

Abstract
The adrenal cortex is a dynamic tissue responsible for the synthesis of steroid hormones, including mineralocorticoids, glucocorticoids, and androgens in humans. Advances have been made in understanding the role of adrenocortical stem/progenitor cell populations in cortex homeostasis and self-renewal. Recently, large molecular profiling studies of adrenocortical carcinoma (ACC) have given insights into proteins and signaling pathways involved in normal tissue homeostasis that become dysregulated in cancer. These data provide an impetus to examine the cellular pathways implicated in adrenocortical disease and study connections, or lack thereof, between adrenal homeostasis and tumorigenesis, with a particular focus on stem and progenitor cell pathways. In this review, we discuss evidence for stem/progenitor cells in the adrenal cortex, proteins and signaling pathways that may regulate these cells, and the role these proteins play in pathologic and neoplastic conditions. In turn, we also examine common perturbations in adrenocortical tumors (ACT) and how these proteins and pathways may be involved in adrenal homeostasis.
Keywords: Adrenal cortex, Stem cells, Progenitor cells, Adrenocortical adenoma, Adrenal tumor, Adrenocortical carcinoma
1. Introduction
The adrenal glands are bilateral endocrine organs positioned above the kidneys. The highly dynamic gland is composed of an outer mesenchymal capsule, underneath which lies the adrenal cortex, and an inner adrenal medulla. The cortex and medulla are separate tissues that carry out disparate functions: the former is responsible for synthesis and secretion of steroid hormones and the latter is a neuroendocrine tissue that produces catecholamines. In the human adrenal, the cortex can be divided into three different zones: the outer zona glomerulosa (zG) that secretes mineralocorticoids; the zona fasciculata (zF) that produces glucocorticoids; and the innermost zona reticularis (zR) that synthetizes adrenal androgens. In the mouse and rat adrenal, the zR is absent.
Over recent decades, several groups have demonstrated that dysregulation of the signaling pathways involved in organogenesis and homeostasis of the adrenal cortex plays a central role in human adrenocortical disease. Conversely, the elucidation of the molecular pathogenesis of different types of genetic disorders that exhibit adrenocortical manifestations has allowed a better understanding of adrenal cortex homeostasis, building a paradigm that integrates physiologic and pathologic processes. The insulin-like growth factor (IGF), Wnt, hedgehog (HH), and protein kinase A (PKA) signaling pathways play major roles during both embryonic development and homeostasis, and have pathogenic roles in a variety of inherited and acquired adrenocortical disorders. Additionally, telomere protection and maintenance is involved in both adrenocortical function and dysfunction. Genetically engineered animal models specifically built to scrutinize the importance of these pathways have provided further evidence regarding their roles in the long-term maintenance and differentiation of stem and progenitor cell populations in the adrenal cortex (see Fig. 1). More recently, molecular profiling studies of large cohorts of patients with adrenocortical carcinoma (ACC) (Assié et al., 2014; Juhlin et al., 2014; Pinto et al., 2015), particularly the recently published data from the TCGA project in ACC (Zheng et al., 2016), have identified novel molecules and signaling pathways that are commonly perturbed in adrenocortical malignancy. These studies have provided further clues regarding the molecular processes that regulate adrenal growth, differentiation, and self-maintenance and how they become deregulated.
Fig. 1. Homeostatic paracrine and endocrine signaling pathways in the adrenal cortex.
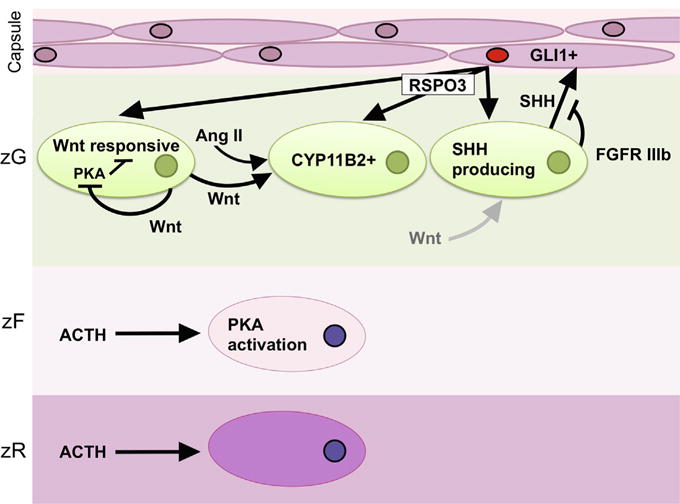
Sonic hedgehog (SHH) ligand is produced by clusters of cells in the zG (SHH producing cells), which serve as a stem/progenitor population for the embryonic and postnatal adrenal cortex. SHH acts on capsular cells, causing GLI1 expression and activation (GLI1+). Signaling downstream of FGFR IIIb in subcapsular zG cells is thought to reduce SHH signaling in the embryonic mouse. GLI1+ cells are also putative stem/progenitors that populate the adrenal cortex embryonically and post-natally in the mouse. Wnt responsive cells are those with active canonical Wnt signaling. WNT4 ligand, an effector and presumptive target of canonical Wnt signaling, acts on zG cells that secrete aldosterone in response to angiotensin II (Ang II) levels. WNT4 expression increases CYP11B2 expression and aldosterone levels. In the absence of WNT4 or in the presence of increased ACTH stimulation, PKA inhibits canonical Wnt signaling. RSPO3 ligand potentiates Wnt signaling, and is necessary for SHH, WNT4, and CYP11B2 expression both embryonically and in the post-natal adrenal. The majority of RSPO3 is produced by capsular GLI1+ cells. Known mechanisms of RSPO3 are mediated by the presence of Wnt ligands, though significant involvement of specific Wnts on SHH-producing cells (indicated by the grey arrow) has not been demonstrated in the adrenal cortex. In the zF, ACTH stimulates PKA activity, which is associated with growth and cortisol production in ACTs.
While a number of prior theories have been put forth regarding the maintenance of adrenocortical growth, recent evidence has defined peripheral capsular and subcapsular cell populations as critical mediators of homeostatic repopulation of the whole adrenal cortex. Pioneering experiments of unilateral rat adrenal enucleation, removing the inner content of the adrenal and leaving behind only the capsule and a layer of cells underneath the capsule, documented complete regeneration of the adrenal cortex. These experiments provide evidence that the stem cell niche is contained in the capsular and/or subcapsular cell compartments (Ingle and Higgins, 1938). Moreover, cell labeling studies support the hypothesis of progressive centripetal displacement of adrenocortical cells throughout life. Cells proliferate in the region under the capsule and are displaced centripetally towards the inner corticomedullary boundary where they undergo apoptosis (Vinson, 2003).
The adrenal capsule has also recently been proposed to serve as a niche for adult adrenocortical stem and progenitor cells located within and/or underneath the capsule (Freedman et al., 2013; King et al., 2009). Several studies by different research groups have indicated that the capsule contains adult stem/progenitor cells nurtured by a population of subcapsular cells that maintain the capsular niche (Huang et al., 2010; Kim et al., 2009; Vidal et al., 2016; Wood and Hammer, 2011) and these subcapsular cells may serve as stem/progenitor cells as well (King et al., 2009). While the available data suggest stem and progenitor characteristics in certain cell populations, the current challenge is still to identify adrenocortical stem cell markers, both for embryonic stem cells involved in the gland’s development, and for adult stem cells, which maintain homeostatic processes.
There is also evidence supporting a hypothesis that repopulation is a zone-specific process. It is well established that zG-specific growth can be stimulated by serum potassium and angiotensin to maintain electrolytic balance, whereas the zF is stimulated by ACTH to produce glucocorticoids. Zone-specific proliferation can be reconciled with centripetal displacement by acknowledgment that there are likely both endocrine mechanisms that rely on expansion of transit amplifying cells and additionally organ-specific homeostatic mechanisms that rely on repopulation by descendants of capsular and subcapsular stem and progenitor cells. These separate mechanisms would be necessary for gland maintenance and allowing a specific zone to expand to respond to increased needs for selective steroidogenesis.
Ongoing research continues to uncover critical pathways involved in adrenocortical homeostasis, hyperplasia, and neoplasia and the connections that can be made between these processes. In particular, discoveries of the underpinnings of the genetic syndromes that predispose patients to adrenocortical adenomas (ACAs) and/or carcinomas, such as Beckwith-Widemann syndrome (BWS) and Carney complex (CNC), have allowed insights to signaling pathways central to both neoplasia and homeostasis (Mussa et al., 2016b; Stratakis et al., 1996). This review will focus on the contribution of cell signaling pathways as it pertains to stem/progenitor biology and neoplasia, especially cancer.
2. SHH signaling
Sonic hedgehog (SHH), together with Indian hedgehog (IHH) and Desert hedgehog (DHH) are secreted ligands of the hedgehog family of morphogenic proteins. The signaling pathway activated by these ligands has been shown to be crucial for stem cell biology and proper organogenesis. Perturbation of any aspect of the system is often detrimental for the organism (Bambakidis and Onwuzulike, 2012). In the adrenal gland, Shh is the only detected ligand. Shh-producing cells are consistently located in the peripheral cortex across species with notable caveats. In the mouse adrenal these cells appear as discrete clusters embedded within the peripheral zG, whereas the rat Shh-expressing cells form a continuous layer called the undifferentiated zone (zU) immediately under the zG cells (Ching and Vilain, 2009; Guasti et al., 2011; Huang et al., 2010; King et al., 2009). Human SHH-expressing cells are organized in sparse subcapsular zG clusters, similar to mice (Boulkroun et al., 2011). Interestingly, adrenocortical Shh-producing cells possess steroidogenic potential as reflected by expression of both Sf1 and Star, yet absence of expression of any of the other enzymes necessary for the terminal reactions of either mineralocorticoid or glucocorticoid production (Laufer et al., 2012).
Shh signaling is activated when the Shh-expressing cells signal to non-steroidogenic cells (Sf1-negative) embedded within the adrenal capsule. Shh ligands bind to the cell-surface receptor patched homolog 1 (Ptc1) on these Shh-responsive cells. Shh binding relieves Ptch1-mediated inhibition of the signal transducer smoothened homolog (Smo), thereby activating downstream signaling through the zinc finger glioma-associated oncogene family (Gli) of transcription factors. The Gli family is composed of Gli1, Gli2, and Gli3, and when activated, promotes the transcription of Hh target genes (Ching and Vilain, 2009; Finco et al., 2015; Huang et al., 2010; King et al., 2009).
The role of Shh pathway in adrenal gland development and homeostasis has recently been investigated. Capsular Gli1-expressing cells are descendants of fetal adrenal cells (Wood et al., 2013). During adrenal development, the Gli1-expressing cells give rise to the undifferentiated Shh-expressing cells of the cortex and their descendent steroidogenic cells of the three cortical zones (King et al., 2009). However, there is a marked decrease in the contribution of Gli1+ cells to the homeostatic cortical replenishment of the adult gland. Instead, the Shh-expressing cells become the main contributors to adult cortical cell maintenance. Lineage tracing studies that mark the cells expressing Shh and their descendants reveal that while Shh-expressing cells are restricted to the outer cortex, these cells and their descendants form clonal, radial stripes that descend into the cortex over time, populating both the zG and the zF (King et al., 2009). These studies indicate a potential role for the Shh-expressing cells as a stem/progenitor population of the adrenal cortex. Indeed, global or adrenal-specific loss of Shh results in smaller adrenal primordium and hypoplastic adrenal cortices, respectively (Ching and Vilain, 2009; Huang et al., 2010; King et al., 2009). Moreover, defects in HH signaling have also been implicated in human diseases. Pallister-Hall syndrome (Hall et al., 1980), a congenital syndrome caused by truncation mutations of GLI3, is associated with varying degrees of adrenal failure (Jamsheer et al., 2012). Although recently debated (Laufer et al., 2012), adrenal agenesis has also been reported in a mouse model of Pallister-Hall syndrome carrying a Gli3 mutation (Böse et al., 2002) providing further support for a major role of Shh signaling in adrenal stem and progenitor cell biology and in homeostatic adrenocortical maintenance.
SHH signaling plays a key role in tumor biology in many organs and tissues, including skin (Li et al., 2011), prostate (Gonnissen et al., 2013), lung (Bermudez et al., 2013), pancreas (Thayer et al., 2003), and bladder (Shigemura and Fujisawa, 2015). While molecular data from the TCGA ACC cohort (https://gdc-portal.nci.nih.gov/projects/TCGA-ACC) do not support a global role of SHH signaling pathway in ACC, increased expression of several HH-associated genes has been observed in a subset of samples with ZNRF3 loss-of-function mutations (see Wnt section, below) (AM Lerario. unpublished observation). An additional study by Gomes and colleagues (Gomes et al., 2014) has observed increased mRNA levels of PTCH1, SMO, GLI3 and SUFU (a negative regulator of the Hh pathway) in a cohort of 13 ACCs, compared to normal adrenal glands. Moreover, treatment of H295R human ACC cells with the SMO inhibitor cyclopamine inhibits cell proliferation, (Werminghaus et al., 2014); furthermore, cyclopamine treatment of H295A human ACC cells diminishes viability and leads to a decrease in β-catenin expression and nuclear localization (Gomes et al., 2014). Together these data support a crosstalk between Hh- and Wnt pathways that may contribute to adrenocortical homeostasis and/or cancer.
Analyses focused on the molecular phenotype of aldosterone producing adrenocortical adenomas (APAs) reveal dramatically increased SHH expression in hyperplastic peritumoral zG cells. Moreover, hierarchical clustering and principal component analysis in APAs compared to control adrenal glands demonstrates an increased expression of genes involved in SHH signaling and well-described transcriptional targets of the SHH pathway (Boulkroun et al., 2011). Whether these expression patterns contribute to increased cell proliferation or are etiologic for APA formation remains unclear.
The absence of a strong SHH signaling signature in ACCs does not preclude, a priori, an involvement of SHH in tumor initiation and progression, as elegantly shown in muscle-invasive bladder cancer. Although precursor lesions originate from Shh-expressing basal stem cells (Shin et al., 2014), Shh expression is not detectable in the resultant tumor. We therefore speculate that certain molecular features (perhaps SHH and/or GLI activity) that contribute to the genetic signature of the cell of origin of an ACC are lost in the process of malignant transformation.
3. Wnt/β-catenin signaling
Canonical Wnt signaling is extensively implicated in homeostatic adrenocortical function, and deregulation is involved in both adrenal adenomas and cancer. Canonical Wnt signaling, also referred to as Wnt/β-catenin signaling, is characterized by β-catenin stabilization downstream of Wnt ligand binding to Frizzled cell surface receptors. Subsequently, β-catenin is translocated into the nucleus and co-activates transcriptional targets (for reviews see Clevers and Nusse, 2012; Kahn, 2014). Other types of Wnt signaling are broadly referred to as non-canonical, a term that encompasses different Wnt-dependent, β-catenin-independent signaling cascades, which includes the planar cell polarity and the Wnt-calcium pathways. In the absence of Wnt ligands, β-catenin nuclear localization and subsequent transcriptional activity is inhibited by the destruction complex, which includes AXIN, APC, GSK3β, and CK1 proteins. The destruction complex binds and phosphorylates free cytoplasmic β-catenin, signaling for ubiquitination and degradation. Upon the binding of a Wnt ligand to the Frizzled receptor and a LRP5/6 co-receptor, the destruction complex is inactivated, allowing cytoplasmic accumulation of β-catenin and nuclear translocation. In the nucleus, β-catenin forms complexes with TCF/LEF transcription factors and activates target gene transcription.
The Wnt signaling pathway is notable for its complex regulatory mechanisms. In addition to the availability of ligands, there are several other layers of regulation, including different types of secreted frizzled receptor inhibitors and autocrine regulation of Wnt ligands. More recently, an important regulatory mechanism mediated by the ZNRF3 and RNF43 proteins was described (Hao et al., 2012; Koo et al., 2012). These E3 ubiquitin ligases inactivate the Frizzled receptors by promoting their internalization. However, in the presence of R-spondins (RSPOs), a family of secreted proteins, this inhibitory mechanism is suppressed. RSPOs bind to leucinerich repeat containing G protein-coupled receptors 4, 5 and 6 (LGR4/5/6), forming a complex that sequesters ZNRF3 and RNF43, ultimately allowing the persistence of Frizzled receptors at the plasma membrane (Zebisch et al., 2013).
A connection between Wnt pathway deregulation and adrenal disease was first established based on observations made on familial adenomatous polyposis (FAP) families. This autosomal dominant inherited disorder is caused by inactivating mutations in APC, a gene that encodes a structural protein of the destruction complex. These patients develop several adenomatous colonic polyps that progress to cancer by age 35–40 (Jasperson and Burt, 1993). Among several other less frequent tumor types, around 13% of these patients develop adrenal masses (Smith et al., 2000), suggesting a role for canonical Wnt signaling in adrenocortical tumorigenesis. In fact, later studies have shown that although APC inactivating mutations are rare in sporadic adrenocortical tumors (ACTs), CTNNB1 (β-catenin) activating mutations are fairly common among both ACAs and ACCs (Assié et al., 2014; Giordano et al., 2009; Pinto et al., 2015; Ronchi et al., 2016; Zheng et al., 2016). Notably, activating mutations in CTNNB1 alter protein phosphorylation, preventing subsequent degradation, and potentiating nuclear transcriptional activity. Previous studies have shown that activating CTNNB1 mutations (affecting the phosphorylation residues in exon 3) occur in over 20% of both ACA and ACC (Heaton et al., 2012; Pinto et al., 2015; Tissier et al., 2005; Zheng et al., 2016). More recently, recurring homozygous deletions or inactivating mutations in ZNRF3 were identified in approximately 20% of ACCs by three different studies (Assié et al., 2014; Juhlin et al., 2014; Zheng et al., 2016). Interestingly, ZNRF3 alterations are mutually exclusive with CTNNB1 and APC mutations, consistent with an activating effect of ZNRF3 loss on canonical Wnt signaling. Taken together, mutations affecting Wnt pathway components (ZNRF3, CTNNB1, and APC), occur in approximately 40% of ACCs, and represent the most frequently altered pathway (Zheng et al., 2016). The landscape of activating Wnt/β-catenin mutations in pediatric ACT also reveals frequent mutations in β-catenin, occurring in approximately 20% of tumors. ZNRF3 deletions and mutations, while observed in adult ACC, are not seen in pediatric ACC (Pinto et al., 2015).
Canonical Wnt/β-catenin signaling has been linked to abnormal growth not only in adrenal malignancy but also in cases of ACAs. A study of APAs shows Wnt/β-catenin signaling activation charac-terized by a lack of activating β-catenin mutations but down-regulation of SFRP2, a negative Wnt regulator (Berthon et al., 2014). Though Wnt/β-catenin signaling has been linked to aldosterone production in APAs and adrenal homeostasis, most ACAs with Wnt/β-catenin pathway mutations are non-functioning (Berthon et al., 2014; Tissier et al., 2005). Activating β-catenin mutations were also found in macronodules (nodules >1 cm) but not micronodules (nodules <1 cm) formed in patients with primary pigmented nodular adrenocortical disease (PPNAD), suggesting that elevated Wnt/β-catenin signaling participates primarily in the increased growth and not the increased steroidogenesis in this disorder (Gaujoux et al., 2008; Tadjine et al., 2008).
The effects of constitutively active β-catenin expression in adrenocortical cells have been investigated both in cell lines and in mouse models. H295R human ACC cells harbor an activating β-catenin mutation. Knockdown of β-catenin in these cells causes cell cycle arrest and increased cell death in vitro, and complete tumor regression in vivo (Gaujoux et al., 2013; Salomon et al., 2015). Small molecule antagonism of the TCF/β-catenin complex also results in H295R growth inhibition (Doghman et al., 2008). In mice with loss of adrenal Apc, β-catenin is stabilized and Wnt signaling is over-activated in the adrenal cortex. At 6 weeks, these mice exhibit adrenal cortical hyperplasia, with some developing microscopic and macroscopic adenomas after 45 weeks. Interestingly, the largest macroscopic adrenal adenoma showed 10-fold higher Igf2 expression compared to controls (Heaton et al., 2012). A mouse model ectopically expressing β-catenin in the mouse adrenal cortex resulted in increased expression of aldosterone and ectopic expression of Cyp11b2 (zG functional marker) (Berthon et al., 2010), supporting that β-catenin promotes aldosterone production. Interestingly, most tumors at a later timepoint did not overproduce aldosterone, unlike tumors at earlier stages (Berthon et al., 2010). Additionally, these mice developed adrenal hyperplasia, dysplasia, progressive loss of zF expression marker Akr1b7, and increasingly disorganized vascularization. At later ages, adrenals from two mice with constitutively active β-catenin presented with features of carcinoma, including local invasion. The data indicate a causative role for β-catenin in adrenal tumorigenesis, though the broad range of phenotypes seen supports a need for additional mutations or epigenetic alterations for carcinoma development.
The prevalence and importance of Wnt pathway alterations in adrenal diseases suggest a prominent role of this pathway in development and homeostasis. Several lines of evidence, including in vitro experiments and transgenic animal models corroborate this hypothesis. Homeostatic adrenal stem/progenitor cell maintenance, proliferation, adrenal zonation, and steroidogenesis are all regulated by canonical Wnt/β-catenin signaling. Nuclear TCF/LEF activity, indicative of activated canonical Wnt/β-catenin signaling, is present in patches of the zG at three weeks postnatally in the mouse (Kim et al., 2008), and at six weeks canonical Wnt appears to be activated in many murine zG cells (Walczak et al., 2014). In agreement with mouse studies, β-catenin histochemistry in the human zG reveals some cells with nuclear localization, indicating there are likely select cells with active canonical signaling (Shaikh et al., 2015).
Disruption of canonical Wnt/β-catenin signaling in Sf1-expressing cells reveals that Wnt signaling is critical for cortical maintenance in both embryonic and adult mouse adrenals. In mouse adrenal development, when β-catenin expression is lost in all steroidogenic cells, adrenals become aplastic are entirely absent by embryonic day (E) 18.5 (Kim et al., 2008). β-catenin inactivation in a subset of steroidogenic cells in the adult mouse adrenal cortex causes decreased cortical proliferation, and progressive cortical thinning and disorganization (Kim et al., 2008). Consistent with histological adrenal failure, ACTH levels are also elevated in these mice with β-catenin loss. The adult adrenal phenotype suggests that partial loss of β-catenin-expressing cells depletes adrenal stem cells, causing progressive failure of adrenal maintenance. In support of this hypothesis, cells with TCF/LEF dependent transcriptional activity, i.e. ‘Wnt-responsive cells,’ include most hedgehog-producing cells and zG cells (steroidogenic and non-steroidogenic) in the mouse (Walczak et al., 2014). While Wnt-responsive cells only form 30% of proliferating cells in the adrenal cortex (Walczak et al., 2014), studies in transgenic adrenals expressing constitutively active β-catenin indicate canonical Wnt signaling can have both cell-autonomous and non-cell-autonomous effects on adrenocortical proliferation (Berthon et al., 2010).
The Wnt4 ligand appears to be particularly involved in maintaining homeostasis and canonical Wnt signaling in the adrenal, and is produced by cells located in the subcapsular/zG region in mice (Heikkila et al., 2002; Vidal et al., 2016). Homozygous loss of Wnt4 in mice results in reduced postnatal adrenal weight, reduced zG area, and reduced aldosterone secretion at birth (Heikkila et al., 2002; Vidal et al., 2016). This suggests that Wnt4 in particular plays a role in zG development and steroidogenesis. Though Wnt4 has been identified as a non-canonical Wnt ligand in other tissues (De, 2011; Sugimura and Li, 2010), in mice with homozygous loss, cortical expression of canonical Wnt target genes Axin2 and Lef1 are decreased, indicating that Wnt4 contributes to canonical Wnt signaling in the mouse adrenal cortex (Drelon et al., 2016; Heikkila et al., 2002). In human patients, a WNT4 homozygous loss-of-function mutation presents with severe developmental defects including adrenal hypoplasia (Mandel et al., 2008). The marked increase in severity of the human WNT4 mutation compared to that in mice, which exhibit no striking difference in volume or morphology of the adrenal glands at birth (Heikkila et al., 2002), suggests that mouse models may be inadequate to reflect the actions of specific human Wnt ligands.
In addition to promoting steroidogenesis, recent evidence indicates that canonical Wnt signaling mediates both establishment of zonation and maintenance of the capsular stem/progenitor cell niche in the adrenal cortex. LGR5 is a key receptor that amplifies Wnt signaling and is highly expressed in both human and mouse zG (as compared to the zF) (Shaikh et al., 2015; Vidal et al., 2016). Rspo3 and Rspo1, ligands known to activate Lgr5, are expressed by capsular cells in the adult mouse (Vidal et al., 2016). Rspo3 appears to be especially crucial: it is expressed in Gli1-expressing stem/progenitor cells in the mouse adrenal capsule from E14.5 onwards, and knockout affects both development of the adrenal cortex and the adult adrenal. Rspo1 knockout on the other hand causes no obvious adrenal phenotype in the mouse.
In the embryonic mouse adrenal, Rspo3 loss causes loss of proliferation, reduction in steroidogenic (Sf1+) cells and loss of Axin2, Wnt4, and Cyp11b2 expression, indicating + necessity for canonical Wnt signaling and aldosterone steroidogenesis. Shh and Gli1 expression are also lost in the embryonic adrenal. In the adult mouse, Rspo3 loss causes reduced adrenal mass, particularly of the zG and capsule, and loss of Shh, Wnt4, and Axin2 expression. These findings establish that Rspo3 expression is key for prenatal and postnatal tissue maintenance and zonation, specifically zG differentiation. Because Rspo3 was found to control Shh signaling, these effects are likely mediated at least in part by capsular stem/progenitor cells characterized by Hh signaling. The authors postulate the effects following loss of Rspo3 lead to a lack of steroidogenic differentiation of stem/progenitor cells, causing a reduced adrenal cortex. While both Rspo3 and Shh loss lead to capsular thinning, the authors note that Shh −/− mice do not lose zG differentiation or zonation despite developing hypoplastic adrenals with lower postnatal corticosterone levels and increased ACTH levels (Huang et al., 2010). This supports that Gli1+ adrenal stem/progenitor cells are controlled by Wnt signaling, and that Wnt signaling has further effects on the zG. The effects of reduced Rspo3 on the subcapsular population of putative stem/progenitor cells in this model has yet to be investigated.
Contradictory results published by Shaikh et al. report that RSPO3 treatment of H295R human ACC cells and human adrenal primary cells resulted in a reduction of aldosterone secretion, as did LGR5 expression. These differences may be due to dissimilarities between humans and mice or may be consequence of in vitro conditions versus in vivo conditions. LGR5 expression in H295R cells also activated AP1/Jun signaling, however this pathway did not affect aldosterone levels (Shaikh et al., 2015), indicating that LGR5 may be involved in non-canonical Wnt signaling in addition to canonical Wnt signaling in the adrenal cortex.
4. FGF signaling
Fibroblast growth factors (FGFs) are a family of eighteen ligands that signal through four FGF receptors (FGFRs) and are involved in development, homeostasis, repair processes, and cancer (Carter et al., 2015). The effects of FGF signaling are specific to cell context and can occur through the activation of several pathways, such as Ras/MAPK, which is linked to cell proliferation and differentiation, PI3K–AKT, an anti-apoptotic pathway, and PKC, which controls cell migration (reivewed by Turner and Grose, 2010). Studies in mice reveal that FGF signaling is essential for adrenal development; knockout of Fgfr2 in mice results in embryonic adrenal hypoplasia or agenesis (Guasti et al., 2013; Kim et al., 2007). Additionally, FGF signaling is known to interact with Hh signaling in various contexts (Mao et al., 2009; Naski et al., 1998; Zhang et al., 2009) and studies by Guasti et al. support that FGF signaling through the Fgfr2 IIIb isoform may negatively regulate Shh signaling, as evidenced by an increase in Gli1+ cells in Fgfr2 IIIb knockout adrenals at E15.5 (Guasti et al., 2013). Fgfr2 IIIb is expressed by subcapsular cells in the mouse (Guasti et al., 2013), further supporting that cells expressing this receptor may overlap with Shh-producing cells, which maintain the stem/progenitor niche in the capsule (Huang et al., 2010) and also serve as stem/progenitor cells (King et al., 2009).
Embryonically, loss of Fgfr2 IIIb causes reduced cortical proliferation (Guasti et al., 2013) and additional studies support that FGF signaling is involved in adrenal growth in adulthood. Fgf2 stimulates increased proliferation in preparations of adult rat adrenal capsule and subcapsular cells following unilateral adrenalectomy, suggesting that Fgf2 mediates compensatory adrenal growth. Authors also show that Fgf2 is localized in a gradient with strongest staining in the zG, and minimal capsule expression (Basile and Holzwarth, 1993). FGF is a potent adrenal mitogen and has been found to increase adrenocortical cell migration as well (Gospodarowicz et al., 1977; Gospodarowicz and Handley, 1975). Transplants of primary zF cells show increased angiogenesis and decreased necrosis when they are cotransplanted with NIH3T3 cells that secrete FGF, as compared to transplants without NIH3T3 cells (Thomas et al., 1997; Thomas and Hornsby, 1999). Altogether, these studies support that FGFs play a variety of important roles in adrenal homeostasis and maintenance and suggest a role in stem/progenitor biology.
FGFs and their receptors have also been implicated in ACC biology. While analyses of the TCGA ACC cohort have not revealed enrichment of FGFs or FGFRs in an ACC subgroup, microarray and other expression profiling have revealed FGFR1 and FGFR4 enrichment in ACCs compared to ACAs or normal tissue (Brito et al., 2012; de Fraipont et al., 2005; Laurell et al., 2009). Additionally, FGFR4 has been reported to be increased 21-fold in pediatric adrenocortical tumors (ACTs) compared to normal adrenal cortex tissues (West et al., 2007). Causality of FGF signaling in adrenocortical tumorigenesis has not been investigated.
5. cAMP/PKA signaling
The cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling pathway is pivotal for adrenocortical growth and maintenance, and it is essential for glucocorticoid and androgen production. Mutations in PKA signaling proteins and regulators have been discovered in both ACAs and ACCs and are the subject of ongoing research. Activation of the cAMP/PKA pathway occurs after the binding of adrenocorticotropic hormone (ACTH) to the melanocortin 2 receptor (MC2R), a 7-transmembrane G-protein coupled receptor (GPCR) that activates Gsα protein to promote the generation of cAMP from ATP following adenylate cyclase stimulation. cAMP binds to the two regulatory (R) subunits of the heterotetrameric enzyme PKA, freeing the two catalytic (C) subunits of PKA. Active PKA phosphorylates CREB (cAMP response element binding protein) and other target substrates responsible for the activation of the transcription of cAMP-responsive genes, including many of the steroidogenic enzymes that define the differentiated state of an adrenocortical cell. The level of intracellular cAMP is also regulated by phosphodiesterases (PDEs) such as PDE8B and PDE11A, that hydrolyze and thereby decrease the cAMP concentration (Boikos and Stratakis, 2007). Four regulatory (RIα, RIβ, RIIα, RIIβ) and four catalytic (Cα, Cβ, Cγ, Prkx) PKA subunit isoforms have been described (Taylor et al., 2013).
Mutations in the cAMP-PKA signaling pathway play etiologic roles in several endocrine tumors and syndromes. These pathologies indicate that increased PKA activation stimulates growth and glucocorticoid production in the adrenal cortex. Somatic mutations in the Guanine Nucleotide Binding Protein Alpha Stimulating 1 (GNAS1) gene, which encodes for the stimulatory G protein subunit Gsα, result in constitutive stimulation of adenylate cyclase and subsequent PKA activation. Post-zygotic mosaicism for activating GNAS1 mutations are the cause of the McCune-Albright Syndrome, a disease characterized by café-au-lait spots, multifocal bone fibrous dysplasia and several endocrine hyperfunction syndromes, including cortisol-producing bilateral adrenocortical hyperplasia (Weinstein et al., 1991). In addition, somatic activating GNAS1 mutations have been described in isolated ACTH-independent macronodular adrenal hyperplasia (AIMAH) (Fragoso et al., 2003), also renamed as primary macronodular hyperplasia (PMAH) (Berthon et al., 2015), and cortisol producing adenomas (Almeida et al., 2012; Goh et al., 2014; Sidhu et al., 2013).
More commonly, mutations in both regulatory and catalytic PKA subunits are observed in both familial and sporadic cortisol-producing adrenocortical diseases. 10% of primary pigmented nodular adrenocortical disease (PPNAD) cases carry inactivating mutations of the PRKAR1A gene, which encodes for the PKA regulatory subunit Iα (Yates et al., 2013). PPNAD is characterized by numerous, pigmented, microscopic adrenal nodules and ACTH-independent Cushing syndrome (Berthon et al., 2015). While PPNAD may occur sporadically, it is often associated with Carney complex type 1 (CNC1) (Kirschner et al., 2000), an autosomal dominant inherited multiple endocrine neoplasia syndrome characterized by spotty skin pigmentation, schwannomas, cardiac myxomas and endocrine abnormalities including somatomamotroph hyperplasia (Carney et al., 1985; Correa et al., 2015; Rothenbuhler and Stratakis, 2010). About two thirds of CNC patients have a PRKAR1A heterozygous germline mutation and more than 126 different mutations have been described (Kirschner et al., 2000; Stratakis et al., 1996). Inactivation of PRKAR1A in vitro promotes PKA activity and cAMP production (Cazabat et al., 2014). PRKAR1A is essential for life as demonstrated in Prkar1a deficient mice that die during embryonic life (Amieux and McKnight, 2002). However, inducible genetic ablation or reduction of expression of Prkar1a in a number of model systems partially phenocopies Carney complex (Griffin et al., 2004; Kirschner et al., 2005). Targeted inactivation of Prkar1a in the adrenal gland leads to increased proliferation, bilateral hyperplasia, and autonomous glucocorticoid production (Sahut-Barnola et al., 2010).
Constitutive PKA activity in the mouse and human adrenal cortex is associated with altered Wnt/β-catenin signaling, another fundamental regulator of adrenal homeostasis (Almeida et al., 2012, 2010). PKA has been shown to activate canonical Wnt signaling in vitro through multiple mechanisms including the phosphorylation of GSK3β (which leads to β-catenin stabilization) (Espiard et al., 2014). However, in mice with constitutively active β-catenin, loss of PKA catalytic unit Prkaca accelerates tumorigenesis (Drelon et al., 2016). Moreover, loss of the PKA regulatory subunit Prkar1a partially rescues adrenal phenotypes caused by constitutively active β-catenin, indicating that PKA activity inhibits Wnt/β-catenin signaling. Lastly, mouse models reveal that WNT4 counteracts PKA-mediated signaling (and PKA-mediated inhibition of canonical Wnt signaling) in the zG (Drelon et al., 2016).
In contrast, in human PPNAD nodules, increased nuclear accumulation of β-catenin and increased expression of genes associated with the canonical Wnt pathway is often observed (Gaujoux et al., 2008). Moreover, somatic mutations of the β-catenin gene CTNNB1 have been found in a subset of PPNAD macronodules (Gaujoux et al., 2008), consistent with cooperatively of these two signaling pathways in neoplastic adrenocortical growth. PPNAD is usually a benign condition, however recent reports have documented malignant transformation in patients with PPNAD and CNC, raising the possibility that abnormalities in PKA signaling may be a predisposing factor for malignant transformation (Anselmo et al., 2012). In fact, a recent molecular profiling study has identified PRKAR1A inactivating somatic mutations in a significant proportion of ACCs (Zheng et al., 2016), and some of these ACCs also harbor alterations in Wnt/β-catenin signaling.
In addition to PRKAR1A mutations in PPNADs, mutations in other regulators of the PKA signaling pathway have been found to be associated with various adrenocortical neoplasms. PRKAR2B protein is conspicuously absent in 50% of cortisol producing adenoma (CPA) samples analyzed by Western blot, despite the absence of genetic mutations in the PRKAR2B gene, perhaps due to protein degradation (Mantovani et al., 2008; Vincent-Dejean et al., 2008). Moreover, abnormal regulation of the PKA pathway has been associated with inactivating mutations in PDE11A and PDE8B, two major regulators of intracellular cAMP levels in the adrenal cortex. Non-pigmented forms of cortisol-producing micronodular hyperplasias and PMAH have been described in patients with germline mutations in these genes. Additionally, somatic mutations have also been described in CPAs (Horvath et al., 2008, 2006; Vezzosi et al., 2012).
More recently, somatic activating mutations in PRKACA (the most highly expressed catalytic PKA isoform in the human adrenal) have been observed in four different CPA cohorts (Beuschlein et al., 2014; Cao et al., 2014; Goh et al., 2014; Sato et al., 2014). Activating mutations in the PRKACA protein have been identified only in cortisol producing adenomas, and it is clinically associated with younger age at diagnosis, smaller tumors, and manifest Cushing syndrome (Di Dalmazi et al., 2014; Goh et al., 2014; Sato et al., 2014).
After the identification of the somatic PRKACA hotspot mutations in CPAs, germline amplifications in the catalytic subunits of PKA have been described in patients with cortisol-producing hyperplasias and adenomas. Forlino and colleagues reported genomic amplifications of PRKACB, which encodes for the catalytic isoform Cβ, associated with Carney complex with non-adrenal manifestations (Forlino et al., 2014). Similarly, Carney and colleagues reported five patients with ACTH-independent Cushing Syndrome, including familial cases, in patients with amplifications of PRKACA (Carney et al., 2015).
In addition to alterations in core constituents of the PKA pathway, molecular defects in upstream genes can also induce abnormal activation of the pathway. In contrast to activating mutations in the TSH receptor, which are a common cause of Plummer syndrome, activating mutations of the MC2R have been rarely found in ACTH-independent adrenal Cushing syndrome (Espiard et al., 2014). On the other hand, ectopic expression of different classes of GPCRs (termed illicit receptors) are frequently described in bilateral macronodular adrenal hyperplasia (BMAH) and are thought to play a role in the abnormal activation of the PKA pathway (Lacroix, 2009). More recently, intra-adrenal ACTH production has also been described as a cause of BMAH (Louiset et al., 2013).
In summary, increased activation of the PKA pathway by several different molecular defects leads to different forms of adrenocortical hormonal hyperactivity and abnormal growth, highlighting the central role of the cAMP/PKA pathway in steroidogenesis, homeostatic maintenance, and pathologic proliferation.
6. Telomeres, TERT, and TERF2 expression
Most vertebrate chromosomes encode repetitive nucleotide sequences at their ends that facilitate chromosomal stability, known as telomeres (Chong et al., 1995; Zhong et al., 1992). When somatic cells divide, the end of the chromosome cannot be replicated by leading strand DNA synthesis, referred to as the end replication problem. There is loss of telomeric DNA with every replication, progressive shortening of telomeres and ultimately initiation of senescence. This shortening can be averted by the enzyme telomerase in stem cells, germ cells, or in neoplastic cells with an acquired capacity for unrestricted clonal expansion (Thomson et al., 1998). Telomerases are composed of two subunits, a protein component TERT (Telomerase Reverse Transcriptase) with reverse transcriptase activity, and a ribonucleotide subunit TERC (Telomerase RNA Component), which serves as a template for telomere elongation (Greider and Blackburn, 1987; Morin, 1989; Shay, 2016). Telomerase prevents shortening of telomeres by providing a template of TTAGGG sequences at the end of the telo-mere, allowing extension in the 5′-3′ direction. The activity of telomerase is considered a marker of human stem cells, and telomerase expression is found by radioactive in situ method in the outer cortex of the adrenal, labeling a putative adrenal stem cell population (Else, 2009; Yashima et al., 1998). Moreover, studies in various mouse models (Bayne et al., 2008) reveal that telomere length is longest in the peripheral cortex, suggesting that telomerase is active in a population of the zG.
Telomeres serve an additional function – telomeres protect chromosomes from nuclease activity and DNA damage response. Telomere DNA is double stranded, with a terminal 3′ single strand that folds back forming a T-loop, preventing its utilization as substrate by nuclear enzymes (de Lange, 2004). Telomere protection is facilitated by a complex of proteins called the “shelterin” complex or the “telosome” (de Lange, 2005), with components that either directly bind telomeric DNA or are involved in protein-protein interactions.
To prevent accumulation of genomic aberrations, telomeres that are too short or lack components of the shelterin complex trigger cell senescence or apoptosis through the DNA-damage signaling pathway and consequent p53 activation (Else et al., 2009). Mouse models that are TERC- and p53-deficient display an increased tumor incidence compared to TERC-deficient, p53 wild-type mice (Chin et al., 1999), indicating a role for telomere dysfunction in promoting carcinogenesis. Telomere shortening and potential dysfunction have also been observed in patients with Li-Fraumeni syndrome, a hereditary genetic condition most commonly caused by mutations of the TP53 gene, and characterized by predisposition to several cancers, included ACC. Patients carrying TP53 mutations and associated malignancies have shorter telomeres compared to carriers with no cancer (Tabori et al., 2007), consistent with an association between telomere shortening and the development of malignancies. To further investigate the role of telomeres in adrenal tumors, Else and colleagues (Else et al., 2008) studied the telomere length maintenance mechanism (TMM) in adrenocortical tumor samples and normal adrenal cortices, and found that all malignant tissues had at least one TMM, while benign tumors and normal adrenal cortices had no TMMs. The analysis of the ACC cell lines SW13, RL251, NCI-H295R, NCI-H295A also showed TMMs (Else, 2009).
It is well established that telomere dysfunction leads to genetic instability and promotes human carcinoma, whereas subsequent reactivation of telomere protective mechanisms promotes escape of senescence and subsequently immortality (Shay and Wright, 2011). Recently, genomic characterization of adrenocortical carcinomas (ACCs) performed by international networks (Assié et al., 2014; Pinto et al., 2015; Zheng et al., 2016) have identified TERT (5p15.33) as one of the candidate driver genes of ACC, with recurrent high-level amplifications occurring in 15% of cases (Zheng et al., 2016). TERF2 (Telomeric Repeat Binding Factor 2, 16q22.1) is also found with recurrent focal amplification (7% of cases) (Zheng et al., 2016). TERT expression is higher in ACCs characterized by whole genomic doubling (WGD), and these tumors exhibit shorter telomeres compared to normal samples and to the non-WGD group, suggesting that TERT is required for maintaining telomere length in these cancers (Zheng et al., 2016).
To date, there are few studies designed to explore the role of telomeres in adrenocortical pathogenesis. In mice, recessive mutation of Acd, a gene widely expressed in adult tissues and during development that encodes for a component of the shelterin protein complex, leads to a phenotype similar to the human adrenal hypoplasia congenita (AHC), with hypoplastic adrenals, adrenocortical dysplasia, and lack of proper adrenocortical zonation. Additionally, Acdacd/acd mice show infertility, reduced body size, and other abnormal morphologic features (Keegan et al., 2005). At a cellular level, these same mice show telomere de-protection, but not telomere shortening (Else et al., 2009). Overall, the phenotypic features suggest stem/progenitor cell failure in the adrenal cortex, probably due to induction of telomere dysfunction and consequent activation of p53-signaling pathways (Else et al., 2009). Homozygous loss of p53 in mice homozygous for Acdacd/acd partially rescues the adrenal cortex, with complete restoration of adrenal size and partial restoration of adrenal architecture, cellular and nuclear size. However, Acdacd/acd mice with p53 loss develop numerous tumors, including adrenocortical carcinomas in 5% of mice (Else et al., 2009). These findings demonstrate a contribution of telomere deprotection to adrenocortical tumorigenesis and genomic instability.
Telomere maintenance, telomerase, and the shelterin complex have clear roles in adrenal homeostasis, with telomerase expression and increased telomere length found in populations of putative adrenal stem/progenitor cells. Telomere maintenance pathways also appear to be necessary in ACCs, indicating that this key stem cell pathway is frequently “hijacked” to promote cancer cell survival. This does not necessarily indicate cell-of-origin, however; telomerase activity is commonly re-activated or upregulated in a cell undergoing malignant progression following a process of telomere shortening, and is maintained at levels higher than in somatic stem cells (Shay and Wright, 2011). Most, but not all ACCs have telomeres shorter than matched normal samples and TERT amplification, TERT promoter mutations, and TERF2 amplification did not correlate with telomere length (Zheng et al., 2016).
7. IGF2 signaling and the 11p15 locus
Insulin-like growth factor 2 (IGF2) is a gene regulated by the imprinted human 11p15 locus that is implicated in both adrenal development and cancer. IGF2 is part of the IGF family of ligands, however, only IGF2 is highly expressed in the human fetal adrenal (Hedborg et al., 1994; Rainey et al., 2001; Voutilainen and Miller, 1988) and is the most common and most highly up-regulated transcript in human ACC (Heaton et al., 2012). The secreted ligand IGF2 binds IGF1 receptor (IGF-1R) and insulin receptor isoform A (IR-A). Both receptors are receptor tyrosine kinases that, when activated, promote cell growth and survival through MAPK and/or PI3K–AKT signaling pathways. In mice, Igf2 expression is essential for overall embryonic and fetal growth and development (Burns and Hassan, 2001), as evidenced in mice without Igf signaling that fail to develop an adrenal cortex (Pitetti et al., 2013). While IGF2 is involved in establishing the fetal adrenocortical cells that are precursors for the adult adrenal cortex and the associated homeostatic niche of the adrenocortical stem/progenitor cells, there is a paucity of data on the role of IGF2 in the postnatal adrenal cortex. IGF2 expression drops rapidly postnatally in the human (Belgorosky et al., 2009), suggesting that high IGF2 expression in ACC indicates a reactivation of fetal/embryonic programs. Interestingly, postnatal IGF1R immunohistochemical staining is strongest in the subcapsular zG cells, and shows increased expression at <3 months of age and again at adrenarchy (Belgorosky et al., 2009). Moreover, studies of microdissected adrenocortical zones (3 males and 1 female, ages 29–46) indicates that IGF2 expression is similarly highest in the zR (GEO accession GSE68889)(Nishimoto et al., 2015). Whether IGF2, or other genes at the 11p15 locus contribute to stem and progenitor cell biology in the adrenal cortex, particularly prenatally, is unclear.
In addition to expression studies in normal adrenal tissue, studies utilizing adrenal glands isolated from patients harboring a variety of human diseases have provided valuable insight into the postnatal roles of IGF2, and draw attention to the roles of other imprinted genes at the 11p15 locus in adrenocortical homeostasis. An imprinted gene refers to a gene in which only one parental copy is expressed. IGF2 and also H19, a long non-coding RNA, are imprinted genes inversely regulated by methylation of imprinting control region 1 (ICR1) (also called imprinting center 1, IC1, or differentially methylated region 1, DMR1) at the human 11p15 locus (mouse chromosome 7). Disruption of this locus occurs in sporadic ACC in pediatric and adult populations as well as in cases of Beckwith–Wiedemann syndrome (BWS), a fetal overgrowth disease associated with pediatric ACC (Assié et al., 2014; Mussa et al., 2016b; Pinto et al., 2015; Zheng et al., 2016).
BWS manifests as a pre- and post-natal overgrowth disorder with congenital malformations and predisposition to tumor development. BWS is molecularly characterized by alterations at chromosome 11p15 that involve mutation or deletion of imprinted genes, methylation alterations, or paternal uniparental disomy (UPD) (reviewed by Mussa et al., 2016b). Aberrations frequently result in a gain of imprinting (hypermethylation) at the ICR1 locus, either by aberrant methylation of the maternal chromosome, or paternal UPD, which results in overexpression of IGF2. Notably, patients with BWS have pathognomonic adrenal cytomegaly and are at over a 600 increased risk of childhood neoplasms, including an increased risk of ACC (DeBaun and Tucker, 1998; Mussa et al., 2016a). It is curious that, in contrast to BWS, the IMAGe syndrome is caused by specific gain-of-function mutations in the maternal CDKN1C gene, which encodes the cell cycle regulator p57Kip2 and (like IGF2) is located within the chromosome 11p15 locus (Eggermann et al., 2014; Kato et al., 2014). This rare disorder involves congenital adrenal hypoplasia, as well as intrauterine growth restriction, metaphyseal congenital adrenal hypoplasia and genital anomalies, from which the acronym IMAGe is derived.
A recent study of BWS patients has found that only paternal UPD, as compared to other BWS genotypes, is associated with ACC specifically (Mussa et al., 2016a) suggesting that other imprinted loci in addition to IGF2 on chromosome 11 are important contributors to pediatric ACC risk. Supporting these observations, mouse models engineered to overexpress Igf2 recapitulate some features of Beckwith Widemann syndrome, but do not show adrenal tumors (Eggenschwiler et al., 1997; Sun et al., 1997). In an analysis of 37 pediatric ACTs, while only two patients were diagnosed with BWS, 100% of cases exhibited overexpression of IGF2 with 91% loss of heterozygosity at 11p due to maternal allele loss. Analysis of SNVs led authors to also conclude that loss of heterozygosity was an early event in pediatric adrenocortical tumorigenesis (Pinto et al., 2015). These observations strongly suggest a causal role for 11p15 alterations in pediatric ACC, albeit not routinely due to BWS.
IGF2 expression is increased 7–200 fold in approximately 85% of adult ACCs (Heaton et al., 2012; Zheng et al., 2016). In contrast, IGF2 is overexpressed in only 5% of adult ACAs (Heaton et al., 2012) and has therefore been speculated to be an initiating lesion in carcinogenesis. Despite the near-ubiquitous IGF2 overexpression in both adult and pediatric ACCs, mouse models of Igf2 overexpression have not resulted in a robust malignant phenotype. Three different models have revealed that while overexpression of Igf2 in the adrenal cortex can result in adrenocortical hyperplasia, Igf2 overexpression alone is not sufficient for neoplastic transformation (Drelon et al., 2012; Weber et al., 1999). While Igf2 overexpression in combination with constitutively active Wnt/β-catenin signaling leads to a small number of carcinomas in the mouse adrenal cortex, the low penetrance argues for the need for additional somatic genetic and epigenetic alterations in adrenocortical carcinogenesis (Drelon et al., 2012; Heaton et al., 2012).
Numerous cancer therapies have been developed to inhibit IGF2-mediated signaling, and a number of compounds have been tested for efficacy in ACC. In contrast to preclinical and in vitro successes (Barlaskar et al., 2009; Guillaud-Bataille et al., 2014), clinical trials of many IGF2-targeted therapies have been unable to significantly induce ACC tumor regression as defined by RECIST criteria in adequate numbers of patients (Haluska et al., 2010; Lerario et al., 2014). Partial response in a small number of patients indicates that a subset of patients may benefit from therapy, however the molecular characteristics that define these cases are unknown. Other pathways may supersede IGF2 stimulation, particularly in the subset of ACCs with a high mutational load reported in the recent TCGA ACC cohort (Zheng et al., 2016). EGFR is also overexpressed in many ACCs, and has been postulated to contribute to lack of response to IGF1R inhibition, with some evidence garnered using cell lines (Xu et al., 2016).
Despite clinical trials and studies in vitro and in vivo, the role of IGF2 in ACC pathogenesis and malignant progression remains unclear. While IGF2 overexpression seems to be an early alteration in the pathogenesis of both pediatric and adult ACCs, studies show it is insufficient for adrenal cortical tumor initiation. A more complete understanding of the functions of IGF2 and the 11p15 locus in adrenal homeostasis in the context of other fundamental signaling pathways could provide us with some knowledge that can be integrated in the study of adrenal carcinogenesis.
8. Concluding remarks
In this review we have highlighted discoveries that have elucidated signaling pathways critical to the stem/progenitor niche in the adrenal cortex and how mutations in these pathways contribute to human disease, most notably human neoplasias. Research has more clearly defined populations of capsular and subcapsular stem/progenitor cells capable of repopulating the adrenal cortex. These cells are characterized by Shh signaling, and more recent developments solidify hypotheses that the stem and progenitor cell niche is both dependent on Wnt signaling and controls Wnt signaling (Vidal et al., 2016). How endocrine regulation with feedforward stimulation elicited by angiotensin II and ACTH intersects with paracrine and autocrine signaling pathways remains to be elucidated. Furthermore, the processes that regulate symmetric and asymmetric division of the stem/progenitor cell are unknown.
In adrenal neoplasia, the cell or cells of origin are unclear, as is the sequence by which mutations contribute to carcinoma (see Fig. 2). Whether adrenocortical stem and progenitor cells are involved and whether steroid hormone secretion is or is not linked to cell of origin remain questions. Interestingly, hypercortisolism is the most common endocrinopathy in ACCs (Zheng et al., 2016) and ACAs (Arnold et al., 2003). ACCs commonly activate cell signaling pathways essential to fetal and embryonic homeostasis, including IGF2 and FGF signaling. ACCs also often activate pathways involved in both embryonic and adult stem/progenitor cell homeostasis, such as canonical Wnt/β-catenin signaling and telomere maintenance programs. In sporadic human cancers, it remains unclear whether a multistep tumor progression model, involving an adenoma to carcinoma sequence, can be applied. Given the low incidence of carcinomas in relation to adenomas (Else et al., 2014), it seems that most adenomas do not achieve malignant potential.
Fig. 2. Schematic representation of potential mechanisms of malignant transformation in the adrenal cortex.
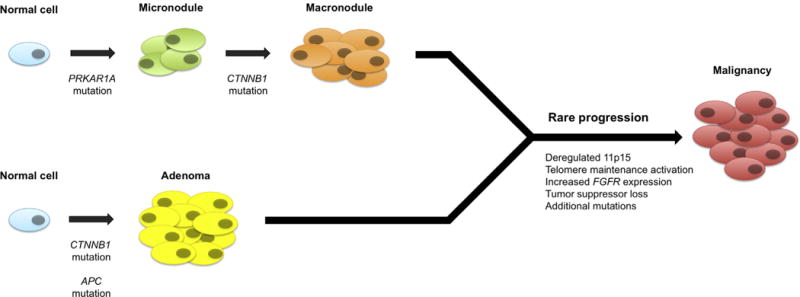
In patients with PPNAD, some of whom harbor germline PRKAR1A mutations, nodules in rare instances become malignant. Macronodules are characterized by the acquisition of additional CTNNB1 mutations. Activating CTNNB1 mutation occurs in ~20% of both ACAs and ACCs, but because ACAs occur much more commonly than ACCs, the occurrence of transforming mutations is extremely rare. In patients with FAP, germline loss of function mutations in APC predispose patients to adrenal masses bilaterally, which may also progress to malignancy. Deregulated 11p15, including overexpression of IGF2, activation of telomere maintenance mechanisms, and increased FGFR expression are additional alterations found in subsets of ACCs but not in ACAs.
Acknowledgments
We thank Antonio M. Lerario for assisting with preparation of the manuscript. This work was supported by the National Institutes of Health (NIH) research grant (RO1 DK062027 to G.D.H.). M.K.P. was supported by a NIH training grant (Cancer Biology Training Program T32 CA009676), the University of Michigan Cancer Center, the University of Michigan Cancer Center's Nancy Newton Loeb Fund, and the Spencer Bell Adrenal Cancer Scholar Endowment.
References
- Almeida MQ, Azevedo MF, Xekouki P, Bimpaki EI, Horvath A, Collins MT, Karaviti LP, Jeha GS, Bhattacharyya N, Cheadle C, Watkins T, Bourdeau I, Nesterova M, Stratakis CA. Activation of cyclic AMP signaling leads to different pathway alterations in lesions of the adrenal cortex caused by germ-line PRKAR1A defects versus those due to somatic GNAS mutations. J Clin Endocrinol Metab. 2012;97:E687–E693. doi: 10.1210/jc.2011-3000. http://dx.doi.org/10.1210/jc.2011-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MQ, Muchow M, Boikos S, Bauer AJ, Griffin KJ, Tsang KM, Cheadle C, Watkins T, Wen F, Starost MF, Bossis I, Nesterova M, Stratakis CA. Mouse Prkar1a haploinsufficiency leads to an increase in tumors in the Trp53+/− or Rb1+/− backgrounds and chemically induced skin papillomas by dysregulation of the cell cycle and Wnt signaling. Hum Mol Genet. 2010;19:1387–1398. doi: 10.1093/hmg/ddq014. http://dx.doi.org/10.1093/hmg/ddq014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann N Y Acad Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. http://dx.doi.org/10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, Lyssikatos C, Horvath A, Carney JA, Stratakis CA. A large family with carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab. 2012;97:351–359. doi: 10.1210/jc.2011-2244. http://dx.doi.org/10.1210/jc.2011-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DT, Reed JB, Burt K. Evaluation and management of the incidental adrenal mass. Proc Bayl Univ Med Cent. 2003;16:7–12. doi: 10.1080/08998280.2003.11927882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assié G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, Mannelli M, Mantero F, Papathomas T, De Krijger R, Tabarin A, Kerlan V, Baudin E, Tissier F, Dousset B, Groussin L, Amar L, Clauser E, Bertagna X, Ragazzon B, Beuschlein F, Libé R, de Reyniès A, Bertherat J. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014;46:607–612. doi: 10.1038/ng.2953. http://dx.doi.org/10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- Bambakidis NC, Onwuzulike K. Sonic hedgehog signaling and potential therapeutic indications. Vitam Hormones. 2012:379–394. doi: 10.1016/B978-0-12-394622-5.00017-1. http://dx.doi.org/10.1016/B978-0-12-394622-5.00017-1. [DOI] [PubMed]
- Barlaskar FM, Spalding AC, Heaton JH, Kuick R, Kim AC, Thomas DG, Giordano TJ, Ben-Josef E, Hammer GD. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009;94:204–212. doi: 10.1210/jc.2008-1456. http://dx.doi.org/10.1210/jc.2008-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile DP, Holzwarth MA. Basic fibroblast growth factor may mediate proliferation in the compensatory adrenal growth response. Am J Physiol. 1993;265:R1253–R1261. doi: 10.1152/ajpregu.1993.265.6.R1253. [DOI] [PubMed] [Google Scholar]
- Bayne S, Jones MEE, Li H, Pinto AR, Simpson ER, Liu J. Estrogen deficiency leads to telomerase inhibition, telomere shortening and reduced cell proliferation in the adrenal gland of mice. Cell Res. 2008;18:1141–1150. doi: 10.1038/cr.2008.291. http://dx.doi.org/10.1038/cr.2008.291. [DOI] [PubMed] [Google Scholar]
- Belgorosky A, Baquedano MS, Guercio G, Rivarola MA. Expression of the IGF and the aromatase/estrogen receptor systems in human adrenal tissues from early infancy to late puberty: implications for the development of adre-narche. Rev Endocr Metab Disord. 2009;10:51–61. doi: 10.1007/s11154-008-9105-1. http://dx.doi.org/10.1007/s11154-008-9105-1. [DOI] [PubMed] [Google Scholar]
- Bermudez O, Hennen E, Koch I, Lindner M, Eickelberg O. Gli1 mediates lung cancer cell proliferation and sonic hedgehog-dependent mesenchymal cell activation. PLoS One. 2013;8:1133–1145. doi: 10.1371/journal.pone.0063226. http://dx.doi.org/10.1371/journal.pone.0063226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, Samson-Couterie B, Zennaro MC, Plouin PF, Skah S, Plateroti M, Lefèbvre H, Sahut-Barnola I, Batisse-Lignier M, Assié G, Martinez AM, Bertherat J, Martinez A, Val P. WNT/β-catenin signalling is activated in aldosteroneproducing adenomas and controls aldosterone production. Hum Mol Genet. 2014;23:889–905. doi: 10.1093/hmg/ddt484. http://dx.doi.org/10.1093/hmg/ddt484. [DOI] [PubMed] [Google Scholar]
- Berthon A, Sahut-Barnola I, Lambert-Langlais S, de Joussineau C, Damon-Soubeyrand C, Louiset E, Taketo MM, Tissier F, Bertherat J, Lefrançois-Martinez AM, Martinez A, Val P. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum Mol Genet. 2010;19:1561–1576. doi: 10.1093/hmg/ddq029. http://dx.doi.org/10.1093/hmg/ddq029. [DOI] [PubMed] [Google Scholar]
- Berthon AS, Szarek E, Stratakis CA. PRKACA: the catalytic subunit of protein kinase A and adrenocortical tumors. Front Cell Dev Biol. 2015;3:26. doi: 10.3389/fcell.2015.00026. http://dx.doi.org/10.3389/fcell.2015.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM, Allolio B. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome – NEJM. N Engl J Med. 2014;370:1019–1028. doi: 10.1056/NEJMoa1310359. http://dx.doi.org/10.1056/NEJMoa1310359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boikos SA, Stratakis CA. Molecular genetics of the cAMP-dependent protein kinase pathway and of sporadic pituitary tumorigenesis. Hum Mol Genet. 2007;16:R80–R87. doi: 10.1093/hmg/ddm019. http://dx.doi.org/10.1093/hmg/ddm019. [DOI] [PubMed] [Google Scholar]
- Böse J, Grotewold L, Rüther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129–1135. doi: 10.1093/hmg/11.9.1129. http://dx.doi.org/10.1093/hmg/11.9.1129. [DOI] [PubMed] [Google Scholar]
- Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, Lefebvre H, Louiset E, Jeunemaitre X, Meatchi T, Benecke A, Lalli E, Zennaro MC. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology. 2011;152:4753–4763. doi: 10.1210/en.2011-1205. http://dx.doi.org/10.1210/en.2011-1205. [DOI] [PubMed] [Google Scholar]
- Brito LP, Ribeiro TC, Almeida MQ, De Lima Jorge AA, Soares IC, Latronico AC, Mendonca BB, Fragoso MCBV, Lerario AM. The role of fibroblast growth factor receptor 4 overexpression and gene amplification as prognostic markers in pediatric and adult adrenocortical tumors. Endocr Relat Cancer. 2012;19:88–90. doi: 10.1530/ERC-11-0231. http://dx.doi.org/10.1530/ERC-11-0231. [DOI] [PubMed] [Google Scholar]
- Burns JL, Hassan AB. Cell survival and proliferation are modified by insulin-like growth factor 2 between days 9 and 10 of mouse gestation. Development. 2001;128:3819–3830. doi: 10.1242/dev.128.19.3819. [DOI] [PubMed] [Google Scholar]
- Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, Zhou W, Li X, Zhong X, Lei Y, Su T, Wang H, Jiang Y, Yang L, Wei W, Yang X, Jiang X, Liu L, He J, Ye J, Wei Q, Li Y, Wang W, Wang J, Ning G. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science. 2014;344:913–917. doi: 10.1126/science.1249480. http://dx.doi.org/10.1126/science.1249480. [DOI] [PubMed] [Google Scholar]
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Med Baltim. 1985;64:270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- Carney JA, Lyssikatos C, Lodish MB, Stratakis CA. Germline PRKACA amplification leads to Cushing syndrome caused by 3 adrenocortical pathologic phenotypes. Hum Pathol. 2015;46:40–49. doi: 10.1016/j.humpath.2014.09.005. http://dx.doi.org/10.1016/j.humpath.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015;25:221–233. doi: 10.1016/j.tcb.2014.11.003. http://dx.doi.org/10.1016/j.tcb.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Cazabat L, Ragazzon B, Varin A, Potier-cartereau M, Vandier C, Vezzosi D, Risk-rabin M, Guellich A, Schittl J, Lechene P, Richter W, Nikolaev VO, Zhang J, Bertherat J, Vandecasteele G. Inactivation of the carney complex gene 1 (PRKAR1A) alters spatiotemporal regulation of cAMP and cAMP-dependent protein kinase: a study using genetically encoded FRET-based reporters. Hum Mol Genet. 2014;23:1163–1174. doi: 10.1093/hmg/ddt510. http://dx.doi.org/10.1093/hmg/ddt510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Ching S, Vilain E. Targeted disruption of sonic Hedgehog in the mouse adrenal leads to adrenocortical hypoplasia. Genesis. 2009;47:628–637. doi: 10.1002/dvg.20532. http://dx.doi.org/10.1002/dvg.20532. [DOI] [PubMed] [Google Scholar]
- Chong L, van Steensel B, Broccoli D, Erdjument-Bromage H, Hanish J, Tempst P, de Lange T. A human telomeric protein. Science. 1995;270:1663–1667. doi: 10.1126/science.270.5242.1663. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. http://dx.doi.org/10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol. 2015;173:M85–M97. doi: 10.1530/EJE-15-0209. http://dx.doi.org/10.1530/EJE-15-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Hung. 2011;43:745–756. doi: 10.1093/abbs/gmr079. http://dx.doi.org/10.1093/abbs/gmr079.Advance. [DOI] [PubMed] [Google Scholar]
- de Fraipont F, El Atifi M, Cherradi N, Le Moigne G, Defaye G, Houlgatte R, Bertherat J, Bertagna X, Plouin PF, Baudin E, Berger F, Gicquel C, Chabre O, Feige JJ. Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic Acid microarrays identifies several candidate genes as markers of malignancy. J Clin Endocrinol Metab. 2005;90:1819–1829. doi: 10.1210/jc.2004-1075. http://dx.doi.org/10.1210/jc.2004-1075. [DOI] [PubMed] [Google Scholar]
- de Lange T. T-loops and the origin of telomeres. Nat Rev Mol Cell Biol. 2004;5:323–329. doi: 10.1038/nrm1359. http://dx.doi.org/10.1038/nrm1359. [DOI] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005 doi: 10.1101/gad.1346005. http://dx.doi.org/10.1101/gad.1346005. [DOI] [PubMed]
- DeBaun MR, Tucker Ma. Risk of cancer during the first four years of life in children from the Beckwith-Wiedemann Syndrome Registry. J Pediatr. 1998;132:398–400. doi: 10.1016/s0022-3476(98)70008-3. http://dx.doi.org/10.1016/S0022-3476(98)70008-3. [DOI] [PubMed] [Google Scholar]
- Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, Quinkler M, Rayes N, Tabarin A, Laure Jullié M, Mantero F, Rubin B, Waldmann J, Bartsch DK, Pasquali R, Lohse M, Allolio B, Fassnacht M, Beuschlein F, Reincke M. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab. 2014;99:E2093–E2100. doi: 10.1210/jc.2014-2152. http://dx.doi.org/10.1210/jc.2014-2152. [DOI] [PubMed] [Google Scholar]
- Doghman M, Cazareth J, Lalli E. The T cell factor/beta-catenin antagonist PKF115-584 inhibits proliferation of adrenocortical carcinoma cells. J Clin Endocrinol Metab. 2008;93:3222–3225. doi: 10.1210/jc.2008-0247. http://dx.doi.org/10.1210/jc.2008-0247. [DOI] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Ragazzon B, Tissier F, Bandiera R, Sahut-Barnola I, de Joussineau C, Batisse-Lignier M, Lefrançois-Martinez AM, Bertherat J, Martinez A, Val P. Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044171. http://dx.doi.org/10.1371/journal.pone.0044171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Sahut-Barnola I, Mathieu M, Dumontet T, Rodriguez S, Batisse-Lignier M, Tabbal H, Tauveron I, Lefrançois-Martinez AM, Pointud JC, Gomez-Sanchez CE, Vainio S, Shan J, Sacco S, Schedl A, Stratakis CA, Martinez A, Val P. PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat Commun. 2016;7:12751. doi: 10.1038/ncomms12751. http://dx.doi.org/10.1038/ncomms12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11:3128–3142. doi: 10.1101/gad.11.23.3128. http://dx.doi.org/10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann T, Binder G, Brioude F, Maher ER, Lapunzina P, Cubellis MV, Bergadá I, Prawitt D, Begemann M. CDKN1C mutations: two sides of the same coin. Trends Mol Med. 2014;20:614–622. doi: 10.1016/j.molmed.2014.09.001. http://dx.doi.org/10.1016/j.molmed.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Else T. Telomeres and telomerase in adrenocortical tissue maintenance, carcinogenesis, and aging. J Mol Endocrinol. 2009;43:131–141. doi: 10.1677/JME-08-0189. http://dx.doi.org/10.1677/JME-08-0189. [DOI] [PubMed] [Google Scholar]
- Else T, Giordano TJ, Hammer GD. Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J Clin Endocrinol Metab. 2008;93:1442–1449. doi: 10.1210/jc.2007-1840. http://dx.doi.org/10.1210/jc.2007-1840. [DOI] [PubMed] [Google Scholar]
- Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ, Hammer GD. Adrenocortical carcinoma. Endocr Rev. 2014;35:282–326. doi: 10.1210/er.2013-1029. http://dx.doi.org/10.1210/er.2013-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else T, Trovato A, Kim AC, Wu Y, Ferguson DO, Kuick RD, Lucas PC, Hammer GD. Genetic p53 deficiency partially rescues the adrenocortical dysplasia phenotype at the expense of increased tumorigenesis. Cancer Cell. 2009;15:465–476. doi: 10.1016/j.ccr.2009.04.011. http://dx.doi.org/10.1016/j.ccr.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espiard S, Ragazzon B, Bertherat J. Protein kinase a alterations in adrenocortical tumors. Horm Metab Res. 2014;46:869–875. doi: 10.1055/s-0034-1385908. http://dx.doi.org/10.1055/s-0034-1385908. [DOI] [PubMed] [Google Scholar]
- Finco I, LaPensee CR, Krill KT, Hammer GD. Hedgehog signaling and steroidogenesis. Annu Rev Physiol. 2015;77:105–129. doi: 10.1146/annurev-physiol-061214-111754. http://dx.doi.org/10.1146/annurev-physiol-061214-111754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, Zuffardi O. PRKACB and carney complex. N Engl J Med. 2014:1067–1069. doi: 10.1056/NEJMc1309730. http://dx.doi.org/10.1056/NEJMc1309730. [DOI] [PubMed]
- Fragoso MCBV, Domenice S, Latronico AC, Martin RM, Pereira MAA, Zerbini MCN, Lucon AM, Mendonca BB. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab. 2003;88:2147–2151. doi: 10.1210/jc.2002-021362. http://dx.doi.org/10.1210/jc.2002-021362. [DOI] [PubMed] [Google Scholar]
- Freedman BD, Kempna PB, Carlone DL, Shah MS, Guagliardo NA, Barrett PQ, Gomez-Sanchez CE, Majzoub JA, Breault DT. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell. 2013;26:666–673. doi: 10.1016/j.devcel.2013.07.016. http://dx.doi.org/10.1016/j.devcel.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux S, Hantel C, Launay P, Bonnet S, Perlemoine K, Lefèvre L, Guillaud-Bataille M, Beuschlein F, Tissier F, Bertherat J, Rizk-Rabin M, Ragazzon B. Silencing mutated β-catenin inhibits cell proliferation and stimulates apoptosis in the adrenocortical cancer cell line H295R. PLoS One. 2013;8:1–5. doi: 10.1371/journal.pone.0055743. http://dx.doi.org/10.1371/journal.pone.0055743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux S, Tissier F, Groussin L, Libé R, Ragazzon B, Launay P, Audebourg A, Dousset B, Bertagna X, Bertherat J. Wnt/β-catenin and 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase a signaling pathways alterations and somatic β-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab. 2008;93:4135–4140. doi: 10.1210/jc.2008-0631. http://dx.doi.org/10.1210/jc.2008-0631. [DOI] [PubMed] [Google Scholar]
- Giordano TJ, Kuick R, Else T, Gauger PG, Vinco M, Bauersfeld J, Sanders D, Thomas DG, Doherty G, Hammer G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res. 2009;15:668–676. doi: 10.1158/1078-0432.CCR-08-1067. http://dx.doi.org/10.1158/1078-0432.CCR-08-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kuntsman JW, Korah R, Suttorp AC, Dietrich D, Haase M, Willenberg HS, Stålberg P, Hellman P, Akerström G, Björklund P, Carling T, Lifton RP. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014;46:613–617. doi: 10.1038/ng.2956. http://dx.doi.org/10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes DC, Leal LF, Mermejo LM, Scrideli CA, Martinelli CE, Fragoso MCBV, Latronico AC, Tone LG, Tucci S, Yunes JA, Cardinalli IA, Mastellaro MJ, Brandalise SR, Ramalho F, Moreira AC, Ramalho LN, de Castro M, Antonini SRR. Sonic hedgehog signaling is active in human adrenal cortex development and deregulated in adrenocortical tumors. J Clin Endocrinol Metab. 2014;99:E1209–E1216. doi: 10.1210/jc.2013-4098. http://dx.doi.org/10.1210/jc.2013-4098. [DOI] [PubMed] [Google Scholar]
- Gonnissen A, Isebaert S, Haustermans K. Hedgehog signaling in prostate cancer and its therapeutic implication. Int J Mol Sci. 2013;14:13979–14007. doi: 10.3390/ijms140713979. http://dx.doi.org/10.3390/ijms140713979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodarowicz D, Handley HH. Stimulation of division of Y1 adrenal cells by a growth factor isolated from bovine pituitary glands. Endocrinology. 1975;97:102–107. doi: 10.1210/endo-97-1-102. http://dx.doi.org/10.1210/endo-97-1-102. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D, Ill CR, Hornsby PJ, Gill GN. Control of bovine adrenal cortical cell proliferation by fibroblast growth factor. Lack of effect of epidermal growth factor Endocrinology. 1977;100:1080–1089. doi: 10.1210/endo-100-4-1080. http://dx.doi.org/10.1210/endo-100-4-1080. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–898. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos SG, Robinson-White A, Lenherr SM, Weinberg FD, Claflin ES, Batista D, Bourdeau I, Voutetakis A, Sandrini F, Meoli EM, Bauer AJ, Cho-Chung YS, Bornstein SR, Carney JA, Stratakis CA. A transgenic mouse bearing an antisense construct of regulatory subunit type 1A of protein kinase A develops endocrine and other tumours: comparison with Carney complex and other PRKAR1A induced lesions. J Med Genet. 2004;41:923–931. doi: 10.1136/jmg.2004.028043. http://dx.doi.org/10.1136/jmg.2004.028043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasti L, Candy Sze WC, McKay T, Grose R, King PJ. FGF signalling through Fgfr2 isoform IIIb regulates adrenal cortex development. Mol Cell Endocrinol. 2013;371:182–188. doi: 10.1016/j.mce.2013.01.014. http://dx.doi.org/10.1016/j.mce.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasti L, Paul A, Laufer E, King P. Localization of Sonic hedgehog secreting and receiving cells in the developing and adult rat adrenal cortex. Mol Cell Endocrinol. 2011;336:117–122. doi: 10.1016/j.mce.2010.11.010. http://dx.doi.org/10.1016/j.mce.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud-Bataille M, Ragazzon B, De Reynies A, Chevalier C, Francillard I, Barreau O, Steunou V, Guillemot J, Tissier F, Rizk-Rabin M, René-Corail F, Al Ghuzlan A, Assié G, Bertagna X, Baudin E, Le Bouc Y, Bertherat J, Clauser E. IGF2 promotes growth of adrenocortical carcinoma cells, but its overexpression does not modify phenotypic and molecular features of adrenocortical carcinoma. PLoS One. 2014;9 doi: 10.1371/journal.pone.0103744. http://dx.doi.org/10.1371/journal.pone.0103744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JG, Pallister PD, Clarren SK, Beckwith JB, Wiglesworth FW, Fraser FC, Cho S, Benke PJ, Reed SD. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly–a new syndrome? Part I: clinical, causal, and pathogenetic considerations. Am J Med Genet. 1980;7:47–74. doi: 10.1002/ajmg.1320070110. http://dx.doi.org/10.1002/ajmg.1320070110. [DOI] [PubMed] [Google Scholar]
- Haluska P, Worden F, Olmos D, Yin D, Schteingart D, Batzel GN, Paccagnella ML, De Bono JS, Gualberto A, Hammer GD. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol. 2010;65:765–773. doi: 10.1007/s00280-009-1083-9. http://dx.doi.org/10.1007/s00280-009-1083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H, Mao X, Ma Q, Zamponi R, Bouwmeester T, Finan PM, Kirschner MW, Porter JA, Serluca FC, Cong F. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012;485:195–200. doi: 10.1038/nature11019. http://dx.doi.org/10.1038/nature11019. [DOI] [PubMed] [Google Scholar]
- Heaton JH, Wood Ma, Kim AC, Lima LO, Barlaskar FM, Almeida MQ, Fragoso MCBV, Kuick R, Lerario AM, Simon DP, Soares IC, Starnes E, Thomas DG, Latronico AC, Giordano TJ, Hammer GD. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and β-catenin. Am J Pathol. 2012;181:1017–1033. doi: 10.1016/j.ajpath.2012.05.026. http://dx.doi.org/10.1016/j.ajpath.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedborg F, Holmgren L, Sandstedt B, Ohlsson R. The cell type-specific IGF2 expression during early human development correlates to the pattern of overgrowth and neoplasia in the Beckwith-Wiedemann syndrome. Am J Pathol. 1994;145:802–817. [PMC free article] [PubMed] [Google Scholar]
- Heikkila M, Peltoketo H, Leppaluoto J, Ilves M, Vuolteenaho O, Vainio S. Wnt-4 deficiency alters mouse adrenal cortex function, reducing aldosterone production. Endocrinology. 2002;143:4358–4365. doi: 10.1210/en.2002-220275. http://dx.doi.org/10.1210/en.2002-220275. [DOI] [PubMed] [Google Scholar]
- Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libè R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38:794–800. doi: 10.1038/ng1809. http://dx.doi.org/10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet. 2008;16:1245–1253. doi: 10.1038/ejhg.2008.85. http://dx.doi.org/10.1038/ejhg.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CCJ, Miyagawa S, Matsumaru D, Parker KL, Yao HHC. Progenitor cell expansion and organ size of mouse adrenal is regulated by sonic hedgehog. Endocrinology. 2010;151:1119–1128. doi: 10.1210/en.2009-0814. http://dx.doi.org/10.1210/en.2009-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingle DJ, Higgins GM. Autotransplantation and regeneration of the adrenal gland. Endocrinology. 1938;22:458–464. http://dx.doi.org/10.1210/endo-22-4-458. [Google Scholar]
- Jamsheer A, Sowińska A, Trzeciak T, Jamsheer-Bratkowska M, Geppert A, Latos-Bieleńska A. Expanded mutational spectrum of the GLI3 gene substantiates genotype-phenotype correlations. J Appl Genet. 2012;53:415–422. doi: 10.1007/s13353-012-0109-x. http://dx.doi.org/10.1007/s13353-012-0109-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasperson KW, Burt RW. APC-associated Polyposis Conditions, GeneRe-views(®) University of Washington; Seattle: 1993. [Google Scholar]
- Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, Nelson-Williams C, Bäckdahl M, Suttorp A-C, Haase M, Choi M, Schlessinger J, Rimm DL, Höög A, Prasad ML, Korah R, Larsson C, Lifton RP, Carling T. Whole-exome Sequencing Characterizes the Landscape of Somatic Mutations and Copy Number Alterations in Adrenocortical Carcinoma. 2014:2014–3282. doi: 10.1210/jc.2014-3282. http://dx.doi.org/10.1210/jc. [DOI] [PMC free article] [PubMed]
- Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. http://dx.doi.org/10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato F, Hamajima T, Hasegawa T, Amano N, Horikawa R, Nishimura G, Nakashima S, Fuke T, Sano S, Fukami M, Ogata T. IMAGe syndrome: clinical and genetic implications based on investigations in three Japanese patients. Clin Endocrinol (Oxf) 2014;80:706–713. doi: 10.1111/cen.12379. http://dx.doi.org/10.1111/cen.12379. [DOI] [PubMed] [Google Scholar]
- Keegan CE, Hutz JE, Else T, Adamska M, Shah SP, Kent AE, Howes JM, Beamer WG, Hammer GD. Urogenital and caudal dysgenesis in adrenocortical dysplasia (acd) mice is caused by a splicing mutation in a novel telomeric regulator. Hum Mol Genet. 2005;14:113–123. doi: 10.1093/hmg/ddi011. http://dx.doi.org/10.1093/hmg/ddi011. [DOI] [PubMed] [Google Scholar]
- Kim AC, Barlaskar FM, Heaton JH, Else T, Kelly VR, Krill KT, Scheys JO, Simon DP, Trovato A, Yang WH, Hammer GD. In search of adrenocortical stem and progenitor cells. Endocr Rev. 2009;30:241–243. doi: 10.1210/er.2008-0039. http://dx.doi.org/10.1210/er.2008-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AC, Reuter AL, Zubair M, Else T, Serecky K, Bingham NC, Lavery GG, Parker KL, Hammer GD. Targeted disruption of beta-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development. 2008;135:2593–2602. doi: 10.1242/dev.021493. http://dx.doi.org/10.1242/dev.021493. [DOI] [PubMed] [Google Scholar]
- Kim Y, Bingham N, Sekido R, Parker KL, Lovell-Badge R, Capel B. Fibroblast growth factor receptor 2 regulates proliferation and Sertoli differentiation during male sex determination. Proc Natl Acad Sci U S A. 2007;104:16558–16563. doi: 10.1073/pnas.0702581104. http://dx.doi.org/10.1073/pnas.0702581104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King P, Paul A, Laufer E. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc Natl Acad Sci U S A. 2009;106:21185–21190. doi: 10.1073/pnas.0909471106. http://dx.doi.org/10.1073/pnas.0909471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. http://dx.doi.org/10.1038/79238. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Kusewitt DF, Matyakhina L, Towns WH, Carney JA, Westphal H, Stratakis CA. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005;65:4506–4514. doi: 10.1158/0008-5472.CAN-05-0580. http://dx.doi.org/10.1158/0008-5472.CAN-05-0580. [DOI] [PubMed] [Google Scholar]
- Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, Clevers H. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–669. doi: 10.1038/nature11308. http://dx.doi.org/10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:245–259. doi: 10.1016/j.beem.2008.10.011. http://dx.doi.org/10.1016/j.beem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Laufer E, Kesper D, Vortkamp A, King P. Sonic hedgehog signaling during adrenal development. Mol Cell Endocrinol. 2012;351:19–27. doi: 10.1016/j.mce.2011.10.002. http://dx.doi.org/10.1016/j.mce.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell C, Velázquez-Fern andez D, Lindsten K, Juhlin C, Enberg U, Geli J, Höög A, Kjellman M, Lundeberg J, Hamberger B, Larsson C, Nilsson P, Bäckdahl M. Transcriptional profiling enables molecular classification of adrenocortical tumours. Eur J Endocrinol. 2009;161:141–152. doi: 10.1530/EJE-09-0068. http://dx.doi.org/10.1530/EJE-09-0068. [DOI] [PubMed] [Google Scholar]
- Lerario AM, Worden FP, Ramm CA, Hasseltine EA, Stadler WM, Else T, Shah MH, Agamah E, Rao K, Hammer GD. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Cancer. 2014;5:232–239. doi: 10.1007/s12672-014-0182-1. http://dx.doi.org/10.1007/s12672-014-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chi S, Xie J. Hedgehog signaling in skin cancers. Cell Signal. 2011;23:1235–1243. doi: 10.1016/j.cellsig.2011.03.002. http://dx.doi.org/10.1016/j.cellsig.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, Libé R, Bram Z, Groussin L, Caron P, Tabarin A, Grunenberger F, Christin-Maitre S, Bertagna X, Kuhn JM, Anouar Y, Bertherat J, Lefebvre H. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med. 2013;369:2115–2125. doi: 10.1056/NEJMoa1215245. http://dx.doi.org/10.1056/NEJMoa1215245. [DOI] [PubMed] [Google Scholar]
- Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, Drugan A, Tiosano D, Gershoni-Baruch R, Choder M, Sprecher E. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. 2008;82:39–47. doi: 10.1016/j.ajhg.2007.08.005. http://dx.doi.org/10.1016/j.ajhg.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani G, Lania AG, Bondioni S, Peverelli E, Pedroni C, Ferrero S, Pellegrini C, Vicentini L, Arnaldi G, Bosari S, Beck-Peccoz P, Spada A. Different expression of protein kinase A (PKA) regulatory subunits in cortisol-secreting adrenocortical tumors: relationship with cell proliferation. Exp Cell Res. 2008;314:123–130. doi: 10.1016/j.yexcr.2007.08.024. http://dx.doi.org/10.1016/j.yexcr.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Mao J, McGlinn E, Huang P, Tabin CJ, McMahon AP. Fgf-dependent Etv4/5 activity is required for posterior restriction of sonic hedgehog and promoting outgrowth of the vertebrate limb. Dev Cell. 2009;16:600–606. doi: 10.1016/j.devcel.2009.02.005. http://dx.doi.org/10.1016/j.devcel.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, Macchiaiolo M, Molinatto C, Baldassarre G, Mariani M, Tarani L, Bedeschi MF, Milani D, Melis D, Bartuli A, Cubellis MV, Selicorni A, Cirillo Silengo M, Larizza L, Riccio A, Ferrero GB. (Epi)genotype–phenotype correlations in Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2016a;24:183–190. doi: 10.1038/ejhg.2015.88. http://dx.doi.org/10.1038/ejhg.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet. 2016b;89:403–415. doi: 10.1111/cge.12635. http://dx.doi.org/10.1111/cge.12635. [DOI] [PubMed] [Google Scholar]
- Naski MC, Colvin JS, Coffin JD, Ornitz DM. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, Nanba K, Rainey WE. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci. 2015;112:E4591–E4599. doi: 10.1073/pnas.1505529112. http://dx.doi.org/10.1073/pnas.1505529112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson RK, Boggs K, Yergeau D, Cheng J, Mulder HL, Manne J, Jenkins J, Mastellaro MJ, Figueiredo BC, Dyer MA, Pappo A, Zhang J, Downing JR, Ribeiro RC, Zambetti GP. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015;6:6302. doi: 10.1038/ncomms7302. http://dx.doi.org/10.1038/ncomms7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitetti JL, Calvel P, Romero Y, Conne B, Truong V, Papaioannou MD, Schaad O, Docquier M, Herrera PL, Wilhelm D, Nef S. Insulin and IGF1 receptors are essential for XX and XY gonadal differentiation and adrenal development in mice. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003160. http://dx.doi.org/10.1371/journal.pgen.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey WE, Carr BR, Wang ZN, Parker CR., Jr Gene profiling of human fetal and adult adrenals. J Endocrinol. 2001;171:209–215. doi: 10.1677/joe.0.1710209. [pii] [DOI] [PubMed] [Google Scholar]
- Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I, Calebiro D, Schwarzmayr T, Appenzeller S, Rubin B, Waldmann J, Scaroni C, Bartsch DK, Mantero F, Mannelli M, Kastelan D, Chiodini I, Bertherat J, Reincke M, Strom TM, Fassnacht M, Beuschlein F. Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. jc2016-1586 J Clin Endocrinol Metab. 2016 doi: 10.1210/jc.2016-1586. http://dx.doi.org/10.1210/jc.2016-1586. [DOI] [PubMed]
- Rothenbuhler A, Stratakis CA. Clinical and molecular genetics of Carney complex. Best Pract Res Clin Endocrinol Metab. 2010;24:389–399. doi: 10.1016/j.beem.2010.03.003. http://dx.doi.org/10.1016/j.beem.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Sahut-Barnola I, de Joussineau C, Val P, Lambert-Langlais S, Damon C, Lefrançois-Martinez AM, Pointud JC, Marceau G, Sapin V, Tissier F, Ragazzon B, Bertherat J, Kirschner LS, Stratakis CA, Martinez A. Cushing’s syndrome and fetal features resurgence in adrenal cortex–specific Prkar1a knockout mice. PLoS Genet. 2010;6:e1000980. doi: 10.1371/journal.pgen.1000980. http://dx.doi.org/10.1371/journal.pgen.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon A, Keramidas M, Maisin C, Thomas M. Loss of β-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget. 2015;6:11421–11433. doi: 10.18632/oncotarget.3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, Yoshida K, Nagata Y, Sato-Otsubo A, Yoshizato T, Suzuki H, Shiozawa Y, Kataoka K, Kon A, Aoki K, Chiba K, Tanaka H, Kume H, Miyano S, Fukayama M, Nureki O, Homma Y, Ogawa S. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science. 2014;344:917–920. doi: 10.1126/science.1252328. http://dx.doi.org/10.1126/science.1252328. [DOI] [PubMed] [Google Scholar]
- Shaikh LH, Zhou J, Teo AED, Garg S, Neogi SG, Figg N, Yeo GS, Yu H, Maguire JJ, Zhao W, Bennett MR, Azizan EAB, Davenport AP, McKenzie G, Brown MJ. LGR5 activates noncanonical Wnt signaling and inhibits aldosterone production in the human adrenal. J Clin Endocrinol Metab. 2015;100:E836–E844. doi: 10.1210/jc.2015-1734. http://dx.doi.org/10.1210/jc.2015-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016;6:584–593. doi: 10.1158/2159-8290.CD-16-0062. http://dx.doi.org/10.1158/2159-8290.CD-16-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Semin Cancer Biol. 2011;21:349–353. doi: 10.1016/j.semcancer.2011.10.001. http://dx.doi.org/10.1016/j.semcancer.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemura K, Fujisawa M. History and epidemiology of antibiotic susceptibilities of Neisseria gonorrhoeae. Curr Drug Targets. 2015;16:272–280. doi: 10.2174/1389450115666141120110724. [DOI] [PubMed] [Google Scholar]
- Shin K, Lim A, Odegaard JI, Honeycutt JD, Kawano S, Hsieh MH, Beachy PA. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat Cell Biol. 2014;16:469–478. doi: 10.1038/ncb2956. http://dx.doi.org/10.1038/ncb2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A, Debelenko L, Misra VK. Infantile adrenocortical tumor with an activating GNAS1 mutation. J Clin Endocrinol Metab. 2013;98:E115–E118. doi: 10.1210/jc.2012-2933. http://dx.doi.org/10.1210/jc.2012-2933. [DOI] [PubMed] [Google Scholar]
- Smith TG, Clark SK, Katz DE, Reznek RH, Phillips RK. Adrenal masses are associated with familial adenomatous polyposis. Dis Colon Rectum. 2000;43:1739–1742. doi: 10.1007/BF02236860. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Carney JA, Lin JP, Papanicolaou DA, Karl M, Kastner DL, Pras E, Chrousos GP. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996;97:699–705. doi: 10.1172/JCI118467. http://dx.doi.org/10.1172/JCI118467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, Li L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res Part C - Embryo Today Rev. 2010;90:243–256. doi: 10.1002/bdrc.20195. http://dx.doi.org/10.1002/bdrc.20195. [DOI] [PubMed] [Google Scholar]
- Sun FL, Dean WL, Kelsey G, Allen ND, Reik W. Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature. 1997;389:809–815. doi: 10.1038/39797. http://dx.doi.org/10.1038/39797. [DOI] [PubMed] [Google Scholar]
- Tabori U, Nanda S, Druker H, Lees J, Malkin D. Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome. Cancer Res. 2007;67:1415–1418. doi: 10.1158/0008-5472.CAN-06-3682. http://dx.doi.org/10.1158/0008-5472.CAN-06-3682. [DOI] [PubMed] [Google Scholar]
- Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic β-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD) Clin Endocrinol (Oxf) 2008;69:367–373. doi: 10.1111/j.1365-2265.2008.03273.x. http://dx.doi.org/10.1111/j.1365-2265.2008.03273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: lessons learned after twenty years. Biochim Biophys Acta - Proteins Proteom. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. http://dx.doi.org/10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Castillo CF, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. http://dx.doi.org/10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Hornsby PJ. Transplantation of primary bovine adrenocortical cells into scid mice. Mol Cell Endocrinol. 1999;153:125–136. doi: 10.1016/s0303-7207(99)00070-2. http://dx.doi.org/10.1016/S0303-7207(99)00070-2. [DOI] [PubMed] [Google Scholar]
- Thomas M, Northrup SR, Hornsby PJ. Adrenocortical tissue formed by transplantation of normal clones of bovine adrenocortical cells in scid mice replaces the essential functions of the animals’ adrenal glands. Nat Med. 1997;3:978–983. doi: 10.1038/nm0997-978. http://dx.doi.org/10.1038/nm0997-978. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagneré AM, René-Corail F, Jullian E, Gicquel C, Bertagna X, Vacher-Lavenu MC, Perret C, Bertherat J. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7627. doi: 10.1158/0008-5472.CAN-05-0593. http://dx.doi.org/10.1158/0008-5472.CAN-05-0593. [DOI] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. http://dx.doi.org/10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Vezzosi D, Libé R, Baudry C, Rizk-Rabin M, Horvath A, Levy I, René-Corail F, Ragazzon B, Stratakis CA, Vandecasteele G, Bertherat J. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J Clin Endocrinol Metab. 2012;97:E2063–E2069. doi: 10.1210/jc.2012-2275. http://dx.doi.org/10.1210/jc.2012-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal V, Sacco S, Rocha AS, da Silva F, Panzolini C, Dumontet T, Doan TMP, Shan J, Rak-Raszewska A, Bird T, Vainio S, Martinez A, Schedl A. The adrenal capsule is a signaling center controlling cell renewal and zonation through Rspo3. Genes Dev. 2016;30:1389–1394. doi: 10.1101/gad.277756.116. http://dx.doi.org/10.1101/gad.277756.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent-Dejean C, Cazabat L, Groussin L, Perlemoine K, Fumey G, Tissier F, Bertagna X, Bertherat J. Identification of a clinically homogenous subgroup of benign cortisol-secreting adrenocortical tumors characterized by alterations of the protein kinase A (PKA) subunits and high PKA activity. Eur J Endocrinol. 2008;158:829–839. doi: 10.1530/EJE-07-0819. http://dx.doi.org/10.1530/EJE-07-0819. [DOI] [PubMed] [Google Scholar]
- Vinson GP. Adrenocortical zonation and ACTH. Microsc Res Tech. 2003;61:227–239. doi: 10.1002/jemt.10331. http://dx.doi.org/10.1002/jemt.10331. [DOI] [PubMed] [Google Scholar]
- Voutilainen R, Miller WL. Developmental and hormonal regulation of mRNAs for insulin-like growth factor II and steroidogenic enzymes in human fetal adrenals and gonads. DNA. 1988;7:9–15. doi: 10.1089/dna.1988.7.9. http://dx.doi.org/10.1089/dna.1988.7.9. [DOI] [PubMed] [Google Scholar]
- Walczak EM, Kuick R, Finco I, Bohin N, Hrycaj SM, Wellik DM, Hammer GD. Wnt-signaling inhibits adrenal steroidogenesis by cell-autonomous and non-cell-autonomous mechanisms. me20141060 Mol Endocrinol. 2014;28 doi: 10.1210/me.2014-1060. http://dx.doi.org/10.1210/me.2014-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, Fottner C, Schmidt P, Brodowski KMH, Gittner K, Lahm H, Engelhardt D, Wolf E. Postnatal overexpression of insulin-like growth factor II in transgenic mice is associated with adrenocortical hyperplasia and enhanced steroidogenesis. Endocrinology. 1999;140:1537–1543. doi: 10.1210/endo.140.4.6660. http://dx.doi.org/10.1210/endo.140.4.6660. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune–albright syndrome. N Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. http://dx.doi.org/10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- Werminghaus P, Haase M, Hornsby PJ, Schinner S, Schott M, Malendowicz LK, Lammers BJ, Goretzki PE, Müller-Mattheis V, Giessing Markus, Willenberg HS. Hedgehog-signaling is upregulated in non-producing human adrenal adenomas and antagonism of hedgehog-signaling inhibits proliferation of NCI-H295R cells and an immortalized primary human adrenal cell line. J Steroid Biochem Mol Biol. 2014;139:7–15. doi: 10.1016/j.jsbmb.2013.09.007. http://dx.doi.org/10.1016/j.jsbmb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- West AN, Neale GA, Pounds S, Figueredo BC, Galindo CR, Pianovski MAD, Oliveira Filho AG, Malkin D, Lalli E, Ribeiro R, Zambetti GP. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67:600–608. doi: 10.1158/0008-5472.CAN-06-3767. http://dx.doi.org/10.1158/0008-5472.CAN-06-3767. [DOI] [PubMed] [Google Scholar]
- Wood MA, Acharya A, Finco I, Swonger JM, Elston MJ, Tallquist MD, Hammer GD. Fetal adrenal capsular cells serve as progenitor cells for steroidogenic and stromal adrenocortical cell lineages in M. musculus. Development. 2013;140:4522–4532. doi: 10.1242/dev.092775. http://dx.doi.org/10.1242/dev.092775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Hammer GD. Adrenocortical stem and progenitor cells: unifying model of two proposed origins. Mol Cell Endocrinol. 2011;336:206–212. doi: 10.1016/j.mce.2010.11.012. http://dx.doi.org/10.1016/j.mce.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Qi Y, Xu Y, Lian J, Wang X, Ning G, Wang W, Zhu Y. Co-inhibition of EGFR and IGF1R synergistically impacts therapeutically on adrenocortical carcinoma. Oncotarget. 2016;2:1–12. doi: 10.18632/oncotarget.8827. http://dx.doi.org/10.18632/oncotarget.8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashima K, Maitra A, Rogers BB, Timmons CF, Rathi A, Pinar H, Wright WE, Shay JW, Gazdar AF. Expression of the RNA component of telomerase during human development and differentiation. Cell Growth Differ. 1998;9:805–813. [PubMed] [Google Scholar]
- Yates R, Katugampola H, Cavlan D, Cogger K, Meimaridou E, Hughes C, Metherell L, Guasti L, King P. Adrenocortical development, maintenance, and disease. Curr Top Dev Biol. 2013;106:239–312. doi: 10.1016/B978-0-12-416021-7.00007-9. http://dx.doi.org/10.1016/B978-0-12-416021-7.00007-9. [DOI] [PubMed] [Google Scholar]
- Zebisch M, Xu Y, Krastev C, MacDonald BT, Chen M, Gilbert RJC, He X, Jones EY. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat Commun. 2013;4:2787. doi: 10.1038/ncomms3787. http://dx.doi.org/10.1038/ncomms3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Verheyden JM, Hassell JA, Sun X. FGF-regulated etv genes are essential for repressing Shh expression in mouse limb buds. Dev Cell. 2009;16:607–613. doi: 10.1016/j.devcel.2009.02.008. http://dx.doi.org/10.1016/j.devcel.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assié G, Morozova O, Akbani R, Shih J, Hoadley KA, Choueiri TK, Waldmann J, Mete O, Robertson AG, Wu HT, Raphael BJ, Shao L, Meyerson M, Demeure MJ, Beuschlein F, Gill AJ, Sidhu SB, Almeida MQ, Fragoso MCBV, Cope LM, Kebebew E, Habra MA, Whitsett TG, Bussey KJ, Rainey WE, Asa SL, Bertherat J, Fassnacht M, Wheeler DA, Hammer GD, Giordano TJ, Verhaak RGW. Comprehensive Pan-Genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016;29:723–736. doi: 10.1016/j.ccell.2016.04.002. http://dx.doi.org/10.1016/j.ccell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Shiue L, Kaplan S, de Lange T. A mammalian factor that binds telomeric TTAGGG repeats in vitro. Mol Cell Biol. 1992;12:4834–4843. doi: 10.1128/mcb.12.11.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]