

Natural inhibitors of Cas9 pretend to be DNA and block target binding, and using them in human cells can reduce off-target events.
Abstract
CRISPR (clustered regularly interspaced short palindromic repeats)–Cas9 gene editing technology is derived from a microbial adaptive immune system, where bacteriophages are often the intended target. Natural inhibitors of CRISPR-Cas9 enable phages to evade immunity and show promise in controlling Cas9-mediated gene editing in human cells. However, the mechanism of CRISPR-Cas9 inhibition is not known, and the potential applications for Cas9 inhibitor proteins in mammalian cells have not been fully established. We show that the anti-CRISPR protein AcrIIA4 binds only to assembled Cas9–single-guide RNA (sgRNA) complexes and not to Cas9 protein alone. A 3.9 Å resolution cryo–electron microscopy structure of the Cas9-sgRNA-AcrIIA4 complex revealed that the surface of AcrIIA4 is highly acidic and binds with a 1:1 stoichiometry to a region of Cas9 that normally engages the DNA protospacer adjacent motif. Consistent with this binding mode, order-of-addition experiments showed that AcrIIA4 interferes with DNA recognition but has no effect on preformed Cas9-sgRNA-DNA complexes. Timed delivery of AcrIIA4 into human cells as either protein or expression plasmid allows on-target Cas9-mediated gene editing while reducing off-target edits. These results provide a mechanistic understanding of AcrIIA4 function and demonstrate that inhibitors can modulate the extent and outcomes of Cas9-mediated gene editing.
INTRODUCTION
Phage-encoded inhibitors of CRISPR (clustered regularly interspaced short palindromic repeats)–Cas bacterial immune systems evolved to enable phage escape from destruction in bacterial cells (1) and have the potential to control CRISPR-Cas enzymes that are deployed for gene editing applications in various cell types (2, 3). Determination of the molecular basis for Cas9 inhibition could shed light on the evolutionary “arms race” between phage and bacteria and suggest new approaches to regulating genome editing in eukaryotic cells. The 87–amino acid anti–CRISPR-Cas9 protein AcrIIA4 is notable in both respects: It inhibits multiple Cas9 proteins, including the widely used Cas9 ortholog from Streptococcus pyogenes, and it blocks Cas9-mediated gene editing in human cells (3).
RESULTS
To investigate the molecular basis for AcrIIA4-mediated Cas9 inhibition, we first tested whether recombinant AcrIIA4 protein interacts directly with S. pyogenes Cas9 (SpyCas9) (Fig. 1A). Purified AcrIIA4 was incubated with SpyCas9 in the presence or absence of a single-guide RNA (sgRNA) that assembles with Cas9 to provide sequence-specific DNA recognition (4). Size exclusion chromatography showed that AcrIIA4 binds to SpyCas9 only in the presence of sgRNA (Fig. 1, B and C, and fig. S1), implying that AcrIIA4 recognizes either a hybrid surface created by the sgRNA and Cas9 or a solely protein surface created upon sgRNA-triggered conformational rearrangement (5, 6). Furthermore, the AcrIIA4-bound Cas9-sgRNA complex displayed a similar proteolytic digestion pattern to that of the Cas9-sgRNA complex alone, suggesting that binding of AcrIIA4 does not alter the Cas9-sgRNA conformation (fig. S2).
Fig. 1. AcrIIA4 binds to the SpyCas9-sgRNA complex.
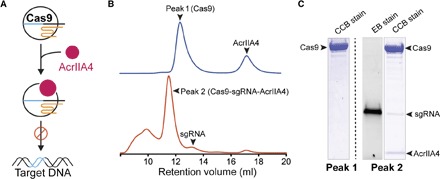
(A) A cartoon depiction of Cas9 protein loaded with the sgRNA binding to AcrIIA4 (pink). Cas9-sgRNA complexed with AcrIIA4 is unable to bind to the target DNA. (B) Size exclusion chromatogram of SpyCas9-sgRNA in the presence or absence of sgRNA after preincubation with AcrIIA4. Relevant peaks are indicated with arrowheads. (C) Coomassie (CCB)– and ethidium bromide (EB)–stained polyacrylamide gel showing the comigration of AcrIIA4 with Cas9 in the presence of gRNA.
To further elucidate the detailed molecular basis of AcrIIA4-mediated inhibition of Cas9 activity, we performed cryo–electron microscopy (cryo-EM) single-particle analysis on a SpyCas9-sgRNA complex bound to AcrIIA4. Cryo-EM images were collected on a Krios microscope using zero-loss energy-filtered imaging and a K2 direct electron detector. After unsupervised three-dimensional (3D) classification of 840,000 particle images, refinement of a class containing 285,600 particles resulted in an EM reconstruction of the SpyCas9-sgRNA-AcrIIA4 complex with an overall resolution of 3.9 Å (fig. S3 and S4). Subsequent local 3D classification that was focused on Cas9’s HNH nuclease domain ultimately yielded two EM reconstructions, one at 3.9 Å resolution where the HNH was poorly resolved because of flexibility (reconstruction 1; obtained as a combination of two 3D classes) (Fig. 2A and fig. S5A) and one with better-defined density for the HNH domain at an average resolution of 4.5 Å (reconstruction 2) (figs. S4 and S5A; see Materials and Methods and fig. S3). In both cryo-EM reconstructions, the density for the HNH domain of Cas9 was weaker than for the rest of the structure, consistent with the previously observed conformational plasticity of the HNH nuclease domain in the pretargeting state. An atomic model for AcrIIA4 was built from reconstruction 1. The EM density map displays excellent main-chain connectivity and side-chain densities for almost all residues of AcrIIA4 (Fig. 2B and fig. S5). We then built an atomic model for the Cas9-sgRNA complex based on the crystal structure of SpyCas9-sgRNA [Protein Data Bank (PDB) ID: 4ZT0] and refined the entire SpyCas9-sgRNA-AcrIIA4 model in real space to good stereochemistry (Fig. 2C and table S1). Overall, Cas9 bound to AcrIIA4 resembles the pretarget state rather than the DNA-bound state (fig. S6).
Fig. 2. Architecture of the SpyCas9-sgRNA in complex with AcrIIA4.
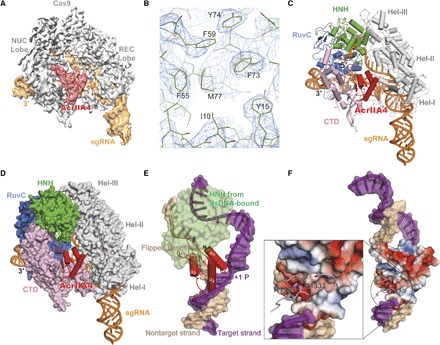
(A) Cryo-EM reconstruction of the AcrII4-bound SpyCas9. The electron density map was contoured at high-threshold levels showing distinct features for each subunit. (B) Representative cryo-EM density for AcrIIA4 with the refined model superimposed. (C) The atomic model of SpyCas9-sgRNA-ArcrIIA4. AcrIIA4 (red) and sgRNA (orange) are shown in a ribbon diagram. (D) Surface representation showing AcrIIA4 binding to the PAM-recognition cleft. (E) Superposition with Cas9–sgRNA–dsDNA (double-stranded DNA) structure (PDB ID: 5F9R). For clarity, Cas9 is omitted except the HNH domain. Target and nontarget DNA strand is colored purple and beige, respectively. (F) Electrostatic surface potential of AcrIIA4 showing that AcrIIA4 fits perfectly into the major groove of PAM duplex and that its surface acts as a dsDNA mimic. The inset shows that the PAM recognition residues (R1333 and R1334) are largely buried in an acidic pocket within AcrIIA4.
De novo model building demonstrated that AcrIIA4 binds to Cas9 with a 1:1 stoichiometry and comprises a three-stranded antiparallel β sheet flanked by one α helix at the N-terminal end and two α helices at the C-terminal end (α1β1β2β3α2α3). Superposition of the AcrIIA4-bound Cas9 structure with DNA-bound Cas9 revealed that AcrIIA4 sits exactly on the protospacer adjacent motif (PAM)–interacting cleft formed between the α-helical recognition (REC) lobe and the nuclease lobe (Fig. 2D and fig. S6). AcrIIA4 completely occupies the PAM binding pocket and thus blocks DNA recognition through contacts between the β3 strand of AcrIIA4 and Cas9 PAM-binding residues (R1333 and R1335) (Fig. 2, E and F). In addition, AcrIIA4 wedges into the DNA melting region immediately upstream of the PAM sequence and sits on top of +1 phosphate on the target strand and the flipped nucleotides on the nontarget strand, indicating that AcrIIA4 could also prevent DNA binding/unwinding. Consistent with this DNA-mimicking binding mode, AcrIIA4 is extremely acidic (Fig. 2F). AcrIIA4 also occupies the same space as the DNA-bound HNH domain and the linker connecting the HNH and RuvC domains (Fig. 2E), suggesting that AcrIIA4 could block the HNH movement required for catalysis.
Collectively, our structural studies show that AcrIIA4 is a highly acidic DNA mimic that blocks target DNA recognition through multiple mechanisms: (i) competitive inhibition of PAM binding; (ii) inhibition of DNA unwinding upstream of the PAM sequence; and (iii) inactivation of HNH domain movement from the inactive to the active conformation. These structural findings help explain the effectiveness of AcrIIA4 as an inhibitor of Cas9-mediated DNA cleavage and cell-based genome editing. Previous studies revealed the importance of Cas9’s interactions with targeted DNA, mediated via PAM in an interaction preceding base pairing between the sgRNA and its target (7). Transient Cas9-sgRNA association with PAM sequences in DNA is thought to enable a rapid target sequence search. By mimicking the structure and electrostatic properties of the DNA PAM sequence, AcrIIA4 might compete for initial DNA binding and thereby prevent target recognition and cleavage (Fig. 2, E to G).
To determine the functional effects of AcrIIA4 binding to Cas9-sgRNA complexes, we subjected DNA substrates with a target sequence to Cas9-sgRNA–catalyzed DNA cleavage assays. A linearized plasmid possessing a target sequence and PAM was incubated with a combination of Cas9, sgRNA, and AcrIIA4, and the products were resolved by gel electrophoresis. Cas9-sgRNA alone completely cut the target DNA within 5 min, whereas AcrIIA4 limited cleavage at even later time points (Fig. 3A). To confirm that AcrIIA4 would also inactivate Cas9 at low enzyme and inhibitor concentrations, we performed similar experiments with a radiolabeled DNA target for increased sensitivity (Fig. 3B). Near-stoichiometric concentrations of AcrIIA4 inhibited DNA cleavage when titrated against 10 nM Cas9-sgRNA complex, indicating an apparent dissociation constant of less than 10 nM. These data show that AcrIIA4 functions as a robust Cas9 “off switch” that can inhibit most Cas9 activity at low concentrations.
Fig. 3. AcrIIA4 inhibits DNA cleavage in vitro.
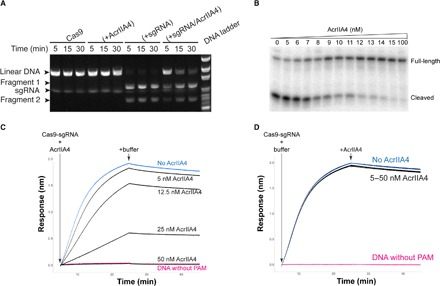
(A) Time-course cleavage assay using linearized plasmid template containing a 20–base pair λ1 DNA target sequence and a 5′-TGG-3′ PAM motif showing that AcrIIA4 inhibits SpyCas9-mediated endonuclease activity. (B) AcrIIA4 inhibits SpyCas9 cleavage of radiolabeled target DNA in vitro. SpyCas9–crRNA (CRISPR RNA)–tracrRNA (trans-activating crRNA) complex (10 nM) was preincubated with increasing concentrations (0 to 100 nM) of AcrIIA4. Substoichiometric synthetic oligonucleotide duplexes (2.5 nM) bearing a radiolabel at the 5′ end of the complementary strand were introduced for 6-min cleavage reactions. Reactions were resolved by denaturing polyacrylamide gel electrophoresis and visualized by phosphorimaging. (C) AcrIIA4 inhibits dCas9-sgRNA binding to a DNA target but does not affect target release, as measured by BLI. Preincubation in increasing concentrations of AcrIIA4 with dCas9-sgRNA reduces the on-rate of association with a DNA target relative to no inhibitor (blue). Maximal inhibition is identical to dCas9-sgRNA added to target DNA with a PAM mutation (pink). (D) Addition of increasing concentrations of AcrIIA4 with the preformed dCas9-sgRNA-DNA complex has no effect on the off-rate of dissociation.
The structure of Cas9 bound to AcrIIA4 suggested that the inhibitor competes for the initial DNA recognition event of PAM binding. However, we previously found that Cas9 binds so tightly to target DNA that its off-rate is negligible (7, 8). This implies an unusual nonequilibrium mode of inhibition, in which AcrIIA4 requires access to Cas9-sgRNA before formation of the Cas9-sgRNA-DNA complex. To test this prediction, we used biolayer interferometry (BLI) to measure the binding of catalytically inactivated Cas9 (dCas9) to a DNA target in the presence of an inhibitor. These experiments were performed under stoichiometric binding conditions in which Cas9 was present at concentrations greater than the dissociation constant of the Cas9-AcrIIA4 interaction. Preincubation of Cas9 with AcrIIA4 markedly inhibited Cas9 binding to DNA (on-rate) in a dose-dependent fashion, including complete prevention of target engagement (Fig. 3C and fig. S7). An electrophoretic mobility shift assay (EMSA) further confirmed that AcrIIA4 does not affect sgRNA loading into Cas9 protein alone (fig. S8) but does inhibit Cas9-sgRNA complex binding to a target DNA (fig. S9). Allowing the Cas9-sgRNA-DNA complex to form and then adding AcrIIA4 had no effect on Cas9’s release of target DNA (off-rate). This implies that AcrIIA4 is only able to access and inhibit Cas9 before DNA binding. These order-of-addition results are consistent with both the structure of AcrIIA4 bound to Cas9 and Cas9’s extremely slow dissociation from DNA. Together, these data support a mechanism for AcrIIA4 inhibition in which the inhibitor blocks Cas9’s ability to bind and cut target DNA by obscuring the PAM-interacting domain.
To determine the ability of AcrIIA4 to regulate gene editing in human cells, we first used human K562 erythroleukemia cells stably expressing a chromosomally integrated blue fluorescent protein (BFP) reporter (8). Nucleofection of Cas9-sgRNA (Cas9 RNP) complexes targeting BFP resulted in the loss of BFP fluorescence in almost all cells, as measured by flow cytometry (fig. S10). Simultaneous delivery of Cas9 RNP and AcrIIA4 protein inhibited Cas9-mediated gene targeting by up to 80% (Fig. 4A, blue symbols, and fig. S10). Simultaneous delivery of AcrIIA4 encoded in a plasmid inhibited gene editing to a lesser extent, possibly because of a delay in expression of the inhibitor from a plasmid relative to immediate nucleofection of Cas9 RNP (Fig. 4A, green symbols, and figs. S10 and S11). This implied that the order-of-addition effects that we observed in biophysical experiments may play a role during gene editing. To test this idea, we nucleofected the AcrIIA4-encoding plasmid 24 hours before introducing Cas9 RNP into cells. The presence of the AcrIIA4-encoding plasmid 24 hours before Cas9 RNP introduction inhibited Cas9-mediated gene targeting to a far greater extent than was observed during co-introduction and to a similar extent as observed during co-introduction of AcrIIA4 protein and Cas9 RNP (Fig. 4B and fig. S12). We found that addition of AcrIIA4 6 hours after Cas9 RNP reduced editing by ~50%, demonstrating the use of inhibitors in revealing in vivo gene editing kinetics (Fig. 4C and fig. S13). In sum, controlling the timing of AcrIIA4 inhibition in human cells strongly affects the frequency of gene editing at a given locus.
Fig. 4. Timed delivery of AcrIIA4 differentially inhibits on- and off-target genome editing in human cells.
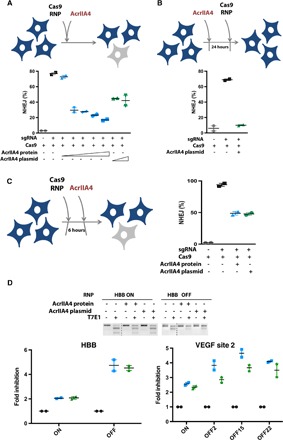
(A) Simultaneous delivery of Cas9 RNP and AcrIIA4 inhibits Cas9-mediated gene targeting in human cells. K562 cells with a chromosomally integrated BFP (BFP-K562) were nucleofected with Cas9 RNP and AcrIIA4 protein [Cas9/AcrIIA4 (molar ratio), 1:0.5, 1:1, 1:2, 1:3, and 1:5] or plasmid (0.7 and 2.8 μg). Nonhomologous end joining (NHEJ) frequencies are quantified by the loss of BFP expression in BFP-K562 cells 96 hours after nucleofection via flow cytometry. Data are presented as means ± SEM from at least two biological replicates. (B) Administration of AcrIIA4 before Cas9 RNP completely inhibits Cas9-mediated gene targeting. BFP-K562 cells were nucleofected with AcrIIA4 plasmid (0.7 μg) 24 hours before Cas9 RNP delivery. Data are presented as means ± SEM from at least two biological replicates. (C) Delivery of AcrIIA4 after introduction of Cas9 RNP yields intermediate inhibition of Cas9 activity. BFP-K562 cells were nucleofected with AcrIIA4 protein [Cas9/AcrIIA4 (molar ratio), 1:5] or plasmid (0.7 μg) 6 hours after Cas9 RNP delivery. Data are presented as means ± SEM from at least two biological replicates. (D) Proper timing of AcrIIA4 delivery diminishes off-target editing events while largely retaining on-target editing. K562 cells were nucleofected with either HBB or EMX1 targeting Cas9 RNP 6 hours before AcrIIA4 protein [Cas9/AcrIIA4 (molar ratio), 1:5] or plasmid (0.7 μg) delivery (18). Representative T7 endonuclease I assay for visualization of HBB on- and off-target editing. Bottom: Inhibition of editing by AcrIIA4 at on- and off-target sites of HBB and VEGFA, as measured by amplicon sequencing.
Using a given sgRNA, Cas9 may target both an on-target site and off-target loci (9–12). However, several lines of evidence suggest that off-target sites may be bound without being immediately cleaved (13–15). Cas9 displaced from uncleaved sites (for example, by cellular factors) would be available for inhibition by AcrIIA4. Because inhibitor timing experiments suggested that at least 50% of on-target Cas9 gene editing takes place within the first 6 hours (Fig. 4C), we asked whether off-target editing could be reduced by properly timed addition of an inhibitor.
We examined on- and off-target editing using sgRNAs targeting β-globin (HBB) and vascular endothelial growth factor A (VEGFA) loci. The off-target sites for both the HBB and VEGF site 2 guides have been previously described (10, 16, 17). Notably, the HBB guide is of therapeutic interest for editing the causative mutation of sickle cell disease but has an off-target site that is nearly identical to the on-target site. We edited the HBB and VEGFA loci using Cas9 RNPs with and without preintroduction of the AcrIIA4 protein or plasmid. Consistent with experiments at the BFP locus, we found that timed addition of AcrIIA4 retained substantial levels of on-target editing. For VEGFA, three representatives off-target sites spanning a range of off-target propensities were selected from the off-target sites previously discovered by GUIDE-seq (18). The primary HBB off-target site has also been previously described (16, 17). T7E1, TIDE (Tracking of Indels by DEcomposition) analysis, and amplicon next-generation sequencing all showed that timed addition of AcrIIA4 can significantly abolish off-target editing and greatly increase the fidelity of Cas9 RNP at both HBB and VEGFA loci (Fig. 4D and fig. S14). Off-target editing at all sites were greatly reduced by timed addition of AcrIIA4 regardless of their frequency. This suggests that Cas9 inhibitors have the potential to be useful in reducing off-target events during research and therapeutic applications.
DISCUSSION
The recent and rapid expansion of the Cas9 toolkit for gene editing applications has lacked an inducible off switch to prevent undesired gene editing. Newly discovered protein inhibitors, encoded by bacteriophages, provide an attractive solution to this problem because these proteins are small and function well in human cells (3). Here, we demonstrate that AcrIIA4, the most potent SpyCas9 inhibitor in human cells, acts as a DNA mimic to block PAM recognition. The deployment of DNA mimics to inhibit a DNA binding protein is an elegant solution that is often deployed in the phage-host arms race. A recent structural study revealed that a class 1 anti-CRISPR (AcrF2) uses a similar strategy (19). Furthermore, phage-encoded restriction enzyme inhibitor proteins have long been known to mimic the DNA target (20). Our results further show that Cas9 inhibitors either delivered as protein or expressed from a plasmid can modulate the efficacy of gene editing at multiple loci in human cells. Although preaddition of inhibitor almost completely abolishes overall gene editing, timed addition of inhibitor after initiating Cas9-sgRNA–based gene editing can adjust the amount of time that Cas9 is active in the nucleus, thereby selectively limiting off-target editing. We anticipate that Cas9 inhibitors could be broadly useful in situations where precise control of either on- or off-target gene editing is desirable, such as during allele-specific therapeutic editing.
MATERIALS AND METHODS
Protein expression and purification
SpyCas9 was purified as previously described (21). Phage-encoded anti-CRISPR AcrIIA4 (3) was subcloned using Clontech’s In-Fusion cloning system into a pGEX-6P-1 expression vector with a glutathione S-transferase (GST) affinity tag and a PreScission Protease cleavage site at the N terminus. The GST-AcrIIA4 fusion protein was overexpressed in Escherichia coli strain BL21 (Novagen) with overnight induction of 0.25 mM isopropyl-β-d-thiogalactopyranoside at 18°C. The soluble fractions from the lysates were purified by affinity chromatography using Glutathione Sepharose 4B resin. The recovered proteins were then digested with PreScission Protease to remove the GST tag and further fractionated by ion exchange column (Hitrap Q), followed by an additional gel filtration chromatography step (HiLoad 16/60 Superdex 200, GE Healthcare) using the storage buffer [50 mM Hepes (pH 7.5), 200 mM NaCl, 5 mM dithiothreitol (DTT), and 10% glycerol]. The purified AcrIIA4 protein was snap-frozen in liquid nitrogen and stored at −80°C.
Analytical size exclusion chromatography
Analytical size exclusion chromatography was conducted on an ÄKTA purification system (GE Healthcare). SpyCas9 protein was loaded onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with a buffer containing 30 mM Hepes (pH 7.5), 200 mM NaCl, and 5 mM DTT. The SpyCas9-sgRNA-AcrIIA4 sample was prepared at a molar ratio of 1:1.6:2.0 for 30 min at room temperature before loading onto the gel filtration column. Eluates were monitored by ultraviolet absorbance at 260 and 280 nm. For clarity, only spectra at 280 nm were shown in the figures.
Limited proteolysis
Limited proteolysis experiment was conducted at room temperature. SpyCas9 with or without AcrIIA4 and the purified SpyCas9-sgRNA complex in the absence or presence of AcrIIA4 were mixed with Elastase (Roche) at a 1:120 (w/w) ratio and incubated at room temperature. Aliquots were taken at noted time points and immediately quenched by the addition of equal amount of 2× SDS–polyacrylamide gel electrophoresis loading dye (Bio-Rad). Samples were further boiled for 5 min at 95°C and then resolved by 4 to 20% Tris-Glycine Mini Gels (Bio-Rad).
Electrophoretic mobility shift assay
For gel shift assay with sgRNA binding, 5 fmol of P32-radiolabeled sgRNA was mixed with 0, 5, 10, 20, 50, and 100 nM SpyCas9 in the absence or presence of AcrIIA4 in a 20-μl reaction containing 30 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM DTT, and 5% (v/v) glycerol. For gel shift assay with DNA binding, 0, 5, 10, 20, 50, and 100 nM purified SpyCas9-sgRNA complex preassembled with or without AcrIIA4 were mixed with 2.5 fmol of P32-radiolabeled dsDNA. Binding reactions were performed for 30 min at room temperature before adding 5 μl of 15% (w/v) Ficoli and 0.2% (w/v) Orange G loading dye. The samples were run on an 8% (w/v) nondenaturing tris–glycine–polyacrylamide gel (37.5:1 acrylamide/bisacrylamide) at 4°C in 0.5× tris-borate EDTA electrophoresis buffer. After electrophoresis, the gels were dried and visualized by phosphorimaging.
Cryo-EM microscopy
SpyCas9-sgRNA-AcrIIA4 complexes in a buffer containing 30 mM tris (pH 8.0), 150 mM NaCl, 20 mM EDTA, 5 mM DTT, and 0.1% glycerol were used for cryo-EM sample preparation. Immediately after glow-discharging the grid for 14 s using a Solaris plasma cleaner, 3.6-μl droplets of the sample (~2 μM) were placed onto C-flat grids with 1.2-μm holes and 1.3-μm spacing between holes (Protochips Inc.). The grids were rapidly plunged into liquid ethane using an FEI Vitrobot Mark IV maintained at 8°C and 100% humidity, after being blotted for 4.5 s with a blot force of 10. Data were acquired using an FEI Titan Krios transmission electron microscope (at the Howard Hughes Medical Institute Cryo-EM Shared Resource at Janelia Research Campus) operated at 300 keV, at a nominal magnification of ×29,000 (1.04 Å pixel size), and with defocus ranging from −1.5 to −3.0 μm. A total of ~2435 micrographs were recorded using SerialEM on a Gatan K2 Summit direct electron detector operated in counting mode. We collected a 6-s exposure fractionated into 30,200-ms frames with a dose of 7.69 e− Å−2 s−1.
Image processing and reconstruction
The 27 frames (we skipped the first frame and the last two frames) of each image stack in the superresolution model were aligned, decimated, summed, and dose-weighted using MotionCor2 (22). Contrast transfer function (CTF) values of the summed micrographs were determined using CTFFIND4 and then applied to dose-weighted summed micrographs for further processing. Initial particle picking to generate template images was performed using EMAN2 (23, 24). About 40,000 particles were selected and then imported into Relion2.0 for reference-free 2D classification (25). Particle picking for the complete data set was carried out using Gautomatch (www.mrc-lmb.cam.ac.uk/kzhang/) with templates generated in the previous 2D classification. About 840,000 particles were selected in total. Using the published sgRNA-bound Cas9 structure (EMD-3276) (12) low-pass–filtered to 60 Å as a reference, we performed 3D classification using Relion2.0 into seven classes and selected the best model with the largest number of particles (~285,600) for further processing. The other six classes likely represented unassembled complexes and junk. Three-dimensional refinement with a soft mask produced structures with good Euler angle distribution and resulted in reconstruction at 3.9 Å resolution after postprocessing. In this reconstruction, the HNH domain was quite noisy because of flexibility. We carried out a second round of 3D classification into three classes: The first one (~97,000 particles) did not have a clear HNH density, the second (~88,000 particles) displayed clear density for the HNH domain, and the third one showed anisotropic particle distribution and was discarded (~100,000 particles). Three-dimensional refinement for the combination of first and second classes produced a structure at 3.9 Å (reconstruction 1). Three-dimensional refinement for the second class alone produced a structure at 4.5 Å (reconstruction 2) with improved HNH density. These two reconstructions were used for atomic model building. All the soft masks used in refinement were generated via a four-step script edited by X. Li (Tsinghua University School of Medicine, Beijing). The local resolution was calculated using the ResMap application (26). We used Relion to automatically calculate B-factors and apply them to the postprocessed maps. B-factors were around −200 Å−2. All reported resolutions are based on the gold-standard 0.143 FSC (Fourier Shell Correlation) criterion using two independent half maps.
Model building and refinement
To generate a complete model, the crystal structure of SpyCas9-sgRNA (PDB ID: 4ZT0) was first fitted into the refined 3D reconstruction map using UCSF Chimera (27) and then manually rebuilt in Coot to fit the density. Some missing amino acids in 4ZT0 were also built in our model. For the AcrIIA4 part, the secondary structure prediction and several amino acids with large side chains (R8, Y15, 30L, 31I, 32I, 33R, 41Y, 42V, 55F, 59F, 63W, 67Y, 75Y, and 80I) were used for registering the sequence length and building the model ab initio in Coot (28). To improve backbone geometry, the atomic model of the SpyCas9-sgRNA-AcrIIA4 model was subjected to PHENIX real-space refinement (global minimization and atomic displacement parameter refinement) with Ramachandran, rotamer, and nucleic acid restraints. The final model was validated using MolProbity (29). All statistics of the data processing and structure refinement of the SpyCas9-sgRNA-AcrIIA4 complex were summarized in table S1. Structural analysis was performed in Coot (28), and figures were prepared using PyMOL (Schrödinger LLC) and UCSF Chimera.
Inhibition of SpyCas9 activity with radiolabeled target DNA and linearized plasmid
This assay was adapted from the oligonucleotide cleavage assay reported by Jinek et al. (4). Sixty-mer oligonucleotide target DNA was purchased from Integrated DNA Technologies (see table S2). The complementary strand, which hybridizes to crRNA, was end-labeled with 32P using T4 polynucleotide kinase (NEB) and [γ-32P]–adenosine triphosphate (PerkinElmer) in accordance with the recommendations of the enzyme’s manufacturer. Unincorporated nucleotides were removed using the ssDNA/RNA Clean & Concentrator kit (D7011, Zymo Research). Labeled and unlabeled oligonucleotides were annealed in the presence of NEB Cas9 buffer [20 mM Hepes, 100 mM NaCl, 5 mM MgCl2, and 0.1 mM EDTA (pH 6.5)] after a 2-min heat denaturation at 95°C. Before cleavage reactions, 10 nM SpyCas9 (NEB) and 20 nM crRNA-tracrRNA complex (Alt-R, Integrated DNA Technologies) were preincubated with various concentrations of AcrIIA4 for 15 min at 37°C in NEB Cas9 buffer. Single–time point reactions were initiated upon the addition of 2.5 nM target DNA. After 6-min incubation at 37°C, reactions were quenched, separated, and imaged as described (4). For plasmid cleavage assay, 600 nM SpyCas9 with or without AcrIIA4 in the absence or presence of sgRNA was used, and the following cleavage products by SpyCas9-sgRNA were resolved on 1% agarose gel and stained with SYBR Safe (Life Technologies).
Biolayer interferometry
We measured dCas9 binding to DNA using BLI (8). Streptavidin-coated BLI probes were used to monitor association and disassociation of dCas9 RNP to biotinylated DNA that had been bound to the probe surface. A 55-mer biotinylated target oligonucleotide and its complement were purchased from Integrated DNA Technologies and annealed to form a dsDNA target (see the Supplementary Materials for sequences). An sgRNA targeting the oligonucleotide was in vitro transcribed (http://dx.doi.org/10.17504/protocols.io.hdrb256) and combined with purified dCas9 protein [University of California, Berkeley (UC Berkeley) QB3 MacroLab, Berkeley, CA] to form RNP. All experiments were conducted at 27°C in a 9:1 mixture of BLI running buffer [20 mM tris (pH 7), 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.01% Tween, and heparin (50 μg/ml)] and protein buffer (10 mM Hepes, 2.5% glycerol, 75 mM KCl, and 0.5 mM DTT). For prebinding inhibition experiments, RNP was then combined with purified AcrIIA4 protein to reach the specified concentration and incubated for 30 min at room temperature. The stages of the BLI experiments were then conducted as follows: 1-min rinse in buffer, 2-min incubation with 100 nM target dsDNA, 2-min baseline measurement and equilibration, 20-min association with 25 nM dCas9 RNP (with AcrIIA4 in the case of the prebinding experiments), and then 20-min disassociation in buffer (with AcrIIA4 in the case of postbinding experiments). BLI data were processed in R by the subtraction of average response from reference samples (in which DNA is loaded onto probes but not exposed to dCas9 or AcrIIA4), subtraction of baseline response at the beginning of the association step, and removal of the small jump discontinuity upon transition from association to disassociation buffer.
Cell line
Previously constructed K562 cells stably expressing a genomically integrated BFP reporter was used (8) and verified mycoplasma-free by Lonza MycoAlert LT-07 (Lonza). All cells were cultured in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1% nonessential amino acids, and 2 mM GlutaMAX.
Nucleofection for editing experiments
Cas9 RNP synthesis was carried out as previously described (17, 30). Briefly, sgRNA was synthesized by assembly polymerase chain reaction (PCR) and in vitro transcription (IVT). A substrate template for T7 RNA polymerase was assembled by PCR with Phusion High-Fidelity DNA polymerase (NEB) from a variable 57- to 59-nucleotide (nt) primer containing the T7 promoter, variable sgRNA guide sequence, and the first 15 nt of the nonvariable region of the sgRNA (T7FwdVar primers, 10 nM; table S2) and an 83-nt primer containing the reverse complement of the invariant region of the sgRNA (T7RevLong, 10 nM), along with amplification primers (T7FwdAmp and T7RevAmp, 200 nM each). Assembled template was used as a substrate for IVT by T7 RNA polymerase using HiScribe T7 High Yield RNA Synthesis Kit (NEB). Resulting transcription products were treated with deoxyribonuclease I (NEB), and RNA was purified by homemade solid-phase reversible immobilization (SPRI) beads (comparable to Beckman-Coulter AMPure beads). Thirty picomoles of Cas9-NLS (nuclear localization sequence) (UC Berkeley QB3 MacroLab, Berkeley, CA) was mixed slowly into Cas9 buffer [20 mM Hepes (pH 7.5), 150 mM KCl, 1 mM MgCl2, 10% glycerol, and 1 mM tris(2-carboxyethyl)phosphine] containing 36 pmol of sgRNA. The resulting 5-μl mixture was incubated for 10 min to allow RNP formation. For conditions concerning simultaneous delivery of Cas9 RNP and AcrIIA4, AcrIIA4 protein or plasmid was added to the RNP mixture and incubated for an additional 5 min. K562 cells (2 × 10−5) were harvested, washed once with phosphate-buffered saline (PBS), and resuspended in 15 μl of SF nucleofection buffer (Lonza). Five microliters of RNP mixture (with or without AcrIIA4) and 15 μl of cell suspension were combined and added into Lonza 4D Nucleocuvette strips and were nucleofected with program FF-120. Prewarmed medium (200 μl) was added to each nucleocuvette, and electroporated cells were transferred to culture dishes. Electroporation of AcrIIA4 alone was carried out by mixing AcrIIA4 protein or plasmid into 20-μl cell suspension in buffer SF, adding the mixture into Lonza 4D Nucleocuvette strips, nucleofecting with program FF-120, adding 200 μl of prewarmed medium, and transferring to culture dishes. Editing outcomes were measured 4 days after nucleofection by flow cytometry, T7E1 assay, TIDE analysis, and next-generation amplicon sequencing.
Flow cytometry
K562-BFP cells were collected 4 days after nucleofection and directly analyzed for fluorescence using Attune NxT Flow Cytometer (Thermo Fisher Scientific). Viable cells were gated on size and shape using forward and side scatter. BFP fluorescence was collected in the VL1 channel using a 440/50 band-pass filter. Flow cytometry data were analyzed using FlowJo (FlowJo).
Construction of 3X-FLAG AcrIIA4 plasmid
AcrIIA4 was amplified from plasmid pJH376 (3) with primer RB_AcrIIA4 Fwd 1 and RV1 (table S2). Initial PCR products were amplified with a second set of primers (RB_AcrIIA4 Fwd 3 and RB_AcrIIA4 RV3 3XFLAG) containing the 3X FLAG sequence and restriction sites (Bam HI and Eco RI) that correspond to the vector that was used to amplify the same region. The amplified DNA and the plasmid pJH376 were subject to enzyme digestion for 1 hour at 37°C and ligated with T4 DNA ligase (NEB M0202). Transformants were obtained using DH5α competent cells (Thermo Fisher Scientific), and sequences were verified through Sanger sequencing by UC Berkeley DNA Sequencing Facility.
Immunoblotting
AcrIIA4 was immunoblotted with K562 cells expressing either control (untagged AcrIIA4) or AcrIIA4-3XFLAG. Whole-cell extracts were prepared from 1× radioimmunoprecipitation assay lysis buffer (Millipore). Extracts were clarified by centrifugation at 15,000g for 15 min at 4°C, and protein concentrations were determined by Pierce BCA (bicinchoninic acid) assay (Thermo Fisher Scientific). Eight micrograms of whole-cell extract was separated on precast 4 to 12% bis tris protein gel (Invitrogen) and transferred to a nitrocellulose membrane. Membranes were blocked in PBS–0.05% Tween 20 (PBST) containing 5% nonfat dry milk and incubated overnight at 4°C with primary antibody [FLAG M2 (F1804, Sigma) and glyceraldehyde-3-phosphate dehydrogenase (14C10, Cell Signaling)] diluted in PBST–5% nonfat dry milk. Membranes were subsequently washed with PBST and incubated with the appropriate IRDye 680RD and IRDye 800CW secondary antibody (LI-COR Biosciences) diluted in PBST–5% nonfat dry milk. Images were detected using the Odyssey Systems (LI-COR Biosciences).
Analysis of editing by T7 endonuclease I and TIDE analysis
Edited K562 cells were harvested 4 days after nucleofection, and genomic DNA was isolated using the QuickExtract DNA Extraction system (Epicentre). PCRs were carried out with 100 ng of genomic DNA using appropriate primers (table S2) and PrimeSTAR GXL Polymerase (Clontech) under standard protocol (30 cycles at 98°C for 10 s, 57°C for 15 s, 68°C for 60 s). PCR products were purified using homemade SPRI beads. For T7E1 assay, 200 ng of purified PCR products was subjected to denaturation/annealing and subsequent T7E1 (NEB) digestion. Digested PCR products were resolved on a 2% agarose gel. For TIDE assay, 50 ng of purified PCR products was mixed with 5 pM primer in a final volume of 15 μl, and samples were subjected to Sanger sequencing (Quintara Biosciences). Sequencing chromatograms were analyzed by TIDE (31), and indel frequencies were determined by the addition of significant insertions and deletions (P < 0.05).
Amplicon next-generation sequencing
Genomic DNA (100 ng) from edited K562 cells was amplified at on- and off-target sites using stubbed primers listed in table S2. Three off-target sites (off-target sites 2, 16, and 22) for VEGFA site 2 were selected from the list of off-targets previously reported (18). PCR products were SPRI-cleaned, followed by amplification of 20 to 50 ng of the first PCR product in a second nine-cycle PCR using Illumina-compatible primers [table S2; primers designed and purchased through the Vincent J. Coates Genomics Sequencing Laboratory (GSL) at UC Berkeley], generating indexed amplicons of an appropriate length for NGS (Next-Generation Sequencing). Libraries from 100 to 200 pools of edited cells were pooled and submitted to the GSL for paired-end 300-cycle processing using a version 3 Illumina MiSeq sequencing kit (Illumina Inc.) after quantitative PCR measurement to determine molarity.
Next-generation sequencing data analysis
Twenty million MiSeq reads were converted to fastq format and simultaneously demultiplexed using Bcl2fastq version 2.18 (Illumina Inc.) and further analyzed using a custom analysis workflow written in Python. Each sample contained >100,000 reads. Any read containing an indel within a window of 12 to 16 bases around the predicted cut site was called an “indel,” and remaining reads were called “unedited.” Percentage of NHEJ was calculated by indel read count/(indel read count + unedited read count).
Supplementary Material
Acknowledgments
We thank R. Huang and Z. Yu for expert EM assistance, A. Chintangal for computer support, and the members of the Bondy-Denomy, Corn, Doudna, and Nogales laboratories for helpful discussions and critical reading of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Funding: F.J. is a Merck Fellow of the Damon Runyon Cancer Research (DRG-2201-14). J.E.C. is supported by the Li Ka Shing Foundation and the Heritage Medical Research Institute. J.E.C. receives funding from AstraZeneca, Pfizer, the Li Ka Shing Foundation, and the Heritage Medical Research Institute. J.S. is supported by the National Institute on Aging of the NIH under award number T32 AG000266. J.B.-D. and B.J.R. are supported by the University of California, San Francisco Program for Breakthrough in Biomedical Research, funded in part by the Sandler Foundation, and an NIH Office of the Director Early Independence Award (DP5-OD021344). J.A.D. receives funding from HHMI, the NIH, the NSF, Roche, Pfizer, the Paul Allen Foundation, and the Keck Foundation. J.A.D. and E.N. are Investigators of the Howard Hughes Medical Institute (HHMI). This work was supported in part by HHMI. Author contributions: J.S., F.J., J.-J.L., E.N., J.B.-D., J.E.C., and J.A.D. conceived and designed the experiments. J.S., F.J., J.-J.L., N.L.B., B.J.R., and S.H.B. carried out experiments. J.S., F.J., J.-J.L., E.N., J.B.-D., J.E.C., and J.A.D. wrote the paper. Competing interests: J.E.C. is a cofounder of Spotlight Therapeutics and Peregrine Biotechnology (neither of these companies are currently working with Cas9 inhibitors) and is a scientific advisor to Mission Therapeutics. J.E.C. is a consultant for Intellia Therapeutics, Editas Medicine, and CRISPR Therapeutics (none of the results in this article were discussed with any of these organizations). J.A.D. is a cofounder of Editas Medicine, Intellia Therapeutics, and Caribou Biosciences and a scientific advisor to Caribou, Intellia, eFFECTOR Therapeutics, and Driver. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. EM-derived maps and atomic coordinates of the Cas9-sgRNA-AcrIIA4 structure have been deposited in the EM Databank and the PDB with accession codes EMD-874 and 5VZL, respectively. The EM data were collected in the EM facility of HHMI Janelia Research Campus.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1701620/DC1
fig. S1. Gel filtration of Cas9 complexes with AcrIIA4.
fig. S2. Exposed region analysis of SpyCas9 at AcrIIA4-free and AcrIIA4-bound states.
fig. S3. Cryo-EM of Cas9 ribonucleoprotein particles.
fig. S4. Classification and refinement workflow.
fig. S5. Atomic modeling.
fig. S6. Model comparison between AcrIIA4-bound and DNA-bound SpyCas9-sgRNA complexes.
fig. S7. Biological replicate data for BLI data shown in Fig. 3C.
fig. S8. EMSA of increasing concentrations of Cas9 binding to sgRNA in the absence or presence of AcrIIA4.
fig. S9. EMSA of Cas9-sgRNA binding to a target DNA with and without AcrIIA4.
fig. S10. Representative flow cytometry data used to create Fig. 4A.
fig. S11. Western blot of AcrIIA4-3XFLAG expression.
fig. S12. Representative flow cytometry data used to create the graph shown in Fig. 4B.
fig. S13. Representative flow cytometry data used to create the graph shown in Fig. 4C.
fig. S14. Quantification of on- and off-target editing at HBB, as measured by TIDE analysis.
table S1. Data collection and model refinement statistics.
table S2. Oligonucleotides used in this study.
REFERENCES AND NOTES
- 1.Bondy-Denomy J., Pawluk A., Maxwell K. L., Davidson A. R., Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawluk A., Amrani N., Zhang Y., Garcia B., Hidalgo-Reyes Y., Lee J., Edraki A., Shah M., Sontheimer E. J., Maxwell K. L., Davidson A. R., Naturally occurring off-switches for CRISPR-Cas9. Cell 167, 1829–1838.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauch B. J., Silvis M. R., Hultquist J. F., Waters C. S., McGregor M. J., Krogan N. J., Bondy-Denomy J., Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell 168, 150–158.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jinek M., Jiang F., Taylor D. W., Sternberg S. H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S., Kaplan M., Iavarone A. T., Charpentier E., Nogales E., Doudna J. A., Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimasu H., Ran F. A., Hsu P. D., Konermann S., Shehata S. I., Dohmae N., Ishitani R., Zhang F., Nureki O., Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sternberg S. H., Redding S., Jinek M., Greene E. C., Doudna J. A., DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L., Corn J. E., Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Fu Y., Foden J. A., Khayter C., Maeder M. L., Reyon D., Joung J. K., Sander J. D., High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mali P., Aach J., Stranges P. B., Esvelt K. M., Moosburner M., Kosuri S., Yang L., Church G. M., CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pattanayak V., Lin S., Guilinger J. P., Ma E., Doudna J. A., Liu D. R., High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiani S., Chavez A., Tuttle M., Hall R. N., Chari R., Ter-Ovanesyan D., Qian J., Pruitt B. W., Beal J., Vora S., Buchthal J., Kowal E. J. K., Ebrahimkhani M. R., Collins J. J., Weiss R., Church G., Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 12, 1051–1054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sternberg S. H., LaFrance B., Kaplan M., Doudna J. A., Conformational control of DNA target cleavage by CRISPR–Cas9. Nature 527, 110–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X., Scott D. A., Kriz A. J., Chiu A. C., Hsu P. D., Dadon D. B., Cheng A. W., Trevino A. E., Konermann S., Chen S., Jaenisch R., Zhang F., Sharp P. A., Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cradick T. J., Fine E. J., Antico C. J., Bao G., CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeWitt M. A., Magis W., Bray N. L., Wang T., Berman J. R., Urbinati F., Heo S.-J., Mitros T., Muñoz D. P., Boffelli D., Kohn D. B., Walters M. C., Carroll D., Martin D. I. K., Corn J. E., Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 8, 360ra134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Thapar V., Wyvekens N., Khayter C., Iafrate A. J., Le L. P., Aryee M. J., Joung J. K., GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury S., Carter J., Rollins M. F., Golden S. M., Jackson R. N., Hoffmann C., Nosaka L., Bondy-Denomy J., Maxwell K. L., Davidson A. R., Fischer E. R., Lander G. C., Wiedenheft B., Structure reveals mechanisms of viral suppressors that intercept a CRISPR RNA-guided surveillance complex. Cell 169, 47–57.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walkinshaw M. D., Taylor P., Sturrock S. S., Atanasiu C., Berge T., Henderson R. M., Edwardson J. M., Dryden D. T. F., Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol. Cell 9, 187–194 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Jiang F., Zhou K., Ma L., Gressel S., Doudna J. A., A Cas9–guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Scheres S. H. W., Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 579, 125–157 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen V. B., Arendall III W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S., Staahl B. T., Alla R. K., Doudna J. A., Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkman E. K., Chen T., Amendola M., van Steensel B., Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1701620/DC1
fig. S1. Gel filtration of Cas9 complexes with AcrIIA4.
fig. S2. Exposed region analysis of SpyCas9 at AcrIIA4-free and AcrIIA4-bound states.
fig. S3. Cryo-EM of Cas9 ribonucleoprotein particles.
fig. S4. Classification and refinement workflow.
fig. S5. Atomic modeling.
fig. S6. Model comparison between AcrIIA4-bound and DNA-bound SpyCas9-sgRNA complexes.
fig. S7. Biological replicate data for BLI data shown in Fig. 3C.
fig. S8. EMSA of increasing concentrations of Cas9 binding to sgRNA in the absence or presence of AcrIIA4.
fig. S9. EMSA of Cas9-sgRNA binding to a target DNA with and without AcrIIA4.
fig. S10. Representative flow cytometry data used to create Fig. 4A.
fig. S11. Western blot of AcrIIA4-3XFLAG expression.
fig. S12. Representative flow cytometry data used to create the graph shown in Fig. 4B.
fig. S13. Representative flow cytometry data used to create the graph shown in Fig. 4C.
fig. S14. Quantification of on- and off-target editing at HBB, as measured by TIDE analysis.
table S1. Data collection and model refinement statistics.
table S2. Oligonucleotides used in this study.