

Abstract
Mesodermal cells signal to neighboring epithelial cells to modulate their proliferation in both normal and disease states. We adapted a C. elegans organogenesis model to enable a genome-wide mesodermal-specific RNAi screen and discovered 39 factors in mesodermal cells that suppress the proliferation of adjacent ‘Ras pathway-sensitized’ epithelial cells. These candidates encode components of protein complexes and signaling pathways that converge on the control of chromatin dynamics, cytoplasmic polyadenylation and translation. Stromal fibroblast-specific deletion of candidate mouse orthologs of several candidates resulted in the hyperproliferation of mammary gland epithelium. Furthermore, a 33-gene signature of human orthologs was selectively enriched in the tumor stroma of breast cancer patients and depletion of these factors from normal human breast fibroblasts increased proliferation of co-cultured breast cancer cells. This cross-species approach identified unanticipated regulatory networks in mesodermal cells with growth suppressive function, exposing the conserved and selective nature of mesodermal-epithelial communication in development and cancer.
INTRODUCTION
Temporal and spatial regulation of signaling from mesodermal cells to epithelial cells is required during embryonic and adult development (Wiseman and Werb, 2002). Mesodermal signaling pathways used to instruct epithelial cell fate and proliferation have been well defined in the context of the developing vulva of C. elegans. During vulval organogenesis, mesodermal tissues play an inductive role by releasing EGF ligand (LIN-3) from the gonad-derived anchor cell, which binds and activates the EGF receptor (LET-23) in adjacent epithelial vulval precursor cells (VPCs) (Hill and Sternberg, 1992). Additional Wnt ligands from multiple tissues including muscles and neurons (Myers and Greenwald, 2007), as well as lateral Notch signaling within VPCs (Chen and Greenwald, 2004), provide spatial orientation cues that cooperate with EGF signaling to appropriately instruct cell divisions that result in the 22-cells of the vulva (Figure S1A; Eisenmann et al., 1998). Disruption of Ras, Notch, or Wnt signaling alters patterning of VPCs and can lead to increased (Multivulva) VPC proliferation (reviewed in Sternberg, 2005).
Mesodermal cells can respond to extreme but normal physiological signals in mammals, such as wound healing, inflammation, organ regeneration and pregnancy (Nelson and Bissell, 2006), and sometimes be inappropriately co-opted in diseases such as cancer (Mueller and Fusenig, 2004; Schedin, 2006). Accordingly, increases in mesodermal cells at the site of a tumor are associated with poor survival of breast, prostate, pancreatic and colon cancer patients (reviewed in Paulsson and Micke, 2014). Cancer-associated fibroblasts (CAFs) represent a major component of the tumor microenvironment, which is also comprised of adipocytes, immune and endothelial cells surrounded by an extra-cellular matrix rich in growth factors, cytokines and enzymatic activities. Classic xenograft experiments demonstrated that co-injection of CAFs enhanced the initiating capacity and aggressiveness of prostate tumor cells (Olumi et al., 1999). Experiments in the last decade suggested that increased PI3K or decreased p53, TGF-β and Notch signaling in stromal fibroblasts collaborates with genetic alterations in tumor cells to incite a number of molecular, cellular and histopathological changes associated with malignant progression (Addadi et al., 2010; Bhowmick et al., 2004; Bronisz et al., 2012; Carstens et al., 2014; Hill et al., 2005; Hu et al., 2012; Pickard et al., 2012; Trimboli et al., 2009). Despite accumulating clinical and experimental evidence highlighting the importance of the tumor stroma in promoting cancer initiation, progression and therapeutic resistance, the identification of mesodermal derived factors that influence the critical hallmarks of tumor cell behavior remains a major challenge (Hu et al., 2005).
Here we exploited the well-defined vulval developmental program and RNAi technology in C. elegans to systematically query and identify mesodermal derived factors and signaling pathways used to suppress proliferation of epithelial cells sensitized with cancer-relevant mutations. Central to this approach was the introduction of two key genetic modifications into worms that allowed non-epithelial cells to be specifically targeted by RNAi and epithelial cells to be sensitized to Ras pathway perturbations. Using this system we performed a genome-wide RNAi screen and identified 39 factors that function non-autonomously to suppress the proliferation of Ras-sensitized epithelial cells, with minimal impact on the proliferation of normal or Wnt pathway-sensitized cells. These candidate genes encode histone variants and components of protein complexes known to converge on the control of chromatin dynamics, cytoplasmic polyadenylation and translation. We then generated mice with conditional alleles of three representative candidates (Tlk1, Tlk2 and Sympk) and show that their fibroblast-specific ablation results in dramatic mammary ductal cell hyper-proliferation in the absence of overt hyper-proliferation in other organs. Moreover, depletion of stromal factors from normal human breast fibroblasts enhanced the proliferation of breast cancer cell lines. In summary, this cross-species approach identified key molecular machineries in mesodermal/stromal cells that suppress the proliferation of adjacent epithelial cells, highlighting the conserved and selective nature of mesodermal-epithelial cell communication during extreme but physiologically sensitized settings in vivo.
RESULTS
Generation of mesoderm-specific RNAi and Ras-sensitized C. elegans
In C. elegans, RNAi precursor molecules freely mobilize across tissues (Jose and Hunter, 2007). Thus, we developed strains that restricted RNAi sensitivity to mesodermal tissues (Figure 1A). To this end, we used RNAi-resistant animals (RNAi-Defective-1, rde-1(−); (Qadota et al., 2007; Tabara et al., 1999) and reinstated RNAi competency selectively back in the mesodermal cells surrounding VPCs (anchor cell, somatic gonad and muscle) by re-expressing wild-type rde-1 from well-characterized promoters (ACEL∷Δpes-10, ddr-2 and myo-3, respectively). Using these promoters to drive gfp expression, we confirmed anchor cell-, somatic gonad- and muscle-expression (Figure 1B, S1B). ddr-2 also drives expression in some neurons and intestinal cells, but not epithelial cells. In our hands, ddr-2 represented the best reagent that lacked epithelial expression but exhibited robust and broad somatic gonad expression among the 40 gfp reporter strains we tested that had similarly annotated expression patterns in Wormbase.org. In addition, a loss-of-function mutation in rrf-3 (rrf-3(−)) was introduced into worms containing all three rde-1 transgenes to enhance general RNAi sensitivity (for simplicity, we term this strain mesoderm-rde; Figure S1C; (Simmer et al., 2002)).
Figure 1. Mesoderm-specific RNAi in C. elegans containing a gap-1(−) mutation.
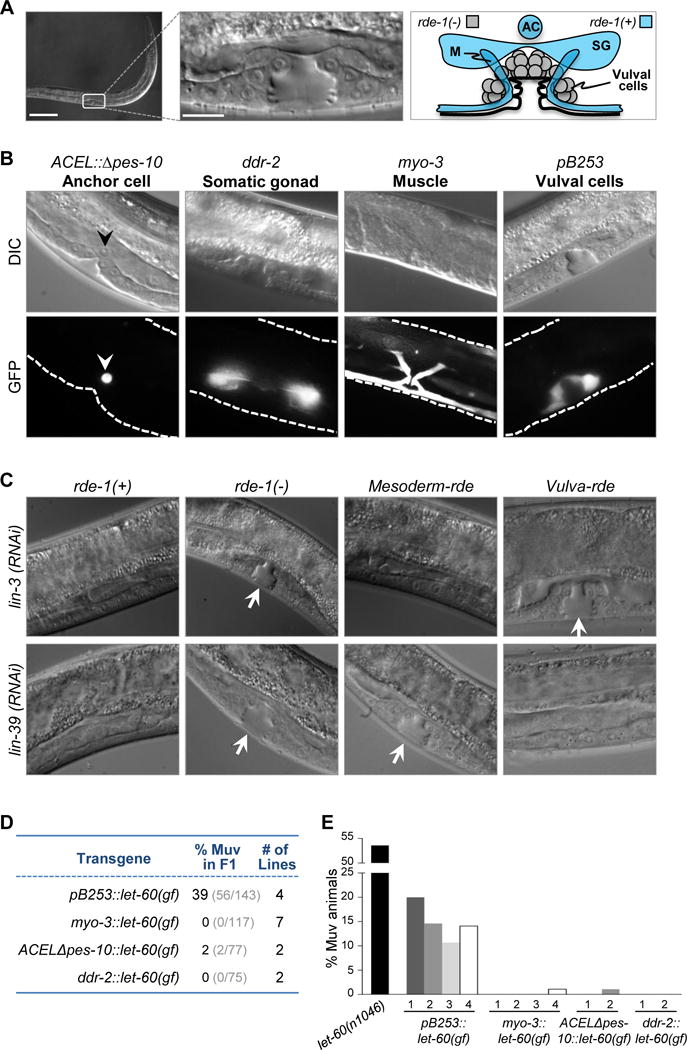
(A) Differential interference contrast (DIC) images of wild-type C. elegans at mid-larval L4 stage and a schematic diagram of a normal vulva. (bar = 100 μm, high-mag. bar = 10 μm) AC, anchor cell; SG, somatic gonad; M, muscle. Diagram of tissues that are RNAi resistant (rde-1(−)) are indicated in gray; tissues that have been reconstituted with rde-1(+) are indicated in blue. (B) DIC (top) and fluorescence (bottom) images of transgenic worms expressing GFP driven by promoters active in the anchor cell (arrowhead), somatic gonad, muscle, and vulval cells. Dashed lines,worm body boundaries. (C) DIC images of wild-type rde-1 worms (rde-1(+)), rde-1 mutants (rde-1(−)), rde-1(−) mutants with mesoderm-specific RNAi (mesoderm-rde), and rde-1(−) mutants with VPC-specific RNAi (vulva-rde) following lin-3 and lin-39 RNAi. Arrows, normal vulva structures. (D) F1 individuals with tissue-specific expression of let-60(gf) constructs were evaluated for the Muv phenotype. The unc-119(+) allele was used as marker to indicate successful transmission of transgenes. The number of stable transgenic lines generated for each construct is indicated. (E) Offspring for the indicated number of transgenic lines (each bar represents a single independent line; >70 animals were analyzed for each line) were evaluated for the Muv phenotype. Homozygous let-60(n1046) mutants were used for comparison. See also Figure S1 and Table S3.
Mesoderm-specific RNAi sensitivity in mesoderm-rde worms was validated using two methods. First, we tested the consequences of RNAi against lin-3 (epidermal growth factor-like ligand) and lin-39 (homeobox transcription factor), two genes essential for vulval induction known to function in the anchor cell and VPCs, respectively. As expected, control RNAi-resistant rde-1(−) animals were unaffected by lin-3(RNAi) and lin-39(RNAi), whereas RNAi-competent rde-1(+) animals exhibited a Vulvaless (Vul) phenotype in response to both treatments (Figure 1C). In mesoderm-rde animals, lin-3(RNAi) but not lin-39(RNAi) preferentially resulted in a Vul phenotype (Figure 1C and S1D). We also reinstated RNAi-competency in VPCs by expressing wild-type rde-1 from a VPC-specific, synthetic promoter that contains regulatory elements from lin-31 (pB253, Figure 1B). In this case, lin-39(RNAi) but not lin-3(RNAi) resulted in a Vul phenotype (Figure 1C). The mild lin-39(RNAi) phenotype in mesoderm-rde animals could be due to leaky RNAi sensitivity in the VPCs, or due to the expression and functions of lin-39 in cells other than the VPCs (Wagmaister et al., 2006). Thus, we used a second established, sensitive assay to test the ability of each tissue-specific promoter to induce a Multivulva (Muv) phenotype when directing the expression of constitutively activated Ras (let-60(gf)) (Myers and Greenwald, 2005). Consistent with previous findings, the VPC-specific expression of activated Ras resulted in a Muv phenotype (Figure 1D, E). In contrast, when driven by the mesoderm promoters, the activated Ras transgene failed to induce ectopic VPC cell divisions, suggesting that these promoters display non-detectable activity in the VPCs with this orthogonal approach. Collectively, these results demonstrated restricted RNAi sensitivity in mesoderm-rde worms.
Next, we introduced a gap-1(−) mutation that would sensitize VPCs to ectopic cell divisions upon receiving additional mesodermal signals. Several observations prompted us to consider gap-1(−) as a ‘sensitizing’ mutation (Figure 2A). Ras GTPase activating protein (GAP) genes are frequently lost in human cancer and function as tumor suppressors (NF1, DAB2IP, RASAL1, RASAL2, RASA1, RASA2 and IQGAP2) (Arafeh et al., 2015; Holzel et al., 2010; Liu et al., 2013; Maertens and Cichowski, 2014; McLaughlin et al., 2013; Min et al., 2010). In C. elegans, a gap-1 loss-of-function allele (gap-1(−)) leads to ectopic Ras signaling, as measured by an in vivo Ras reporter (Figure S1E, egl-17p∷cfp-lacZ; (Yoo et al., 2004)). The level of Ras activation attained in gap-1(−) worms, however, is insufficient to promote ectopic VPC divisions and consequently, the vulva appears anatomically normal (Hajnal et al., 1997; Hopper et al., 2000). Importantly, gap-1(−) can collaborate with additional oncogenic mutations to induce excessive VPC divisions and a Muv phenotype (Hopper et al., 2000; Yoo et al., 2004). Based on these previous observations we hypothesized that gap-1(−) would synergize with the RNAi mediated depletion of critical mesodermal factors to elicit a Muv phenotype. Thus, we intercrossed gap-1(−) and mesoderm-rde worms to generate the final strain to be used in the genome-wide RNAi screen.
Figure 2. Genome-wide screen for the identification of mesodermal factors that induce a Muv phenotype.
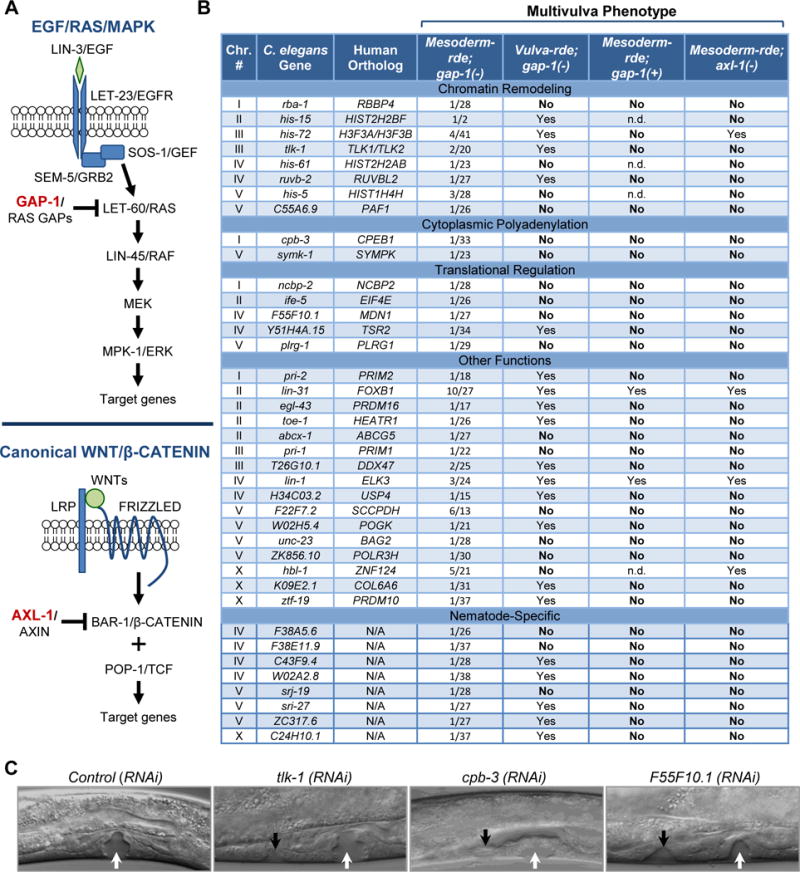
(A) Schematic representation highlighting the inhibitory roles of GAP-1 and AXL-1 in EGF/RAS/MAPK and canonical WNT/β-CATENIN signaling, respectively. (B) Summary of Muv phenotypes after RNAi-mediated depletion of identified genes in mesoderm-rde;gap-1(−), vulva-rde;gap-1(−), mesoderm-rde;gap-1(+) and mesoderm-rde;axl-1(−) animals. ‘YES’ ≥ 1 and ‘NO’ = 0 Muv out of approximately 40 worms. n.d., not determined due to developmental deficiencies. Chr., chromosome. (C) Representative DIC images of Muv in mesoderm-rde;gap-1(−) worms at mid-L4 stage following mesodermal RNAi. White and black arrows, normal and ectopic vulva structures, respectively. See also Figure S2.
Genome-wide screen in C. elegans identifies mesodermal factors that suppress epithelial cell proliferation
The Ahringer E. coli RNAi library containing 16,757 clones, which account for approximately 86% of the C. elegans genome (Kamath et al., 2003), was used to systematically feed mesoderm-rde;gap-1(−) larvae on a gene-by-gene basis (Figure S1F). Ectopic vulval proliferation results in ventral protrusions in the adult. We thus used these protrusions (easily visualized by standard light microscopy) as an indication of additional vulval cell proliferation. RNAi resulting in overt vulval defects in at least two of three independent replicates were classified as candidates (Figure S2A). The testing of over 16,000 C. elegans genes identified 483 mesodermal candidates that when depleted by mesoderm-specific RNAi, led to vulval defects. To discriminate ectopic VPC divisions from other tissue alterations that could lead to vulval protrusions, differential interference contrast (DIC) microscopy was used to examine fourth stage larvae at the cellular level. Evaluation of the 483 candidate RNAi using this secondary screen resulted in 39 mesodermal products that when depleted, promoted ectopic VPC divisions (Figure 2B, C and S2B).
Several conclusions may be drawn from the identity of these 39 genes. First, 31 of the 39 (79%) genes have 33 mammalian orthologs (his-72 and tlk-1 have two orthologs each), whereas only 47% of all genes in the RNAi library have a mammalian ortholog (Figure S1F). This level of enrichment for mammalian orthologs was highly significant (P<0.0001), suggesting that mesodermal signaling networks revealed by this screen are conserved from nematodes to humans. Second, the vast majority of the 39 candidates were never identified by previous whole body RNAi screens using non-sensitized C. elegans, highlighting the mesoderm-specific nature of our screen and suggesting distinct aspects of mesoderm-epithelium communication in the control of cell proliferation. Also of note, the screen did not recover known genes that restrict lin-3/EGF gene expression, such as synMuv genes. This was not completely unexpected, however, given that some synMuv genes have their effects in dpy-7-expressing epidermal cells (Myers and Greenwald, 2005) and our screen strain was not targeting this particular tissue type. Finally, there was a striking enrichment for factors that control chromatin remodeling, cytoplasmic polyadenylation and translation (Figure 2B and S2B). Mesodermal factors related to chromatin remodeling included orthologs to a histone H3 variant (H3F3A, H3F3B), three additional histones (HIST2H2BF, HIST2H2AB and HIST1H4H), tousled-like kinase that phosphorylates histone H3 (TLK1, TLK2) (Carrera et al., 2003), and key chromatin binding factors (RBBP4, PAF1 and RUVBL2). Factors related to cytoplasmic polyadenylation and translation included two components (SYMPK and CPEB1) of a complex that regulates the translation of specific subsets of mRNAs by modulating their poly(A) tail length (D’Ambrogio et al., 2013), and five components of the protein translation machinery (NCBP2, EIF4E, MDN1, TSR2 and PLRG1). The convergence of mesodermal hits on a few selected processes is remarkable, involving multiple components of the same protein complex or with the same general function.
Cell-type and gap-1(−)-dependent functions of mesodermal gene candidates
To determine whether the 39 candidate genes exclusively function in mesodermal cells or also function in VPCs to suppress ectopic cell divisions, we examined the effects of depleting candidates in mesoderm-rde;gap-1(−) versus vulva-rde;gap-1(−) worm strains. This comparison showed that depletion of 20 out of the 39 gene products (51%) from the VPC compartment also induced excessive cell divisions and a Muv phenotype (Figure 2B), which included two genes with established roles in VPCs during development (Tan et al., 1998). These findings suggest that the other 19 genes function exclusively in mesodermal cells to suppress ectopic VPC divisions. Intriguingly, the 19 mesoderm-specific genes have functions enriched for cytoplasmic polyadenylation (cpb-3 and symk-1) and translation control (ncbp-2, ife-5, F55F10.1, plrg-1). This analysis stratifies factors that function exclusively in mesodermal cells from factors that also have a cell autonomous role in cell fate and proliferation control decisions.
We next tested whether suppression of ectopic VPC division by the 39 genes was dependent or independent of the gap-1(−) sensitizing mutation. To this end, we compared depletion of each candidate in mesoderm-rde animals with an intact (gap-1(+)) or mutant gap-1 allele. As listed in Figure 2B, depletion of only 2 genes (lin-31 and lin-1) led to ectopic VPC divisions in gap-1(+) worms, suggesting that the vast majority of the 39 mesodermal factors suppress excessive VPC proliferation only in Ras pathway-sensitized worms and not in the context of normal development.
Finally, we examined whether these mesodermal factors collaborate with other growth control pathways in VPCs beyond Ras by examining the consequence of depleting the 39 genes in mesoderm-rde worms containing an axl-1(−) mutation. The human orthologs of axl-1, AXIN1 and AXIN2 are part of the complex that targets β-catenin for degradation (Figure 2A). Importantly, loss of axl-1 in worms activates Wnt signaling in VPCs but is insufficient to cause ectopic VPC divisions; however, when combined with other activating mutations in the Wnt pathway, axl-1(−) leads to a Muv phenotype (Oosterveen et al., 2007). We found that in this Wnt-sensitized background, mesodermal depletion of only 4 of the 39 genes resulted in a Muv phenotype (Figure 2B). In addition, a separate genome-wide screen with the mesoderm-rde;axl-1(−) strain identified only distinct factors whose depletion resulted in a Muv phenotype (data not shown).These findings demonstrate that depletion of individual mesodermal genes is unlikely to simply create a microenvironment that is generally permissive for cell proliferation. Rather, we suggest that their depletion from mesodermal tissues generates productive signals that cooperate with defined mutations and/or growth-regulatory pathways in neighboring epithelial VPCs.
Mesodermal factors suppress ectopic Ras signaling in sensitized VPCs
Developmental programs often use diverse signaling inputs that converge on distinct components of the same pathway to exacerbate a defined signaling output. Thus, given that GAP-1 is a negative regulator of the Ras/MAPK pathway, we initially investigated whether depletion of each of the 39 mesodermal factors might promote excessive VPC proliferation through a common underlying mechanism involving hyper-activation of this pathway. To this end, an in vivo reporter (egl-17p∷cfp-lacZ) was used to quantify Ras/MAPK signaling in response to RNAi treatment of mesoderm-rde animals with either an intact or mutant gap-1 allele. In control gap-1(+) larvae, reporter activity was exclusively restricted to the 1° VPC lineage after one division of P6.p (Figure 3A, B). As expected, approximately 60% of gap-1(−) larvae exhibited low levels of reporter activity in presumptive 2° VPC lineages at this same time point (Figure 3A, C). Depletion of most mesodermal genes in the gap-1(+) background also led to low ectopic Ras activity in presumptive 2° VPC lineages (Figure 3B), albeit to varying extents. Importantly, depletion of most mesodermal genes in the gap-1(−) sensitized background resulted in a further increase in the frequency and intensity of reporter activity in presumptive 2° and 3° VPC lineages (Figure 3A, C). These findings show that mesodermal depletion of the 39 genes augments Ras activity beyond the levels attained in gap-1(−) sensitized worms, and suggest that combinatorial activation of Ras signaling may be a common mechanism by which their depletion from mesodermal cells promotes ectopic division of gap-1(−) vulval epithelial cells.
Figure 3. Depletion of mesodermal factors induces ectopic Ras signaling in VPC lineages.
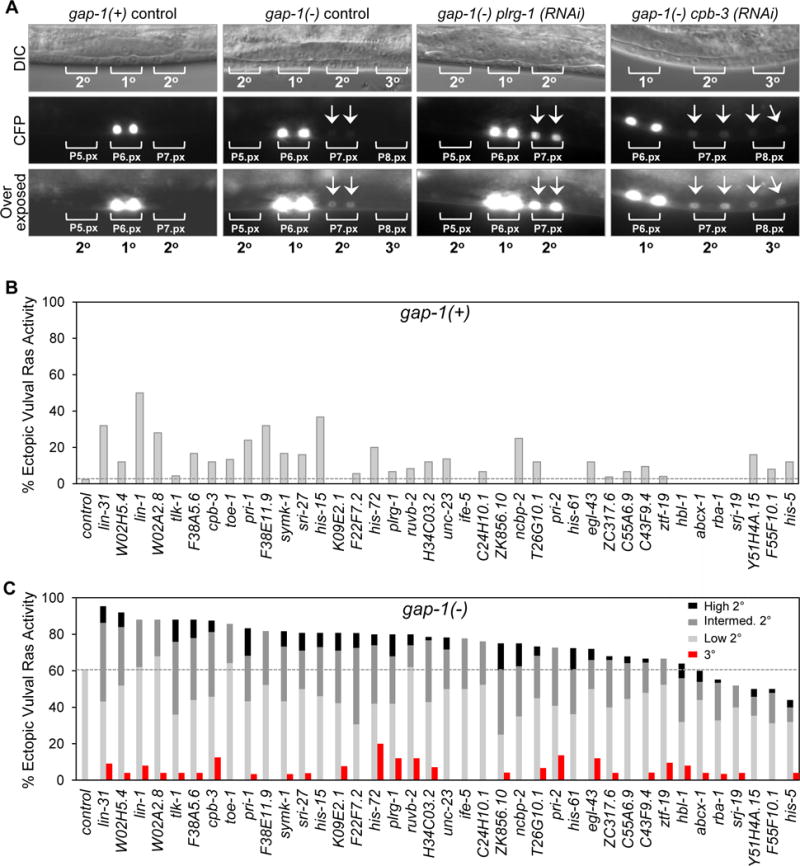
(A) Representative DIC and fluorescence images of non-sensitized (gap-1(+)) and sensitized (gap-1(−)) worms expressing a LET-60/RAS-dependent transcriptional reporter. Bottom panels are overexposed to better visualize CFP expression in P5.px, P7.px and P8.px cells. (B) Percentage of gap-1(+) animals with ectopic vulval Ras activity only in presumptive 2° lineage cells following gene-specific or empty vector (control) RNAi. n=44 (control); n>25 (each gene). (C) Percentage of gap-1(−) animals with ectopic vulval Ras activity in both 2° and 3° lineage cells (red bars) or only in presumptive 2° lineage cells (gray bars) following gene-specific or empty vector (control) RNAi. CFP intensity in presumptive 2° lineage cells was scored as high, intermediate (intermed.) and low. n=99 (control); n>25 (each gene). See also Figure S1.
Fibroblast-specific ablation of Tlk1, Tlk2 and Sympk in mice promotes proliferation of mammary ductal epithelial cells
Sequence analysis of the 39 candidate genes across species showed that the mesodermal factors identified above are conserved in mice and humans (Figure 2B and S1F), suggesting similar mesodermal-epithelial communication networks may be at play in the control of cellular proliferation in mammals. Thus, we considered whether representative mammalian orthologs of two C. elegans candidate genes (tlk-1 and symk-1) involved in chromatin remodeling and cytoplasmic polyadenylation could function to regulate cell proliferation in a cell non-autonomous manner in the mouse.
Tousled-like kinase 1 and 2 (Tlk1 and Tlk2) are the two related mouse orthologs of the C. elegans tlk-1 gene. TLK1 and TLK2 constitute a family of serine/threonine kinases that phosphorylate the histone chaperone ASF1 and histone H3 to coordinately regulate chromatin assembly and remodeling (Carrera et al., 2003; Klimovskaia et al., 2014; Li et al., 2001; Sillje and Nigg, 2001; Sillje et al., 1999). To rigorously evaluate their roles in vivo, we used standard homologous recombination approaches to introduce conditional LoxP-flanked Tlk1 and Tlk2 alleles into the mouse genome (Tlk1Neo and Tlk2Neo, respectively; Figure 4A, B). Southern blot and PCR genotyping confirmed correct integration of Tlk1Neo and Tlk2Neo into mouse embryonic stem cells (ES; Figure 4C, D). Targeted ES cells were then used to generate mice carrying the germ-line transmitted Tlk1Neo and Tlk2Neo alleles, and positive offspring were bred with Actin-Flpe mice to remove the Frt-flanked neomycin selection cassette and yield mice with the conditional Tlk1F and Tlk2F alleles (Figure 4C, D).
Figure 4. In vivo deletion of Tlk1 and Tlk2 in mouse mammary fibroblasts increases proliferation of adjacent ductal epithelium.
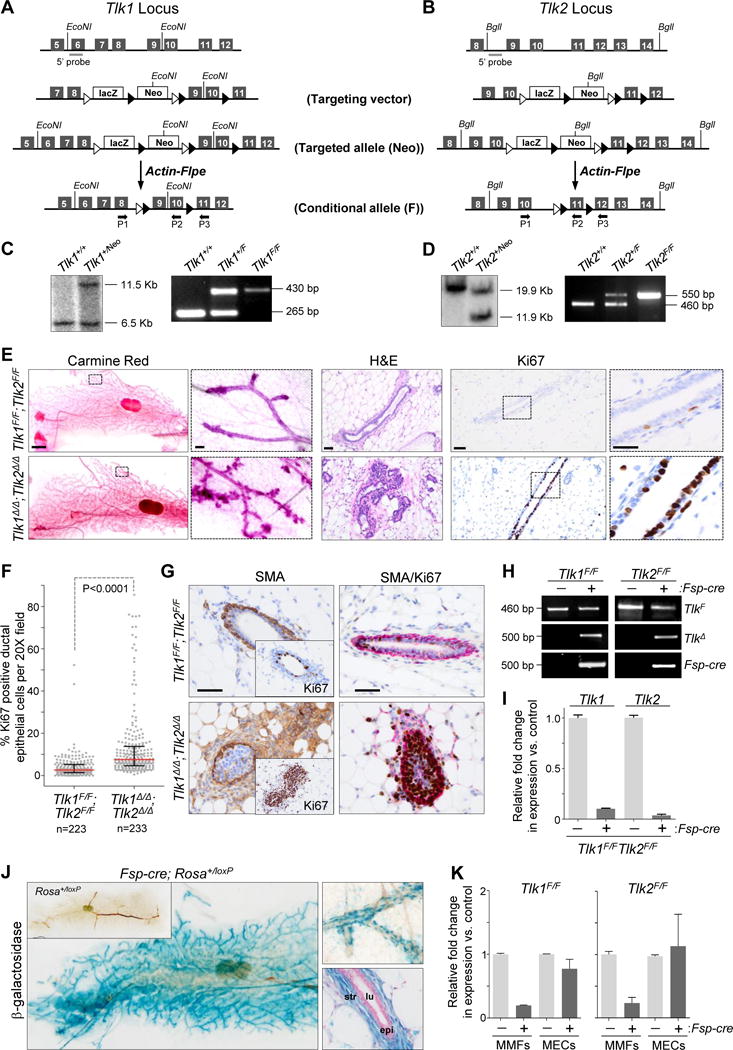
(A, B) Schematic overview of the targeting vector used to generate the Tlk1 and Tlk2 conditional knock-out mice. Line 1: partial genomic locus of mouse Tlk1 or Tlk2 gene with exons represented as dark gray boxes and Southern probe as light gray bar. Line 2: targeting vector with exons 9 and 10 in Tlk1 and exon 11 in Tlk2 flanked by loxP elements (black triangles), the LacZ reporter cassette and neomycin (Neo) resistance cassette flanked by FRT sites (white triangles). Line 3: targeted genomic locus (Neo). Line 4: Neo cassette was removed by Actin-Flpe mediated recombination (Flox (F)). Black arrows P1–P3 indicate location of genotyping primers. Diagram not to scale. (C, D) Southern blot (left panels) of ES cells confirming targeting of Tlk1 or Tlk2 genomic loci respectively. PCR genotyping of genomic DNA (right panels) from WT (Tlk1+/+ or Tlk2+/+) heterozygous (Tlk1+/F or Tlk2+/F) and homozygous (Tlk1F/F or Tlk2F/F) mice with primers P1/P2 following deletion of Neo cassette. (E) Left: bright field images of whole mount inguinal mammary glands from 10-week-old Tlk1F/F;Tlk2F/F(control) and Fsp-cre;Tlk1F/F;Tlk2F/F (Tlk1Δ/Δ;Tlk2Δ/Δ) mice stained with carmine red (bar = 2000 μm). Boxes indicate area of magnification in adjacent panel (bar = 100 μm). Middle: H&E stained sections of opposite inguinal gland (bar = 50 μm). Right: representative images of sections of inguinal mammary glands immuno-stained for the proliferation marker Ki67 (brown) and counterstained with hematoxylin (blue) (bar = 50 μm). Boxes indicate area of magnification in adjacent panel (bar = 20 μm). (F) Percent Ki67 positive epithelial cells per 20X field (dots) of tissue sections described in (E). Red bar, median; black bars, interquartile range; pooled counts from 6 control mice or 5 Fsp-cre mice. P value: Mann-Whitney test of medians. (G) Left: Representative images of inguinal mammary gland sections immuno-stained for smooth muscle actin (SMA; brown) and counterstained with hematoxylin (blue) (bar = 50 μm). Insets represent consecutive sections immuno-stained with Ki67 (brown) and counterstained with hematoxylin (blue). Right: Representative images of inguinal mammary gland sections immuno-stained for smooth muscle actin (SMA; red) and Ki67 (brown); counterstained with hematoxylin (blue) (bar = 50 μm). (H) PCR genotyping of mammary fibroblasts derived from Tlk1F/F and Tlk2F/F mice with or without Fsp-cre to detect the Flox allele (TlkF; P1/P2 primers), the deletion product formed after Fsp-cre-mediated recombination (TlkΔ; P1/P3 primers) and Fsp-cre. (I) qRT-PCR for Tlk1 or Tlk2 on cDNA derived from fibroblasts isolated from Tlk1F/F;Tlk2F/F mammary glands of control (−) and Fsp-cre positive (+) mice. Expression was normalized to Gapdh. (J) Mammary gland wholemounts from Fsp-cre;RosaloxP/+ and RosaloxP/+ (left, inset) mice stained for β-galactosidase. Higher (10×) magnification of a wholemount gland (top-right) and a cross section stained for β-galactosidase and counterstained with nuclear fast red (bottom-right). Str, stromal fibroblasts; lu, lumen; epi, epithelial cells. (K) qRT-PCR for Tlk1 expression (left) on cDNA derived from Tlk1F/F mammary fibroblasts (MMFs) or mammary epithelial cells (MECs) lacking (−) or containing (+) the Fsp-cre transgene. qRT-PCR for Tlk2 expression (right) on cDNA derived from Tlk2F/F mammary fibroblasts (MMFs) or mammary epithelial cells (MECs) lacking (−) or containing (+) the Fsp-cre transgene. Expression was normalized to Gapdh. See also Figure S3, S4 and Table S3.
The Fsp-cre transgene (Trimboli et al., 2009) was used to evaluate the role of TLK1 and TLK2 in stromal fibroblasts of the mouse. Fsp-cre transgene expression in fibroblasts is turned on beginning at E15.5 of mouse embryonic development (Trimboli et al., 2009; Trimboli et al., 2008). By interbreeding Fsp-cre; Tlk1F/F and Tlk2F/F mice we generated cohorts of control Tlk1F/F;Tlk2F/F and experimental Fsp-cre;Tlk1F/F;Tlk2F/F (Tlk1Δ/Δ;Tlk2Δ/Δ) mice. Both Tlk1F/F;Tlk2F/F and fibroblast-deleted Tlk1Δ/Δ;Tlk2Δ/Δ ten-week-old mice appeared externally normal and necropsies revealed normal anatomy with fully developed organs (data not shown). However, whole-mount preparations of inguinal mammary glands from Tlk1Δ/Δ;Tlk2Δ/Δ females displayed increased tertiary branching and alveolar clustering when compared to similarly estrous cycle staged Tlk1F/F;Tlk2F/F mammary glands (Figure 4E). When the individual Tlk1 or Tlk2 genes were ablated in fibroblasts (Tlk1Δ/Δ and Tlk2Δ/Δ, respectively), we observed a similar hyper-proliferative phenotype as when both genes were simultaneously deleted, albeit less pronounced (Figure S3D, E). Immunohistochemistry (IHC) with the proliferation marker Ki67 revealed increased mammary duct epithelial cell proliferation in Tlk1Δ/Δ;Tlk2Δ/Δ females (Figure 4E, F). Interestingly, Tlk1Δ/Δ;Tlk2Δ/Δ glands had increased SMA-positive fibroblasts, which almost always surrounded the Ki67-positive ducts (Figure 4G and S3A) and are typically only observed around terminal end-buds of wild-type mammary glands, indicating that loss of Tlk1 and Tlk2 leads to a fibroblast phenotype reminiscent of cancer-associated fibroblasts (CAFs) (Paulsson and Micke, 2014). IHC with cleaved caspase-3 specific antibody showed no difference in programmed cell death between the two cohorts (Figure S3B, C). Likewise, phosphor-ERK staining (indicative of RAS pathway activity) was similar between cohorts (Figure S3F).
Previous cell-type marker analysis using a β-galactosidase Rosa26LoxP reporter allele showed specific Fsp-cre transgene expression in stromal fibroblasts surrounding the mammary epithelial ducts, with virtually no expression in cytokeratin-positive epithelial cells, F4/80-positive macrophages or CD31-positive endothelial cells (Trimboli et al., 2009; Trimboli et al., 2008). Here we report minimal expression in other organs such as lung, liver, stomach and intestine (Figure S4A; perhaps contributing to the mammary-specificity of the phenotype) with widespread expression in skin (Figure S4B; precluding analysis of fibroblast-specific effects). Indeed, the β-galactosidase Rosa26LoxP reporter allele (Soriano, 1999) showed cre-mediated recombination specifically in stromal fibroblasts surrounding the mammary epithelial ducts (Figure 4J and S4C). Cre-mediated recombination was validated by PCR genotyping of genomic DNA and by quantitative real time PCR (qRT-PCR) of mRNA isolated from purified Tlk1Δ/Δ;Tlk2Δ/Δ mammary fibroblasts (Figure 4H, I). Moreover, qRT-PCR of mRNA from purified mammary fibroblasts and epithelial cells confirmed the fibroblast-specific reduction in Tlk1 and Tlk2 expression (Figure 4K).
The mouse ortholog of the C. elegans symk-1 gene, Symplekin (Sympk), encodes a scaffold protein that tethers target mRNAs to components of the cytoplasmic polyadenylation complex (Barnard et al., 2004), including CPEB1, which was also identified by our screen. The SYMPK protein complex binds to the 3′ untranslated region of a select class of mRNAs containing a consensus UUUUA1–3U sequence and regulates their translation through polyadenylation (activation) and deadenylation (repression) of poly(A) tails (D’Ambrogio et al., 2013). Using a similar strategy as described above, we generated mice with a targeted Sympk allele (SympkNeo/Neo; Figure 5A, B), which were bred with Actin-Flpe mice to yield offspring with a conditional LoxP-flanked Sympk allele (SympkF/F; Figure 5A, C). To evaluate the consequences of Sympk ablation in stromal fibroblasts of the mouse, Fsp-cre and SympkF/F mice were interbred to generate cohorts of control SympkF/F and Fsp-cre;SympkF/F (SympkΔ/Δ) mice. Ten-week-old SympkF/F and SympkΔ/Δ mice were viable and appeared externally normal without any obvious defects in organogenesis, including in the gross morphology of mammary glands (Figure S5A). However, IHC with Ki67 showed increased ductal epithelial cell proliferation in mammary glands from SympkΔ/Δ females relative to similarly estrous cycle staged SympkF/F females (Figure S5A, B). By six months of age, mammary glands from SympkΔ/Δ females displayed ectopic tertiary branching, increased alveolar clustering and excessive mammary epithelial cell proliferation when compared to control mice (Figure 5D, E). As in Tlk1Δ/Δ;Tlk2Δ/Δ females, there was no difference in cleaved caspase-3 (CC3) (Figure S5C–F) nor phosphor-ERK levels (Figure S5G) between SympkΔ/Δ and control mammary glands. Fibroblast-specific cre-mediated recombination was confirmed by PCR genotyping of genomic DNA and qRT-PCR of mRNA from isolated mammary fibroblasts and epithelial cells from SympkΔ/Δ female mice (Figure 5F–H). Thus, fibroblast-specific deletion of Tlk1, Tlk2 and Sympk in mice promoted the hyper-proliferation of adjacent mammary duct epithelial cells. From these observations we conclude that in the absence of TLK and SYMPK, the mammary gland is innately sensitive to ectopic stromal signaling, possibly reflecting the extreme hormone responsiveness of this organ.
Figure 5. In vivo deletion of Sympk in mouse mammary fibroblasts increases proliferation of adjacent ductal epithelium.
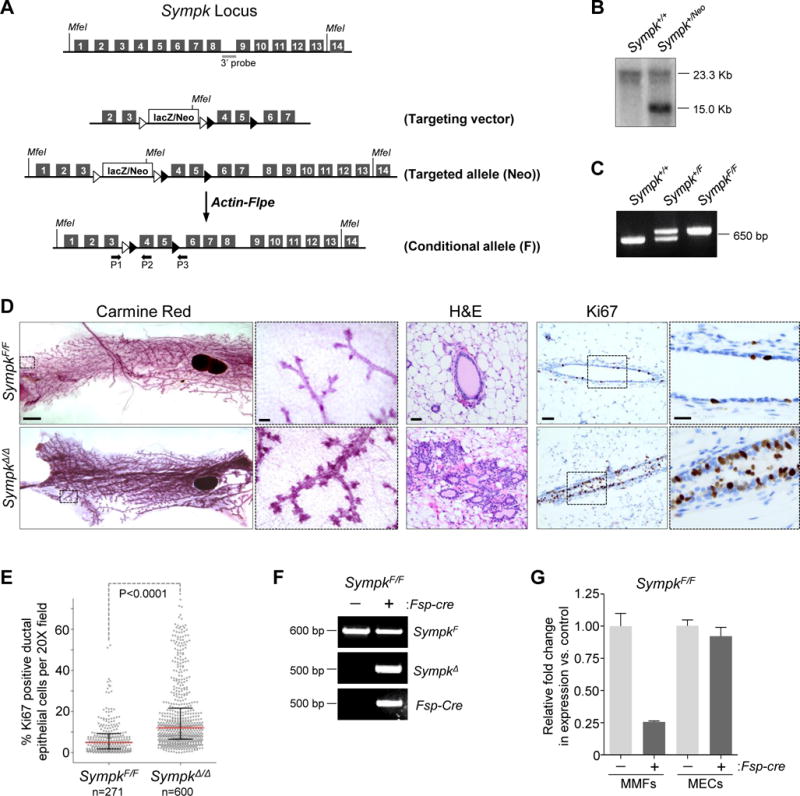
(A) Schematic overview of the targeting vector used to generate a Symplekin (Sympk) conditional knock-out mouse. See Figure 4 legend for key. (B) Southern blot of genomic DNA confirming targeting of Sympk genomic locus. (C) PCR genotyping of genomic DNA from WT (Sympk+/+) heterozygous (Sympk+/F) and homozygous (SympkF/F) mice with primers P1/P2 following deletion of Neo cassette. (D) Left: bright field images of whole mount inguinal mammary glands from 6-month-old SympkF/F or Fsp-cre;SympkF/F (SympkΔ/Δ) mice stained with carmine red (bar = 1000 μm). Boxes indicate area of magnification in adjacent panel (bar = 100 μm). Middle: H&E stained sections of opposite inguinal gland (bar = 50 μm). Right: representative images of sections of inguinal mammary gland from immuno-stained for the proliferation marker Ki67 (brown) and counterstained with hematoxylin (blue) (bar = 50 μm). Boxes indicate area of magnification in adjacent panel (bar = 20 μm). (E) Percent Ki67 positive epithelial cells per 20X field (dots) of tissue sections described in (D). Red bar, median; black bars, interquartile range; pooled counts from 5 control mice or 9 mice for the Fsp-cre group. P value: Mann-Whitney test of medians. (F) PCR genotyping of mammary fibroblasts derived from SympkF/F mice with or without Fsp-cre to detect Flox allele (F; P1/P2), deletion product formed after Fsp-cre-mediated recombination (SympkΔ; P1/P3) and Fsp-cre. (G) qRT-PCR for Sympk expression on cDNA derived from SympkF/F mammary fibroblasts (MMFs) or mammary epithelial cells (MECs) lacking (−) or expressing (+) the Fsp-cre transgene. Expression was normalized to Gapdh. See also Figure S4, S5 and Table S3.
The C. elegans-derived expression signature is associated with breast cancer stroma
To explore the relevance of our findings to human cancer, we utilized the C. elegans-derived 33-human ortholog signature to query the transcriptome profiles of stromal and epithelial compartments of both tumor and normal tumor-adjacent tissues that are derived from HER2-positive breast cancer patients (Table S4). Specifically, we laser capture microdissected (LCM) tumor-epithelium (n=40) and tumor-associated stroma (n=39), as well as normal-epithelium (n=14) and adjacent normal-stroma (n=14; at least 2 cm distant from tumor) from resected tumor tissue (Figure 6A and Table S4). RNA was then isolated from LCM samples and expression evaluated using an Agilent platform (Finak et al., 2008; Trimboli et al., 2009). We then queried these compartment-specific mRNA expression data sets with our C. elegans-derived 33-ortholog signature. The heatmap shown in Figure 6B represents the expression of the 33-ortholog signature in normal-stroma and tumor-stroma samples ranked along the x-axis according to the rank-sum across genes (see Methods; the complete analysis of the expression data will be published elsewhere). Compared to 10,000 permutations of 33 randomly selected genes, the 33-ortholog signature could effectively distinguish human tumor-stroma from adjacent normal-stroma (P=0.001). In contrast, a parallel analysis of expression profiles derived from LCM tumor and normal epithelium compartments (n=40 tumor epithelium; n=14 normal epithelium) failed to distinguish tumor epithelium from normal epithelium (P=0.482; Figure S6). We also note while some orthologs were down-regulated as expected from the C. elegans data (since depletion resulted in the Muv phenotype), many were up-regulated, possibly in response to proliferation of neighboring epithelial cells in an ineffective attempt to suppress proliferation. Furthermore, some of these genes may have evolved different functions over time or might be influenced by specific genetic interactions and therefore be varied across a patient population when compared to a homogenous population of worms.
Figure 6. The human ortholog signature distinguishes breast tumor stroma from adjacent normal stroma.
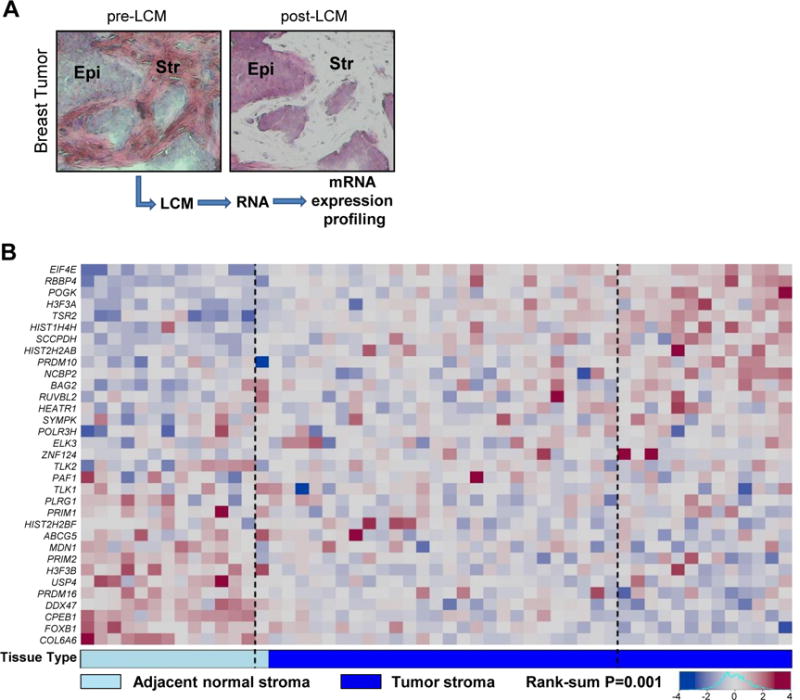
(A) Representative images showing pre- and post-laser capture microdissected (LCM) breast tumor stroma. Epi, epithelium; Str, stroma. (B) Heatmap displaying RNA expression of the 33 human orthologs in normal (n=14) and tumor stroma (n=39) from breast cancer patients. The boundaries (dashed lines) represent the 95% distribution limits for rank-sums of 10,000 randomly generated samples, where patient samples outside these boundaries have more extreme rank-sums than expected by chance. The rank-sum P value indicates the ability of the gene signature to distinguish normal from tumor stroma in breast cancer patients. See also Figure S6 and Table S4.
Taken together, we conclude that the C. elegans-derived expression signature is selectively represented in tumor-stroma of human HER2-positive breast cancer. Whether this stromal signature is also represented in other breast cancer subtypes remains to be determined.
Depletion of human orthologs from stromal fibroblasts enhances tumor cell proliferation
The selective representation of C. elegans-derived expression signature in breast cancer stroma may reflect stroma composition alterations during cancer progression and consequent gene expression changes. We thus intended to directly test the functionality of these identified genes in suppressing epithelial proliferation. Fibroblasts represent a major cellular component of breast tumor stroma. Therefore, we isolated primary breast fibroblasts from normal tissue (10 cm distant from the tumor) of breast cancer patients (n=44; see Methods). RNA was then isolated from these ‘normal’ fibroblasts and expression of the 33 human orthologs was confirmed using NanoString assays (Table S1). A representative fibroblast cell line with intermediate expression levels of the 33 genes was immortalized and depleted for each ortholog individually by shRNA lentiviral transduction; expression of 22 out of the 33 human orthologs was effectively reduced (Table S3, Figure S7A). We then assessed the consequence of their depletion on the proliferation of co-cultured immortalized non-transformed breast epithelial cells (MCF10A) and a panel of well-characterized breast cancer cells (MDA-MB-468, MDA-MB-231, BT474, MCF-7, and T47D; Figure S7C) using an established 3-dimensional (3D) assay (Figure 7A). In these assays, shRNA-expressing breast fibroblasts were co-embedded with normal or breast cancer cells expressing DsRed within a laminin-rich basement membrane extract in a 96-well plate format (Sasser et al., 2007a; Sasser et al., 2007b; Studebaker et al., 2008). Generally, tumor cell lines proliferated equally well when cultured with or without control fibroblasts, with MCF7 and T47D proliferating marginally better when cultured with control fibroblasts (Figure S7B). However, as shown by the heat maps in Figure 7B, the proliferation of MDA-MB-468, T47D and MCF7 cells was significantly enhanced by fibroblasts depleted for most stromal genes, when compared to fibroblasts treated with control shRNA (P<0.05, also see Table S2). The other two breast cancer cell lines (MDA-MB-231 and BT474) were less responsive, and MCF10A cells were completely non-responsive to co-cultured depleted fibroblasts. Examples of growth curves for MCF10A and each tumor cell line co-cultured with control fibroblasts or fibroblast depleted for four representative candidate stromal genes are shown in Figure 7C. These results demonstrate the selective effect of candidate gene-depleted fibroblasts on cancer cells versus non-transformed cells. Collectively, the data suggests that the core pathways used in mesoderm-epithelium communication are conserved from worms to humans and can be co-opted during development of human cancer.
Figure 7. Depletion of human orthologs in normal breast fibroblasts enhances breast cancer cell proliferation.
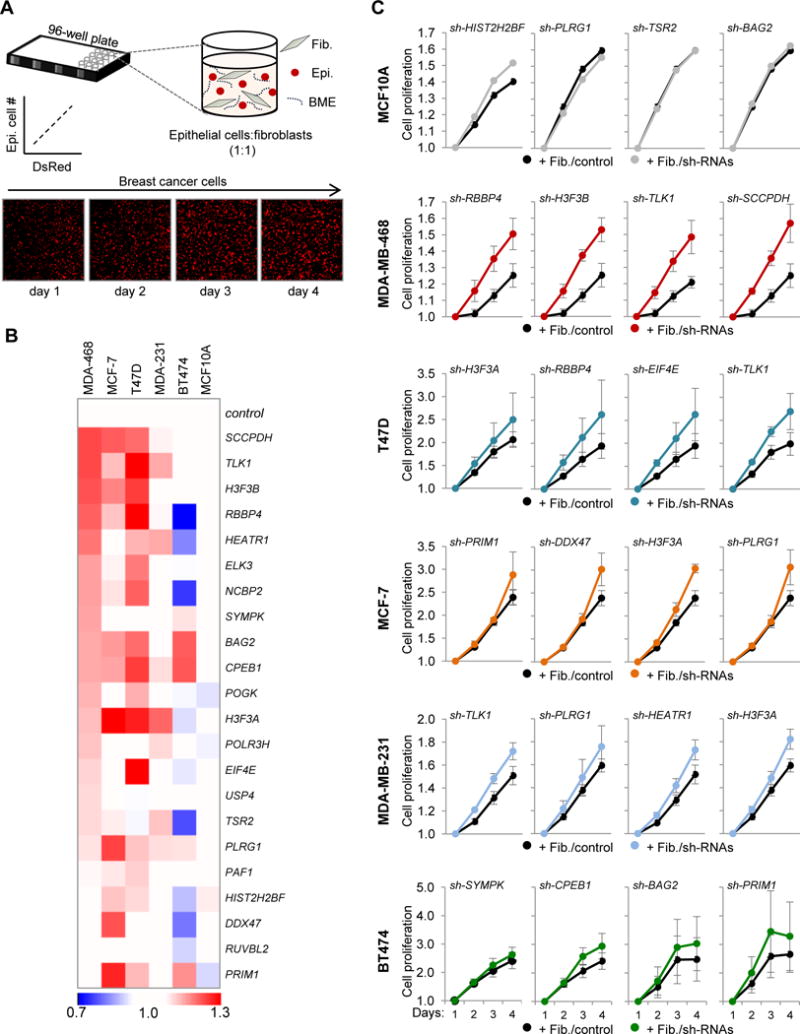
(A) Schematic representation of the 3D proliferation assay. DsRed-positive human breast epithelial cells (Epi.) were co-cultured with human breast fibroblasts (Fib.) at a 1:1 ratio in basement membrane extract (BME). Epithelial cell number correlates with DsRed fluorescence. Images of MCF-7 breast cancer cells are shown over four days. (B) Heatmap summarizing the effects of fibroblast-specific depletion of each stromal factor on the proliferation of co-cultured normal epithelial cells (MCF10A) or a panel of breast cancer cell lines. Co-culture results are presented relative to a non-silencing control shRNA at day 4 of culturing (breast cancer cells, n=3 independent experiments with each experiment performed in triplicate; MCF10A cells, n=2 independent experiments with each experiment performed in triplicate). (C) Growth curves of normal epithelial cells and breast cancer cells co-cultured for 4 days with fibroblasts expressing non-silencing (Fib./control) or stromal factor-specific shRNAs (Fib./sh-RNAs); the examples shown represent co-cultures with fibroblasts treated with factor-specific shRNAs that promoted tumor cell proliferation. Data are presented as average ±SEM. See also Figure S7, Table S1 and Table S2.
DISCUSSION
Mesodermal cells respond to a variety of extreme but physiological conditions as a mechanism to communicate with other cell types and help maintain normal tissue function (Nelson and Bissell, 2006). In cancer, stromal fibroblasts embedded within the tumor microenvironment provide neighboring cell types, including tumor cells, critical signals that facilitate the initiation and progression of carcinoma. However, the relevant stromal signals remain ill defined, as are the pathways that regulate them. Utilizing RNAi technology and a C. elegans vulval organogenesis system we identified 39 candidates involved in specialized forms of chromatin assembly and remodeling, mRNA processing and translation control that suppress the proliferation of adjacent sensitized epithelial cells. Fibroblast-specific knock-out models of three representative mammalian orthologs show that TLK1, TLK2 and SYMPK function in a cell non-autonomous manner to suppress mammary epithelial cell proliferation in the mouse. Importantly, expression profiling of laser capture microdissected cell compartments from human-derived normal and tumor tissue showed dysregulated expression of the related 33-ortholog signature selectively in the tumor-stroma of breast cancer patients. We suggest that these conserved candidates are utilized by mesodermal cells to communicate with and restrict the proliferative capacity of neighboring epithelial cells sensitized with a Ras pathway activating mutation.
Mesodermal components converge on the control of chromatin dynamics and translation
The level of convergence identified by this RNAi screen on mesodermal components of the same pathway, molecular processes or protein complexes was remarkable and unanticipated. In general, these candidate factors are conserved constituents of macromolecular machineries that control chromatin dynamics or cytoplasmic polyadenylation and translation.
Tousled like kinase (Tlk) genes were first identified in plants and encode conserved nuclear serine-threonine kinases that impact flowering and leaf morphology (Sillje et al., 1999). TLKs phosphorylate histone H3 and ASF1A/B, two DNA-replication dependent and independent histone chaperones that facilitate chromatin assembly (Carrera et al., 2003; Klimovskaia et al., 2014). The scaffolding protein SYMPLEKIN (SYMPK) is a core component of a cytoplasmic polyadenylation complex that includes CPEB1 (also identified in this screen) and CPSF (Barnard et al., 2004). This complex regulates the translation of target mRNAs through polyadenylation (activation) and deadenylation (repression) of poly(A) tails (D’Ambrogio et al., 2013) in diverse biological contexts (Chang et al., 2012; D’Ambrogio et al., 2013; Fernandez-Miranda and Mendez, 2012; Si et al., 2003). Our analysis of fibroblast-specific mouse knockouts and human cell lines validated the hypothesis that chromatin and translation regulation in fibroblasts represent conserved pathways used to communicate with epithelial cells sensitized during development (mammary) or by oncogenic mutations (cancer).
It is notable that none of the 39 genes identified by this RNAi screen encoded secreted factors. We interpret this result to mean that depletion of any single mesodermal secreted factor is insufficient to significantly increase VPC proliferation; rather, altering regulatory hubs that simultaneously control a host of factors likely has a stronger impact on the proliferation of adjacent cells. This was not completely unexpected as this was a depletion screen seeking to identify factors with growth-suppressive properties and secreted growth suppressive factors have yet to be described in the C. elegans vulva development literature. Understanding how these stromal factors and their downstream effectors in normal and activated fibroblasts impact signaling of neighboring epithelial cells will be an exciting future challenge.
Sensitivity of normal mammary ducts to stromal signals
Why the stromal-specific ablation of TLK1, TLK2 and SYMPK has an impact on the epithelium of the mammary gland specifically is not entirely clear, albeit subtle changes in cell proliferation in other organs may have been missed by visual detection. It is also unclear why the mammary gland, unlike the worm vulva, is responsive to stromal-specific deletion of TLK1, TLK2 and SYMPK in the absence of prior ‘sensitization’ by Ras pathway alterations. The mammary gland, unlike most other organs, is under the intense influence of hormonal regulation and perpetually poised to expand, contract or remain quiescent when appropriately instructed. The remodeling of the mammary ductal network during postnatal development and subsequently during estrous cycling and pregnancy involves close stromal-epithelial interactions (Hennighausen and Robinson, 2005; Richert et al., 2000; Robinson, 2007; Sternlicht, 2006; Sternlicht et al., 2006; Wiseman and Werb, 2002). Thus, the unique sensitivity of mammary ducts to stromal fibroblast signaling through TLK1, TLK2 and SYMPK may be due to the strong periodic hormonal influence, which has been linked to the priming of epithelial cells to growth factor induced Ras-MAPK signaling (Lange et al., 1999; Skildum et al., 2005) although other signaling pathways may also be involved. We cannot rule out, however, that differential Fsp-cre transgene expression across organs is a contributing factor to the mammary specific phenotype observed in mice. How these stromal factors are regulated and how the receiving epithelial cells integrate stromal signals into proliferative responses are important mechanistic questions that remain to be addressed.
Specificity in mesodermal-epithelial communication
This work also revealed exquisite signaling specificity of mesodermal factors in different genetic backgrounds. The observation that mesoderm-specific depletion of these factors did not impact gap-1(+) worms or worms with an axl-1(−) sensitizing mutation argues that factor-depleted mesoderm-derived signals can only engage sensitized gap-1(−) VPCs to ectopically enter the cell cycle, but not VPCs sensitized by other-pathway (Wnt) alterations. The analysis of Ras-reporter worms suggests that this level of specificity involves, at least in part, the re-engagement of Ras pathway signaling beyond the levels exhibited in gap-1(−) worms. Human cell co-culture systems also showed a differential impact of fibroblast-specific ablation of candidate factors on “normal” and individual tumor cell lines, where distinct genetic landscapes in tumor cells may determine their responses to the depletion of stromal factors (Figure 7). Thus, productive stroma-tumor interactions may be predicated by the specific genetic alteration present in epithelial cells. While the details may differ between worms, mice and humans, it would appear that the signaling axes utilized for mesoderm-epithelium communication are functionally conserved. In summary, this cross-species approach begins to chart a road map of the components and networks used by mesodermal tissues to signal and control epithelial cell proliferation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gustavo Leone (leoneg@musc.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
C. elegans Strains and Maintenance
All C. elegans strains were maintained under standard conditions at 20°C and listed below: N2 wild-type, AH12 gap-1(ga133), KN611 axl-1(tm1095), rde-1(ne219), NL2099 rrf-3(pk1426), CM1859 unc-119(e2498); guEx1252[myo-3p∷gfp; unc-119(+)], CM1866 unc-119(e2498); guEx1259[ddr-2p∷gfp; unc-119(+)], CM1869 unc-119(e2498); guEx1261[ACEL-Δpes-10∷gfp; unc-119(+)], CM1871 unc-119(e2498); guEx1263[pB253(lin-31p)∷gfp; unc-119(+)], CM2073 rde-1(ne219); rrf-3(pk1426); guIs37[myo-3p∷rde-1(+); ddr-2p∷rde-1(+); ACEL-Δpes-10∷rde-1(+); sur-5p∷gfp; unc-119(+)], CM2129 rde-1(ne219); rrf-3(pk1426); gap-1(ga133); guIs37[myo-3p∷rde-1(+); ddr-2p∷rde-1(+); ACEL-Δpes-10∷rde-1(+); sur-5p∷gfp; unc-119(+), GS3582 unc-4(e120); arIs92[egl-17∷NLS-cfp-lacZ; unc-4(+); ttx-3∷gfp], CM2307 unc-4(e120); gap-1(ga133); arIs92[egl-17∷NLS-cfp-lacZ; unc-4(+); ttx-3∷gfp], CM2423 unc-119(e2498); rde-1(ne219); rrf-3(pk1426); gap-1(ga133); guIs39[lin-31p∷rde-1(+); sur-5p∷gfp; unc-119(+)].
C. elegans Tissue-Specific RNAi Strain Construction
First-strand cDNA for rde-1 was synthesized from total RNA isolated from N2 wild-type animals, and cDNA for let-60(gf) was synthesized from total RNA isolated from MT2124 let-60(n1046), with Superscript II Reverse Transcriptase Kit (Invitrogen) following the manufacturer’s protocol. let-60(gf) cDNA was amplified as a single product, and cloned directly into the plasmids (described below). The 5′ (~1 kb) and 3′ fragments (~2 kb) of the full-length rde-1(+) cDNA were individually amplified by PCR using Pfu DNA Polymerase Kit (Stratagene). The PCR primers used are listed in Table S3. The PCR products were digested with restriction enzymes and sequentially inserted into the pBluescript II KS(+) phagemid vector (Agilent Technologies; 212207). The full-length rde-1(+) cDNA was then excised using KpnI and SacI and cloned into the pPD49.26 C. elegans expression vector from the Fire Lab Kit (Addgene). All constructs were sequence-verified to ensure no mutations were introduced into cDNA sequences. Tissue-specific promoters were chosen based on previous studies (myo-3 (Myers and Greenwald, 2007); ACEL (Hwang and Sternberg, 2004)) or identified by examining approximately forty reporter strains with GFP expression detected in the somatic gonad (ddr-2; http://gfpweb.aecom.yu.edu/index). The regulatory sequences were amplified by PCR using Pfu DNA Polymerase Kit with N2 genomic DNA as templates. PCR primers used are listed in Table S3. The PCR products (myo-3 and ddr-2) were digested with restriction enzymes and cloned into pPD95.69 vector upstream of the nls∷gfp reporter cassette. ACEL sequences were cloned into pPD107.94 vector upstream of the truncated Δpes-10 minimal promoter, and the fusion ACEL-Δpes-10 promoter was cloned into pPD95.69 vector upstream of the nls∷gfp reporter cassette. All resultant promoter∷gfp reporters were utilized to generate transgenic strains to verify the expression pattern of corresponding promoters. Promoter sequences were then individually cloned into pPD49.26 containing rde-1(+) cDNA to drive tissue-specific rde-1(+) cDNA expression. Similar methods were used to achieve rde-1(+) cDNA expression restricted to vulval cells (Vulva-rde; gap-1(−) strain) by cloning rde-1 cDNA into the NotI site in pB253 vector (Myers and Greenwald, 2007; Tan et al., 1998). To produce the let-60(gf) constructs with the three mesodermal promoters, the rde-1(+) cDNA was excised and replaced with the let-60(gf) cDNA to produce the let-60(gf) constructs (using NheI and SacI for ddr-2, and KpnI and SacI for myo-3 and ACEL-Δpes-10). For the promoter in pB253, let-60(gf) cDNA was inserted downstream of the promoter, using BglII and NotI sites. The C. elegans strains with tissue-specific RNAi were generated by microinjection of plasmids containing rde-1 cDNA downstream of tissue-specific promoters into the germline of hermaphrodite rde-1(ne219) mutant animals using standard methods (Mello et al., 1991) with unc-119 as the selection marker. F1 progeny without uncoordinated movements were selected. Individual F2 worms were isolated to establish independent lines. Transgenic strains were sensitized with a gap-1(ga133) mutation to promote elevated VPC Ras/MAPK signaling. let-60(gf) transgenes were generated using 50 ng/ul of experimental DNA and 15 ng/ul unc-119(+) DNA, and evaluated as extrachromosomal arrays. rde-1(+) extrachromosomal transgene arrays were integrated into chromosomes with γ-irradiation at the Ohio State University Reactor Lab as previously described (http://wormlab.caltech.edu/protocols/).
Generation of Conditional Mouse Alleles and Maintenance
Animals were used in compliance with federal and Ohio State University Laboratory Animal Resources regulations and housed under normal conditions. Standard homologous recombination was used to target the conditional Tlk1 and Tlk2 alleles into hybrid Bl57/129SV ES cells using targeting vectors purchased from EUCOMM- IMPC (IKMC project number: 71713 and 79307 respectively) and injected into E3.5 C57BL/6 blastocysts. Sympk frozen embryos (C57BL/6N) were obtained from EMMA (EM:05086), the distributor for WSTI and generated using EUCOMM ES clone EPD0069_2_H09. All were transferred into foster mothers and the resulting chimeras were bred with National Institutes of Health Black Swiss female mice, and agouti offspring were genotyped by PCR and confirmed by Southern blot analysis. (Figure 4A–D, Figure 5A–C). Female mice 10–24 weeks of age were used for all studies.
Cell Lines
All breast cancer cells were maintained in a humidified incubator at 37°C and 5% CO 2 in RPMI 1640 growth medium (Life Technologies) containing 10% fetal bovine serum (Thermo Scientific HyClone), 2 mmol/L L-glutamine, 100 units/ml penicillin, and 100 g/ml streptomycin (Life Technologies). All DsRed-positive human breast cancer cells were previously generated (Sasser et al., 2007a; Sasser et al., 2007b; Studebaker et al., 2008). MCF10A breast epithelial cells were maintained in standard MCF10A growth medium: DMEM/F12 (Life Technologies) containing 5% heat-inactivated horse serum (Life Technologies), 20 ng/ml recombinant human EGF (PeproTech), 0.5 g/ml hydrocortisone (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 10 g/ml insulin (Sigma-Aldrich), 100 units/ml penicillin, and 100 g/ml streptomycin (Life Technologies). Cell lines were authenticated at University of Arizona Genetic Core by autosomal STR typing, using the Promega PowerPlex16HS assay.
Primary Cultures
Human Breast Fibroblasts
Primary human breast fibroblasts were isolated from human breast tissue as previously described (Orimo et al., 2005) with minor modifications. Tissue specimens were collected at The Ohio State University Wexner Medical Center (Columbus, OH) in accordance with protocols approved by the research ethics committee. Tissue biopsies from grossly normal human breast tissue specimens >10 cm from breast tumors were received on ice, minced into small fragments, and digested in dissociation buffer comprised of DMEM growth medium (Life Technologies) containing 10% fetal bovine serum (Thermo Scientific HyClone), 100 units/ml penicillin, 100 g/ml streptomycin (Life Technologies), and 2 mg/ml collagenase III (Worthington Biochemical Corp.) for sixteen to eighteen hours at 37°C. Digested tissue was allowed to separate by gravity to remove tissue debris, and supernatants were collected to pellet fibroblasts. The fibroblast pellets were washed and cultured in DMEM containing 10% fetal bovine serum and 25 g/ml plasmocin (InvivoGen) for one week. Fibroblasts were subsequently maintained in DMEM growth medium containing 10% fetal bovine serum, 100 units/ml penicillin, 100 g/ml streptomycin. For immortalization, 4×105 HEK293T cells per well were plated into 6-well plates overnight. The following day, subconfluent HEK293T cells were co-transfected with pCL-10A1 plasmid (Imgenex), which encodes HIV-1 Gag, Pol, Tet, and Rev proteins and pBABE-hygro-hTERT plasmid (Addgene plasmid 1773) using jetPEI DNA transfection reagent (Polyplus Transfection) according to the manufacturer’s protocol to generate VSV-G pseudotyped lentivirus. Viral supernatants were 0.45 m filtered and used to infect primary human breast fibroblasts with hTERT for immortalization. Fibroblasts were selected with hygromycin for ten days to generate stable hTERT-positive, immortalized human breast fibroblasts.
Culturing MECs and MMFs
Thoracic mammary glands were isolated and minced into 1- to 2-mm fragments using microdissection scissors in 0.15% collagenase III (Worthington Biochemical Corp.) overnight in a humidified incubator at 37°C and 5% CO2. The following day, digested glands were vigorously resuspended by pipetting (30–40×) in 10 mL and subjected to 12 min of gravity separation. Supernatants were removed and fibroblasts were isolated from them by centrifugation (1200 rpm for 5 min). Fresh medium was added to the sedimented epithelial cell containing organoids for four consecutive rounds of gravity separation (10 min each). Epithelial cells (MECs) and fibroblasts (MMFs) were maintained in DMEM/F12 (Life Technologies) containing 10% heat-inactivated fetal-bovine serum (Life Technologies), 20 ng/ml recombinant EGF (MECs only, PeproTech), 0.5 g/ml hydrocortisone (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 10 g/ml insulin (Sigma-Aldrich), 100 units/ml penicillin, and 100 g/ml streptomycin (Life Technologies) with either 0.04 mM or 1.05 mM CaCl2, respectively.
Human Samples
Breast tumor/stroma specimens used for expression profiling and associated clinical data (Table S4) were collected at McGill University Health Center (Montreal, Canada) between 1999 and 2013 in accordance with protocols approved by the research ethics committee. Written consent was obtained on an individual basis for all patients participating in this study.
METHOD DETAILS
C. elegans RNAi Screen and Microscopy
The genome-wide screen utilized the Ahringer E. coli RNAi library (Source Bioscience LifeSciences) targeting 16,757 C. elegans genes (~86% of genome) and was performed by feeding the CM2129 strain (see above) individual E. coli clones expressing double-stranded RNA. RNAi clones from 384-well library plates were inoculated into 96-well deep-well plates (VWR) with Luria broth (LB) medium (Invitrogen) containing 50 μg/mL carbenicillin (Sigma-Aldrich) for 16 hours at 37°C. E. coli were then seeded onto 12-well plates (Corning) with standard nematode growth medium (NGM) agar but containing 2× normal peptone, and containing 1 mM IPTG (Lab Scientific) and 25 μg/mL carbenicillin and incubated for 24 hours at room temperature. Synchronized embryos were seeded onto these plates and incubated for 4 days at 20°C. For the primary RNAi screen, approximately 50 animals were scored for abnormal vulva-associated morphologies using an Olympus SZ60 dissection microscope. Each RNAi clone was tested in three independent replicates, and genes that promoted aberrant morphologies upon stromal RNAi in at least two of three replicates were considered primary candidate ‘hits’. All primary candidate ‘hits’ were individually re-evaluated using the same 12-well plate RNAi platform, but plates were incubated at 20°C for 48 hours and approximately 30 L4 stage worms were selected for imaging. DIC images were captured with a Spot RT Monochrome digital camera (Diagnostic Instruments) using a Zeiss Axioskop2 microscope to identify the final 39 mesodermal candidate genes whose depletion induced multivulva in ≥ 1 animal. RNAi clones were sequence-verified to ensure the identity of each target gene.
Vulval Ras/MAPK Signaling Assay
The GS3582 strain expressing an egl-17p∷LacZ-CFP fluorescent reporter transgene (Yoo et al., 2004) was used to determine how whole-worm RNAi-mediated depletion of the 39 identified genes affects Ras/MAPK signaling in vulval cells. The same 12-well plate RNAi platform described above was utilized to evaluate synchronized L3 worms at two rounds of VPC division following RNAi5. DIC images were captured with a Hamamatsu ORCA-ER digital camera using a Zeiss Axioplan2 microscrope. The same system was used to capture corresponding CFP fluorescent images with one-second exposures.
Whole-Mount Staining of Mammary Glands
Inguinal mammary glands were spread on glass slides, fixed with Carnoy’s (60% EtOH, 10% acetic acid, 30% chloroform) for 12 hr at 4°C, rehydrated and stained with carmine alum (Sigma) for 12 hr at 4°C, dehydrated through ethanols and xylenes, mounted and coverslipped.
Histopathology and IHC
Inguinal mammary glands for were fixed in 10% neutral-buffered formalin solution for 48 hr and transferred to 70% ethanol. Tissues were processed, embedded in paraffin, cut in 5 m sections on positively charged slides, de-paraffinized, rehydrated, and stained with H&E. For immunohistochemistry, all sections were stained using a Bond Rx autostainer (Leica). Briefly, slides were baked at 65°C for 15min and automated software performed dewaxing, rehydration, antigen retrieval, blocking, primary antibody incubation, post primary antibody incubation, detection (DAB), and counterstaining using Bond reagents (Leica). Samples were then removed from the machine, dehydrated through ethanols and xylenes, mounted and coverslipped. Antibodies for the following markers were diluted in antibody diluent (Leica): rabbit antibodies- Ki67 (1:200, Abcam), CC3 (1:800, Cell Signaling Technology), pERK (1:400, Cell Signaling Technology).
qRT-PCR Analysis
Total RNA was extracted from MMFs or MECs using Trizol (Invitrogen) following the manufacturer’s protocol. Reverse transcription of 1 g of total RNA was performed by combining 1 L of SuperScript III reverse transcriptase (Invitrogen, Life Technologies Corp.), 4 L of 5× buffer, 0.5 L of 100 M oligo(dT) primer, 0.5 L of 25 mM deoxyribonucleotide (dNTP), 1.0 L of 0.1 M DTT, 1.0 L of RNase inhibitor (Roche), and water up to a volume of 20 L. Reactions were incubated for 60 min at 50°C and then diluted ten-fold with 180 L of water. qRT–PCR was performed using StepOne Plus (Applied Biosystems) qPCR machine. Each PCR contained 1.5 L of cDNA template and primers at a concentration of 100 nM in a final volume of 20 L of SYBR Green reaction mix (Bio-Rad). Each PCR yielded only the expected amplicon, as shown by the melting temperature profiles of the final products and by gel electrophoresis. Reactions were performed in triplicates, and relative amounts of cDNA were normalized to GAPDH. Primer sequences are listed in Table S3.
Breast Tumor and Stroma Total RNA Expression Profiling
Tissue samples were collected within 30 minutes after surgery, embedded in TissueTek OCT (Somagen, Edmonton, Alberta, Canada) and stored in liquid nitrogen until use. Frozen specimens were cryo-sectioned (10 m thickness) and stained using the Arcturus HistoGene LCM Frozen Section Staining Kit (Life Technologies) before LCM was performed. All tissues included in this study were re-examined by a clinical pathologist dedicated to the project. Tissue specimens were laser capture microdissected using a PixCell IIe LCM system (Arcturus) within three hours following tissue staining. Total RNA was extracted using the PicoPure RNA isolation kit (Life Technologies) as per the manufacturer’s directions. Subsequently, 0.5 to 1 ng of total RNA was subjected to two rounds of T7 linear amplification using the Arcturus RiboAmp HS Plus kit (Life Technologies) and labeled with Cy3 dye according to the manufacturer’s procedure. Prior to microarray hybridizations, amplified products were quantified using a spectrophotometer (Nanodrop, Wilmington, Delaware, USA) and subjected to BioAnalyzer assay for quality control (Agilent Technologies, Santa Clara, California, USA). SurePrint G3 Human Gene Expression 8×60K v1 microarrays (Agilent Technologies, product G4112A) were used for all experiments. RNA samples were subjected to fragmentation followed by 18 h hybridization, washing, and scanning (Agilent Technologies, model G2505B) according to the manufacturer’s protocol. Samples were hybridized against Cy5-labelled Universal Human Reference RNA (Stratagene, Jolla, California, USA).
Human Mammary Fibroblast Candidate Gene Expression Analysis
Total RNA was isolated from primary human breast fibroblasts using TRIzol reagent (Life Technologies) according to the manufacturer’s protocol followed by RNA clean up using a RNA clean-up and concentration kit (Norgenbiotek). Total RNA was submitted to NanoString Technologies for mRNA quantification of candidate genes by the nCounter® gene expression analysis system.
Human Lentivirus Production and shRNA Transduction
Optimal shRNA-mediated target gene knockdown efficiency was initially determined by lentiviral transduction of HEK293T cells with three to six individual gene-specific GIPZ shRNA-miR plasmids (Thermo Scientific) per candidate gene. The single shRNA plasmid demonstrating the most efficient target gene knockdown was utilized for subsequent infection of human breast fibroblasts. 4×105 HEK293T cells per well were plated into 6-well plates overnight. The following day, subconfluent HEK293T cells were triple-transfected with the pCMVΔR8.91 packaging plasmid (encoding HIV-1 Gag, Pol, Tet, and Rev proteins), pMD2.G (encoding vesicular stomatitis virus (VSV)-G enevolpe protein; both plasmids kindly provided by Dr. Jose Silva), and gene-specific GIPZ shRNA-miR plasmid using jetPEI™ DNA transfection reagent (Polyplus Transfection) according to the manufacturer’s protocol to generate VSV-G pseudotyped lentivirus. Fresh medium was added the following day, and 48 hours post-transfection, HEK293T cell medium was filtered and added to 1×105 human breast fibroblasts at 70% confluence in a 6-well plate two times over two consecutive days. 72 hours post-infection, fibroblasts were monitored for GFP expression and treated with 0.75 g/ml puromycin for three to five days to select for shRNA-positive cells. RNA was isolated as described above and complementary DNA (cDNA) was generated with SuperScript III reverse transcriptase kit reagents (Invitrogen), 200 ng total RNA, and oligo(dT)20. Real-time quantitative PCR was used to validate target gene knockdown following lentiviral shRNA transduction, using human RT-qPCR primers in Table S3.
3-Dimensional Co-Culture Proliferation Assay
A 3-dimensional (3D) breast fibroblast-cancer cell co-culture proliferation assay was performed as previously described and all DsRed-positive human breast cancer cells were previously generated (Sasser et al., 2007a; Sasser et al., 2007b; Studebaker et al., 2008). MCF10A cells were infected with lentivirus expressing pLVX-DsRed-Express2-C1 and DsRed-positive cells were enriched by flow cytometry based on DsRed fluorescence. This DsRed fluorescence-based quantitative growth assay measures DsRed fluorescence, which correlates precisely with breast cancer cell number. Stable, DsRed-positive human breast cancer cells (triple-negative MDA-MB-231, MDA-MB-468; estrogen receptor (ER)-positive MCF-7, T47D; HER2-positive BT474) or normal, immortalized mammary epithelial cells (MCF10A) and human breast fibroblasts were trypsinized from standard culture plates and co-cultured in 3 mg/ml Cultrex® basement membrane extract (Trevigen) constituted in serum-free, phenol red-free RPMI 1640 growth medium in black-walled 96-well tissue culture plates. Co-cultures were set up in triplicate and included a scrambled shRNA fibroblast co-culture control each set up day. Plates were read daily on a SpectraMax M2 fluorescence microplate reader (Molecular Devices), and autofluorescence of basement membrane extract alone was subtracted from each well. Data were normalized to fluorescence intensities of each co-culture on day one to adjust for initial cancer cell number.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphs/heatmaps were generated and statistical analyses were performed using GraphPad and R. No statistical analysis was used to predetermine sample size. Sample sizes, statistical tests and p-values are indicated in the text, figures and figure legends.
Human Ortholog Enrichment
The C. elegans sequence IDs of the 39 identified genes were entered into WormMart (http://www.wormbase.org), selecting Sequence Name, Homologous Protein, Homolog Species, and Homolog BLASTP evalue from the Attributes list. The number with human orthologs (Homolog BLASTP evalue <= 0.00001) were tallied. The same process for C. elegans sequence IDs from all RNAi tested in the screen was performed. A Fisher’s exact test was used to determine if the frequency of C. elegans genes with a human ortholog differed between the two gene lists.
Co-Culture Data Analysis
The growth rate data were matched from gene-specific shRNA and non-silencing control shRNA within the replicates and by culturing time (days). Wilcoxon signed-rank test (one tail only) was performed to determine if the human ortholog-depleted fibroblasts significantly enhance the proliferation of co-cultured epithelial cells compared to control fibroblasts. A fold change cutoff of 1.1 at day 4 from gene-specific to control shRNA was applied. All analyses were performed with the R statistical programming language (www.R-project.org).
IHC Quantification
Multispectral Analysis of dual-Color IHC/eosin stained samples were imaged using the PerkinElmer’s Vectra® multispectral slide analysis system. For each stained mammary section, multispectral images of the entire slide were captured manually for each 20X field containing ductal structures. For quantification of the DAB staining, the multispectral images were reviewed and analyzed using inForm® Tissue Finder software. A pattern recognition algorithm was used for processing as follows: (1) trainable tissue segmentation to segment ductal epithelial cells and (2) segmentation of the DAB-positive cells within this population from eosin-stained nuclei. Automatic quantification of these populations was used to calculate percent positive cells. For pERK quantification, automated cell segmentation of the pERK-positive tissue category was used to locate the subcellular compartments and scored by binning the spectrally unmixed DAB signal into four categories depending on the staining intensity (0+, 1+, 2+, and 3+), providing data in percent. The H-score, which ranges from 0 to 300, was calculated using the following formula: [1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+)]. Thus, H-score measures staining intensity as well as percentage of positive cells in a given cellular compartment.
DATA AND SOFTWARE AVAILABILITY
The breast tumor and stroma expression analysis dataset has been deposited in the NCBI Gene Expression Omnibus (GEO) and is publicly available under accession number: GSE83591.
Supplementary Material
Table S1, related to Figure 7. Human Ortholog mRNA expression in primary breast fibroblasts.
Table S2, related to Figure 7. P values associated with each human breast fibroblast-breast cancer cell co-culture.
Table S3, related to Figures 1, 4, 5, 7, S7. Oligonucleotide sequences of primers used for cloning, mouse genotyping, real-time quantitative PCR and lentiviral shRNA targets.
Table S4, related to Figure 6, S6. Summary of clinical information for Her2+ breast cancer patients associated with laser capture microdissected tumor samples studies and breast cancer patients associated with normal samples.
Highlights.
Mesodermal suppressors of epithelial proliferation identified in C. elegans
Mesoderm-epithelial signaling components are highly conserved across species
Depletion of candidates in murine fibroblasts causes mammary hyper-proliferation
Human orthologs of identified genes are altered in breast cancer stroma
In Brief.
Liu, Dowdle, Khurshid, Sullivan et al. identify mesodermal factors in C. elegans that suppress growth of adjacent epithelial cells. These candidates are shown to have conserved function in murine contexts, and to be altered in human breast cancer stroma.
Acknowledgments
We thank Dr. Iva Greenwald (Columbia University) for kindly providing pB253 plasmid, Dr. David Baillie (Simon Fraser University) for GFP reporter C. elegans strains, and Dr. Hendrik C. Korswagen (Hubrecht Institute) for the axl-1(tm1095) C. elegans strain. We are thankful to Dr. Anita Hopper and Dr. Denis Guttridge for critical suggestions. Some C. elegans strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the Nucleic Acid Core Shared Resources at Ohio State University, and the Breast Cancer Functional Genomics Group at McGill University. This work was funded by an NIH P01 CA097189 grant (M.C.O., G.L., M.P., M.H., J.D.P, S.A.F), and grants to G.L (OSUCCC Pelotonia Award), H.M.C. (NSF IOB-0949489 and NSF/NIH DMS-1361251), M.P. (Axe Banque de tissus et de données of the Réseau de Recherche en Cancer of the Fonds de recherche du Québec - Santé, with the assistance of the Quebec Breast Cancer Foundation; Terry Fox Research Institute). H.L., J.A.D., K.R., M.M., M.L., M.E.H. and C.S. were recipients of Pelotonia Fellowships. N.J.S. and J.M. were supported by NIH T32 fellowship (5 T32 CA106196-9). None of the authors of this manuscript have a financial interest related to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.M.C. and G.L. designed and supervised the current study, analyzed data, and helped write the manuscript. M.C.O. provided critical advice in experimental design and analyzed data. H.L., J.A.D., S.K. and N.J.S. equally contributed to the design and performance of the experiments, collected and analyzed data, and helped write the manuscript. K.R., M.M., P.D., E.W., K.T., M.L., M.E.H., K.E., C.S., M.J.S., S.W.C., S.S. and R.K designed and performed experiments and collected and analyzed data. X.T., J.M., S.A.F., J.D.P., S.M.I.S advised and performed statistical and bioinformatic analyses. E.M. and C.L.S. advised and helped write the manuscript. Y.L., S.M., and L.D.Y. established human breast fibroblast cell lines. N.B., A.T., M.H., and M.P. advised and performed laser capture microdissection and gene expression profiling.
References
- Addadi Y, Moskovits N, Granot D, Lozano G, Carmi Y, Apte RN, Neeman M, Oren M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer research. 2010;70:9650–9658. doi: 10.1158/0008-5472.CAN-10-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, Wilmott JS, Gartner JJ, Di Pizio A, Winograd-Katz S, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47:1408–1410. doi: 10.1038/ng.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard DC, Ryan K, Manley JL, Richter JD. Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell. 2004;119:641–651. doi: 10.1016/j.cell.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F, et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2012;14:159–167. doi: 10.1038/ncb2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera P, Moshkin YM, Gronke S, Sillje HH, Nigg EA, Jackle H, Karch F. Tousled-like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes Dev. 2003;17:2578–2590. doi: 10.1101/gad.276703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens JL, Shahi P, Van Tsang S, Smith B, Creighton CJ, Zhang Y, Seamans A, Seethammagari M, Vedula I, Levitt JM, et al. FGFR1-WNT-TGF-beta signaling in prostate cancer mouse models recapitulates human reactive stroma. Cancer Res. 2014;74:609–620. doi: 10.1158/0008-5472.CAN-13-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Zhang C, Cao Y. Expression and distribution of symplekin regulates the assembly and function of the epithelial tight junction. Histochem Cell Biol. 2012;137:319–327. doi: 10.1007/s00418-011-0906-z. [DOI] [PubMed] [Google Scholar]
- Chen N, Greenwald I. The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell. 2004;6:183–192. doi: 10.1016/s1534-5807(04)00021-8. [DOI] [PubMed] [Google Scholar]
- D’Ambrogio A, Nagaoka K, Richter JD. Translational control of cell growth and malignancy by the CPEBs. Nat Rev Cancer. 2013;13:283–290. doi: 10.1038/nrc3485. [DOI] [PubMed] [Google Scholar]
- Eisenmann DM, Maloof JN, Simske JS, Kenyon C, Kim SK. The beta-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development. 1998;125:3667–3680. doi: 10.1242/dev.125.18.3667. [DOI] [PubMed] [Google Scholar]
- Fernandez-Miranda G, Mendez R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res Rev. 2012;11:460–472. doi: 10.1016/j.arr.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- Hajnal A, Whitfield CW, Kim SK. Inhibition of Caenorhabditis elegans vulval induction by gap-1 and by let-23 receptor tyrosine kinase. Genes Dev. 1997;11:2715–2728. doi: 10.1101/gad.11.20.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 2005;6:715–725. doi: 10.1038/nrm1714. [DOI] [PubMed] [Google Scholar]
- Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Hill RJ, Sternberg PW. The gene lin-3 encodes an inductive signal for vulval development in C. elegans. Nature. 1992;358:470–476. doi: 10.1038/358470a0. [DOI] [PubMed] [Google Scholar]
- Holzel M, Huang S, Koster J, Ora I, Lakeman A, Caron H, Nijkamp W, Xie J, Callens T, Asgharzadeh S, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell. 2010;142:218–229. doi: 10.1016/j.cell.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper NA, Lee J, Sternberg PW. ARK-1 inhibits EGFR signaling in C. elegans. Mol Cell. 2000;6:65–75. [PubMed] [Google Scholar]
- Hu B, Castillo E, Harewood L, Ostano P, Reymond A, Dummer R, Raffoul W, Hoetzenecker W, Hofbauer GF, Dotto GP. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell. 2012;149:1207–1220. doi: 10.1016/j.cell.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- Jose AM, Hunter CP. Transport of sequence-specific RNA interference information between cells. Annu Rev Genet. 2007;41:305–330. doi: 10.1146/annurev.genet.41.110306.130216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Klimovskaia IM, Young C, Stromme CB, Menard P, Jasencakova Z, Mejlvang J, Ask K, Ploug M, Nielsen ML, Jensen ON, et al. Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat Commun. 2014;5:3394. doi: 10.1038/ncomms4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange CA, Richer JK, Horwitz KB. Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol. 1999;13:829–836. doi: 10.1210/mend.13.6.0290. [DOI] [PubMed] [Google Scholar]
- Li Y, DeFatta R, Anthony C, Sunavala G, De Benedetti A. A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene. 2001;20:726–738. doi: 10.1038/sj.onc.1204147. [DOI] [PubMed] [Google Scholar]
- Liu D, Yang C, Bojdani E, Murugan AK, Xing M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J Natl Cancer Inst. 2013;105:1617–1627. doi: 10.1093/jnci/djt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul. 2014;55:1–14. doi: 10.1016/j.jbior.2014.04.002. [DOI] [PubMed] [Google Scholar]
- McLaughlin SK, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, Beroukhim R, Polyak K, Brown M, Kuperwasser C, et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell. 2013;24:365–378. doi: 10.1016/j.ccr.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- Myers TR, Greenwald I. lin-35 Rb acts in the major hypodermis to oppose ras-mediated vulval induction in C. elegans. Dev Cell. 2005;8:117–123. doi: 10.1016/j.devcel.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Myers TR, Greenwald I. Wnt signal from multiple tissues and lin-3/EGF signal from the gonad maintain vulval precursor cell competence in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:20368–20373. doi: 10.1073/pnas.0709989104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterveen T, Coudreuse DY, Yang PT, Fraser E, Bergsma J, Dale TC, Korswagen HC. Two functionally distinct Axin-like proteins regulate canonical Wnt signaling in C. elegans. Dev Biol. 2007;308:438–448. doi: 10.1016/j.ydbio.2007.05.043. [DOI] [PubMed] [Google Scholar]
- Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–68. doi: 10.1016/j.semcancer.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Pickard A, Cichon AC, Barry A, Kieran D, Patel D, Hamilton P, Salto-Tellez M, James J, McCance DJ. Inactivation of Rb in stromal fibroblasts promotes epithelial cell invasion. EMBO J. 2012;31:3092–3103. doi: 10.1038/emboj.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, Inoue M, Hikita T, Koppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173. doi: 10.1016/j.gene.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richert MM, Schwertfeger KL, Ryder JW, Anderson SM. An atlas of mouse mammary gland development. J Mammary Gland Biol Neoplasia. 2000;5:227–241. doi: 10.1023/a:1026499523505. [DOI] [PubMed] [Google Scholar]
- Robinson GW. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 2007;8:963–972. doi: 10.1038/nrg2227. [DOI] [PubMed] [Google Scholar]
- Sasser AK, Mundy BL, Smith KM, Studebaker AW, Axel AE, Haidet AM, Fernandez SA, Hall BM. Human bone marrow stromal cells enhance breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3D tumor environments. Cancer Lett. 2007a;254:255–264. doi: 10.1016/j.canlet.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Sasser AK, Sullivan NJ, Studebaker AW, Hendey LF, Axel AE, Hall BM. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007b;21:3763–3770. doi: 10.1096/fj.07-8832com. [DOI] [PubMed] [Google Scholar]
- Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6:281–291. doi: 10.1038/nrc1839. [DOI] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- Sillje HH, Nigg EA. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr Biol. 2001;11:1068–1073. doi: 10.1016/s0960-9822(01)00298-6. [DOI] [PubMed] [Google Scholar]
- Sillje HH, Takahashi K, Tanaka K, Van Houwe G, Nigg EA. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999;18:5691–5702. doi: 10.1093/emboj/18.20.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–1319. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19:327–339. doi: 10.1210/me.2004-0306. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sternberg PW. Vulval development. WormBook. 2005:1–28. doi: 10.1895/wormbook.1.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD. Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2006;8:201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74:365–381. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studebaker AW, Storci G, Werbeck JL, Sansone P, Sasser AK, Tavolari S, Huang T, Chan MW, Marini FC, Rosol TJ, et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008;68:9087–9095. doi: 10.1158/0008-5472.CAN-08-0400. [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Tan PB, Lackner MR, Kim SK. MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell. 1998;93:569–580. doi: 10.1016/s0092-8674(00)81186-1. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- Wagmaister JA, Miley GR, Morris CA, Gleason JE, Miller LM, Kornfeld K, Eisenmann DM. Identification of cis-regulatory elements from the C. elegans Hox gene lin-39 required for embryonic expression and for regulation by the transcription factors LIN-1, LIN-31 and LIN-39. Dev Biol. 2006;297:550–565. doi: 10.1016/j.ydbio.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Bais C, Greenwald I. Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science. 2004;303:663–666. doi: 10.1126/science.1091639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 7. Human Ortholog mRNA expression in primary breast fibroblasts.
Table S2, related to Figure 7. P values associated with each human breast fibroblast-breast cancer cell co-culture.
Table S3, related to Figures 1, 4, 5, 7, S7. Oligonucleotide sequences of primers used for cloning, mouse genotyping, real-time quantitative PCR and lentiviral shRNA targets.
Table S4, related to Figure 6, S6. Summary of clinical information for Her2+ breast cancer patients associated with laser capture microdissected tumor samples studies and breast cancer patients associated with normal samples.