

This article summarizes the evidence for the impact of BRAF mutations on treatment outcome of anti‐EGFR monoclonal antibodies. Based on a review of literature, eight meta‐analyses were included in this study, which consistently show that patients with BRAF mutations have a lack of treatment benefit of anti‐EGFR monoclonal antibodies. Considering the quality and quantity of available evidence, current guidelines may be revised.
Keywords: BRAF, RAS, Cetuximab, Panitumumab, Predictive biomarker, Anti‐epidermal growth factor receptor
Abstract
Recently, the American Society of Clinical Oncology (ASCO) and the European Society for Medical Oncology (ESMO) recommended that patients with epidermal growth factor receptor (EGFR)‐expressing metastatic colorectal cancer could be treated with anti‐EGFR monoclonal antibodies (mAbs) cetuximab and panitumumab only in absence of Rat‐Sarcoma (RAS) mutations. In addition to the previously established biomarker Kirsten rat sarcoma viral oncogene homolog (KRAS) exon 2, cumulative evidence also shows that patients whose tumors harbor KRAS exons 3 or 4 and neuroblastoma rat‐sarcoma viral oncogene homolog (NRAS) exons 2, 3, and 4 mutations are found unlikely to benefit from anti‐EGFR treatment.
In line with the resistance of RAS mutated (mt) tumors, treatment response in BRAFmt tumors may also be altered given their important role in the EGFR signaling pathway. However, BRAF is not recommended as predictive biomarker yet because the evidence for the impact of BRAF mutations on treatment outcome is considered insufficient.
This article summarizes the evidence for the impact of BRAF mutations on treatment outcome of anti‐EGFR mAbs. Based on a review of literature, eight meta‐analyses were included that consistently show that patients with BRAF mutations have a lack of treatment benefit of anti‐EGFR mAbs. After discussing the quality and quantity of available evidence, we conclude that evidence is stronger than suggested by ESMO and ASCO. Additionally, we highlight that the quality of evidence for BRAF is even higher than for extended RAS as a biomarker. We therefore advise ESMO and ASCO to reconsider BRAF status as a predictive biomarker for response.
Implications for Practice.
In metastatic colorectal cancer (mCRC), therapy with anti‐epidermal growth factor receptor (EGFR) monoclonal antibodies cetuximab and panitumumab is indicated in absence of RAS mutations. Cumulative evidence shows that patients with BRAF mutations, who comprise 10% of the mCRC population, do not benefit from anti‐EGFR‐antibody treatment. Although guidelines state that evidence for BRAF as a predictive marker is insufficient, we highlight that the quality and quantity of evidence is higher than suggested. We therefore encourage the use of BRAF as a predictive marker in order to exclude patients from therapy for whom limited treatment benefit is expected.
Introduction
The RAS‐RAF‐MEK‐ERK (MAPK) pathway plays a pivotal role in the regulation of cell proliferation, survival, and differentiation. Constitutive activation of this pathway is frequently observed in human cancers and is associated with high rates of cancer cell proliferation. Within the MAPK pathway, RAS, RAF, MEK, and ERK are key proteins in signal transduction. In tumor cells, the MAPK pathway is often constitutively activated by gain‐of‐function mutations in one of the signaling proteins including but not limited to RAS and RAF. In colorectal cancer (CRC), activation of the MAPK pathway is often a result of mutations in the RAS family protein Kirsten rat sarcoma viral oncogene homolog (KRAS), which is found in 40% of the patients [3], [4]. Mutations occur most frequently in exon 2 (36%) and less frequently in exons 3 (2%) and 4 (2%). In addition to KRAS, neuroblastoma rat sarcoma viral oncogene homolog (NRAS) mutations occur in about 3% [3], [4]. KRAS and NRAS are very closely related, although their biological roles are slightly different. Whereas functional KRAS is essential for cell survival, NRAS is not required. Therefore, KRAS gain‐of‐function mutations may have a larger impact on tumor growth and proliferation compared with NRAS mutations [5].
The first effector protein of RAS is RAF, comprising c‐RAF1, BRAF, and ARAF. Of these, BRAF has the most important biological function and is also most frequently mutated [6]. BRAF and RAS mutations are mutually exclusive, which highlights their functional importance [7]. Gain‐of‐function mutations in exon 15 result in the BRAFV600E variant in about 10% of the CRC population and induce constitutive MAPK‐pathway activation [5], [8]. Other mutations that occur less frequently include the variants G469V (<0.1%), D594G (<0.3%), and K601E (unknown frequency) [6]. All mutations lead to constitutive activation of downstream proteins within the MAPK‐pathway independent of upstream activation signals, yet the p.V600E variant is the strongest activator [8], [9]. BRAF mutations in CRC occur most frequently in tumors originating from the appendix and the ascending and transverse colon, defined as right‐sided tumors [10], [11], [12].
In up to 90% of colorectal tumors, epidermal growth factor receptor (EGFR) is overexpressed, which renders EGFR an attractive drug target [13], [14], [15], [16]. Upon binding of its ligands, including epidermal growth factor, betacellulin, epiregulin, and neuregulins, cell proliferation and growth are induced primarily through the MAPK and PI3K/AKT signaling pathways (Fig. 1) [17].
Figure 1.

Schematic overview of the MAPK signaling pathway. Adapted with permission from van Geel et al. [66].
Abbreviations: EGFR, epidermal growth factor receptor; mAb, monoclonal antibody; MAPK, RAS‐RAF‐MEK‐ERK.
Cetuximab (Erbitux® [14], [18]) and panitumumab (Vectibix® [15], [19]) are anti‐EGFR monoclonal antibodies (mAbs) that exert their antitumor effect through inhibition of EGFR signaling. Both drugs are registered for the treatment of EGFR‐expressing metastatic CRC (mCRC) after failure of first‐ and/or second‐line therapies.
In line with the biological mechanism, several trials showed that the effects of anti‐EGFR treatment are decreased when mutations downstream of EGFR are present that cause MAPK‐pathway activation independent of EGFR signaling such as mutations in KRAS (in 40%) and BRAF (in 10%) [1], [3], [4], [5], [20]. It is now generally accepted that mutations in KRAS exon 2 diminish treatment response when anti‐EGFR mAbs are given as a single agent or combined with chemotherapy [1], [14], [15], [21]. More recently, several retrospective analyses showed that not only KRAS exon 2 mutations but also KRAS exons 3 and 4 and NRAS exons 2, 3, and 4 mutations are predictive biomarkers [1], [2], [7], [22], [23], [24], [25], [26], [27]. Treatment guidelines for metastastatic CRC now recommend upfront RAS testing before start of anti‐EGFR mAb therapy [1], [2] in order to exclude patients with mutated RAS from therapy with these agents.
BRAF mutations could have comparable effects on anti‐EGFR mAb treatment response as RAS mutations. BRAFV600E gain‐of‐function mutations comprise 80%–96% of all BRAF mutations and occur in about 10% of CRC patients [4], [6], [28]. Although several meta‐analyses indicate that BRAF status may be a predictive biomarker for treatment efficacy [4], [29], [30], [31], [32], the use of BRAF status as a predictive biomarker is not recommended yet because evidence is considered less convincing than the evidence for RAS mutations [1], [2].
This manuscript describes the evidence that is available for BRAF mutations as a predictive biomarker for response to anti‐EGFR mAbs in mCRC. We will discuss the load and quality of clinical evidence for the impact of BRAF mutations on anti‐EGFR mAb treatment outcomes and argue why this can be considered convincing enough to include BRAF mutation status in the panel of upfront mutation tests in anti‐EGFR mAb therapy.
Evidence for BRAF Mutations as a Predictive Biomarker
A PubMed search was performed to collect meta‐analyses that included data of BRAF mutated (mt) patients and BRAF wild‐type (wt) patients and survival outcome of treatment with the anti‐EGFR mAbs cetuximab or panitumumab using the following terms: (molecular testing OR mutation) AND (BRAF OR RAF) AND survival AND EGFR AND “colorectal cancer” AND meta‐analysis (full methods available in the supplemental online Appendix 1). Eight meta‐analyses were identified that report on the overall response rate (ORR), progression‐free survival (PFS), or overall survival (OS) of BRAFmt patients treated with anti‐EGFR mAbs cetuximab or panitumumab as single agents or combined with chemotherapy. Four of these were considered high‐quality reviews, and only these will be extensively discussed in this section [4, 29–31]. The results of all meta‐analyses are summarized in Table 1.
Table 1. Overview of meta‐analyses, which show hazard ratios for PFS, OS, and odds ratios for ORR on anti‐epidermal growth factor receptor monoclonal antibody therapy by BRAF status for the KRASwt group and (°) for the KRAS unselected group.
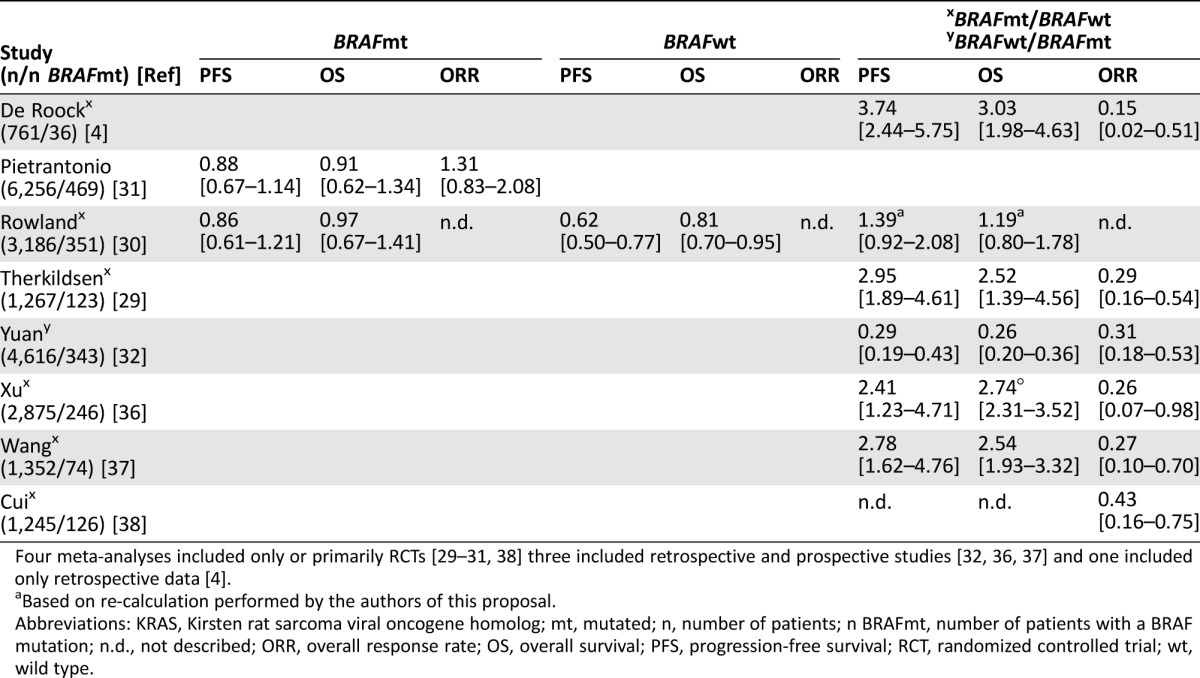
Four meta‐analyses included only or primarily RCTs [29–31, 38] three included retrospective and prospective studies [32, 36, 37] and one included only retrospective data [4].
Based on re‐calculation performed by the authors of this proposal.
Abbreviations: KRAS, Kirsten rat sarcoma viral oncogene homolog; mt, mutated; n, number of patients; n BRAFmt, number of patients with a BRAF mutation; n.d., not described; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; RCT, randomized controlled trial; wt, wild type.
De Roock et al. [4] comprehensively analyzed the relationship between different pathway mutations and treatment response and survival in mCRC patients treated with cetuximab combined with chemotherapy. The authors collected tumor samples and clinical data from 11 European investigators who had published data on cetuximab‐treated mCRC patients. Finally, 761 tumor samples were analyzed for BRAF status (screened for the mutations p.D594G, p.V600E, p.V600M, and p.K601E). In 36 patients, a BRAF mutation was found, being mostly p.V600E (n = 35) and one p.D594G. In a selection of patients without KRAS mutations, it was found that BRAFmt patients (n = 24) had a significantly lower ORR (8.3%) compared with BRAFwt patients (n = 326, ORR 38%; OR 0.15 [95% confidence interval {CI} 0.02–0.51]) and shorter PFS (hazard ratio {HR} 3.74 [95% CI 2.44–5.75]) and OS (HR 3.03 [95% CI 1.98–4.63]). The association between disease control and BRAF status was significant in multivariate analysis (adjusted OR BRAFmt vs. BRAFwt 0.059; p < .0001), as was the association with KRAS, NRAS, and PIK3CA exon 20. Still, 2 out of 24 patients had a response to treatment despite BRAF mutations. The authors report that one of these had a p.D594G mutation that leads to weaker activation of the MAPK pathway compared with p.V600E mutations [33]. The other responder had a low copy number of BRAFV600Emt genes that may explain the sensitivity to cetuximab. The authors conclude that the response rate of 24.4% in an unselected population could be increased to 36.3% in a KRASwt population and further to 38.4% in KRASwt and BRAFwt patients [4]. Another 1.5% ORR improvement could be achieved by NRAS testing according to their results. This study highlighted the importance of BRAF in addition to KRAS status in treatment with anti‐EGFR mAbs. Several meta‐analyses have been performed to confirm the findings of De Roock et al.
Pietrantonio et al. [31] performed a meta‐analysis of randomized controlled trials (RCTs) to examine the effect of anti‐EGFR mAbs on PFS, OS, and ORR in BRAFmt/KRASwt advanced CRC. Nine phase III trials and one phase II trial were included that compared anti‐EGFR mAbs as monotherapy or added to chemotherapy with chemotherapy or best supportive care in advanced KRASwt CRC. In total, these comprised 6,256 patients on first‐line (six trials) and second‐line treatment (two trials) or who were chemo‐refractory (two trials). The authors show that patients with BRAFmt CRC (n = 469) do not have a significant benefit in PFS (HR PFS benefit 0.88 [95% CI 0.67–1.14]), OS (HR OS 0.91 [95% CI 0.62–1.34]), or ORR (OR 1.31 [95% CI 0.83–2.08]) from treatment with anti‐EGFR mAbs. All mutations comprised p.V600E mutations except for 21 patients (13%) in the trial from Smith et al. who had the p.D594G mutation [34]. The response rate of patients with BRAFmt varied from 10.8% to 52.2% on anti‐EGFR mAbs compared with 6.4% to 40% on chemotherapy. Based on these results, BRAFmt patients seem to have modest responses to anti‐EGFR mAbs, yet, overall, a significant response rate and survival benefit is lacking in this population [31]. A drawback of this meta‐analysis is that a comparison with BRAFwt patients has not been made.
Rowland et al. [30] reviewed RCTs that evaluated the effect of BRAF mutations on treatment benefit (OS and PFS) from anti‐EGFR mAbs for KRAS exons 2 and 3 wt metastatic CRC mCRC. All of the included trials have also been reviewed by Pietrantonio et al. [31]. However, Rowland et al. excluded the trials by Tveit et al. [35] and Stintzing et al. [24], probably because the former did not provide data on PFS and OS and the latter had bevacizumab with FOLFIRI as control treatment instead, which did not meet the inclusion criteria. The review by Rowland et al. thus differs from Pietrantonio et al. by inclusion criteria but moreover by their statistical tests [30], [31].
Seven articles covering eight RCTs were included in which 3,168 KRASwt patients were treated with cetuximab or panitumumab (four studies each) added to chemotherapy or with chemotherapy alone. About 11% of the tumors harbored a BRAF mutation (n = 351), of which 94% (n = 330) were p.V600E mutations and 6% (n = 21) were p.D594G mutations [34]. Rowland et al. not only reported outcomes for the BRAFmt subgroup but also compared BRAFmt patients with BRAFwt patients. Results show a lack of PFS benefit in the BRAFmt group (HR PFS benefit 0.86 [95% CI 0.61–1.21]), whereas patients with BRAFwt had significant benefit (HR PFS benefit 0.62 [95% CI 0.50–0.77]) from addition of anti‐EGFR mAbs to chemotherapy. The interaction test (PFS HR BRAFmt/PFS HR BRAFwt) showed a close to significant difference (p = .07). For OS, BRAFmt patients (HR 0.97 [95% CI 0.67–1.41]) also had no benefit, whereas BRAFwt patients (HR 0.81 [95% CI 0.70–0.95]) had significantly improved OS. The interaction test for OS was not significant (p = .43). For both PFS and OS, the difference between BRAFmt and BRAFwt patients was bigger in the second‐line setting, showing a very strong trend towards significance (interaction test PFS p = .05; OS p = .38). The authors conclude that based on the nonsignificant interaction test values, the effect of BRAF mutations on PFS and OS cannot be confirmed [30].
However, it is of great importance to note that the interaction test for PFS was very close to significance. To allow proper interpretation of the p value (.07), CIs of the interaction test value should be taken into account. However, these were not provided by the authors. Based on our re‐estimation, as described in the methods section, the 95% CI for the interaction on PFS should range from 0.92 to 2.08. This underscores that there is a high chance that anti‐EGFR mAbs have a different effect on PFS in BRAFmt patients. To summarize, these results confirm a significant lack of PFS benefit of anti‐EGFR mAbs treatment in BRAFmt patients, which is relevantly though not significantly different from the BRAFwt patients.
Another meta‐analysis was performed by Therkildsen et al. [29], who reviewed the impact of alterations in KRAS other than exon 2, NRAS, BRAF, PIK3CA, and PTEN on clinical benefit of anti‐EGFR treatment combined with chemotherapy in KRAS exon 2 wt patients in 21 RCTs and one nonrandomized trial. Among 1,267 patients treated with cetuximab or panitumumab in first to greater than fourth setting mostly, BRAF mutations were detected in 123 patients, all of which were p.V600E except for one p.K601E variant. BRAFmt patients were found to have significantly lower ORR (OR ORR 0.29 [95% CI 0.16–0.54]) and shorter PFS (HR 2.95 [95% CI 1.89–4.61]) and OS (HR 2.52 [95% CI 1.39–4.56]) compared with BRAFwt patients. The authors also report that the response rate can be increased from 37.6% on average in KRASwt selected patients to 39% in KRASwt/BRAFwt selected patients.
In our opinion, these meta‐analyses provide high‐quality clinical evidence for the lack of efficacy of anti‐EGFR mAb treatment on response and survival endpoints for patients with BRAFmt tumors. Supporting evidence can be found in four meta‐analyses that included survival endpoints based on retrospective studies, mainly [32], [36], [37], or that included response rate as an endpoint only (Table 1) [38].
Evidence for BRAF Compared with RAS
Initially, Erbitux® and Vectibix® were registered for patients with EGFR‐expressing metastatic CRC only in presence of KRAS exon 2 wt. This was based on seven pivotal trials with cetuximab that show that the KRAS exon 2 mutated population had no benefit on primary endpoints ORR, PFS, or OS [14], [15], [39]
This is supported by a review of five RCTs that was performed by the American Society of Clinical Oncology (ASCO) [21]. The authors showed that all five trials, comprising 2,095 patients, consistently detected a lack of benefit from treatment with anti‐EGFR mAbs in 720 patients with KRAS exon 2 mutations in terms of PFS and ORR (Table 2), whereas KRAS exon 2 wt patients did have significant benefit.
Table 2. Summary of the results from five randomized clinical trials which report on the effects of KRAS exon 2 mutationsa.
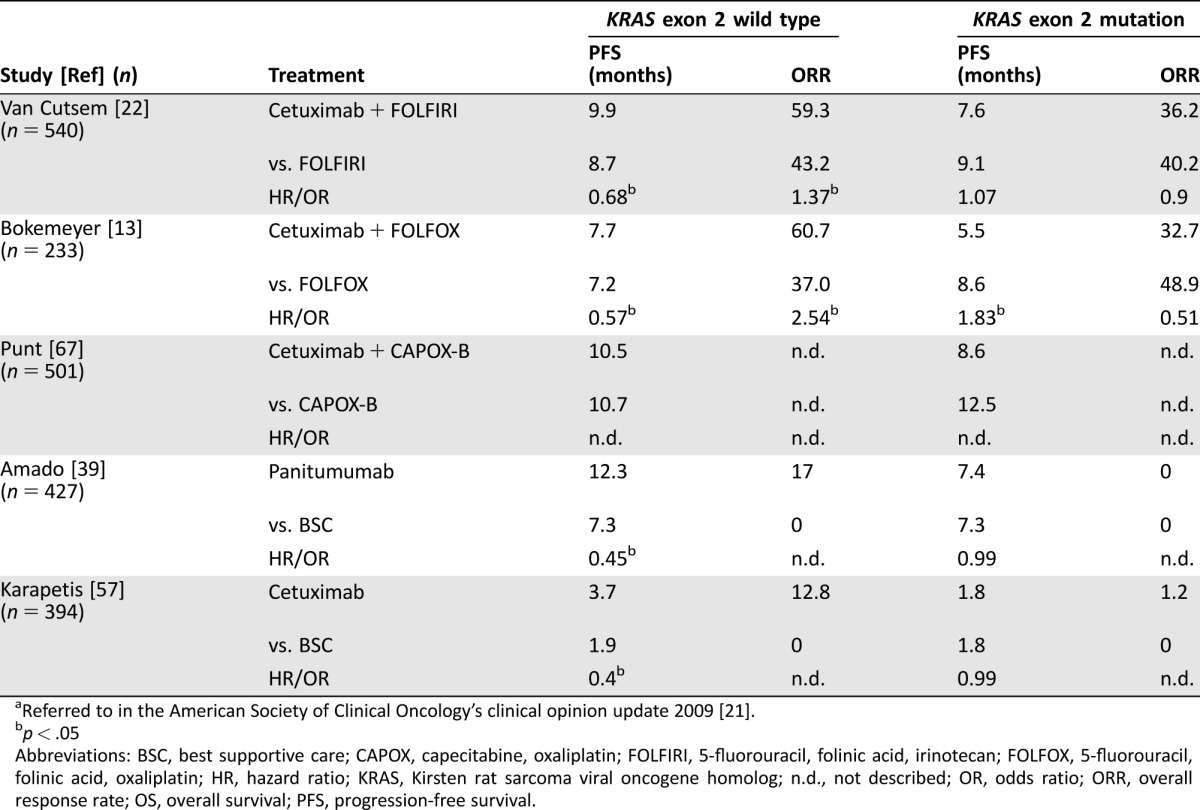
Referred to in the American Society of Clinical Oncology's clinical opinion update 2009 [21].
p < .05
Abbreviations: BSC, best supportive care; CAPOX, capecitabine, oxaliplatin; FOLFIRI, 5‐fluorouracil, folinic acid, irinotecan; FOLFOX, 5‐fluorouracil, folinic acid, oxaliplatin; HR, hazard ratio; KRAS, Kirsten rat sarcoma viral oncogene homolog; n.d., not described; OR, odds ratio; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival.
In October 2015, ASCO recommended to extend upfront testing of KRAS exon 2 with KRAS exons 3 and 4 and NRAS exons 2, 3, and 4 [1]. In July 2016, the European Society for Medical Oncology (ESMO) supported this “extended RAS” testing in their consensus guideline on the management of metastatic CRC [2]. In Europe, these findings were incorporated in the product labels of Erbitux® and Vectibix®, which state that the benefit‐risk ratio of treatment is negative for patients with KRAS or NRAS exons 2, 3, or 4 mutations [18], [19].
As supportive evidence for KRAS exons 2, 3, and 4 testing, ASCO referred to ten [40], [41], [42], [43], [44], [45], [46], [47], [48], [49] meta‐analyses and two [50], [51] health technology assessment reports that reviewed 137 primary studies with 19,543 patients. Table 3 summarizes findings of these ten meta‐analyses. In all studies, a statistically significant PFS benefit in patients without KRAS exons 2, 3, or 4 mutations was found, whereas benefit was not significant in KRAS exons 2, 3, and 4 mutants. The effects of KRAS mutations on OS were less consistent. Five out of the 13 trials detected no statistically significant difference in OS between KRASmt and KRASwt patients treated with anti‐EGFR mAbs. This is mainly due to lack of consistent OS benefit when adding anti‐EGFR mAbs to standard of care in the overall and KRASwt population. In KRASwt patients, OS benefit was detected in only 6 out of 13 trials [45], [46], [47], [49], [50], [51].
Table 3. Summary of the data from meta‐analyses addressing the effect of KRAS exons 2, 3, and 4 mutations referred to in the American Society of Clinical Oncology's clinical opinion update 2015 [1].
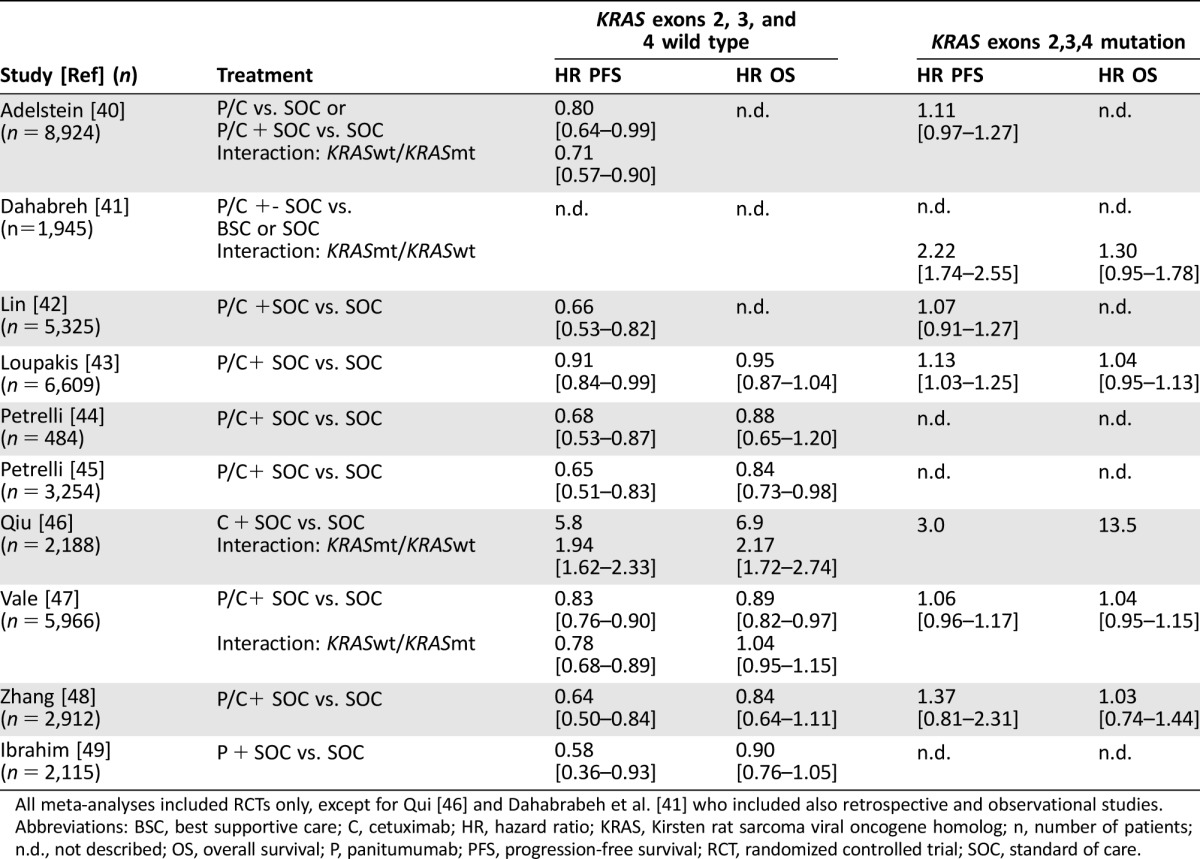
All meta‐analyses included RCTs only, except for Qui [46] and Dahabrabeh et al. [41] who included also retrospective and observational studies.
Abbreviations: BSC, best supportive care; C, cetuximab; HR, hazard ratio; KRAS, Kirsten rat sarcoma viral oncogene homolog; n, number of patients; n.d., not described; OS, overall survival; P, panitumumab; PFS, progression‐free survival; RCT, randomized controlled trial; SOC, standard of care.
In contrast to the evidence for KRAS exons 2, 3, and 4, only five articles report on the impact of KRAS exons 3 and 4 and NRAS mutations by itself (Table 3) [2], [7], [22], [25], [26], [27]. Although these five trials consistently show that patients with RAS mutations do not have significant treatment benefit, it should be noted that this is based on a small group with RAS mutations other than KRAS exon 2. The subgroup of KRAS exons 3 and 4 and NRAS exons 2, 3, and 4 mutated patients comprises only 10%–20% of the study populations, which were 360 patients in total. To increase the power, most studies merge patients with any RAS mutation into one or two groups. This supports extended RAS testing but does not provide evidence on the effect of KRAS exons 3 and 4 and NRAS mutations by itself. In addition, the detected lack of OS benefit in the RASmt subgroup is of limited value in three out of five trials because the RASwt group did not have OS benefit either, and no significant interaction has been confirmed.
Despite these limitations, evidence was considered convincing enough by ASCO and ESMO to recommend upfront KRAS and NRAS exons 2, 3, and 4 mutation testing, so that only patients whose tumors do not harbor mutations in these exons will be given anti‐EGFR mAb therapy [1], [2].
Discussion
Strong Evidence for Impact of BRAFmt on Anti‐EGFR mAb Treatment Outcome
ASCO's and ESMO's most recent guidelines for the treatment of mCRC posit that there is currently insufficient evidence to recommend BRAF mutations as a biomarker for response to anti‐EGFR therapy [52]. ESMO's guideline refers to three clinical trials [53], [54], [55] and two meta‐analysis [30], [31], which show conflicting results. The authors of the guideline suggest that the evidence for BRAF mutations as a predictive biomarker for anti‐EGFR therapy in later lines is accumulating, but the role in earlier treatment lines is uncertain [2]. It is therefore recommended that BRAF status is used as a prognostic marker or as a selection tool for clinical trials only. Based on our literature review, the evidence for the use of BRAF as a predictive marker may be stronger than suggested.
All eight meta‐analyses that were reviewed, covering first‐line and second‐line settings (supplemental online Table 1), consistently show that BRAFmt patients do not have significant benefit from anti‐EGFR mAbs in terms of ORR, PFS, and OS [30], [31], and when compared with BRAFwt patients, they have significantly less ORR, PFS, and OS benefit (Table 1) [4], [29], [32], [36], [37]. A significant interaction between BRAF and outcome has been confirmed in five meta‐analyses [4], [29], [32], [36], [37]. Because both cetuximab and panitumumab were registered mainly based on PFS benefit [7], [18], [19], [22], [25], [26], [27], the detected lack of PFS benefit in BRAFmt patients should warrant the use in this population.
Only Rowland et al. reported a nonsignificant interaction between the BRAFwt and BRAFmt group on both PFS and OS, although the result for PFS was close to significant (p = .07). This finding should be interpreted in context with the power to detect significant differences. Results of Rowland et al. are based on a group of 3,096 patients. Although this is one of the three most extensive meta‐analyses, it provides a power of 19% [56], whereas a sample size of 6,500 patients is required to detect a significant interaction effect with a power of 80% (calculation provided in methods section) [30].
Overall, the load of strong clinical evidence for BRAF mutations as a biomarker is based on a population of 628 patients with BRAF mutations [4], [29], [31]. In comparison, the evidence for KRAS exons 3 and 4 and NRAS exons 2, 3, and 4 comes from only five studies with 360 patients with RAS mutations other than KRAS exon 2 [7, 22]. The lack of treatment benefit in this group guided ASCO's and ESMO's clinical opinion on extended RAS testing, while a significant interaction between KRAS exons 3 and 4/NRAS exons 2, 3, and 4 mutant and wt patients has not been confirmed. Based on this, the evidence for the impact of BRAF mutations on efficacy of anti‐EGFR mAb treatment as a single agent or combined with chemotherapy can be considered stronger than for KRAS and NRAS mutations.
Biologically, it is possible that BRAFmt tumors respond to anti‐EGFR treatment due to tumor heterogeneity, low copy numbers, and/or a varying potency of BRAF mutations to activate the MAPK pathway. As an example, the p.V600E mutation is a strong pathway activator, whereas the p.D594G is less activating and may still allow responses.
Risks of Using BRAF as a Predictive Marker
If upfront molecular testing of BRAF will be applied, it is expected that 10% of the mCRC population will be identified as BRAFmt and will be excluded from anti‐EGFR mAb therapy. The most important risk of the use of BRAF as a predictive biomarker lies in withholding BRAFmt patients from a potentially effective treatment with anti‐EGFR mAbs. In BRAFmt patients, response rates of 8.3% [4] to 18% [36] have been reported compared with 38% [4] to 42.4% [36] in BRAFwt patients. Although a direct effect of anti‐EGFR mAbs cannot be ruled out, the responses may also be induced by backbone chemotherapy, which was administered in the majority of trials and which induced response rates of 13%–40% [31]. This is supported by the finding that responses rates in BRAFmt patients are higher when anti‐EGFR mAbs are combined with chemotherapy (ORR 8.3%–18% [4], [36]) compared with monotherapy (ORR 1.2% [57]). As a comparison, it should also be noted that in patients with KRAS exon 2 mutations relevant response rates of 33%–36% [13], [54] have been observed on anti‐EGFR treatment added to chemotherapy (Table 2) as well as incidental responses on monotherapy [18]. Yet, this did not hinder implementation of KRAS as a biomarker.
Biologically, it is possible that BRAFmt tumors respond to anti‐EGFR treatment due to tumor heterogeneity, low copy numbers, and/or a varying potency of BRAF mutations to activate the MAPK pathway [8]. As an example, the p.V600E mutation is a strong pathway activator, whereas the p.D594G is less activating and may still allow responses [58], [59]. It is unlikely that this plays a big role in the study results as described in this review because the majority of patients had BRAFV600E mutations. However, it may explain specific cases of responders. Importantly, preclinical and clinical evidence shows that BRAF mutations may sensitize tumors to anti‐EGFR treatment when combined with targeted agents such as BRAF and MEK inhibitors [60], [61], [62]. The potential of these combinations is currently studied in clinical trials [62], [63]. The use of BRAF as predictive marker as discussed in this review therefore only applies to anti‐EGFR monotherapy or combined with chemotherapy.
Limitations
The robustness of evidence for BRAF mutations as a predictive biomarker is limited by some factors.
Firstly, compared with KRAS, BRAF mutations may have a less pronounced predictive effect. Whereas patients with KRAS exons 2, 3, and 4 mutations have 10%–30% higher risk of progression during treatment with anti‐EGFR mAbs compared with control treatment (Tables 3 and 4), the effect of BRAF mutations seems smaller. This is based on PFS HRs with a wider CI in patients with BRAFmt on anti‐EGFR therapy (Table 1) compared with patients with KRAS exons 2, 3, and 4 mutations (Table 3). However, it should be noted that the evidence for extended RAS testing was based on groups in which patients with KRAS exons 3 and 4 and NRAS exons 2, 3, and 4 mutations were merged. The effect of these mutations are even more convincing if KRAS exon 2 mutations are also included (any RAS mutation, Table 4). Merging all RAS mutations is needed to improve the power of the study. However, when comparing the load of evidence for extended RAS with the load of evidence for BRAF, it should be taken into account that the group of BRAFmt patients is always analyzed separately, resulting in a less evident result. Merging this group with RASmt patients would improve the power, but the relevance is questionable because of the different biology of the mutations.
Table 4. Overview of RCTs showing hazard ratios for PFS and OS stratified by RAS statusa.

Referred to in the American Society of Clinical Oncology's clinical opinion updates of 2009 and 2015 [1], [21].
Abbreviations: B, bevacizumab; C, cetuximab; FOLFIRI, folinic acid + fluorouracil + irinotecan; FOLFOX, folinic acid + fluorouracil + oxaliplatin; KRAS, Kirsten rat sarcoma viral oncogene homolog; n, population evaluable for RAS status; n.d., not described; NRAS, neuroblastoma rat sarcoma viral oncogene homolog; OS, overall survival; P, panitumumab, PFS, progression‐free survival; RAS, rat sarcoma viral oncogene homolog.
Secondly, the impact of patient selection by BRAF status on OS remains uncertain. Because even in RASwt and BRAFwt patients, the OS benefit of anti‐EGFR mAbs is not consistently confirmed among different studies (Table 4); OS seems an unreliable endpoint to assess the predictive value of BRAF for outcomes on anti‐EGFR therapy. Therefore, the effects on PFS should guide decision‐making instead of effects on OS. In addition, uncertainties about the predictive value of BRAF may be a result of other predictive biomarkers beyond KRAS and BRAF. Recent evidence highlights the importance of primary sidedness on CRC prognosis and response [12]. Right‐ and left‐sided tumors have a different biological origin, resulting in different molecular characteristics. While left‐sided tumors are associated with EGFR overexpression, right‐sided tumor more often carry BRAF mutations [10], [12] and are associated with poorer response and a shorter survival independent of treatment [11], [64]. Although sidedness has been identified as an independent biomarker [11], the association between sidedness, KRAS and BRAF status, and response to anti‐EGFR mAbs is still to be clarified.
Moreover, cumulating evidence shows that in absence of a confirmed BRAF mutation, a similar gene expression profile can be present, referred to as BRAF‐like tumors. BRAF‐like tumors have comparable characteristics as BRAFmt tumors, leading to treatment resistance by constitutive MAPK‐pathway activation independent of EGFR signaling. Future clinical validation studies should reveal whether the evidence for BRAF status as a predictive biomarker could become stronger by including BRAF‐like gene signatures [65].
Future clinical validation studies should reveal whether the evidence for BRAF status as a predictive biomarker could become stronger by including BRAF‐like gene signatures
Conclusion
Recent guidelines recommend upfront extended RAS testing in mCRC patients in order to exclude patients with KRAS exons 2, 3, and 4 and NRAS exons 2, 3, and 4 mutations from therapy with anti‐EGFR mAbs. As outlined in this review, the evidence for BRAF testing is of an even higher level than the evidence for extended RAS testing. Across all studies, no ORR, PFS, or OS benefit could be detected in any BRAFmt subgroup. Moreover, significant interactions of BRAF status with treatment outcome have been observed. This review highlights that despite limitations in power and effect size, the current evidence should be enough to draw conclusions.
Based on consistent lack of benefit of anti‐EGFR mAb therapy in BRAFmt patients, it is advised that anti‐EGFR mAb therapy is excluded for these patients. The authors therefore encourage ASCO and ESMO to reconsider BRAF as a predictive biomarker, as this will help in selecting patients for whom maximum treatment benefit is expected.
See http://www.TheOncologist.com for supplemental material available online.
Author Contributions
Conception/Design: Jan H. M. Schellens, Emilie M. J. van Brummelen
Collection and/or assembly of data: Jan H. M. Schellens, Emilie M. J. van Brummelen
Data analysis and interpretation: Emilie M. J. van Brummelen, Anthonius de Boer, Jos H. Beijnen, Jan H. M. Schellens
Manuscript writing: Jan H. M. Schellens, Emilie M. J. van Brummelen
Final approval of manuscript: Emilie M. J. van Brummelen, Anthonius de Boer, Jos H. Beijnen, Jan H. M. Schellens
Disclosures
The authors indicated no financial relationships.
Supplementary Information
References
- 1. Allegra CJ, Rumble RB, Hamilton SR et al. Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti‐epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol 2016;34:179–185. [DOI] [PubMed] [Google Scholar]
- 2. Van Cutsem E, Cervantes A, Adam R et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386–1422. [DOI] [PubMed] [Google Scholar]
- 3.Welcome Trust Sanger Institute. Cosmic: Cancer Browser. Available at https://cancer.sanger.ac.uk/cosmic/browse/tissue#sn=large_intestine&ss= all&hn=all&sh=all&in=t&src=tissue&all_data=n. Accessed August 18, 2016.
- 4. De Roock W, Claes B, Bernasconi D et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy‐refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 2010;11:753–762. [DOI] [PubMed] [Google Scholar]
- 5. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003;3:11–22. [DOI] [PubMed] [Google Scholar]
- 6. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015;16:281–298. [DOI] [PubMed] [Google Scholar]
- 7. Douillard JY, Oliner KS, Siena S et al. Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369:1023–1034. [DOI] [PubMed] [Google Scholar]
- 8. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- 9.Stover D. BRAF c. 1781A>G (D594G) mutation in colorectal cancer. My Cancer Genome. Available at 2015.Available at https://www.mycancergenome.org/content/disease/colorectal-cancer/braf/148/. Accessed March 30, 2017.
- 10. Missiaglia E, Jacobs B, D'Ario G et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol 2014;25:1995–2001. [DOI] [PubMed] [Google Scholar]
- 11. Tejpar S, Stintzing S, Ciardiello F et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild‐type metastatic colorectal cancer: Retrospective analyses of the CRYSTAL and FIRE‐3 trials. JAMA Oncol: 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciombor KK, Goldberg RM. Primary tumor sidedness as prognostic and predictive biomarker in metastatic colorectal cancer. JAMA Oncol 2016. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 13. Bokemeyer C, Bondarenko I, Makhson A et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first‐line treatment of metastatic colorectal cancer. J Clin Oncol 2009;27:663–671. [DOI] [PubMed] [Google Scholar]
- 14.Bristol‐Myers Squibb. Product Label Erbitux (cetuxiumab). 2009. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125084s0228lbl.pdf. Accessed August 4, 2016.
- 15.Amgen Inc. Product Label Vectibix (panitumumab). 2006. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125147s080lbl.pdf. Accessed August 4, 2016.
- 16. Saif MW. Colorectal cancer in review: The role of the EGFR pathway. Expert Opin Investig Drugs 2010;19:357–369. [DOI] [PubMed] [Google Scholar]
- 17. Normanno N, De Luca A, Bianco C et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006;366:2–16. [DOI] [PubMed] [Google Scholar]
- 18.European Medicines Agency. Erbitux (cetuximab) Eur. Public Assess. Rep. 2015. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000558/WC500029119.pdf. Accessed March 30, 2017.
- 19.European Medicines Agency. Vectibix (panitumumab) Eur. Public Assess. Rep. 2015. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000741/WC500047710.pdf. Accessed March 30, 2017.
- 20. Armaghany T, Wilson JD, Chu Q et al. Genetic alterations in colorectal cancer. Gastrointest Cancer Res 2012;5:19–27. [PMC free article] [PubMed] [Google Scholar]
- 21. Allegra CJ, Jessup JM, Somerfield MR et al. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti‐epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091–2096. [DOI] [PubMed] [Google Scholar]
- 22. Van Cutsem E, Lenz HJ, Köhne CH et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015;33:692–700. [DOI] [PubMed] [Google Scholar]
- 23. Sorich MJ, Wiese MD, Rowland A et al. Extended RAS mutations and anti‐EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta‐analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21. [DOI] [PubMed] [Google Scholar]
- 24. Stintzing S, Modest DP, Rossius L et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE‐3): A post‐hoc analysis of tumour dynamics in the final RAS wild‐type subgroup of this randomised open‐label phase 3 trial. Lancet Oncol 2016;17:1426–1434. [DOI] [PubMed] [Google Scholar]
- 25. Bokemeyer C, Köhne CH, Ciardiello F et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer 2015;51:1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peeters M, Oliner KS, Price TJ et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second‐line treatment for metastatic colorectal cancer. Clin Cancer Res 2015;21:5469–5479. [DOI] [PubMed] [Google Scholar]
- 27. Schwartzberg LS, Rivera F, Karthaus M et al. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild‐type KRAS exon 2 metastatic colorectal. J Clin Oncol 2014;32:2240–2247. [DOI] [PubMed] [Google Scholar]
- 28. Barras D. BRAF mutation in colorectal cancer: An update. Biomark Cancer 2015;7:9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Therkildsen C, Bergmann TK, Henrichsen‐Schnack T et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti‐EGFR treatment in metastatic colorectal cancer: A systematic review and meta‐analysis. Acta Oncol 2014;53:852–864. [DOI] [PubMed] [Google Scholar]
- 30. Rowland A, Dias MM, Wiese MD et al. Meta‐analysis of BRAF mutation as a predictive biomarker of benefit from anti‐EGFR monoclonal antibody therapy for RAS wild‐type metastatic colorectal cancer. Br J Cancer 2015;112:1888–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pietrantonio F, Petrelli F, Coinu A et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta‐analysis. Eur J Cancer 2015;51:587–594. [DOI] [PubMed] [Google Scholar]
- 32. Yuan ZX, Wang XY, Qin QY et al. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti‐EGFR monoclonal antibodies: A meta‐analysis. PLoS One 2013;8:e65995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wan PT, Garnett MJ, Roe SM et al. Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell 2004;116:855–867. [DOI] [PubMed] [Google Scholar]
- 34. Smith CG, Fisher D, Claes B et al. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy ± cetuximab. Clin Cancer Res 2013;19:4104–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tveit KM, Guren T, Glimelius B et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first‐line treatment of metastatic colorectal cancer: The NORDIC‐VII Study. J Clin Oncol 2012;30:1755–1762. [DOI] [PubMed] [Google Scholar]
- 36. Xu Q, Xu AT, Zhu MM et al. Predictive and prognostic roles of BRAF mutation in patients with metastatic colorectal cancer treated with anti‐epidermal growth factor receptor monoclonal antibodies: A meta‐analysis. J Dig Dis 2013;14:409–416. [DOI] [PubMed] [Google Scholar]
- 37. Wang Q, Hu W, Song QB et al. BRAF V600E mutation as a predictive factor of anti‐EGFR monoclonal antibodies therapeutic effects in metastatic colorectal cancer: A meta‐analysis. Chinese Med Sci J 2014;29:197–203. [DOI] [PubMed] [Google Scholar]
- 38. Cui D, Cao D, Yang Y et al. Effect of BRAF V600E mutation on tumor response of anti‐EGFR monoclonal antibodies for first‐line metastatic colorectal cancer treatment: A meta‐analysis of randomized studies. Mol Biol Rep 2014;41:1291–1298. [DOI] [PubMed] [Google Scholar]
- 39. Amado RG, Wolf M, Peeters M et al. Wild‐type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626–1634. [DOI] [PubMed] [Google Scholar]
- 40. Adelstein BA, Dobbins TA, Harris CA et al. A systematic review and meta‐analysis of KRAS status as the determinant of response to anti‐EGFR antibodies and the impact of partner chemotherapy in metastatic colorectal cancer. Eur J Cancer 2011;47:1343–1354. [DOI] [PubMed] [Google Scholar]
- 41. Dahabreh IJ, Terasawa T, Castaldi PJ et al. Systematic review: Anti–epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med 2011;154:37–49. [DOI] [PubMed] [Google Scholar]
- 42. Lin AY, Buckley NS, Lu AT et al. Effect of KRAS mutational status in advanced colorectal cancer on the outcomes of anti‐epidermal growth factor receptor monoclonal antibody therapy: A systematic review and meta‐analysis. Clin Colorectal Cancer 2011;10:63–69. [DOI] [PubMed] [Google Scholar]
- 43. Loupakis F, Cremolini C, Salvatore L et al. Clinical impact of anti‐epidermal growth factor receptor monoclonal antibodies in first‐line treatment of metastatic colorectal cancer: Meta‐analytical estimation and implications for therapeutic strategies. Cancer 2012;118:1523–1532. [DOI] [PubMed] [Google Scholar]
- 44. Petrelli F, Barni S. Resectability and outcome with anti‐EGFR agents in patients with KRAS wild‐type colorectal liver‐limited metastases: A meta‐analysis. Int J Colorectal Dis 2012;27:997–1004. [DOI] [PubMed] [Google Scholar]
- 45. Petrelli F, Borgonovo K, Cabiddu M et al. Cetuximab and panitumumab in KRAS wild‐type colorectal cancer: A meta‐analysis. Int J Colorectal Dis 2011;26:823–833. [DOI] [PubMed] [Google Scholar]
- 46. Qiu LX, Mao C, Zhang J et al. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: A meta‐analysis of 22 studies. Eur J Cancer 2010;46:2781–2787. [DOI] [PubMed] [Google Scholar]
- 47. Vale CL, Tierney JF, Fisher D et al. Does anti‐EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta‐analysis. Cancer Treat Rev 2012;38:618–625. [DOI] [PubMed] [Google Scholar]
- 48. Zhang L, Ma L, Zhou Q. Overall and KRAS‐specific results of combined cetuximab treatment and chemotherapy for metastatic colorectal cancer: A meta‐analysis. Int J Colorectal Dis 2011;26:1025–1033. [DOI] [PubMed] [Google Scholar]
- 49. Ibrahim EM, Abouelkhair KM. Clinical outcome of panitumumab for metastatic colorectal cancer with wild‐type KRAS status: A meta‐analysis of randomized clinical trials. Med Oncol 2011;28:S310–S317. [DOI] [PubMed] [Google Scholar]
- 50. Hoyle M, Crathorne L, Peters J et al. The clinical effectiveness and cost‐effectiveness of cetuximab (mono‐ or combination chemotherapy), bevacizumab (combination with non‐oxaliplatin chemotherapy) and panitumumab (monotherapy) for the treatment of metastatic colorectal cancer after first‐line chemotherapy (review of technology appraisal No.150 and part review of technology appraisal No. 118): A systematic review and economic model. Health Technol Assess 2013;17:1–237. [DOI] [PMC free article] [PubMed]
- 51.Health Quality Ontario. KRAS testing for anti‐EGFR therapy in advanced colorectal cancer: An evidence‐based and economic analysis. Ont Health Technol Assess Ser 2010;10:1–49. [PMC free article] [PubMed]
- 52.OncLive. New Molecular Testing Guidelines Pending for Colorectal Cancer. 2015. Available at http://www.onclive.com/publications/oncology-live/2015/july-2015/new-molecular-testing-guidelines-pending-for-colorectal-cancer. Accessed August 19, 2016.
- 53. Seymour MT, Brown SR, Middleton G et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild‐type, fluorouracil‐resistant advanced colorectal cancer (PICCOLO): A prospectively stratified randomised trial. Lancet Oncol 2013;14:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van Cutsem E, Köhne CH, Láng I et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first‐line treatment for metastatic colorectal cancer: Updated Analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011;29:2011–2019. [DOI] [PubMed] [Google Scholar]
- 55. Peeters M, Oliner KS, Parker A et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 2013;19:1902–1912. [DOI] [PubMed] [Google Scholar]
- 56. Core Team R. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. 2014. Available at http://www.r-project.org/. Accessed March 30, 2017.
- 57. Karapetis CS, Khambata‐Ford S, Jonker DJ et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–1765. [DOI] [PubMed] [Google Scholar]
- 58. Perrone F, Lampis A, Orsenigo M et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 2008;20:84–90. [DOI] [PubMed] [Google Scholar]
- 59. Pegram MD, Borges VF, Ibrahim N et al. Phase I dose escalation pharmacokinetic assessment of intravenous humanized anti‐MUC1 antibody AS1402 in patients with advanced breast cancer. Breast Cancer Res 2009;11:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prahallad A, Sun C, Huang S et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100–103. [DOI] [PubMed] [Google Scholar]
- 61. Yaeger R, Cercek A, O'Reilly EM et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF‐mutant metastatic colorectal cancer patients. Clin Cancer Res 2015;21:1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tabernero J, Van Geel R, Kyrre Guren T et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF‐mutant colorectal cancer (BRAFm CRC) J Clin Oncol 2016;34:3544a. [Google Scholar]
- 63. Atreya CE, van Cutsem E, Bendell JC et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti‐EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J Clin Oncol 2015;33:103a. [Google Scholar]
- 64. Venook A, Niedwedzwiecki D, Innocenti F, et al. Impact of primary tumor location (1°) on overall survival (OS) and progression‐free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34:3504a. [Google Scholar]
- 65. Vecchione L, Gambino V, Raaijmakers J et al. A vulnerability of a subset of colon cancers with potential clinical utility. Cell 2016;165:317–330. [DOI] [PubMed] [Google Scholar]
- 66. van Geel RM, Beijnen JH, Bernards R et al. Treatment individualization in colorectal cancer. Curr Colorectal Cancer Rep 2015;11:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tol J RCPC. Randomized phase III study of capecitabine, oxaliplatin, and bevacizumab with or without cetuximab in advanced colorectal cancer (ACC), the CAIRO2 study of the Dutch Colorectal Cancer Group (DCCG). J Clin Oncol 2008;26:LBA4011a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.