that expressional downregulation of AJ proteins represents an important mechanism of epithelial barrier disruption during intestinal inflammation.
Dysfunction of E-cadherin-based cell-cell adhesions in the inflamed gut may also be associated with post-translation modification of AJ proteins. For example, mice deficient in receptor protein tyrosine phosphatase sigma demonstrated enhanced intestinal permeability, developed IBD-like intestinal symptoms, and had increased susceptibility to enteric infection [55]. Molecular abnormalities identified in these animals involved increased phosphorylation of E-cadherin and β-catenin in the intestinal epithelium, which is likely to destabilize AJs. Interestingly, genetic polymorphism of this receptor tyrosine phosphatase is linked to the development of UC [46]. On the other hand, tyrosine phosphorylation of β-catenin was found to be dramatically increased in tissue samples of CeD patients [56]. Further studies are warranted to determine the impact of post-translational modification of different AJ proteins on the disruption of the gut barrier during mucosal inflammation.
Molecular mechanisms that mediate the decreased expression of epithelial AJ proteins in inflamed intestinal mucosa remain poorly characterized. Generally, increased production of proinflammatory cytokines such as TNFα and IFNγ is considered as the major upstream event leading to the disruption of the gut barrier [10, 57]. However, the roles of these cytokines in AJ disassembly and downregulation of AJ proteins in model epithelia remain controversial [15]. Since different inflammatory mediators have additive roles in enhancing epithelial permeability, the defects of AJ organization observed in IBD patients are likely to be driven by a complex inflammatory milieu composed of multiple cytokines, chemokines, free radicals, and lipid mediators.
In contrast to a large body of literature that focuses on the mechanisms of E-cadherin downregulation in cancer, little is known about similar mechanisms during intestinal inflammation. A handful of studies suggests that inflammatory stimuli may downregulate expression of epithelial AJ proteins on transcriptional, translational, and post-translational levels. For example, the diminished transcription of E-cadherin mRNA could be due to promoter methylation of the E-cadherin gene. Indeed, hypermethylation of the E-cadherin promoter was observed in patients with chronic UC and UC-associated colon cancer [58, 59]. This was accompanied by decreased E-cadherin expression. Since hypermethylation of the E-cadherin promoter is more abundant in UC patients with dysplastic lesions of the intestinal epithelium as compared to non-dysplastic mucosa, such epigenetic blockage of E-cadherin expression is likely to be limited to late stages of inflammation and its progression into colorectal cancer.
Another possible mechanism for the decreased expression of AJ proteins involves inhibition of their translation. Indeed, translation of E-cadherin and p120-catenin is decreased in inflammatory breast cancer [60] and cytokine-exposed model pancreatic epithelium [54]. These effects are associated with defects in the translation initiation machinery. On the other hand, impaired protein translation is reported in colonic epithelial cells treated with IFNγ and TNFα [61]. Therefore, it is tempting to speculate that impaired protein translation may be responsible for decreased expression of E-cadherin and some catenins in the inflamed intestinal mucosa.
Expression of E-cadherin and other AJ proteins could be also regulated on the post-translational level. One reported mechanism involves proteolytic degradation of E-cadherin. E-cadherin degradation was observed during colonization of intestinal epithelium by enteric pathogens such as Bacteroides fragilis and Campylobacter jejuni [62, 63], or following neutrophil infiltration in the inflamed mucosa [64-66]. Invading bacteria and activated immune cells release different proteases that cause shedding of the E-cadherin ectodomain, thereby decreasing the concentration of adhesion competent protein at the plasma membrane. It is noteworthy that proteolytic cleavage is not a unique feature of E-cadherin, since TJ and desmosomal proteins are also cleaved during mucosal inflammation and infection [65, 66]. This makes it difficult to establish the relative contribution of proteolytic degradation of individual junctional proteins in the disruption of the gut barrier.
3.2. Decreased expression of TJ proteins during intestinal inflammation
In contrast to the relatively less-studied alterations in the AJ organization, a large body of literature documents the abnormal molecular composition of epithelial TJs during intestinal inflammation and infection. Among these abnormalities, the most notable are ‘claudin switching,’ the decreased expression of non-claudin transmembrane proteins, and the loss of ZO-1 from the cytosolic plaque of TJs. Claudin switching is manifested by the altered balance between different members of the claudin protein family in the gut [5, 23]. Specifically, major intestinal inflammatory diseases such CD, UC, CeD, and IBS are associated with the decreased expression of sealing claudins, primarily claudin 3, 4, 5, 7, and 8 [5, 37, 39, 67]. Since sealing claudins are essential for the formation of ion selective TJ pores, their downregulation would be expected to compromise the integrity of the gut barrier that in turn would accelerate the development of mucosal inflammation. This suggestion is supported by a recent study of mice with intestinal epithelial specific knockout of claudin-7 [68]. These knockout animals demonstrated increased intestinal permeability, defects in TJ morphology, and they developed spontaneous inflammation with mucosal ulceration. It remains to be determined if the loss of other sealing claudins in the intestinal epithelium recapitulates the above phenotypes of claudin-7 knockout mice.
The production of different cytokines is thought to be responsible for the downregulation of sealing claudins in inflamed intestinal mucosa. Indeed, TNFα, IFNγ, interleukins (IL) 1 IL6, IL9, and IL23 have been found to decrease claudin expression and increase epithelial permeability in vitro and in vivo [10, 57, 69-73]. These cytokines interact with their enterocyte receptors and induce intracellular signaling cascades resulting in the altered activity of various transcriptional factors in the nucleus. Recent studies identified several transcriptional factors (Hopx, Hnf4α, Klf4, Tcf712, Hif1β) that regulate claudin expression during differentiation of normal colonic cell monolayers and under conditions that mimic low oxygen availability in the gut [74, 75]. However, it has not yet been determined if the dysfunction of one of these transcriptional factors contributes in the decreased expression of different claudin proteins in the inflamed mucosa.
The ability of microRNAs (miRs) to regulate claudin expression and epithelial barrier permeability during intestinal inflammation is an emerging area of research. miRs are short strand non-coding RNA molecules that post-transcriptionally control expression of a large variety of genes. miRs bind to the targeted mRNAs resulting in either their degradation or inhibition of translation. A disbalance in intestinal miRs has been recently implicated in the development of IBD [76]. Some of these effects could be related to the regulation of the gut barrier, since mice with intestinal-epithelial deletion of a miR-processing protein Dicer demonstrated severe defects of barrier integrity [77]. Evidence suggest that induction of different miRs can be responsible for the decreased expression of intestinal epithelial claudins during mucosal inflammation. Specifically, miR-29 suppresses claudin-1 level [78], miR-93 inhibits claudin-3 [71], and miR-223 downregulates claudin-8 expression [73]. Of note, claudins are not the only targets for different miRs at epithelial junctions, since this mechanism was also implicated in the expressional regulation of occludin and ZO-1 [79, 80].
Another aspect of the claudin switching involves the induction of channel-forming leaky claudins-2 and 15 in the inflamed intestinal mucosa. While claudin-15 overexpression is a specific feature of CeD [25], upregulation of claudin-2 is a common manifestation of different types of gastrointestinal inflammation [81-83]. Since claudin-2 is a pore-forming claudin, its induction is expected to increase ionic permeability of the gut, thereby potentially triggering or exaggerating mucosal inflammation. Furthermore, claudin-2 was shown to displace sealing claudin-4 from TJs, further weakening the paracellular barrier [84]. Surprisingly, targeted overexpression of claudin-2 in the intestinal epithelium increased barrier permeability, but did not induce spontaneous inflammation in mice [85]. Conversely, these transgenic animals developed immune tolerance and were protected from experimental colitis. Such protective effects of claudin-2 overexpression was associated with increased expression of the transforming growth factor (TGF)-β, which has strong immunomodulator activity [85]. Interestingly, induction of TGF-β appears to be a common compensatory mechanism that suppresses either spontaneous inflammation or experimental colitis in different animal models with increased gut permeability [86, 87].
Molecular mechanisms mediating the upregulation of claudin-2 expression during mucosal inflammation have been extensively investigated. IL-13 and IL-6 are powerful inductors of this TJ protein in cultured intestinal epithelial cells [81]. The cytokines stimulate claudin-2 promoter activity via at least two distinct signaling cascades involving PI3 kinase and MEK/ERK. A recent study proposed a novel mechanism that controls the integrity of epithelial barrier by modulating the cellular level of claudin-2 protein [88]. This study showed that starvation-induced autophagy enhances epithelial barrier by triggering selective degradation of claudin-2. Since defective autophagy has been attributed to IBD, it is tempting to speculate that such autophagy inhibition can attenuate claudin-2 degradation, thereby enhancing its expression in the inflamed mucosa.
In addition to the decreased levels of sealing claudins, expression of other epithelial TJ proteins such as JAM-A, occludin, and ZO-1 is frequently downregulated during intestinal inflammation [43, 89, 90]. Functional consequences of JAM-A downregulation have been demonstrated by studies of knockout animals, whereas the pathophysiologic implications of occludin and ZO-1 depletion remain unclear. Indeed, experiments involving mice with total knockout of JAM-A have implicated JAM-A in controlling permeability of the intestinal epithelial barrier to ions and large uncharged molecules [86]. Furthermore, when compared to wild-type controls, JAM-A-null mice shown a significantly exaggerated inflammatory response and increased mortality during dextran sodium sulfate (DSS)-induced colitis [86]. In contrast, mice with total knockout of occludin did not demonstrate detectable defects of the intestinal epithelial barrier structure and permeability [91]. Little is known regarding the roles of mammalian ZO proteins in vivo due to early embryonic lethality of ZO-1 null mice [92]. Of note, a Vibrio cholerae toxin that binds to and displace ZO-1 from TJs was shown to increase paracellular permeability and trigger junctional disassembly in intestinal cell monolayers in vitro and in rabbit small intestine in vivo [93]. This data indicates the important role of ZO-1 in controlling structure and function of the gut barrier.
4. Altered trafficking of AJ/TJ proteins in the inflamed gut
Decreased expression of AJ and TJ proteins is not the only mechanism that underlies functional defects of epithelial junctions in the inflamed intestinal mucosa, where junctional proteins are frequently redistributed from the cell-cell contact into cytoplasmic compartments [25, 94, 95]. Vesicle trafficking plays a key role in the establishment and remodeling of epithelial junctions at the plasma membrane. A steady-state vesicle trafficking of AJ and TJ proteins occurs even in confluent epithelial cell monolayers with fully-assembled junctions. This is a cyclic process consisting of two opposite phases: the exocytic delivery of newly-synthesized or recycled junctional proteins to the cell surface and the removal of protein components of mature AJ and TJ via endocytosis [96]. An equilibrium of these two trafficking events results in proper assembly and functioning of epithelial junctions, whereas disbalanced trafficking of AJ and TJ proteins results in junctional disassembly (Fig. 2). Disruption of AJs and TJs could be induced by either increased endocytosis or decreased exocytosis of junctional proteins (Fig. 2). Evidence suggest that both events may contribute to defective junctional architecture and functions in the inflamed or injured intestinal mucosa.
Figure 2. Vesicle trafficking mechanisms that regulate the integrity and disruption of the intestinal epithelial barrier.
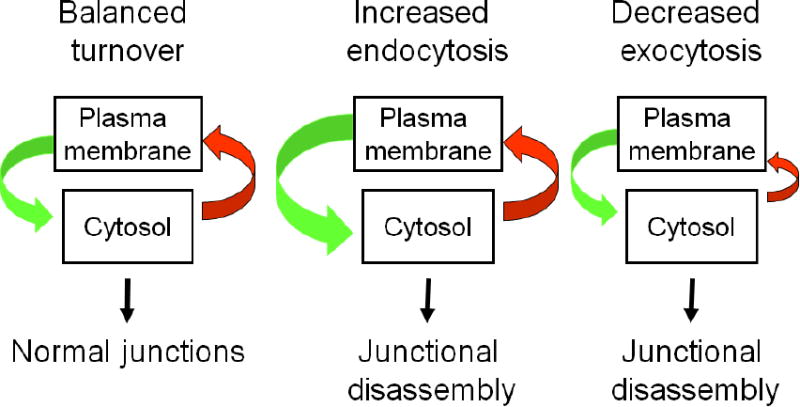
The diagram depicts a balanced vesicle trafficking cycle that regulates proper assembly of epithelial junction in normal intestinal mucosa. Altering this cycle by either increasing endocytosis or inhibiting exocytosis of junctional proteins results in AJ and TJ disassembly.
4.1. Increased endocytosis of junctional proteins during intestinal inflammation
The roles of AJ and TJ endocytosis in the disruption of the mucosal barrier in IBD was originally suggested more than a decade ago [97]. However, the mechanisms that regulate internalization of junctional proteins in normal and inflamed gut remain poorly understood. Since it is difficult to investigate vesicle trafficking in whole animals, the majority of studies in this field were performed using model in vitro systems, by exposing epithelial cell monolayers to inflammatory mediators and pathogens. Generally, plasma membrane proteins can be internalized via multiple endocytosis pathways, among which clathrin-mediated endocytosis, lipid raft/clathrin-mediated endocytosis, and micropinocytosis have been the most intensively characterized. Limited data are available regarding the endocytic pathways that mediate internalization of intestinal epithelial AJ proteins. For example, internalization of E-cadherin and p120-catenin was found to be accelerated by extracellular calcium-depletion [98] and by epithelial cell exposure to either IFNγ [99] or trefoil factor [100]. Clathrin-mediated endocytosis is the most common pathway of E-cadherin endocytosis [15]. The affinity to this pathway is probably determined by the reported interactions of classical cadherins with clathrin adaptors such as AP-2 and Numb [15, 101]. IFNγ-induced internalization of E-cadherin requires its ubiquitination, which is mediated by Src tyrosine kinase and an ubiquitin ligase, Hakai [99]. Interestingly, internalized E-cadherin is not targeted for immediate degradation and is accumulated in a cytoplasmic storage compartment [98, 99]. This stored protein is likely to be reused to rapidly re-build cell-cell adhesions during epithelial restitution.
Three major endocytic pathways have been shown to mediate TJ internalization in intestinal epithelial monolayers. Examples include clathrin-dependent endocytosis of the entire TJ complex in calcium-depleted cells [98], lipid raft/caveolae-mediated endocytosis of occludin triggered by F-actin-depolymerizing drugs [102], and micropinocytosis of transmembrane TJ proteins in IFNγ-exposed epithelial cells [103]. Additionally, lipid raft-dependent endocytosis appears to mediate TNFα-induced increase in permeability of the intestinal epithelial barrier in vivo [104]. There are several explanations for such a diversity of the internalization pathways that drive TJ disassembly. One possibility is the relative abundance of the particular endocytosis machinery in different types of epithelial cells. A second possibility is that different extracellular stimuli activate distinct internalization pathways at the plasma membrane. Finally, despite the common belief that TJs become internalized as a whole entity, this junctional complex could first be dissociated and then individual TJ proteins could be internalized via different endocytic pathways. This possibility is supported by a special link between occludin and lipid raft/caveolae dependent endocytosis. Indeed, occludin and caveolin-1 colocalize and form a complex at normal TJs and following junctional disassembly [105, 106]. Furthermore, occludin is able to affect caveolin-1 recruitment to lipid rafts [107], whereas either pharmacologic disruption of lipid rafts or caveolin-1 knockdown was shown to inhibit cytokine-stimulated occludin endocytosis in vitro and in vivo [104, 108].
Which upstream molecular events become activated by inflammatory stimuli to trigger TJ disassembly? Recent live cell imaging demonstrated that IFNγ/TNFα-induced disruption of TJs in model intestinal epithelial cells is accompanied by the increased mobility of claudin-4 and redistribution of this protein from TJs to the lateral plasma membrane [84]. While this increased mobility of claudin-4 is indicative of junctional destabilization, its role in endocytosis of TJ proteins remains unclear. Other early events of stimuli-induced TJ disassembly involve specific post-translational modification of junctional proteins, which could be essential for their binding to endocytic adaptors. Specifically, occludin and claudin-1 were found to be ubiquitinated by different ubiquitin ligases, leading to TJ disassembly and internalization [109, 110]. Furthermore, several members of the claudin family became phosphorylated in the colonic mucosa during DSS colitis-induced breakdown of the intestinal barrier [111]. While phosphorylation of some claudin proteins is known to affect their dynamics during TJ disassembly [112], the relationships between such post-translational modification and claudin endocytosis in inflamed mucosa remain to be elucidated.
It should be noted that the increased internalization of AJ and TJ proteins during gut inflammation may not be a specific process limited to apical junctions, but rather a bystander effect of the general increase in intensity of epithelial endocytosis. Indeed, it has been realized for a long time that enterocytes of IBD patients have an increased ability to internalize luminal antigens [113-115]. This phenomenon, observed in both CD and UC patients, is called ‘rapid antigen uptake into the cytosol of enterocytes’ (RACE). RACE is a characteristic feature of both inflamed and non-inflamed intestinal mucosa of IBD patients, and it was proposed to play an important role in the disease by increasing transcytosis of luminal antigens and their presentation to the lamina propria immune system [114]. Since TJs and AJs are located in the close proximity to the apical plasma membrane, the increased formation and internalization of apical membrane vesicles may destabilize junctional complexes and shift their trafficking cycle toward endocytosis. While molecular mechanisms of RACE and its relationships with endocytosis of epithelial junctions remain to be established, a possible link between these processes highlights multiple benefits of therapeutic inhibition of apical endocytosis in IBD. Such inhibition may attenuate mucosal inflammation by blocking both transcellular and paracellular routes of luminal antigen and pathogen penetration in the gut.
4.2. Decreased exocytosis of junctional proteins during intestinal inflammation
Exocytosis of AJ and TJ proteins involves their vesicle-mediated transport from the cell interior to the plasma membrane. This event represents an interplay of two different processes: delivery of newly-synthesized junctional proteins from the endoplasmic reticulum (ER) and the Golgi, and recycling of previously internalized molecular components of AJs and TJs [15, 96]. Exocytic trafficking involves interactions of carrier vesicles with different intracellular compartments, and it is regulated by different adaptor and signaling proteins. For example, the initial tethering of vesicles to the targeted membrane compartment is regulated by a family of Rab small GTPases, whereas the ultimate fusion of two phospholipid membranes is driven by the SNARE (Soluble N-ethylmaleimide-sensitive factor Associated Receptor) proteins [116]. Recent studies shed light on some stages of junctional protein exocytosis in the intestinal mucosa and suggest that trafficking defects may significantly contribute to the disruption of the gut barrier during mucosal inflammation. One study targeted a reticulon-4B/NOGO-B protein, which is essential for formation of ER tubules [117]. Downregulation of reticulon-4B in intestinal epithelial cell monolayers attenuated the establishment of the paracellular barrier and decreased expression of E-cadherin, α-catenin, and occludin. Importantly, reticulon-4B expression was found to be downregulated in the intestinal mucosa of CD patients and IL-10 knockout mice that develop spontaneous colitis [117]. These results highlight a possible contribution of impaired ER structure in the disruption of epithelial junctions during intestinal inflammation. Significance of the ER-Golgi trafficking in the establishment of the intestinal epithelial barrier was also demonstrated by studies targeting an important regulator of vesicle fusion, soluble NSF-attachment protein alpha (αSNAP). αSNAP controls multiple vesicle trafficking events, most notable, trafficking between the ER and the Golgi and within the Golgi complex [118]. Downregulation of αSNAP in model intestinal epithelium had profound effects on cell monolayers by disrupting the barrier integrity, inducing AJ and TJ disassembly and selectively downregulating expression of E-cadherin and p120-catenin [119]. Expression and functions of αSNAP during tissue inflammation have not been investigated, and this could be an important topic for future studies. The roles of the Golgi integrity in normal assembly of intestinal epithelial junctions is further highlighted by experiments with either pharmacologic or genetic fragmentation of the Golgi, which caused AJ and TJ disassembly and disruption of the epithelial barrier [119, 120]. It is noteworthy that the described abnormalities of the ER and the Golgi structure are not an artefact of the described in vitro systems. Profound vacuolarization and fragmentation of the ER and the Golgi have been observed both in inflamed and non-inflamed intestinal mucosa of UC patients [2]. This phenomenon was attributed to the well-recognized induction of the ER stress during intestinal inflammation, and one of its functional consequences is likely to be interrupted exocytosis of junctional proteins.
Not only the supply of junctional proteins via the biosynthetic pathway, but also their recycling appeared to be essential for the integrity of the intestinal epithelial barrier. Recent studies highlighted the role of Rab11-positive recycling endosomes. Inhibition of this vesicular compartment was crucial for cholera toxin-dependent disruption of the intestinal barrier in Drosophila, an isolated murine ileal loop, and cultured human intestinal epithelial cells [121]. Interestingly, ablation of Rab11a in the mouse intestinal epithelium triggered IBD-like gut inflammation without disruption of the epithelial barrier by altering cellular distribution and signaling by pattern-recognition receptors [122]. The recycling compartment in polarized epithelial cells is composed of different endosomal populations that specialize in the trafficking of different proteins. For example, Rab13 containing endosomes were implicated in the selective trafficking of intestinal epithelial TJ, whereas Rab8-positive endosomes were essential for E-cadherin exocytosis [123]. Of note, Rab13 expression was found to be decreased in the intestinal mucosa of CD patients [124], however, the impact of such Rab13 depletion on the diseases pathogenesis remains unknown.
5. Cytoskeletal abnormalities and junctional disassembly
Association of apical junctions with the cortical actin cytoskeleton is critical for the integrity and plasticity of the gut barrier [35, 125]. While AJs are connected to the prominent circumferential F-actin belt, TJs interact with more disorganized apical F-actin bundles (Fig. 3). Interestingly, the biochemical features of the associated actin filaments may significantly influence stability and dynamics of apical junctions. For example, TJs appear to be associated with stable filaments composed of γ-cytoplasmic actin, whereas AJs uniquely bind to more dynamic filaments polymerized by the β-cytoplasmic actin isoform [126]. A number of studies have shown that different actin-depolymerizing drugs rapidly increased paracellular permeability and induced AJ and TJ disassembly [127]. These data highlight association with the actin cytoskeleton as crucial mechanism regulating integrity and barrier function of apical junctions. How does interaction with the actin cytoskeleton regulate junctional architecture and remodeling? At least three different mechanisms have been suggested. The first mechanism involves clustering and stabilization of junctional proteins at the specific regions of the lateral plasma membrane. The second mechanism involves inhibition of AJ/TJ protein endocytosis. The third mechanism involves transduction of mechanical forces by junction-associated F-actin bundles to drive stimuli-induced assembly and disassembly of AJ and TJ. Importantly, disruption of the perijunctional actin cytoskeleton has been observed in model intestinal epithelial cell monolayers exposed to different inflammatory mediators [128, 129], and in tissue samples of human patients with mucosal inflammation [125, 130]. Therefore, it is important to understand whether such cytoskeletal abnormalities are essential for the dysfunctions of the intestinal epithelial barrier during mucosal inflammation.
Figure 3. Regulation of epithelial apical junctions by the actin cytoskeleton.
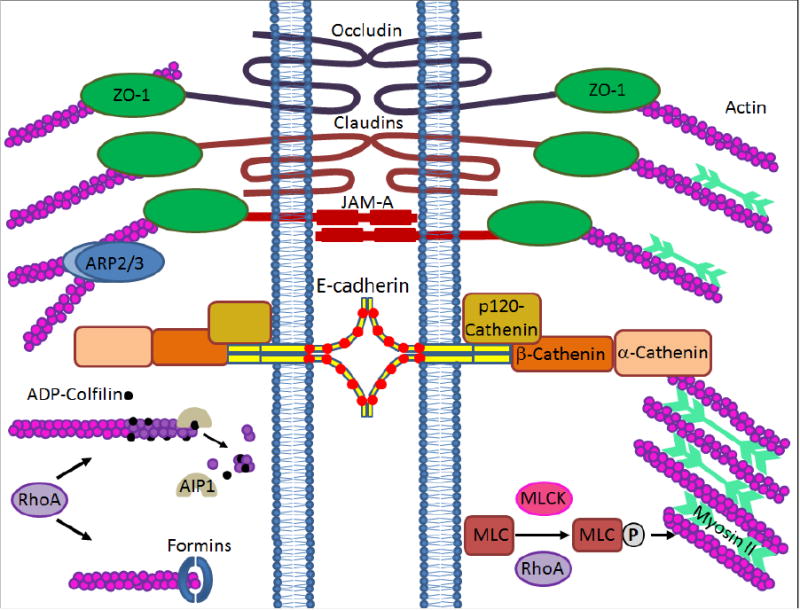
The diagram shows association of epithelial AJs and TJs with the cortical actin cytoskeleton. Major actin-binding proteins, cytoskeletal motors, and signaling molecules that are known to regulate intestinal epithelial junctions are presented.
5.1 Actin filament turnover regulates assembly and permeability of the intestinal epithelial barrier
In epithelial cells, assembly and dynamics of actin filaments is regulated by a large number of ABPs, and motor proteins. The architecture and dynamics of the actin cytoskeleton depend on actin filament turnover, and filament interactions with non-muscle myosin II (NM II). The F-actin turnover ensures the polarized growth and movement of actin filaments, and it involves the addition of monomeric actin to one (barbed) filament end coupled with the removal of monomeric actin from the opposite (pointed) end. F-actin turnover accelerates during disruption and assembly of epithelial junctions [131, 132]. However, this process occurs even in stationary confluent cell monolayers where it is involved in the maintenance of normal intestinal epithelial barrier [133]. F-actin turnover is regulated by various ABPs responsible for accelerating either the polymerization or depolymerization steps. The filament polymerization is primarily mediated by the actin related protein (Arp) 2/3 complex and formin proteins, which are responsible for the assembly of branched and linear actin filaments respectively. Depolymerization of actin filaments is primarily driven by members of the actin depolymerizing factor (ADF)/cofilin family [134]. Limited studies that investigated the roles of actin filament turnover in the regulation of the gut barrier focused on the Arp2/3 complex and ADF/cofilin proteins. For example, an early report suggested the involvement of Arp2/3-dependent actin polymerization in the assembly of AJs and TJs in model epithelial cell monolayers [131]. In contrast, a recent study of mice with intestinal epithelial specific knockout of the Arp2/3 complex component, ArpC3, revealed that this complex is dispensable for the assembly of normal apical junctions [135]. Instead, ArpC3 knockout animals developed defects of endolysosomal system and displayed inhibited nutrient absorption resulting in their early postnatal death. Generation of the inducible knockdown of the Arp2/3 complex in the intestinal epithelium is needed to test the roles of this F-actin regulator in the disruption and restitution of the gut barrier during inflammation.
Intestinal epithelium, which expresses both ADF and cofilin-1, is one of few tissues with high expression of two actin-depolymerizing proteins [136]. Despite their close homology, these regulators of F-actin turnover appear to play non-redundant functions in the assembly of apical junctions and the formation of the epithelial barrier in vitro [136]. Of note, mutant corn mice with spontaneous total knockout of ADF show neither increased barrier permeability nor disrupted apical junctions in the intestinal epithelium [136]. This most likely reflects functional compensation from the intestinal cofilin-1. However, ADF-null mice demonstrate exaggerated barrier breakdown, cell apoptosis, and mucosal inflammation during experimental colitis. This data suggests that experimental colitis may trigger a robust remodeling of the epithelial actin cytoskeleton, which requires ADF activity and cannot be compensated by cofilin-1. Interestingly, mice deficient in another regulator of F-actin dynamics, cortactin, also did not develop spontaneous mucosal inflammation, but were more susceptible to experimental colitis [137]. Together, these results emphasize protective role of the efficient actin filament turnover that either limits epithelial damage or accelerates restitution of injured and inflamed intestinal mucosa.
Additional evidence implicating ADF/cofilin proteins in the regulation of model intestinal epithelial barrier is provided by a recent study that focuses on the actin-interacting protein (Aip)-1. Aip1 (also known as WDR1) accelerates ADF/cofilin-dependent F-actin turnover by recruiting these proteins to actin filaments [134]. Aip1 was found to selectively localize at TJs in cultured intestinal epithelial cells and to be enriched at apical junctions in mouse colonic mucosa [133]. Furthermore, Aip1 depletion increased epithelial permeability and attenuated AJ and TJ assembly by impairing architecture of the perijunctional F-actin belt [133]. Interestingly, mice with inactivating mutations of Aip1 develop a spontaneous inflammatory response associated with activation of the inflammasome and cytokine secretion [138]. It would be important to examine how conditional depletion of Aip1 in the intestinal epithelium affects the integrity of the gut barrier and the development of mucosal inflammation in vivo.
5.2. Non-muscle myosin II as a master regulator of apical junction integrity and remodeling in the gut
Perijunctional actin filaments associate with NM II, a key molecular motor that uses the chemical energy of ATP hydrolysis to create mechanical forces that shape the architecture and drive the remodeling of the actin cytoskeleton [125, 127]. NM II functions as a molecular ensemble of two heavy chains, two essential light chains, and two regulatory myosin light chains (RMLC) [139]. Activated NM II heavy chains simultaneously interact with actin filaments and with each other creating large linear myofibrils. Such heavy chain interactions determine two major activities of this cytoskeletal motor. One activity involves the sliding of actin filaments against each other, thereby driving actin filament contraction and relaxation. Another NM II activity involves the cross-linking (bundling) of actin filaments, which result in the assembly of thick actomyosin bundles [139].
Despite the large number of publications that focus on myosin-dependent disruption of the gut barrier during mucosal inflammation, the roles of this motor protein at the intestinal-epithelial junctions remain incompletely understood. A current dogma in the field postulates that increased phosphorylation of RMLC results in NM II activation and increased actomyosin contractility, which drives TJ disassembly in the inflamed intestinal mucosa. This mechanism is supported by solid in vitro and in vivo evidence, and it was extensively discussed in previous reviews [4, 35, 125]. However, there are several reasons to believe that such a narrow focus on RLMC phosphorylation is not sufficient to paint a comprehensive picture of NM II functions at epithelial junctions, which are dependent on NM II heavy chains. Indeed, NM II heavy chains determine all functional properties of this cytoskeletal motor—most notably, ATP hydrolysis, actin filament binding, and myofibril formation. Furthermore, RMLC does not selectively regulate NM II activity, but has additional cellular functions. For example, several studies reported promiscuous interactions of RMLC with different partners, such as the heavy chains of unconventional myosin classes 14, 15, 18, and 19, and well as non-myosin proteins, the bile acid transporter, and calponin [140]. Hence, functional effects of altered RMLC phosphorylation should be considered with caution. Of note, a recent study reveals that NM II heavy chains robustly interact with membrane phospholipids resulting in the displacement of RMLC from the myosin complex [141]. These molecular events may have some impact on membrane associated structures such as epithelial junctions. Finally, a variety of regulatory mechanisms specifically target NM II heavy chains. They include heavy chain phosphorylation and binding to different scaffolding proteins such as shroom, anillin, and septins.
Several studies used pharmacologic and genetic inhibition of NM II heavy chains to examine their functions in model intestinal epithelial cell monolayers [131, 132, 142]. These studies yielded the following major conclusions. First, NM II motor activity is important for the maintenance of normal barrier properties of epithelial monolayers. Second, NM II plays a key role in regulating junctional remodeling by driving two opposite phases of this process: junctional assembly and reassembly. Third, NM IIA heavy chain, but not the closely related NM IIB or NM IIC heavy chain isoforms, acts as critical regulator of AJ/TJ functions and remodeling in vitro. The physiological relevance of these conclusions was tested in a recent study examining the intestinal epithelial specific knockout of NM IIA in mice [87]. This study revealed that loss of NM IIA is sufficient to increase permeability of the gut barrier and to trigger low-scale intestinal inflammation in vivo. Such inflammation did not become a full scale colitis due to a TGF-β-dependent compensatory response that resulted in mucosal accumulation of antibacterial immunoglobulin A [87]. Importantly, deletion of NM IIA in the intestinal epithelium increased animal sensitivity to DSS colitis, which was associated with marked epithelial erosion and further exacerbation of the barrier breakdown [87]. Taken together, this data suggests that a balanced activity of perijunctional NM IIA mediates the integrity of normal intestinal epithelial barrier. When junction-associated NM IIA is either overactivated or inhibited, these opposite events result in barrier disruption. Importantly, the described study of the mice with intestinal epithelial specific knockout of NM IIA suggests that abnormal organization of the epithelial actomyosin cytoskeleton plays a causal role in the development of intestinal inflammation.
Many previous studies identified extracellular mediators and signaling cascades that stimulate activity of intestinal epithelial NM II during mucosal inflammation. However, little is known about the roles and mechanisms of NM IIA inhibition in gastrointestinal diseases. Different mechanisms control the activity and cellular distribution of NM IIA motors in vitro. These mechanisms include expressional regulation of this cytoskeletal motor, its chaperon-assisted folding, specific phosphorylation of NM IIA heavy chain, as well as heavy chain interactions with myosin-binding proteins. Additional studies are needed to elucidate which of these mechanisms regulate functions of intestinal epithelial NM IIA in healthy gut and during intestinal inflammation.
5.3. Signaling events that regulate remodeling of the perijunctional cytoskeleton in the inflamed gut
Molecular architecture and dynamics of the perijunctional cytoskeleton is regulated by different signaling events, which are originated either from epithelial junctions, cell-matrix adhesions, or different cell surface receptors. In well-differentiated polarized cell monolayers, such signaling promotes the assembly of prominent actomyosin bundles that support junctional integrity and tightness of the paracellular barrier. However, signaling cascades initiated by inflammation and other external stressors may trigger profound remodeling of the apical actin cytoskeleton leading to junctional disassembly. The Rho family of small GTPases plays key roles in regulating cytoskeletal architecture and dynamics. These GTPase cycle through the active, GTP-bound and inactive, GDP-bound forms, and upon activation, interact with and stimulate different downstream kinases and accessory proteins. While actions of all three major members of this family, namely Rho, Rac1, and Cdc42, have been implicated in the control of epithelial junctions, the majority of studies addressed the roles of Rho GTPase in the regulation of barrier integrity in normal and inflamed intestinal mucosa [143, 144]. Rho activation affects the actin cytoskeleton by different mechanisms that include stimulation of NM II activity via downstream Rho-associated kinase (ROCK), as well as alterations of actin filament turnover. The latter effect is determined by the Rho-ROCK control of ADF/cofilin-dependent depolymerization and Rho-formin dependent control of polymerization of actin filaments. Rho itself is tightly regulated by various guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) that activate and inactivate Rho, respectively.
The first study involving Rho inhibition in model intestinal epithelial cell monolayers demonstrated that inactivation of this GTPase results in selective disruption of TJ integrity and breakdown of the paracellular barrier [145]. Later, it was shown that RhoA is activated during AJ/TJ disassembly in calcium-depleted epithelial cells and that inhibition of Rho or ROCK is sufficient to prevent junctional breakdown in this experimental system [146]. Consistent with these results, the bacterial cytotoxic necrotizing factor that is known to stimulate activity of Rho, Rac1, and Cdc42 profoundly disrupted a model intestinal epithelial barrier by inducing selective TJ disassembly [105]. These early in vitro studies suggest that intestinal epithelial TJ integrity depends on the fine balance of Rho activity: where both too little of active Rho and too much of active Rho have detrimental effects on the TJ structure and barrier integrity. It should be noted that the Rho GTPase subfamily is composed by highly homologous members (RhoA, RhoB, and RhoC) that play unique and redundant roles in different cellular responses [147]. Since the described pharmacological approaches modulate activity of all these Rho isoforms, future studies that use more specific tools are required to examine specific roles of RhoA, RhoB, and RhoC at intestinal epithelial junctions.
Do Rho GTPases play roles in the regulation of the gut barrier during intestinal inflammation in vivo? Published data regarding intestinal RhoA activity in clinical and experimental IBD are sparse and inconsistent. One study reported a robust RhoA activation in the inflamed intestinal mucosa of CD patients and mice with trinitrobenzoic acid-induced colitis [148]. However, a more recent study suggested that RhoA is inactivated in the intestinal epithelium of IBD patients [149]. Such inactivation is not caused by the expressional downregulation of this GTPase, but rather by its defective prenylation, which depletes a membrane-bound pool of RhoA. Remarkably, intestinal epithelial-specific deletion of either RhoA or an enzyme that mediates Rho prenylation resulted in epithelial injury and spontaneous intestinal inflammation [149]. On the other hand, acute pharmacological inhibition of ROCK had profound anti-inflammatory effects and attenuated experimental murine colitis [148]. Furthermore, deletion of two different Rho GAP proteins, Arhgap17 and myosin IXb, which should increase Rho activity, had adverse effects on the intestinal epithelial homeostasis [150, 151]. Thus, Arhgap17-deficient mice displayed increased gut permeability and higher sensitivity to DSS colitis (Lee ST 2016). Consistent with RhoA activation, myosin IXb depletion resulted in the increased RMLC phosphorylation in intestinal epithelium, which was accompanied by mucosal injury, AJ disassembly, and gut leakiness (Hogan PD 2016). Collectively, these results suggest a dual role of RhoA in the regulation of the intestinal epithelial barrier in vivo. An intermediate activity of this GTPase is essential for maintenance of the intestinal epithelial homeostasis and preservation of the normal gut barrier. Shift of this balance toward either inactivation or activation of RhoA by inflammatory or infection agents may trigger profound junction disassembly and barrier disruption, thereby perpetuating mucosal inflammation.
Little is known about the roles of Rac1 and Cdc42 in the regulation of the intestinal epithelial barrier in vivo. The intestinal epithelial-specific knockout of Cdc42 in mice increased barrier permeability in parallel to inducing multiple abnormalities of mucosal homeostasis, such as defective Paneth cell differentiation, crypt hyperplasia, and microvilli inclusion [152]. Furthermore, total knockout of a Rac1 GEF (called ‘dedicator of cytokinesis 2’) in mice increased animal susceptibility to enteric infection and exaggerated mucosal inflammation [153]. These results suggest that a functional crosstalk between different Rho GTPases is essential for the maintenance of normal intestinal epithelial homeostasis. Future studies are needed to fully elucidate causes and consequences of altered epithelial Rho, Rac1, and Cdc42 signaling during intestinal inflammation.
6. Conclusion
Disruption of the intestinal epithelial barrier is increasingly recognized as a crucial mechanism of various inflammatory and immune disorders. This barrier dysfunction culminates from a complex cross-talk between different cells and signaling molecules in the gut resulting in altered molecular composition of epithelial TJs and AJs, and increased passage of different molecules through opened intercellular spaces. Recent studies revealed that compromised functions of epithelial junctions are not a specific, limited effect of the mucosal inflammation. This phenomenon is a consequence of the global changes in intestinal epithelial cell homeostasis in the inflamed gut that involve transcriptional reprogramming of the cells and alterations in their cytoskeletal architecture and membrane trafficking. We have already gained significant knowledge of some of these homeostatic changes, but we have just scratched the surface in understanding the others. Unravelling the complexity of the mechanisms that mediate intestinal epithelial barrier dysfunction is essential for the development of pharmacological strategies that may prevent barrier disruption or accelerate its restoration during mucosal inflammation. Such strategies are likely to combine small-molecule inhibitors of endocytosis, modulators of the F-actin dynamics and/or NM IIA contractility, as well as substances that stimulate expression of different AJ and TJ proteins. Future studies are needed to test the feasibility and benefits of this multimodal ‘therapeutic’ protection of the gut barrier in different inflammatory disorders.
highlights.
Increased permeability of the gut barrier contributes to the development of various inflammatory and immune disorders.
Disruption of the gut barrier is mediated by disassembly of epithelial adherens and tight junctions.
Increased endocytosis and/or attenuated exocytosis of junctional proteins mediate epithelial barrier breakdown in inflamed gut
An interplay between actin filament turnover and myosin II activity regulates integrity and functions of intestinal epithelial junctions.
Acknowledgments
The author thanks Dr. Kevin Hogan for editing the manuscript. We apologize to our colleagues whose studies were not cited due to space limitation. This work was supported in part by funding from the NIH: RO1 DK108278 (A.I.I.) and P30 CA016059 (Cancer Center Support Grant).
List of Abbreviations
- ABP
actin-binding protein
- ADF
actin-depolymerizing factor
- Aip1
actin-interacting protein 1
- Arp
actin-related proteins
- CAR
coxsakievirus and adenovirus receptor
- CD
Crohn's disease, CeD, celiac disease
- EM
electron microscopy
- ER
endoplasmic reticulum
- IBD
inflammatory bowel disease
- IBS
inflammatory bowel symptoms
- IFNγ
interferon γ
- IL
interleukin, JAM-A, junctional adhesion molecule A
- MARVEL
MAL and related proteins for vesicle trafficking and membrane link
- _NM II
non-muscle myosin II
- RACE
rapid antigen uptake into the cytosol of enterocytes
- RMLC
regulatory myosin light chain
- ROCK
Rho-associated kinase
- siRNA
small interfering RNA
- TAMP
tight junction-associated MARVEL proteins
- TEER
transepithelial electrical resistance
- TNFα
tumor necrosis factor α
- TJs
tight junctions
- UC
ulcerative colitis
- ZO
zonula occludens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol. 1998;60:143–159. doi: 10.1146/annurev.physiol.60.1.143. [DOI] [PubMed] [Google Scholar]
- 2.McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:100–113. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189. doi: 10.1186/s12876-014-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 5.Barmeyer C, Schulzke JD, Fromm M. Claudin-related intestinal diseases. Semin Cell Dev Biol. 2015;42:30–38. doi: 10.1016/j.semcdb.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Halpern MD, Denning PW. The role of intestinal epithelial barrier function in the development of NEC. Tissue Barriers. 2015;3:e1000707. doi: 10.1080/21688370.2014.1000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandl K, Schnabl B. Is intestinal inflammation linking dysbiosis to gut barrier dysfunction during liver disease? Expert Rev Gastroenterol Hepatol. 2015;9:1069–1076. doi: 10.1586/17474124.2015.1057122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winer DA, Luck H, Tsai S, Winer S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016;23:413–426. doi: 10.1016/j.cmet.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Luissint AC, Parkos CA, Nusrat A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions. Cell Junction Remodeling, and Mucosal Repair, Gastroenterology. 2016;151:616–632. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onyiah JC, Colgan SP. Cytokine responses and epithelial function in the intestinal mucosa. Cell Mol Life Sci. 2016;73:4203–4212. doi: 10.1007/s00018-016-2289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blander JM. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J. 2016;283:2720–2730. doi: 10.1111/febs.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornick S, Tawiah A, Chadee K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers. 2015;3:e982426. doi: 10.4161/21688370.2014.982426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanov AI, Naydenov NG. Dynamics and regulation of epithelial adherens junctions: recent discoveries and controversies. Int Rev Cell Mol Biol. 2013;303:27–99. doi: 10.1016/B978-0-12-407697-6.00002-7. [DOI] [PubMed] [Google Scholar]
- 16.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 17.Hardy RG, Brown RM, Miller SJ, Tselepis C, Morton DG, Jankowski JA, Sanders DS. Transient P-cadherin expression in radiation proctitis; a model of mucosal injury and repair. J Pathol. 2002;197:194–200. doi: 10.1002/path.1092. [DOI] [PubMed] [Google Scholar]
- 18.Rikitake Y, Mandai K, Takai Y. The role of nectins in different types of cell-cell adhesion. J Cell Sci. 2012;125:3713–3722. doi: 10.1242/jcs.099572. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka-Okamoto M, Hori K, Ishizaki H, Itoh Y, Onishi S, Yonemura S, Takai Y, Miyoshi J. Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J Cell Sci. 2011;124:2231–2240. doi: 10.1242/jcs.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov AI. Structure and regulation of intestinal epithelial tight junctions: current concepts and unanswered questions. Adv Exp Med Biol. 2012;763:132–148. doi: 10.1007/978-1-4614-4711-5_6. [DOI] [PubMed] [Google Scholar]
- 23.Capaldo CT, Nusrat A. Claudin switching: Physiological plasticity of the Tight Junction. Semin Cell Dev Biol. 2015;42:22–29. doi: 10.1016/j.semcdb.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Lu Z, Ding L, Lu Q, Chen YH. Claudins in intestines: Distribution and functional significance in health and diseases. Tissue Barriers. 2013;1:e24978. doi: 10.4161/tisb.24978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barmeyer C, Fromm M, Schulzke JD. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflugers Arch. 2017;469:15–26. doi: 10.1007/s00424-016-1914-6. [DOI] [PubMed] [Google Scholar]
- 26.Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. 2014;36:157–165. doi: 10.1016/j.semcdb.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raleigh DR, Marchiando AM, Zhang Y, Shen L, Sasaki H, Wang Y, Long M, Turner JR. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell. 2010;21:1200–1213. doi: 10.1091/mbc.E09-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuse M, Izumi Y, Oda Y, Higashi T, Iwamoto N. Molecular organization of tricellular tight junctions. Tissue Barriers. 2014;2:e28960. doi: 10.4161/tisb.28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zihni C, Balda MS, Matter K. Signalling at tight junctions during epithelial differentiation and microbial pathogenesis. J Cell Sci. 2014;127:3401–3413. doi: 10.1242/jcs.145029. [DOI] [PubMed] [Google Scholar]
- 30.Luissint AC, Nusrat A, Parkos CA. JAM-related proteins in mucosal homeostasis and inflammation. Semin Immunopathol. 2014;36:211–226. doi: 10.1007/s00281-014-0421-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 32.Ivanov AI, Young C, Den Beste K, Capaldo CT, Humbert PO, Brennwald P, Parkos CA, Nusrat A. Tumor suppressor scribble regulates assembly of tight junctions in the intestinal epithelium. Am J Pathol. 2010;176:134–145. doi: 10.2353/ajpath.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741–754. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight Junction Pore and Leak Pathways: A Dynamic Duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das P, Goswami P, Das TK, Nag T, Sreenivas V, Ahuja V, Panda SK, Gupta SD, Makharia GK. Comparative tight junction protein expressions in colonic Crohn's disease, ulcerative colitis, and tuberculosis: a new perspective. Virchows Arch. 2012;460:261–270. doi: 10.1007/s00428-012-1195-1. [DOI] [PubMed] [Google Scholar]
- 37.Goswami P, Das P, Verma AK, Prakash S, Das TK, Nag TC, Ahuja V, Gupta SD, Makharia GK. Are alterations of tight junctions at molecular and ultrastructural level different in duodenal biopsies of patients with celiac disease and Crohn's disease? Virchows Arch. 2014;465:521–530. doi: 10.1007/s00428-014-1651-1. [DOI] [PubMed] [Google Scholar]
- 38.Landy J, Al-Hassi HO, Ronde E, English NR, Mann ER, Bernardo D, Ciclitira PJ, Clark SK, Knight SC, Hart AL. Innate immune factors in the development and maintenance of pouchitis. Inflamm Bowel Dis. 2014;20:1942–1949. doi: 10.1097/MIB.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 39.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jankowski JA, Bedford FK, Boulton RA, Cruickshank N, Hall C, Elder J, Allan R, Forbes A, Kim YS, Wright NA, Sanders DS. Alterations in classical cadherins associated with progression in ulcerative and Crohn's colitis. Lab Invest. 1998;78:1155–1167. [PubMed] [Google Scholar]
- 41.Kosovac K, Brenmoehl J, Holler E, Falk W, Schoelmerich J, Hausmann M, Rogler G. Association of the NOD2 genotype with bacterial translocation via altered cell-cell contacts in Crohn's disease patients. Inflamm Bowel Dis. 2010;16:1311–1321. doi: 10.1002/ibd.21223. [DOI] [PubMed] [Google Scholar]
- 42.Perry I, Tselepis C, Hoyland J, Iqbal TH, Scott D, Sanders SA, Cooper BT, Jankowski JA. Reduced cadherin/catenin complex expression in celiac disease can be reproduced in vitro by cytokine stimulation. Lab Invest. 1999;79:1489–1499. [PubMed] [Google Scholar]
- 43.Wilcz-Villega E, McClean S, O'Sullivan M. Reduced E-cadherin expression is associated with abdominal pain and symptom duration in a study of alternating and diarrhea predominant IBS. Neurogastroenterol Motil. 2014;26:316–325. doi: 10.1111/nmo.12262. [DOI] [PubMed] [Google Scholar]
- 44.Zhang C, Liu LW, Sun WJ, Qin SH, Qin LZ, Wang X. Expressions of E-cadherin, p120ctn, beta-catenin and NF-kappaB in ulcerative colitis. J Huazhong Univ Sci Technolog Med Sci. 2015;35:368–373. doi: 10.1007/s11596-015-1439-9. [DOI] [PubMed] [Google Scholar]
- 45.Consortium UIG, Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, Nimmo ER, Massey D, Blaszczyk K, Elliott T, Cotterill L, Dallal H, Lobo AJ, Mowat C, Sanderson JD, Jewell DP, Newman WG, Edwards C, Ahmad T, Mansfield JC, Satsangi J, Parkes M, Mathew CG, C Wellcome Trust Case Control. Donnelly P, Peltonen L, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Craddock N, Deloukas P, Duncanson A, Jankowski J, Markus HS, Mathew CG, McCarthy MI, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Samani N, Trembath RC, Viswanathan AC, Wood N, Spencer CC, Barrett JC, Bellenguez C, Davison D, Freeman C, Strange A, Donnelly P, Langford C, Hunt SE, Edkins S, Gwilliam R, Blackburn H, Bumpstead SJ, Dronov S, Gillman M, Gray E, Hammond N, Jayakumar A, McCann OT, Liddle J, Perez ML, Potter SC, Ravindrarajah R, Ricketts M, Waller M, Weston P, Widaa S, Whittaker P, Deloukas P, Peltonen L, Mathew CG, Blackwell JM, Brown MA, Corvin A, McCarthy MI, Spencer CC, Attwood AP, Stephens J, Sambrook J, Ouwehand WH, McArdle WL, Ring SM, Strachan DP. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muise AM, Walters TD, Glowacka WK, Griffiths AM, Ngan BY, Lan H, Xu W, Silverberg MS, Rotin D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn's disease. Gut. 2009;58:1121–1127. doi: 10.1136/gut.2008.175117. [DOI] [PubMed] [Google Scholar]
- 47.Vieira AF, Paredes J. P-cadherin and the journey to cancer metastasis. Mol Cancer. 2015;14:178. doi: 10.1186/s12943-015-0448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermuller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–228. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- 49.Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol. 2012;7:35–60. doi: 10.1146/annurev-pathol-011811-120905. [DOI] [PubMed] [Google Scholar]
- 50.Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–1207. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 51.Schneider MR, Dahlhoff M, Horst D, Hirschi B, Trulzsch K, Muller-Hocker J, Vogelmann R, Allgauer M, Gerhard M, Steininger S, Wolf E, Kolligs FT. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS One. 2010;5:e14325. doi: 10.1371/journal.pone.0014325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smalley-Freed WG, Efimov A, Burnett PE, Short SP, Davis MA, Gumucio DL, Washington MK, Coffey RJ, Reynolds AB. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 120:1824–1835. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27:7551–7559. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naydenov NG, Baranwal S, Khan S, Feygin A, Gupta P, Ivanov AI. Novel mechanism of cytokine-induced disruption of epithelial barriers: Janus kinase and protein kinase D-dependent downregulation of junction protein expression. Tissue Barriers. 2013;1:e25231. doi: 10.4161/tisb.25231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muise AM, Walters T, Wine E, Griffiths AM, Turner D, Duerr RH, Regueiro MD, Ngan BY, Xu W, Sherman PM, Silverberg MS, Rotin D. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol. 2007;17:1212–1218. doi: 10.1016/j.cub.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 56.Ciccocioppo R, Finamore A, Ara C, Di Sabatino A, Mengheri E, Corazza GR. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am J Clin Pathol. 2006;125:502–511. doi: 10.1309/DTYR-A91G-8R0K-TM8M. [DOI] [PubMed] [Google Scholar]
- 57.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–871. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azarschab P, Porschen R, Gregor M, Blin N, Holzmann K. Epigenetic control of the E-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer. 2002;35:121–126. doi: 10.1002/gcc.10101. [DOI] [PubMed] [Google Scholar]
- 59.Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001;48:367–371. doi: 10.1136/gut.48.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silvera D, Arju R, Darvishian F, Levine PH, Zolfaghari L, Goldberg J, Hochman T, Formenti SC, Schneider RJ. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol. 2009;11:903–908. doi: 10.1038/ncb1900. [DOI] [PubMed] [Google Scholar]
- 61.Hu S, Ciancio MJ, Lahav M, Fujiya M, Lichtenstein L, Anant S, Musch MW, Chang EB. Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology. 2007;133:1893–1904. doi: 10.1053/j.gastro.2007.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elmi A, Nasher F, Jagatia H, Gundogdu O, Bajaj-Elliott M, Wren B, Dorrell N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell Microbiol. 2016;18:561–572. doi: 10.1111/cmi.12534. [DOI] [PubMed] [Google Scholar]
- 63.Wu S, Rhee KJ, Zhang M, Franco A, Sears CL. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma-secretase-dependent E-cadherin cleavage. J Cell Sci. 2007;120:1944–1952. doi: 10.1242/jcs.03455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, Taggart C, Pacheco Y, Devouassoux G, Bentaher A. Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respir Res. 2016;17:129. doi: 10.1186/s12931-016-0449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Butin-Israeli V, Houser MC, Feng M, Thorp EB, Nusrat A, Parkos CA, Sumagin R. Deposition of microparticles by neutrophils onto inflamed epithelium: a new mechanism to disrupt epithelial intercellular adhesions and promote transepithelial migration. FASEB J. 2016;30:4007–4020. doi: 10.1096/fj.201600734R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nava P, Kamekura R, Nusrat A. Cleavage of transmembrane junction proteins and their role in regulating epithelial homeostasis. Tissue Barriers. 2013;1:e24783. doi: 10.4161/tisb.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oshima T, Miwa H, Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol. 2008;23(Suppl 2):S146–150. doi: 10.1111/j.1440-1746.2008.05405.x. [DOI] [PubMed] [Google Scholar]
- 68.Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. 2012;142:305–315. doi: 10.1053/j.gastro.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amasheh M, Grotjohann I, Amasheh S, Fromm A, Soderholm JD, Zeitz M, Fromm M, Schulzke JD. Regulation of mucosal structure and barrier function in rat colon exposed to tumor necrosis factor alpha and interferon gamma in vitro: a novel model for studying the pathomechanisms of inflammatory bowel disease cytokines. Scand J Gastroenterol. 2009;44:1226–1235. doi: 10.1080/00365520903131973. [DOI] [PubMed] [Google Scholar]
- 70.Gerlach K, McKenzie AN, Neurath MF, Weigmann B. IL-9 regulates intestinal barrier function in experimental T cell-mediated colitis. Tissue Barriers. 2015;3:e983777. doi: 10.4161/21688370.2014.983777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haines RJ, Beard RS, Jr, Chen L, Eitnier RA, Wu MH. Interleukin-1beta Mediates beta-Catenin-Driven Downregulation of Claudin-3 and Barrier Dysfunction in Caco2 Cells. Dig Dis Sci. 2016;61:2252–2261. doi: 10.1007/s10620-016-4145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haines RJ, Beard RS, Jr, Eitner RA, Chen L, Wu MH. TNFalpha/IFNgamma Mediated Intestinal Epithelial Barrier Dysfunction Is Attenuated by MicroRNA-93 Downregulation of PTK6 in Mouse Colonic Epithelial Cells. PLoS One. 2016;11:e0154351. doi: 10.1371/journal.pone.0154351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H, Chao K, Ng SC, Bai AH, Yu Q, Yu J, Li M, Cui Y, Chen M, Hu JF, Zhang S. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016;17:58. doi: 10.1186/s13059-016-0901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lili LN, Farkas AE, Gerner-Smidt C, Overgaard CE, Moreno CS, Parkos CA, Capaldo CT, Nusrat A. Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4-alpha cascades. Tissue Barriers. 2016;4:e1214038. doi: 10.1080/21688370.2016.1214038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, Kendrick AA, Kelly CJ, Bayless AJ, Kominsky DJ, Campbell EL, Kuhn KA, Furuta GT, Colgan SP, Glover LE. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell. 2015;26:2252–2262. doi: 10.1091/mbc.E14-07-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalla R, Ventham NT, Kennedy NA, Quintana JF, Nimmo ER, Buck AH, Satsangi J. MicroRNAs: new players in IBD. Gut. 2015;64:504–517. doi: 10.1136/gutjnl-2014-307891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, Kaestner KH. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology. 2010;139:1654–1664, 1664 e1651. doi: 10.1053/j.gastro.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou Q, Costinean S, Croce CM, Brasier AR, Merwat S, Larson SA, Basra S, Verne GN. MicroRNA 29 targets nuclear factor-kappaB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology. 2015;148:158–169. doi: 10.1053/j.gastro.2014.09.037. e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang H, Rao JN, Wang JY. Posttranscriptional Regulation of Intestinal Epithelial Tight Junction Barrier by RNA-binding Proteins and microRNAs. Tissue Barriers. 2014;2:e28320. doi: 10.4161/tisb.28320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luettig J, Rosenthal R, Barmeyer C, Schulzke JD. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers. 2015;3:e977176. doi: 10.4161/21688370.2014.977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88:1110–1120. doi: 10.1038/labinvest.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang YG, Wu S, Xia Y, Sun J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS One. 2013;8:e58606. doi: 10.1371/journal.pone.0058606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Capaldo CT, Farkas AE, Hilgarth RS, Krug SM, Wolf MF, Benedik JK, Fromm M, Koval M, Parkos C, Nusrat A. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell. 2014;25:2710–2719. doi: 10.1091/mbc.E14-02-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahmad R, Chaturvedi R, Olivares-Villagomez D, Habib T, Asim M, Shivesh P, Polk DB, Wilson KT, Washington MK, Van Kaer L, Dhawan P, Singh AB. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014;7:1340–1353. doi: 10.1038/mi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naydenov NG, Feygin A, Wang D, Kuemmerle JF, Harris G, Conti MA, Adelstein RS, Ivanov AI. Nonmuscle Myosin IIA Regulates Intestinal Epithelial Barrier in vivo and Plays a Protective Role During Experimental Colitis. Sci Rep. 2016;6:24161. doi: 10.1038/srep24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015;290:7234–7246. doi: 10.1074/jbc.M114.597492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coeffier M, Gloro R, Boukhettala N, Aziz M, Lecleire S, Vandaele N, Antonietti M, Savoye G, Bole-Feysot C, Dechelotte P, Reimund JM, Ducrotte P. Increased proteasome-mediated degradation of occludin in irritable bowel syndrome. Am J Gastroenterol. 2010;105:1181–1188. doi: 10.1038/ajg.2009.700. [DOI] [PubMed] [Google Scholar]
- 90.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, Saitou M, Tsukita S, Fromm M. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 92.Katsuno T, Umeda K, Matsui T, Hata M, Tamura A, Itoh M, Takeuchi K, Fujimori T, Nabeshima Y, Noda T, Tsukita S, Tsukita S. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell. 2008;19:2465–2475. doi: 10.1091/mbc.E07-12-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fasano A, Fiorentini C, Donelli G, Uzzau S, Kaper JB, Margaretten K, Ding X, Guandalini S, Comstock L, Goldblum SE. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96:710–720. doi: 10.1172/JCI118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bergmann KR, Liu SX, Tian R, Kushnir A, Turner JR, Li HL, Chou PM, Weber CR, De Plaen IG. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am J Pathol. 2013;182:1595–1606. doi: 10.1016/j.ajpath.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spindler V, Meir M, Vigh B, Flemming S, Hutz K, Germer CT, Waschke J, Schlegel N. Loss of Desmoglein 2 Contributes to the Pathogenesis of Crohn's Disease. Inflamm Bowel Dis. 2015;21:2349–2359. doi: 10.1097/MIB.0000000000000486. [DOI] [PubMed] [Google Scholar]
- 96.Fletcher SJ, Iqbal M, Jabbari S, Stekel D, Rappoport JZ. Analysis of occludin trafficking, demonstrating continuous endocytosis, degradation, recycling and biosynthetic secretory trafficking. PLoS One. 2014;9:e111176. doi: 10.1371/journal.pone.0111176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ivanov AI, Nusrat A, Parkos CA. The epithelium in inflammatory bowel disease: potential role of endocytosis of junctional proteins in barrier disruption. Novartis Found Symp. 2004;263:115–124. [PubMed] [Google Scholar]
- 98.Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–188. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smyth D, Leung G, Fernando M, McKay DM. Reduced surface expression of epithelial E-cadherin evoked by interferon-gamma is Fyn kinase-dependent. PLoS One. 2012;7:e38441. doi: 10.1371/journal.pone.0038441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Durer U, Hartig R, Bang S, Thim L, Hoffmann W. TFF3 and EGF induce different migration patterns of intestinal epithelial cells in vitro and trigger increased internalization of E-cadherin. Cell Physiol Biochem. 2007;20:329–346. doi: 10.1159/000107519. [DOI] [PubMed] [Google Scholar]
- 101.Yang Y, Chen L, Tian Y, Ye J, Liu Y, Song L, Pan Q, He Y, Chen W, Peng Z, Wang R. Numb modulates the paracellular permeability of intestinal epithelial cells through regulating apical junctional complex assembly and myosin light chain phosphorylation. Exp Cell Res. 2013;319:3214–3225. doi: 10.1016/j.yexcr.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 102.Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell. 2005;16:3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- 104.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci. 2003;116:725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 106.Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113(Pt 10):1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- 107.Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci. 123:2844–2852. doi: 10.1242/jcs.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schwarz BT, Wang F, Shen L, Clayburgh DR, Su L, Wang Y, Fu YX, Turner JR. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology. 2007;132:2383–2394. doi: 10.1053/j.gastro.2007.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raikwar NS, Vandewalle A, Thomas CP. Nedd4-2 interacts with occludin to inhibit tight junction formation and enhance paracellular conductance in collecting duct epithelia. Am J Physiol Renal Physiol. 299:F436–444. doi: 10.1152/ajprenal.00674.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Takahashi S, Iwamoto N, Sasaki H, Ohashi M, Oda Y, Tsukita S, Furuse M. The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J Cell Sci. 2009;122:985–994. doi: 10.1242/jcs.040055. [DOI] [PubMed] [Google Scholar]
- 111.Li J, Li YX, Chen MH, Li J, Du J, Shen B, Xia XM. Changes in the phosphorylation of claudins during the course of experimental colitis. Int J Clin Exp Pathol. 2015;8:12225–12233. [PMC free article] [PubMed] [Google Scholar]
- 112.Twiss F, Oldenkamp M, Hiemstra A, Zhou H, Matheron L, Mohammed S, de Rooij J. HGF signaling regulates Claudin-3 dynamics through its C-terminal tyrosine residues. Tissue Barriers. 2013;1:e27425. doi: 10.4161/tisb.27425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kersting S, Bruewer M, Schuermann G, Klotz A, Utech M, Hansmerten M, Krieglstein CF, Senninger N, Schulzke JD, Naim HY, Zimmer KP. Antigen transport and cytoskeletal characteristics of a distinct enterocyte population in inflammatory bowel diseases. Am J Pathol. 2004;165:425–437. doi: 10.1016/S0002-9440(10)63308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pravda J. Crohn's disease: evidence for involvement of unregulated transcytosis in disease etio-pathogenesis. World J Gastroenterol. 2011;17:1416–1426. doi: 10.3748/wjg.v17.i11.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Soderholm JD, Streutker C, Yang PC, Paterson C, Singh PK, McKay DM, Sherman PM, Croitoru K, Perdue MH. Increased epithelial uptake of protein antigens in the ileum of Crohn's disease mediated by tumour necrosis factor alpha. Gut. 2004;53:1817–1824. doi: 10.1136/gut.2004.041426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burgoyne RD, Morgan A. Secretory granule exocytosis. Physiol Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 117.Rodriguez-Feo JA, Puerto M, Fernandez-Mena C, Verdejo C, Lara JM, Diaz-Sanchez M, Alvarez E, Vaquero J, Marin-Jimenez I, Banares R, Menchen L. A new role for reticulon-4B/NOGO-B in the intestinal epithelial barrier function and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;308:G981–993. doi: 10.1152/ajpgi.00309.2014. [DOI] [PubMed] [Google Scholar]
- 118.Naydenov NG, Harris G, Brown B, Schaefer KL, Das SK, Fisher PB, Ivanov AI. Loss of soluble N-ethylmaleimide-sensitive factor attachment protein alpha (alphaSNAP) induces epithelial cell apoptosis via down-regulation of Bcl-2 expression and disruption of the Golgi. J Biol Chem. 2012;287:5928–5941. doi: 10.1074/jbc.M111.278358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Naydenov NG, Brown B, Harris G, Dohn MR, Morales VM, Baranwal S, Reynolds AB, Ivanov AI. A membrane fusion protein alphaSNAP is a novel regulator of epithelial apical junctions. PLoS One. 2012;7:e34320. doi: 10.1371/journal.pone.0034320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu M, Yang S, Qiu Y, Chen G, Wang W, Xu C, Cai W, Sun L, Xiao W, Yang H. Par-3 modulates intestinal epithelial barrier function through regulating intracellular trafficking of occludin and myosin light chain phosphorylation. J Gastroenterol. 2015;50:1103–1113. doi: 10.1007/s00535-015-1066-z. [DOI] [PubMed] [Google Scholar]
- 121.Guichard A, Cruz-Moreno B, Aguilar B, van Sorge NM, Kuang J, Kurkciyan AA, Wang Z, Hang S, Pinetonn de Chambrun GP, McCole DF, Watnick P, Nizet V, Bier E. Cholera toxin disrupts barrier function by inhibiting exocyst-mediated trafficking of host proteins to intestinal cell junctions. Cell Host Microbe. 2013;14:294–305. doi: 10.1016/j.chom.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu S, Nie Y, Knowles B, Sakamori R, Stypulkowski E, Patel C, Das S, Douard V, Ferraris RP, Bonder EM, Goldenring JR, Ip YT, Gao N. TLR sorting by Rab11 endosomes maintains intestinal epithelial-microbial homeostasis. EMBO J. 2014;33:1882–1895. doi: 10.15252/embj.201487888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamamura R, Nishimura N, Nakatsuji H, Arase S, Sasaki T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol Biol Cell. 2008;19:971–983. doi: 10.1091/mbc.E07-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ohira M, Oshitani N, Hosomi S, Watanabe K, Yamagami H, Tominaga K, Watanabe T, Fujiwara Y, Maeda K, Hirakawa K, Arakawa T. Dislocation of Rab13 and vasodilator-stimulated phosphoprotein in inactive colon epithelium in patients with Crohn's disease. Int J Mol Med. 2009;24:829–835. doi: 10.3892/ijmm_00000300. [DOI] [PubMed] [Google Scholar]
- 125.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baranwal S, Naydenov NG, Harris G, Dugina V, Morgan KG, Chaponnier C, Ivanov AI. Nonredundant roles of cytoplasmic beta- and gamma-actin isoforms in regulation of epithelial apical junctions. Mol Biol Cell. 2012;23:3542–3553. doi: 10.1091/mbc.E12-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ivanov AI. Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci. 2008;13:6662–6681. doi: 10.2741/3180. [DOI] [PubMed] [Google Scholar]
- 128.Musch MW, Walsh-Reitz MM, Chang EB. Roles of ZO-1, occludin, and actin in oxidant-induced barrier disruption. Am J Physiol Gastrointest Liver Physiol. 2006;290:G222–231. doi: 10.1152/ajpgi.00301.2005. [DOI] [PubMed] [Google Scholar]
- 129.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Noth R, Lange-Grumfeld J, Stuber E, Kruse ML, Ellrichmann M, Hasler R, Hampe J, Bewig B, Rosenstiel P, Schreiber S, Arlt A. Increased intestinal permeability and tight junction disruption by altered expression and localization of occludin in a murine graft versus host disease model. BMC Gastroenterol. 2011;11:109. doi: 10.1186/1471-230X-11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell. 2005;16:2636–2650. doi: 10.1091/mbc.E05-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol Biol Cell. 2004;15:2639–2651. doi: 10.1091/mbc.E04-02-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lechuga S, Baranwal S, Ivanov AI. Actin-interacting protein 1 controls assembly and permeability of intestinal epithelial apical junctions. Am J Physiol Gastrointest Liver Physiol. 2015;308:G745–756. doi: 10.1152/ajpgi.00446.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 135.Zhou K, Sumigray KD, Lechler T. The Arp2/3 complex has essential roles in vesicle trafficking and transcytosis in the mammalian small intestine. Mol Biol Cell. 2015;26:1995–2004. doi: 10.1091/mbc.E14-10-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang D, Naydenov NG, Feygin A, Baranwal S, Kuemmerle JF, Ivanov AI. Actin-Depolymerizing Factor and Cofilin-1 Have Unique and Overlapping Functions in Regulating Intestinal Epithelial Junctions and Mucosal Inflammation. Am J Pathol. 2016;186:844–858. doi: 10.1016/j.ajpath.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Citalan-Madrid AF, Vargas-Robles H, Garcia-Ponce A, Shibayama M, Betanzos A, Nava P, Salinas-Lara C, Rottner K, Mennigen R, Schnoor M. Cortactin deficiency causes increased RhoA/ROCK1-dependent actomyosin contractility, intestinal epithelial barrier dysfunction, and disproportionately severe DSS-induced colitis. Mucosal Immunol. 2017 doi: 10.1038/mi.2016.136. [DOI] [PubMed] [Google Scholar]
- 138.Kim ML, Chae JJ, Park YH, De Nardo D, Stirzaker RA, Ko HJ, Tye H, Cengia L, DiRago L, Metcalf D, Roberts AW, Kastner DL, Lew AM, Lyras D, Kile BT, Croker BA, Masters SL. Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1beta. J Exp Med. 2015;212:927–938. doi: 10.1084/jem.20142384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heissler SM, Sellers JR. Four things to know about myosin light chains as reporters for non-muscle myosin-2 dynamics in live cells. Cytoskeleton (Hoboken) 2015;72:65–70. doi: 10.1002/cm.21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu X, Shu S, Billington N, Williamson CD, Yu S, Brzeska H, Donaldson JG, Sellers JR, Korn ED. Mammalian Nonmuscle Myosin II Binds to Anionic Phospholipids with Concomitant Dissociation of the Regulatory Light Chain. J Biol Chem. 2016;291:24828–24837. doi: 10.1074/jbc.M116.739185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ivanov AI, Bachar M, Babbin BA, Adelstein RS, Nusrat A, Parkos CA. A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS ONE. 2007;2:e658. doi: 10.1371/journal.pone.0000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Citalan-Madrid AF, Garcia-Ponce A, Vargas-Robles H, Betanzos A, Schnoor M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers. 2013;1:e26938. doi: 10.4161/tisb.26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zihni C, Terry SJ. RhoGTPase signalling at epithelial tight junctions: Bridging the GAP between polarity and cancer. Int J Biochem Cell Biol. 2015;64:120–125. doi: 10.1016/j.biocel.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nusrat A, Giry M, Turner JR, Colgan SP, Parkos CA, Carnes D, Lemichez E, Boquet P, Madara JL. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia. Proc Natl Acad Sci U S A. 1995;92:10629–10633. doi: 10.1073/pnas.92.23.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Samarin SN, Ivanov AI, Flatau G, Parkos CA, Nusrat A. Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol Biol Cell. 2007;18:3429–3439. doi: 10.1091/mbc.E07-04-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schaefer A, Reinhard NR, Hordijk PL. Toward understanding RhoGTPase specificity: structure, function and local activation. Small GTPases. 2014;5:6. doi: 10.4161/21541248.2014.968004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Segain JP, Raingeard de la Bletiere D, Sauzeau V, Bourreille A, Hilaret G, Cario-Toumaniantz C, Pacaud P, Galmiche JP, Loirand G. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: evidence in Crohn's disease and experimental colitis. Gastroenterology. 2003;124:1180–1187. doi: 10.1016/s0016-5085(03)00283-x. [DOI] [PubMed] [Google Scholar]
- 149.Lopez-Posadas R, Becker C, Gunther C, Tenzer S, Amann K, Billmeier U, Atreya R, Fiorino G, Vetrano S, Danese S, Ekici AB, Wirtz S, Thonn V, Watson AJ, Brakebusch C, Bergo M, Neurath MF, Atreya I. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J Clin Invest. 2016;126:611–626. doi: 10.1172/JCI80997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hegan PS, Chandhoke SK, Barone C, Egan M, Bahler M, Mooseker MS. Mice lacking myosin IXb, an inflammatory bowel disease susceptibility gene, have impaired intestinal barrier function and superficial ulceration in the ileum. Cytoskeleton (Hoboken) 2016;73:163–179. doi: 10.1002/cm.21292. [DOI] [PubMed] [Google Scholar]
- 151.Lee SY, Kim H, Kim K, Lee H, Lee S, Lee D. Arhgap17, a RhoGTPase activating protein, regulates mucosal and epithelial barrier function in the mouse colon. Sci Rep. 2016;6:26923. doi: 10.1038/srep26923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melendez J, Liu M, Sampson L, Akunuru S, Han X, Vallance J, Witte D, Shroyer N, Zheng Y. Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology. 2013;145:808–819. doi: 10.1053/j.gastro.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu Z, Man SM, Zhu Q, Vogel P, Frase S, Fukui Y, Kanneganti TD. DOCK2 confers immunity and intestinal colonization resistance to Citrobacter rodentium infection. Sci Rep. 2016;6:27814. doi: 10.1038/srep27814. [DOI] [PMC free article] [PubMed] [Google Scholar]