

Abstract
Nitrogenase enzymes are used by microorganisms for converting atmospheric N2 to ammonia, which provides an essential source of N atoms for higher organisms. The active site of the molybdenum-dependent nitrogenase is the unique carbide-containing iron-sulfur cluster called the iron-molybdenum cofactor (FeMoco). On the FeMoco, N2 binding is suggested to occur at one or more iron atoms, but the structures of the catalytic intermediates are not clear. In order to establish the feasibility of different potential mechanistic steps during biological N2 reduction, chemists have prepared iron complexes that mimic various structural aspects of the iron sites in FeMoco. This reductionist approach gives mechanistic insight, and also uncovers fundamental principles that could be used more broadly for small molecule activation. Here, we review recent results and highlight directions for future research. In one direction, synthetic iron complexes have now been shown to bind N2, break the N-N triple bond, and produce ammonia catalytically. Carbon and sulfur based donors have been incorporated into the ligand spheres of Fe-N2 complexes to show how these atoms may influence the structure and reactivity of the FeMoco. Hydrides have been incorporated into synthetic systems, which can bind N2, reduce some nitrogenase substrates, and/or reductively eliminate H2 to generate reduced iron centers. Though some carbide-containing iron clusters are known, none yet have sulfide bridges or high-spin iron atoms like the FeMoco.
Graphical Abstract
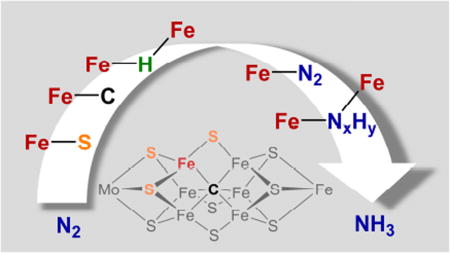
1. INTRODUCTION
Nitrogen is an essential element for all life, and the supply of biologically available nitrogen limits the productivity of many terrestrial and marine ecosystems.1–3 The most plentiful source of nitrogen is N2, but this molecule has low reactivity. Biological fixation of N2 to ammonia,4–8 a bioavailable source of nitrogen, is performed by diazotrophic microorganisms.9–11 These specialized microorganisms can be free-living or in symbiotic relationships with certain plants (e.g. legumes) and animals (e.g. termites).12,13 Before the invention of the Haber-Bosch process for industrial ammonia production, most nitrogen atoms in living organisms originated from these microorganisms.14,15 They have the only type of enzymes that can perform the key N2-reducing reaction, and these are termed nitrogenase enzymes. Elucidation of the mechanism of biological N2 fixation by nitrogenases is a great challenge in bioinorganic chemistry. Chemists aim to explain the mechanism of N2 reduction in nature, and are also excited about the opportunity to harness the power of nitrogenase for other applications.
N2 reduction by nitrogenases occurs at metal clusters, with the most well-characterized being the iron-molybdenum cofactor (FeMoco) (Fig. 1).6,16,19–21 FeMoco contains seven Fe and one Mo that are connected by bridging sulfides and arranged around a central carbide. The biosynthesis of FeMoco from two Fe4S4 clusters, an additional S atom, and a C atom initially forms an Fe8S9C carbide cluster as an intermediate.22,23 Substitution of an apical Fe for Mo/homocitrate is followed by the transfer of the cluster to the apo form of the catalytic MoFe protein.
Figure 1.

Structure of the resting state FeMoco from high-resolution X-ray crystallography.16 The oxidation states of the iron atoms shown here are from X-ray absorption and anomalous dispersion studies.17,18 The belt iron sites are colored red.
Alternative nitrogenases use V or Fe in place of Mo, and are less efficient at N2 fixation than the Mo-dependent enzyme.24–26 X-ray structures of the alternative nitrogenases are not yet available, but biochemical and genetic studies indicate that the structures of their active site cofactors are similar to the FeMoco.24,25,27 Spectroscopic studies further support the idea that the iron-vanadium cofactor (FeVco) contains an interstitial carbide like the FeMoco.28,29 The shared cluster type in various nitrogenases, and studies on the activity and spectroscopy of variants of the FeMo enzyme that derive from point mutations,30–33 indicate that the belt iron sites (indicated in red in Fig. 1) are the site of N2 binding and reduction.6,32,34
Well-defined synthetic “model” systems are often used to provide insight into the feasibility of different mechanisms and structures within enzymes.35,36 However, the chemistry of iron complexes in a coordination environment like the Fe sites in the FeMoco was unknown until a number of recent advances. Here we discuss the implications of recent progress in synthetic iron complexes supported by ligand scaffolds that coordinate to iron through carbon and/or sulfur, as well as iron hydrides that are relevant to the mechanism of N2 reduction.6 Reviews discussing N2 reduction by Mo systems, by Fe systems with P and N ligands, and by other metals are available elsewhere.37–44
2. COORDINATION CHEMISTRY OF FEMOCO
Recent attention in the nitrogenase modeling community has focused on the belt Fe sites (see Fig. 1), each of which has pseudotetrahedral 3S-1C coordination in the resting state. X-ray crystal structures are known for nitrogenases from both Azotobacter vinelandii and Clostridium pasterianum to resolutions of better than 1.1 Å, and the atomic positions in the FeMoco in each were determined to high precision (~0.02 Å).16,45 The Fe-S bond lengths are all the same within ± 0.03 Å, suggesting that none of them are protonated. The FeMoco is mixed-valent in the resting state with both Fe2+ and Fe3+ centers, which couple to give an S = 3/2 ground state.6,7 A combination of X-ray absorption spectroscopy and computations show that the oxidation states are Fe2+3Fe3+4Mo3+ in the resting state.17,46,47 Spatially-resolved anomalous dispersion refinement of the structure recently confirmed this picture.18 These data suggest the oxidation state assignments indicated in Figure 1, and the three sites with higher electron density are on the side of the cofactor that abuts two positively charged arginine residues. Thus, even though the FeMoco has near three-fold symmetry in the core bonding, there is asymmetry imposed by the local polarity in the protein.
Though this detailed characterization of the resting state geometric and electronic structure is a major achievement, the binding of substrates occurs only after conversion of the FeMoco to other, more reduced forms that have not been structurally characterized.6 The cofactor is connected to the protein through only two linkages, and there are many precedents for structural rearrangements in synthetic iron-sulfur clusters,48,49 and therefore the Fe-C-S cluster structure is likely to rearrange during the catalytic cycle. Binding of N2 to Fe sites is implicated most strongly (see above), so most chemists have considered that breaking or elongation of the bonds to belt Fe atoms could accompany or precede substrate binding. Consequently, N2 binding proposals include either elongation/dissociation of an Fe-C bond (e.g. 1, Fig. 2),50–53 or Fe-S bond cleavage with protonation of a sulfide (e.g. 2, Fig. 2).54–58 Supporting 1, extended X-ray absorption fine structure (EXAFS) and nuclear resonance vibrational spectroscopy (NRVS) studies indicate the lengthening of Fe-C bond in FeMoco upon binding of propargyl alcohol.59 In Fe-phosphine model complexes (described below) Fe-C bond elongation has been observed upon reduction and N2 binding.60,61 Supporting 2, Fe–S bond cleavage in FeMoco is experimentally supported by the observation of sulfide replacement by CO in a structure of CO-treated nitrogenase,62 by Fe–S cleavage in smaller Fe–S clusters,63,64 and by observation of Fe-S cleavage with N2 binding in a Fe model complex.65 Another plausible geometry for N2 binding is endo coordination, where N2 is positioned close to three additional iron atoms (3, Fig. 2).6,66 Other hypotheses for N2 binding modes include η2 coordination and bridging modes.57,66 End-on binding of N2 has been proposed on the basis of ENDOR studies of species trapped during N2 reduction by nitrogenase, which show coupling to only one type of N environment.67
Figure 2.

Possible FeMoco intermediates during reduction of N2. Additional potential protonation sites and cluster charges are not shown.
N2 reduction by Mo-dependent nitrogenase always releases at least one equivalent of H2 per N2 reduced.68,69 The dependence of the N2 reduction rate on [N2] suggests the limiting stoichiometry in eq 1 (Pi = inorganic phosphate-containing products).7,68,70 Note that this stoichiometric reaction is highly exergonic because of the consumption of so much ATP.71,72 The overall energy efficiency of N2 reduction is low, which reflects the challenging kinetic problems for performing this multi-step reaction under ambient conditions.
Mechanistic data on Mo-nitrogenase are commonly rationalized using the Thorneley–Lowe kinetic scheme, which was derived from global fitting of data from kinetic studies on the enzymatic reaction.70,73–76 In this scheme, intermediates are named En, where n represents the number of electrons added to the resting state. Importantly, the kinetic data indicate that N2 binding does not take place until 3–4 electrons have been added. A series of EPR/cryoannealing studies confirm this model, and specifically show that E4 can reversibly bind N2 to form a spectroscopically observed intermediate.77,78 Figure 3 shows a version of the Thorneley-Lowe scheme with binding of N2 at the E4 state.
Figure 3.

Thorneley-Lowe scheme for N2 reduction.
One frustrating aspect of nitrogenase research is that none of the redox potentials for En transformations are known.79 The electrons are supplied by the “Fe protein” which has an iron-sulfur cluster whose potential is influenced by the binding of ATP, ADP, and the catalytic protein.80 The Fe protein is the only reductant known that leads to N2 reduction, and addition of the Fe protein in the absence of substrates inevitably leads to production of H2, rendering the electrochemistry irreversible.7 However, the use of the same re-ductant for all eight electrons is an important constraint, implying that there is not excessive charge buildup on the cofactor.6,81,82 This feature has traditionally been explained by assuming that one proton is added per electron, as shown in Fig. 3. This is an example of proton-coupled electron transfer (PCET), where the negative charge of each electron is balanced by the positive charge on a proton.83,84
ENDOR studies suggest that the E4 state (presumably protonated to give E4H4) has two bridging hydrides, implying that there are two additional protons that reside on the sulfide bridges.85–91 One potential structure for E4H4 that satisfies the experimental constraints is shown as 4 in Figure 2.88 Interestingly, the 4H+/4e− can be lost from E4 as two molecules of H2 to regenerate the resting state, even in frozen solution.87,90 The fact that H2 is a competitive inhibitor of N2 reduction by the enzyme, that D2 can be incorporated into certain substrates,7,78 and that higher N2 pressures lead to higher concentrations of E4H2N2,77 support the idea that reversible H2 loss is an integral part of binding of N2 by nitrogenase.6,70,77,78 Although this reductive elimination of H2 “wastes” two reducing equivalents, it might be the only way under biological constraints to create a reduced iron site that can binds N2 strongly.55,77 Fryzuk and Quadrelli have reviewed the ability of hydrides to store electrons for N2 activation without formal reduction of the metal.81,82 Interestingly, the reductive elimination of H2 does not occur until N2 binding, as shown by the fact that H2/D2 exchange only occurs in the presence of N2.7
After N2 binding, multistep delivery of electrons and protons to N2 could give a range of potential intermediates. Two mechanistic pathways, distal and alternating,34 are commonly invoked as possibilities (Fig. 4), and are distinguished by the location of protons on the N2 unit and by the timing of ammonia release.6 An alternating pathway for N2 reduction on iron is supported by computational, spectroscopic, and trapping studies on the enzyme.34,92–97 The alternating mechanism implicates hydrazine (H2N–NH2) and diazene (HN=NH) complexes as intermediates, consistent with the observations that these two molecules are substrates for nitrogenase.93,95–97 Though Fig. 4 shows intermediates with only one iron center, many of the NxHy species along the alternating pathway could be bridging (see below), so more than one iron center could well be involved in the reduction. In addition, hybrid mechanisms could involve intermediates from both alternating and distal pathways, by addition of protons or transfer of H atoms in intermediates.61,98,99
Figure 4.

Potential intermediates in N2 reduction on iron. All steps except NH3 release include input of H+ and e−.
Nitrogenases are capable of reducing a variety of other unsaturated compounds including CO and CO2.100 Vanadium-dependent nitrogenase is especially efficient for reductive coupling of CO to make C-C bonds,101,102 a process related to Fischer-Tropsch synthesis.103 Even isolated cofactors display the ability to form C-C bonds from CO.104 The coordination chemistry of FeMoco with CO and CO2 is relevant to understanding the mechanism behind these potentially useful processes and aids the study of CO inhibition of the enzyme, and has been discussed in detail elsewhere.62,105
3. SYNTHETIC IRON-SULFUR CLUSTERS
Our description of synthetic compounds will start with the most obvious feature of FeMoco: that it is an iron-sulfur (Fe-S) cluster. Fe-S clusters are found in a range of different kinds of enzymes and electron-transfer proteins.106,107 In order to understand their properties, Fe-S clusters have been synthesized, often through assembly from simple starting materials.48,49,108 Artificial clusters of many sizes and topologies are now accessible, and heterometals can be incorporated into clusters. However, no Fe-S cluster with a carbide has yet been synthesized by chemists.
Ohki, Tatsumi, and coworkers reported several examples of iron-sulfur clusters that have a topology reminiscent of FeMoco but with a central sulfide (Fig. 5).109–111 Fe8S7 cluster 6 was formed from the reaction of coordinatively unsaturated dinuclear Fe2+ complex 5 with S8 in toluene.109 In this remarkable self-assembly process, some Fe2+ centers in the starting material are oxidized to give a Fe2+5Fe3+3 cluster. The Fe centers in 6 are arranged in two Fe4S3 units that are connected by three thiolate bridges and a central µ6-sulfide. A Mo2Fe6S9 cluster which has two cuboidal MoFe3(µ3-S)3 units joined via a central µ6-S atom was reported by Holm.112 Selectively joining MoFe3S3 and Fe4S3 subclusters in structures similar to FeMoco remains a challenge. It is likely that comparison of symmetrical and unsymmetrical clusters would provide insight into the role of Mo in the FeMoco.
Figure 5.

Structures of synthetic iron-sulfur clusters. Dmp = 2,6-(mesityl)2C6H3. Ar = 2,4,6-iPr3C6H2.
A related Fe8-sulfur cluster (8) that contains an oxygen atom as the central atom has been synthesized by the reaction of the coordinatively unsaturated dinuclear iron(II) thiolate/alkoxide complex 7 with water and sulfur in toluene (Fig. 5).111 The oxygen atom in this cluster of five Fe2+ and three Fe3+ forms a bridge between two Fe4S3 fragments. One of the exciting aspects of this cluster is the presence of two coordinatively unsaturated Fe atoms that are weakly bound to mesityl rings. Though this cluster does not react with 1 atm of N2, its reduced forms have not yet been reported and might have heightened N2 reactivity.
Clusters related to 6 but having one of the bridging thiolates replaced with an alkoxide or an amide are also known.109,111 Although they do not contain Mo, the synthesis of clusters in Fig. 5 demonstrates the stability of cluster structures similar to FeMoco. They are further relevant to the proposed structure of the cofactor in all- iron nitrogenase, and to the Fe8S9C cluster that is formed during biosynthesis of FeMoco.22,23
Despite remarkable achievements in the synthesis of complex Fe-S clusters, N2 binding to an iron-sulfide cluster has never been reported.48,49,113–115 Recently, a synthetic iron-sulfide cluster was combined with cofactor-deficient nitrogenase, which formed an artificial enzyme that reduced acetylene, but not N2.116 Pairing of synthetic and biological components has substantial promise as a strategy for identifying the essential components of the nitrogenase mechanism.
4. Fe-N2 COMPLEXES WITH SULFUR LIGANDS
It is well-known that the σ interaction of N2 with metals is weak, and that π-backbonding from filled metal d orbitals into the π* orbitals of N2 is most influential.38 Iron-N2 complexes have been isolated in a number of systems where the iron atom has the +2 oxidation state or lower.39,40 However, only a few synthetic complexes are known that have both sulfur and N2 ligands on the same iron center (Fig. 6).117 Since these resemble feasible structures of crucial N2-bound species in nitrogenase catalytic cycle, they have been important topics of inquiry.
Figure 6.

Fe-N2 complexes with thioether ligands. BArF4− = tetrakis(3,5-trifluoromethylphenyl)borate.
The Peters group reported a series of thioether ligated Fe-N2 complexes with tetradentate ligands that are conceptually related to their previous tris(phosphine) complexes.118 A ligand with one thioether and two phosphine donors yielded the cationic paramagnetic (S = 1) complex 10 (Fig. 6). The N2 ligand in 10 is lost under reduced pressure, and this lability is consistent with its high N-N stretching frequency of 2156 cm−1. Reduction of 10 to with potassium/graphite (KC8), Na(Hg), or CoCp2 resulted in N2 loss, cleavage of the S-C(alkyl) bond, bridging S, and/or thioether dissociation. These undesired reactions illustrate common difficulties that arise during attempts to stabilize N2 complexes using sulfur-based ligands.
When a ligand with two thioethers and one phosphine arm was used, the resulting iron(II) complex did not bind N2. However, partial reduction of this complex with 0.5 equiv of Cr(C6H6)2 resulted in the mixed valent species 11 and 12, which have bridging N2 (Fig. 6).118 Compounds 10–12 have N2 bound to Fe that is ligated by sulfur donors, and their distorted trigonal-bipyramidal geometries model the potential binding mode 1 in FeMoco (Fig. 2).
Interestingly, a mononuclear Fe-N2 species 13 with two thioether donors can be obtained by introduction of a hydride ligand at the iron center (Fig. 6).118 Since metal-N2 interactions are so dependent on backbonding, it is reasonable that the more electron rich Fe in 13 is better able to bind N2. A similar iron-N2 hydride complex 14 was obtained using a ligand with a single thioether donor. Thus, it appears that strong-field hydride ligands can facilitate N2 binding to Fe, corresponding to one possible role for hydrides in the FeMoco.119
Thiolate donors, which are negatively charged, are better suited to model the anionic sulfides in FeMoco. Peters recently described the first example of a thiolate-iron-N2 species, with Fe1+Fe1+, Fe1+Fe2+, and Fe2+Fe2+ in binuclear thiolate bridged iron complexes that are additionally coordinated by phosphine and silyl donors (15, Fig. 7).120 The anionic bis(N2) species produces 1.8 ± 0.3 equiv of ammonia when treated with excess KC8 and [H(OEt2)2]BArF4, and also can catalyze the disproportionation of N2H4 to NH3 and N2. Furthermore, the same ligand scaffold enables formation of Fe1+Fe2+ and Fe2+Fe2+ iron-hydride species having an Fe-N2 fragment (16, Fig. 7).
Figure 7.

N2 binding to iron using a thiolate donor.
In order to create an environment consisting only of donors present in the FeMoco, the bis(thiolate) ligand platform in 17 was introduced (Fig. 8).65 Reduction of tris(thiolate) iron(II) complex 17 to a formal iron(0) oxidation state at low temperature resulted in the loss of one thiolate, and formation of a terminal N2 complex 18 that features partial coordination of the central arene with Fe-C distances of 2.04 and 2.24 Å (Fig. 8). The iron in 18 thus has close coordination of two S and one C, as in the proposed N2 binding mode 2 for FeMoco (Fig. 2). Breaking an Fe-S bond concomitant with N2 binding during the formation of 18 suggests that breaking an Fe–S bond is a chemically reasonable route to N2 binding in FeMoco. Furthermore, 18 has a quite activated N2 ligand, as evidenced by N–N stretching frequency of 1880 cm−1. This shows that thiolates give electron rich, high-spin iron centers that backbond well into the π* orbital of N2, though N2 is lost readily from 18 at room temperature.
Figure 8.

N2 binding to an iron–sulfur–carbon site. Ar = 2,4,6-iPr3C6H2.
Future work is necessary to advance the chemistry of biomimetic complexes with S donors. First, it will be advantageous in the future to incorporate carbides and additional iron atoms in a complex with a S-dominated ligand sphere. In addition, use of sulfides rather than thiolates is a challenge that will require careful cluster design. Ideally, it will be possible to design S/C ligand spheres that enable direct comparison of hypotheses 1 and 2 (Fig. 2 above).
5. CARBIDE COMPLEXES, AND Fe-N2 COMPLEXES WITH CARBON LIGANDS
The carbide in the FeMoco comes from the methyl group of the sulfonium ion S-adenosylmethionine, which is thought to be transferred to a cluster sulfur, and subsequently subjected to abstraction of H atoms.121–123 The carbide is neither exchanged nor lost from the cofactor during catalysis.124 The fact that there is cellular machinery devoted to the synthesis of this unusual cage structure implies that the carbide may play a functional role in N2 reduction. Some possibilities for this functional role include enforcement of an appropriate geometry in the active site, stabilization of the cluster (note that carbides are present in steel125),124 transient formation/cleavage of Fe-C bonds,50–53 and reversible formation of C-H bonds. Recent computational studies propose that the carbide could even engage in covalent bonding with the N atoms during the N2 reduction.126,127
Synthetic iron carbide clusters are also known, though in these cases the carbides come from very different sources.128–144 In a six-iron example, the Fe6 cluster [Me4N]2[Fe6C(CO)16] (19, Fig. 9a) was studied in the early 1970’s as a model of possible intermediates in the iron-catalyzed Fischer-Tropsch process.129,130 In collaboration with DeBeer, we recently reported more extensive crystallographic characterization of 19, as well as X-ray emission studies that elucidated the electronic structure of the iron-carbide core and demonstrated the delocalized orbitals that give rise to Fe-C bond-ing.145 Oxidation of 19 leads to related Fe5 and Fe4 carbide/carbonyl clusters.134,142 One transformation with particular relevance to the FeMoco mechanism is that [Fe4C(CO)12]2− (21, Fig. 9b) can be oxidized in the presence of H2 to give neutral HFe4(CH)(CO)12 (22).134,136,139,146 This product can also be synthesized through double protonation of the cluster.134,140 The oxidative addition of H2 is the microscopic reverse of reductive elimination from a hydride-iron cluster, and suggests that reversible formation of C-H bonds in the FeMoco should be considered as a possibility.
Figure 9.

Structures and reactivity of synthetic iron-carbide complexes.
It should be borne in mind that there are several differences between these synthetic carbide clusters and the FeMoco. First, none of these carbide clusters have the trigonal prismatic geometry of the FeMoco. Second, the π-acidic CO ligands in the synthetic iron-carbide clusters stabilize Fe1+ and Fe0 oxidation levels, as opposed to Fe3+ and Fe2+ centers that are present in the enzyme. Finally, the strong-field CO ligands in the synthetic clusters make them diamagnetic, which contrasts to the sulfide-supported, high-spin iron sites in the enzyme.47 Thus, the behavior of these CO-supported clusters may not be representative of the FeMoco environment. The synthesis of iron-carbide clusters in weak-field environments (preferably with S donors) is needed in order to predict the chemical behavior of Fe atoms in the FeMoco.
In the above carbonyl clusters, the carbide is derived from reduction of a coordinated CO, and thus it is probable that new synthetic strategies will be needed for non-carbonyl clusters. Several µ-carbido complexes with iron are known, although their chemistry has not been explored extensively.147–153 The diamagnetic tetra-phenylporphyrinato complex 20 with a linear Fe=C=Fe bridge was theoretically predicted and synthesized in the early 1980’s (Fig. 9a).147,148,154 The carbide in 20 originates from CI4 in a one-step reaction, suggesting that this route might be applicable to other carbides in non-carbonyl environments.
Because of the difficulty in designing appropriate clusters, simpler carbon-based donors have been used to make mononuclear complexes. One compelling choice of C-donor is the N-heterocyclic carbene (NHC), which can form stable complexes with iron.155 However, NHC-stabilized Fe-N2 complexes are rare.156 Peters reported that a two-coordinate (CAAC)2Fe complex 23 [CAAC = cyclic (alkyl)(amino)carbene] binds N2 at low temperature to form a putative three-coordinate N2 complex 24 (Fig. 10).157 It can be reduced to Fe(-I) in the heterobimetallic bridging N2 complex 25, which has a low N-N stretching frequency of 1850 cm−1. Three-coordinate Fe-N2 complexes are rare and have previously been observed using bulky β-diketiminate ligands.158–160 Treatment of 25 with silyl chlorides functionalizes N2 to give iron silyldiazenido complexes 26. Catalytic reduction of N2 to ammonia can be achieved using KC8 and [H(OEt2)2]BArF4 catalyzed by 23 at −95 °C, with yields of up to 3.3 ± 1.1 equivalents of NH3 per iron.157 Thus, the electron-rich CAAC ligands and the low coordination number are particularly effective for N2 activation. Other Fe-N2 complexes supported by chelating NHC ligand frameworks are also known (e.g. 27a–c, Fig. 11), but have not been studied as extensively.161–164
Figure 10.

N2 binding and functionalization using CAAC ligands. Ar = 2,6-iPr2C6H3. R = Me, Et.
Figure 11.

N2 binding to Fe using NHC ligands.
Alkyl ligands have also been used as carbide mimics. In a systematic set of studies, Peters described trigonal pyramidal complexes of a tris(phosphine) platform with different alkyl ligands placed in one axial position (Fig. 12). First, the Fe1+ complex 28 was reduced to give an Fe0 complex with N2 binding trans to the carbon ligand (29, Fig. 12a).60 Upon reduction and N2 binding, the Fe–C distance in 28 (2.153(2) Å) elongated to 2.254(5) Å. Treatment of the complex with a silyl triflate at low temperature affords four-coordinate Fe diazenido complex 30, with Fe–C = 2.116(1) Å. These show the great flexibility of the Fe–C bond, supporting model 1 for N2 binding in FeMoco (Fig. 2).
Figure 12.

Fe-N2 complexes with trans carbon and N2 ligands.
A related system with aryl linkers gave N2 complexes having three different oxidation states (31, Fig. 12b).61 Fe-C bond lengthening from 2.081(3) Å in the cationic to 2.165(2) Å in the anionic complex again showed the flexibility of the Fe-C bond, and the anionic complex is capable of catalytic ammonia production (47 equiv/Fe).165
Ohki and coworkers reported a coordinatively unsaturated iron complex 32, from solutions of which diiron-N2 complex 33 crystalizes under N2 atmosphere (Fig. 13).166 Complex 33 contains a Fe-N2 moiety in a solely carbon-based ligand environment, with Cp*, NHC, and an alkyl donor. The N2 in 33 is held weakly, as indicated by reversion to 32 upon dissolving, and by a high N-N stretching frequency of 2126 cm−1.
Figure 13.

N2 binding to Fe using multiple carbon-based ligands.
Further examples of C-ligated Fe-N2 complexes include bis(imino)pyridine stabilized complexes with alkyl and aryl ligands167–169 and also bis-cyclometalated CPC and CNC pincer supported complexes,170,171 as well as N2 complexes stabilized by η6-interaction with arenes.171–173 In the long run, all of these complexes will add to our knowledge about the influence of donor power and geometry on N2 activation by iron.
6. Fe-H COMPLEXES WITH SULFUR AND N2 LIGANDS
As described above, the E4H4 state of nitrogenase has been shown by ENDOR spectroscopy to have two H atoms with direct bonds to iron (Fe-hydrides).86–89 In principle, these hydrides could be either terminal (Fe-H) or bridging (Fe-H-Fe), and ab initio studies of a simplified model have evaluated the relative energies of these different binding modes.174 The ENDOR spectra of E4H4 show an intrinsic T tensor (coupling between the unpaired spin and the 1H spin) that is rhombic.6,86 Analogous ENDOR experiments on isolable Fe-H compounds with terminal hydrides showed an axial tensor, while bridging hydrides gave a rhombic tensor.6,175,176 This combination of experiments validated the idea that that the signals from the FeMoco-hydride (“E4”) are most consistent with bridging hydrides in E4H4.6 However, the active form might have transient terminal hydrides, as is currently believed for hydrogenase enzymes based on synthetic modeling studies.177
Most isolated iron-hydride species have low-spin electronic configurations, whereas the weak-field environment of the iron atoms in the FeMoco is electronically different. This motivates the preparation of new iron hydrides in weak-field ligand coordination environments, particularly those having sulfur-based ligands. We developed low-coordinate Fe(µ-H)2Fe dimers that have weak-field lig-and environments, and these react with nitrogenase-relevant substrates such as alkynes, CO2, cyanide, and azide (but only react with N2 upon irradiation).178–180 Murray recently reported Fe3(µ-H)3 clusters stabilized by a trinucleating β-diketiminate scaffold, which react with CO2 but not other nitrogenase substrates.181 However, none of the above Fe-hydride species contained S donors.
Iron-hydride-thiolate complexes without CO or P ligands are ra-re.182,183 Qu and coworkers recently reported diiron hydrides in a sulfur and carbon rich environment, which are bridged by bis(thiolate) and stabilized by Cp* ligands (34 and 35, Fig. 14).183
Figure 14.

Bridging iron hydrides in sulfur- and carbon-rich environment.
A recent report from our group introduced the sulfide and hydride bridged diiron complex 36 (Fig. 15a), which models a potential coordination mode for hydrides in FeMoco as depicted in 4 (Fig. 2 above).182 Hydride complex 36 reacted with CO2 to give bridging formate complex 37, which is relevant to CO2 reduction by nitrogenase.184–186 Complex 16b from above,120 which has a terminal hydride (Fig. 15b), also reduces CO2 to give a thiolate bridged formate (38). Therefore, both bridging and terminal hydrides appear to be active in S-supported Fe-H complexes, at least for CO2 reduction.
Figure 15.

Iron hydrides with sulfur ligands that reduce CO2.
Formation of N2 complexes within a iron-sulfide-hydride framework is particularly relevant to the proposed reductive elimination of two hydrides from E4H4. Reduction of 36 by 2 e− under N2 resulted in the formation of a diiron(0)–N2 complex.182 Liberation of H2 is observed, suggesting a parallel to the reductive elimination in the Thorneley-Lowe scheme. However, 36 has only one hydride and therefore cannot reductively eliminate H2 in an intramolecular reaction. In addition, the low yields of the N2 complex and H2 (~25%) makes the significance of H2 evolution in this system uncertain.
Studies of N2 binding to Fe complexes with multiple hydrides are more relevant to N2 binding from 4 (Fig. 2). Though well-precedented with other metals,81,82 such studies are rare with Fe complexes.187–189 The Peters group recently reported the mixed-valent Fe2+(µ-H)2Fe1+ complex 40, which displays 106-fold enhancement of N2 binding affinity over Fe2+(µ-H)2Fe2+ species 39 (Fig. 16).119 Treatment of 39 with excess KC8 and [(Et2O)2H]-BArF4 resulted in production of 1.4 ± 0.5 mol equiv NH3. This is the first example of bridging hydrides in an Fe-N2 complex, which is relevant to N2 binding by E4H4. The authors suggest a model in which the role of hydrides may be to increase the ligand field at iron, stabilizing a low-spin electronic configuration that is preferred for N2 binding. However, many iron-N2 complexes with highly activated N2 ligands have been observed in high-spin complexes of iron, so the need for low-spin Fe is not clear.65,158–160,190 Overall, more studies are needed, particularly on S-supported systems, to draw firmer conclusions about the dependence of N2 binding and H2 elimination on the spin state of iron.
Figure 16.

Binding of N2 in the presence of bridging hydrides.
7. Fe-NxHy COMPLEXES WITH SULFUR LIGANDS
N2 binding and reduction is now well established with iron complexes in P and N ligand environments. Recent highlights include cleavage of N2 to two nitrides (41) and catalytic systems for conversion of N2 to ammonia by mononuclear model complexes, the most successful being 42 (Fig. 17).61,157,191,192,41,44,165 Peters has shown that phosphine ligands provide excellent platforms for functionalization of N2 on iron,60,193,194 and enable characterization of potential N2 reduction intermediates Fe-NxHy,98,99,165,194–196 including systems that feature a coordinated hydride.40,42,43 Ammonia production by tripodal trisphosphine ligands with an axial donor (Si, C, or B) increases in the order Si < C < B.165 The higher degree of Fe-heteroatom bond flexibility in 42 could be crucial for stabilizing the variety of N2 reduction intermediates in the catalytic cycle.61,192,193,197 In a stoichiometric study using the B-based ligand, an Fe≡N–NH2 intermediate was characterized that models the distal mechanism for N2 reduction (Fig. 4).98 Subsequent study with a Si-based ligand postulated a pathway for N2 reduction that includes Fe=N–NH2 and Fe-NH2NH2 species.99 However, sulfur-supported complexes are needed to more closely resemble the iron environment in the FeMoco.
Figure 17.

Recent examples of N2 activation on iron with N- and P-based supporting ligands.
Sellmann and coworkers characterized hydrazine, diazene, and NH3 complexes using multidentate thioether/thiolate ligands.198,199 The trans-N2H2 ligand in complex 43 is bound to two iron centers and stabilized by hydrogen bonding interactions with S-ligands, a scenario that is feasible in FeMoco (Fig. 18). However, no N2 complex was accessible in this system. More generally, no reported system with S donors has yet been observed to bind N2 and to bind partially reduced NxHy species. Additionally, none of these S-based systems has been reported to perform N2 reduction, though some are capable of catalytic reduction of hydrazine.200,201
Figure 18.

Fe-NxHy species in sulfur-rich environments. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
In a notable contribution, Qu and coworkers used a thiolate-bridged diiron system that binds cis-diazene (44), and this complex can be protonated to give N2H3 species 45 (Fig. 18).58 Upon treatment with 2 equivalents of a reducing reagent and protonation, 45 is transformed into the bridging amide complex 46. These reactions are relevant because transformation of N2 to ammonia might be mediated by at least two neighboring Fe atoms through a series of NxHy bridged intermediates.66 This Cp*/thiolate supported system is capable of hydrazine reduction as well.58
Binding of nitrogenase-relevant NxHy intermediates to Fe complexes with sulfides, rather than thiolates, is rare. Holland and coworkers reported a diiron sulfide species that is coordinatively unsaturated, to which N2H4 and a substituted hydrazido ligand can bridge in a µ-η1:η1 fashion (47, Fig. 18).202,203 Complexes of substituted hydrazines have greater stability in this system,203 and one mixed-valence system was studied using ENDOR for comparison of hyperfine parameters to nitrogenase intermediates.204 However, this diiron sulfide system has not yet been observed to bind N2 without loss of sulfide. More model complexes are needed that have the combination of sulfur rich ligand environments, N2 binding, and the ability to stabilize various Fe-NxHy intermediates.
8. CONCLUSION AND OUTLOOK
At the turn of the century, synthetic modeling of nitrogenase was based on molybdenum, because of its demonstrated ability to reduce N2 to ammonia.37 However, emerging spectroscopy, crystallography, and biosynthesis of the enzyme has suggested iron binding of N2 instead. The broader acceptance of the iron hypothesis has crucially depended on advances in the understanding of synthetic iron complexes. Most notably, it has been shown that iron species are capable of reducing N2 to NH3, including several cases with catalytic reduction of N2 noted above. Equally influential has been the realization that other hypothetical features of FeMoco intermediates, such as high-spin iron-hydrides, iron-diazenes and iron-hydrazines are feasible structures in the presence of sulfides and other sulfur-based donors. Spectroscopic studies on these species provide signatures for use in identifying intermediates, and their reactivity shows what reactions are reasonable to expect on the FeMoco.
Many of the initial advances in N2 reduction have been enabled by N and P based ligands, even though FeMoco iron sites use S and C donors. Thus, the next stage in nitrogenase modeling is to incorporate S and C donors into the model iron complexes. Although preparation of an exact model of the FeMoco would be an amazing synthetic achievement, perfect structural fidelity is not needed for deep insight. For example, analogous systems with and without S and C ligands enable us to understand the influence of these ligand types on the FeMoco reactivity and spectroscopy. Further, there is a need for systems that allow observation of the elementary steps that link NxHy species. These use proton-coupled electron transfer (PCET),83,205 which has not yet been studied in the context of nitrogen reduction. The addition of single electrons and protons as part of the Thorneley-Lowe scheme suggests that PCET is crucial for nitrogenase catalysis, as noted above.6,7,24 There have only been a few studies on PCET for any iron-sulfur clusters.64,206–208 Studies on PCET to N2, N2H2, N2H4, and other fragments of relevance to N2 reduction are needed,99,209 because such reactions are involved in the part of the nitrogenase mechanism spanning the E4 to E8 levels. We also note that systems that engage in PCET often involve hydrogen bonds,210 but to our knowledge there are not yet any examples of hydrogen bonding to an N2 unit in a transition-metal complex.
Another priority is the understanding of reversible H2 loss with N2 binding. Reductive elimination of H2 simultaneously generates an open site and two reducing equivalents on the metal, both of which contribute to N2 binding ability.81,82 Interestingly, the available evidence on nitrogenase indicates that H2 loss, though reversible, occurs only upon addition of N2. This observation suggests that the enzyme might generate an iron-H2 complex that undergoes an associative ligand exchange reaction to bind N2. This idea was also recently suggested by the kinetics of H2 loss upon photolyzing nitrogenase.88 The reductive elimination of H2 provides an ideal way for the FeMoco to store reducing equivalents while leaving the iron sites in reasonable oxidation states. Though two electrons are “wasted” when H2 is lost, the need for nitrogen fixation makes this a worthwhile bargain of thermodynamics for kinetics. (In vivo, it is also likely that the “lost” H2 can be reabsorbed for recovery of some of the energy.) Synthetic iron compounds that exchange H2 and N2 have been studied in phosphine-containing systems,211,212 and should be extended to sulfur-containing environments. Also, a biomimetic strategy of H2 loss may enable chemists to forego the use of very strong acids and reducing agents during the catalytic reduction of N2.165
This Perspective has highlighted these and other areas in which synthetic chemistry will continue to enrich our understanding of the fascinating process of reducing N2.
Acknowledgments
Nitrogen research in the Holland lab has been generously supported by the National Institutes of Health (GM065313).
Footnotes
The authors declare no competing financial interest.
References
- 1.Vitousek PM, Howarth RW. Biogeochemistry. 1991;13:87. [Google Scholar]
- 2.Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, Schlesinger WH, Tilman DG. Ecol. Applic. 1997;7:737. [Google Scholar]
- 3.Falkowski PG. Nature. 1997;387:272. [Google Scholar]
- 4.Leigh GJ. The World’s Greatest Fix: A History of Nitrogen and Agriculture. Oxford University Press; New York: 2004. [Google Scholar]
- 5.Holland PL. In: Comprehensive Coordination Chemistry II. McCleverty J, Meyer TJ, editors. Vol. 8. Elsevier; Oxford: 2004. p. 569. [Google Scholar]
- 6.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC. Chem. Rev. 2014;114:4041. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgess BK, Lowe DJ. Chem. Rev. 1996;96:2983. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 8.Ribbe MW, editor. Nitrogen Fixation. Humana Press; New York: 2011. [Google Scholar]
- 9.Zehr JP, Jenkins BD, Short SM, Steward GF. Environ. Microbiol. 2003;5:539. doi: 10.1046/j.1462-2920.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- 10.Gaby JC, Buckley DH. Environ. Microbiol. 2011;13:1790. doi: 10.1111/j.1462-2920.2011.02488.x. [DOI] [PubMed] [Google Scholar]
- 11.Boyd E, Peters JW. Front. Microbiol. 2013;4 doi: 10.3389/fmicb.2013.00201. Article 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpenter EJ, Capone DG. Chapter 4 - Nitrogen Fixation in the Marine Environment. In: Capone DG, Bronk DA, Mulholland MR, Carpenter EJ, editors. Nitrogen in the Marine Environment (2nd Edition) Academic Press; San Diego: 2008. p. 141. [Google Scholar]
- 13.Ohkuma M, Noda S, Kudo T. Appl. Environ. Microbiol. 1999;65:4926. doi: 10.1128/aem.65.11.4926-4934.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galloway JN, Dentener FJ, Capone DG, Boyer EW, Howarth RW, Seitzinger SP, Asner GP, Cleveland CC, Green PA, Holland EA, Karl DM, Michaels AF, Porter JH, Townsend AR, Vöosmarty CJ. Biogeochemistry. 2004;70:153. [Google Scholar]
- 15.Galloway JN, Leach AM, Bleeker A, Erisman JW. Phil. Trans. R. Soc. B. 2013;368:20130120. doi: 10.1098/rstb.2013.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjornsson R, Lima FA, Spatzal T, Weyhermuller T, Glatzel P, Bill E, Einsle O, Neese F, DeBeer S. Chem. Sci. 2014;5:3096. [Google Scholar]
- 18.Spatzal T, Schlesier J, Burger E-M, Sippel D, Zhang L, Andrade SLA, Rees DC, Einsle O. Nat. Commun. 2016;7 doi: 10.1038/ncomms10902. Article 10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 21.Einsle O. J. Biol. Inorg. Chem. 2014;19:737. doi: 10.1007/s00775-014-1116-7. [DOI] [PubMed] [Google Scholar]
- 22.Ribbe MW, Hu Y, Hodgson KO, Hedman B. Chem. Rev. 2014;114:4063. doi: 10.1021/cr400463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Ribbe MW. Biochim. Biophys. Acta. 2013;1827:1112. doi: 10.1016/j.bbabio.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eady RR. Chem. Rev. 1996;96:3013. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 25.Masepohl B, Schneider K, Drepper T, Müller A, Klipp W. Alternative Nitrogenases. In: Leigh GJ, editor. Nitrogen Fixation at the Millennium. Elsevier; Amsterdam: 2002. p. 191. [Google Scholar]
- 26.Zhao Y, Bian S-M, Zhou H-N, Huang J-F. J. Integr. Plant Biol. 2006;48:745. [Google Scholar]
- 27.Hu Y, Ribbe MW. J. Biol. Inorg. Chem. 2015;20:435. doi: 10.1007/s00775-014-1225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fay AW, Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. J. Am. Chem. Soc. 2010;132:12612. doi: 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rees JA, Bjornsson R, Schlesier J, Sippel D, Einsle O, DeBeer S. Angew. Chem. Int. Ed. 2015;54:13249. doi: 10.1002/anie.201505930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benton PMC, Laryukhin M, Mayer SM, Hoffman BM, Dean DR, Seefeldt LC. Biochemistry. 2003;42:9102. doi: 10.1021/bi034595x. [DOI] [PubMed] [Google Scholar]
- 31.Seefeldt LC, Dance IG, Dean DR. Biochemistry. 2004;43:1401. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 32.Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DR. Acc. Chem. Res. 2005;38:208. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 33.Sarma R, Barney BM, Keable S, Dean DR, Seefeldt LC, Peters JW. J. Inorg. Biochem. 2010;104:385. doi: 10.1016/j.jinorgbio.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seefeldt LC, Hoffman BM, Dean DR. Annu. Rev. Biochem. 2009;78:701. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holm RH, Solomon EI. Chem. Rev. 2004;104:347. doi: 10.1021/cr0206364. [DOI] [PubMed] [Google Scholar]
- 36.Ibers JA, Holm RH. Science. 1980;209:223. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 37.Schrock RR. Acc. Chem. Res. 2005;38:955. doi: 10.1021/ar0501121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKay BA, Fryzuk MD. Chem. Rev. 2004;104:385. doi: 10.1021/cr020610c. [DOI] [PubMed] [Google Scholar]
- 39.Crossland JL, Tyler DR. Coord. Chem. Rev. 2010;254:1883. [Google Scholar]
- 40.Hazari N. Chem. Soc. Rev. 2010;39:4044. doi: 10.1039/b919680n. [DOI] [PubMed] [Google Scholar]
- 41.MacLeod KC, Holland PL. Nature Chem. 2013;5:559. doi: 10.1038/nchem.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoenkhoen N, de Bruin B, Reek JNH, Dzik WI. Eur. J. Inorg. Chem. 2015;2015:567. [Google Scholar]
- 43.Köthe C, Limberg C. Z. Anorg. Allg. Chem. 2015;641:18. [Google Scholar]
- 44.Nishibayashi Y. Inorg. Chem. 2015;54:9234. doi: 10.1021/acs.inorgchem.5b00881. [DOI] [PubMed] [Google Scholar]
- 45.Zhang L-M, Morrison CN, Kaiser JT, Rees DC. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015;71:274. doi: 10.1107/S1399004714025243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bjornsson R, Neese F, Schrock R, Einsle O, DeBeer S. J. Biol. Inorg. Chem. 2015;20:447. doi: 10.1007/s00775-014-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris TV, Szilagyi RK. Inorg. Chem. 2011;50:4811. doi: 10.1021/ic102446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SC, Holm RH. Chem. Rev. 2004;104:1135. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 49.Lee SC, Lo W, Holm RH. Chem. Rev. 2014;114:3579. doi: 10.1021/cr4004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holland PL. Can. J. Chem. 2005;83:296. [Google Scholar]
- 51.MacBeth CE, Harkins SB, Peters JC. Can. J. Chem. 2005;83:332. [Google Scholar]
- 52.Hinnemann B, Nørskov JK. J. Am. Chem. Soc. 2004;126:3920. doi: 10.1021/ja037792s. [DOI] [PubMed] [Google Scholar]
- 53.Dance I. J. Am. Chem. Soc. 2007;129:1076. doi: 10.1021/ja0644428. [DOI] [PubMed] [Google Scholar]
- 54.Hallmen PP, Kästner J. Z. Anorg. Allg. Chem. 2015;641:118. [Google Scholar]
- 55.Varley JB, Wang Y, Chan K, Studt F, Norskov JK. Phys. Chem. Chem. Phys. 2015;17:29541. doi: 10.1039/c5cp04034e. [DOI] [PubMed] [Google Scholar]
- 56.Kästner J, Blöchl PE. J. Am. Chem. Soc. 2007;129:2998. doi: 10.1021/ja068618h. [DOI] [PubMed] [Google Scholar]
- 57.Schimpl J, Petrilli HM, Blöchl PE. J. Am. Chem. Soc. 2003;125:15772. doi: 10.1021/ja0367997. [DOI] [PubMed] [Google Scholar]
- 58.Li Y, Li Y, Wang B, Luo Y, Yang D, Tong P, Zhao J, Luo L, Zhou Y, Chen S, Cheng F, Qu J. Nature Chem. 2013;5:320. doi: 10.1038/nchem.1594. [DOI] [PubMed] [Google Scholar]
- 59.George SJ, Barney BM, Mitra D, Igarashi RY, Guo Y, Dean DR, Cramer SP, Seefeldt LC. J. Inorg. Biochem. 2012;112:85. doi: 10.1016/j.jinorgbio.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rittle J, Peters JC. Proc. Natl. Acad. Sci. U.S.A. 2013;110:15898. doi: 10.1073/pnas.1310153110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Creutz SE, Peters JC. J. Am. Chem. Soc. 2014;136:1105. doi: 10.1021/ja4114962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC. Science. 2014;345:1620. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alwaaly A, Dance I, Henderson RA. Chem. Commun. 2014;50:4799. doi: 10.1039/c4cc00922c. [DOI] [PubMed] [Google Scholar]
- 64.Saouma CT, Morris WD, Darcy JW, Mayer JM. Chem. Eur. J. 2015;21:9256. doi: 10.1002/chem.201500152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Čorić I, Mercado BQ, Bill E, Vinyard DJ, Holland PL. Nature. 2015;526:96. doi: 10.1038/nature15246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dance I. Dalton Trans. 2012;41:4859. doi: 10.1039/c2dt00049k. [DOI] [PubMed] [Google Scholar]
- 67.Barney BM, Lukoyanov D, Igarashi RY, Laryukhin M, Yang T-C, Dean DR, Hoffman BM, Seefeldt LC. Biochemistry. 2009;48:9094. doi: 10.1021/bi901092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simpson F, Burris R. Science. 1984;224:1095. doi: 10.1126/science.6585956. [DOI] [PubMed] [Google Scholar]
- 69.Rivera-Ortiz JM, Burris RH. J. Bacteriol. 1975;123:537. doi: 10.1128/jb.123.2.537-545.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorneley RNF, Lowe DJ. Met. Ions Biol. 1985;7:221. [Google Scholar]
- 71.Kurnikov IV, Charnley AK, Beratan DN. J. Phys. Chem. B. 2001;105:5359. [Google Scholar]
- 72.Studt F, Tuczek F. Angew. Chem. Int. Ed. 2005;44:5639. doi: 10.1002/anie.200501485. [DOI] [PubMed] [Google Scholar]
- 73.Lowe DJ, Thorneley RNF. Biochem. J. 1984;224:877. doi: 10.1042/bj2240877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thorneley RNF, Lowe DJ. Biochem. J. 1984;224:887. doi: 10.1042/bj2240887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thorneley RNF, Lowe DJ. Biochem. J. 1984;224:895. doi: 10.1042/bj2240895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thorneley RNF, Lowe DJ. Biochem. J. 1984;224:903. doi: 10.1042/bj2240903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lukoyanov D, Yang Z-Y, Khadka N, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2015;137:3610. doi: 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Z-Y, Khadka N, Lukoyanov D, Hoffman BM, Dean DR, Seefeldt LC. Proc. Natl. Acad. Sci. U.S.A. 2013;110:16327. doi: 10.1073/pnas.1315852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.A change in the visible absorption spectrum of FeMoco is seen at −480 mV: Watt GD, Burns A, Lough S, Tennent DL. Biochemistry. 1980;19:4926. doi: 10.1021/bi00562a035..
- 80.Christiansen J, Dean DR, Seefeldt LC. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001;52:269. doi: 10.1146/annurev.arplant.52.1.269. [DOI] [PubMed] [Google Scholar]
- 81.Ballmann J, Munha RF, Fryzuk MD. Chem. Commun. 2010;46:1013. doi: 10.1039/b922853e. [DOI] [PubMed] [Google Scholar]
- 82.Jia H-P, Quadrelli EA. Chem. Soc. Rev. 2014;43:547. doi: 10.1039/c3cs60206k. [DOI] [PubMed] [Google Scholar]
- 83.Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem. Rev. 2012;112:4016. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 85.Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Acc. Chem. Res. 2013;46:587. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2005;127:6231. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 87.Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1451. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lukoyanov D, Khadka N, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2016;138:1320. doi: 10.1021/jacs.5b11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2010;132:2526. doi: 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lukoyanov D, Yang Z-Y, Duval S, Danyal K, Dean DR, Seefeldt LC, Hoffman BM. Inorg. Chem. 2014;53:3688. doi: 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thorneley RNF, Eady RR, Lowe DJ. Nature. 1978;272:557. [Google Scholar]
- 92.Lukoyanov D, Dikanov SA, Yang Z-Y, Barney BM, Samoilova RI, Narasimhulu KV, Dean DR, Seefeldt LC, Hoffman BM. J. Am. Chem. Soc. 2011;133:11655. doi: 10.1021/ja2036018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lukoyanov D, Yang Z-Y, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5583. doi: 10.1073/pnas.1202197109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barney BM, Lukoyanov D, Yang T-C, Dean DR, Hoffman BM, Seefeldt LC. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17113. doi: 10.1073/pnas.0602130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barney BM, McClead J, Lukoyanov D, Laryukhin M, Yang T-C, Dean DR, Hoffman BM, Seefeldt LC. Biochemistry. 2007;46:6784. doi: 10.1021/bi062294s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barney BM, Laryukhin M, Igarashi RY, Lee H-I, Dos Santos PC, Yang T-C, Hoffman BM, Dean DR, Seefeldt LC. Biochemistry. 2005;44:8030. doi: 10.1021/bi0504409. [DOI] [PubMed] [Google Scholar]
- 97.Barney BM, Yang T-C, Igarashi RY, Dos Santos PC, Laryukhin M, Lee H-I, Hoffman BM, Dean DR, Seefeldt LC. J. Am. Chem. Soc. 2005;127:14960. doi: 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- 98.Anderson JS, Cutsail GE, Rittle J, Connor BA, Gunderson WA, Zhang L, Hoffman BM, Peters JC. J. Am. Chem. Soc. 2015;137:7803. doi: 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rittle J, Peters JC. J. Am. Chem. Soc. 2016;138:4243. doi: 10.1021/jacs.6b01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Seefeldt LC, Yang Z-Y, Duval S, Dean DR. Biochim. Biophys. Acta. 2013;1827:1102. doi: 10.1016/j.bbabio.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee CC, Hu Y, Ribbe MW. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rofer-DePoorter CK. Chem. Rev. 1981;81:447. [Google Scholar]
- 104.Lee CC, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2012;51:1947. doi: 10.1002/anie.201108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scott AD, Pelmenschikov V, Guo Y, Yan L, Wang H, George SJ, Dapper CH, Newton WE, Yoda Y, Tanaka Y, Cramer SP. J. Am. Chem. Soc. 2014;136:15942. doi: 10.1021/ja505720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beinert H, Holm RH, Münck E. Science. 1997;277:653. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 107.Rouault TA, editor. Iron-Sulfur Clusters in Chemistry and Biology. Walter de Gruyter; Berlin: 2014. [Google Scholar]
- 108.Ohki Y, Tatsumi K. Z. Anorg. Allg. Chem. 2013;639:1340. [Google Scholar]
- 109.Ohki Y, Ikagawa Y, Tatsumi K. J. Am. Chem. Soc. 2007;129:10457. doi: 10.1021/ja072256b. [DOI] [PubMed] [Google Scholar]
- 110.Hashimoto T, Ohki Y, Tatsumi K. Inorg. Chem. 2010;49:6102. doi: 10.1021/ic100692v. [DOI] [PubMed] [Google Scholar]
- 111.Ohta S, Ohki Y, Hashimoto T, Cramer RE, Tatsumi K. Inorg. Chem. 2012;51:11217. doi: 10.1021/ic301348f. [DOI] [PubMed] [Google Scholar]
- 112.Zhang Y, Zuo J-L, Zhou H-C, Holm RH. J. Am. Chem. Soc. 2002;124:14292. doi: 10.1021/ja0279702. [DOI] [PubMed] [Google Scholar]
- 113.Lee SC, Holm RH. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3595. doi: 10.1073/pnas.0630028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Henderson RA. Binding Substrates to Synthetic Fe–S-Based Clusters and the Possible Relevance to Nitrogenases. In: Weigand W, Schollhammer P, editors. Bioinspired Catalysis. Wiley-VCH Verlag GmbH & Co. KGaA; 2014. p. 289. [Google Scholar]
- 115.Yao W, Gurubasavaraj PM, Holland PL. All-Ferrous Iron– Sulfur Clusters. In: Rabinovich D, editor. Molecular Design in Inorganic Biochemistry. Vol. 160. Springer; Berlin: 2014. p. 1. [Google Scholar]
- 116.Tanifuji K, Lee CC, Ohki Y, Tatsumi K, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2015;54:14022. doi: 10.1002/anie.201507646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bart SC, Lobkovsky E, Bill E, Wieghardt K, Chirik PJ. Inorg. Chem. 2007;46:7055. doi: 10.1021/ic700869h. [DOI] [PubMed] [Google Scholar]
- 118.Takaoka A, Mankad NP, Peters J. C. J. Am. Chem. Soc. 2011;133:8440. doi: 10.1021/ja2020907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rittle J, McCrory CCL, Peters JC. J. Am. Chem. Soc. 2014;136:13853. doi: 10.1021/ja507217v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Creutz SE, Peters J. C. J. Am. Chem. Soc. 2015;137:7310. doi: 10.1021/jacs.5b04738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wiig JA, Hu Y, Lee CC, Ribbe MW. Science. 2012;337:1672. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lancaster KM, Hu Y, Bergmann U, Ribbe MW, DeBeer S. J. Am. Chem. Soc. 2013;135:610. doi: 10.1021/ja309254g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wiig JA, Hu Y, Ribbe MW. Nat. Commun. 2015;6 doi: 10.1038/ncomms9034. Article 8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wiig JA, Lee CC, Hu Y, Ribbe MW. J. Am. Chem. Soc. 2013;135:4982. doi: 10.1021/ja401698d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Godec M, Večko Pirtovšek T, Šetina Batič B, McGuiness P, Burja J, Podgornik B. Sci. Rep. 2015;5:16202. doi: 10.1038/srep16202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McKee ML. J. Phys. Chem. A. 2016;120:754. doi: 10.1021/acs.jpca.5b10384. [DOI] [PubMed] [Google Scholar]
- 127.Rao L, Xu X, Adamo C. ACS Catal. 2016;6:1567. [Google Scholar]
- 128.Braye EH, Dahl LF, Hubel W, Wampler DL. J. Am. Chem. Soc. 1962;84:4633. [Google Scholar]
- 129.Churchill MR, Wormald J, Knight J, Mays MJ. J. Am. Chem. Soc. 1971;93:3073. [Google Scholar]
- 130.Churchill MR, Wormald J. J. Chem. Soc, Dalton Trans. 1974:2410. [Google Scholar]
- 131.Gourdon A, Jeannin Y. Organometallics. 1986;5:2406. [Google Scholar]
- 132.Bogdan PL, Sabat M, Sunshine SA, Woodcock C, Shriver DF. Inorg. Chem. 1988;27:1904. [Google Scholar]
- 133.Rossell O, Seco M, Segalés G, Alvarez S, Pellinghelli MA, Tiripicchio A, de Montauzon D. Organometallics. 1997;16:236. [Google Scholar]
- 134.Tachikawa M, Muetterties EL. J. Am. Chem. Soc. 1980;102:4541. [Google Scholar]
- 135.Tachikawa M, Sievert AC, Muetterties EL, Thompson MR, Day CS, Day VW. J. Am. Chem. Soc. 1980;102:1725. [Google Scholar]
- 136.Beno MA, Williams JM, Tachikawa M, Muetterties EL. J. Am. Chem. Soc. 1981;103:1485. [Google Scholar]
- 137.Boehme RF, Coppens P. Acta Crystallogr., Sect. B. 1981;37:1914. [Google Scholar]
- 138.Bradley JS, Ansell GB, Leonowicz ME, Hill EW. J. Am. Chem. Soc. 1981;103:4968. [Google Scholar]
- 139.Davis JH, Beno MA, Williams JM, Zimmie J, Tachikawa M, Muetterties EL. Proc. Natl. Acad. Sci. U.S.A. 1981;78:668. doi: 10.1073/pnas.78.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Holt EM, Whitmire KH, Shriver DF. J. Organomet. Chem. 1981;213:125. [Google Scholar]
- 141.Wadepohl H, Braga D, Grepioni F. Organometallics. 1995;14:24. [Google Scholar]
- 142.Tachikawa M, Geerts RL, Muetterties EL. J. Organomet. Chem. 1981;213:11. [Google Scholar]
- 143.Muetterties EL. Catal. Rev. Sci. Eng. 1981;23:69. [Google Scholar]
- 144.Zanello P. Structure and Electrochemistry of Transition Metal Carbonyl Clusters with Interstitial or Semi-Interstitial Atoms: Contrast between Nitrides or Phosphides or Carbides. In: Gielen M, Willem R, Wrackmeyer B, editors. Unusual Structures and Physical Properties in Organometallic Chemistry. John Wiley & Sons; Chichester: 2002. [Google Scholar]
- 145.Delgado-Jaime MU, Dible BR, Chiang KP, Brennessel WW, Bergmann U, Holland PL, DeBeer S. Inorg. Chem. 2011;50:10709. doi: 10.1021/ic201173j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Beno MA, Williams JM, Tachikawa M, Muetterties EL. J. Am. Chem. Soc. 1980;102:4542. [Google Scholar]
- 147.Mansuy D, Lecomte JP, Chottard JC, Bartoli JF. Inorg. Chem. 1981;20:3119. [Google Scholar]
- 148.Goedken VL, Deakin MR, Bottomley LA. J. Chem. Soc., Chem. Commun. 1982:607. [Google Scholar]
- 149.Rossi G, Goedken VL, Ercolani C. J. Chem. Soc, Chem. Commun. 1988:46. [Google Scholar]
- 150.Kienast A, Bruhn C, Homborg H. Z. Anorg. Allg. Chem. 1997;623:967. [Google Scholar]
- 151.Kienast A, Galich L, Murray KS, Moubaraki B, Lazarev G, Cashion JD, Homborg HJ. Porphyrins Phthalocyanines. 1997;1:141. [Google Scholar]
- 152.Galich L, Kienast A, Hückstädt H, Homborg H. Z. Anorg. Allg. Chem. 1998;624:1235. [Google Scholar]
- 153.Knauer W, Beck W. Z. Anorg. Allg. Chem. 2008;634:2241. [Google Scholar]
- 154.Tatsumi K, Hoffmann R, Whangbo M-H. J. Chem. Soc., Chem. Commun. 1980:509. [Google Scholar]
- 155.Riener K, Haslinger S, Raba A, Högerl MP, Cokoja M, Herrmann WA, Kühn FE. Chem. Rev. 2014;114:5215. doi: 10.1021/cr4006439. [DOI] [PubMed] [Google Scholar]
- 156.Ohki Y, Seino H. Dalton Trans. 2016;45:874. doi: 10.1039/c5dt04298d. [DOI] [PubMed] [Google Scholar]
- 157.Ung G, Peters JC. Angew. Chem. Int. Ed. 2015;54:532. doi: 10.1002/anie.201409454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J. Am. Chem. Soc. 2001;123:9222. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]
- 159.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J. Am. Chem. Soc. 2006;128:756. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 160.Dugan TR, MacLeod KC, Brennessel WW, Holland PL. Eur. J. Inorg. Chem. 2013;2013:3891. doi: 10.1002/ejic.201300187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Danopoulos AA, Wright JA, Motherwell WB. Chem. Commun. 2005:784. doi: 10.1039/b415562a. [DOI] [PubMed] [Google Scholar]
- 162.Pugh D, Wells NJ, Evans DJ, Danopoulos AA. Dalton Trans. 2009:7189. doi: 10.1039/b907141p. [DOI] [PubMed] [Google Scholar]
- 163.Danopoulos AA, Pugh D, Smith H, Saβmannshausen J. Chem. Eur. J. 2009;15:5491. doi: 10.1002/chem.200900027. [DOI] [PubMed] [Google Scholar]
- 164.Yu RP, Darmon JM, Hoyt JM, Margulieux GW, Turner ZR, Chirik PJ. ACS Catal. 2012;2:1760. doi: 10.1021/cs300358m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Del Castillo TJ, Thompson NB, Peters JC. J. Am. Chem. Soc. 2016 doi: 10.1021/jacs.6b01706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ohki Y, Hatanaka T, Tatsumi K. J. Am. Chem. Soc. 2008;130:17174. doi: 10.1021/ja8063028. [DOI] [PubMed] [Google Scholar]
- 167.Fernández I, Trovitch RJ, Lobkovsky E, Chirik PJ. Organometallics. 2008;27:109. [Google Scholar]
- 168.Scott J, Vidyaratne I, Korobkov I, Gambarotta S, Budzelaar PHM. Inorg. Chem. 2008;47:896. doi: 10.1021/ic701643d. [DOI] [PubMed] [Google Scholar]
- 169.Tondreau AM, Milsmann C, Patrick AD, Hoyt HM, Lobkovsky E, Wieghardt K, Chirik PJ. J. Am. Chem. Soc. 2010;132:15046. doi: 10.1021/ja106575b. [DOI] [PubMed] [Google Scholar]
- 170.Liu N, Li X, Sun H. J. Organomet. Chem. 2011;696:2537. [Google Scholar]
- 171.Bartholomew ER, Volpe EC, Wolczanski PT, Lobkovsky EB, Cundari TR. J. Am. Chem. Soc. 2013;135:3511. doi: 10.1021/ja311021u. [DOI] [PubMed] [Google Scholar]
- 172.Rose RP, Jones C, Schulten C, Aldridge S, Stasch A. Chem. Eur. J. 2008;14:8477. doi: 10.1002/chem.200801071. [DOI] [PubMed] [Google Scholar]
- 173.Sunada Y, Imaoka T, Nagashima H. Organometallics. 2013;32:2112. [Google Scholar]
- 174.Dance I. Biochemistry. 2006;45:6328. doi: 10.1021/bi052217h. [DOI] [PubMed] [Google Scholar]
- 175.Kinney RA, Saouma CT, Peters JC, Hoffman BM. J. Am. Chem. Soc. 2012;134:12637. doi: 10.1021/ja303739g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chiang KP, Scarborough CC, Horitani M, Lees NS, Ding K, Dugan TR, Brennessel WW, Bill E, Hoffman BM, Holland PL. Angew. Chem. Int. Ed. 2012;51:3658. doi: 10.1002/anie.201109204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang W, Nilges MJ, Rauchfuss TB, Stein M. J. Am. Chem. Soc. 2013;135:3633. doi: 10.1021/ja312458f. [DOI] [PubMed] [Google Scholar]
- 178.Smith JM, Lachicotte RJ, Holland PL. J. Am. Chem. Soc. 2003;125:15752. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]
- 179.Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. J. Am. Chem. Soc. 2008;130:6624. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dugan TR, Bill E, MacLeod KC, Brennessel WW, Holland PL. Inorg. Chem. 2014;53:2370. doi: 10.1021/ic4013137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee Y, Anderton KJ, Sloane FT, Ermert DM, Abboud KA, García-Serres R, Murray LJ. J. Am. Chem. Soc. 2015;137:10610. doi: 10.1021/jacs.5b05204. [DOI] [PubMed] [Google Scholar]
- 182.Arnet NA, Dugan TR, Menges FS, Mercado BQ, Brennessel WW, Bill E, Johnson MA, Holland PL. J. Am. Chem. Soc. 2015;137:13220. doi: 10.1021/jacs.5b06841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang D, Li Y, Wang B, Zhao X, Su L, Chen S, Tong P, Luo Y, Qu J. Inorg. Chem. 2015;54:10243. doi: 10.1021/acs.inorgchem.5b01508. [DOI] [PubMed] [Google Scholar]
- 184.Rebelein JG, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2014;53:11543. doi: 10.1002/anie.201406863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yang Z-Y, Moure VR, Dean DR, Seefeldt LC. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19644. doi: 10.1073/pnas.1213159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee CC, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2015;54:1219. doi: 10.1002/anie.201410412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Helleren CA, Henderson RA, Leigh GJ. J. Chem. Soc., Dalton Trans. 1999:1213. [Google Scholar]
- 188.Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ, Rowley AT. J. Chem. Soc., Dalton Trans. 1993:3041. [Google Scholar]
- 189.Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ. J. Organomet. Chem. 1990;391:C41. [Google Scholar]
- 190.Stoian SA, Vela J, Smith JM, Sadique AR, Holland PL, Münck E, Bominaar EL. J. Am. Chem. Soc. 2006;128:10181. doi: 10.1021/ja062051n. [DOI] [PubMed] [Google Scholar]
- 191.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Moret M-E, Peters JC. J. Am. Chem. Soc. 2011;133:18118. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lee Y, Mankad NP, Peters JC. Nature Chem. 2010;2:558. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Saouma CT, Kinney RA, Hoffman BM, Peters JC. Angew. Chem. Int. Ed. 2011;50:3446. doi: 10.1002/anie.201006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Saouma CT, Müller P, Peters JC. J. Am. Chem. Soc. 2009;131:10358. doi: 10.1021/ja903967z. [DOI] [PubMed] [Google Scholar]
- 197.Moret M-E, Peters JC. Angew. Chem. Int. Ed. 2011;50:2063. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sellmann D, Soglowek W, Knoch F, Moll M. Angew. Chem. Int. Ed. EngI. 1989;28:1271. [Google Scholar]
- 199.Sellmann D, Sutter J. Acc. Chem. Res. 1997;30:460. [Google Scholar]
- 200.Chang Y-H, Chan P-M, Tsai Y-F, Lee G-H, Hsu H-F. Inorg. Chem. 2014;53:664. doi: 10.1021/ic402108w. [DOI] [PubMed] [Google Scholar]
- 201.Chen Y, Zhou Y, Chen P, Tao Y, Li Y, Qu J. J. Am. Chem. Soc. 2008;130:15250. doi: 10.1021/ja805025w. [DOI] [PubMed] [Google Scholar]
- 202.Stubbert BD, Vela J, Brennessel WW, Holland PL. Zeitschrift für anorganische und allgemeine Chemie. 2013;639:1351. doi: 10.1002/zaac.201300163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J. Am. Chem. Soc. 2004;126:4522. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]
- 204.Lees NS, McNaughton RL, Gregory WV, Holland PL, Hoffman BM. J. Am. Chem. Soc. 2008;130:546. doi: 10.1021/ja073934x. [DOI] [PubMed] [Google Scholar]
- 205.Huynh MHV, Meyer TJ. Chem. Rev. 2007;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Albers A, Demeshko S, Dechert S, Saouma CT, Mayer JM, Meyer F. J. Am. Chem. Soc. 2014;136:3946. doi: 10.1021/ja412449v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Saouma CT, Pinney MM, Mayer JM. Inorg. Chem. 2014;53:3153. doi: 10.1021/ic403131p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Albers A, Bayer T, Demeshko S, Dechert S, Meyer F. Chem. Eur. J. 2013;19:10101. doi: 10.1002/chem.201301760. [DOI] [PubMed] [Google Scholar]
- 209.Pappas I, Chirik PJ. J. Am. Chem. Soc. 2015;137:3498. doi: 10.1021/jacs.5b01047. [DOI] [PubMed] [Google Scholar]
- 210.Warren JJ, Mayer JM. Biochemistry. 2015;54:1863. doi: 10.1021/acs.biochem.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tyler DR. Z. Anorg. Allg. Chem. 2015;641:31. [Google Scholar]
- 212.Lee Y, Kinney RA, Hoffman BM, Peters JC. J. Am. Chem. Soc. 2011;133:16366. doi: 10.1021/ja207003m. [DOI] [PMC free article] [PubMed] [Google Scholar]