

Abstract
1. INTRODUCTION
The development of biodegradable polymers represents a revolution in medicine spanning over 50 years and leading to significant biotechnological advancements in drug delivery, biomaterials, tissue engineering, and medical device development while bringing together chemists, engineers, biologists, and physicians in a unique and collaborative manner. The time frame of this revolution mirrors discoveries of more potent therapeutics in the form of peptides, proteins, nucleic acids, and other bioactive molecules. The short half-lives of many of these modern therapeutics, in addition to the nonspecific distribution and toxicity of previously identified small molecule drugs, has been a major driving force for the development of polymeric drug delivery platforms. The successful clinical translation of the earlier macro- and micro-drug-delivery systems has led to the evolution of controlled-release nanodrug delivery platforms that are capable of overcoming pharmacological limitations with substantial advantages over conventional dosage forms. Investigations and discoveries in synthetic methodologies, fabrication methods, and mathematical models for studying the mechanisms of controlled drug release have led to the ability to create tunable polymeric nanoparticle (NP) drug delivery systems capable of localized and sustained delivery, facilitating improvements in the therapeutic index of drugs. The ability to control the release of therapeutics and the extremely versatile nature of polymeric drug delivery platforms offer numerous important advantages (Figure 1).
Figure 1.

Key features of polymeric drug delivery systems (DDS).
Starting for the most part in the 1960s, polymeric drug delivery has flourished since the pioneering works of notable researchers in the field, including Folkman, Langer, Higuchi, Roseman, Peppas, Heller, Ringsdorf, and Speiser.1–18 We have witnessed an evolution in drug delivery capabilities from the initial use of controlled-release polymers in macroscopic drug depots, implants, and suture materials to injectable microscale controlled drug delivery systems, such as microparticles, to nanoscale drug delivery, which may progressively represent a bigger part of the pipeline of pharmaceutical companies over the next few decades.19,20 To date we have seen the first bench-to-bedside translation of targeted and tunable controlled release polymeric NPs for small molecule drugs, from initial proof-of-concept in vitro,21,22 to successful in vivo investigations,23,24 which have laid the foundation for human testing and ongoing phase II clinical trials for multiple cancer types.25 The emergence of controlled-release polymeric NPs has fostered novel investigations of synthetic methodologies, bioconjugation techniques, and a rapid proliferation of scientific publications addressing the use of target-site activated chemical reactions that trigger changes in NP structure, shape, chemistry, and degradation rates. The ability to tune the physicochemical properties of polymeric NPs and to incorporate targeting elements into their design has allowed new generations of controlled-release polymeric NPs to navigate the complex and chemically rich in vivo environment.
With our improved understanding of biological processes in diseased states, the design of controlled-release polymeric NP drug delivery systems has evolved from classical release mechanisms to the utilization of local biochemical changes in aberrant disease states to trigger and activate drug release. In diseases such as cancer an inevitable shift in homeostatic chemical equilibrium occurs, such as amplified or triggered enzymatic activity, a change toward acidic pH, reductive or oxidative states, or an increase in reactive oxygen species.33 These differential biochemical signatures can be exploited for the development of more precise therapies and offer ample opportunity for polymer design, rendering further control over the site-specificity and kinetics of drug release. In addition to exogenously triggered drug release, endogenously controlled physical parameters, such as local induction of thermal, electrical, ultrasound, or magnetic energy, can also be used to trigger various responsive components of drug delivery systems. As a result, to maximize the utility of degradable polymers in drug delivery, interest is growing in adding biologically responsive elements to the overall polymer design, to achieve more biologically controlled therapeutic outcomes.
The overall aim and scope of this review is to provide an informative account of physical, chemical, and biological parameters that can be harnessed to gain full spatiotemporal control of drug delivery. We will initially introduce the properties of commonly utilized degradable polymers used in drug delivery and then offer a retrospective account of the evolution of controlled-release polymers and how pioneering work in the field evolved to produce the current application of nanotechnology to medicine and healthcare (nanomedicine) as related to polymeric NPs. We will then focus on the various mechanisms for controlling drug release from polymeric NPs. We conclude by discussing the challenges of activatable controlled-release polymeric NPs and provide an outlook for these systems. Since polymers have been most widely used for controlled-release drug delivery, this review focuses on polymeric nanomedicines; however, a number of excellent reviews have also been published on different types of stimuli-responsive NPs developed using liposomes, micelles, silica NPs, and metal oxides.37–43 Furthermore, many of the concepts and parameters discussed herein can also be applied to the spatiotemporal controlled delivery of diagnostic and theranostic agents, which have been presented elsewhere.44–50
2. BIODEGRADABLE POLYMERS IN CONTROLLED RELEASE DRUG DELIVERY
Natural biodegradable materials have a long history of use in medicine, dating back over 3000 years when ancient Egyptians used plant fibers, hair, tendons, and wool fibers as suture material.51–53 The need to develop safer biodegradable sutures and to improve on commonly used catgut sutures led to investigations of synthetic biodegradable polymers.56 The earlier applications of synthetic biodegradable polymers dates back to the 1960s and 1970s, when the polyesters poly(glycolic acid) (PGA), poly(D,L-lactic acid) (PLA), and poly(D,L-lactic-co-glycolic acid) (PLGA) were developed for use as biodegradable sutures.60 Since then, the use of degradable polymers in drug delivery applications has become prominent due to their biocompatibility and degradability properties, as they can break down inside the body to produce nontoxic natural byproducts such as water and carbon dioxide, and are thereby easily eliminated.62 Although much of the pioneering work in controlled-release systems was conducted using nondegradable polymers, degradable and biodegradable polymers are the preferred choice for the development of polymeric drug delivery systems, and the terms biodegradable, bioabsorbable, bioeliminable, and bioerodible are often used interchangeably to describe polymers such as PLGA and PLA. These terms have recently been defined by the IUPAC.64 It is important to note that the term “biodegradable” has been defined according to the IUPAC as the break down of polymers due to cellular or in vivo biological actions.64 For the purposes of drug delivery, many of the degradable polymers utilized are commonly referred to as biodegradable, even though the mechanism of degradation is not biological and in most cases is hydrolysis driven. Given that the majority of degradable polymers used in drug delivery applications are based on hydrolyzable ester bonds and evidence for their biological breakdown in vivo is limited or conflicting, in this review we refer to these polymers using the correct terminology of “degradable polymers” based on the IUPAC definition (in vivo degradation resulting solely from hydrolysis by the water present in tissues and organs, referred to as hydrolysis or hydrolytic degradation).64
2.1. General Polymer Properties
Degradable polymers can be hydrolyzed in vivo and are usually classified according to whether their source is natural or synthetic. Commonly used polymers include the α-hydroxy acids, polyanhydrides, and natural sugars such as chitosan, in addition to many other types (Figure 2). Synthetic degradable polymers are favored in tissue-engineering or drug delivery applications, as they have less batch-to-batch variability and immunogenicity as compared to degradable polymers from natural sources.79 Polymers can be rendered degradable through the inclusion of labile ester, anhydride, and amide chemical linkages, which are susceptible to common degradation mechanisms involving hydrolysis or enzymatic cleavage leading to the gradual scission (breaking of chemical bonds) of polymer chains. Drug release from degradable polymers can be governed by (1) surface erosion of the polymer matrix, (2) cleavage of polymer bonds at the surface or within the bulk of the matrix, or (3) diffusion of the physically entrapped drug. However, drug release is often a result of a simultaneous combination of all three.12
Figure 2.
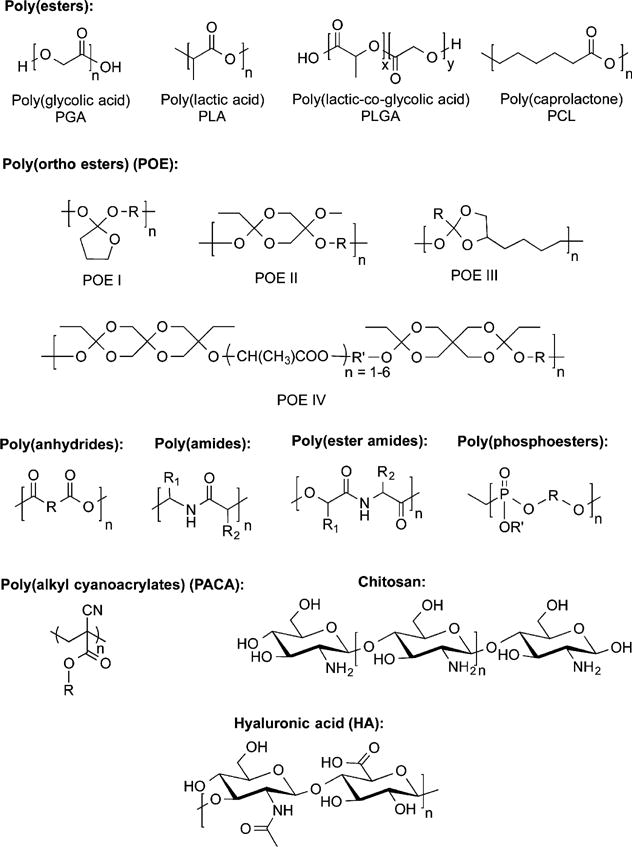
Examples of common degradable and biodegradable polymers with representative monomer units used in drug delivery.
The chemical nature of the monomer is crucial to the overall design of degradable polymers, as the polymer breakdown products in the body ultimately influence the overall toxicity and biocompatibility of the polymers. The degree of polymerization describes the number of repetitive monomeric units, which is usually greater than 100 for high molecular weight (MW) polymers.80 Polymer weight can be determined according to the weight-average (Mw) or the number-average (Mn).81,82 The polydispersity index (PDI) of the system is indicated by the ratio of Mw/Mn, with values close to 1 representing monodisperse systems.82,83 Although the term PDI is widely used, this term has now been replaced by the IUPAC with “dispersity”, as denoted by Đ.84 Numerous studies have been carried out demonstrating the ramifications of these parameters on drug release from polymeric drug delivery systems.85–93 Degradable polymers (Figure 2) are attractive materials for the design of drug delivery systems, tissue-engineering scaffolds, implants, and surgical materials.94–96 PLGA is the most utilized of the degradable polymers, due its long history of clinical use and favorable controlled-release and degradation behavior.
PLGA can be used for the entrapment of most types of therapeutics with a wide range of MWs and can be fabricated into particles of various sizes and shapes.97 PLGA’s drug release capabilities can be tuned by varying properties such as MW, ratio of lactide to glycolide, and drug concentration. For example, the extra methyl group in the side chain of PLA makes this polymer more hydrophobic compared to PGA, with an increase in the PLA content leading to less water absorption and therefore slower degradation rates.98 Parameters such as crystallinity, glass transition temperature (Tg), solubility, and MW can influence the rate and release behavior of incorporated drug molecules, and an understanding of key parameters that influence polymer matrix properties is important in order to improve biodegradation and drug release behavior, and these concepts are discussed in subsequent sections.
2.1.1. Polymer Crystallinity
The partial alignment of polymer molecular chains can contribute to polymer crystallization, affecting polymer physical and chemical properties. Polymer crystallinity describes the degree of crystalline regions within a polymer sample in relation to amorphous regions and is an important concept in drug delivery, since only amorphous regions are permeable and therefore accessible to water molecules.99 Polymer mechanical strength, swelling, hydrolytic, and biodegradation rates are dependent on the degree of crystallinity, which in turn is governed by the nature of the monomers.
When the polymer chain has a stereoregular structure (regular monomer repeat units), linear polymers of high MW can rearrange into crystallites.100 Crystalline domains are separated by amorphous regions, as polymers never reach 100% crystallinity and are therefore semicrystalline. Crystallinity, which can be determined using X-ray diffraction methods, is a factor that can affect the release rate of drugs from polymeric drug delivery systems and is influenced by both the size and orientation of the polymer chains. A high degree of crystallinity causes slower drug release states when low MW polymers are used, but at high MWs (when porosity is higher), crystallinity has less impact on drug release.80
Due to the asymmetric α-carbon, PLA can be described as having D- or L- stereochemical centers (or R or S, respectively), giving rise to two enantiomeric forms of PDLA or PLLA, with PLGA being generally described as poly(D,L-lactic-co-glycolic acid) with D- and L-lactic acid equally distributed. PLLA is highly crystalline and PDLA is completely amorphous due to disordered polymer chains, while PGA is highly crystalline due to the lack of methyl groups on the side chain. In the case of copolymerization of PLA and PGA (which yields PLGA), the degree of crystallinity and amorphousness depend on the ratio of the monomers; e.g., a 50:50 ratio of lactide to glycolide results in an amorphous polymer, with increases in the lactic acid content leading to a more crystalline polymer.101 Studies have shown that the rate of drug release is higher in polyesters with a low degree of crystallinity because of higher macromolecular chain mobility.102,103
2.1.2. Polymer Glass Transition
Another factor that influences polymer physicochemical properties and is related to crystallinity is the glass transition temperature of polymers. The type of polymer and temperature dictate whether the amorphous region is in a “glasslike” or “rubberlike” state, and this depends on the polymer’s glass transition state Tg (the temperature at which the rubbery state to glassy state transition occurs) and can be determined using differential scanning calorimetry.80 Below the Tg, the polymer is in a glassy state; i.e., it has limited mobility and low diffusion rates. Above the Tg, the polymer is in a rubbery state, which facilitates higher mass transfer rates of water and drug molecules throughout the matrix.104 For effective design of degradable polymers in drug delivery applications, a balance between amorphous and crystalline states is necessary, as these parameters have direct effects on the degree of mechanical toughness and rate of drug release. For this reason, polymeric NPs are usually prepared from copolymers that contain both hydrophobic and hydrophilic segments, which make physical properties such as drug-release rates more predictable.105 The Tg of polymers such as PLGA decreases with a reduction in its lactide monomer content and MW, and several studies have investigated the effects of Tg on the rate of drug release.98,106–109
2.1.3. Polymer Hydrophilicity and Hydrophobicity
Solubility is a key concept in the design of degradable polymeric drug delivery systems and is dependent on the chemical nature, structure, and degree of crystallinity within the polymer. Polymer hydrophobicity generally increases with MW, with an increase in backbone branching resulting in more-water-soluble polymers.104 When the polymer used is hydrophobic in nature, drug release is controlled by surface erosion, and when there is a balance between hydrophobic and hydrophilic functionalities in the polymer backbone, degradation can occur from within the bulk of the polymeric system.110,111
For macro- and microscale polymeric drug delivery systems, the degree of hydrophilicity of the constituent polymers is important, although because physical properties at the nanoscale such as a very high surface-to-volume ratio render colloids of polymeric NPs stable in aqueous conditions, a wide variety of hydrophobic polymers may be utilized.111 Furthermore, the blending of hydrophilic polymers with hydrophobic polymers can increase pore formation along with an increase in the rate of polymer degradation and drug release. Polymer composition is highly important in the solubility of the polymer. For example, increasing the glycolic acid portion of PLGA, which renders the polymer more hydrophilic, can result in faster degradation rates.112,113 The solubility of the monomer is therefore a critical factor in the rate of drug release from polymeric drug delivery systems.114,115
2.1.4. Polymer Molecular Weight
The physical properties of polymers, such as Tg, solubility, viscosity, crystallinity, mechanical strength, and degradation rate, are related to the polymer’s MW, with low-MW polymers degrading more rapidly.100 The MW of degradable polymers has a significant impact on the drug-release profile from NPs and can influence the biological properties of polymeric drug delivery systems, such as elimination, phagocytosis, and biological activity.108,115–119 For example, estradiol-loaded NPs made with low-MW PLGA (14 500–45 000 Da) were found to exhibit zero-order release kinetics, compared to higher-MW (85 000–213 000 Da) PLGA, which exhibited a square root of time (Higuchi’s pattern) dependent release.120 Furthermore, drug Cmax in the plasma was shown to be polymer MW dependent (higher for the lower-MW polymers).
By using a range of PLGA MW polymers and PEG polymers, Valencia et al. were able to synthesize a library of 45 polymer NPs with varying sizes and surface hydrophilicities.117 These NPs were then screened for macrophage uptake effects on the basis of fluorescence detection of internalized NPs, confirming that NP size was a key determinant of macrophage uptake (over PEG surface density), with smaller NPs made from lower-MW PLGAs exhibiting the least amount of uptake. In general, low-MW polymers produce smaller NPs, resulting in altered drug release kinetics, enhanced blood circulation times, reduced accumulation in organs such as the liver and the spleen, and therefore enhanced biological efficacy due to longer duration of drug exposure.
The rate of drug release from polymeric drug delivery systems correlates with the mechanical properties of the polymer; high-MW polymers have a low elastic modulus, producing a relatively nondeformable matrix that limits the number of pore-forming channels. Low-MW polymers have a high elastic modulus, and the matrix is more deformable, causing pores to expand as a result of osmotic pressure.119
2.2. Poly(esters)
2.2.1. PLGA Polymers
Polyesters are polymers with ester bond linkages in the carbon backbone. The aliphatic polyesters include PGA, PLA, and PLGA and are the most extensively investigated degradable polymers to date (Figure 2).121–125 PLGA is synthesized from ring-opening polymerization (ROP) of cyclic lactide and glycolide monomers126 and degrades via hydrolysis of its ester bonds in water.127 A diverse range of monomers can be used in the synthesis of aliphatic polyesters, and polycondensation of difunctional monomers yields low-MW polymers. ROP is used for high-MW polymers.126 The simplest linear aliphatic polyester is PGA, and due to its high crystallinity (45–55%), it has poor solubility in most organic solvents.88 PGA has a Tg of 35–40 °C and therefore strong mechanical properties.88 The low solubility and fast degradation into glycolic acid makes PGA a poor choice for polymeric NP drug delivery applications;88 therefore, copolymers of PGA such as PLGA have been prepared for drug delivery applications.
The polycondensation of D- or L-lactic acid leads to the formation of PLA (Figure 2), which is a mixture of D,L-lactide (the natural isomer is L-lactide). PLA is hydrophobic due to the presence of the methyl side group,80 and properties of PLA can be changed by tuning D- and L-isomer racemization.88 The equivalent PLLA polymer from L-lactide is semicrystalline, whereas the PDLA polymer from D,L-lactide is amorphous. This in turn leads to changes in mechanical strength and degradation rates, with PLLA being hard and transparent with a Tg of 53 °C and PDLA having a Tg of 55 °C and low mechanical strength.88 The degradation rate of PLA is influenced by its crystallinity, and since the degradation rate of PLLA is lower than that of PGA, copolymers of lactide and glycolide began to be investigated for sutures and implant materials. This led to the development of PLGA, which can be obtained in a variety of MWs and lactide-to-glycolide ratios (L/G); hence, when the ratio of L:G monomer decreases, so does the degradation rate of PLGA.88 For example, the degradation times of PLGA polymer with 50:50, 75:25, and 85:15 (L:G) were shown to be 1–2, 4–5, and 5–6 months in aqueous conditions.88 Degradable drug delivery systems using PLGA and their precise controlled-release mechanisms will be discussed in subsequent sections. Ease of synthesis of poly(esters) by ROP or condensation polymerization and low toxicity has led to their commercial availability and popularity as degradable polymers. Radiolabeling studies of PLGA NP degradation have shown that eventually all the polymer is degraded in vivo and cleared via respiration from the lungs.128
2.2.2. PLGA Copolymers
To improve the in vivo circulation and biocompatibility of PLGA NPs, block copolymers of PLGA and PEG (PLGA–PEG) were developed.129–132 PLGA–PEG diblocks (AB) or PLGA–PEG–PLGA (ABA) copolymers are used to formulate NPs or thermogels, respectively. Due to the hydrophilicity of PEG, these chains can orient themselves toward the surface of polymeric NPs and lead to increased hydration. For in vivo applications and prolonged circulation, the surface of NPs is frequently coated with PEG, leading to neutral NP surface charges and minimization of bioadhesion and immunological response as a result of sterically inhibiting both electrostatic and hydrophobic interactions of plasma components with NPs.133–135 The solubility of PEG also allows for various targeting ligands to be conjugated to the distal end, therefore leading to targeted NPs.129,136–137 Triblock polyester ABA polymers where the A and B blocks are conjugated via ester linkages can form highly viscous gels at physiological temperatures, leading to temperature responsive degradable polymers.137 Advantages of polyester ABA and BAB type copolymers forming hydrogels have been extensively discussed elsewhere.138,139
Polyester–PEG copolymers are favorable block polymers for the generation of NPs with PEGylated surfaces, in addition to NP surface bioconjugation with various reactive PEG polymers, and PEG is currently a clinically validated polymer.19 Interestingly, a few studies to date have demonstrated anti-PEG IgG responses with immunologic memory in humans.19,140,141 However further studies are required to fully elucidate these findings.140,142
2.2.3. Polycaprolactones
Another commonly utilized degradable aliphatic polyester used in drug delivery applications is poly(caprolactone) (PCL) (Figure 2), which is made from the ROP of ε-caprolactone using tin octoate catalyst. PCL has good solubility and a very low Tg (approximately −60 °C), making it a semirigid material at room temperature. To date, PCL has found more uses in tissue engineering as scaffold matrix material than in particulate-based drug delivery.88 One reason for this is its lack of solubility and extremely slow degradation rates (2–3 years for pure PCL); however, modification of PCL with other polymers (such as block polymer synthesis or blending with PLGA and PLA) have led to improvements in its degradation and reactivity.143–145 The synthesis of PCL block copolymers leads to amphiphilic structures and different mechanical and physical properties (tuned by varying the ratio of the blocks, etc.). Hydrophilic block segments of either PEG, poly(acrylic acid) (PAA), poly(2-ethyl-2-oxazoline) (PEtOz), poly(N-isopropylacrylamide) (PNIPAAm), or poly(N,N-dimethylamino-2-ethyl methacrylate) (PDMAEMA) conjugated to the hydrophobic PCL segment have been used to formulate micelles.146 For example, RGD-functionalized PCL–PEG diblock copolymers were used to formulate doxorubicin-loaded micelles for targeting drug delivery.147 There are numerous examples of di- and triblock polymers synthesized with PCL polymers, and further examples of PCL diblock micelles used in controlled-release drug delivery applications have been reviewed.146
2.2.4. Poly(alkyl cyanoacrylates)
A class of degradable polyesters are poly(alkyl cyanoacrylates) (PACAs), which are synthesized from alkyl cyanoacrylate monomers, and their use in drug delivery applications was pioneered by Couvreur and Speiser in the late 1970s.148,149 The presence of two highly reactive electron-withdrawing groups on the alkyl cyanoacrylate monomers leads to rapid polymerization reactions.150 In general, these polymerization reactions are conducted in aqueous solutions, with the hydroxide ion acting as the initiator.150 The average MWs of these polymers are between 5 and 10 kDa to allow for full biodegradation and elimination of the monomers. Interestingly, fast anionic polymerization by biomolecules in the skin is causative of the tissue adhesive properties of liquid skin plasters and band aids that are based on alkyl cyanoacrylates.150 PACAs can be synthesized from alkyl cyanoacetate via the Knoeveagel condensation reaction leading to oligomers or via a thermal depolymerization reaction of these oligomers.151 Diblock and triblock copolymers with PEG and PACA blocks have also been synthesized using zwitterionic polymerization.151 The progression of NPs made using PACAs has been most advanced up to now, and these passively targeted NPs are currently in phase III clinical trials for the treatment of resistant liver cancer.152
2.3. Poly(ortho esters)
Developed as synthetic polymers for use as sutures in surgery in the 1970s, poly(ortho esters) (POEs) can be divided into four classes (POE I–IV, Figure 2), usually have three geminal ether bonds, and are hydrophobic surface-eroding polymers.88,153 POEs typically release drugs through diffusion mechanisms and are degradable; drug release is initiated by hydrolysis of polymer chains on the outer shell of the matrix.154 It has been shown that hydrolysis rates may be accelerated via the addition of acidic excipients.155 Furthermore, basic excipients have the ability to stabilize the bulk polymer but diffuse out of the surface region, encouraging surface erosion.156 Therefore, adjusting the acidity/basicity allows for some degree of control of temporal release. Although orthoester bonds are quite reactive in nature, polymers with these linkers are highly hydrophobic, with limited water penetrability. POE I hydrolyzes rapidly in water to produce γ-butyrolactone, which converts to γ-hydroxybutyric acid, and therefore, its use in drug delivery applications has been discontinued.157 POE II has a low degree of water sorption and a Tg of 22 °C (if based on 1,6-hexanediol) and is therefore also hydrophobic.157 POE III is a semisolid at room temperature, which provides the advantage of drug mixing without the need to use solvents or high temperatures.157 POE III is less hydrophobic than POE II. The extremely slow erosion rates of POE I–III have prevented this class from being investigated nearly as much as the POE IV class, which has been shown to have significant potential for use in bioerodible drug delivery systems when lactic and glycolic monomers are instilled in the polymer backbone.158 POE IV incorporates a mono- or diglycolide segment that is incorporated in the polymer backbone. This leads to the generation of lactic or glycolic acid monomers, which catalyze the hydrolysis of further ester linkages in the polymer. The degradation rate of POE IV is highly tunable, since it can be varied by controlling the nature of the diol R group and latent acid diols (and their relative proportions). POE IV has been used to deliver payloads such as small molecule analgesics and nucleic acids.159
2.4. Poly(anhydrides)
Poly(anhydrides) generally consist of poly(sebacic acid), poly(adipic acid), and poly(terphthalic acid), among others.160 Poly(anhydrides) undergo surface erosion, in which release kinetics is dependent on properties of the drug payload and intrinsic dissolution rates (Figure 2).161 In the case of a hydrophobic drug, the degradation of the polyanhydrides is the main factor in release, whereas in the case of a hydrophilic drug, solute transport is dependent on the concentration gradient between the delivery system and surrounding medium.161 However, the degradation of the poly(anhydride) bond also depends on the chemistry of the polymer backbone, which can be tuned by varying the nature of the monomer by up to 6 orders of magnitude.162 Both drug solubility and the dissolution rate of the polymer need to be considered when investigating poly(anhydride) drug delivery.163 Poly(anhydrides) have been fabricated into micro- or NPs for intravenous, oral, and aerosol drug delivery,164 as well as for vaccine, protein, and chemotherapeutics delivery.165–168
2.5. Poly(amides)
The most frequently utilized poly(amides) for drug delivery are poly(amino acids) (Figure 2). Their synthesis has already been extensively researched,169 which makes them an ideal mature platform to build upon. Poly(amino acids) are typically used to deliver low-MW drugs, are relatively nontoxic,170 and are typically cleaved by enzymes as they are stable to hydrolysis.171 The degradation rates of poly(amino acids) are dependent on the hydrophilicity of the amino acids that make up the polymer.172,173 Polyamides are generally semi-crystalline. Biodegradation can be accelerated by using side groups such as benzyl, hydroxyl, or methyl groups during copolymerization. Most poly(amino acids) are made up of a single type of amino acid and include the most widely used poly(amino acids): poly(γ-glutamic acid) and poly(L-lysine).174–177
Poly(γ-glutamic acid) is a water-soluble degradable poly(amide) that exists in both D- and L-optically active forms and is a highly functionalizable polymer. The carboxylate side chain can be used for covalent attachment of functional moieties and drug molecules.88 Polymeric NPs composed of poly(γ-glutamic acid) have been developed for the delivery of chemoimmunotherapies,178 chemotherapeutics,179 and therapeutic proteins.180,181
The major applications of poly(L-lysine)-based polymers has been in gene delivery, where the highly positively charged amino groups can interact with negatively charged siRNA or DNA chains.182 These peptides are also useful for facilitating endosomal escape due to their buffering capacity, although their highly cationic nature renders them toxic and further modifications are often required to mask their charge.174,183
2.6. Poly(ester amides)
Poly(ester amides) (PEAs) are polymers that have both ester and amide linkages on their backbones (Figure 2), yielding favorable mechanical and biological properties with enzyme-catalyzed biodegradability.184–187 Invented in the late 1970s,184–187 the combination of the favorable properties of polyesters and polyamides with poly(amino acids) has produced amino acid-based PEAs with promise for biomedical and pharmaceutical applications.184–187 Typically, the PEA backbone consists of nontoxic building blocks such as α-amino acids, fatty diols, and aliphatic dicarboxylic acids.188 If hydrophobic amino acids are used (e.g., Phe, Leu), the resulting amino acid-based PEAs are hydrophobic and water insoluble.185 Due to the alternation of amide and ester bonds in this type of polymer, the rate of polymer degradation under hydrolytic mechanisms is slow, and most of the reported faster degradation rates are catalyzed by enzymes.189 These polymers are also highly crystalline, and their degradation rates can be modified by controlling the Phe:Gly ratios. The poly(β-amino esters) (PBAEs) are synthesized via the Michael addition reaction of diester diacrylates with primary and secondary amines and have led to the generation of large libraries of PBAEs with various backbones, which has facilitated the high throughput screening of polymers for gene delivery.190–193
2.7. Poly(phosphoesters)
Poly(phosphoesters) are degradable and biocompatible polymers with utility in drug delivery and tissue engineering (Figure 2).194,195 The pentavalency of the phosphorus atom allows for the conjugation of a range of side chains, such as proteins or small drugs. The conjugation of different side chains also allows for the tuning of the physicochemical properties of the polymers.196 Since poly(phosphoesters) are structurally similar to nucleic acid biopolymers, they biodegrade through hydrolysis and enzymatic digestion and are excellent classes of polymers for nucleic acid delivery due to the conjugation of charged groups to the phosphate side chain.197,198 Poly(phosphoesters) can be synthesized by ROP, condensation, and addition polymerizations,88 and their fast degradation rates produce nontoxic products. For drug delivery applications, poly(phosphoesters) can be copolymerized with polyethers and polyesters, and they are gaining attention as biomaterials.196,199–202
2.8. Naturally Occurring Biodegradable Polymers
Naturally occurring biodegradable polymers have been used in drug delivery applications, due to their abundance in nature and biocompatibility, and include protein-based polymers, such as collagen, albumin, gelatin, and polysaccharides, such as agarose, alginate, carrageenan, hyaluronic acid (HA), dextran, chitosan, and cyclodextrins.203 In particular, natural polyscaccharides have been widely used in tissue engineering and bioscaffold designs, in addition to NP fabrication for drug delivery. Although highly biodegradable in nature, a limitation of natural polymers is their batch-to-batch variability and broad MW distributions, making them less attractive than synthetic polymers that are more reproducible and versatile. Two widely used polysaccharide-based polymers for drug delivery applications are chitosan and hyaluronic acid polymers, which are further discussed below.
2.8.1. Chitosan
Biodegradable polysaccharides have shown potential in drug delivery applications. One major polysaccharide with broad utility is chitosan (Figure 2), which is derived from the chitin found naturally in crustacean exoskeleton.204 Deacetylation of chitin produces randomly repeating units of D-glucosamine and N-acetylglucosamine, with the degree of deacetylation being related to chitosan’s crystallinity and degradation rates.205 Chitosan is highly insoluble in water (due to its crystalline nature) and must be solubilized in dilute acid solutions prior to use. Chitosan has been extensively used for the development of oral drug delivery systems.206,207 It is broken down by lysozyme and chitosan polymers with low degrees of acetylation and can remain in vivo for several months.88 Chitosan’s degradation can be accelerated by disrupting the significant network of hydrogen bonding, through the inclusion of bulky side groups. Major applications of chitosan involve wound dressing and healing (where it has also demonstrated anti-inflammatory and antibacterial properties),208,209 gene delivery due to its highly positive charge,210 oral delivery,211 and pulmonary drug delivery due to its mucoadhesive properties.212,213 Additionally, chitosan has been fabricated into numerous NPs for drug delivery applications.214,215 The ease with which the side groups can be modified and deacetylated and its blending with a variety of other polymers make chitosan a versatile and bioactive polymer.216 The utility of chitosan for the development of a range of polymeric NPs for drug delivery applications (and in particular for oral delivery) has been previously extensively discussed.215–219
2.8.2. Hyaluronic Acid-Based Polymers
HA is a naturally occurring linear polysaccharide polymer composed of D-glucuronic acid and N-acetyl-D-glucosamine disaccharide.220 HA has been utilized for a wide range of applications, since it has highly favorable properties, including being biodegradable, biocompatible, nontoxic, and nonimmunogenic. It has been used in various surgical, tissue engineering, and drug delivery applications.221 Since the past decade HA has been used to develop numerous HA drug or biologic conjugates and hydrogel depot drug delivery systems. Bioconjugation of HA to therapeutics improves drug solubility, PK (pharmacokinetics), and clearance.222 The pKa of HA carboxyl groups is between 3 and 4, rendering the polymer anionically charged at neutral pH.223 Therefore, HA is highly hydrophilic, and due to its ability to absorb water, it can expand up to 1000 times its solid volume, leading to a loose, hydrated network.224 This property makes HA an attractive material for hydrogel-based drug delivery. HA also has intrinsic targeting ability, since it can selectively interact with the CD44 receptor, termed the hyaluronan receptors for endocytosis (HARE), and this has been used as an active targeting strategy for delivery of anticancer drugs.225,226 The conjugation of HA to various drugs, peptides, and proteins has been the prominent application of HA polymers to date.223
3. EVOLUTION OF CONTROLLED-RELEASE POLYMERS
Controlled release of drugs from polymeric drug delivery systems is typically achieved by regulation of the rates of polymer biodegradation and drug diffusion out of the polymer matrix. Starting from the pioneering work led by Judah Folkman on the use of silicone rubber as a controlled-release matrix, whereby anesthetic gases encapsulated in a reservoir were shown to diffuse through the porous silicon membrane,227 the controlled drug delivery field can be chronicled in evolving phases, where we have seen the implementation of macroscopic controlled drug delivery devices and implants evolve into microscopic degradable polymer depot drug delivery systems pioneered by Robert Langer, leading to the current nanoscopic era of NP-based controlled drug release.6 Ultimately, each drug delivery scale has inherent advantages and disadvantages and the choice of scale relies highly on the biological end points required. Table 1 summarizes the main differences between macro-, micro-, and nanoscaled drug delivery systems, and these concepts are further described in the following sections.
Table 1.
Comparison of Macro-, Micro-, and Nanosized Drug Delivery Systems (DDS)
| Polymeric DDS | Advantages | Disadvantages |
|---|---|---|
| Macroscale | • Highly tuneable and controlled drug release achievable (over days, weeks, months and years) | • Original DDS are non-biodegradable (in particular the earlier silicone based contraceptive depots) |
| • Large size allows comprehensive mathematical and empirical modelling of drug release (with wide body of literature available being the earliest established polymeric DDS) | • Majority of approved devices require insertion and removal procedures by trained personnel therefore creating patient compliance issues | |
| • Large size facilitates higher payloads and continuous controlled release of therapeutics | • In some cases surgical procedures are required | |
| • Reliable DDS for many critical applications including contraceptives and drug eluting stents with numerous marketed products | • Possibility of foreign body reactions | |
| • Prolonged release eliminates or reduces the need for repeat dosing | • Possibility of fibrosis encapsulation of DDS | |
| • Pulsatile or constant or zero-kinetics rate of drug release achievable | • Tissue scarring | |
| • For certain applications it can eliminate the need for injections | • Limited biological applications due to lack of systemic circulation and transport of DDS | |
| • Drug reservoir can be achieved at desired anatomic site | • Difficult to reach consensus on how to benchmark and clinically assess macroscale DDS | |
| Microscale | • Tuneable controlled release of drugs (hours, days, months) | • Large size limits in vivo circulation time and biodistribution leading to rapid clearance post systemic administration |
| • Large utility for local depot-release drug delivery applications | • Can lead to agglomeration and clotting | |
| • Wide range of payloads possible (proteins, peptides, small drugs) | • Applications mostly limited to subcutaneous and intramuscular injections due to rapid systemic clearance | |
| • Sustained and controlled release of small and macromolecules up to months (after local injection) | • DDS cannot traverse biological barriers due to large size and remains at local site of injections | |
| • Targeting of DDS to specific organs, tissues and cells possible | • Limited cellular uptake and subcellular applications due to large size | |
| • Biodegradable and in vivo degradable | ||
| • Standardized and facile fabrication of polymeric microparticles | ||
| • Higher drug loading and more control over drug release achievable (compared to nanoscale DDS) | ||
| Nanoscale | • High surface-to-volume ratios, allowing for maximal surface binding to targets and the inclusion of high number of ligands or other molecules on the surface of nanoparticles | • For oncology applications, reliance focused on the EPR effect in patients, which is variable and remains unpredictable among patients and tumors |
| • Small size facilitates long systemic circulation times and higher tissue penetration | • Smaller nanoparticles prone to renal clearance (<10 nm) | |
| • Colloidal nature facilitates accumulation in tumors and inflamed tissues through enhanced permeation and retention (EPR) effects | • Larger nanoparticles prone to increased uptake by key organs such as liver, lung and spleen | |
| • Highly tunable and decoupled nanoparticle pharmacokinetics and drug pharmacokinetics | • Small size can lead to off-target effects as nanoparticles can easily accumulate in other locations | |
| • Can be optimized to bypass the reticuloendothelial and blood brain barrier systems more effectively | • Size dependent toxicity of nanocarriers can limit dosing amounts | |
| • Amenable to cellular uptake mechanisms due to small size | • Consensus on size related biological outcomes often difficult to reach due to the variety of nanomaterials investigated across numerous laboratories | |
| • Effective for subcellular targeting and drug delivery applications | ||
| • Small size of NPs allows for facile sterilization via filtration, removing the need for complete aseptic manufacturing processes and therefore facilitating ease of fabrication | ||
| • Nanoscale phenomena enabled triggered control of drug release via directed exogenous stimuli (see section 5) |
3.1. Evolution of Macroscale Polymeric Drug Delivery Systems
Drug incorporation into solid polymers began in the 1950s for agricultural research, with extensions of this work for medicine beginning in the mid-1960s.12 Macroscale polymeric drug delivery devices were originally developed to achieve spatiotemporal control of drug delivery from a local drug depot device. This allowed for a range of payloads (such as small molecule drugs, proteins, and bioactive agents) to be delivered in a controlled manner at the site of treatment. Drugs are generally released from macroscale polymeric DDS via one of three mechanisms: (1) diffusion-controlled release, (2) drug-carrier affinity, or (3) degradation of the matrix material. Though the release rate of the therapeutic payload can be driven by any one of these mechanisms, it usually involves all three.228
Earlier drug delivery devices were mostly made of non-degradable polymers, including polyurethanes, silicone rubber, and poly(ethylene-co-vinyl acetate) (PEVA). Drug transport in these nondegradable systems was primarily driven by diffusion; thus, these polymers were used to devise reservoir or drug depot devices.127 Release rates from reservoir-based devices can be controlled by changing the thickness and degree of permeability of the rate-controlling membrane (RCM).229 For matrix-based systems, drug release is diffusion driven and can be affected by concentration gradients, diffusion distance, and amount of matrix swelling.
Following the demonstration of controlled release with silicone polymers, several macroscale drug delivery products were developed for contraceptive purposes, treatment of glaucoma, and drug eluting skin patches.4 In fact, delivery of steroids and their derivatives for contraception has been one of the most widely studied applications of controlled-release polymers, and among the first such systems was Norplant, approved by the FDA in 1990 (Figure 3–1).1 Norplant is an upper-arm-implanted contraceptive formed of six silicone tube capsules, each 2.4 mm × 34 mm long and containing 36 mg of the progestin levonorgestrel.2 The hormone is released at 3.8 pg/cm length/day, and the implant is effective for 5 years after implantation. This product has since been replaced by an improved version (Norplant II), which was approved by the FDA in 1996.232 This implant uses polysiloxanes consisting of a backbone of inorganic Si–O–Si units, and drug release was controlled mainly by diffusion at a constant linear rate. Another example of an implantable contraceptive controlled-release device is Implanon.233 However, one limitation of these DDS is that since the silicone capsules are nondegradable, they must be removed after drug release is complete.
Figure 3.
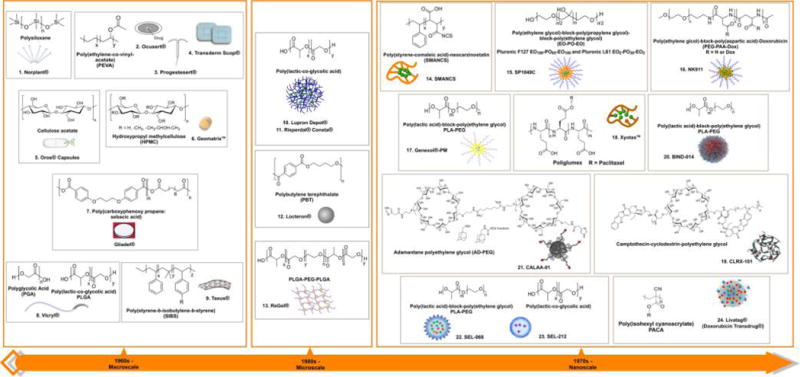
Evolution of polymeric drug delivery systems spanning over 50 years. 1960s: the macroscopic era of polymeric drug delivery. (1) Norplant is an upper-arm-implantable polymeric drug delivery depot for contraceptive use [active pharmaceutical ingredient (API): progestin levonorgestrel];1 (2) Ocusert is a drug-eluting polymeric eye implant for the treatment of glaucoma or ocular hypertension (API: pilocarpine);2 (3) Progestesert is a T-shaped polymeric IUD for contraceptive use (API: progesterone);3 (4) Transderm Scop is a polymer-based skin-patch drug delivery system for the treatment of motion sickness (API: scopolamine);26 (5) OROS is a polymeric capsule system for oral delivery (numerous APIs, including salbutamol, nifedipine, hydromorphone, verapamil, paliperidone, nifedipine);2,27,28 (6) Geomatrix is a polymeric tablet system for oral delivery (API: diclofenac, molsidomine, zileuton, nisoldipine, alfuzosin hydrochloride, ropinirole, paroxetine, levodopa, and benserazide);29,30 (7) Gliadel is an implantable polymeric disk for the treatment of glioblastoma multiforme [API: bis(2-chloroethyl)nitrosourea (BCNU)];31,32 (8) Vicryl is a degradable surgical suture;34 Taxus is a polymer-coated metal drug-eluting stent for prevention of restenosis (API: antiproliferative drugs such as paclitaxel).35,36 1980s: the microscopic era of polymeric drug delivery. (10) Lupron Depot is a polymeric microparticle for the treatment of prostate, breast, ovarian and endometrial cancers and also for endometriosis, infertility and benign prostatic hypertrophy (API: leuprolide acetate; a luteinizing hormone releasing hormone (LHRH) agonist);55 (11) Risperdal Consta is an antipsychotic, PLGA based polymeric microsphere, which is a long-acting injectable approved for the treatment of schizophrenia and the maintenance treatment of Bipolar I Disorder (API: risperidone);230,231 (12) Locteron is a polymeric microparticle for the treatment of hepatitis C (API: interferon-α2b);57 (13) ReGel is a polymeric drug depot delivery system for the treatment of various diseases (API: small molecules, peptides, and proteins).58,59 1970s: the nanoscopic era of polymer drug delivery. (14) Neocarzinostatin conjugated to styrene-maleic anhydride for cancer therapy (API: neocarzinostatin);61 (15) SP1049C is a polymeric micelle for the treatment of advanced adenocarcinoma of the esophagus and esophageal junction (API: doxorubicin);63 (16) NK911 is a polymeric micelle for the treatment of a range of cancers (API: doxorubicin);126 b (17) Genexol-PM (or Cynviloq) is a polymeric micelle formulation for the treatment of a range of cancers (API: paclitaxel);65 (18) Xyotax is a polymeric NP for the treatment of advanced non-small-cell lung cancer (API: paclitaxel);66,67 (19) CRL-101 is a polymeric NP for renal cell carcinoma and ovarian and rectal cancer (API: camptothecin);68–73 (20) BIND-014 is a polymeric NP targeted to prostate-specific membrane antigen (PSMA) for the treatment of castration-resistant prostate cancer, non-small cell lung cancer, cervical, bladder, and head and neck cancers (API: doxetaxel);74,75 (21) CALAA-01 is a targeted polymeric NP for cancer treatment (API: siRNA);121,76 (22) SEL-068 is a targeted polymeric NP, which is an antismoking vaccine (API: nicotine antigen, T-helper-cell peptide, and adjuvant);121,77,78 (23) SEL-212 is a polymeric NP, is designed to be the first non‑immunogenic biologic therapy for the treatment of refractory and tophaceous gout (API: rapamycin co-administered with pegsiticase, clinical trials identifier: NCT02648269); (24) Livatag is a polymeric NP for the treatment of liver cancer (API: doxorubicin).152
Among other macro-DDS marketed by the pioneering drug delivery company ALZA are Ocusert (releasing the antiglaucoma drug pilocarpine) and Progestesert (an intrauterine device for the release of progesterone).3,234 These macroscale controlled drug delivery devices are composed of PEVA, which allows drugs to be released at a constant rate with zero-order kinetics.235 PEVA shows a slow release over a long period of time and good biocompatibility.236,237 Polymer permeability (and thus release rates) can be altered by varying the copolymer ratios.238 The diffusivity of the polymer matrix is dependent on the crystallinity of the polymer, and it has been shown that increasing the crystallinity reduces the diffusivity.238 Ocusert was the first controlled-release polymer drug delivery system to be used in the clinic (Figure 3–2).2 Placed in the lower eyelid, this implant delivered the active ingredient pilocarpine at a steady rate over a one-week period, which led to an improvement over daily eye drop administration of pilocarpine and resulted in fewer side effects.239 Progestesert was a T-shaped intrauterine (IUD) macroscale drug delivery device also composed of PEVA, capable of delivering 65 pg/day of progesterone for 1 year (Figure 3–3).3 Transderm Scop (Figure 3–4) was the first skin-patch drug delivery system that also used PEVA as the RCM to deliver 1 mg of scopolamine over 3 days for the treatment of motion sickness.26
In addition to the aforementioned in situ macroscale drug delivery devices, controlled-release polymeric systems for oral drug delivery to the gastrointestinal (GI) tract were also developed beginning in the 1980s.4 For example, OROS (osmotic controlled-release oral delivery system, Figure 3–5) is a pulsatile-release oral delivery capsule that has a permeable outer shell with small laser-drilled holes, which allows water to enter via osmotic pressure during its trafficking through the GI tract.2,27,28 This action in turn pushes the active drug through the opening with controlled zero-order and flat PK. This system can be further tuned to release the drug within certain regions of the GI tract. The RCM in these systems consists of cellulose acetate, which maintains a constant rate of water diffusion into the capsule while an equal volume of the drug liquid or suspension is forced out.240 A small amount of low-MW PEG may also be used to initiate water diffusion.
Another controlled-release drug delivery platform uses hydroxylpropyl methylcellulose (HPMC) as a highly swellable hydrophilic polymer to modulate drug release and is marketed as the Geomatrix system (Figure 3–6).29 The combination of polymer layers with different swelling, gelling, and erosion rates results in a controlled rate of drug release, and this system is tunable toward a range of drug payloads and release properties.241
Controlled-release polymers have also been used to develop wafer DDS. Poly(carboxyphenoxy propane: sebacic acid) was used to develop Gliadel, a degradable polymeric disklike implant, secreting carmustine for the treatment of glioblastoma multiforme (brain cancer) (Figure 3–7). The polymer and drug are formulated into polymeric disks, which are placed into the brain after surgery and tumor removal.31,32
As discussed earlier, during the 1960s and 1970s the most widely used class of degradable polymers, the poly(hydroxyl acids), were originally developed to make sutures. PGA was first synthesized as a degradable suture. This polymer was then further optimized through the addition of lactic acid to produce PLGA and was marketed as Vicryl suture by Ethicon Inc. (Figure 3–8).34 Poly(alkyl cyanoacrylates) have also been used as surgical glue (Superglue) for over 40 years.148 Controlled-release polymers have also found use as coatings on bare metal stents for the controlled-release of antirestenotic drugs. Taxus, one such device approved in both Europe and the United States in 2004, uses poly(styrene-b-isobutylene-b-styrene) (SIBS) to control paclitaxel elution from a metal stent (Figure 3–9). These devices are currently being developed to incorporate degradable polymer coatings such as PLGA and PLA to minimize side effects.
Controlled polymeric drug release evolved with macroscale DDS, and we have seen the successful translation of numerous products to the clinic.228 Although some early products incorporating controlled-release drug delivery are still on the market in the form of drug-eluting depots, stents, and implants, the successful translation of these systems is not without its challenges (Table 1). Regulatory approval, the high cost of preclinical to clinical translation of these systems (in particular for biologics delivery), and patient compliance are hurdles that still need to be more effectively addressed. The need to remove drug depot implants and improve patient compliance led to the investigation of microscale DDS.
3.2. Evolution of Microscale Polymeric Drug Delivery Systems
In the 1970s the pioneering work of Langer and Folkman demonstrated the capability of polymers to release macromolecular therapeutic proteins in a controlled manner.6 The need for pulsatile release of hormones to resemble physiological levels was one motivation for developing tunable controlled-release systems, and the realization of the therapeutic potency of biologic drugs alongside their short half-lives in vivo spurred the investigation of microscale controlled-release DDS for systemic administration.
In the late 1970s and early 1980s, a PLGA microparticle system was developed that was capable of controlled release of luteinizing hormone-releasing hormone (LHRH) for up to 1 month for the treatment of prostate cancer.86,242 The plasma half-life of LHRH is 2.9 h; however, as the polymer slowly degrades, therapeutic levels of this antitumor peptide can be maintained for up to 3 months.243 Later, Decapentyl LP, a version of this microparticle, was subsequently developed for the treatment of prostate cancer and approved for clinical use in Europe in 1986.54 This was the first degradable microparticle drug delivery system approved for human use and is still available today as Lupron Depot (Figure 3–10).55
A highly successful long acting PLGA microsphere is Risperdal Consta (Figure 3–11). This PLGA based microsphere contains the antipsychotic drug risperidone and is an intramuscular formulation, with improved efficacy in the treatment of patients with schizophrenia.230 In the 1990s, the development of the ProLease process for the fabrication of homogeneous batches of drug-loaded PLGA microparticles using a low-temperature ultrasonic spraying technique facilitated the development of numerous drug-loaded PLGA microparticles.244 Further microparticles were developed using poly(butylene terephthalate) (PBT) for the delivery of interferon-α2b (Locteron, Figure 3–12).57
The first thermally responsive, degradable, controlled-release polymeric drug depot delivery systems were developed by Kim and co-workers using diblock and triblock copolymers of PLGA–PEG, which were termed ReGel (Figure 3–13).58 A major advantage of PLGA as the controlled-release polymer of choice is that its physicochemical properties have been widely studied and its release profiles can be easily tuned on the basis of MW and the lactide:glycolide monomer ratios.62 Drug release can be controlled from hours to months, and the blending of other polymers in the formulation can further control drug release. The versatility of PLGA allows for a wide range of payloads—from small-molecule drugs to peptides and proteins—to be encapsulated. The in vivo degradation of polymers such as PLGA therefore facilitated sustained release with tunable dosing without the need for surgical procedures.
3.3. Evolution of Nanoscale Polymeric Drug Delivery Systems
The ability to manufacture and control the assembly of polymers to nanoscale dimensions, combined with growing interest in applying nanotechnology to medicine, drove the further downsizing of controlled-release DDS from macro- or microscale products to the nanoscale.245 Indeed, the clinical success of the initially developed microparticles validated the concept of controlled release from polymers and set the stage for the era of polymeric controlled-release NPs. Driven by the fast pace of innovation and emerging successes of NP-based drug delivery, the phenomenal interest and investment in nanomedicine research and development is set to progressively improve the landscape of controlled-release drug delivery applications.246–248
Beginning with pioneering discoveries with the earlier nanomedicines (liposomes and polymer–drug conjugates)249–254 we have seen how the ability to modify the surface of these particles using the inert hydrophilic polymer PEG (a process termed PEGylation) facilitated the widespread use of these nanoscale DDS in vivo and led to the current astonishing pace of preclinical NP development.132 Prior to these events, the development of antibody technologies and bioconjugation techniques facilitated the creation of NPs specifically targeted toward disease antigens.253,255 In addition to antibodies, antibody fragments, peptides, aptamers (Apts), sugars, and small molecules have also been used to create targeted NPs.253,255–257 Specific targeting of polymeric NPs allows for their differential spatial localization within the body, minimizing the drug payload’s off-target adverse effects. Among the different approaches to NP targeting are “passive” and “active” targeting.258 Passive targeting refers to the preferential accumulation of NPs (bearing no affinity ligands) at active sites and is directly related to the inherent biophysicochemical properties of the NP (size, shape, charge, flexibility, etc.).19 Active targeting describes the mode of action of NPs with surface modification to incorporate affinity ligands with specificity to disease cells and tissues.19
Initially, nanoscale polymeric drug release systems involved the covalent conjugation of drugs to pendant groups on the polymer backbone, such as conjugation to poly(hydroxypropyl methacrylamide) (PHPMA) polymers.259,260 Here the drugs were bonded to the polymer via tetrapeptide linkages that were degradable by cathepsin B.259 These drug–polymer conjugates were then further targeted using ligands such as sugar molecules.259 Conjugation of the anticancer peptide neocarzinostatin to styrene-maleic anhydride (SMANCS, Figure 3–14) in 1984 by Maeda et al. led to the establishment of the enhanced permeation and retention (EPR) effect theory by Maeda, who observed that these colloidal macromolecular drug conjugates accumulated in tumors due to “leaky” vasculature.61 As a result of these breakthrough findings, passive targeting of NPs to tumors has been widely exploited in oncology applications.61
Another important development in the field was the synthesis of PEGylated block copolymers to formulate polymer micelle NPs.261 Cabral and Kataoka synthesized A–B block copolymers of PEG block polymers conjugated to hydrophobic amino acid blocks.262 These block copolymers could self-assemble into PEGylated polymeric micelles at very low critical micelle concentrations (cmc), with their hydrophobic cores loaded with small-molecule hydrophobic drugs.263 The drugs could be loaded either physically or by direct conjugation to the amino acid pendant groups of the polymer backbone. The terminal distal ends of the PEG polymers were also conjugated to targeting ligands, creating targeted NPs.263,264 Around the same period, Pluronic triblock polymers were also developed.265 Together these developments led to the clinical translation of a number of passively targeted polymer micelle NPs, including SP1049C (Figure 3–15), NK911 (Figure 3–16), Genexol-PM (Figure 3–17), and others, which are currently in clinical trials for cancer treatment.63,266,267
SP1049C is a Pluronic polymeric micelle NP composed of a doxorubicin (DOX)-entrapping hydrophobic core and a hydrophilic tail and is currently undergoing phase II studies in patients with metastatic cancer of the esophagus and esophageal junction who have been found refractive to standard chemotherapy treatments.63 SP1049C was shown to be efficacious in bypassing p-glycoprotein-mediated drug resistance.268 Patients were treated with a single dose of SP1049C (75 mg/m2 DOX) given as an intravenous infusion every 3 weeks.63 The results of this study and preclinical studies demonstrated superior antitumor efficacy for SP1049C when compared to free DOX.63 In a similar manner, we have seen the development of NK911, a micellar NP comprising PEG, DOX, and poly(aspartic acid), and Genexol-PM, a paclitaxel (Ptxl)-encapsulated PEG–PLA micelle formulation currently in clinical development for various cancers in USA and Europe, and clinically approved in South Korea in 2007.267,269–271 Genexol-PM does not require the use of the toxic Cremphor EL excipient for drug solubility and has therefore led to decreased toxicity and an increase in Ptxl maximum tolerated dose (MTD) for breast cancer therapy.272,273 Genexol-PM administration demonstrated increased treatment–response rates when given to patients who were not responsive to standard taxane therapy with Ptxl/carboplatin therapies, further suggesting improved outcomes for multidrug resistant (MDR) cases. Xyotax (Ptxl–poliglumex, Figure 3–18), also a passively targeted polymeric NP in which Ptxl is conjugated to poly(L-glutamic acid), was shown to preferentially target ovarian tumors.66,67 Another example of a passively targeted polymeric NP undergoing phase trials is CRLX-101 (previously known as IT-101, Figure 3–19), a camptothecin–cyclodextrin polymer conjugate that has shown prolonged circulation times and slow drug release kinetics in vivo, in preclinical and clinical studies.68–72,274 This NP formulation is being investigated as both a monotherapy and in combination with other clinically approved therapeutics for antiangiogenic and drug-resistance therapy.70,73 By encapsulating camptothecin within these polymeric NPs, the systemic toxicity of the drug is improved. In this manner, many promising drugs that do not meet toxicity, stability, or solubility requirements can be revisited once entrapped within polymeric NPs.
Work led by Farokhzad and co-workers established single step self-assembly techniques for-the-first in human clinical translation of targeted polymeric nanoparticles. Initial proof-of-concept in vitro demonstrated the effective nanoengineering of targeted Apt NPs for the first time, and subsequent targeting of prostate cancer cells over-expressing the prostate-specific membrane antigen (PSMA) receptor with up to 77-fold increase in binding compared to non-targeted controls.21 The efficacy of these NPs was further investigated in preclinical models where tumor growth was monitored up to 109 days and results revealed that a single administration of Apt NPs containing docetaxel (Dtxl) was significantly effective at tumor size reduction compared to controls, showing almost complete tumor reduction and 100% survival (compared to 57% for non-targeted NPs and 14% for Dtxl alone).23 This study presented improvements in the therapeutic index of Dtxl, in addition to that of Ptxl,24 and paved the path for clinical translation of this polymeric platform in humans. Next, employing a modular self-assembly approach using pre-functionalized polymeric materials, libraries of targeted NPs that varied narrowly from each other in their biophysicochemical properties were developed by BIND Therapeutics, which led to the translation of the most optimal hit; BIND-014-the first targeted and controlled release polymeric NP for cancer chemotherapy to reach clinical development.25 BIND-014, is a PSMA-targeted Dtxl-encapsulated polymeric NP composed of PLA-PEG (Figure 3–20). PSMA is a transmembrane protein overexpressed on the surface of prostate cancer cells and tumor-associated neovasculature of virtually all solid tumours.275,276 BIND-014 is capable of delivering up to 10 times more Dtxl to tumors compared to an equivalent dose of free Dtxl in multiple animal models,74 and is currently undergoing clinical trials for castration-resistant prostate cancer, non-small cell lung cancer, cervical, bladder, and head and neck cancers.75 However, up to now as evidenced by initial clinical trial results, these first-generation targeted polymeric NPs have in some cases demonstrated activity against tumors that is not materially differentiated from their parent drug, underscoring the need for patient selection (i.e. those with high EPR). Ascertaining EPR in patients is no trivial task, however research efforts towards investigating companion diagnostics is helping to shed light on this complex problem. For example, we have recently shown that magnetic resonance imaging (MRI) could be a useful tool to identify patients susceptible to higher NP accumulation in tumors.277 We investigated the use of a clinically established 30 nm magnetic NP contrast agent for its potential to predict colocalization of PLGA-PEG NPs to tumors in mice using MRI. The magnetic MRI contrast agent was able to predict the colocalization of the polymeric NPs with >85% accuracy and circulation within the microvasculature with >95% accuracy, despite their markedly different sizes and compositions. Computational analysis of NP transport enabled predictive modeling of polymeric NP distribution based on imaging data and identified key parameters governing intratumoral NP accumulation and macrophage uptake. Using MRI we could accurately predict initial treatment response and drug accumulation in a preclinical efficacy study using a paclitaxel-encapsulated NP in tumor-bearing mice. These approaches yielded valuable insight into the in vivo kinetics of NP distribution and suggested that clinically relevant imaging modalities and agents, can be used to select patients with high EPR for treatment with therapeutic polymeric NPs (or similar agents).277
CALAA-01 is the first targeted polymeric NP to enter the clinic for siRNA delivery (Figure 3–21).279 The CALAA-01 NP consists of siRNA that reduces the expression of the M2 subunit of ribonucleotide reductase (R2), cyclodextrin containing polymer (CDP) for siRNA condensation, adamantine-PEG (AD-PEG) for steric stabilization, and adamantine-PEG conjugated to human Tf (AD-PEG–Tf) to target the TfR overexpressed on the surface of most cancer cells.19,280 CALAA-01 employs a unique two-vial formulation strategy, which allows for the rapid self-assembly of the NP (50–70 nm) delivery system components (CDP, AD-PEG, AD-PEG–Tf) with siRNA at the point of care.279,121 This formulation is also capable of high siRNA payload delivery and endosomal pH-triggered (<6.0) release of siRNA once NPs are endocytosed.121,76
SEL-068 is a first-in-class synthetic and integrative targeted polymeric NP vaccine to reach clinical development for the treatment of chronic conditions, such as smoking addiction (Figure 3–22). SEL-068 is fabricated using degradable PLGA and PLA–PEG polymers and contains nicotine as antigen, T-helper-cell peptides, and TLR agonists as adjuvants and is currently under development for smoking cessation and relapse prevention.121,77 The encapsulation and controlled release of the synthetic TLR agonist by the polymeric NP matrix minimizes systemic inflammatory cytokine response, leading to an improvement in the overall safety of this novel adjuvant-containing vaccine. The administration of SEL-068, which is based on modular self-assembly NP technology,24 results in high antinicotine antibody concentrations and high antinicotine antibody affinity. This leads to the sequestration of nicotine molecules in the circulation and largely blocks central nervous system exposure, thereby diminishing the addictive effects of nicotine.121 SEL-068 is the first targeted, controlled-release polymeric vaccine delivery NP to enter the clinic and is based on the synthetic vaccine particle (SVPs) technology developed by Selecta Biosciences. These SVPs can effectively coencapsulate both antigen and adjuvants capable of cellular immunity and robust humoral responses.78
Currently, a passively targeted poly(isohexyl cyanoacrylate) polymeric NP termed Livatag (doxorubicin Transdrug) is undergoing phase III clinical trials for hepatocellular carcinoma (Figure 3–23).152 This degradable polymeric NP developed by BioAlliance Pharma encapsulates doxorubicin and demonstrated high antitumor activity against MDR protein-overexpressing hepatocellular carcinomas in vitro and in vivo.152 In one phase II trial, Livatag led to 88.9% survival rate after 18 months of treatment, whereas 54.5% survival rate was observed in patients with the current transarterial chemoembolization treatment.152 Currently, this polymeric nanoscale DDS has progressed the furthest in phase trials (currently undergoing phase III investigation).152
Numerous targeted polymeric NPs are currently under investigation for various drug delivery applications; in particular, polymeric NPs are attractive platforms for the targeted and controlled release of antigens and adjuvants in nanoimmunotherapy applications as well as for the delivery of anti-inflammatory agents.281–284 Targeted polymeric NPs engineered to deliver inflammation resolving biological therapeutics have also been recently developed that can target inflammatory sites in a spatiotemporal manner, facilitating the tempering of inflammation using controlled-release proresolving mediators.285–287
3.4. The EPR Effect
Many of the polymeric NPs discussed in this section exploit the so-called EPR effect mentioned previously for tumor penetration and accumulation. In the context of controlled-release drug behavior within the intratumoral environment, important variables include NP design, the nature of the drug, and the diffusivity of both the NP and encapsulated drug throughout the vasculature, in addition to patient tumor heterogeneity and the lack of or predisposition to the EPR effect. As more patient data from clinical trials using both active and passively targeted NPs accumulates, the issues of tumor heterogeneity and patient response variability become more significant. It is therefore important to acknowledge not only that the EPR effect is a complex phenomenon, shaped by many biological variables in the tumor microenvironment, but also that our understanding of this effect is constantly evolving.
Preclinical research has begun to address EPR limitations by investigating normalization of tumor vessels prior to NP administration, in addition to utilizing companion diagnostics using approved NP imaging contrast agents to assess EPR beforehand.258,289 Tumor hallmarks, such as hypoxic gradients, increased interstitial pressure, and patient tumor heterogeneity, and lack of or predisposition to the EPR effect point to major gaps in this theory, and these concepts are discussed in detail elsewhere.258,289 We have also previously investigated the benefits of companion diagnostics as an EPR effect indicator and also showed the influence of tumor-associated macrophages on polymeric NP accumulation within tumors.277,278
3.5. Fabrication Techniques of Polymeric Nanoparticles
The progression from macro- to micropolymeric DDS was facilitated due to new fabrication methodologies that were able to produce uniformly sized drug-loaded polymeric microparticles using low temperature casting techniques, which established the ProLease process.290 This in turn facilitated the development of injectable controlled-release polymeric systems for the delivery of a wide range of therapeutics, including macromolecular biologics.228
The ability to increase the half-life of protein drugs by conjugating the inert and lipophilic polymer PEG to their surface led to the further miniaturization of controlled-release polymeric systems to the nanoscale, whereby the same PEGylation principles were applied to the development of polymeric NPs for in vivo use.19 Initially, this progress was motivated by the transition from macro- and depot-based drug delivery systems to injectable microparticle drug delivery systems, including albumin or galactose microspheres that were injected systemically for imaging studies.132 However, due to their large micrometer sizes, these particles were cleared from circulation rapidly, significantly hindering their use.132 Therefore, in order to facilitate longer circulation times in vivo, nanospheres with PEGylated surfaces were developed.132
Numerous methods have been developed for the encapsulation of therapeutics into polymeric NPs, the choice of which depends on the polymer and drug properties. Either bottom-up or top-down techniques have been employed; bottom-up methods include emulsion, interfacial polymerization, and precipitation polymerization. These polymerization methods employ a monomer as the initiator, and although highly defined NPs with precise size and shapes are obtainable, this technique is limited by the fact that the polymeric materials are not biodegradable and synthetic reagents may be retained as residues. One way around this is to use presynthesized and characterized polymers with added functionalities of PEG or targeting moieties; these are top-down preparations, a commonly used method for preparing NPs via the self-assembly of block copolymers, which can occur during nanoprecipitation (also referred to as solvent displacement), emulsification/solvent evaporation, and salting-out methods.149,291,292 The NP fabrication route is often dependent on the physicochemical properties of the drug molecules, along with NP size and loading requirements.19 Nanoprecipitation, oil-in-water (O/W) emulsification–solvent evaporation (single emulsion), and water-in-oil-in-water (W/O/W) emulsification–solvent evaporation (double emulsion) are three of the most commonly utilized methods for preparing a variety of polymeric NPs.19,293 In general, a number of considerations should be taken into account for the preparation of polymeric NPs and these include solvent choice, the solubility of the drugs (e.g., log Po/w), the mixing time of the aqueous and organic solvents, the type of surfactant used, the concentration of polymer in the organic solution, and the ratio of organic to aqueous solution, in addition to other factors.293–295 Newer approaches, such as supercritical technology, electrospraying, premix membrane emulsification, and aerosol flow reactor methods, have also been developed and are discussed elsewhere.293 The most common methods of preparing polymeric NPs for drug delivery applications are described in more detail in subsequent sections.
3.5.1. Prolease Technique
The ProLease technique is a process whereby microspheres are produced at very cold temperatures with bioactive polymers entrapped, which in turn facilitates high retention of biological activity. After the polymer and payload mixture is atomized into a vessel, cold liquefied gas or liquid causes the polymer droplets to immediately freeze, and the solvent in the droplets is extracted, leading to hardened spherical microparticles.290 The need to maintain the integrity of biological drugs, such as proteins, led to the development of this technique, which was initially utilized for the entrapment of recombinant human growth hormone (rhGH).244 The goal was to create slow-releasing polymeric microspheres that could be injected once a month instead of daily or triweekly. The ProLease method is suitable for microsphere fabrication, since it uses low temperatures to preserve the protein integrity and nonaqueous based (the protein is not subjected to oil–water interface where it can denature) entrapment, which leads to high protein encapsulation efficiencies. This microsphere formulation was composed of PGA (50:50) and termed Nutropin Depot, and it was approved by the FDA in 1999 for growth hormone deficiencies.296 This technique has limitations in that it is not easily applicable to other types of proteins, the use of liquid nitrogen on a large-scale may not be feasible, and organic solvents are still required during the process.
3.5.2. Nanoprecipitation/Solvent Displacement
Using the inherent solubility and gelling properties of polymers, NPs may be spontaneously formed whereby the addition of a solution of polymers and drugs to a nonsolvent phase leads to phase separation and therefore nanoprecipitation, salting out, or coacervation of the polymers into NPs. For example, in nanoprecipitation the polymeric constituents and drug are dissolved in an organic solvent that is miscible with water, and this solution is then added dropwise to an aqueous (nonsolvent) solution under stirring.297 The precipitation of the hydrophobic polymer instantaneously leads to the self-assembly of core–shell-like spheres, which are the most energetically stable structures, and during this process the drug can also become entrapped within the polymeric core.295 Following this the organic solvent is evaporated either by reduced-pressure evaporation or by further stirring of the solution (if the solvent is relatively volatile). The instantaneous formation of NPs is governed by the Marangoni principle, which describes interfacial interactions between liquid phases.298 Hydrophobic–PEG diblock polymers are formulated into NPs using the nanoprecipitation technique, leading to NPs with a hydrophobic core that entraps the drugs, surrounded by a hydrophilic outer shell.294 Dtxl-loaded NPs were prepared using nanoprecipitation, whereby the hydrophobic drug Dtxl was mixed and coprecipitated with the diblock polymer poly(lactic-co-glycolic acid)-polyethylene glycol-carboxylic acid (PLGA–PEG–COOH).299 The resulting NPs had a hydrophobic core composed of PLGA, wherein Dtxl was encapsulated, and a hydrophilic shell composed of PEG. Nanoprecipitation is a facile technique that is amenable to scaleup and requires only mild stirring under minimal sheer stress, leading to small-sized NPs (<100 nm) obtained using this technique. A limitation of this technique is poor entrapment of hydrophilic drugs (which can remain in the aqueous phase),293 lower entrapment efficiencies as compared to emulsion-based methods, and residual organic solvent impurities.294 Recent advances in which the aqueous water phase (nonsolvent) is replaced with organic solvents, such as methanol and ethanol, has facilitated the use of this technique for hydrophilic drugs also.293
3.5.3. Emulsion Techniques
Emulsion methods can be divided into water-in-oil (W/O), oil-in-water (O/W), and double emulsion (W/O/W). Following formation of an emulsion, the mixture is homogenized and can be used to prepare micro- or nanosized particles.300 The type of emulsification method utilized ultimately depends upon the properties of the polymer, drug, and also the degree of miscibility of the organic (oil) solvent with the water phase.
3.5.3.1. Single Emulsion
The single emulsion technique O/W requires the drug to be soluble in a water-immiscible organic solvent. In this method, the polymer and the drug are dissolved in a volatile water-immiscible solvent such as dichloromethane or ethyl acetate, and the organic phase is emulsified under intense shear stress into an aqueous phase containing appropriate amounts of a surfactant, such as sodium cholate or poly(vinyl alcohol) (PVA). The organic solvent is allowed to evaporate, allowing the self-assembly of NPs.301 The O/W emulsification technique is suitable for entrapping hydrophobic drugs and generally results in higher drug loading and encapsulation efficiency compared to nanoprecipitation, as well as achieving complete solvent removal. However, it requires an additional input of energy such as sonication or homogenization and the resulting NPs are often larger than those obtained through nanoprecipitation.301
3.5.3.2. Double Emulsion
Double emulsion (W/O/W) is generally used for encapsulation of hydrophilic drugs and proteins. In this method, the drug is dissolved in a small volume of an aqueous phase together with a surfactant and this is emulsified in an organic phase containing the polymer. The W/O emulsion formed is then dispersed in a larger volume of an aqueous phase with or without surfactant to form the double W/O/W emulsion. Finally, the solution undergoes evaporation of the remaining organic solvent, yielding NPs.301 This method normally yields NPs with larger size than nanoprecipitation or O/W methods, with moderate drug loading and encapsulation efficiency.294
3.5.4. Electrospraying Methods
Electrospraying methods (also referred to as electrohydrodynamic techniques) refer to methods that use electrostatic forces to fabricate nano- or microsized particles of various sizes and shapes, in addition to fibers.302 In this technique, electrostatic forces are used as the driving force for fabrication using an electrically charged fluid jet. Here a solution of polymer and drug dissolved in a conductive solvent is used, and by adjusting the concentration and processing parameters, including flow rate and voltage, the continuous charged jet can be broken into small droplets with defined sizes and shapes. The advantages of these methods are the ability to obtain high loading efficiencies, narrow particle-size distributions, and facile particle synthesis due to single-step processing. Electrospraying techniques have been used to produce layer-by-layer NPs consisting of PLGA–DOX/PLA–Ptxl/PLGA layers designed to have low initial burst release. The release rates of each drug could be controlled by changing the polymer concentrations, flow rate, particle size, and shell thickness, leading to zero-order release profiles.302 Although currently not amenable to industrial scaleup, their use for the fabrication of multidrug DDS is highly valuable.
3.5.5. Microfluidics
Microfluidics involves the manipulation of nanoliter volumes in microscale fluidic channels, and in the past few years, applications of microfluidics have expanded from conventional chemical and biological analysis to other fields, such as chemical reactions, biochemical assays, and cell handling.303 Soft lithography using PDMS as a method for fabricating prototype devices has allowed the simple fabrication of pneumatically activated valves and rapid mixers, providing the ability to mix reagents rapidly and to provide homogeneous reaction environments. The continuous variability of reaction conditions, temperature control, and addition of reagents at precise time intervals are some of the key features that have made microfluidic systems useful for the synthesis of NPs.304 Furthermore, synthesis carried out in microchannels allows for in-line characterization,305 feedback control,306 and high-throughput continuous synthesis,307 which potentially enables screening and optimization of libraries of NPs with varying properties. Polymeric NPs prepared by bulk synthesis tend to have variable physicochemical properties (size, surface composition, and drug loading) due to the inability to control the mixing of precursors.308 Using hydrodynamic flow focusing, polymeric NPs exhibiting narrow size distributions compared to bulk synthesis have been prepared in a reproducible manner.308,309 For polymeric NPs prepared through microfluidics, higher drug encapsulation without increase in NP size has been observed.308 Another method to prepare NPs takes advantage of the rapid mixing microenvironment that occurs in microdroplets formed inside microfluidic channels.310 For instance, cross-linked alginate NPs were synthesized in a microchannel using aqueous alginate droplets as templates, followed by the shrinkage of the drops. This method exhibited remarkable control over the NP properties, specifically, size and size distribution.304,311 Given the volume of research currently involving the microfluidic synthesis of NPs, it is expected that as more therapeutic NPs reach the clinical stage, the need for improved synthesis methods would also increase, at which point microfluidic technologies may become an important tool in the development of NPs.
4. CLASSICAL MECHANISMS OF DRUG RELEASE
For polymeric systems, “drug release” typically refers to how a drug molecule is transported from a starting position in a polymeric matrix to the polymer matrix’s outer surface and, finally, how it is released into the surrounding environment.247 Drug molecules can be transported out of drug delivery systems via diffusion through water-filled pores, a process governed by random movements of the drug and driven by chemical potential gradients and convection produced by osmotic pressure. In addition to diffusion, drug molecules can be released from the polymer matrix by erosion, which leads to pore formation and erosion effects and can be observed after an initial diffusion-controlled lag period. This section discusses classical drug release mechanisms based on drug diffusion, polymer degradation, and erosion.
The main controlled-release mechanisms can be summarized as (A) drug diffusion through water-filled pores, (B) diffusion through the polymer matrix, (C) osmotic pumping, and (D) erosion (Figure 4).312
Figure 4.
Drug release mechanisms from polymeric NPs: (A) diffusion through water filled pores, (B) diffusion through the polymer matrix, (C) osmotic pumping, and (D) erosion. Adapted from ref 62.
4.1. Drug Diffusion through Water Filled Pores
This process describes the random movement of drug molecules driven by a chemical potential gradient that is approximated by the concentration gradient. In degradable polymeric systems, the drug release rate is controlled by diffusion through a network of pores, with evolving structures as the polymer matrix also degrades. Water is immediately absorbed by polymeric NPs and is a faster process than drug release. The water that occupies the polymer matrix leads to water-filled pores over time, and the size of the pores becomes larger and more numerous, eventually leading to large enough pores to facilitate drug release.313
4.2. Drug Diffusion through the Polymer Matrix
In this scenario, drug molecules simply diffuse out of the polymer matrix. In nondegradable drug delivery reservoirs or depots, diffusion is the main driving force for drug release, with rate of release remaining constant and not affected by concentration gradients but by properties of the polymeric membrane (permeability and thickness).127
4.3. Osmotic Pumping
A further way of drug transport through water-filled pores is convection driven. The influx of water into a nonswelling system is caused by osmotic pressure, and drug transport as a result of this force is referred to as osmotic pumping. The design and creation of osmotically driven DDS using osmogens and semipermeable membranes is an actively investigated field, and is discussed in detail elsewhere.314
4.4. Erosion
4.4.1. Surface Erosion
Surface erosion occurs when polymers degrade starting at the matrix/scaffold surface, slowly reducing the size of the matrix/scaffold, from the exterior toward the interior,171 and occurs when the rate of erosion is greater than the rate of water penetration in the bulk polymer.79 Surface erosion is ideal for many drug delivery applications, as erosion kinetics (and therefore drug release) is controllable and reproducible. Moreover, the slow water permeation rate is ideal for drug delivery because water-vulnerable drugs are protected.171
4.4.2. Bulk Erosion
Bulk erosion occurs when water penetrates the bulk of the polymer, which results in homogeneous degradation of the entire matrix.315 The rate at which water permeates into the bulk is greater than the rate of erosion,171 and as a result, the polymers within the bulk of the matrix are likely to be hydrolyzed. Bulk erosion is less predictable than surface erosion and does not protect drugs from the environment, making it a suboptimal mechanism for controlled drug delivery.171
4.5. A Note on Zero-Order Kinetics
The drive to achieve controlled zero-order kinetics of drug release motivated the development of controlled-release devices and has seen degradable polymers play a major role in the implementation of drug-eluting microspheres, medical devices, and, more recently, NPs. Drug concentrations below the minimum effective concentration or above the MTD result in ineffective treatment or toxicity, respectively. Zero-order release kinetics ensures a steady plasma concentration of the drug within the therapeutic range (Figure 5).
Figure 5.
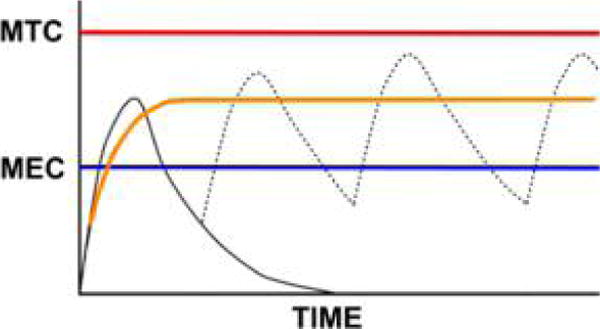
Plasma drug concentrations should remain within the therapeutic range to be both effective and well-tolerated. The therapeutic range lies between the minimum effective concentration (MEC) and above the maximum toxic concentration (MTC). In the case of rapidly absorbed and eliminated drugs, a single dose leads to a fast rise and fall in drug concentration (black solid curve). Frequent dosing at regular intervals leads to oscillating drug concentrations in the plasma (black solid curve followed by a dotted curve), and the drug concentration can fall outside of the therapeutic range for significant periods. Zero-order release (orange curve) leads to constant drug concentration in plasma (after an initial peak) and can be optimized to lie between the MEC and the MTC.
Controlled-release systems offer both temporal and spatial drug release control and can protect the therapeutic cargo. There are a number of considerations when designing drug delivery formulations, such as the mechanisms involved in the release of the drug. Ultimately, in addition to controlling the rate and location of drug release, controlled-release formulations can aid in patient compliance by minimizing dosing regimens. For effective treatment of disease, a prolonged release mechanism can maintain the therapeutic dose for longer periods of time, minimizing both underexposure and the risk of toxicity from overexposure.316
It is of note that in addition to zero-order drug delivery, oral pulsatile drug delivery systems have also been developed that can deliver drugs according to the circadian rhythm of the body, where the drug is released as a pulse after a lag time.317 The release profile for these systems follows a sigmoid curve after a lag time. The benefits of pulsatile drug release can be utilized in circumstances where continuous drug release is not beneficial or when chronopharmacological effects are needed (requiring nocturnal drug release) or when drugs follow a first-pass effect.317 These other methods of controlling drug release rate may also be beneficial in cases when constant dosing results in receptor down-regulation/desensitization and biological tolerance.
4.6. Drug Release Profile
The classical drug release profile is a good starting point in any discussion of controlled-release mechanisms. As mentioned earlier, zero-order release profiles are idealized states of drug release, and although preferred, a zero-order release profile is not representative of drug release from drug delivery systems (in particular, polymeric NPs). The most common drug release profile from polymeric drug delivery systems is in fact a triphasic profile (Figure 6) and is typically observed for macromolecular drug delivery systems with heterogeneous degradation behavior.62,318 The release rate can sometimes also be biphasic when the particles are smaller. Phase I of a triphasic release profile is referred to as the burst release effect and is the rapid release of drug molecules close to the surface or near the water layer (burst release is explained in more detail in section 4.7).62 Phase II of the release profile is a slow release phase governed by slow drug diffusion through the polymer matrix or through existing pores and is simultaneous with polymer hydrolysis and degradation.62 Phase III can be a faster release phase, as erosion begins.
Figure 6.
Model drug release profiles: open squares, burst and rapid phase II; filled circles, triphasic release with short phase II; crosses, burst and zero-order release; filled diamonds, triphasic release; dashes, biphasic release without burst release. Reprinted with permission from ref 62. Copyright 2011 Elsevier B.V.
Drug release from PLGA micro- or nanosystems shows a triphasic pattern. The initial burst release effect is due to the rapid release of surface-bound drug molecules, the second phase (steady controlled release) is related to diffusion and hydrolysis, and the third phase is as a result of bulk erosion.62 Studies have shown that concentration gradients and the shape of the drug delivery device are dominant factors in drug release, and later release is governed by the rate of polymer degradation.98 Whether the payload is hydrophobic or hydrophilic also affects release: hydrophobic drugs produce a zero-order release rate, whereas hydrophilic drugs display a triphasic pattern.127 However, it is important to note that because numerous phenomena make up the different release phases, caution must be taken in making general inferences from release profiles, particularly as the beginning and end of each phase are not always obvious.62
The release behavior of polymeric drug delivery systems depends on a wide range of physicochemical parameters and should be investigated on a case-by-case basis. For example, though a slow second phase or lag phase could suggest a densely packed polymer system with low porosity, this effect could also be the result of pore closure, polymer–drug interactions, or drug–drug interactions.62,319 Furthermore, the second burst release or a fast phase III could be attributed to erosion effects, but they could also be the result of polymer disintegration or cracks in the matrix. The release profile of polymeric drug delivery systems is useful primarily to ascertain the sustained release rate. Ultimately, many investigations of drug release profiles for controlled-release systems have been conducted in vitro, i.e., outside of physiological conditions. In order to more accurately predict and evaluate treatment outcomes in vivo, more intelligent methods of real-time monitoring of drug release (i.e., separation from NP carrier) are needed and efforts in this direction have recently begun.320–322
4.7. Burst Release Effect
In controlled-release polymeric drug delivery systems, when the delivery system first becomes immersed in the release medium, a rapid and short release of drug is observed, followed by a stable “plateau” profile. The former is usually referred to as the initial “burst release” (Figure 7), with low-MW drugs, peptides, and proteins having higher propensities for burst release as a result of osmotic pressures.323 Although under certain circumstances an initial sharp release of the therapeutic could be desirable, it is often unpredictable with uncontrollable duration and dose.323 For example, if pulsatile delivery is required, burst release can be triggered by rapid changes in the local environment. However, for the most part, avoiding the burst release effect is desirable to minimize any initial toxicity associated with a high dose. Huang and Brazel have addressed a number of factors and experimental observations of burst release in monolithic polymer-controlled drug delivery systems and suggested methods of controlling burst release, and Table 2 gives an overview of these findings.323
Figure 7.
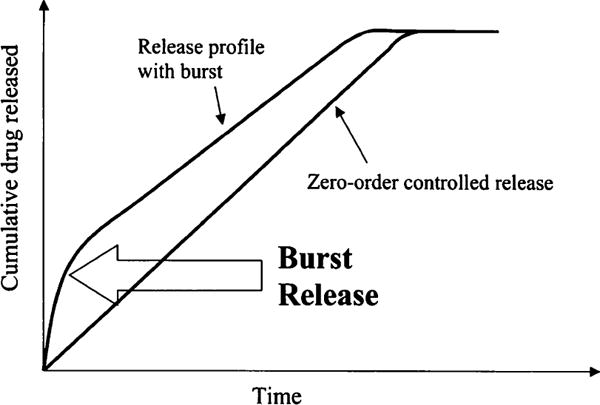
Burst release effect. Reprinted with permission from ref 323. Copyright 2001 Elsevier B.V.
Table 2.
Summary of Burst Release Effects
| Causes of burst release | Favorable situations for burst release | Disadvantages of burst release |
|---|---|---|
| • Storage conditions leading to drug diffusion to the surface and burst release due to placement in release medium | • Rapid treatment, i.e., wound treatment | • Mechanisms poorly understood |
| • Morphology and porosity of the drug delivery system | • Targeted delivery | • Uncontrollable duration |
| • Heterogenous polymer matrices and manufacturing defects | • Pulsatile release | • Difficult to control the released dose |
| • Percolation-limited diffusion | • Local or systemic toxicity (as a result of high local drug dose) | |
| • Post-drug-loading via equilibrium partitioning and the use of highly concentrated drug solutions | • Loss of drug quantity, leading to higher costs | |
| • Diffusion and transport of drug molecules during fabrication | • Shorter release profiles | |
| • Drying and rehydration processes | • Requirement of further dosing |
A number of causes for burst release in drug delivery systems have been put forward, including (1) device storage conditions during which the drug has diffused to the surface of the device membrane and bursts outward once placed in the release medium; (2) slow polymer gel formation in injectable hydrogels, i.e., when the polymer precursors are injected for reversible sol–gel transition purposes and a slow precursor setting allows some drug leakage; (3) heterogeneous polymer matrices and manufacturing defects; (4) percolation-limited diffusion; (5) drug loading by equilibrium partitioning when highly concentrated drug solutions are used; (6) the diffusion and migration of drug molecules during fabrication; and (7) drying processes leading to burst release effects as water molecules move to the gel surface, carrying drug molecules via convection and leading to higher drug concentrations at the surface of the carrier.324 Strategies to avoid the burst effect have included the addition of a drug-free outer-layer coating to the drug delivery system.323 For example, the multiple coating of polycation layers of either poly(L-lysine HBr) or poly(vinyl amide) on alginate beads was shown to prevent the burst release of proteins, leading to a more controlled release.323
In the same study, it was also observed that the burst effect could be reduced with the use of higher-MW polymers to hinder the release of the proteins.323 Nonuniform drug loading into polymer matrices has also been shown to minimize the burst release, as higher drug concentrations away from the surface avoid the formation of a rubbery gel layer.323 However, achieving nonuniform drug distribution in polymer matrices is not a trivial task. Decreasing the hydrophilicity of the polymer matrix can also affect burst release by minimizing water entry into the system, which changes the polymer pore sizes, decreasing the mobility of the protein payload.323 Multiple factors affect burst release properties and should be investigated on a case-by-case basis, including the morphology of the polymer system, the initial monomer concentration, the hydrophobicity of the polymer pendant groups, the MW of the polymer chain, and the nature and size of the payload, as well as the fabrication method, drying processes, and diffusion distances of the polymer matrix.
4.8. Drug Release from Aliphatic Poly(esters)
Poly(esters) such as PLGA or PLA tend to undergo bulk erosion, but drug release is not limited to degradation alone.236,325 The two main mechanisms involved in PLGA drug delivery systems are diffusion and degradation/erosion. Initially, concentration gradients and the shape of the drug delivery device dominate drug release. Degradation of the polymer matrix begins to predominate in the later stages of drug release, and the release kinetics becomes more “traditional”, i.e., controlled by degradation/erosion.62,325 Controlled-release mechanisms involving PLGA polymers have been addressed in detail and will be summarized here on the basis of previous reports.62
A wide range of physicochemical properties affect drug release from PLGA drug delivery systems (Figure 8). In the first instance, water absorption leads to pore formation in the polymer matrix on a faster time scale than drug release. Over time, absorption causes the polymer to swell, creating pores large enough for drug transport.312 The hydrolysis of ester bonds in PLGA polymers takes place immediately following water contact and leads to glycolic and lactic acid production, which further catalyzes hydrolysis. Ester bond scission is an autocatalytic process that leads to heterogeneous degradation within the polymer matrix that is driven by an acid gradient.62
Figure 8.

General factors effecting drug release kinetics from polymeric DDS.
PLGA undergoes bulk erosion due to rapid hydration, and the dissolution of the polymer degradation products creates even more pores. These pores then grow in size as further contact with water leads to hydrolysis and local acid gradients further catalyze polymer degradation. Smaller pores coalesce to form larger pores, a process controlled by the mobility of the polymer chains, which is in turn dependent on the Tg. The more glasslike the matrix, the slower the hydrolysis and therefore water absorption.312 In addition to pore formation, other factors influencing drug release include drug dissolution, polymer–drug interactions, and drug–drug interactions.
Ultimately, water absorption in PLGA-based drug delivery systems (and therefore the rate of drug release) are governed by a number of parameters, which include (1) the MW of polymer (typically PLGA is between 50 and 150 kDa), (2) the lactide-to-glycolide ratio (ranges between L:G 50:50 and 100:0), (3) end-group capping (usually ester or acid), (4) surrounding release buffer/medium, (5) pH and osmolality, (6) nature of the payload, (7) additives (surfactants, salts, etc.), (8) the size and shape of the system, and (9) the density and porosity of the system. A low MW and low L:G ratio and uncapped polymer end groups make for a more hydrophilic polymer, which increases the amount of water absorption and therefore hydrolysis and erosion rates. Often divalent cation or basic anion salts used in stabilizing proteins can also affect pore formation and hydrolysis reactions. Plasticizing or surfactant additives also influence drug release by affecting water absorption, hydrolysis rates, and plasticizing or leading to crystallinity within the polymer matrix.312 The mass of drug encapsulated can also affect drug release rates, as it is directly related to how much space remains in the matrix for water absorption. The size of the drug delivery system also influences release, as larger particles increase the pH gradient. Shape also affects drug release, as the ratio of surface area to volume is an important parameter facilitating more water contact. Temperature is important, too, as the mobility of the polymer chains increases at higher temperatures. Salts and additives in the surrounding medium can also affect drug release by driving osmolality. As a result, drug release rates in vitro and in vivo are different, with the latter more likely to see faster rates of polymer degradation and drug release and shorter lag phases. Finally, surface interactions of the drug delivery particles with proteins and cells of the immune system in vivo can also affect drug release.326
4.9. Mathematical Models of Polymeric Drug Release
Mathematical modeling of drug release aims to predict drug release rates and drug diffusion behavior from drug delivery systems. This information in turn aids in the optimization of the release kinetics and assertion of the physical mechanisms involved in drug transport, which is facilitated by comparing experimental data with mathematical models. Mathematical models can shed light on the effect of various parameters, such as shape, size, and composition of the drug delivery system, on the overall drug release rate. The accurate prediction of drug release can ultimately improve overall therapeutic efficacy and drug safety profiles. Ultimately, it is envisaged that the systemic use of mathematical models to predict drug release rates and behavior can lower costs and experimental times, leading to more effective drug formulations and more precise dosing regimens.
A range of mathematical models have been proposed and used to predict and explain drug release from polymeric drug delivery systems and can be either empirical/semiempirical or mechanistic in nature. From the two, mechanistic models are designed by taking into account biophysicochemical properties and can provide insight into drug diffusion, degradation, and erosion. Mechanistic models involve the concept of diffusivity and are based on Fick’s law, which relates diffusive flux to concentration under the assumption of a steady state—with flux going from areas of high concentration to areas of low concentration and this magnitude being proportional to the concentration gradient and describing the behavior of solutes—and Fick’s second law predicting how diffusion leads to concentrations to vary with time. As a result, drug delivery devices commonly fall into one of two categories that describe solute diffusion: Fickian and non-Fickian. Fickian diffusion occurs when the polymer relaxation time (tr) is greater than the solvent diffusion time (td). Non-Fickian diffusion occurs when tr ≈ td and cannot be modeled with Fick’s laws of diffusion.327
The goal of modeling the release process is to gain a deeper understanding of the release mechanisms of specific material systems.127 Despite the known thresholds for modeling each system, discrepancies have been shown between theoretical mathematical models and experimental data, as many different factors interact to produce drug release. Furthermore, current models are not sufficient to describe complex drug delivery devices, which may contain multiple material constituents, or responsive drug release mechanisms involving interactions among intricate environmental conditions.127 Fu and Kao have recently summarized mathematical modeling concepts for nondegradable and degradable polymers from the last 25 years in a recent review. In the next section we highlight and discuss some of these mathematical models.127
4.9.1. Diffusion-Based Mathematical Models
Diffusion occurs due to the random molecular motion of drugs and involves concentration gradients. Drugs can move out through permeation or through movement via pores and channels. Drug release from a simple slablike device can be modeled from a derivation of Fick’s second law of diffusion127,328
| (1) |
where Mt is the sum of drug released at time t, with M0 being the total of the drug-loaded mass, D is the diffusion coefficient, and h is the device thickness. This equation holds true as an early time approximation model, remaining accurate for up to 60% of the total release, 0 ≤ Mt/M0 ≤ 0.6. The late-time approximation holds true for the final stages of release, 0.4 ≤ Mt/M0 ≤ 1.0, and is expressed as follows:329
| (2) |
Equations 1 and 2 share the same parameters. This model assumes that the dimension and physical properties do not change as the drug is released; for example, there is no loss of bulk polymer material or degradation.127 The diffusion coefficient can be determined through nuclear magnetic resonance (NMR) or fluorescence correlation spectroscopy.330,331 Studies have shown that both the early- and late-time approximations correlate with experimental data, implying that the dominant release mechanism in these studies was Fickian diffusion.332,333 For example, eqs 1 and 2 were used to predict successfully diffusion coefficients of proteins entrapped within peptide hydrogel scaffolds.332 In another study, the diffusion coefficient of a small-molecule asthma drug was measured in PAA–PEG hydrogels.333 In both of these studies, good correlation between experimental data and the Fickian diffusion model was achieved. Some limitations exist when the drug delivery device is heterogeneous in structure (i.e., composed of different materials or layers), there are moving boundary conditions, non-Fickian diffusion, or when dealing with ionic species, and therefore, the diffusion coefficient cannot be considered to be constant throughout the system.
4.9.2. Mathematical Models for Polymer Dissolution
Polymer dissolution refers to a polymer releasing its drug payload into a thermodynamically compatible medium or solvent.334 Drug release from dissolution-based systems can be controlled by solute diffusion as well as polymer dissolution.335 In a model allowing for solute migration and a swelling polymer rubber phase in one-dimensional systems, such as slabs, films, or disks the equation for accumulative drug release is:
| (3) |
Variables A and B are designated as
| (4) |
| (5) |
where l is the initial crystalline polymer thickness, D represents the solvent diffusion coefficient value, Dd is the drug diffusion coefficient, v1* and vd* are the fixed fractions of the solvent and the drug (respectively) at the polymer crystalline-rubber transition, v1,eq and vd,eq are the fixed fractions of the solvent and drug (respectively) at the polymer rubber phase-solvent phase equilibrium, kd is the disentanglement rate of the polymer chain, and t is time. An increase in drug release was observed during simulation with larger diffusion coefficients. Additionally, non-Fickian case II behavior was observed as A/B approached zero;336 thus, the model holds true for Fickian diffusion, case II diffusion, and the transition mechanism between the phases. This model has been used to represent polymer dissolution, as there is good correlation between experimental data and the underlying equations.127 The release of cimetidine hydrochloride from a PVA tablet was shown to be driven via a Fickian diffusion process using this model, and good agreement between eq 3 and experimental data was observed.335
4.9.3. Mathematical Models for Polymer Erosion
Surface-eroding polymers can be represented using Hopfenberg’s model, where zero-order surface release of the drug determines the rate-limiting step.127 The following equation holds true for spheres, cylinders, and slabs
| (6) |
where Mt and M∞ are the cumulative drug release at time t and infinite time, k0 is the erosion rate constant, c0 is the initial concentration of the drug, a is the radius of the cylinder/sphere (or half the thickness of the slab), and n is a shape factor where n = 3 for a sphere, n = 2 for a cylinder, and n = 1 for a slab. However, predicted values using this model for a cylindrical tablet did not correlate well with experimental values.336 The next model was developed for drug release from an erodible polymer matrix, which takes into consideration axial and radial erosion factors
| (7) |
where ka is the radial erosion rate constant, kb is the axial erosion rate constant, a0 and b0 are the tablet’s starting radius and thickness (respectively), and C0 is the initial concentration of drug in the matrix. Under specific conditions, where ka ≈ kb, the model accurately described drug release from a cylindrical tablet.337 A unified model to represent both surface and bulk erosion has been developed. In this model, diffusion reaction equations are combined with dissolution and pore formation mechanisms to determine drug release. The presence of water within a polymeric matrix can be described using
| (8) |
where Cw is the concentration of water with respect to time, the diffusivity of water within the polymer matrix is denoted by Dw, k represents the degradation rate constant, with Mw representing the MW of the polymer.338 The hydrolytic breakdown of polymer bonds in the polymer matrix has been defined according to eq 9. It is assumed that the diffusion of the polymer components is not a major factor before the beginning of erosion, when most drugs will have begun to be released.339,340
| (9) |
Equations 8 and 9 share the same parameters. The dissolution of the drug can be modeled using a second-order rate expression.341 The change in solvent concentration with respect to time must be considered in this expression, which can be seen in the equation below:
| (10) |
Here kdis denotes the intrinsic dissolution rate constant, the normalized solid drug concentration in the polymer matrix is represented by CSn, with CAn being the difference between the aqueous agent concentration and its limit of solubility (CAmx), and finally, CWn is the normalized concentration of water. The next model accounts for drug concentration in a polymer matrix with respect to position and time, using Fick’s second law as well as the dissolution rate expression336
| (11) |
where Deff is the effective diffusivity term, and the other parameters are shared with eq 10. Deff is dependent on the polymer matrix porosity (ε) as well as the diffusivity of the drug through the matrix (DA): Deff = DAε. If one integrates the total normalized concentration of drug in the matrix over all space, it will yield the cumulative fraction of remaining drug in the polymer matrix in time.
| (12) |
Equation 12 shares the same parameter set as eqs 10 and 11. Furthermore, one can calculate the fraction of drug released, R(t), using the simple equation below:
| (13) |
The polymer matrix porosity depends on both time and space and follows a cumulative normal distribution model based on the MW or degradation rate distribution of said polymer336
| (14) |
where the variance is σ2 and is based on the polymer matrix crystallinity as well as the corresponding distribution of degradation rates. Mw,r is the MW of the polymer matrix during release, and Mw is the MW of the polymer.
4.9.4. Multicomponent Mathematical Models
Mathematical models for more complex multipolymer blends have been developed.342 Release kinetics modeled for each polymer, such as PLGA and PCL, were modeled and developed, and their models were then combined. The following expression represents the drug-release kinetics of PCL
| (15) |
where the first term models the initial burst phase, followed by the second term, which models the diffusion-controlled release, with a lack of degradation term for PCL. Mt is the proportion of drug released at time t, M∞ is the proportion of drug released at time infinity, and ϕb,PCL and ϕd,PCL are the fraction of drug release through burst phase and diffusion, respectively, where (ϕb,PCL + ϕd,PCL) = 1. PLGA is modeled similarly but also includes a degradation term
| (16) |
where the first term models the burst release, the second term describes relaxation-induced drug dissolution release, and the third term represents diffusion-controlled drug release. Equation 16 shares the same parameters as eq 15 but is specific for PLGA. The next model represents the drug release from blends of PCL and PLGA
| (17) |
where fPCL and fPLGA are the fractions of drug that partition into and are also released (from PCL and PLGA, respectively), the sum of which is 1.342
As seen by the variety of models described above, it is clear that no single model can accurately describe the release kinetics for each type of polymer. This is the result of multiple factors being involved with drug release mechanisms and their interactions. The most relevant mathematical model should be considered with any assumptions or limitations (material composition and structure of drug delivery system; physicochemical properties of the drug; shape, size, dimension, and geometry of the device; etc.), prior to application to experimental data, by taking into consideration the physicochemical properties of the polymeric carrier and drug. Further detailed mathematical models have also been discussed elsewhere.127,328,334,343,344
5. EXOGENEOUSLY TRIGGERED DRUG RELEASE
Stimuli-responsive NPs can be engineered to release their therapeutic cargo on “cue” according to specific cellular or extracellular stimuli triggered via chemical, biochemical, or physical means (Figure 9). This triggered release in turn can lead to changes in the nanocarrier structure or chemistry, leading to the release of the therapeutic payload in a particular biological environment.
Figure 9.
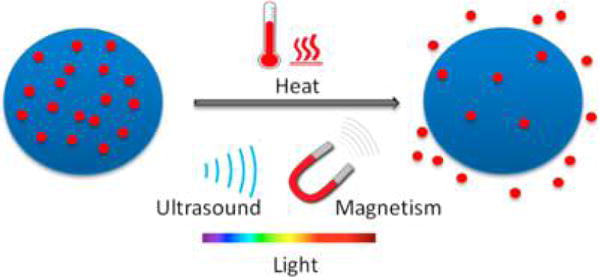
Exogenously triggered drug release by stimuli such as heat, ultrasound, magnetic energy, and light.
If the drug is entrapped within polymeric NPs, release can be triggered by causing structural changes such as polymer degradation, deshedding of surface layers, and charge switching; on the other hand, drug molecules covalently conjugated to the polymer backbone can be released by breaking the linking bonds. Building on our understanding of classical drug release from polymeric NPs (diffusion, surface erosion, and degradation), further manipulation of the site and rate of drug release as a function of the nanocarrier composition is now a widely expanding field of polymeric NP drug delivery, providing further opportunities for controlling drug release. In this review, stimuli that result due to inherent chemical biological pathologies of disease (such as pH, reactive oxygen species, elevated enzyme levels, etc.) are referred to as “endogenous stimuli”, whereas stimuli that result via manipulation from outside the body (such as heat, light, electrical, or ultrasound induction) are referred to as “exogenous stimuli”. A major development of stimuli-responsive drug delivery systems has been to minimize systemic toxicities and unfavorable drug–plasma interactions and to dose and treat disease more efficiently. In the following sections, we will discuss thermoresponsive, light-responsive, and ultrasound-responsive controlled-release polymeric NP systems, since these external triggers have been widely utilized in drug delivery applications.
5.1. Thermoresponsive Drug Release
Temperature-sensitive polymers have been investigated to specifically release their payloads during an induced narrow temperature range, after the delivery of a hyperthermic stimulus at the target tissue. This type of stimuli-responsive drug release can be facilitated since thermoresponsive polymers can change their physical and chemical properties in response to heat. Several studies have highlighted the abnormal temperatures in tumors and other inflammatory diseases as a direct result of abnormal blood flow, leukocyte infiltration, a high rate of metabolic activity, and a high rate of cell proliferation in diseased tissues.345,346 Temperature differences between normal and tumor tissue have also been used to diagnose early-stage tumors and malignancy.347–349 Besides these intrinsic temperature variations, larger temperature changes can be induced artificially at specific locations by applying heat from an external source. This formed the basis for hyperthermia treatment, which exploits the higher sensitivity of tumor tissues to high temperatures as compared to normal tissues.350 According to the National Cancer Institute of the NIH, “Hyperthermia is a type of cancer treatment in which body tissue is exposed to high temperatures (up to 113 °F) to damage and kill cancer cells”.351 Both intrinsic tumor temperature variations and externally induced hyperthermic temperature changes offer attractive stimuli for the site-specific delivery of chemotherapeutic agents.
Polymers that undergo conformational changes in response to temperature variation have been widely studied for the development of thermoresponsive nanocarriers since the initial report of the use of poly(N-isopropylacrylamide) (PNIPAM) by Scarpa et al. in the 1960s. Conformational changes that translate into volume changes in thermoresponsive polymers originate from the switching of polymer hydration states. The most widely used thermoresponsive polymers are hydrophilic below a certain temperature and become hydrophobic above a specific temperature.352 The temperature at which this phase transition occurs is referred to as the lower critical solution temperature (LCST).352 Below the LCST, polymers are hydrated with an extended chain conformation (soluble), and above the LCST, they are dehydrated with a collapsed chain conformation (insoluble) (Figure 10).353 When assembled into a nanocarrier system, these thermoresponsive polymers release their payload through a change in their hydration state and volume, either in response to the intrinsic temperature variations in the diseased region or in response to externally applied heat. The response to temperature change is generally sharp and allows for the delivery of a payload in a spatiotemporal manner.38,354,355 The temperature range at which the nanocarrier responds can be tuned by modulating the balance between hydrophilic and hydrophobic moieties of the constituting polymer. The ideal range in which a thermoresponsive nanocarrier should release its payload is between 37 and 42 °C, in order to minimize any toxic affects due to protein denaturation above the latter temperature.33 In the following section, we discuss widely explored classes of thermoresponsive polymers for polymeric drug delivery applications.
Figure 10.
Illustration of a thermoresponsive polymer undergoing a transition from its coil structure (soluble/expanded) to a globule structure (insoluble/collapsed) in an aqueous environment. Reprinted with permission from ref 356. Copyright 2012 Elsevier Ltd.
5.1.1. Poly(N-isopropylacrylamide) vs Poly(N-vinylcaprolactam) Thermoresponsive Polymers
Poly(N-isopropylacrylamide) (polyNIPAAm) is the most widely studied thermoresponsive polymer. It possesses a very sharp LCST of 32 °C, which is well below normal body temperature.357 This implies that polyNIPAAm is not suitable for the fabrication of drug delivery nanocarriers, since it would not exhibit thermoresponsiveness in the desired temperature range (37–42 °C). PolyNIPAAm is a type II thermoresponsive polymer, as its LCST does not depend on polymer MW.357 The LCST of such a polymer can be modulated by incorporating a hydrophilic component, which increases the LCST, or a hydrophobic component, which decreases the LCST. Efforts have been made to modulate the LCST of polyNIPAAm by preparing copolymers of hydrophobic comonomers and have yielded a range of polyNIPAAm-based thermoresponsive materials with LCST in the desired temperature range, and this topic has been exhaustively covered in other excellent reviews.352,358–361 Although a variety of polyNIPAAm-based thermoresponsive drug delivery systems with optimum physiochemical characteristics have been published in the literature, their biological safety and thus relevance for in vivo medical applications still remain a matter of concern. The acrylamide monomer used for the synthesis of polyNIPAAm is known for its neurotoxicity, which would require rigorous purification procedures before the product could be used for in vivo applications. In addition to the monomer’s toxicity, polyNIPAAm itself is not fully biocompatible, and in vivo experiments on mice have revealed considerable systemic toxicity.362 Thus, despite its ideal thermoresponsive properties, polyNIPAAm’s utility is limited by its toxicity. However, knowledge gained from the extensive numbers of investigations on polyNIPAAm is being applied to the development of alternative thermoresponsive materials. Poly(N-vinylcaprolactam) (polyNVCL) is considered a promising alternate thermoresponsive polymer,363 since it possesses a LCST similar to that of polyNIPAAm.364
Unlike polyNIPAAm, polyNVCL is a type I thermoresponsive polymer, as its LCST can be tuned between 32 and 50 °C by altering the polymer MW (18 000–150 000 g/mol), decreasing the LCST while increasing both MW and concentration.357,364,365 Attempts have also been made to control the LCST by preparing random and block copolymers. Preliminary data on the biocompatibility of polyNVCL has revealed its superiority over polyNIPAAm.366,367 PolyNVCLs are gaining interest as thermoresponsive polymers since they have exhibited low in vitro cytotoxicity.363 However, there are no studies on their biocompatibility in vivo. The hydrophilic cyclic amide side groups bonded to the C–C backbone are one reason for the lowered toxicity of these polymers, and in contrast to PNIPAAm, hydrolysis of polyNVCL does not lead to the production of toxic amine side products.363 The low cytotoxity of polyNVCL was demonstrated by a number of studies, and one particular study showed polyNVCL to be nontoxic to Caco-2 and Calu-3 cell lines up to 10 mg mL−1.366 These studies should help pave the path for further in vivo toxicity studies in order to fully harness the advantages of polyNVCL-type polymers in drug delivery.
5.1.2. Oligoethylene Glycol Thermoresponsive Materials
5.1.2.1. Pendant Group Approach with Methacrylates and Methacrylamide
In addition to the antifouling nature of PEG, a variety of PEG- and oligoethylene glycol (OEG)-based materials exhibiting thermal responsiveness have been reported.352 Acrylates and methacrylates with OEG (OEGAs and OEGMAs) groups represent another emerging category of polymers with interesting thermoresponsive properties in solution, such as adjustable LCST and PEG-like biocompatibility.352 The seminal work of Lutz et al. demonstrated the superior thermoresponsive properties of a copolymer derived from OEGMAs compared to polyNIPAAM.368 A number of comprehensive reviews on the impact of the backbone hydrophobicity (acrylate vs methacrylate), the side chain hydrophobicity (originating from the number of ethylene oxide repeat units and its architecture), the chemical nature of comonomer (backbone and pendant group), and the nature of the backbone end group on the thermoresponsive characteristics of the resulting material have been published previously.356,369,370 A variety of thermoresponsive materials derived from OEMAs and OEFMAs have also been reported in the literature, although polymeric NPs have not been developed using these polymers and their utility remains to be seen for drug delivery applications.371 In addition, the hydrocarbon backbone of these materials, which would be the residual material after in vivo hydrolysis of the pendant ester or amide linkages, may impair their potential as candidates for the development of thermoresponsive NPs.
5.1.3. Thermoresponsive Degradable Polymers
From a clinical translational perspective, polymers employed for development of thermoresponsive drug delivery should be both biocompatible and degradable. If the polymeric material employed is not degradable, the MW and the associated physicochemical characteristics of the thermoresponsive polymer should be optimized to allow it to pass through various biological barriers and be excreted from the body after delivering the payload. Despite the extensive literature on tuning thermoresponsive behavior, little attention has been paid to assessing the postdelivery fate of the polymers. This issue is of particular concern when thermoresponsive delivery systems are derived from polymers synthesized from methacrylate and methacrylamide monomers. After the hydrolysis of their pendant groups, these polymers can leave behind hydrocarbon chains, which are not easily metabolized in biological systems. The drug delivery systems derived from the aliphatic polyesters, polycarbonates, polysaccharides, and polyamides are of specific interest because their degradable nature enables their use in a number of real-life clinical applications for tissue engineering and drug delivery. Since many of these polymers have been approved by the FDA, a thermoresponsive drug delivery system derived from these polymers is more amenable to clinical translation.
5.1.3.1. Polyesters
A number of attempts have been made to produce thermoresponsive materials based on degradable materials. Like the OEGAs and OEGMAs, the pendant-group approach has also been applied to the aliphatic polyesters. Jiang et al. reported a successful pendant group approach that involved the tin (2-ethylhexanoate)2-catalyzed ROP of an alkynyl-group-substituted lactide-based monomer (meso/rac-3,6-di-2-propynyl-1,4-dioxane-2,5-dione).372 The percentage of the pendant alkynyl groups can be controlled by carrying out a copolymerization reaction with a certain amount of lactide monomer. In addition, these researchers also successfully demonstrated a block copolymerization approach. By controlling the ratio between hydrophilic OEG-N3 and 1-azidodecane, the authors obtained poly(propargyl glycolide) (PPGL) with a precisely controlled LCST (referred to as a cloud point) between 25 and 65 °C (Figure 11). Although this report did not discuss the potential of fabricating thermoresponsive nanocarriers, it provided a solid foundation for future developments.
Figure 11.
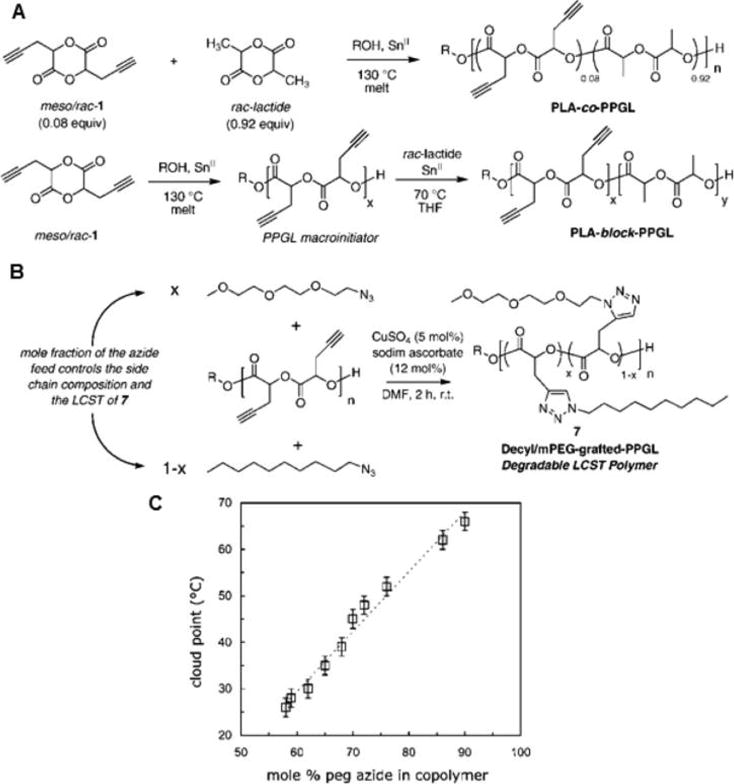
(A) Synthesis of lactide-based random and block copolymers with pendant alkyne groups using Sn(II)-catalyzed ROP. (B) Synthesis of a polyglycolide-based thermoresponsive polymer via azide–alkyne click reaction. (C) Thermoresponsive nature of synthesized polymers demonstrated by measuring cloud point at 450 nm as a function of the mole percent of PEG chains grafted to a poly(propargylglycolide) homopolymer. Reprinted with permission from ref 372. Copyright 2008 American Chemical Society.
Recently, Rainbolt et al. applied the pendant group approach to the development of a stimuli-responsive poly(ε-caprolactone) system.373 As mentioned previously, ε-caprolactones are biocompatible aliphatic polyesters that exhibit a slow biodegradability profile and are suitable for drug delivery systems that require sustained release over several days. By controlling the ratio between the monomers γ-2-[2-(2-methoxyethoxy)ethoxy]-ethoxy-ε-caprolactone (MEEECL) and γ-(2-methoxyethoxy)-ε-caprolactone (MECL) and by varying the number of pendant ethylene oxide units, the authors were able to tune the LCST of the resulting polyester between 30 and 50 °C (Figure 12). Though this report also included the fabrication of NPs, data regarding the thermoresponsiveness of the resulting NPs, encapsulation of the payload, and thermoresponsive release were not presented. A number of polymeric NPs using combinations of caprolactone and other biologically suitable polymers have been reported to date for sustained delivery of a wide range of small-molecule drugs.374–377
Figure 12.
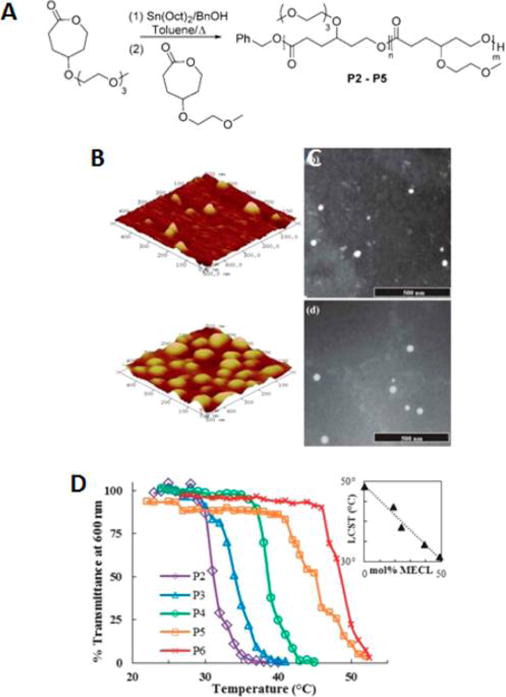
(A) Synthesis of diblock copolymers from γ-substituted caprolactone monomers. (B) 3D tapping-mode AFM images showing polymeric micelles on a mica substrate: (top) high caprolactone content polymer and (bottom) low caprolactone content polymer. (C) TEM images of the same polymeric micelles stained with 1% phosphotungstic acid, respectively. (D) Thermoresponsive nature of micelles revealed by determining the percent transmittance at 600 nm for aqueous solutions of polymers with different mole percents of γ-(2-methoxyethoxy)-3-caprolactone (MECL) (P2, 49 mol %; P3, 39 mol %; P4, 24 mol %; P5, 19 mol %; P6, 0 mol %) as a function of temperature. The inset shows the decrease in LCST as a function of mole percent of MECL. Reprinted with permission from ref 373. Copyright 2013 Royal Society of Chemistry.
5.1.3.2. Polycarbonates
Aliphatic polycarbonates are also attractive biocompatible and degradable materials for the development of nanomedicines.378 In contrast to polyesters, which exhibit bulk erosion degradation in vivo, polycarbonates are characterized by surface erosion degradation, and (also unlike polyesters) they do not produce acidic byproducts.379–382 The ROP of cyclic carbonates, such as trimethylene carbonates, is the preferred route to synthesize polycarbonates and their copolymers with aliphatic polyesters.
Extensive efforts have been made to synthesize polycarbonates with a variety of end and pendant groups for the precise tuning of their physical properties for biomedical applications.383–390 A considerable advantage offered by polycarbonates over polyesters is the wide range of pendant groups that can be accessed by using an appropriately functionalized monomer or by postpolymerization functionalization.378,391 Thermoresponsive polycarbonates via the pendant group approach have been produced.392 Kim et al. employed a hydrophobic hydrocarbon and hydrophilic PEG pendant group combination to produce a therapeutic nanocarrier based on a thermoresponsive nanostructured polycarbonate block copolymer.393 Methyltrimethyl-carbonate (MTC)-based monomers with ethyl (MTC-C2), dodecyl (MTC-C12), and PEG (MTC-PEG) were employed (Figure 13).393 Using PEG substituents with different MWs and a combination of different degrees of polymerization (DPs) for the three monomers, the LCST of the resulting thermoresponsive polycarbonate block copolymers (TRCm-a,b,c, where m represents the PEG MW, and a, b, and c represent the DPs of MTC-C2, MTC-C12, and MTC-PEG, respectively) could be tuned between 36 and 53 °C in PBS. One micelle formulation, TRC350-10,30,60, exhibited an LCST of 36 °C and was shown to self-assemble into nanostructures (Figure 13). The size of the NPs increased as the temperature was raised above the LCST, due to the increase in the hydrophobicity that resulted in aggregation. The Ptxl-loaded NPs exhibited temperature-dependent release kinetics, with faster release above the LCST. The authors suggested that there was a collapse of the core–shell architecture when the temperature was raised above the LCST, which would explain this faster release. However, the release kinetics was very slow, taking 7 days to release ∼60% of the loaded drug at a temperature above the LCST. The slow release kinetics may be suitable for a range of controlled-release applications. In vitro cytotoxicity studies with HepG2 and HEK293 cells showed TRC350-10,30,60 to have minimal toxicity after 48 h incubation either at 32 or 37 °C.393 In HepG2 cells the anticancer activity of paclitaxel-loaded TRC350-10,30,60 micelles was superior at 37 °C (above the LCST) compared to free paclitaxel, and this was also reflected in the lower IC50 observed at 37 vs 32 °C for this micelle (the IC50 value of free paclitaxel did not change). This study demonstrates that drug release and cellular uptake of these micelles are higher at body temperature in vitro.
Figure 13.
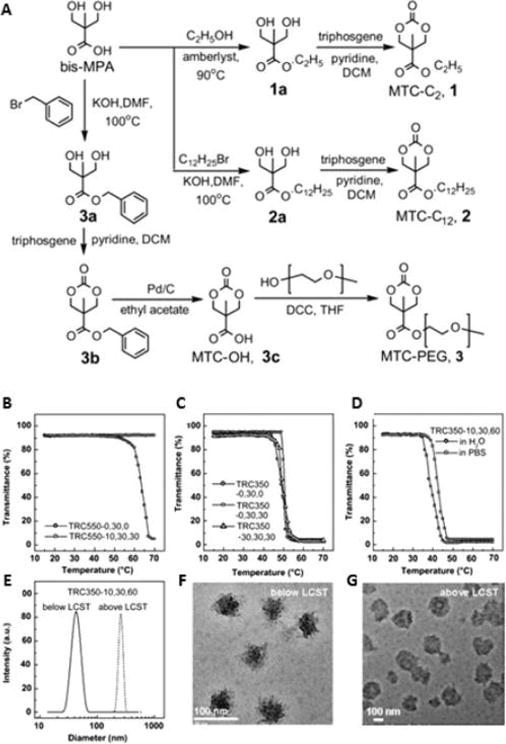
(A) Reaction scheme for the synthesis of cyclic carbamate monomers with different alkyl- and PEG-based pendant groups. (B–D) Temperature responsiveness of block copolymers demonstrated by measuring percent transmittance at 500 nm as a function of temperature. (E) DLS size distribution. TEM images of NPs obtained from polymer TRC350-10,30,60 below (F) and above (G) the LCST. Reprinted with permission from ref 393. Copyright 2011 Elsevier Ltd.
5.1.3.3. Pluronic-Based Thermoresponsive Polymers
Given their biocompatible and degradable nature, polyesters with OEG pendant groups are interesting materials for thermoresponsive nanomedicines. In addition to the pendant group approach, the block copolymer approach has also been explored for the development of thermoresponsive biocompatible and degradable nanocarriers. Akin to PEG, Pluronic block copolymers,394 also known as poloxamers, are another class of thermoresponsive polymers widely favored for their biocompatibility.395 Pluronic block copolymers are made up of hydrophilic antifouling poly(ethylene oxide) (PEO, A) and hydrophobic thermoresponsive poly(propylene oxide) (PPO, B) blocks in an ABA block copolymer arrangement (PEO–PPO–PEO). The PEO blocks also possess terminal hydroxyl groups. The FDA has approved several members of this class of polymers for pharmaceutical applications. In addition to their relevance for the development of nanocarriers, Pluronic block copolymers are also known to modify cellular responses. A thorough investigation has established the ability of Pluronic unimers to sensitize MDR cancer cells.396–406 The ability of a Pluronic block copolymer to modify biological response was shown to diminish upon micellization, which suggests the critical role of Pluronic unimers in sensitizing MDR cells.406,407 Toward the goal of developing nanocarriers with tunable properties, Pluronic block polymers are conveniently accessible in a wide range of MWs and PEO-to-PPO ratios, which have been exploited for modulating the cloud point and cmc during nanocarrier fabrication.395,397 Nanocarriers with a hydrophobic PPO core and a hydrophilic PEO shell are formed above the cmc or LCST. The ability of the PPO core to encapsulate a variety of hydrophobic payloads and respond to temperature changes has been demonstrated in the literature and can also be modulated by varying the PPO MW. Dissolving the Pluronic and payload at lower temperatures and subsequently raising the temperature above the cmc generally leads to the fabrication of payload-encapsulated nanocarriers. A variety of Pluronic-based nanomedicine formulations are being investigated, and the most notable having been clinically evaluated is SP1049C, as discussed previously (Figure 3–15).408
Nanocarriers derived purely from Pluronic possess soft cores and are thermodynamically stable, unlike the kinetically stable nanocarriers derived from block copolymers with hard hydrophobic segments.409 The physical stability of Pluronic nanocarriers is highly dependent on their concentration and environmental temperature. This has limited their in vivo applications, as the nanocarriers dissociate upon dilution as the concentrations drop below the cmc during systemic circulation. A number of chemical and physical strategies are being explored to improve their systemic stability while their thermoresponsive nature is preserved. In a series of studies, He and co-workers developed Pluronic F127 with terminal functional groups reactive toward amines by reacting hydroxyl end groups with succinic anhydride or 4-nitrophenyl chloroformate (4-NPC).410–412 The terminal reactive groups were then used to carry out a cross-linking reaction with multiple amino groups attached to macromolecules, namely, chitosan or polyethylene imine,413 using a unique emulsification/solvent evaporation method that led to the formation of thermally responsive nanocapsules (Figure 14). These nanocapsules developed by combining succinic anhydride for activation and chitosan for cross-linking changed dramatically in size, in response to a change in temperature: from >200 nm at 4 °C to ∼25 nm at 37 °C.410 The authors loaded the therapeutic payloads into the swollen nanocapsules via a co-soaking process at 4 °C. Freeze-drying and then heating to 55 °C, followed by dialysis at 37 °C, ensured the removal of any free nonencapsulated payload. The thermoresponsive release of the payload was elicited by employing a cold-shock treatment (4 °C) during the release test performed at 37 °C. After 3 h at 37 °C, release of the payload was negligible. The cold shock administered at this stage led to a release of ∼94% of the total payload. This induced drastic swelling in the nanocapsules, increasing their permeability and eliciting a burst release of the payload. The volume expansion as a result of the cold shock at 4 °C was further shown to help the nanocapsulate mechanically break, escape the endosome/lysosome system, and efficiently release the payload in the cytosol of MCF-7 cells, and cell viability was observed to be more than 99%.410
Figure 14.
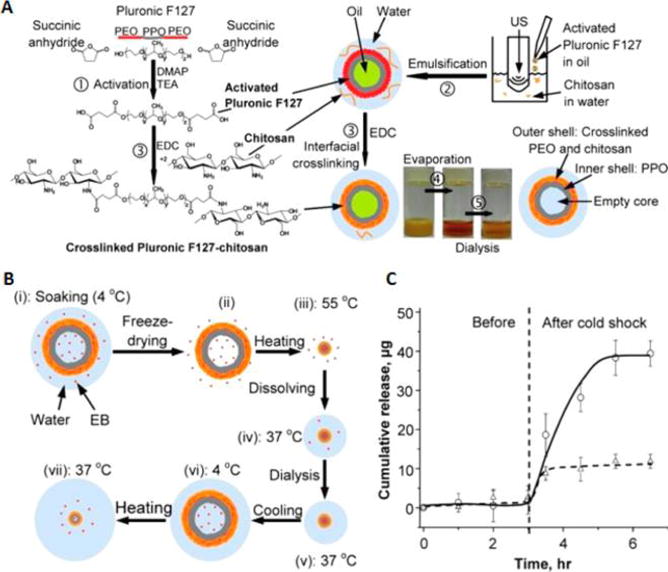
(A) Synthesis of cross-linked Pluronic F127–chitosan and illustration of nanocapsule fabrication. Photos of dichloromethane-based oil-in-water emulsion before and after rotary evaporation of oil phase and after dialysis to remove unreacted components. (B) Depiction of payload encapsulation and release into the cross-linked Pluronic F127–chitosan NPs. (C) Thermoresponsive payload release profile of cross-linked Pluronic F127–chitosan NPs. Reprinted with permission from ref 410. Copyright 2010 American Chemical Society.
Besides chemical cross-linking strategies, physical hybrids derived from a combination of Pluronic and other materials have also been reported in the literature as thermoresponsive nanocarriers. Our group recently reported a hybrid therapeutic protein delivery system referred to as “thermosponge nanoparticle platform (TNP)” derived from Pluronic F127 and terminally charged PLGA or PLA (Figure 15).414 The empty and protein-loaded TNPs were fully characterized for their physiochemical characteristics and stability, and the core–shell nature of the TNPs was evident from the transmission electron microscopy (TEM) images (Figure 16). Size investigations by dynamic light scattering (DLS) revealed a decrease in the size of TNPs when the temperature was increased from 4 to 37 °C.
Figure 15.
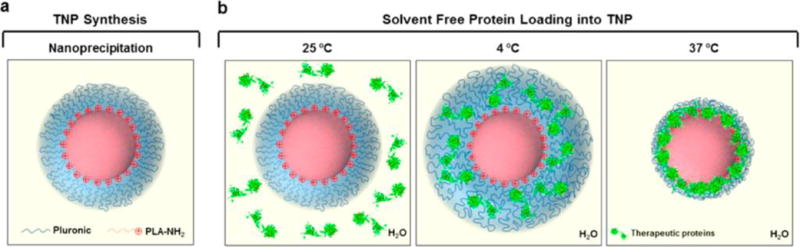
Thermosponge nanoparticles for protein delivery. (a) Thermosponge nanoparticle (TNP) prepared through a one-step nanoprecipitation method using terminally charged PLA-NH2 and Pluoronic F127. (b) Solvent-free loading of proteins into the TNPs through thermoresponsive swellings and electrostatic interactions between the charged core and protein payload. Reprinted with permission from ref 414. Copyright 2014 American Chemical Society.
Figure 16.
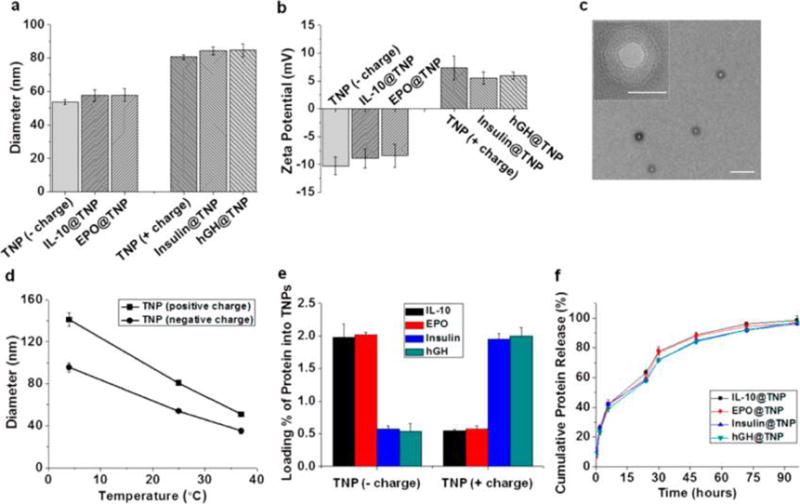
Characterizations of the TNPs: Hydrodynamic diameters (a) and surface charges of the empty and protein-loaded TNPs (b). TEM image of TNPs (outer scale bar = 500 nm, inset scale bar = 50 nm) (c). Temperature-induced swelling and deswelling as a function of temperature (d). Therapeutic proteins loaded into both negatively and positively charged TNPs (e). Cumulative in vitro release of the therapeutic proteins from TNPs in PBS buffer at 37 °C (f). Reprinted with permission from ref 414. Copyright 2014 American Chemical Society.
By exploiting the electrostatic charge on the core surface, we were able to load the protein payloads by cooling a cosuspension of the TNP and protein from 25 to 4 °C, subsequently warming it to 37 °C (temperature for shell contraction). The synergic effect of the core surface charge and thermoresponsive volume variations enabled the loading of negatively (insulin and human growth hormone/hGH) and positively (IL-10 and erythropoietin/EPO) charged therapeutic proteins with high loading efficiency and capacity. The loaded proteins retained their full bioactivity, while the TNPs significantly increased their half-lives and systemic exposure in mice. To test the in vivo efficacy of the TNPs, a mouse model of contact dermatitis was used to assess the anti-inflammatory effects of the IL-10-loaded TNPs. Mice treated with these NPs were shown to have less edema and myeloid infiltration than mice injected with free-IL-10. In this study, the temperature-responsive behavior of Pluronic was used to load proteins into NPs without the need for organic solvents.
The terminal hydroxyl groups of Pluronic block copolymers can also be explored for the initiation of ROP of cyclic dilactone monomers to prepare Pluronic–polyester block polymers.415 In a recent report by Guo et al., Pluronic F127 (F127) was employed as a macroinitiator to produce thermoresponsive F127–PLA (FP) block copolymers (Figure 17).416 One of the end groups was reacted with N,N′-carbonyldiimidazole (CDI), while the other was used for ROP of the lactide. The CDI-activated end group was reacted with a diamine and then with an active ester derivative of folic acid, producing a folate-conjugated FP (FA–FP) block copolymer. FA–FP can be used for active targeting to folate receptors, which are typically overexpressed in a variety of tumors. Through a nanoprecipitation process, NPs with a PLA core and F127 shell were prepared from the FA–FP block copolymer with different DPs of the PLA block (Figure 17).
Figure 17.
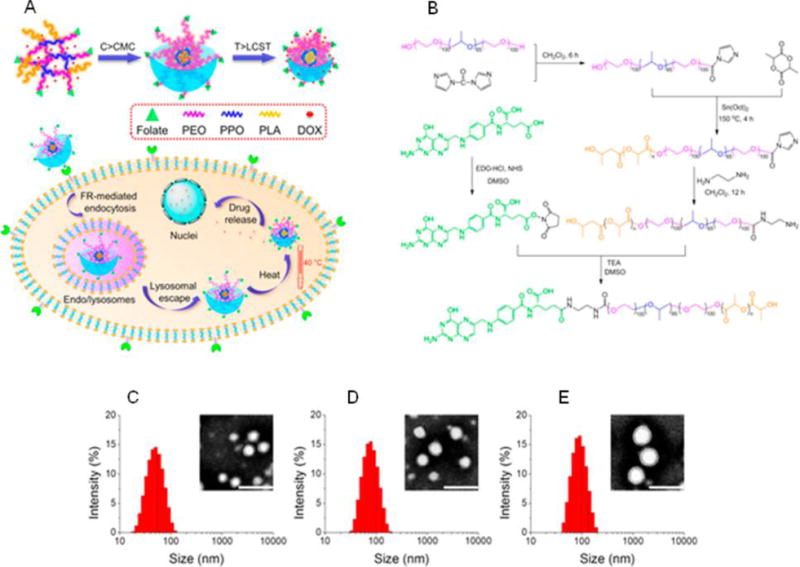
(A) Illustration of thermoresponsive nanocarriers for temperature-controlled drug release. (B) Synthesis of folate-conjugated block copolymer of F127 and PLA. TEM images (scale bar 100 nm) and particle size (DLS) of thermoresponsive NPs obtained from FP with different DPs of PLA: (C) DP = 50, (D) DP = 100, and (E) DP = 200. Reprinted with permission from ref 416. Copyright 2014 American Chemical Society.
Interestingly, the thermoresponsive nature of the NPs, as reflected by the decrease in particle size and increase in absorbance of the particle suspension with the increase in temperature from 25 to 55 °C, can be tuned by varying the DPs of the PLA block. The DOX-loaded NPs showed a clear thermoresponsive drug release, particularly the NPs derived from the FP with the PLA DP of 100 (FP100) (Figure 18). At 37 °C, FP100 showed a very slow release with a burst release at 40 °C. The collapse of the hydrophilic shell at a temperature above the LCST resulted in a fast payload release. The LCST of FP100 NPs is reported to be between 37 and 40 °C, so they would remain stable under normothermia, whereas they would rapidly release their payload under mild hyperthermia, making them interesting nanocarrier candidates for tumor targeting. Conjugating them to FA markedly increased the uptake of FA–FP100 NPs by HeLa cells expressing the folate receptor when compared to the FA lacking FP100 NPs. The LCST of the FA–FP100 was not much different than that of FP100. The researchers also reported that the efficient thermoresponsive payload release translated into the lowering of the IC50 value from 1.45 ± 0.03 μg/mL at 37 °C to 0.28 ± 0.02 μg/mL at 40 °C.
Figure 18.
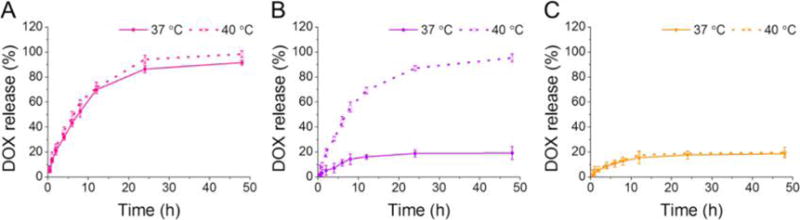
Thermoresponsive release of DOX at 37 and 40 °C from micelles of FP with PLA of different DPs; FP50 with PLA DP = 50 (A), FP100 with PLA DP = 100 (B), and FP200 with PLA DP = 200 (C). Reprinted with permission from ref 416. Copyright 2014 American Chemical Society.
Successful in vivo application of thermoresponsive nanocarriers would require a straightforward method of heating the tumor region. Induction of a magnetic field has the potential to be the most clinically effective way of tumor-targeted heating. Magnetic hyperthermia usually uses magnetite (Fe3O4) NPs (referred to as superparamagnetic iron oxide NPs or SPIONs) that are coated or encapsulated in the thermoresponsive polymer.
When an alternating magnetic field (AMF) is applied, the SPIONs generate heat, attributable to magnetic hysteresis loss and Néel relaxation.33 In a magnetically responsive system, either a released payload or generated heat is responsible for providing the intended therapeutic effect. In the former scenario, the application of AMF is expected to release the payload in response to the generated heat. Near-infrared (NIR) radiation can also be employed to induce localized heating via photothermal conversion, i.e., converting energy into heat.417 This is frequently achieved using gold nanorods or nanoshells engineered to a certain diameter or thickness that can respond to incoming NIR and generate heat, which in turn can be exploited as a stimulus to trigger payload release from temperature-responsive nanocarriers. NIR-triggered payload release can also be facilitated by the disintegration of bonds that link the payload to the carrier, known as photocleaving, or the decay of a capping agent that blocks the payload via the photothermal effect. Frequently, theranostic agents that use NIR heat-triggered drug release also take advantage of the heat to stimulate photohyperthermia or enable photodynamic therapies.
Although gaining considerable interest as responsive materials in drug delivery, the majority of polymers used to develop thermally responsive drug delivery systems have not as yet been extensively investigated in the body, and until these systems are more thoroughly tested for their biocompatibility, they are mostly confined to preclinical development. It is important to investigate the toxicity of the constituent monomers in these systems, since these components are most likely causative of any toxicity arising in vivo.
5.2. Light-Responsive Drug Release
Nanocarriers capable of undergoing physical or chemical changes in response to light irradiation are attractive for designing safe treatment regimens that offer spatiotemporal control over the release of encapsulated therapeutic payloads. A number of chemical and physical processes can be initiated simply by light irradiation at a specific wavelength. The light- or radiation-triggered processes can be either reversible or irreversible and involve formation or cleavage of bonds, interconversion of isomers (e.g., cis–trans), switching of electrostatic charge, and rearrangement of chemical reactions. Incorporation of the functional groups that interact with light and undergo the aforementioned transitions in the polymers (in the backbone or as pendant groups) has been explored for the development of light-responsive polymeric materials.
Nanocarriers fabricated from light-responsive polymers can be triggered to disintegrate and release the encapsulated payload via light induction. The clean and convenient nature of light-triggered processes has stimulated tremendous interest, particularly for application in a variety of biomedical fields. The suitability of a certain light-triggered process for a targeted biomedical application depends on the radiation wavelength required. In the context of light-responsive nanomedicines, the light required should be benign to normal tissues, show minimal absorption and interaction with the biological components, and offer substantial tissue penetration for in vivo applications. Radiation of shorter wavelengths, such as γ-rays or X-rays, offer high energy and are used in radiotherapy to damage the DNA of (and kill) cancerous cells.418 However, since the high energy of γ-rays and X-rays can damage normal tissues; they cannot be used as a stimulus for developing light-responsive nanomedicines. Moving from the high- to lower-energy end of the electromagnetic spectrum, UV radiation is next in line. A number of functional groups responsive to UV radiation can be incorporated into polymers, and such materials have been widely demonstrated to undergo physical and chemical changes upon UV irradiation.419
5.2.1. UV-Light-Resposive Controlled Release
Azobenzene is known to undergo UV light (340–380 nm) triggered transition from apolar trans to polar cis isomeric forms, referred to as photoisomerization. The transition is reversed either upon storage in the dark or by irradiation with visible light (420–490 nm). Upon irradiation with UV light, the change in hydrophilicity and the transition from trans to cis conformation can effect disintegration of the nanocarriers derived from polymers bearing azobenzene groups, thus affecting the payload release. Building on original investigations by Sánchez and co-workers,420 Blasco et al.421,422 have reported amphiphilic linear dendritic block copolymers (LDBCs) with the azobenzene units presented at the periphery of a fourth-generation 2,2-di-(hydroxymethyl)propionic acid (bis-MPA)-based dendron (Figure 19). The substituents on the azobenzene units were shown to influence the self-assembling behavior of the resulting LDBCs. Unlike the 4-cynanoazobenzene-substituted LDBCs, the 4-isobutyloxyazobenzene-substituted LDBCs formed stable NPs in water that were able to encapsulate both hydrophilic and hydrophobic payloads. The release of the dye molecules, which acted as payloads, was triggered by irradiation with UV radiation wavelengths facilitated by trans–cis photoisomerization of azobenzene moieties.
Figure 19.
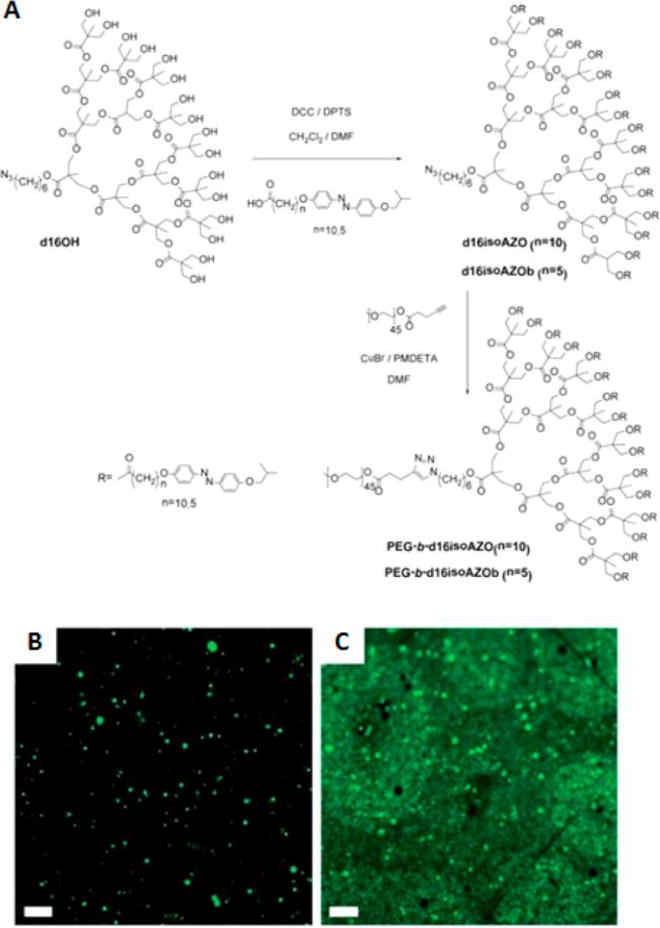
(A) Synthesis of light-responsive linear–dendritic block copolymers. Fluorescence microscopy images of the loaded linear dendritic block copolymer (PEG-b-d16isoAZO) vesicles. Vesicle suspension without irradiation (B) and after 1 h of irradiation at 365 nm and 2.6 mW cm2 (C). Scale bar = 5 mm. Reprinted with permission from ref 421. Copyright 2013 Royal Society of Chemistry.
Spiropyran (SP) is also known to undergo photoisomerization from a hydrophobic closed SP form to a hydrophilic zwitterionic open merocyanine (MC) form upon UV irradiation. The transition can be reversed by irradiation with visible light.423,424 Although a variety of SP-based materials exploit the switching of SP–MC states to create light-responsive systems for a variety of applications,425,426 incorporation of SPs into degradable and biocompatible polymers for the development of light-responsive nanomedicines has been rarely explored.427,428 Besides azobenzene and SP, materials based on cinnamic acid, cinnamic ester, and coumarin are also capable of responding to UV radiation.429–431 Under UV-triggered dimerization, these functional groups undergo photoinduced cross-linking of polymer NPs. Furthermore, 2-diazo-1,2-naphthoquinone (DNQ) [which transforms to hydrophilic 3-indenecarboxylic acid (3IC) via the Wolf rearrangement], o-nitrobenzyl ester, coumarinyl ester, and pyrenylmethyl ester groups (which undergo cleavage) have also been explored for the development of UV-radiation-responsive materials. Despite several known UV-radiation-responsive physical and chemical transformations that can trigger payload release, the medical applications of UV-light-responsive nanocarriers are limited by the high energy of UV radiation (which is harmful to human tissues) and their insufficient tissue penetration, as well as their absorption by biological components.432,433
5.2.2. NIR-Light-Responsive Controlled Release
Since radiation with longer wavelengths has reduced scattering and relatively benign biological effects, deeper penetration into human tissues is possible, and these wavelengths can be exploited for developing light-reponsive nanomedicines.434 Among radiation wavelengths longer than UV, NIR radiation (750–1000 nm) offers the distinct advantages of being benign and penetrating deeper into tissues.434,435 UV-radiation-triggered processes can also be initiated by NIR radiation via multiple-photon absorption (such as two-photon absorption) and the upconversion process. Liu et al.436 reported the use of a two-photon absorption process for fabricating NIR-responsive DOX-loaded polymeric micelles derived from DNQ-grafted dextran (Dex-DNQ) (Figure 20). The Wolff rearrangement of hydrophobic DNQ hydrophilic 3-IC upon NIR irradiation (808 nm) was demonstrated to enhance the intracellular delivery of DOX. The cell viability of HepG2 cancer cells was significantly inhibited upon incubation with DOX-loaded Dex-DNQ micelles and irradiation with NIR light.
Figure 20.
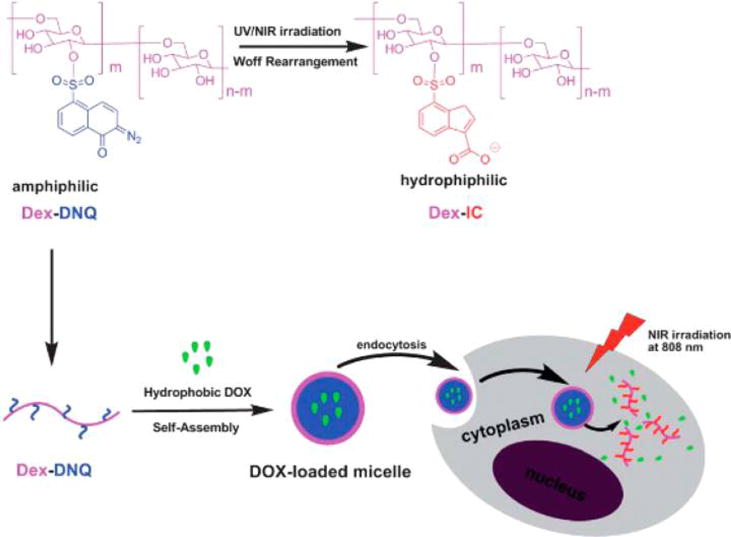
Self-assembly and light-induced Wolff rearrangement of a dextran-graft-(2-diazo-1,2-naphthoquinone) amphiphilic copolymer for fabrication of NIR-responsive DOX-loaded micelles and illustration of light-responsive DOX release. Reprinted with permission from ref 436. Copyright 2012 Royal Society of Chemistry.
de Gracia Lux et al. created two different polymers with the same backbone but different terminal groups.437 Both terminal groups responded to UV radiation (through a one-photon process) and NIR (through a two-photon process), where the backbone consisted of a quinone-methide structure. This backbone is unique, as once the end group is cleaved by incoming radiation, it degrades into its monomers in a domino-like fashion. After forming NPs with Nile Red encapsulated, UV radiation elicited a burst release of Nile Red, while NIR radiation gave a much more gradual release. This study highlighted the fact that despite the success of the two-photon process, its practical application is impeded by the materials’ slow response when the UV-radiation-triggered process is stimulated via two-photon absorption using lower-energy NIR radiation. Furthermore, the two-photon process is much less efficient and required longer irradiation periods, which can sometimes be inconvenient. An alternative process for triggering a UV-induced reaction with NIR radiation involves the use of upconversion NPs (UCNPs). UCNPs are capable of absorbing several NIR light photons (because of their lanthanide dopants) and emitting a single photon of wavelength in the UV range.438 Almutairi and co-workers applied the upconversion process for triggering the NIR-induced degradation of a polyester bearing o-nitrobenzyl pendant groups, with a backbone made up of the diol monomer with self-immolative quinone-methide moieties (Figure 21). The system has been previously demonstrated to show an uncaging in response to UV (350 nm) or two-photon NIR (750 nm) via cleavage of 4,5-dimethoxy-2-nitrobenzyl carbamate pendant groups with the formation of 4,5-dimethoxy-2-nitrosobenzaldehyde. This cleavage induces the cyclization of the remaining diamine pendant groups, which ultimately disassembles the unstable quinone-methide moieties.439 By loading the highly luminescent core–shell NaYF4:Yb·Tm, the UCNPs absorb in the NIR region (980 nm) while strongly emitting in the UV region (which overlaps with the absorbance of photocleavable o-nitrobenzyl groups). Coumarin 153 (C153), coloaded as a model drug payload, was shown to release upon NIR irradiation with release kinetics controllable by varying the power of radiation between 250 and 1000 mW. Though the UCNPs-induced upconversion process is an interesting strategy, extensive biocompatibility and biodegradability have yet to be established for these materials before their clinical application is possible.
Figure 21.
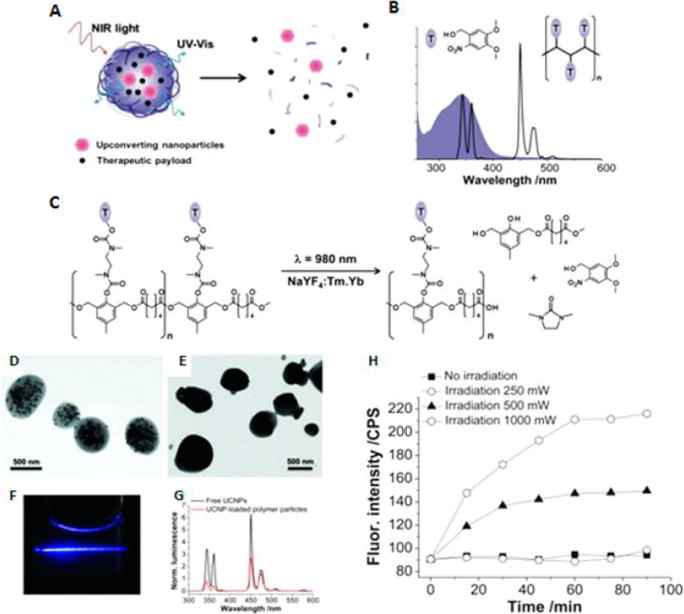
(A) Light-triggered degradation and payload release from light-sensitive NPs via upconversion phenomenon. (B) Overlap between the UV-emission of NaYF4:Yb·Tm UCNPs (unshaded) and absorption (shaded blue) of o-nitrobenzyl (ONB) groups. (C) Schematic illustration of NIR-induced degradation of polyester bearing ONB pendant groups via upconversion. TEM images of polycresol NPs with (D) and without (E) UCNPS. (F) Optical image of the luminescence emitted from UCNP-loaded polymer NPs exposed to a 980 nm laser. (G) Luminescence of free and polymer-loaded UCNPs. (H) C153 release as a function of NIR irradiation time (pulsed laser light, 980 nm). Reprinted with permission from ref 440. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.
The upconversion phenomenon provides remote access to a wealth of UV-induced photochemical and photophysical processes. However, when using NIR radiation there is an upper limit (around 950 nm) above which water and lipids begin to absorb the incoming radiation,433 which will affect the specificity of the NIR-light-responsive treatment regimen. Furthermore, the in vivo toxicity and fate of the caging groups that are being used to impart the light responsiveness are as yet unknown. NIR radiation is also commonly employed for inducing a photothermal effect in combination with gold nanorods of certain dimensions. The heat generated can be exploited as a stimulus to trigger payload release from a temperature-responsive nanocarrier system, as described earlier in the section related to temperature-responsive nanomedicines.
5.3. Ultrasound-Responsive Controlled Drug Release
Ultrasound (US), which is defined as high-frequency pressure waves produced by mechanical oscillations in response to an alternating current applied across piezoelectric materials, has been widely used for imaging purposes in medicine. The frequencies of US used for medical applications fall over the threshold of the human-audible low-frequency pressure wave range (>20 kHz).441,442 US waves are classified as low-, medium-, and high-frequency when their frequency falls in the ranges <1, 1–5, and 5–10 MHz, respectively.443 US can penetrate centimeters deep into tissue in a noninvasive manner and can be focused with high intensity on a single point. Low-frequency US (20–200 kHz) in particular does not damage or overly heat the tissues, though it is difficult to focus because it produces a larger focus point. US with frequencies higher than 1 MHz possesses relatively poor capability of penetrating into tissues because of the higher scattering that results with higher attenuation, which is proportional to the increase in frequency. The attenuation of high-frequency US traveling through tissue is proportionally transformed into heat, which can be damaging to the tissues. The penetration depth of US with a frequency of 200 kHz is 10 times higher than that of 2 MHz US, whereas US with a frequency of 5 MHz does not penetrate at all.444 Interestingly, the higher-frequency US can be focused on small volumes. A high-intensity focused US (HIFU; frequency range 0.8–3.5 MHz) beam can target tumors and can harmlessly penetrate the skin and most other tissues. HIFU was primarily developed for the treatment of a variety of tumors.445
The interaction of US with tissue fluids can have thermal or nonthermal effects. The thermal effects of US are associated with the transfer of acoustic energy to the tissues, and this effect increases with power density and the focus of the US. The nonthermal effects of US originate mainly from cavitating bubbles. At low acoustic pressures, the interaction of air bubbles with US makes them oscillate in response to the negative and positive pressure cycles. The air bubbles oscillate stably (stable or noninertial oscillation) and undergo compression and rarefaction at low acoustic amplitudes, and the frequency of their oscillation resonates with the frequency of the applied US. An increase in acoustic pressure results in nonlinear and violent oscillations that ultimately collapses/destroys the bubbles (referred to as collapse, inertial, or transient cavitation).446,447 The collapse of bubbles via inertial cavitation can generate shock waves and a temperature increase in the surrounding medium. The resultant high shear force has been demonstrated to enhance drug uptake by cells (in vivo and in vitro), by temporarily increasing the membrane permeability, and can also disassemble nanocarriers, triggering the release of therapeutic payloads.360,448
Our wealth of knowledge on the interaction of US with biological systems, the development of sophisticated US-producing and -focusing equipment, and its benign nature make US a very attractive stimulus for spatiotemporal control over payload release by nanocarriers. For fabricating US-responsive NPs, the development of polymeric materials that respond to US stimulus is the biggest challenge. Husseini, Pitt, Rapoport, and other co-workers have extensively explored micelles derived from Pluronic P105, NanoDeliv (P105 micelles stabilized via cross-linking), and some PEG–polyester block copolymers as US-responsive drug delivery nanocarriers.447,449–454 These researchers have addressed questions related to the mechanisms of drug release from these micelles in response to US stimuli and established various parameters associated with US, such as controlling the release kinetics, the frequency of US used, the nature of treatment (i.e., continuous or pulse wave), the duration of exposure to US, and the intensity and power density of US. Most of the micelles studied in these reports fall into the category of US-responsive nanocarriers that upon application of US, release their payload by simple diffusion without any degradation of the polymer’s constituents.
Zhang et al. investigated the HIFU-triggered release of hydrophobic Nile Red dye encapsulated in degradable and biocompatible PLA-b-PEG block copolymer NPs.455 The dye release depended upon time as well as HIFU intensity (Figure 22). This release was irreversible, and as predicted, the disassembled polymer NPs were unable to reassemble into NPs and re-encapsulate the payload. This led the authors to propose that treatment with HIFU caused polymer degradation (Figure 22). They further substantiated their claim by showing a decrease in polymer MW during exposure to HIFU (1.1 MHz, 200 W), thus confirming that PLA-b-PEG can be used as an US-degradable system for the development of US-responsive polymeric NPs.
Figure 22.
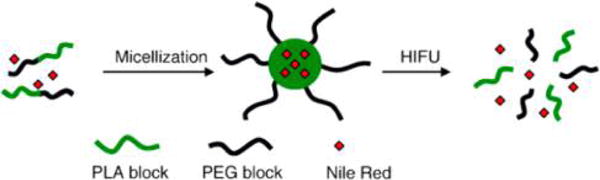
Release of loaded Nile Red from a PEG-b-PLA micelle upon exposure to HIFU. Reprinted with permission from ref 455. Copyright 2009 Elsevier B.V.
Recently, increasing attention is being given to the development of nanomedicines using US-responsive degradable polymers. The recently coined concept of mechanophores—functionalities incorporated into polymer molecules enabling them to respond to applied stress—offers novel paths for generation of stress-responsive materials.456–460 This has led to the investigation of mechanolabile functional groups that may cleave in response to the applied US.460,461 One such example is 2-tetrahydropyranyl methacrylate (THPMA), a constituent of the PEG–THPMA block copolymer, which has been demonstrated to hydrolyze in response to HIFU.462 Building upon this work, Xuan et al. reported the hydrolysis of THPMA units of an amphiphilic block copolymer in response to HIFU (1.1 MHz, 100 W).463 The hydrolysis of THPMA units increased the hydrophilicity of the block copolymer, which in turn increased the LCST from 25 to 42 °C. The self-assembled NPs fabricated from the amphiphilic block copolymer were also shown to disassemble in response to the applied HIFU.
Xia and co-workers further established ester and disulfide bonds as US responsive mechanophores.464,465 Focus on the disulfide bond as a mechanophore emerged because of its low dissociation energy (ES–S ∼ 268 kJ mol−1) and longer bond length (∼2.03 Å) compared to the C–C bond.466 A block copolymer of PEG and PLA, in which the two blocks were connected through a disulfide bond (PEG–S–S–PLA) was shown to undergo degradation as reflected by a decrease in MW (Figure 23).464 The NPs derived from the self-assembly of PEG–S–S-PLA block copolymer were also observed to disintegrate when subjected to treatment with HIFU (80 W). Disulfide bonds are established reduction-responsive moieties; however, the release of the pyrene loaded into the PEG–S–S–PLA was more efficient under HIFU treatment compared to the release induced by the reducing agent. Furthermore, PLA is constituted by aliphatic ester linkages, which are prone to hydrolysis under US treatment and are likely to contribute strongly to polymer degradation.
Figure 23.
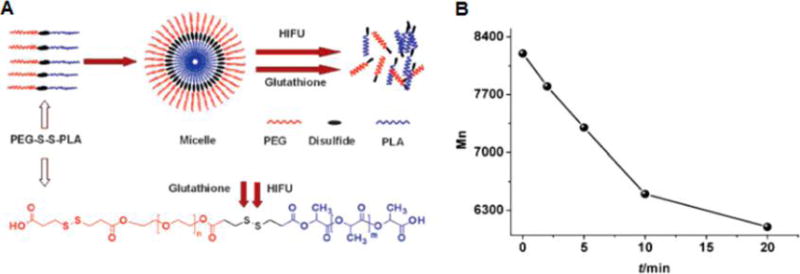
(A) Illustration of redox and HIFU responsiveness of PEG–S–S–PLA micelles. (B) Decrease in the MW of the polymer (as determined by GPC) as a function of HIFU exposure time. Reprinted with permission from ref 464. Copyright 2010 Royal Society of Chemistry.
The suitability of ester bonds was particularly well established by preparing block copolymers of PEG and polypropylene glycol (PPG), where the two blocks were connected via an ester linkage (PEG–COO–PPG).465 The series of polymers produced in this work also included block copolymers that contained sulfides (PEG–COO–S–PPG) or disulfides (PEG–COO–SS–PPG) in addition to the ester linkage. The sulfide bond in PEG–COO–S–PPG is not responsive to reduction, but the release of pyrene loaded in the NPs was as responsive to HIFU as PEG–COO–PPG. Although these studies established the ester linkage as a potential US-responsive mechanophore, the suitability of the disulfide bond as an independent US-responsive mechanophore has yet to be established.
In summary, US is a promising stimulus for spatiotemporal control over the delivery of therapeutic payloads loaded into nanocarriers. Previous reports have established the foundation for more detailed investigations, particularly for the development of degradable and biocompatible polymers that may exhibit efficient degradation and release when subjected to US. Development of new mechanophores that might be incorporated into degradable and biocompatible polymeric systems may further lead to interesting US-responsive polymeric NPs for controlled-release drug delivery applications. In addition to exogenous stimuli, nanocarriers can also be designed to release therapeutic agents in response to a variety of endogenous stimuli (pH, reducing agents, reactive oxygen species, and enzymes). Endogenous stimuli are of particular interest, as they are specific to disease-related microenvironmental and pathological changes.
6. ENDOGENOUSLY TRIGGERED DRUG RELEASE
A range of internal stimuli, such as changes in pH, redox state, and ionic content within tissues and cells, can be utilized for the development of chemically triggered drug release from polymeric NPs (Figure 24). For example, solid tumors can have acidic pH environments (pH 6–7) that can be used to trigger chemical changes in polymer bonds, leading to increased drug release. Subcellular compartments also offer various low-pH environments postuptake of NPs (endosome, lysosome, cytosol, etc.), which can cause either surface layer shedding or conformational changes in polymeric NPs, leading to site-specific and increased drug release. In this section we will discuss a range of chemical triggers than can be harnessed for the control of drug release from polymeric NPs.
Figure 24.
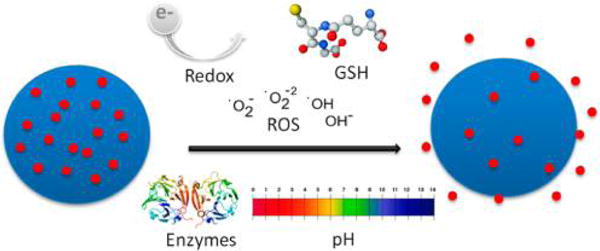
Internally triggered drug release by stimuli such as redox potential and reactive oxygen species (ROS), enzymes, and pH.
6.1. Redox-Responsive Controlled Release
The gradient between intracellular and extracellular concentrations of reducing agents as an endogenous stimulus has been used for controlling drug release. Glutathione tripeptide (γ-glutamyl-cysteinyl-glycine, GSH) is a common biological reducing agent with an intracellular concentration of ∼2–10 mM and an extracellular concentration of ∼2–20 μM.467 Compared to normal cells, the gradient of GSH concentration is 4 times higher in tumor cells.468–471 The higher intracellular GSH concentration is maintained by the NADPH and glutathione reductases and is dependent on NADH/NAD+, NADPH/NADP+, and thioredoxinred/thioredoxinox redox mechanisms.472–474 The reducing enzyme γ-interferon-inducible lysosomal reductase (GILT), along with the cysteine reducing agent and iron (kept in a reduced state caused by the acidic and thiol-rich environment of the lysosomes), contributes to this high intracellular reducing environment.475 This remarkably high intracellular reducing potential constitutes an excellent endogenous stimulus for designing nanocarriers that are programmed for the intracellular release of therapeutic agents.
Disulfide (–SS–) and diselenide (–SeSe–) bonds have been recognized as redox-sensitive bonds that are reduced to thiols (–SH) and selenols (–SeH) exclusively by the intracellular reducing environment (Figure 25).469,476–478 Consequently, a variety of degradable polymers bearing bioreducible disulfide or diselenide bonds are under investigation for developing reduction-responsive nanomedicines.476,479
Figure 25.
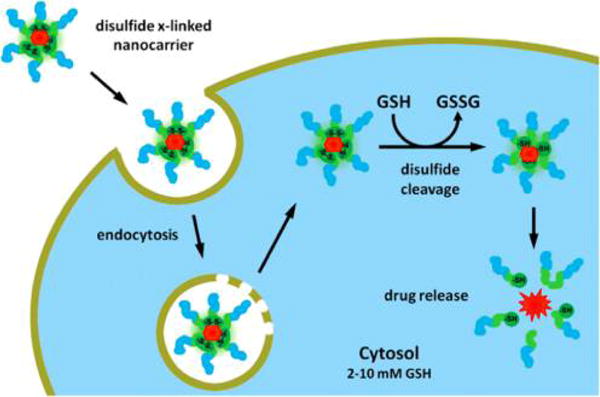
Redox responsive carriers can be made using reducible –SS– bonds. Reprinted with permission from ref 480. Copyright 2012 Elsevier B.V.
A balance between the hydrophobicity and hydrophilicity of amphiphilic polymers is necessary for the fabrication of stable payload-encapsulating nanocarriers. Disturbing this balance, whether physically or chemically, can lead to a disruption of the micelles, which affects the release of the payload. Pendant groups with a specific physical and chemical nature are necessary to fabricate stable micelles. Pendant groups can be connected to the main chain of polymers via bioreducible functional groups. Such micelles would disassemble under a reducing environment. A variety of polysaccharide-based polymers, such as hyaluronic acid,481,482 chitosan oligosaccharides,483 and carboxymethyl dextran,484 have been functionalized with bioreducible micelle-disassembling functionalities for fabrication of reduction-responsive nanocarriers. Li et al. synthesized hyaluronic acid–deoxycholic acid (HA-ss-DOCA) conjugates for redox-sensitive release of Ptxl, and micelles formed from this conjugate were shown to disassemble in the presence of 20 mM glutathione (Figure 26).481 Both in vitro toxicity studies with MDA-MB-231 cells and in vivo xenograft tumor experiments confirmed differential and enhanced therapeutic efficacy.
Figure 26.
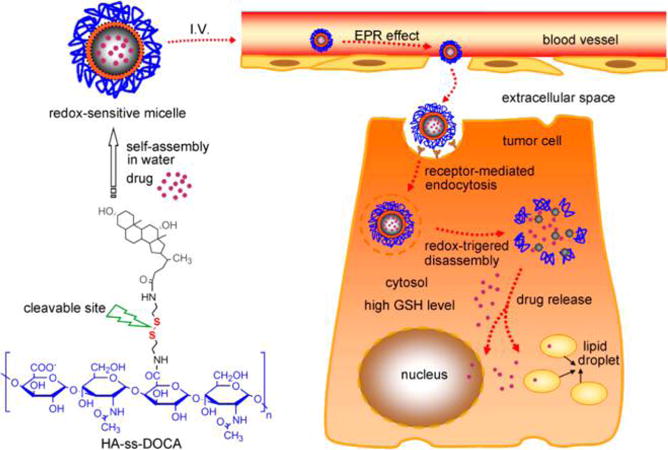
Illustration of self-assembly of reduction-responsive drug-loaded HA-ss-DOCA into redox-sensitive micelles, their intracellular trafficking pathway, and drug release. The cleavage of the disulfide bond in the intracellular reducing environment results in disconnection of pendant DOCA groups and disassembly of HA-ss-DOCA micelles, releasing the drug in the cytoplasm. Reprinted with permission from ref 481. Copyright 2012 Elsevier Ltd.
The release of Ptxl from these micelles was shown to be GSH concentration dependent and was found to be optimal at 20 mM GSH, which is similar to the intracellular environment of tumor cells. Of note is that with these micellar systems, drug release profiles are short, with, for instance, 90% of the Ptxl payload of the HA-ss-DOCA NPs being released within 24 h, therefore limiting their applications for more prolonged drug release requirements of up to multiple days. Interestingly, in the same study, Li et al. demonstrated the disintegration of the micelles after GSH incubation using AFM (Figure 27).
Figure 27.
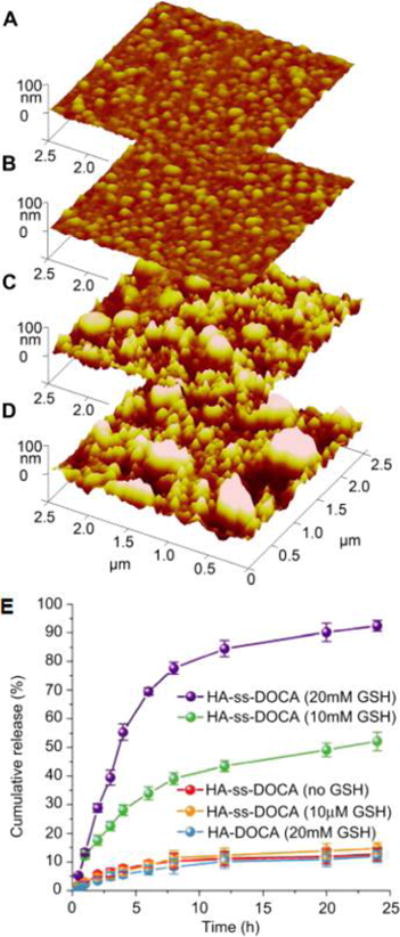
Characterization of redox-sensitive NPs. AFM images showing morphological changes of the micelles: (A) no GSH, (B) with 10 μM GSH, and (C) 10 mM and (D) 20 mM GSH after 24 h incubation. (E) Cumulative triggered-release profile of Ptxl in the presence of GSH at different concentrations. Reprinted with permission from ref 481. Copyright 2012 Elsevier Ltd.
Nucleic acid delivery is another area where redox-responsive polymers (Figure 28) have been utilized since charge complexation with cationic polymers such as poly(disulfide amine), disulfide-containing poly(amido amine), and histidine-polycations have shown potential in gene therapy.485–487
Figure 28.
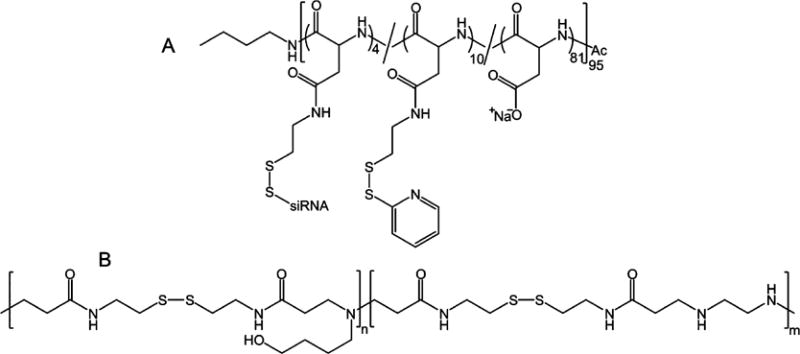
Examples of cationic polymers with –SS– bonds as pendant groups (A) or in the main chain (B) for development of redox-triggered nucleic acid delivering nanocarriers. Reprinted with permission from refs 486 (Copyright 2011 Springer.) and 488 (Copyright 2010 Elsevier Ltd.).
Positioning a bioreducible group at the junction of hydrophilic and hydrophobic blocks of amphiphilic linear block copolymers is also a commonly explored strategy in redox-triggered systems. Micelles fabricated from such amphiphilic block copolymers are constituted by hydrophobic payload-encapsulating cores connected to their hydrophilic shells through a bioreducible moiety. In the intracellular reducing environment, the shells are readily “shedded”, leading to a sudden exposure of the payload-containing cores to the intracellular hydrolytic environment, which accelerates the payload release via core degradation. These micelles are commonly referred to as reduction-responsive “shell-sheddable”489 drug delivery nanocarriers, originally developed by Zhong and co-workers using shell-sheddable degradable block copolymers.476 In earlier work, they reported a block copolymer of PEG and PCL (PEG–SS–PCL) with a disulfide bond located at the junction of the two blocks (Figure 29).490 The self-assembled micelles were able to load DOX and exhibited fast quantitative release in 10 h under a reducing environment comparable to intracellular reducing agent concentrations (10 mM dithiothreitol, DTT), whereas the release was markedly slower in the absence of a reducing agent. Slow release was also reported in the presence of 10 mM DTT for a PEG–PCL polymer lacking a disulfide bond (Figure 29).
Figure 29.
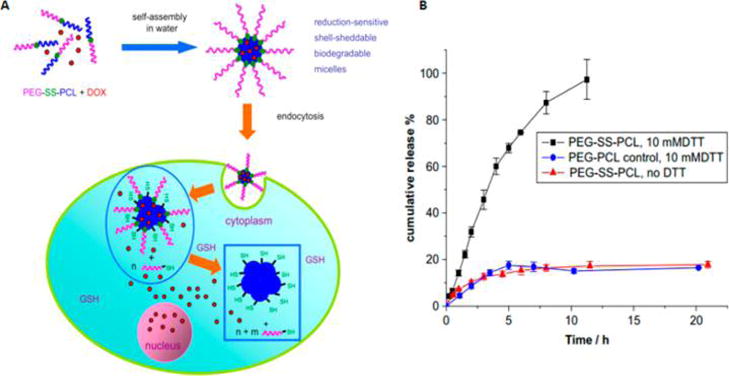
(A) Illustration of DOX-loaded shell-sheddable PEG–SS–PCL micelles fabrication and intracellular reduction-responsive drug release. (B) Cumulative release of DOX from PEG–SS–PCL micelles under different reducing environments. Reprinted with permission from ref 490. Copyright 2009 Elsevier Ltd.
In a subsequent study, Zhong and co-workers systematically established the impact of PEG–SS–PCL content on the reduction responsiveness of micelles fabricated from a mixture of reducible PEG–SS–PCL and a nonreducible PEG–PCL.491 The DOX payload release rate increased with the weight percent of PEG–SS–PCL content. The micelles containing 30, 50, 70, and 90 wt % of PEG–SS–PCL exhibited cumulative DOX release of 29.4, 42.7, 77.9, and 86.9%, respectively. Using a combination of bioreducible PEG–SS–PCL and galactose-conjugated, reduction-insensitive PEG–PCL,492 reduction-responsive micelles capable of delivering payloads to the nucleus of HepG2 cells that overexpress the asialoglycoprotein receptor (ASGP-R) have been developed. In addition to polyesters, the “shell-sheddable” concept has also been applied to polycarbonates, where a PEG and polycarbonate were conjugated through a disulfide-bearing linker.493 The polycarbonate employed was laterally functionalized with pH-sensitive trimethoxybenzylidene acetals, and the resulting micelles exhibited dual reduction and pH-responsive payload-release behavior. In addition to linear block copolymers, star-shaped molecular architecture has been explored for fabrication of reduction-responsive shell-sheddable micelles exhibiting efficient release exclusively in response to an intracellular-mimicking reducing environment.494,495 Together these studies emphasize the role of disulfide bonds in the reduction-responsive release of payloads.
Shell-sheddable micelles with a PEG shell and a core constituted by other degradable polymers, including PLA,496 poly(ε-benzyloxycarbonyl-L-lysine),497 and poly(γ-benzyl L-glutamate),498 have also been fabricated. Besides a PEG shell, other hydrophilic analogs of PEG have been employed for fabrication of reduction-responsive shell-sheddable micelles. These include poly(ethyl ethylene phosphate) (PEEP)499–501 and dextran.502 Diselenide-based reduction-responsive shell-sheddable micelles have been less fully explored. Wang and co-workers have reported branched polyethylene amines linked to PEG chains via diselenide linkages (PEG–SeSe–PEI) for reduction-responsive intracellular DNA release.503 The diselenide-based system was compared with the PEG–PEI system lacking diselenide bonds and with the PEG–SS–PEI system containing disulfide bonds. The comparison revealed that PEG–SeSe–PEI/DNA polyplexes were more efficient in their capability for endosomal escape. When compared to the disulfide-based reduction-responsive systems, the better performance of the diselenide-based system may be attributed to the greater size of the selenium atom and its lower electronegativity, which results in lower bond energies of the C–S and C–Se bonds.479
Hydrophilic–hydrophobic block copolymers with disulfide bonds distributed throughout the polymer blocks are another widely pursued approach for fabricating reduction-responsive nanocarriers. Micelles fabricated from such block copolymers have cores rich in bioreducible –SS– or –SeSe– bonds and tend to completely dismantle through polymer disintegration under the intracellular reducing environment, accelerating the payload release. Engbersen and co-workers have pioneered the synthesis of bioreducible linear polyamido amines (PAA), particularly for nonviral gene delivery.504–506 The synthesis involved a Michael addition between N,N′-cystaminebis(acrylamide) (CBA) and the primary amine monomers.504 Besides PAA, bioreducible poly(β-amino esters) containing –SS– bonds in the backbone repeat units have also been synthesized by Xing and co-workers,507,508 who used an amine-terminated PEG to impart amphiphilic character to these polymers. DOX-encapsulated micelles derived from the poly(β-amino esters) containing –SS– bonds demonstrated a reduction-responsive payload release and a higher cytotoxicity for HepG2 tumor cells when compared to reduction-insensitive micelles. A concept was recently employed by Green and colleagues509,510 to synthesize poly(β-amino esters) with different pendant functional groups exhibiting reduction-responsive siRNA delivery (Figure 30). By controlling the ratio between different monomers, these researchers tuned the physical properties of the polymers to produce nanocarriers capable of efficient gene knockdown (91 ± 1%) in primary human glioblastoma cells and superior performance compared to commercial Lipofectamine 2000.
Figure 30.
(A) Synthesis of reduction-responsive poly(β-amino esters). (B) Self-assembly of reduction-responsive poly(β-amino esters) into siRNA-loaded NPs and their gene-knockdown efficiency. Reprinted from ref 509. Copyright 2014 American Chemical Society.
Cross-linking of the micellar core is employed to improve the stability of micelles under dilution during systemic circulation without impairing the payload-release behavior of the nanocarriers. Cross-linking via bioreducible linker molecules (Figure 31) offers systemic stability, but it also ensures a fast release of payloads only in response to the intracellular reducing environment. Development of strategies for the synthesis of degradable polymers with appropriate pendant groups is essential for fabricating core-cross-linked nanocarriers. In a recent attempt, Li and co-workers fabricated a block copolymer constituted by hydrophilic PEG and hydrophobic poly(ester carbonate) with pendant bromo groups.511 The bromo groups were transformed into azide groups via postpolymerization modification. The azide groups were then used to cross-link the core using the terminal alkyne groups of propargyl 3,3′-dithiopropionate via the azide–alkyne click reaction during the NP fabrication. Paclitaxel was loaded into these particles in an in situ manner. Ptxl-encapsulated core-cross-linked NPs exhibited a burst release of Ptxl under conditions mimicking the intracellular environment, while the release rate was very slow in a nonreducing environment.
Figure 31.
(A) Synthesis of cross-linkable polyester carbonate and fabrication of reduction-responsive core-cross-linked micelles (CCL). The nanocarriers exhibited reduction-responsive paclitaxel (here abbreviated PTX) release (B) and cell viability (C). Reprinted with permission from ref 511. Copyright 2015 Royal Society of Chemistry.
Attempts have also been made to synthesize polymers with pendant thiol groups, which can be transformed into dilsulfide cross-links via oxidation. Jing and co-workers reported a block copolymer of PEG and poly(ester carbonate) bearing pendant thiol groups (Figure 32).512 The free thiol groups were oxidized to cross-link the self-assembled micelles, rendering them stable even in DMF. DOX-loaded core-cross-linked NPs exhibited a faster reduction-responsive release rate compared to a slower release rates in a nonreducing environment. The reduction responsiveness affect was also demonstrated in MCF-7 cells. In addition to thiols, both lipoyl513,514 and cyclic disulfide units515,516 have also been employed to fabricate various core-cross-linked reduction-responsive nanocarriers. As well as core cross-linking, shells or interface cross-linking has also been explored in the fabrication of reduction-responsive nanocarriers. A Y-shaped ABC2 triblock copolymer based on PEG, poly-L-glutamate (PLG), and (PLA)2 has also been reported.517 These fabricated micelles were cross-linked at the interface of PEG and PLA by reacting pendant carboxylic acid groups from the middle PLG block using cystamine as a cross-linker. The reduction-responsive release of the encapsulated DOX from these cross-linked micelles proved superior to that of the non-cross-linked micelles.
Figure 32.
(A) Synthesis of polycarbonate with pendant thio groups. (B) Cumulative release of DOX from the cross-linked mPEG-b-P(LA-co-MTCSH) micelles at varying concentrations of glutathione. Reprinted with permission from ref 512. Copyright 2012 Royal Society of Chemistry.
6.2. Oxidation-Responsive Drug Release
Reactive oxygen species (ROS) are partially reduced metabolites of oxygen and include superoxide anion, hydroxyl radical, hydrogen peroxide, peroxynitrile, and hypochlorous acid (HOCl).518–520 While normal ROS levels are necessary for homeostasis, high levels in certain pathological conditions can damage cells by causing oxidation of lipids, proteins, and DNA. Localized high levels of ROS have been found in a number of pathological conditions, including neurological and inflammatory diseases, infections, cancer, atherosclerosis, and diabetes.521–532 This has triggered interest in developing functional polymers for the fabrication of nanocarriers capable of payload release in response to ROS. Certain polymers respond to ROS through the oxidation of functional groups, such as sulfides,533,534 or selenides,535 increasing their solubility in water. Polymers can also undergo physical degradation in response to ROS if they contain oxidizable functionalities such as thioketals, boronic esters,536–539 or (oligo)prolines.522,540,541 Both scenarios lead to the disassembly of the micelles fabricated from ROS-responsive polymers and include a mechanism for ROS-triggered payload release. Chen and co-workers have recently reported a linear PEG-based poly(β-thioether ester) with a backbone made up of alternating hydrophilic PEG and hydrophobic β-thioether ester segments via thiolene polymerization.542 The polymer was temperature-responsive, and a water suspension of the collapsed form of the polymer at elevated temperatures became completely soluble when treated with H2O2 as an oxidizing agent. NMR spectroscopy revealed the transformation of sulfide bonds into sulfoxide and sulfones after treatment with H2O2, which increased the polymer’s hydrophilicity and water solubility. The fabricated micelles also exhibited an accelerated payload release in an oxidizing environment.
Wilson et al. synthesized a poly(1,4-phenyleneacetone dimethylene thioketal) (PPADT) with ROS-sensitive thioketal linkages for the localized delivery of siRNA to diseased intestinal tissue (Figure 33).543 Thioketal bonds are optimal for this application, as they are acid-, base-, and protease-stable and can facilitate highly selective release of therapeutics to inflamed tissues in the intestine. In this study, levels of TNF-α mRNA were reduced in response to the delivery of the thioketal NPs loaded with siRNA against the proinflammatory cytokine TNF-α, leading to controlled gene silencing after oral administration in a murine colitis model.
Figure 33.
ROS-sensitive polymer (PPADT) complexes with siRNA/DOTAP against TNF-α to form thioketal nanoparticles (TKNs); scale bar represents 1.5 μm (a). The TNF-α-TKNs undergo degradation at the sites of inflammation where phagocytes produce high levels of ROS, releasing their TNF-α-siRNA (b). An acetal exchange reaction was used to synthesize PPADT (c); PTSA = p-toluenesulfonic acid. Reprinted with permission from ref 543. Copyright 2010 Nature Publishing Group.
6.3. pH-Responsive Drug Release
Appreciable pH variations at the systemic, tissue, and cellular level have been identified in certain pathological conditions. For instance, the extracellular environment of tumor tissues is notably more acidic (pH 6.2–6.9) than normal tissues and systemic blood pH.544–546 As tumor cells proliferate at an abnormally high rate, lack of nutrients leads to a high rate of glycolysis and accumulation of lactic acid, lowering the environmental pH. This pH gradient has been explored as an endogenous stimulus for the development of nanocarriers capable of delivering their payloads selectively in response to an acidic tumor environment. In addition, the acidic environment of intracellular compartments (endosomes and lysosomes, pH 4.0–6.0) has also been explored for designing pH-responsive nanocarriers programmed for intracellular release of payloads.547,548 pH-responsive nanocarriers can be fabricated from polymers that switch between hydrophilic (swollen) and hydrophobic (collapsed) states as a result of protonation and deprotonation of labile functional groups (e.g., amine and carboxylic acid groups) in response to changes in environmental pH. Switching from a hydrophobic to a hydrophilic state leads to the solubilization of the polymer and the disassembly of the micellar architecture of the nanocarrier system in an aqueous environment. Consequently, this offers a mechanism for pH-responsive payload release.
Polymers with amine groups have been widely explored for the fabrication of pH-responsive nanocarriers, where the protonation of amine increases polymer hydrophilicity and micellar disassembly.549 Amine groups are also known to efficiently neutralize the acidic endosomal environment via protonation. This increases endosomal pH, triggering the ionic transport of the endosome into the lumen, where they ultimately swell and burst, releasing the internalized NPs (referred to as the proton-sponge effect).550 Commonly available polyamines, such as polyethyleneimine, are toxic because of the high pKa (∼9) of their amine groups, which can induce membrane lysis at both acidic and physiological pH.413 However, the amine groups can be protected by acid-sensitive functional groups, which in an acidic endosomal environment cleave and expose the amine groups, triggering endosomal escape (via the proton-sponge effect) and releasing the payload into the cytoplasm. Alternatively, incorporation of low-pKa heterocycles in place of primary amines as pendant groups has also been reported to reduce the toxicity of the polymer system.551 β-Carboxylic amides are neutral or slightly negatively charged under physiological conditions and are known to hydrolyze under acidic pH and become free amines. Polymers with β-carboxylic amides have been shown to encapsulate cationic payloads and efficiently release them in response to an acidic extracellular tumor environment or the endosomal/lysosomal intracellular pH.552–555 The positive charge acquired during the transformation from β-carboxylic amide to free amine may also help in endosomal escape. Chen et al. recently reported a strategy for the protection of the pendant amine groups on methoxy poly-(ethylene glycol)-block-poly(L-lysine) (mPEG-b-PLL) with β-carboxylic amides by employing different anhydrides, including succinic anhydride (SA), cis-cyclohexene-1,2-dicarboxylic anhydride (CDA), cis-aconitic anhydride (CA), and dimethylmaleic anhydride (DMMA) (Figure 34).556 The pendant group’s negative charge was imparted to the polymer through ROP, which was employed to encapsulate the positively charged DOX. The order of acid sensitivity was also demonstrated to influence the DOX release kinetics, offering an avenue for adjusting the pH responsiveness of nanocarrier systems for on-demand intracellular delivery (Figure 34).
Figure 34.
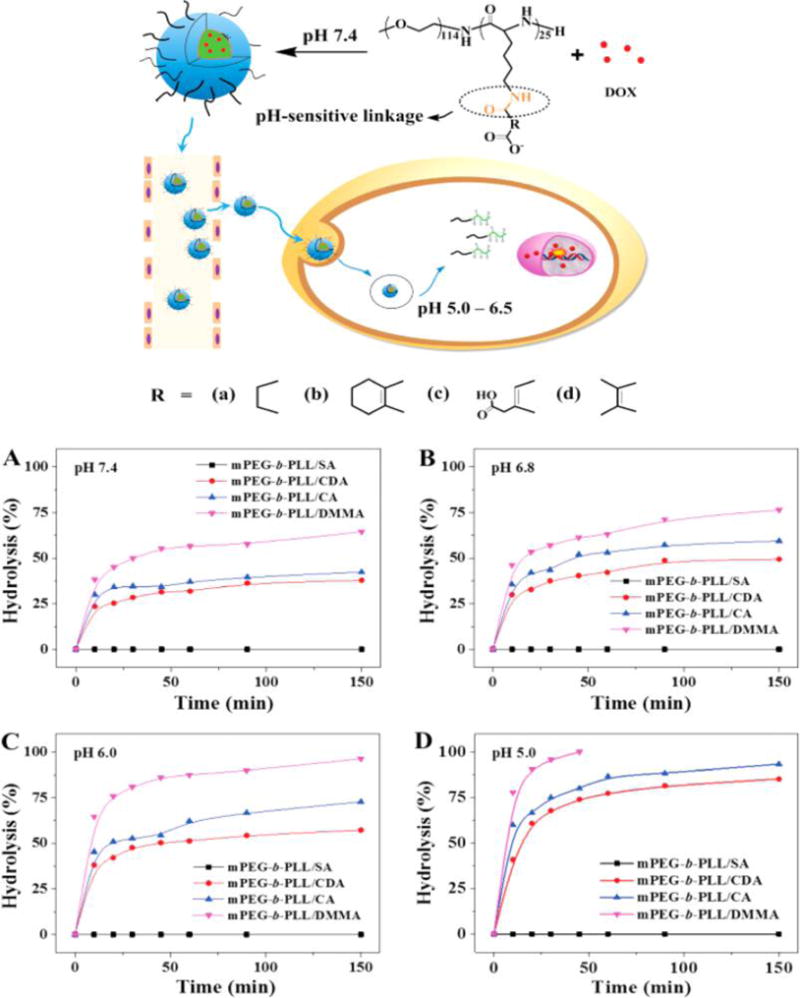
(Top) Illustration of DOX-loaded pH-responsive nanocarrier fabricated from polymers with various pH-sensitive β-carboxylic amide pendant groups. (A–D) Evaluation of pH-responsive degradation kinetics of nanocarriers fabricated from polymers with different β-carboxylic amide pendant groups. Reprinted with permission from ref 556. Copyright 2015 Royal Society of Chemistry.
In addition to β-carboxylic amide, a wide variety of polymers bearing other acid-cleavable functional groups have been reported. Acetal, orthoester, hydrazone, and oxime bonds and boronic acid esters are the most commonly explored acid-sensitive functionalities.547,557,558 For instance, pH-labile acetal or ketal linkages have been introduced into the polymer backbone (linear or branched) and at the junction between hydrophilic and hydrophobic block copolymers.559–565 Recently, Ni and colleagues reported a three-armed star-block copolymer architecture constituted by poly(ethylene glycol) methoxy (mPEG) and poly(ε-caprolactone) (PCL) that are linked via acetal groups (Figure 35).413 The star polymer was shown to self-assemble into spherical or rod-shaped micelles, the morphology of which could be controlled by varying the polymer concentration. Both the star polymer and the micelles were found to be stable under neutral pH but disintegrated under acidic pH. DOX-encapsulating micelles exhibited pH-responsive release kinetics, and in vitro experiments on HeLa cells revealed efficient intracellular delivery of DOX.
Figure 35.
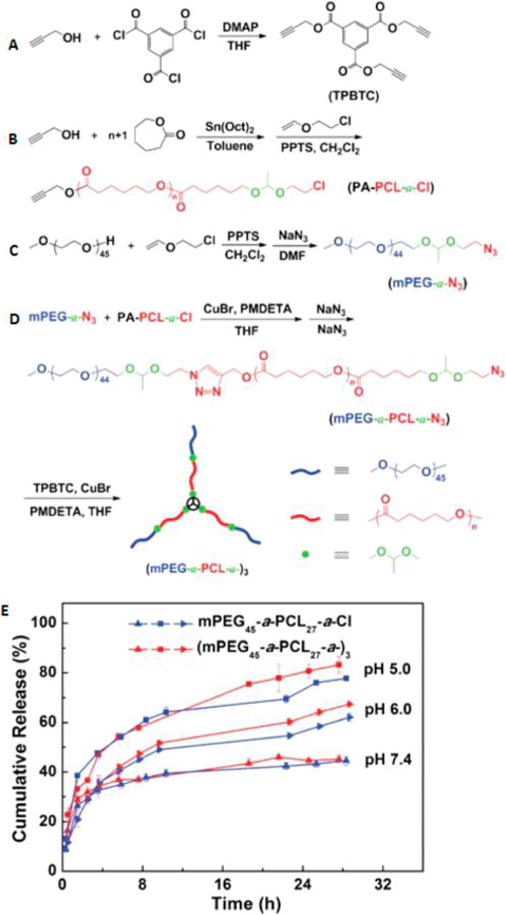
(A–D) Synthesis of acid-cleavable star-block copolymer. (E) In vitro cumulative release of DOX from DOX-loaded pH-sensitive micelles fabricated from star-block copolymer. Reprinted with permission from ref 413. Copyright 2015 Royal Society of Chemistry.
There are few examples of poly(ortho ester)-based pH-responsive degradable polymers, primarily because of the limited number of synthetic strategies available for the synthesis of these types of polymers.558,566 Polymers with main-chain oxime linkages are also rare. However, Zhu and co-workers successfully developed oxime linkages placed at the junction of PEG and PCL in the triblock copolymer, with PEG blocks flanking the PCL block. DOX-loaded micelles were made from this polymer and showed fast release kinetics under acidic conditions.567 A polymer (based on PCL, polyurethane, and PEG) with hydrazone linkages connecting the individual blocks of the polymers was also reported. pH-responsive disintegration and payload release were demonstrated for Ptxl-loaded micelles derived from this polymer. In addition to incorporating pH-labile linkages into the main chain polymers, drug molecules have also been conjugated to the polymer chains as pendant groups via pH-labile linkages, e.g., hydrazones568–570 and boronic acid esters.571,572 Cheng et al. recently conjugated DOX to PLA as pendant groups via an acid-sensitive Schiff base linkage (Figure 36).573 The resulting micelles exhibit fast release kinetics under acidic conditions.
Figure 36.
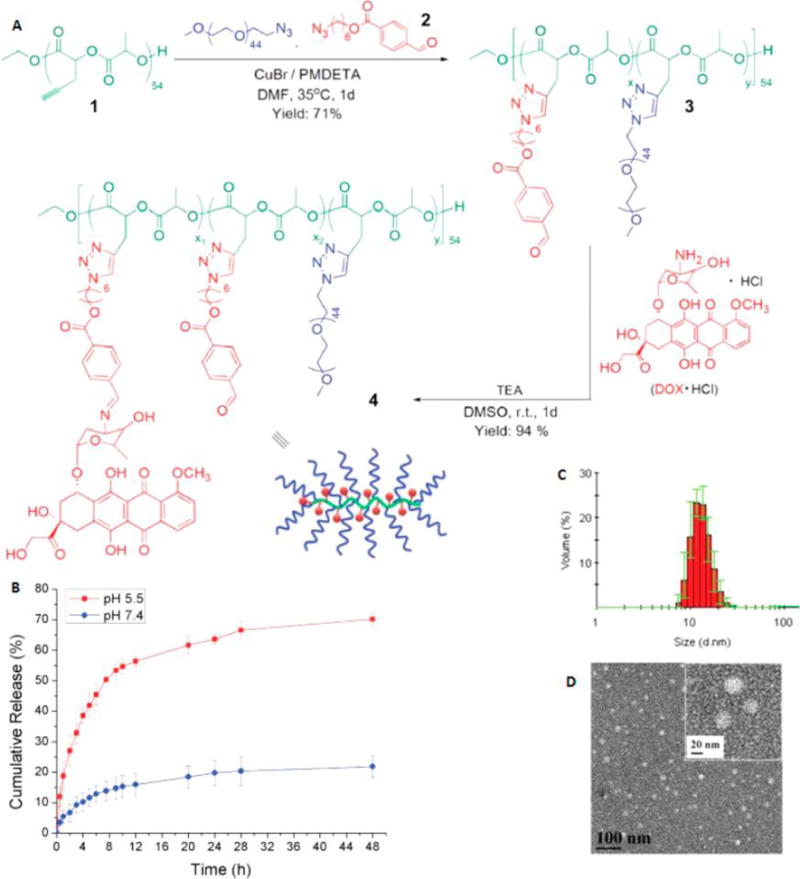
(A) Synthesis of DOX-conjugated PLA with pH-sensitive Schiff base connections. (B) pH-responsive release of DOX from micelles fabricated from polymer 4. Size of pH-sensitive micelles determined by DLS (C) and corresponding TEM images (D). Reprinted with permission from ref 573. Copyright 2015 Royal Society of Chemstry.
The pH differences within the tumor and endosomal environment were exploited in a study where the imidazole group was utilized as an ionizable group in the tumor environment (Figure 37). The authors developed pH-responsive magnetic nanogrenades (PMNs) by coassembly of small iron oxide NPs and two ligands bearing catechol groups for surface binding to the iron oxide NPs and imidazole for pH sensitivity. The design of these NPs allowed for a two-stage pH activation, initially in the tumor, which facilitated cellular uptake and then endo/lysosomal pH-dependent theranostic activity.
Figure 37.
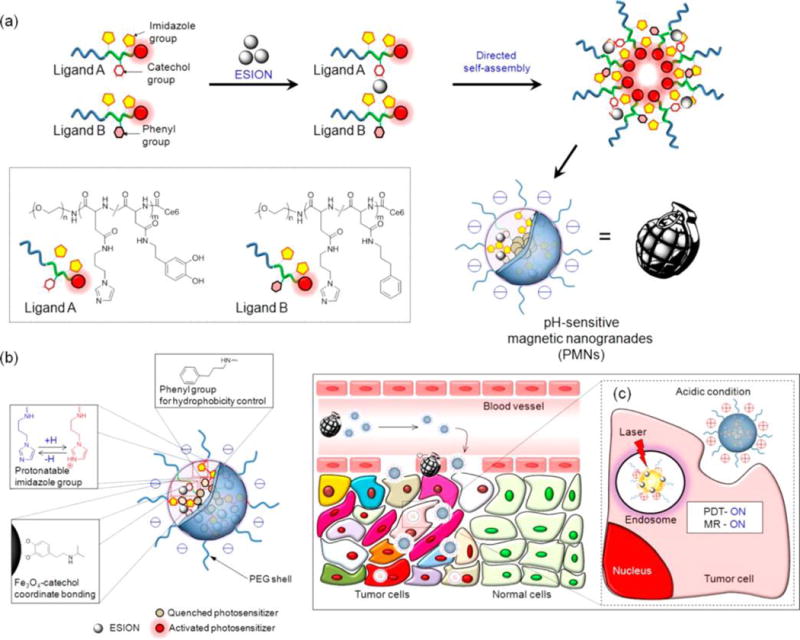
Tumor-pH-responsive magnetic nanogrenades (PMNs). (a) Ligand-directed self-assembly of iron oxide NPs. (b, c) PMNs change their surface charge from negative to positive in response to tumor extracellular pH (pH ∼6.8), leading to cellular uptake and cellular internalization. Within the endosome, the low pH (∼5.5) causes disassembly of the PMNs, leading to increased imaging signal and also photodynamic therapy. Reprinted with permission from ref 574. Copyright 2014 American Chemical Society.
PLGA NPs with pH-sensitive moieties for nanoimmunotherapy applications have been developed that are capable of rapid intracellular antigen release once taken up in antigen-presenting cells.575 In this study, PLGA NPs were loaded with ammonium bicarbonate (NH4HCO3), which led to increased release of the coencapsulated antigen (ovalbumin, OVA) (Figure 38). The hypothesis in this study was that protons within the endosomes and lysosomes of dendritic cells react with the ammonium bicarbonate to release NH3 and CO2, which destabilized the NPs.
Figure 38.
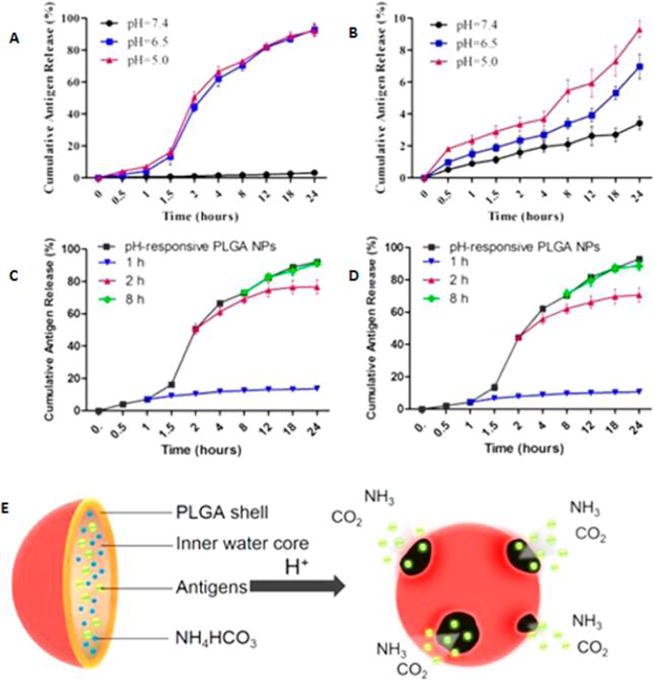
Cumulative in vitro release of OVA from (A) pH-responsive PLGA NPs and (B) non-pH-responsive PLGA NPs incubated at various pH ranges. In vitro release of antigens from pH-responsive PLGA NPs after incubation with proton scavenger for 1, 2, and 8 h at (C) pH 5.0 and (D) pH 6.5 at 37 °C. (E) Illustration of pH-responsive PLGA NPs and the release mechanism. Reprinted with permission from ref 575. Copyright 2015 American Chemical Society.
The development of layer-by-layer (LbL) NPs responsive to pH changes has become an actively investigated field whereby pH variations lead to disruptive mechanisms on LbL NPs.576,577 Polymeric NPs fabricated via the LbL approach are comprised of weak polyelectrolytes that become pH responsive due to protonation/deprotonation of charged groups.578 Using PLGA-coated poly(L-lysine) and dextran sulfate-coated multilayers, Hammond and co-workers showed that LbL can lead to stabilized drug delivery and reduced liver accumulation and improvements in PK.
The acidic environment of bacterial infections generated due to anaerobic fermentation and inflammation was also recently exploited in a study by Radovic-Moreno et al. for the specific delivery of antibiotics to Gram-positive and -negative bacteria.579 In this study, the authors used PLGA-b-polyhistidine-b-PEG triblock copolymers as charge-switching elements, which allowed the polymeric NPs to interact with the negatively charged bacterial cell surfaces.
The use of pH differences between healthy and disease states in addition to extracellular and intracellular states has led to extensive investigations of pH-responsive polymers for controlled drug release and is also the subject of numerous other published works.580–586
7. ENZYME-TRIGGERED DRUG RELEASE
Enzymes play vital roles in every biological and metabolic process in the body, and as a result, regulation of enzyme activity is highly important in disease states. The dysregulation of enzyme levels can be related to various disease pathologies and can be exploited for the design of responsive polymers in controlled-release drug delivery. For example, the concentration and activity of certain enzymes have been shown to be up-regulated during inflammatory processes and in tumors.587 A highly useful feature of enzyme-responsive polymers is the exceptional selectivity with which reactions can be achieved, since enzymes have extremely high reactivity for their substrates.588,589 Enzyme-responsive polymers can undergo changes via enzyme catalysis, leading to disintegration, dissociation, or self-assembly, and morphological transitions of the parent NPs as facilitated by electrostatic interactions, hydrogen bonding, van der Waals forces, and solubility interactions. As a result, a wide range of polymers with enzyme bioactive moieties have been developed for drug delivery applications.361,590–592
The selective bioactivity of enzymes can be useful in tumor-targeting and drug release applications whereby cleavage of bonds can lead to site-specific binding to antigens or drug release, improving drug pharmacokinetics and toxicty.593,594 The instillation of site-specific enzyme-triggered functionalities on polymers can lead to cleavage of surface NP layers, exposing tumor-specific ligands/chemical properties that can lead to the enhanced accumulation of NPs within tumors or cancer cell uptake.594 These enzyme-active moieties can either be incorporated into the main polymer chain or be part of the side chain groups, and these moieties can often be peptides, DNA, or synthetic sequences. The cleavage of the enzyme-specific sequences can lead to drug release as a result of changes in the structure and amphiphilicity of constituent NP polymers. Enzyme-responsive NPs can also be prepared via supramolecular polymer assembly without covalent conjugation. An alternative method for developing enzyme-responsive NPs is the preparation of enzyme-responsive assemblies based on noncovalent yet electrostatic interactions of the enzyme–substrates with other copolymers.595 Numerous characteristics of enzyme-responsive polymers have also been investigated, including tuning and control of chain topologies, the specific location of the cleavable moiety, length and linkers, and overall chemical structure of the polymer. In terms of the chain topologies of enzyme-responsive polymers, block copolymer, graft copolymer, and star copolymers have been developed to date. In this section, we will discuss the key types of enzymes that can be exploited in enzyme-controlled drug release and recent enzyme-active polymers for controlled-release drug delivery.
7.1. Enzyme-Responsive Polymeric Micelles
The most common method of preparing enzyme-responsive polymers is by covalent conjugation of enzyme-cleavable sequences to the polymer backbone. The main mechanisms of enzyme-responsive NPs involve the cleavage of esters and short peptide sequences by esterases or proteases, and a common cleavable sequence is Gly-Phe-Leu-Gly, which is cleaved in the lysosome by cathepsin B, which is overexpressed in cancer cells.596 Endocytosis of polymeric NPs ultimately leads to the accumulation of these particles within endocytic compartments, and local enzymes can be harnessed for effective drug delivery.548 Cathepsins are one family of enzymes that are found in endosomes and lysosomes and are a class of protease. There are different types of cathespsins and they are also upregulated in tumor cells. Investigations have shown that cathepsin B overexpression is correlated with cancer invasion and metastasis.596 As a result, the proteolytic action of cathepsin B can be harnessed in developing more responsive polymeric NPs that can release their therapeutic payload in a triggered manner, only once inside cancer cells. The most advanced enzyme-responsive polymers are the copolymers of HPMA with the cathepsin B cleavage sequence (Gly-Phe-Leu-Gly), as they have been translated to the clinic, in particular, for the delivery of anticancer drugs, and extensive pioneering studies led by Duncan and co-workers have demonstrated the clinical antitumor activity of these copolymer–anticancer drug conjugates.597–599
The micelle polymeric platform has been the most commonly used polymeric drug delivery platform for the development of enzyme-responsive NPs. For example, enzyme-responsive self-assembled polymeric micelles were developed by Zhang and co-workers for tumor-specific DOX delivery (Figure 39).600 In this study, DOX was complexed with polyGC double-stranded DNA fragments to form an intercalated complex (DOXepolyGC), and this was then combined with cationic gelatin in order to form NPs (termed CPX1). Here the cationic gelatin was used since it can be digested by gelatinase (GA), a matrix metalloproteinase (MMP) enzyme that is highly upregulated in tumors.587 Since there are also high levels of gelatinase present in the liver, they further coated the initial CPX1 core with a pH-sensitive PEG-histamine-modified alginate (PEGeHistamine–alginate) polymer to produce CPX2. The hypothesis here is that once CPX2 reaches the tumors, the acidic tumor microenvironment (pH 6.2–6.7) protonates the His–alginate polymer, conferring a cationic charge to it and therefore leading to the dissociation of NP assembly due to electrostatic repulsion and the release of CPX1 core complexes, which can be further disassembled via the action of tumor gelatinases and deoxyribonuclease (DNase), leading to local DOX release. It is of interest to note that almost no DOX was released from these complexes in the absence of enzymes or lowered pH (Figure 39). This elegant study demonstrates the sequential pH-triggered protonation and enzyme-catalyzed degradation of NP complexes for highly specific and targeted controlled-release drug delivery to tumor tissues. In this study, the protease activity of MMP enzymes was harnessed as they are a class of endopeptidases with widespread use in triggered controlled drug delivery to date. The role of MMPs in cancer metastasis and tumor invasion and cardiovascular disease has been widely reported.601–605 MMPs are a family of over 20 zinc-dependent proteases that cleave extracellular matrices (ECM) such as proteoglycans, glycoproteins, and collagen.606 In particular, MMP-2 and MMP-9 are positively correlated with metastasis and cancer cell invasion.607
Figure 39.
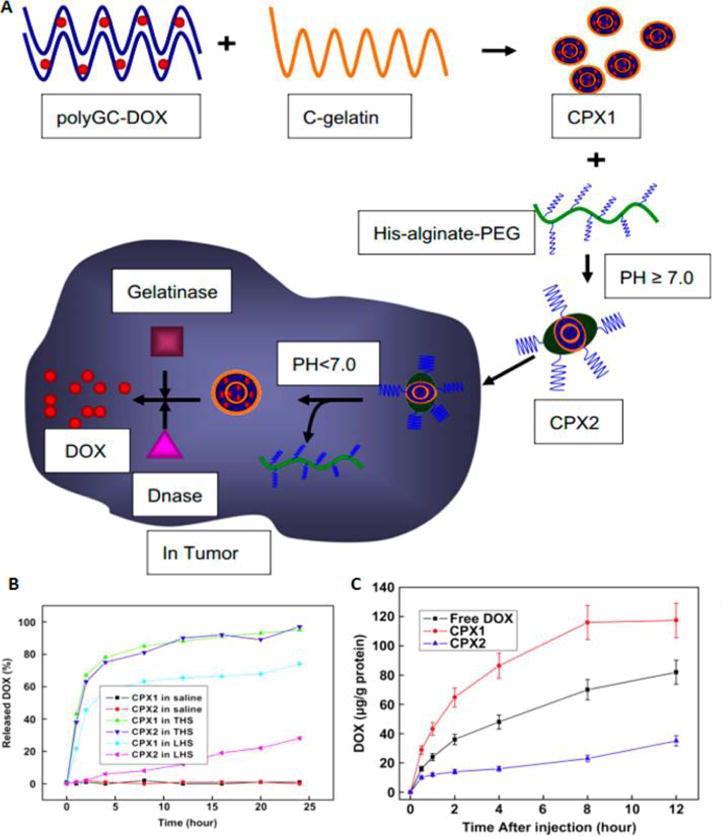
(A) Sequential pH-triggered protonation and enzyme-catalyzed degradation of NP complexes for controlled-release DOX delivery to tumor tissues. (B) DOX release rates in the presence of tumor homogenate (THS) or liver homogenate (LHS). (C) DOX concentrations in the liver after iv injection of DOX-loaded enzyme-degradable NPs (dose of 20 mg/kg of body weight). Reprinted with permission from ref 600. Copyright 2010 Elsevier Ltd.
In one case, the MMP-2-cleavable peptide sequence GPLGVRG was instilled in between poly(ethylene glycol)- and diethylenetriamine-modified poly(aspartamide) to produce the diblock copolymer PEG227-GPLGVRG-PAsp(DET)64 (Figure 40). This polymer was complexed with DNA to form polyplex micelles. Upon MMP-2 cleavage in tumors, the PEG shell layer was shed, which revealed a positive surface for interaction and uptake with cancer cells, respectively.588
Figure 40.
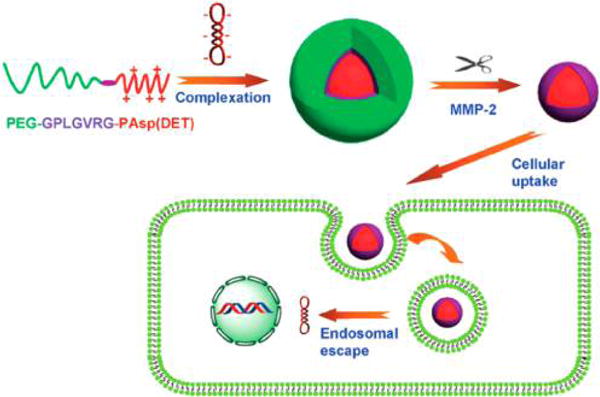
PEG-sheddable micelle polyplexes responsive to MMP-2 leading to enhanced cellular uptake and endosomal escape for gene therapy. Reprinted with permission from ref 588. Copyright 2015 Royal Society of Chemistry.
Further enzyme-responsive polymeric micelles have been developed with azoreductase-responsive polymers. In one study, amphiphilic block copolymers of poly(ethylene glycol)-b-poly(styrene) (PEG-b-PS) with an azobenzene linker were synthesized and used to form micelles in solution.608 Upon exposure to azoreductase, the azobenzene linker is cleaved, leading to the disassembly of the micelle. These micelles can be used for colon-specific drug delivery applications in the intestine, since azoreductase is present in the intestine.
Enzyme-catalyzed triggered drug release has many advantages, such as high efficiency, selectivity, and specificity, and can be achieved under mild physiological conditions. It is interesting to note that currently most of the existing examples involve responsive elements to one specific enzyme type; however, future developments could involve the incorporation of multienzyme-based cascade reactions that can target the release of drugs in a more controlled manner. Recent work using lipids presented the development of multicompartment vesicles, inside of which an engineered multistep enzymatic pathway was carried out (involving three enzymes in the cascade, lactase, glucose oxidase, and horseradish peroxidase).609 In this novel study, each step is isolated, with the products capable of traversing adjacent compartments as aided by transmembrane protein pores. This study demonstrates how signaling cascades using enzymes can be artificially engineered. These principles can be applied to polymeric multienzyme cascades, whereby each substrate released can catalyze the cleavage or disruption of the NP surface layer, leading to the triggered release of drugs in a highly specific manner.
7.2. Hydrogel-Based Enzyme-Responsive Controlled Drug Release
Hydrogels have been widely explored as enzyme-responsive materials, as these materials can change their physical properties in response to enzyme catalysis, which can lead to macroscopic swelling or collapse. This in turn can lead to the release of encapsulated drugs. Hydrogels have been widely investigated in drug delivery, and their triggered gelation, biodegradation, and drug release have been previously discussed.610,611 The work of Ulijn and co-workers has demonstrated the utility of enzymes as biological stimuli for triggering drug release, and the overall design of these systems is presented in Figure 41.612
Figure 41.
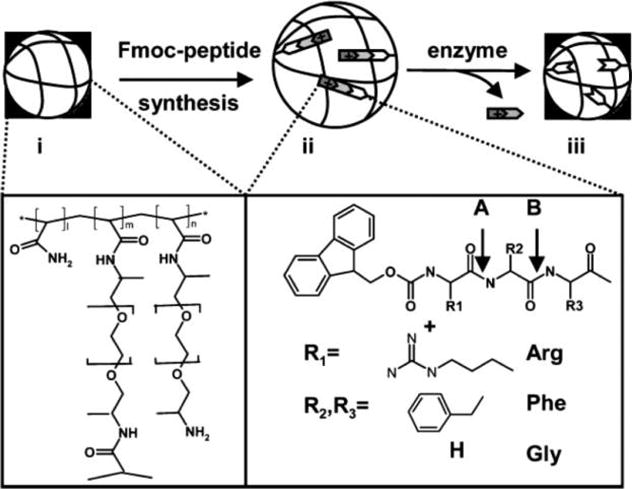
Overall design of enzyme-responsive hydrogels. Acrylamide polymeric backbones cross-linked with PEG800 having pendant PEG linkers with amino functionalities for conjugation. Proteases can cleave at A or B positions, leading to the removal of the charged Arg group, collapse of the bead structure, and reduced molecular assembly. Reprinted with permission from ref 612. Copyright 2005 Royal Society of Chemistry.
In a further study, the same group developed enzyme-responsive synthetic polymeric microparticles that were capable of controlled-release functions (Figure 42).594 PEGA hydrogels copolymerized with acrylamide and PEG, and enzyme cleavable sequences were fabricated. The PEGA polymers were conjugated with peptide zwitterionic enzyme-cleavable peptide linkers. At low pH (5.0), the particles undergo swelling and encapsulate dextran as a model macromolecular payload. An increase in pH regenerates nonswelling particles, causing a reduction in size and leading to payload entrapment. Exposure to enzymes leads to hydrolysis of the peptide sequences and release of the dextran payload due to reswelling.
Figure 42.
(A) Scanning electron micrographs of PEGA hydrogel microparticle. (B) Chemical structure of the polymer (PEGA = copolymer of PEG and acrylamide, with PEG MW being 800) and mechanism of enzyme responsiveness. (C, D) Illustration of pH-responsive payload encapsulation and enzyme-responsive release behavior. Reprinted with permission from ref 594. Copyright 2012 Royal Society of Chemistry.
In summary, although still in its infancy, the field of enzyme-responsive polymeric controlled drug release has produced novel applications for polymeric NPs, and more progress and investigation are still necessary to ascertain the full utility of these systems in the clinic.
8. CONCLUSIONS AND OUTLOOK
During the course of the evolution of polymeric NPs as targeted- and controlled-release therapeutics, we have witnessed the progression of these systems from first-generation passively targeted PEGylated systems for enhanced in vivo circulation to second-generation targeted systems capable of selectively binding to disease targets. Now we are witnessing the next wave of evolution of these systems: third generation NPs capable of triggered drug release in response to various types of external or internal stimuli. In this review, we have discussed and summarized a variety of parameters that affect drug release from these polymeric systems, the wide variety of responsive modalities that can be incorporated into the NP building blocks, and how nature can guide us in the design of “smart” nanomaterials to combat disease.
With the translation of a number of polymeric drug delivery platforms (see Figure 3) to the clinic, the need for smarter, more precise and optimally tuned drug release is critical. The limitations of current polymeric drug delivery systems show that the drug molecule is susceptible to diffusive transport out of the nanocarrier as soon as it has become encapsulated. Therefore, it is clear that methodologies for switching drug release “on” or “off” on cue from polymeric NPs can be advantageous, with direct impact on nanocarrier manufacturing, storage, distribution, and in vivo therapeutic efficacy. In order to enhance the therapeutic potential of toxic chemotherapies and other APIs, it is critical to be able to transport the drug and release it only at the site of disease or therapeutic activity in a spatiotemporal manner. For stimuli-responsive polymeric NP drug delivery to become mainstream, the rigorous testing and validation of responsive polymers and materials is extremely important, in particular, as currently a wide variety of materials are being developed that have not yet been clinically validated. Of course, the responsive modalities and choice of chemistries to instill triggered components within nanocarriers are increasing rapidly; however, it is important to keep in mind the design principles that will ultimately streamline these platforms toward a more facile clinical translation trajectory, with an emphasis on characterization and optimization of the biophysicochemical properties of NPs.
We have seen how mechanistic studies and mathematical modeling have led to our improved understanding of drug release mechanisms. The design of new controlled-release polymeric nanomedicines with predetermined release properties can be optimized through the use of accurate mathematical modeling. It will be highly beneficial if desirable release profile parameters could be theoretically predicted using mathematical and computer modeling in order to know the exact mass transport mechanisms involved in the drug release process and to stimulate the effect of polymer design parameters on release mechanisms and rates a priori. This information can then be used to ascertain the appropriate nature of the payload for the delivery system, the therapeutic dose required, and the optimal size and shape. Although experimental proof is still required and combinatorial polymeric NP library development strategies have led to the identification of optimal polymer and NP characteristics for specific drug delivery, mathematical and computer-based modeling of drug release phenomena can significantly impact drug delivery efforts in the future.
Controlled-release drug delivery has continued to provide healthcare and pharmaceutical innovations and incentives, and led to the development and marketing of a number of macro and micro polymeric drug delivery systems (Figure 3), which in turn have spurred the nanoscale development of polymeric drug delivery NPs. The real impact of the current polymeric NP technologies in clinical trials will be evident in the next few decades; as such, on the basis of our evolving understanding of disease biology and the move toward more personalized therapeutics, the development of the next stage of nanoenabled drug delivery products that can further improve drug performance, patient compliance, and therapeutic efficacy, which are precisely responsive to disease pathology and biochemistry, is highly timely.
Of course, the bench-to-bedside translation of stimuli-responsive NPs is not without its challenges, and their multicomponent designs make their manufacturing and reproducibility a nontrivial task. In addition to manufacturing challenges, and given the fact that not many of the stimuli-responsive materials have been tested in vivo, there are other factors that need to be taken into consideration when designing stimuli-responsive systems. For example, the very endogenous cues that are paramount to the trigged drug release at the site of disease may be highly variable from one patient to another, which may make standardization and benchmarking difficult. By the same token, the use of external stimuli for triggered release will also need careful monitoring and application, since the types of energies and physical stimuli used may cause damage to the surrounding healthy tissue. Although it is encouraging to see that two stimuli-responsive systems—thermosensitive liposomes (ThermoDox) and iron oxide NPs (MagForce)—are in clinical use currently, there is still a lot of ground to cover to fully test the current stimuli-responsive systems being developed, which are mostly at the in vitro proof-of-principle stage. Nevertheless, with effective yet simple nanoengineering and established preclinical and clinical nanomaterial testing protocols, the translation of stimuli-responsive polymeric drug delivery systems is looking positive. Therefore, research and development, at both the academic and pharmaceutical level on smart, stimuli-responsive nanomaterials that can further enhance the potential of the nanomedicines in development today, is not only important but a pivotal step along the evolutionary and revolutionary path of nanotherapeutics to date.
Acknowledgments
This work was supported by the National Cancer Institute (NCI) (grant U54-CA151884), the National Heart, Lung, and Blood Institute (NHLBI) R01 grant (HL127464-01A1), the National Institute of Biomedical Imaging and Bioengineering (NIBIB) R01 grant (EB015419-01), and the David Koch-Prostate Cancer Foundation Award in Nanotherapeutics.
Biographies

Omid Farokhzad is an associate professor at Harvard Medical School (HMS) and a physician–scientist at Brigham and Women’s Hospital (BWH). He is the director of the Laboratory of Nanomedicine and Biomaterials (http://farokhzad.bwh.harvard.edu), a faculty member of the Brigham Research Institute Cancer Research Center, and a member of the Dana Farber/Harvard Cancer Center. He completed his postgraduate clinical and postdoctoral research training at BWH/HMS and the Massachusetts Institute of Technology in the laboratory of Institute Professor Robert Langer, respectively. He received his M.D. and M.A. from the Boston University School of Medicine and his M.B.A. from the Massachusetts Institute of Technology. Omid Farokhzad’s research is focused on the development of therapeutic nanoparticle technologies and the high-throughput combinatorial nanoengineering of multifunctional nanoparticles for medical applications.

Nazila Kamaly is an instructor in the Laboratory of Nanomedicine and Biomaterials at Harvard Medical School (HMS) and Brigham and Women’s Hospital (BWH). She received her combined Bachelor’s and Master’s degrees in medicinal chemistry from University College London and completed her Ph.D. on the development of theranostic nanoparticles at the Department of Chemistry at Imperial College London in 2008. She was a postdoctoral fellow at HMS/BWH and Imperial College London prior to her current position. Her research is focused on the development of multifunctional nanoparticles for therapeutic and imaging applications in a number of diseases. She will be taking up an Associate Professor (Seniorforsker) position in the Department of Nanotech at the Danish Technical University in Denmark next.

Basit Yameen is an instructor in the Laboratory of Nanomedicine and Biomaterials at Harvard Medical School (HMS) and Brigham and Women’s Hospital (BWH). He received his Ph.D. in chemistry from Johannes Gutenberg University of Mainz for his research work on smart materials conducted at the Max Planck Institute for Polymer Research in Mainz, Germany, in 2008. He was then an Alexander von Humboldt Postdoctoral Research Fellow in the Institute for Chemical Technology and Polymer Chemistry at the Karlsruhe Institute of Technology (KIT) in Germany before joining the Laboratory of Nanomedicine and Biomaterials at Harvard Medical School and Brigham and Women’s Hospital. He is currently actively involved in the development of stimuli-responsive payload release and disease-targeted nanoparticles for theranostic applications.

Jun Wu is an instructor in the Laboratory of Nanomedicine and Biomaterials at Harvard Medical School (HMS) and Brigham and Women’s Hospital (BWH). He received his B.S. degree in chemistry from Nanjing University and his M.S. degree in chemistry from Stony Brook University. In 2011, he obtained his Ph.D. degree in biomedical engineering from Cornell University. His research is interdisciplinary and combines polymer chemistry, biomaterials, nanotechnology, drug delivery, and tissue engineering for the development of novel amino acid-based functional polymers and nanostructures for drug delivery and tissue-engineering applications. He will be taking up a Professorship position in the School of Engineering at Sun Yat-Sen University in China next.
Footnotes
Notes
The authors declare the following competing financial interest(s): Dr. Farokhzad declares financial interests in BIND Therapeutics, Selecta Biosciences, Tarveda Therapeutics and Placon Therapeutics, four biotechnology companies developing nanoparticle technologies for medical applications. All other authors declare no conflict of interest.
References
- 1.Cullins VE. Injectable and implantable contraceptives. Curr Opin Obstet Gynecol. 1992;4:536–543. [PubMed] [Google Scholar]
- 2.Langer R. Implantable controlled release systems. Pharmacol Ther. 1983;21:35–51. doi: 10.1016/0163-7258(83)90066-9. [DOI] [PubMed] [Google Scholar]
- 3.Zaffaroni A. Systems for controlled drug delivery. Med Res Rev. 1981;1:373–386. doi: 10.1002/med.2610010404. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Controlled Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J, Long DM. The Use of Silicone Rubber as a Carrier for Prolonged Drug Therapy. J Surg Res. 1964;4:139–142. doi: 10.1016/s0022-4804(64)80040-8. [DOI] [PubMed] [Google Scholar]
- 6.Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi T, Kuramoto R. Study of possible complex formation between macromolecules and certain pharmaceuticals. I. Polyvinylpyrrolidone with sulfathiazole, procaine hydrochloride, sodium salicylate, benzyl penicillin, chloramphenicol, mandelic acid, caffeine, theophylline, and cortisone. J Am Pharm Assoc Sci Ed. 1954;43:393–397. [PubMed] [Google Scholar]
- 8.Theeuwes F, Hussain A, Higuchi T. Quantitative analytical method for determination of drugs dispersed in polymers using differential scanning calorimetry. J Pharm Sci. 1974;63:427–429. doi: 10.1002/jps.2600630325. [DOI] [PubMed] [Google Scholar]
- 9.Roseman TJ, Higuchi WI. Release of medroxyprogesterone acetate from a silicone polymer. J Pharm Sci. 1970;59:353–357. doi: 10.1002/jps.2600590317. [DOI] [PubMed] [Google Scholar]
- 10.Roseman TJ. Release of steroids from a silicone polymer. J Pharm Sci. 1972;61:46–50. doi: 10.1002/jps.2600610106. [DOI] [PubMed] [Google Scholar]
- 11.Peppas NA, Merrill EW. Development of semicrystalline poly(vinyl alcohol) hydrogels for biomedical applications. J Biomed Mater Res. 1977;11:423–434. doi: 10.1002/jbm.820110309. [DOI] [PubMed] [Google Scholar]
- 12.Langer RS, Peppas NA. Present and future applications of biomaterials in controlled drug delivery systems. Biomaterials. 1981;2:201–214. doi: 10.1016/0142-9612(81)90059-4. [DOI] [PubMed] [Google Scholar]
- 13.Heller J. Controlled release of biologically active compounds from bioerodible polymers. Biomaterials. 1980;1:51–57. doi: 10.1016/0142-9612(80)90060-5. [DOI] [PubMed] [Google Scholar]
- 14.Heller J, Himmelstein KJ. Poly(ortho ester) biodegradable polymer systems. Methods Enzymol. 1985;112:422–436. doi: 10.1016/s0076-6879(85)12033-1. [DOI] [PubMed] [Google Scholar]
- 15.Batz HG, Franzmann G, Ringsdorf H. Model reactions for synthesis of pharmacologically active polymers by way of monomeric and polymeric reactive esters. Angew Chem, Int Ed Engl. 1972;11:1103–1104. doi: 10.1002/anie.197211031. [DOI] [PubMed] [Google Scholar]
- 16.Przybylski M, Zaharko DS, Chirigos MA, Adamson RH, Schultz RM, Ringsdorf H. DIVEMA-methotrexate: Immune-adjuvant role of polymeric carriers linked to antitumor agents. Cancer Treat Rep. 1978;62:1837–1843. [PubMed] [Google Scholar]
- 17.Khanna SC, Jecklin T, Speiser P. Bead polymerization technique for sustained-release dosage form. J Pharm Sci. 1970;59:614–618. doi: 10.1002/jps.2600590508. [DOI] [PubMed] [Google Scholar]
- 18.Birrenbach G, Speiser PP. Polymerized micelles and their use as adjuvants in immunology. J Pharm Sci. 1976;65:1763–1766. doi: 10.1002/jps.2600651217. [DOI] [PubMed] [Google Scholar]
- 19.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41:2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-assembled targeted nanoparticles: evolution of technologies and bench to bedside translation. Acc Chem Res. 2011;44:1123–1134. doi: 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- 21.Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64(21):7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 22.Farokhzad OC, Khademhosseini A, Jon S, Hermmann A, Cheng J, Chin C, Kiselyuk A, Teply B, Eng G, Langer R. Microfluidic system for studying the interaction of nanoparticles and microparticles with cells. Anal Chem. 2005;77(17):5453–5459. doi: 10.1021/ac050312q. [DOI] [PubMed] [Google Scholar]
- 23.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hrkach J, Von Hoff D, Mukkaram AM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 26.Apfel CC, Zhang K, George E, Shi S, Jalota L, Hornuss C, Fero KE, Heidrich F, Pergolizzi JV, Cakmakkaya OS, Kranke P. Transdermal scopolamine for the prevention of postoperative nausea and vomiting: a systematic review and meta-analysis. Clin Ther. 2010;32:1987–2002. doi: 10.1016/j.clinthera.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Kendall MJ, Jack DB, Woods KL, Laugher SJ, Quarterman CP, John VA. Comparison of the pharmacodynamic and pharmacokinetic profiles of single and multiple doses of a commercial slow-release metoprolol formulation with a new Oros delivery system. Br J Clin Pharmacol. 1982;13:393–398. doi: 10.1111/j.1365-2125.1982.tb01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta BP, Thakur N, Jain NP, Banweer J, Jain S. Osmotically controlled drug delivery system with associated drugs. J Pharm Pharm Sci. 2010;13:571–588. doi: 10.18433/j38w25. [DOI] [PubMed] [Google Scholar]
- 29.Conte U, Maggi L. Modulation of the dissolution profiles from Geomatrix multi-layer matrix tablets containing drugs of different solubility. Biomaterials. 1996;17:889–896. doi: 10.1016/0142-9612(96)83284-4. [DOI] [PubMed] [Google Scholar]
- 30.Rosen H, Abribat T. The rise and rise of drug delivery. Nat Rev Drug Discovery. 2005;4:381–385. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 31.Brem H, Gabikian P. Biodegradable polymer implants to treat brain tumors. J Controlled Release. 2001;74:63–67. doi: 10.1016/s0168-3659(01)00311-x. [DOI] [PubMed] [Google Scholar]
- 32.Dang W, Daviau T, Brem H. Morphological characterization of polyanhydride biodegradable implant gliadel during in vitro and in vivo erosion using scanning electron microscopy. Pharm Res. 1996;13:683–691. doi: 10.1023/a:1016035229961. [DOI] [PubMed] [Google Scholar]
- 33.Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 34.Horton CE, Adamson JE, Mladick RA, Carraway JH. Vicryl synthetic absorbable sutures. Am Surg. 1974;40:729–731. [PubMed] [Google Scholar]
- 35.Grube E, Silber S, Hauptmann KE, Mueller R, Buellesfeld L, Gerckens U, Russell ME. TAXUS I: six- and twelve-month results from a randomized, double-blind trial on a slow-release paclitaxeleluting stent for de novo coronary lesions. Circulation. 2003;107:38–42. doi: 10.1161/01.cir.0000047700.58683.a1. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrini DO, Gomes VO, Lasevitch R, Smidt L, Azeredo MA, Ledur P, Bodanese R, Sinnott L, Moriguchi E, Caramori P. Efficacy and safety of drug-eluting stents in the real world: 8-year followup. Arq Bras Cardiol. 2014;103:174–182. doi: 10.5935/abc.20140110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Movahedi F, Hu RG, Becker DL, Xu C. Stimuli-responsive liposomes for the delivery of nucleic acid therapeutics. Nanomedicine. 2015;11:1575–1584. doi: 10.1016/j.nano.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Ta T, Porter TM. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J Controlled Release. 2013;169:112–125. doi: 10.1016/j.jconrel.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Xiao K, Zhu W, Deng W, Lam KS. Stimuli-responsive cross-linked micelles for on-demand drug delivery against cancers. Adv Drug Delivery Rev. 2014;66:58–73. doi: 10.1016/j.addr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen YC, Lo CL, Hsiue GH. Multifunctional nanomicellar systems for delivering anticancer drugs. J Biomed Mater Res Part A. 2014;102:2024–2038. doi: 10.1002/jbm.a.34850. [DOI] [PubMed] [Google Scholar]
- 41.Yang P, Gai S, Lin J. Functionalized mesoporous silica materials for controlled drug delivery. Chem Soc Rev. 2012;41:3679–3698. doi: 10.1039/c2cs15308d. [DOI] [PubMed] [Google Scholar]
- 42.Chandra S, Barick KC, Bahadur D. Oxide and hybrid nanostructures for therapeutic applications. Adv Drug Delivery Rev. 2011;63:1267–1281. doi: 10.1016/j.addr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Hola K, Markova Z, Zoppellaro G, Tucek J, Zboril R. Tailored functionalization of iron oxide nanoparticles for MRI, drug delivery, magnetic separation and immobilization of biosubstances. Biotechnol Adv. 2015;33:1162–1176. doi: 10.1016/j.biotechadv.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem Soc Rev. 2013;42:7289–7325. doi: 10.1039/c3cs60048c. [DOI] [PubMed] [Google Scholar]
- 45.Carregal-Romero S, Caballero-Diaz E, Beqa L, Abdelmonem AM, Ochs M, Huhn D, Suau BS, Valcarcel M, Parak WJ. Multiplexed sensing and imaging with colloidal nano- and microparticles. Annu Rev Anal Chem. 2013;6:53–81. doi: 10.1146/annurev-anchem-062012-092621. [DOI] [PubMed] [Google Scholar]
- 46.Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011;44:1029–1038. doi: 10.1021/ar200019c. [DOI] [PubMed] [Google Scholar]
- 47.Muthu MS, Leong DT, Mei L, Feng SS. Nanotheranostics – application and further development of nanomedicine strategies for advanced theranostics. Theranostics. 2014;4:660–677. doi: 10.7150/thno.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han N, Yang YY, Wang S, Zheng S, Fan W. Polymer-based cancer nanotheranostics: retrospectives of multi-functionalities and pharmacokinetics. Curr Drug Metab. 2013;14:661–674. doi: 10.2174/1389200211314060003. [DOI] [PubMed] [Google Scholar]
- 49.Kim TH, Lee S, Chen X. Nanotheranostics for personalized medicine. Expert Rev Mol Diagn. 2013;13:257–269. doi: 10.1586/erm.13.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mura S, Couvreur P. Nanotheranostics for personalized medicine. Adv Drug Delivery Rev. 2012;64:1394–1416. doi: 10.1016/j.addr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Goldenberg IS. Catgut, silk, and silver—The story of surgical sutures. Surgery. 1959;46:908–912. [PubMed] [Google Scholar]
- 52.The history and evolution of surgical sutures. SA Nurs J. 1968;35:21. [PubMed] [Google Scholar]
- 53.Muffly TM, Tizzano AP, Walters MD. The history and evolution of sutures in pelvic surgery. J R Soc Med. 2011;104:107–112. doi: 10.1258/jrsm.2010.100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimamoto T. Pharmaceutical aspects. Nasal and depot formulations of leuprolide. J Androl. 1987;8:S14–S16. [PubMed] [Google Scholar]
- 55.Toguchi H. Pharmaceutical manipulation of leuprorelin acetate to improve clinical performance. J Int Med Res. 1990;18:35–41. doi: 10.1177/03000605900180S107. [DOI] [PubMed] [Google Scholar]
- 56.Laufman H, Rubel T. Synthetic absorable sutures. Surg Gynecol Obstet. 1977;145:597–608. [PubMed] [Google Scholar]
- 57.De Leede LG, Humphries JE, Bechet AC, Van Hoogdalem EJ, Verrijk R, Spencer DG. Novel controlled-release Lemna-derived IFN-alpha2b (Locteron): pharmacokinetics, pharmacodynamics, and tolerability in a phase I clinical trial. J Interferon Cytokine Res. 2008;28:113–122. doi: 10.1089/jir.2007.0073. [DOI] [PubMed] [Google Scholar]
- 58.Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable block copolymers as injectable drug-delivery systems. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 59.Loh XJ, Scherman OA. Polymeric and Self Assembled Hydrogels: From Fundamental Understanding to Applications. Royal Society of Chemistry; Cambridge, UK: 2013. [Google Scholar]
- 60.Kulkarni RK, Moore EG, Hegyeli AF, Leonard F. Biodegradable poly(lactic acid) polymers. J Biomed Mater Res. 1971;5:169–181. doi: 10.1002/jbm.820050305. [DOI] [PubMed] [Google Scholar]
- 61.Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjugate Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 62.Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems–a review. Int J Pharm. 2011;415:34–52. doi: 10.1016/j.ijpharm.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 63.Valle JW, Armstrong A, Newman C, Alakhov V, Pietrzynski G, Brewer J, Campbell S, Corrie P, Rowinsky EK, Ranson M. A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Invest New Drugs. 2011;29:1029–1037. doi: 10.1007/s10637-010-9399-1. [DOI] [PubMed] [Google Scholar]
- 64.Vert M, Doi Y, Hellwich K-H, Hess M, Hodge P, Kubisa P, Rinaudo M, Schué F. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012) Pure Appl Chem. 2012;84:377–410. [Google Scholar]
- 65.Kim HJ, Kim KH, Yun J, Kim SH, Lee SC, Bae SB, Kim CK, Lee NS, Lee KT, Kim DJ, Park SK, Won JH, Hong DS, Park HS, Kim D-J. Phase II Clinical Trial of Genexol(R) (Paclitaxel) and Carboplatin for Patients with Advanced Non-small Cell Lung Cancer. Cancer Res Treat. 2011;43:19–23. doi: 10.4143/crt.2011.43.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boddy AV, Plummer ER, Todd R, Sludden J, Griffin M, Robson L, Cassidy J, Bissett D, Bernareggi A, Verrill MW, Calvert AH. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin Cancer Res. 2005;11:7834–7840. doi: 10.1158/1078-0432.CCR-05-0803. [DOI] [PubMed] [Google Scholar]
- 67.Galic VL, Herzog TJ, Wright JD, Lewin SN. Paclitaxel poliglumex for ovarian cancer. Expert Opin Invest Drugs. 2011;20:813–821. doi: 10.1517/13543784.2011.576666. [DOI] [PubMed] [Google Scholar]
- 68.Schluep T, Hwang J, Cheng J, Heidel JD, Bartlett DW, Hollister B, Davis ME. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 69.Davis ME. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv Drug Delivery Rev. 2009;61:1189–1192. doi: 10.1016/j.addr.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 70.Conley SJ, Baker TL, Burnett JP, Theisen RL, Lazarus D, Peters CG, Clouthier SG, Eliasof S, Wicha MS. CRLX101, an nvestigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res Treat. 2015;150:559–567. doi: 10.1007/s10549-015-3349-8. [DOI] [PubMed] [Google Scholar]
- 71.Eliasof S, Lazarus D, Peters CG, Case RI, Cole RO, Hwang J, Schluep T, Chao J, Lin J, Yen Y, Han H, Wiley DT, Zuckerman JE, Davis ME. Correlating preclinical animal studies and human clinical trials of a multifunctional, polymeric nanoparticle. Proc Natl Acad Sci U S A. 2013;110:15127–15132. doi: 10.1073/pnas.1309566110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weiss GJ, Chao J, Neidhart JD, Ramanathan RK, Bassett D, Neidhart JA, Choi CH, Chow W, Chung V, Forman SJ, Garmey E, Hwang J, Kalinoski DL, Koczywas M, Longmate J, Melton RJ, Morgan R, Oliver J, Peterkin JJ, Ryan JL, Schluep T, Synold TW, Twardowski P, Davis ME, Yen Y. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Invest New Drugs. 2013;31:986–1000. doi: 10.1007/s10637-012-9921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pham E, Birrer MJ, Eliasof S, Garmey EG, Lazarus D, Lee CR, Man S, Matulonis UA, Peters CG, Xu P, Krasner C, Kerbel RS. Translational impact of nanoparticle-drug conjugate CRLX101 with or without bevacizumab in advanced ovarian cancer. Clin Cancer Res. 2015;21:808–818. doi: 10.1158/1078-0432.CCR-14-2810. [DOI] [PubMed] [Google Scholar]
- 74.www.bindbio.com.
- 75.Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. Int J Nanomed. 2014;9:467–483. doi: 10.2147/IJN.S36654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bartlett DW, Davis ME. Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjugate Chem. 2007;18:456–468. doi: 10.1021/bc0603539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.www.selectabio.com.
- 78.Ilyinskii PO, Roy CJ, O’Neil CP, Browning EA, Pittet LA, Altreuter DH, Alexis F, Tonti E, Shi J, Basto PA, Iannacone M, Radovic-Moreno AF, Langer RS, Farokhzad OC, von Andrian UH, Johnston LP, Kishimoto TK. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014;32:2882–2895. doi: 10.1016/j.vaccine.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marin E, Briceno MI, Caballero-George C. Critical evaluation of biodegradable polymers used in nanodrugs. Int J Nanomed. 2013;8:3071–3090. doi: 10.2147/IJN.S47186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Allcock HR, Lampe FW. Contemporary Polymer Chemistry. Prentice-Hall; Englewood Cliffs, NJ: 1981. [Google Scholar]
- 81.Cowie JMG, Arrighi V. Polymers: Chemistry and Physics of Modern Materials. CRC Press; Boca Raton, FL: 2008. [Google Scholar]
- 82.Atkins PW, De Paula J. Atkins’ Physical Chemistry. Oxford University Press; Oxford: 2010. [Google Scholar]
- 83.Young RJ, Lovell PA. Introduction to Polymers. CRC Press; Boca Raton, FL: 2011. [Google Scholar]
- 84.Stepto RFT. Dispersity in Polymer Science. Pure Appl Chem. 2009;81:351–353. [Google Scholar]
- 85.Li H, Hardy RJ, Gu X. Effect of drug solubility on polymer hydration and drug dissolution from polyethylene oxide (PEO) matrix tablets. AAPS PharmSciTech. 2008;9:437–443. doi: 10.1208/s12249-008-9060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12:1–99. doi: 10.1615/critrevtherdrugcarriersyst.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- 87.Asano M, Yoshida M, Omichi H, Mashimo T, Okabe K, Yuasa H, Yamanaka H, Morimoto S, Sakakibara H. Biodegradable poly(DL-lactic acid) formulations in a calcitonin delivery system. Biomaterials. 1993;14:797–799. doi: 10.1016/0142-9612(93)90047-6. [DOI] [PubMed] [Google Scholar]
- 88.Ulery BD, Nair LS, Laurencin CT. Biomedical Applications of Biodegradable Polymers. J Polym Sci, Part B: Polym Phys. 2011;49:832–864. doi: 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Steendam R, van Steenbergen MJ, Hennink WE, Frijlink HW, Lerk CF. Effect of molecular weight and glass transition on relaxation and release behaviour of poly(DL-lactic acid) tablets. J Controlled Release. 2001;70:71–82. doi: 10.1016/s0168-3659(00)00342-4. [DOI] [PubMed] [Google Scholar]
- 90.Ramkissoon-Ganorkar C, Liu F, Baudys M, Kim SW. Effect of molecular weight and polydispersity on kinetics of dissolution and release from ph/temperature-sensitive polymers. J Biomater Sci, Polym Ed. 1999;10:1149–1161. doi: 10.1163/156856299x00739. [DOI] [PubMed] [Google Scholar]
- 91.Yang D, Liu X, Jiang X, Liu Y, Ying W, Wang H, Bai H, Taylor WD, Wang Y, Clamme JP, Co E, Chivukula P, Tsang KY, Jin Y, Yu L. Effect of molecular weight of PGG-paclitaxel conjugates on in vitro and in vivo efficacy. J Controlled Release. 2012;161:124–131. doi: 10.1016/j.jconrel.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 92.Zhang J, Chen XG, Sun GZ, Huang L, Cheng XJ. Effect of molecular weight on the oleoyl-chitosan nanoparticles as carriers for doxorubicin. Colloids Surf B. 2010;77:125–130. doi: 10.1016/j.colsurfb.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 93.Li J, Rothstein SN, Little SR, Edenborn HM, Meyer TY. The effect of monomer order on the hydrolysis of biodegradable poly(lactic-co-glycolic acid) repeating sequence copolymers. J Am Chem Soc. 2012;134:16352–16359. doi: 10.1021/ja306866w. [DOI] [PubMed] [Google Scholar]
- 94.Bat E, Zhang Z, Feijen J, Grijpma DW, Poot AA. Biodegradable elastomers for biomedical applications and regenerative medicine. Regener Med. 2014;9:385–398. doi: 10.2217/rme.14.4. [DOI] [PubMed] [Google Scholar]
- 95.Garg S, Bourantas C, Serruys PW. New concepts in the design of drug-eluting coronary stents. Nat Rev Cardiol. 2013;10:248–260. doi: 10.1038/nrcardio.2013.13. [DOI] [PubMed] [Google Scholar]
- 96.Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev. 2013;42:1147–1235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]
- 97.Champion JA, Katare YK, Mitragotri S. Particle shape: a new design parameter for micro- and nanoscale drug delivery carriers. J Controlled Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers. 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Odian GG. Principles of Polymerization. Wiley; New York: 1991. [Google Scholar]
- 100.Ehrenstein GW. Polymeric Materials: Structure–Properties–Applications. Hanser; Munich: 2001. [Google Scholar]
- 101.Müller WEG, Wang XPD, Schröder HC. Biomedical Inorganic Polymers: Bioactivity and Applications of Natural and Synthetic Polymeric Inorganic Molecules. Springer Science & Business Media; Berlin: 2014. [Google Scholar]
- 102.Karavelidis V, Karavas E, Giliopoulos D, Papadimitriou S, Bikiaris D. Evaluating the effects of crystallinity in new biocompatible polyester nanocarriers on drug release behavior. Int J Nanomed. 2011;6:3021–3032. doi: 10.2147/IJN.S26016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zilberman M. Dexamethasone loaded bioresorbable films used in medical support devices: structure, degradation, crystallinity and drug release. Acta Biomater. 2005;1:615–624. doi: 10.1016/j.actbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 104.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Polymers for drug delivery systems. Annu Rev Chem Biomol Eng. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design, and performance. Crit Rev Ther Drug Carrier Syst. 2004;21:133–193. doi: 10.1615/critrevtherdrugcarriersyst.v21.i3.10. [DOI] [PubMed] [Google Scholar]
- 106.Xu Q, Chin SE, Wang CH, Pack DW. Mechanism of drug release from double-walled PDLLA(PLGA) microspheres. Biomaterials. 2013;34:3902–3911. doi: 10.1016/j.biomaterials.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Omelczuk MO, McGinity JW. The influence of polymer glass transition temperature and molecular weight on drug release from tablets containing poly(DL-lactic acid) Pharm Res. 1992;9:26–32. doi: 10.1023/a:1018967424392. [DOI] [PubMed] [Google Scholar]
- 108.Robinson JR, Lee VHL. Controlled Drug Delivery: Fundamentals and Applications. Marcel Dekker; New York: 1987. [Google Scholar]
- 109.Schliecker G, Schmidt C, Fuchs S, Wombacher R, Kissel T. Hydrolytic degradation of poly(lactide-co-glycolide) films: effect of oligomers on degradation rate and crystallinity. Int J Pharm. 2003;266:39–49. doi: 10.1016/s0378-5173(03)00379-x. [DOI] [PubMed] [Google Scholar]
- 110.Dumitriu S. Polymeric Biomaterials. Marcel Dekker; New York: 2002. [Google Scholar]
- 111.Chin WS, Sow CH, Wee ATS. Science at the Nanoscale: An Introductory Textbook. Pan Stanford; Singapore: 2010. [Google Scholar]
- 112.Lu L, Peter SJ, Lyman MD, Lai HL, Leite SM, Tamada JA, Uyama S, Vacanti JP, Langer R, Mikos AG. In vitro and in vivo degradation of porous poly(DL-lactic-co-glycolic acid) foams. Biomaterials. 2000;21:1837–1845. doi: 10.1016/s0142-9612(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 113.Park TG. Degradation of poly(lactic-co-glycolic acid) microspheres: effect of copolymer composition. Biomaterials. 1995;16:1123–1130. doi: 10.1016/0142-9612(95)93575-x. [DOI] [PubMed] [Google Scholar]
- 114.Kim JM, Seo KS, Jeong YK, Lee HB, Kim YS, Khang G. Co-effect of aqueous solubility of drugs and glycolide monomer on in vitro release rates from poly(D,L-lactide-co-glycolide) discs and polymer degradation. J Biomater Sci, Polym Ed. 2005;16:991–1007. doi: 10.1163/1568562054414676. [DOI] [PubMed] [Google Scholar]
- 115.Zugasti ME, Zornoza A, del Mar Goni M, Isasi JR, Velaz I, Martin C, Sanchez M, Martinez-Oharriz MC. Influence of soluble and insoluble cyclodextrin polymers on drug release from hydroxypropyl methylcellulose tablets. Drug Dev Ind Pharm. 2009;35:1264–1270. doi: 10.1080/03639040902882306. [DOI] [PubMed] [Google Scholar]
- 116.Knopp MM, Olesen NE, Holm P, Langguth P, Holm R, Rades T. Influence of Polymer Molecular Weight on Drug-Polymer Solubility: A Comparison between Experimentally Determined Solubility in PVP and Prediction Derived from Solubility in Monomer. J Pharm Sci. 2015;104:2905–2912. doi: 10.1002/jps.24410. [DOI] [PubMed] [Google Scholar]
- 117.Valencia PM, Pridgen EM, Rhee M, Langer R, Farokhzad OC, Karnik R. Microfluidic platform for combinatorial synthesis and optimization of targeted nanoparticles for cancer therapy. ACS Nano. 2013;7:10671–10680. doi: 10.1021/nn403370e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim H, Fassihi R. Application of binary polymer system in drug release rate modulation. 2. Influence of formulation variables and hydrodynamic conditions on release kinetics. J Pharm Sci. 1997;86:323–328. doi: 10.1021/js960307p. [DOI] [PubMed] [Google Scholar]
- 119.Saltzman WM. Drug Delivery: Engineering Principles for Drug Therapy. Oxford University Press; Oxford: 2001. [Google Scholar]
- 120.Mittal G, Sahana DK, Bhardwaj V, Ravi Kumar MN. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Controlled Release. 2007;119:77–85. doi: 10.1016/j.jconrel.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 121.Wong W, Mooney D. Synthetic Biodegradable Polymer Scaffolds. Birkhauser; Boston: 1997. pp. 51–82. [Google Scholar]
- 122.Lanza R, Langer R, Chick W. Principles of Tissue Engineering. R. G. Landes; Austin, TX: 1997. [Google Scholar]
- 123.Hollinger JO. Biomedical Applications of Synthetic Biodegradable Polymers. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- 124.Shalaby SW. Biomedical Polymers: Designed-to-Degrade Systems. Hanser; Cincinnati, OH: 1994. [Google Scholar]
- 125.Singh L, Kumar V, Ratner BD. Generation of porous microcellular 85/15 poly (DL-lactide-co-glycolide) foams for biomedical application. Biomaterials. 2004;25:2611–2617. doi: 10.1016/j.biomaterials.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 126.Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci, Polym Ed. 2006;17:247–289. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 127.Fu Y, Kao WJ. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin Drug Delivery. 2010;7:429–444. doi: 10.1517/17425241003602259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bazile DV, Ropert C, Huve P, Verrecchia T, Mariard M, Frydman A, Veillard M, Spenlehauer G. Body distribution of fully biodegradable [14C]-poly(lactic acid) nanoparticles coated with albumin after parenteral administration to rats. Biomaterials. 1992;13:1093–1102. doi: 10.1016/0142-9612(92)90142-b. [DOI] [PubMed] [Google Scholar]
- 129.Saffer EM, Tew GN, Bhatia SR. Poly(lactic acid)-poly(ethylene oxide) block copolymers: new directions in self-assembly and biomedical applications. Curr Med Chem. 2011;18:5676–5686. doi: 10.2174/092986711798347324. [DOI] [PubMed] [Google Scholar]
- 130.Kumar N, Ravikumar MN, Domb AJ. Biodegradable block copolymers. Adv Drug Delivery Rev. 2001;53:23–44. doi: 10.1016/s0169-409x(01)00219-8. [DOI] [PubMed] [Google Scholar]
- 131.Gref R, Minamitake Y, Peracchia MT, Domb A, Trubetskoy V, Torchilin V, Langer R. Poly(ethylene glycol)-coated nanospheres: potential carriers for intravenous drug administration. Pharm Biotechnol. 2002;10:167–198. doi: 10.1007/0-306-46803-4_6. [DOI] [PubMed] [Google Scholar]
- 132.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 133.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem, Int Ed. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 134.Torchilin VP. Polymer-coated long-circulating microparticulate pharmaceuticals. J Microencapsulation. 1998;15:1–19. doi: 10.3109/02652049809006831. [DOI] [PubMed] [Google Scholar]
- 135.Torchilin VP, Omelyanenko VG, Papisov MI, Bogdanov AA, Jr, Trubetskoy VS, Herron JN, Gentry CA. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta, Biomembr. 1994;1195:11–20. doi: 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 136.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kissel T, Li Y, Unger F. ABA-triblock copolymers from biodegradable polyester A-blocks and hydrophilic poly(ethylene oxide) B-blocks as a candidate for in situ forming hydrogel delivery systems for proteins. Adv Drug Delivery Rev. 2002;54:99–134. doi: 10.1016/s0169-409x(01)00244-7. [DOI] [PubMed] [Google Scholar]
- 138.Ghahremankhani AA, Dorkoosh F, Dinarvand R. PLGA-PEG-PLGA tri-block copolymers as in situ gel-forming peptide delivery system: effect of formulation properties on peptide release. Pharm Dev Technol. 2008;13:49–55. doi: 10.1080/10837450701702842. [DOI] [PubMed] [Google Scholar]
- 139.Jeong B, Bae YH, Kim SW. In situ gelation of PEG-PLGA-PEG triblock copolymer aqueous solutions and degradation thereof. J Biomed Mater Res. 2000;50:171–177. doi: 10.1002/(sici)1097-4636(200005)50:2<171::aid-jbm11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 140.Yang Q, Lai SK. Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7:655–677. doi: 10.1002/wnan.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schellekens H, Hennink WE, Brinks V. The immunogenicity of polyethylene glycol: facts and fiction. Pharm Res. 2013;30:1729–1734. doi: 10.1007/s11095-013-1067-7. [DOI] [PubMed] [Google Scholar]
- 142.Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Controlled Release. 2013;172:38–47. doi: 10.1016/j.jconrel.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 143.Dash TK, Konkimalla VB. Polymeric modification and its implication in drug delivery: poly-epsilon-caprolactone (PCL) as a model polymer. Mol Pharmaceutics. 2012;9:2365–2379. doi: 10.1021/mp3001952. [DOI] [PubMed] [Google Scholar]
- 144.Lu XL, Sun ZJ, Cai W, Gao ZY. Study on the shape memory effects of poly(L-lactide-co-epsilon-caprolactone) biodegradable polymers. J Mater Sci: Mater Med. 2008;19:395–399. doi: 10.1007/s10856-006-0100-3. [DOI] [PubMed] [Google Scholar]
- 145.Huang MH, Chou AH, Lien SP, Chen HW, Huang CY, Chen WW, Chong P, Liu SJ, Leng CH. Formulation and immunological evaluation of novel vaccine delivery systems based on bioresorbable poly(ethylene glycol)-block-poly(lactide-co-epsilon-cap-rolactone) J Biomed Mater Res, Part B. 2009;90B:832–841. doi: 10.1002/jbm.b.31352. [DOI] [PubMed] [Google Scholar]
- 146.Li Z, Tan BH. Towards the development of polycaprolactone based amphiphilic block copolymers: molecular design, self-assembly and biomedical applications. Mater Sci Eng C. 2014;45:620–634. doi: 10.1016/j.msec.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 147.Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem, Int Ed. 2004;43:6323–6327. doi: 10.1002/anie.200460800. [DOI] [PubMed] [Google Scholar]
- 148.Vauthier C, Dubernet C, Fattal E, Pinto-Alphandary H, Couvreur P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv Drug Delivery Rev. 2003;55:519–548. doi: 10.1016/s0169-409x(03)00041-3. [DOI] [PubMed] [Google Scholar]
- 149.Couvreur P, Kante B, Roland M, Guiot P, Bauduin P, Speiser P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: preparation, morphological and sorptive properties. J Pharm Pharmacol. 1979;31:331–332. doi: 10.1111/j.2042-7158.1979.tb13510.x. [DOI] [PubMed] [Google Scholar]
- 150.Graf A, McDowell A, Rades T. Poly(alkylcyanoacrylate) nanoparticles for enhanced delivery of therapeutics – is there real potential? Expert Opin Drug Delivery. 2009;6:371–387. doi: 10.1517/17425240902870413. [DOI] [PubMed] [Google Scholar]
- 151.Nicolas J, Couvreur P. Synthesis of poly(alkyl cyanoacrylate)-based colloidal nanomedicines. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2009;1:111–127. doi: 10.1002/wnan.15. [DOI] [PubMed] [Google Scholar]
- 152.Reddy LH, Couvreur P. Nanotechnology for therapy and imaging of liver diseases. J Hepatol. 2011;55:1461–1466. doi: 10.1016/j.jhep.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 153.Heller J, Barr J. Poly(ortho esters)—From concept to reality. Biomacromolecules. 2004;5:1625–1632. doi: 10.1021/bm040049n. [DOI] [PubMed] [Google Scholar]
- 154.Heller J, Sparer R, Zentner G, Chasin M, Langer R. Biodegradable Polymers as Drug Delivery Systems. Marcel Dekker; New York: 1990. [Google Scholar]
- 155.Seymour LW, Duncan R, Duffy J, Ng S, Heller J. Poly (ortho ester) matrices for controlled release of the antitumour agent 5-fluorouracil. J Controlled Release. 1994;31:201. [Google Scholar]
- 156.Roskos K, Fritzinger B, Rao S, Armitage G, Heller J. Development of a drug delivery system for the treatment of periodontal disease based on bioerodible poly (ortho esters) Biomaterials. 1995;16:313. doi: 10.1016/0142-9612(95)93259-g. [DOI] [PubMed] [Google Scholar]
- 157.Heller J, Barr J, Ng SY, Shen HR, Schwach-Abdellaoui K, Einmahl S, Rothen-Weinhold A, Gurny R. Poly(ortho esters) – their development and some recent applications. Eur J Pharm Biopharm. 2000;50:121–128. doi: 10.1016/s0939-6411(00)00085-0. [DOI] [PubMed] [Google Scholar]
- 158.Heller J, Barr J, Ng SY, Abdellauoi KS, Gurny R. Poly(ortho esters): synthesis, characterization, properties and uses. Adv Drug Delivery Rev. 2002;54:1015–1039. doi: 10.1016/s0169-409x(02)00055-8. [DOI] [PubMed] [Google Scholar]
- 159.Qi M, Li X, Yang Y, Zhou S. Electrospun fibers of acid-labile biodegradable polymers containing ortho ester groups for controlled release of paracetamol. Eur J Pharm Biopharm. 2008;70:445–452. doi: 10.1016/j.ejpb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 160.Shalaby SW, Allen AL. Biomedical Polymers: Designed-to-Degrade Systems. Hanser; Munich: 1994. [Google Scholar]
- 161.Park ES, Maniar M, Shah JC. Biodegradable polyanhydride devices of cefazolin sodium, bupivacaine, and taxol for local drug delivery: preparation, and kinetics and mechanism of in vitro release. J Controlled Release. 1998;52:179–189. doi: 10.1016/s0168-3659(97)00223-x. [DOI] [PubMed] [Google Scholar]
- 162.Leong KW, Brott BC, Langer R. Bioerodible polyanhydrides as drug-carrier matrices. I: Characterization, degradation, and release characteristics. J Biomed Mater Res. 1985;19:941–955. doi: 10.1002/jbm.820190806. [DOI] [PubMed] [Google Scholar]
- 163.Manoharan C, Singh J. Evaluation of polyanhydride microspheres for basal insulin delivery: Effect of copolymer composition and zinc salt on encapsulation, in vitro release, stability, in vivo absorption and bioactivity in diabetic rats. J Pharm Sci. 2009;98:4237–4250. doi: 10.1002/jps.21741. [DOI] [PubMed] [Google Scholar]
- 164.Kumar N, Langer RS, Domb AJ. Polyanhydrides: an overview. Adv Drug Delivery Rev. 2002;54:889–910. doi: 10.1016/s0169-409x(02)00050-9. [DOI] [PubMed] [Google Scholar]
- 165.Petersen LK, Huntimer L, Walz K, Ramer-Tait A, Wannemuehler MJ, Narasimhan B. Combinatorial evaluation of in vivo distribution of polyanhydride particle-based platforms for vaccine delivery. Int J Nanomed. 2013;8:2213–2225. doi: 10.2147/IJN.S45317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Petersen LK, Sackett CK, Narasimhan B. High-throughput analysis of protein stability in polyanhydride nanoparticles. Acta Biomater. 2010;6:3873–3881. doi: 10.1016/j.actbio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 167.Lesniak MS, Upadhyay U, Goodwin R, Tyler B, Brem H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005;25:3825–3831. [PMC free article] [PubMed] [Google Scholar]
- 168.Pfeifer BA, Burdick JA, Langer R. Formulation and surface modification of poly(ester-anhydride) micro- and nanospheres. Biomaterials. 2005;26:117–124. doi: 10.1016/j.biomaterials.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 169.Nathan A, Kohn J. Amino Acid Derived Polymers. Hanser; New York: 1994. [Google Scholar]
- 170.Anderson JM, Gibbons DF, Martin RL, Hiltner A, Woods R. The potential for poly-alpha-amino acids as biomaterials. J Biomed Mater Res. 1974;8:197–207. doi: 10.1002/jbm.820080320. [DOI] [PubMed] [Google Scholar]
- 171.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 172.Marck KW, Wildevuur CH, Sederel WL, Bantjes A, Feijen J. Biodegradability and tissue reaction of random copolymers of L-leucine, L-aspartic acid, and L-aspartic acid esters. J Biomed Mater Res. 1977;11:405–422. doi: 10.1002/jbm.820110308. [DOI] [PubMed] [Google Scholar]
- 173.Martin EC, May PD, McMahon WA. Amino acid polymers for biomedical applications. I. Permeability properties of L-leucine DL-methionine copolymers. J Biomed Mater Res. 1971;5:53–62. doi: 10.1002/jbm.820050105. [DOI] [PubMed] [Google Scholar]
- 174.Shih IL, Van YT, Shen MH. Biomedical applications of chemically and microbiologically synthesized poly(glutamic acid) and poly(lysine) Mini-Rev Med Chem. 2004;4:179–188. doi: 10.2174/1389557043487420. [DOI] [PubMed] [Google Scholar]
- 175.Lalatsa A, Schatzlein AG, Mazza M, Le TB, Uchegbu IF. Amphiphilic poly(L-amino acids) – new materials for drug delivery. J Controlled Release. 2012;161:523–536. doi: 10.1016/j.jconrel.2012.04.046. [DOI] [PubMed] [Google Scholar]
- 176.Dey RK, Ray AR. Synthesis, characterization, and blood compatibility of polyadmidoamines copolymers. Biomaterials. 2003;24:2985–2993. doi: 10.1016/s0142-9612(03)00122-4. [DOI] [PubMed] [Google Scholar]
- 177.Manocha B, Margaritis A. Production and characterization of gamma-polyglutamic acid nanoparticles for controlled anticancer drug release. Crit Rev Biotechnol. 2008;28:83–99. doi: 10.1080/07388550802107483. [DOI] [PubMed] [Google Scholar]
- 178.Seth A, Heo MB, Lim YT. Poly (gamma-glutamic acid) based combination of water-insoluble paclitaxel and TLR7 agonist for chemo-immunotherapy. Biomaterials. 2014;35:7992–8001. doi: 10.1016/j.biomaterials.2014.05.076. [DOI] [PubMed] [Google Scholar]
- 179.Lollo G, Rivera-Rodriguez GR, Bejaud J, Montier T, Passirani C, Benoit JP, Garcia-Fuentes M, Alonso MJ, Torres D. Polyglutamic acid-PEG nanocapsules as long circulating carriers for the delivery of docetaxel. Eur J Pharm Biopharm. 2014;87:47–54. doi: 10.1016/j.ejpb.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 180.Zhu Y, Akagi T, Akashi M. Self-assembling stereocomplex nanoparticles by enantiomeric poly(gamma-glutamic acid)-poly(lactide) graft copolymers as a protein delivery carrier. Macromol Biosci. 2014;14:576–587. doi: 10.1002/mabi.201300434. [DOI] [PubMed] [Google Scholar]
- 181.Meng L, Ji B, Huang W, Wang D, Tong G, Su Y, Zhu X, Yan D. Preparation of pixantrone/poly(gamma-glutamic acid) nanoparticles through complex self-assembly for oral chemotherapy. Macromol Biosci. 2012;12:1524–1533. doi: 10.1002/mabi.201200137. [DOI] [PubMed] [Google Scholar]
- 182.Tiera MJ, Shi Q, Winnik FM, Fernandes JC. Polycation-based gene therapy: current knowledge and new perspectives. Curr Gene Ther. 2011;11:288–306. doi: 10.2174/156652311796150408. [DOI] [PubMed] [Google Scholar]
- 183.Lochmann D, Jauk E, Zimmer A. Drug delivery of oligonucleotides by peptides. Eur J Pharm Biopharm. 2004;58:237–251. doi: 10.1016/j.ejpb.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 184.Huang SJ, Bansleben DA, Knox JR. Biodegradable polymers: Chymotrypsin degradation of a low molecular weight poly(ester-urea) containing phenylalanine. J Appl Polym Sci. 1979;23:429–437. [Google Scholar]
- 185.Katsarava R, Beridze V, Arabuli N, Kharadze D, Chu CC, Won CY. Amino acid-based bioanalogous polymers. synthesis, and study of regular poly(ester amide)s based on bis(alpha -amino acid) alpha, w-alkylene diesters, and aliphatic dicarboxylic acids. J Polym Sci, Part A: Polym Chem. 1999;37:391–407. [Google Scholar]
- 186.Guo K, Chu CC, Chkhaidze E, Katsarava R. Synthesis and characterization of novel biodegradable unsaturated poly(ester amide)s. J Polym Sci, Part A: Polym Chem. 2005;43:1463–1477. [Google Scholar]
- 187.Deng M, Wu J, Reinhart-King CA, Chu C-C. Synthesis and Characterization of Biodegradable Poly(ester amide)s with Pendant Amine Functional Groups and in Vitro Cellular Response. Biomacromolecules. 2009;10:3037–3047. doi: 10.1021/bm9006437. [DOI] [PubMed] [Google Scholar]
- 188.Diaz A, Katsarava R, Puiggali J. Synthesis, properties and applications of biodegradable polymers derived from diols and dicarboxylic acids: from polyesters to poly(ester amide)s. Int J Mol Sci. 2014;15:7064–7123. doi: 10.3390/ijms15057064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ghaffar A, Draaisma GJ, Mihov G, Dias AA, Schoenmakers PJ, van der Wal S. Monitoring the in vitro enzyme-mediated degradation of degradable poly(ester amide) for controlled drug delivery by LC-ToF-MS. Biomacromolecules. 2011;12:3243–3251. doi: 10.1021/bm200709r. [DOI] [PubMed] [Google Scholar]
- 190.Akinc A, Lynn DM, Anderson DG, Langer R. Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery. J Am Chem Soc. 2003;125:5316–5323. doi: 10.1021/ja034429c. [DOI] [PubMed] [Google Scholar]
- 191.Green JJ, Zugates GT, Langer R, Anderson DG. Poly(beta-amino esters): procedures for synthesis and gene delivery. Methods Mol Biol. 2009;480:53–63. doi: 10.1007/978-1-59745-429-2_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Siegwart DJ, Whitehead KA, Nuhn L, Sahay G, Cheng H, Jiang S, Ma M, Lytton-Jean A, Vegas A, Fenton P, Levins CG, Love KT, Lee H, Cortez C, Collins SP, Li YF, Jang J, Querbes W, Zurenko C, Novobrantseva T, Langer R, Anderson DG. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc Natl Acad Sci U S A. 2011;108:12996–13001. doi: 10.1073/pnas.1106379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Anderson DG, Lynn DM, Langer R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew Chem Int Ed. 2003;42:3153–3158. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 194.Lim YH, Heo GS, Rezenom YH, Pollack S, Raymond JE, Elsabahy M, Wooley KL. Development of a Vinyl Ether-Functionalized Polyphosphoester as a Template for Multiple Postpolymerization Conjugation Chemistries and Study of Core Degradable Polymeric Nanoparticles. Macromolecules. 2014;47:4634–4644. doi: 10.1021/ma402480a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mao HQ, Leong KW. Design of polyphosphoester-DNA nanoparticles for non-viral gene delivery. Adv Genet. 2005;53:275–306. [PubMed] [Google Scholar]
- 196.Zhang F, Zhang S, Pollack SF, Li R, Gonzalez AM, Fan J, Zou J, Leininger SE, Pavia-Sanders A, Johnson R, Nelson LD, Raymond JE, Elsabahy M, Hughes DM, Lenox MW, Gustafson TP, Wooley KL. Improving paclitaxel delivery: in vitro and in vivo characterization of PEGylated polyphosphoester-based nanocarriers. J Am Chem Soc. 2015;137:2056–2066. doi: 10.1021/ja512616s. [DOI] [PubMed] [Google Scholar]
- 197.Zhao Z, Wang J, Mao HQ, Leong KW. Polyphosphoesters in drug and gene delivery. Adv Drug Delivery Rev. 2003;55:483–499. doi: 10.1016/s0169-409x(03)00040-1. [DOI] [PubMed] [Google Scholar]
- 198.Mao HQ, Leong KW. Design of Polyphosphoester-DNA Nanoparticles for Non-Viral Gene Delivery. Adv Genet. 2005;53:275–306. [PubMed] [Google Scholar]
- 199.Liu J, Huang W, Pang Y, Zhu X, Zhou Y, Yan D. Hyperbranched polyphosphates for drug delivery application: design, synthesis, and in vitro evaluation. Biomacromolecules. 2010;11:1564–1570. doi: 10.1021/bm100188h. [DOI] [PubMed] [Google Scholar]
- 200.Wang YC, Tang LY, Sun TM, Li CH, Xiong MH, Wang Wang. Self-assembled micelles of biodegradable triblock copolymers based on poly(ethyl ethylene phosphate) and poly(-caprolactone) as drug carriers. Biomacromolecules. 2008;9:388–395. doi: 10.1021/bm700732g. [DOI] [PubMed] [Google Scholar]
- 201.Lim YH, Tiemann KM, Heo GS, Wagers PO, Rezenom YH, Zhang S, Zhang F, Youngs WJ, Hunstad DA, Wooley KL. Preparation and in vitro antimicrobial activity of silver-bearing degradable polymeric nanoparticles of polyphosphoester-block-poly(L-lactide) ACS Nano. 2015;9:1995–2008. doi: 10.1021/nn507046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Elsabahy M, Zhang S, Zhang F, Deng ZJ, Lim YH, Wang H, Parsamian P, Hammond PT, Wooley KL. Surface charges and shell crosslinks each play significant roles in mediating degradation, biofouling, cytotoxicity and immunotoxicity for polyphosphoester-based nanoparticles. Sci Rep. 2013;3:3313. doi: 10.1038/srep03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5:447–451. doi: 10.1016/s1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 204.Kean T, Roth S, Thanou M. Trimethylated chitosans as nonviral gene delivery vectors: cytotoxicity and transfection efficiency. J Controlled Release. 2005;103:643–653. doi: 10.1016/j.jconrel.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 205.Ren D, Yi H, Wang W, Ma X. The enzymatic degradation and swelling properties of chitosan matrices with different degrees of N-acetylation. Carbohydr Res. 2005;340:2403–2410. doi: 10.1016/j.carres.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 206.Fonte P, Araujo F, Silva C, Pereira C, Reis S, Santos HA, Sarmento B. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnol Adv. 2015;33:1342–1354. doi: 10.1016/j.biotechadv.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 207.Shukla SK, Mishra AK, Arotiba OA, Mamba BB. Chitosan-based nanomaterials: a state-of-the-art review. Int J Biol Macromol. 2013;59:46–58. doi: 10.1016/j.ijbiomac.2013.04.043. [DOI] [PubMed] [Google Scholar]
- 208.Ribeiro MP, Espiga A, Silva D, Baptista P, Henriques J, Ferreira C, Silva JC, Borges JP, Pires E, Chaves P, Correia IJ. Development of a new chitosan hydrogel for wound dressing. Wound Repair Regen. 2009;17:817–824. doi: 10.1111/j.1524-475X.2009.00538.x. [DOI] [PubMed] [Google Scholar]
- 209.Kim IY, Yoo MK, Seo JH, Park SS, Na HS, Lee HC, Kim SK, Cho CS. Evaluation of semi-interpenetrating polymer networks composed of chitosan and poloxamer for wound dressing application. Int J Pharm. 2007;341:35–43. doi: 10.1016/j.ijpharm.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 210.Molinaro R, Wolfram J, Federico C, Cilurzo F, Di Marzio L, Ventura CA, Carafa M, Celia C, Fresta M. Polyethylenimine and chitosan carriers for the delivery of RNA interference effectors. Expert Opin Drug Delivery. 2013;10:1653–1668. doi: 10.1517/17425247.2013.840286. [DOI] [PubMed] [Google Scholar]
- 211.Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S. Oral delivery of therapeutic proteins and peptides: a review on recent development. Drug Delivery. 2013;20:237–246. doi: 10.3109/10717544.2013.819611. [DOI] [PubMed] [Google Scholar]
- 212.Casettari L, Illum L. Chitosan in nasal delivery systems for therapeutic drugs. J Controlled Release. 2014;190:189–200. doi: 10.1016/j.jconrel.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 213.Benediktsdottir BE, Baldursson O, Masson M. Challenges in evaluation of chitosan and trimethylated chitosan (TMC) as mucosal permeation enhancers: From synthesis to in vitro application. J Controlled Release. 2014;173:18–31. [PubMed] [Google Scholar]
- 214.Chen Z, Zhang L, Song Y, He J, Wu L, Zhao C, Xiao Y, Li W, Cai B, Cheng H, Li W. Hierarchical targeted hepatocyte mitochondrial multifunctional chitosan nanoparticles for anticancer drug delivery. Biomaterials. 2015;52:240–250. doi: 10.1016/j.biomaterials.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 215.Du H, Yang X, Zhai G. Design of chitosan-based nanoformulations for efficient intracellular release of active compounds. Nanomedicine. 2014;9:723–740. doi: 10.2217/nnm.14.8. [DOI] [PubMed] [Google Scholar]
- 216.Hu L, Sun Y, Wu Y. Advances in chitosan-based drug delivery vehicles. Nanoscale. 2013;5:3103–3111. doi: 10.1039/c3nr00338h. [DOI] [PubMed] [Google Scholar]
- 217.Luo Y, Wang Q. Recent development of chitosan-based polyelectrolyte complexes with natural polysaccharides for drug delivery. Int J Biol Macromol. 2014;64:353–367. doi: 10.1016/j.ijbiomac.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 218.Ragelle H, Vandermeulen G, Preat V. Chitosan-based siRNA delivery systems. J Controlled Release. 2013;172:207–218. doi: 10.1016/j.jconrel.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 219.Chen MC, Mi FL, Liao ZX, Hsiao CW, Sonaje K, Chung MF, Hsu LW, Sung HW. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv Drug Delivery Rev. 2013;65:865–879. doi: 10.1016/j.addr.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 220.Kogan G, Soltes L, Stern R, Gemeiner P. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett. 2007;29:17–25. doi: 10.1007/s10529-006-9219-z. [DOI] [PubMed] [Google Scholar]
- 221.Sudha PN, Rose MH. Beneficial effects of hyaluronic acid. Adv Food Nutr Res. 2014;72:137–176. doi: 10.1016/B978-0-12-800269-8.00009-9. [DOI] [PubMed] [Google Scholar]
- 222.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, Lee MY, Hoffman AS, Hahn SK. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Controlled Release. 2010;141:2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 223.Chen B, Miller RJ, Dhal PK. Hyaluronic acid-based drug conjugates: state-of-the-art and perspectives. J Biomed Nanotechnol. 2014;10:4–16. doi: 10.1166/jbn.2014.1781. [DOI] [PubMed] [Google Scholar]
- 224.Laurent TC, Fraser JR. Hyaluronan. FASEB J. 1992;6:2397–2404. [PubMed] [Google Scholar]
- 225.Ghosh SC, Neslihan Alpay S, Klostergaard J. CD44: a validated target for improved delivery of cancer therapeutics. Expert Opin Ther Targets. 2012;16:635–650. doi: 10.1517/14728222.2012.687374. [DOI] [PubMed] [Google Scholar]
- 226.Skandalis SS, Gialeli C, Theocharis AD, Karamanos NK. Advances and advantages of nanomedicine in the pharmacological targeting of hyaluronan-CD44 interactions and signaling in cancer. Adv Cancer Res. 2014;123:277–317. doi: 10.1016/B978-0-12-800092-2.00011-3. [DOI] [PubMed] [Google Scholar]
- 227.Folkman J, Long DM, Jr, Rosenbaum R. Silicone rubber: a new diffusion property useful for general anesthesia. Science. 1966;154:148–149. doi: 10.1126/science.154.3745.148. [DOI] [PubMed] [Google Scholar]
- 228.Kearney CJ, Mooney DJ. Macroscale delivery systems for molecular and cellular payloads. Nat Mater. 2013;12:1004–1017. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 229.Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364:328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 230.Second-generation long-acting injectable antipsychotic agents: an overview. DTB. 2012;50:102–105. doi: 10.1136/dtb.2012.08.0127. [DOI] [PubMed] [Google Scholar]
- 231.Swainston Harrison T, Goa KL. Long-acting risperidone: a review of its use in schizophrenia. CNS Drugs. 2004;18(2):113–132. doi: 10.2165/00023210-200418020-00005. [DOI] [PubMed] [Google Scholar]
- 232.Garza-Flores J, Hall PE, Perez-Palacios G. Long-acting hormonal contraceptives for women. J Steroid Biochem Mol Biol. 1991;40:697–704. doi: 10.1016/0960-0760(91)90293-e. [DOI] [PubMed] [Google Scholar]
- 233.Isley M. Implanon: the subdermal contraceptive implant. J Pediatr Adolesc Gynecol. 2010;23:364–367. doi: 10.1016/j.jpag.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 234.Pollack IP, Quigley HA, Harbin TS. The Ocusert pilocarpine system: advantages and disadvantages. South Med J. 1976;69:1296–1298. doi: 10.1097/00007611-197610000-00013. [DOI] [PubMed] [Google Scholar]
- 235.Peppas NA. Historical perspective on advanced drug delivery: how engineering design and mathematical modeling helped the field mature. Adv Drug Delivery Rev. 2013;65:5–9. doi: 10.1016/j.addr.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 236.Biondi M, Ungaro F, Quaglia F, Netti PA. Controlled drug delivery in tissue engineering. Adv Drug Delivery Rev. 2008;60:229–242. doi: 10.1016/j.addr.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 237.Kamalesh S, Tan P, Wang J, Lee T, Kang ET, Wang CH. Biocompatibility of electroactive polymers in tissues. J Biomed Mater Res. 2000;52:467–478. doi: 10.1002/1097-4636(20001205)52:3<467::aid-jbm4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 238.Tallury P, Alimohammadi N, Kalachandra S. Poly(ethyleneco-vinyl acetate) copolymer matrix for delivery of chlorhexidine and acyclovir drugs for use in the oral environment: effect of drug combination, copolymer composition and coating on the drug release rate. Dent Mater. 2007;23:404–409. doi: 10.1016/j.dental.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 239.Friederich RL. The pilocarpine Ocusert: A new drug delivery system. Ann Ophthalmol. 1974;6:1279–1284. [PubMed] [Google Scholar]
- 240.Sareen R, Jain N, Kumar D. An insight to osmotic drug delivery. Curr Drug Delivery. 2012;9:285–296. doi: 10.2174/156720112800389106. [DOI] [PubMed] [Google Scholar]
- 241.Moodley K, Pillay V, Choonara YE, du Toit LC, Ndesendo VM, Kumar P, Cooppan S, Bawa P. Oral drug delivery systems comprising altered geometric configurations for controlled drug delivery. Int J Mol Sci. 2012;13:18–43. doi: 10.3390/ijms13010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Swanson LJ, Seely JH, Garnick MB. Gonadotropin-releasing hormone analogs and prostatic cancer. Crit Rev Oncol Hematol. 1988;8:1–26. doi: 10.1016/s1040-8428(88)80003-9. [DOI] [PubMed] [Google Scholar]
- 243.Parmar H, Phillips RH, Lightman SL, Edwards L. Early tumor exacerbation in patients treated with long acting analogues of gonadotrophin releasing hormone. Br Med J. 1985;291:1645. doi: 10.1136/bmj.291.6509.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Tracy MA. Development and scale-up of a microsphere protein delivery system. Biotechnol Prog. 1998;14:108–115. doi: 10.1021/bp9701271. [DOI] [PubMed] [Google Scholar]
- 245.Kreuter J. Nanoparticles and nanocapsules—New dosage forms in the nanometer size range. Pharm Acta Helv. 1978;53:33–39. [PubMed] [Google Scholar]
- 246.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21:1184–1191. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 247.Langer R. New methods of drug delivery. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 248.Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc Chem Res. 2011;44:1123–1134. doi: 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- 249.Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13:238–252. doi: 10.1016/s0022-2836(65)80093-6. [DOI] [PubMed] [Google Scholar]
- 250.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 251.Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1217. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 252.Bangham AD, Horne RW. Negative Staining of Phospholipids and Their Structural Modification by Surface-Active Agents as Observed in the Electron Microscope. J Mol Biol. 1964;8:660–668. doi: 10.1016/s0022-2836(64)80115-7. [DOI] [PubMed] [Google Scholar]
- 253.Torchilin VP. Immunoliposomes and PEGylated immunoliposomes: possible use for targeted delivery of imaging agents. ImmunoMethods. 1994;4:244–258. doi: 10.1006/immu.1994.1027. [DOI] [PubMed] [Google Scholar]
- 254.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 255.Allen TM, Chonn A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 1987;223:42–46. doi: 10.1016/0014-5793(87)80506-9. [DOI] [PubMed] [Google Scholar]
- 256.Leserman LD, Barbet J, Kourilsky F, Weinstein JN. Targeting to cells of fluorescent liposomes covalently coupled with monoclonal antibody or protein A. Nature. 1980;288:602–604. doi: 10.1038/288602a0. [DOI] [PubMed] [Google Scholar]
- 257.Heath TD, Fraley RT, Papahdjopoulos D. Antibody targeting of liposomes: cell specificity obtained by conjugation of F(ab′)2 to vesicle surface. Science. 1980;210:539–541. doi: 10.1126/science.7423203. [DOI] [PubMed] [Google Scholar]
- 258.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Delivery Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Duncan R, Kopeckova-Rejmanova P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopecek J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of daunomycin and puromycin conjugates in vitro. Br J Cancer. 1987;55:165–174. doi: 10.1038/bjc.1987.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kopecek J, Sprincl L, Lim D. New types of synthetic infusion solutions. I. Investigation of the effect of solutions of some hydrophilic polymers on blood. J Biomed Mater Res. 1973;7:179–191. doi: 10.1002/jbm.820070206. [DOI] [PubMed] [Google Scholar]
- 261.Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K. Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res. 1991;51:3229–3236. [PubMed] [Google Scholar]
- 262.Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Controlled Release. 2014;190:465–476. doi: 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 263.Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Delivery Rev. 2003;55:403–419. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 264.Kabanov AV, Chekhonin VP, Alakhov V, Batrakova EV, Lebedev AS, Melik-Nubarov NS, Arzhakov SA, Levashov AV, Morozov GV, Severin ES, et al. The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles. Micelles as microcontainers for drug targeting. FEBS Lett. 1989;258:343–345. doi: 10.1016/0014-5793(89)81689-8. [DOI] [PubMed] [Google Scholar]
- 265.Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19:1–72. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 266.Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br J Cancer. 2004;91:1775–1781. doi: 10.1038/sj.bjc.6602204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10:3708–3716. doi: 10.1158/1078-0432.CCR-03-0655. [DOI] [PubMed] [Google Scholar]
- 268.Danson S, Ferry D, Alakhov V, Margison J, Kerr D, Jowle D, Brampton M, Halbert G, Ranson M. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br J Cancer. 2004;90:2085–2091. doi: 10.1038/sj.bjc.6601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27:2569–2589. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Park SR, Oh D-Y, Kim D-W, Kim T-Y, Heo DS, Bang Y-J, Kim NK, Kang WK, Kim H-T, Im S-A, Suh J-H, Kim H-K, Kim H-K. A multi-center, late phase II clinical trial of Genexol (paclitaxel) and cisplatin for patients with advanced gastric cancer. Oncol Rep. 2004;12:1059–1064. [PubMed] [Google Scholar]
- 271.https://clinicaltrials.gov/show/NCT00876486.
- 272.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, Rha SY, Lee MY, Ro J. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108:241–250. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 273.Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- 274.Gaur S, Wang Y, Kretzner L, Chen L, Yen T, Wu X, Yuan YC, Davis M, Yen Y. Pharmacodynamic and pharmacogenomic study of the nanoparticle conjugate of camptothecin CRLX101 for the treatment of cancer. Nanomedicine. 2014;10:1477–1486. doi: 10.1016/j.nano.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 275.Elsasser-Beile U, Buhler P, Wolf P. Targeted therapies for prostate cancer against the prostate specific membrane antigen. Curr Drug Targets. 2009;10:118–125. doi: 10.2174/138945009787354601. [DOI] [PubMed] [Google Scholar]
- 276.Slovin SF. Targeting novel antigens for prostate cancer treatment: focus on prostate-specific membrane antigen. Expert Opin Ther Targets. 2005;9:561–570. doi: 10.1517/14728222.9.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH, Yang KS, Laughney AM, Wojtkiewicz G, Kamaly N, et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7(314):314ra183. doi: 10.1126/scitranslmed.aac6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, et al. Tumour-associated macrophages act as a slow-release reservoir of nanotherapeutic Pt(IV) pro-drug. Nat Commun. 2015;6:8692. doi: 10.1038/ncomms9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharmaceutics. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 280.Heidel JD, Liu JY, Yen Y, Zhou B, Heale BS, Rossi JJ, Bartlett DW, Davis ME. Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro. Clin Cancer Res. 2007;13:2207–2215. doi: 10.1158/1078-0432.CCR-06-2218. [DOI] [PubMed] [Google Scholar]
- 281.Conniot J, Silva JM, Fernandes JG, Silva LC, Gaspar R, Brocchini S, Florindo HF, Barata TS. Cancer immunotherapy: nanodelivery approaches for immune cell targeting and tracking. Front Chem. 2014;2:105. doi: 10.3389/fchem.2014.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Craparo EF, Bondi ML. Application of polymeric nanoparticles in immunotherapy. Curr Opin Allergy Clin Immunol. 2012;12:658–664. doi: 10.1097/ACI.0b013e3283588c57. [DOI] [PubMed] [Google Scholar]
- 283.Gadde S, Even-Or O, Kamaly N, Hasija A, Gagnon PG, Adusumilli KH, Erakovic A, Pal AK, Zhang XQ, Kolishetti N, Shi J, Fisher EA, Farokhzad OC. Development of therapeutic polymeric nanoparticles for the resolution of inflammation. Adv Healthcare Mater. 2014;3:1448–1456. doi: 10.1002/adhm.201300688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhang XQ, Even-Or O, Xu X, van Rosmalen M, Lim L, Gadde S, Farokhzad OC, Fisher EA. Nanoparticles containing a liver X receptor agonist inhibit inflammation and atherosclerosis. Adv Healthcare Mater. 2015;4:228–236. doi: 10.1002/adhm.201400337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kamaly N, Fredman G, Subramanian M, Gadde S, Pesic A, Cheung L, Fayad ZA, Langer R, Tabas I, Farokhzad OC. Development and in vivo efficacy of targeted polymeric inflammation-resolving nanoparticles. Proc Natl Acad Sci U S A. 2013;110:6506–6511. doi: 10.1073/pnas.1303377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokhzad O, Tabas I. Targeted nanoparticles containing the proresolving peptide Ac2–26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Soehnlein O. (Re)solving atherosclerosis. Sci Transl Med. 2015;7:275fs7. doi: 10.1126/scitranslmed.aaa5355. [DOI] [PubMed] [Google Scholar]
- 288.Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110:18632–18637. doi: 10.1073/pnas.1318415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Gombotz WR, Healy MS, Brown L. Very low temperature casting of controlled release microspheres. US5019400 A. US Patent. 1991
- 291.Jung T, Breitenbach A, Kissel T. Sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide)s facilitate the preparation of small negatively charged biodegradable nanospheres. J Controlled Release. 2000;67:157–169. doi: 10.1016/s0168-3659(00)00201-7. [DOI] [PubMed] [Google Scholar]
- 292.Allemann E, Leroux JC, Gurny R, Doelker E. In vitro extended-release properties of drug-loaded poly(DL-lactic acid) nanoparticles produced by a salting-out procedure. Pharm Res. 1993;10:1732–1737. doi: 10.1023/a:1018970030327. [DOI] [PubMed] [Google Scholar]
- 293.Grabnar PA, Kristl J. The manufacturing techniques of drug-loaded polymeric nanoparticles from preformed polymers. J Microencapsulation. 2011;28:323–335. doi: 10.3109/02652048.2011.569763. [DOI] [PubMed] [Google Scholar]
- 294.Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385:113–142. doi: 10.1016/j.ijpharm.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 295.Chan JM, Valencia PM, Zhang L, Langer R, Farokhzad OC. Polymeric nanoparticles for drug delivery. Methods Mol Biol. 2010;624:163–175. doi: 10.1007/978-1-60761-609-2_11. [DOI] [PubMed] [Google Scholar]
- 296.Cleland JL, Johnson OL, Putney S, Jones AJS. Recombinant human growth hormone poly(lactic-co-glycolic acid) microsphere formulation development. Adv Drug Delivery Rev. 1997;28:71–84. doi: 10.1016/s0169-409x(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 297.Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55:R1–4. [Google Scholar]
- 298.Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm. 1998;24:1113–1128. doi: 10.3109/03639049809108571. [DOI] [PubMed] [Google Scholar]
- 299.Farokhzad OC, Cheng JJ, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Anton N, Benoit JP, Saulnier P. Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Controlled Release. 2008;128:185–199. doi: 10.1016/j.jconrel.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 301.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Delivery. 2004;1:321–333. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 302.Zamani M, Prabhakaran MP, Ramakrishna S. Advances in drug delivery via electrospun and electrosprayed nanomaterials. Int J Nanomed. 2013;8:2997–3017. doi: 10.2147/IJN.S43575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.DeMello AJ. Control and detection of chemical reactions in microfluidic systems. Nature. 2006;442:394–402. doi: 10.1038/nature05062. [DOI] [PubMed] [Google Scholar]
- 304.Marre S, Jensen KF. Synthesis of micro and nanostructures in microfluidic systems. Chem Soc Rev. 2010;39:1183–1202. doi: 10.1039/b821324k. [DOI] [PubMed] [Google Scholar]
- 305.Fraikin JL, Teesalu T, McKenney CM, Ruoslahti E, Cleland AN. A high-throughput label-free nanoparticle analyser. Nat Nanotechnol. 2011;6:308–313. doi: 10.1038/nnano.2011.24. [DOI] [PubMed] [Google Scholar]
- 306.Gong J, Kim CJ. All-electronic droplet generation on-chip with real-time feedback control for EWOD digital microfluidics. Lab Chip. 2008;8:898–906. doi: 10.1039/b717417a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wang H, Liu K, Chen KJ, Lu Y, Wang S, Lin WY, Guo F, Kamei K, Chen YC, Ohashi M, Wang M, Garcia MA, Zhao XZ, Shen CK, Tseng HR. A rapid pathway toward a superb gene delivery system: programming structural and functional diversity into a supramolecular nanoparticle library. ACS Nano. 2010;4:6235–6243. doi: 10.1021/nn101908e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Karnik R, Gu F, Basto P, Cannizzaro C, Dean L, Kyei-Manu W, Langer R, Farokhzad OC. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008;8:2906–2912. doi: 10.1021/nl801736q. [DOI] [PubMed] [Google Scholar]
- 309.Rhee M, Valencia PM, Rodriguez MI, Langer R, Farokhzad OC, Karnik R. Synthesis of size-tunable polymeric nanoparticles enabled by 3D hydrodynamic flow focusing in single-layer microchannels. Adv Mater. 2011;23:H79–83. doi: 10.1002/adma.201004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Song H, Tice JD, Ismagilov RF. A microfluidic system for controlling reaction networks in time. Angew Chem, Int Ed. 2003;42:768–772. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- 311.Zhao CX, He LZ, Qiao SZ, Middelberg APJ. Nanoparticle synthesis in microreactors. Chem Eng Sci. 2011;66:1463–1479. [Google Scholar]
- 312.Blasi P, D’Souza SS, Selmin F, DeLuca PP. Plasticizing effect of water on poly(lactide-co-glycolide) J Controlled Release. 2005;108:1–9. doi: 10.1016/j.jconrel.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 313.Webber WL, Lago F, Thanos C, Mathiowitz E. Characterization of soluble, salt-loaded, degradable PLGA films and their release of tetracycline. J Biomed Mater Res. 1998;41:18–29. doi: 10.1002/(sici)1097-4636(199807)41:1<18::aid-jbm3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 314.Keraliya RA, Patel C, Patel P, Keraliya V, Soni TG, Patel RC, Patel MM. Osmotic drug delivery system as a part of modified release dosage form. ISRN Pharm. 2012;2012:528079. doi: 10.5402/2012/528079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Park H, Park K, Shalaby WS. Biodegradable Hydrogels for Drug Delivery. CRC Press; Boca Raton, FL: 2011. [Google Scholar]
- 316.Laurencin CT, Langer R. Polymeric controlled release systems: New methods for drug delivery. Clin Lab Med. 1987;7:301–323. [PubMed] [Google Scholar]
- 317.Jain D, Raturi R, Jain V, Bansal P, Singh R. Recent technologies in pulsatile drug delivery systems. Biomatter. 2011;1:57–65. doi: 10.4161/biom.1.1.17717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Berchane NS, Carson KH, Rice-Ficht AC, Andrews MJ. Effect of mean diameter and polydispersity of PLG microspheres on drug release: experiment and theory. Int J Pharm. 2007;337:118–126. doi: 10.1016/j.ijpharm.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 319.Kang J, Lambert O, Ausborn M, Schwendeman SP. Stability of proteins encapsulated in injectable and biodegradable poly(lactide-co-glycolide)-glucose millicylinders. Int J Pharm. 2008;357:235–243. doi: 10.1016/j.ijpharm.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Zhang Y, Yin Q, Yen J, Li J, Ying H, Wang H, Hua Y, Chaney EJ, Boppart SA, Cheng J. Non-invasive, real-time reporting drug release in vitro and in vivo. Chem Commun. 2015;51:6948–6951. doi: 10.1039/c4cc09920f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Redy-Keisar O, Ferber S, Satchi-Fainaro R, Shabat D. NIR Fluorogenic Dye as a Modular Platform for Prodrug Assembly: Real-Time in vivo Monitoring of Drug Release. ChemMedChem. 2015;10:999–1007. doi: 10.1002/cmdc.201500060. [DOI] [PubMed] [Google Scholar]
- 322.Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem, Int Ed. 2006;45:8149–8152. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]
- 323.Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Controlled Release. 2001;73:121–136. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 324.Peppas NA, Narasimhan B. Mathematical models in drug delivery: how modeling has shaped the way we design new drug delivery systems. J Controlled Release. 2014;190:75–81. doi: 10.1016/j.jconrel.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 325.Wang X, Venkatraman SS, Boey FY, Loo JS, Tan LP. Controlled release of sirolimus from a multilayered PLGA stent matrix. Biomaterials. 2006;27:5588–5595. doi: 10.1016/j.biomaterials.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 326.Pearson RM, Juettner VV, Hong S. Biomolecular corona on nanoparticles: a survey of recent literature and its implications in targeted drug delivery. Front Chem. 2014;2:108. doi: 10.3389/fchem.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Crommen JH, Schacht EH, Mense EH. Biodegradable polymers. II. Degradation characteristics of hydrolysis-sensitive poly-[(organo)phosphazenes] Biomaterials. 1992;13:601. doi: 10.1016/0142-9612(92)90028-m. [DOI] [PubMed] [Google Scholar]
- 328.Grassi M, Grassi G. Mathematical modelling and controlled drug delivery: matrix systems. Curr Drug Delivery. 2005;2:97–116. doi: 10.2174/1567201052772906. [DOI] [PubMed] [Google Scholar]
- 329.Ferrero C, Massuelle D, Doelker E. Towards elucidation of the drug release mechanism from compressed hydrophilic matrices made of cellulose ethers. II. Evaluation of a possible swelling-controlled drug release mechanism using dimensionless analysis. J Controlled Release. 2010;141:223–233. doi: 10.1016/j.jconrel.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 330.Park K, Shalaby W, Park H. Biodegradable Hydrogels for Drug Delivery. Technomic Publishing Co.; Lancaster, PA: 1993. [Google Scholar]
- 331.Modesti G, Zimmermann B, Borsch M, Herrmann A, Saalwachter K. Diffusion in Model Networks as Studied by NMR and Fluorescence Correlation Spectroscopy. Macromolecules. 2009;42:4681–4689. doi: 10.1021/ma900614j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Koutsopoulos S, Unsworth LD, Nagai Y, Zhang S. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc Natl Acad Sci U S A. 2009;106:4623–4628. doi: 10.1073/pnas.0807506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Serra L, Domenech J, Peppas NA. Drug transport mechanisms and release kinetics from molecularly designed poly(acrylic acid-g-ethylene glycol) hydrogels. Biomaterials. 2006;27:5440–5451. doi: 10.1016/j.biomaterials.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 334.Narasimhan B. Mathematical models describing polymer dissolution: consequences for drug delivery. Adv Drug Delivery Rev. 2001;48:195–210. doi: 10.1016/s0169-409x(01)00117-x. [DOI] [PubMed] [Google Scholar]
- 335.Narasimhan B, Peppas NA. Molecular analysis of drug delivery systems controlled by dissolution of the polymer carrier. J Pharm Sci. 1997;86:297–304. doi: 10.1021/js960372z. [DOI] [PubMed] [Google Scholar]
- 336.Hopfenberg H. Controlled Release from Erodible Slabs, Cylinders, and Spheres. American Chemical Society; Washington, DC: 1976. pp. 26–32. [Google Scholar]
- 337.Karasulu HY, Ertan G, Kose T. Modeling of theophylline release from different geometrical erodible tablets. Eur J Pharm Biopharm. 2000;49:177–182. doi: 10.1016/s0939-6411(99)00082-x. [DOI] [PubMed] [Google Scholar]
- 338.Rothstein SN, Federspiel WJ, Little SR. A unified mathematical model for the prediction of controlled release from surface and bulk eroding polymer matrices. Biomaterials. 2009;30:1657–1664. doi: 10.1016/j.biomaterials.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Gopferich A, Tessmar J. Polyanhydride degradation and erosion. Adv Drug Delivery Rev. 2002;54:911–931. doi: 10.1016/s0169-409x(02)00051-0. [DOI] [PubMed] [Google Scholar]
- 340.Shah S, Cha Y, Pitt C. Poly (glycolic acid-co-DL-lactic acid): diffusion or degradation controlled drug delivery? J Controlled Release. 1992;18:261–270. [Google Scholar]
- 341.Seader J. Seperation Process Principles. Wiley; New York: 1998. [Google Scholar]
- 342.Lao LL, Venkatraman SS, Peppas NA. Modeling of drug release from biodegradable polymer blends. Eur J Pharm Biopharm. 2008;70:796–803. doi: 10.1016/j.ejpb.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 343.Siepmann J, Gopferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Delivery Rev. 2001;48:229–247. doi: 10.1016/s0169-409x(01)00116-8. [DOI] [PubMed] [Google Scholar]
- 344.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Delivery Rev. 2001;48:139–157. doi: 10.1016/s0169-409x(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 345.Zeisberger EE, Schönbaum EE, Lomax PE. Thermal Balance in Health and Disease: Recent Basic Research and Clinical Progress. Birkhauser; Berlin: 1994. [Google Scholar]
- 346.Hurwitz M, Stauffer P. Hyperthermia, radiation and chemotherapy: the role of heat in multidisciplinary cancer care. Semin Oncol. 2014;41:714–729. doi: 10.1053/j.seminoncol.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 347.Tepper M, Shoval A, Hoffer O, Confino H, Schmidt M, Kelson I, Keisari Y, Gannot I. Thermographic investigation of tumor size, and its correlation to tumor relative temperature, in mice with transplantable solid breast carcinoma. J Biomed Opt. 2013;18:111410. doi: 10.1117/1.JBO.18.11.111410. [DOI] [PubMed] [Google Scholar]
- 348.Yahara T, Koga T, Yoshida S, Nakagawa S, Deguchi H, Shirouzu K. Relationship Between Microvessel Density and Thermographic Hot Areas in Breast Cancer. Surg Today. 2003;33:243–248. doi: 10.1007/s005950300055. [DOI] [PubMed] [Google Scholar]
- 349.Stefanadis C, Markou D, Petraki K, Panagiotakos DB, Fasoulakis C, Kyriakidis A, Papadimitriou C, Toutouzas PK. Increased Temperature of Malignant Urinary Bladder Tumors In Vivo: The Application of a New Method Based on a Catheter Technique. J Clin Oncol. 2001;19:676–681. doi: 10.1200/JCO.2001.19.3.676. [DOI] [PubMed] [Google Scholar]
- 350.Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer. 2008;44:2546–2554. doi: 10.1016/j.ejca.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 351.Hyperthermia in Cancer Treatment. http://www.cancer.gov/about-cancer/treatment/types/surgery/hyperthermia-fact-sheet.
- 352.Roy D, Brooks WL, Sumerlin BS. New directions in thermoresponsive polymers. Chem Soc Rev. 2013;42:7214–7243. doi: 10.1039/c3cs35499g. [DOI] [PubMed] [Google Scholar]
- 353.Calejo MT, Sande SA, Nystrom B. Thermoresponsive polymers as gene and drug delivery vectors: architecture and mechanism of action. Expert OpinDrug Delivery. 2013;10:1669–1686. doi: 10.1517/17425247.2013.846906. [DOI] [PubMed] [Google Scholar]
- 354.Hu J, Zhang G, Ge Z, Liu S. Stimuli-responsive tertiary amine methacrylate-based block copolymers: Synthesis, supramolecular self-assembly and functional applications. Prog Polym Sci. 2014;39:1096–1143. [Google Scholar]
- 355.Kelley EG, Albert JN, Sullivan MO, Epps TH., III Stimuli-responsive copolymer solution and surface assemblies for biomedical applications. Chem Soc Rev. 2013;42:7057–7071. doi: 10.1039/c3cs35512h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Weber C, Hoogenboom R, Schubert US. Temperature responsive bio-compatible polymers based on poly(ethylene oxide) and poly(2-oxazoline)s. Prog Polym Sci. 2012;37:686–714. [Google Scholar]
- 357.Hoogenboom R, Thijs HM, Jochems MJ, van Lankvelt BM, Fijten MW, Schubert US. Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: alternatives to poly(N-isopropylacrylamide)? Chem Commun. 2008:5758–5760. doi: 10.1039/b813140f. [DOI] [PubMed] [Google Scholar]
- 358.Twaites BR, de las Heras Alarcon C, Cunliffe D, Lavigne M, Pennadam S, Smith JR, Gorecki DC, Alexander C. Thermo and pH responsive polymers as gene delivery vectors: effect of polymer architecture on DNA complexation in vitro. J Controlled Release. 2004;97:551–566. doi: 10.1016/j.jconrel.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 359.Wei H, Cheng S-X, Zhang X-Z, Zhuo R-X. Thermosensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog Polym Sci. 2009;34:893–910. [Google Scholar]
- 360.Qiu Y, Luo Y, Zhang Y, Cui W, Zhang D, Wu J, Zhang J, Tu J. The correlation between acoustic cavitation and sonoporation involved in ultrasound-mediated DNA transfection with polyethylenimine (PEI) in vitro. J Controlled Release. 2010;145:40–48. doi: 10.1016/j.jconrel.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 361.de Las Heras Alarcon C, Pennadam S, Alexander C. Stimuli responsive polymers for biomedical applications. Chem Soc Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 362.Shakya AK, Kumar A, Nandakumar KS. Adjuvant properties of a biocompatible thermo-responsive polymer of N-isopropylacrylamide in autoimmunity and arthritis. J R Soc Interface. 2011;8:1748–1759. doi: 10.1098/rsif.2011.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Liu J, Debuigne A, Detrembleur C, Jérôme C. Poly(N-vinylcaprolactam): A Thermoresponsive Macromolecule with Promising Future in Biomedical Field. Adv Healthcare Mater. 2014;3:1941–1968. doi: 10.1002/adhm.201400371. [DOI] [PubMed] [Google Scholar]
- 364.Ramos J, Imaz A, Forcada J. Temperature-sensitive nanogels: poly(N-vinylcaprolactam) versus poly(N-isopropylacrylamide) Polym Chem. 2012;3:852–856. [Google Scholar]
- 365.Meeussen F, Berghmans H, Verbrugghe S, Goethals E, Du Prez F. Phase behaviour of poly(N-vinyl caprolactam) in water. Polymer. 2000;41:8597–8602. [Google Scholar]
- 366.Vihola H, Laukkanen A, Valtola L, Tenhu H, Hirvonen J. Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam) Biomaterials. 2005;26:3055–3064. doi: 10.1016/j.biomaterials.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 367.Liu J, Detrembleur C, Hurtgen M, Debuigne A, De Pauw-Gillet M-C, Mornet S, Duguet E, Jeró̂me C. Reversibly crosslinked thermo- and redox-responsive nanogels for controlled drug release. Polym Chem. 2014;5:77–88. [Google Scholar]
- 368.Lutz J-F, Akdemir O, Hoth A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST- Is the Age of Poly(NIPAM) Over? J Am Chem Soc. 2006;128:13046–13047. doi: 10.1021/ja065324n. [DOI] [PubMed] [Google Scholar]
- 369.Vancoillie G, Frank D, Hoogenboom R. Thermoresponsive poly(oligo ethylene glycol acrylates) Prog Polym Sci. 2014;39:1074–1095. [Google Scholar]
- 370.Lutz JF. Polymerization of oligo(ethylene glycol) (meth)-acrylates: Toward new generations of smart biocompatible materials. J Polym Sci, Part A: Polym Chem. 2008;46:3459–3470. [Google Scholar]
- 371.Lutz JF. Thermo-Switchable Materials Prepared Using the OEGMA-Platform. Adv Mater. 2011;23:2237–2243. [Google Scholar]
- 372.Jiang X, Vogel EB, Smith MR, III, Baker G. “Clickable” Polyglycolides- Tunable Synthons for Thermoresponsive, Degradable Polymers. Macromolecules. 2008;41:1937–1944. [Google Scholar]
- 373.Rainbolt EA, Washington KE, Biewer MC, Stefan MC. Towards smart polymeric drug carriers: self-assembling γ-substituted polycaprolactones with highly tunable thermoresponsive behavior. J Mater Chem B. 2013;1:6532. doi: 10.1039/c3tb21488e. [DOI] [PubMed] [Google Scholar]
- 374.Xu S, Wang W, Li X, Liu J, Dong A, Deng L. Sustained release of PTX-incorporated nanoparticles synergized by burst release of DOXHCl from thermosensitive modified PEG/PCL hydrogel to improve anti-tumor efficiency. Eur J Pharm Sci. 2014;62:267–273. doi: 10.1016/j.ejps.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 375.Cheng Y, Hao J, Lee LA, Biewer MC, Wang Q, Stefan MC. Thermally controlled release of anticancer drug from self-assembled gamma-substituted amphiphilic poly(epsilon-caprolactone) micellar nanoparticles. Biomacromolecules. 2012;13:2163–2173. doi: 10.1021/bm300823y. [DOI] [PubMed] [Google Scholar]
- 376.Safaei Nikouei N, Lavasanifar A. Characterization of the thermo- and pH-responsive assembly of triblock copolymers based on poly(ethylene glycol) and functionalized poly(epsilon-caprolactone) Acta Biomater. 2011;7:3708–3718. doi: 10.1016/j.actbio.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 377.Waknis V, Jonnalagadda S. Novel poly-DL-lactide-polycaprolactone copolymer based flexible drug delivery system for sustained release of ciprofloxacin. Drug Delivery. 2011;18:236–245. doi: 10.3109/10717544.2010.528070. [DOI] [PubMed] [Google Scholar]
- 378.Chen W, Meng F, Cheng R, Deng C, Feijen J, Zhong Z. Advanced drug and gene delivery systems based on functional biodegradable polycarbonates and copolymers. J Controlled Release. 2014;190:398–414. doi: 10.1016/j.jconrel.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 379.Pitt CG, Zhong-wei G. Modification of the rates of the chain cleavage of poly-ε-caprolactone and related polyesters in the solid state. J Controlled Release. 1987;4:283–292. [Google Scholar]
- 380.Fu K, Pack DW, Klibanov AM, Langer R. Visual Evidence of Acidic. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 381.Pego AP, Brouwer LA, van Wachem PB, Poot AA, Grijpma DW, Feijen J, Van Luyn MJA. In vivo behavior of poly(1,3-trimethylene carbonate) and copolymers of 1,3-trimethylene carbonate with D,L-lactide or -caprolactone- Degradation and tissue response. J Biomed Mater Res. 2003;67A:1044–1054. doi: 10.1002/jbm.a.10121. [DOI] [PubMed] [Google Scholar]
- 382.Zhang Z, Kuijer R, Bulstra SK, Grijpma DW, Feijen J. The in vivo and in vitro degradation behavior of poly(trimethylene carbonate) Biomaterials. 2006;27:1741–1748. doi: 10.1016/j.biomaterials.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 383.Mespouille L, Coulembier O, Kawalec M, Dove AP, Dubois P. Implementation of metal-free ring-opening polymerization in the preparation of aliphatic polycarbonate materials. Prog Polym Sci. 2014;39:1144–1164. [Google Scholar]
- 384.Dai S, Xue L, Li Z. Enzymatic Ring-Opening Polymerization of Trimethylene Carbonate with Macrodiol: Synthesis of Block Poly(ester-co-carbonate) for Biomaterial Preparation. ACS Catal. 2011;1:1421–1429. [Google Scholar]
- 385.Normand M, Kirillov E, Carpentier J-F, Guillaume SM. Cyclodextrin-Centered Polyesters: Controlled Ring-Opening Polymerization of Cyclic Esters from β-Cyclodextrin-Diol. Macromolecules. 2012;45:1122–1130. [Google Scholar]
- 386.Bartolucci S, Rella R, Guagliardi A, Raia CA, Gambacorta A, De Rosa M, Rossi M. Malic Enzyme from Archaebacterium Sulfolobus solfuturicus. J Biol Chem. 1987;262:7725–7731. [PubMed] [Google Scholar]
- 387.Dargaville BL, Vaquette C, Peng H, Rasoul F, Chau YQ, Cooper-White JJ, Campbell JH, Whittaker AK. Cross-linked poly(trimethylene carbonate-co-L-lactide) as a biodegradable, elastomeric scaffold for vascular engineering applications. Biomacromolecules. 2011;12:3856–3869. doi: 10.1021/bm201291e. [DOI] [PubMed] [Google Scholar]
- 388.Olsén P, Odelius K, Albertsson A-C. Ring-Closing Depolymerization: A Powerful Tool for Synthesizing the Allyloxy-Functionalized Six-Membered Aliphatic Carbonate Monomer 2-Allyloxymethyl-2-ethyltrimethylene Carbonate. Macromolecules. 2014;47:6189–6195. [Google Scholar]
- 389.Uysal BB, Gunay US, Hizal G, Tunca U. Orthogonal multifunctionalization of aliphatic polycarbonate via sequential Michael addition and radical-thiol-ene click reactions. J Polym Sci, Part A: Polym Chem. 2014;52:1581–1587. [Google Scholar]
- 390.Aguirre-Chagala YE, Santos JL, Huang Y, Herrera-Alonso M. Phenylboronic Acid-Installed Polycarbonates for the pH-Dependent Release of Diol-Containing Molecules. ACS Macro Lett. 2014;3:1249–1253. doi: 10.1021/mz500594m. [DOI] [PubMed] [Google Scholar]
- 391.Tempelaar S, Mespouille L, Coulembier O, Dubois P, Dove AP. Synthesis and post-polymerisation modifications of aliphatic poly(carbonate)s prepared by ring-opening polymerisation. Chem Soc Rev. 2013;42:1312–1336. doi: 10.1039/c2cs35268k. [DOI] [PubMed] [Google Scholar]
- 392.Ajiro H, Takahashi Y, Akashi M. Thermosensitive Biodegradable Homopolymer of Trimethylene Carbonate Derivative at Body Temperature. Macromolecules. 2012;45:2668–2674. [Google Scholar]
- 393.Kim SH, Tan JPK, Fukushima K, Nederberg F, Yang YY, Waymouth RM, Hedrick JL. Thermoresponsive nanostructured polycarbonate block copolymers as biodegradable therapeutic delivery carriers. Biomaterials. 2011;32:5505–5514. doi: 10.1016/j.biomaterials.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 394.Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Controlled Release. 2008;130:98–106. doi: 10.1016/j.jconrel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Alvarez-Lorenzo C, Sosnik A, Concheiro A. PEO-PPO Block Copolymers for Passive Micellar Targeting and Overcoming Multidrug Resistance in Cancer Therapy. Curr Drug Targets. 2011;12:1112–1130. doi: 10.2174/138945011795906615. [DOI] [PubMed] [Google Scholar]
- 396.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv Drug Delivery Rev. 2002;54:759–779. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]
- 397.Bates FS, Hillmyer MA, Lodge TP, Bates CM, Delaney KT, Fredrickson GH. Multiblock polymers: panacea or Pandora’s box? Science. 2012;336:434–440. doi: 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- 398.Batrakova EV, Miller DW, Li S, Alakhov VY, Kabanov AV, Elmquist WF. Pluronic P85 Enhances the Delivery of Digoxin to the Brain—In Vitro and in Vivo Studies. J Pharm Exp Ther. 2001;296:551–557. [PubMed] [Google Scholar]
- 399.Batrakova EV, Li S, Vinogradov SV, Alakhov VY, Miller DW, Kabanov AV. Mechanism of Pluronic Effect on P-Glycoprotein Efflux System in Blood–Brain Barrier—Contributions of Energy Depletion and Membrane Fluidization. J Pharm Exp Ther. 2001;299:483–493. [PubMed] [Google Scholar]
- 400.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discovery. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 401.Regev R, Katzir H, Yeheskely-Hayon D, Eytan GD. Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 2007;274:6204–6214. doi: 10.1111/j.1742-4658.2007.06140.x. [DOI] [PubMed] [Google Scholar]
- 402.Batrakova EV, Li S, Alakhov VYu, Elmquist WF, Miller DW, Kabanov AV. Sensitization of Cells Overexpressing Multidrug-Resistant Proteins by Pluronic P85. Pharm Res. 2003;20:1581–1590. doi: 10.1023/a:1026179132599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Effect of excipients on breast cancer resistance protein substrate uptake activity. J Controlled Release. 2007;124:1–5. doi: 10.1016/j.jconrel.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 404.Yamagata T, Kusuhara H, Morishita M, Takayama K, Benameur H, Sugiyama Y. Improvement of the oral drug absorption of topotecan through the inhibition of intestinal xenobiotic efflux transporter, breast cancer resistance protein, by excipients. Drug Metab Dispos. 2007;35:1142–1148. doi: 10.1124/dmd.106.014217. [DOI] [PubMed] [Google Scholar]
- 405.Minko T, Batrakova EV, Li S, Li Y, Pakunlu RI, Alakhov VY, Kabanov AV. Pluronic block copolymers alter apoptotic signal transduction of doxorubicin in drug-resistant cancer cells. J Controlled Release. 2005;105:269–278. doi: 10.1016/j.jconrel.2005.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Batrakova E, Lee S, Li S, Venne A, Alakhov V, Kabanov A. Alexander Kabanov Fundamental Relationships Between the Composition of Pluronic Block Copolymers and Their Hypersensitization Effect in MDR Cancer Cells. Pharm Res. 1999;16:1373–1379. doi: 10.1023/a:1018942823676. [DOI] [PubMed] [Google Scholar]
- 407.Miller DW, Batrakova EV, Waltner TO, Alakhov VYu, Kabanov AV. Interactions of Pluronic Block Copolymers with Brain Microvessel Endothelial Cells- Evidence of Two Potential Pathways for Drug Absorption. Bioconjugate Chem. 1997;8:649–657. doi: 10.1021/bc970118d. [DOI] [PubMed] [Google Scholar]
- 408.Pitto-Barry A, Barry NPE. Pluronic® block-copolymers in medicine: from chemical and biological versatility to rationalisation and clinical advances. Polym Chem. 2014;5:3291–3297. [Google Scholar]
- 409.Arranja A, Schroder AP, Schmutz M, Waton G, Schosseler F, Mendes E. Cytotoxicity and internalization of Pluronic micelles stabilized by core cross-linking. J Controlled Release. 2014;196:87–95. doi: 10.1016/j.jconrel.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 410.Zhang W, Gilstrap K, Wu L, Bahadur KCR, Moss MA, Wang Q, Lu X, He X. Synthesis and Characterization of Thermally Responsive Pluronic F127 Chitosan Nanocapsules for Controlled Release and Intracellular Delivery of Small Molecules. ACS Nano. 2010;4:6747–6759. doi: 10.1021/nn101617n. [DOI] [PubMed] [Google Scholar]
- 411.Rao W, Zhang W, Poventud-Fuentes I, Wang Y, Lei Y, Agarwal P, Weekes B, Li C, Lu X, Yu J, He X. Thermally Responsive Nanoparticle-Encapsulated Curcumin and Its Combination with Mild Hyperthermia for Enhanced Cancer Cell Destruction. Acta Biomater. 2014;10:831–842. doi: 10.1016/j.actbio.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Zhang W, Rong J, Wang Q, He X. The encapsulation and intracellular delivery of trehalose using a thermally responsive nanocapsule. Nanotechnology. 2009;20:275101. doi: 10.1088/0957-4484/20/27/275101. [DOI] [PubMed] [Google Scholar]
- 413.Hu J, He J, Zhang M, Ni P. Precise modular synthesis and a structure–property study of acid-cleavable star-block copolymers for pH-triggered drug delivery. Polym Chem. 2015;6:1553–1566. [Google Scholar]
- 414.Choi WI, Kamaly N, Riol-Blanco L, Lee IH, Wu J, Swami A, Vilos C, Yameen B, Yu M, Shi J, Tabas I, von Andrian UH, Jon S, Farokhzad OC. A solvent-free thermosponge nanoparticle platform for efficient delivery of labile proteins. Nano Lett. 2014;14:6449–6455. doi: 10.1021/nl502994y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Yu Y, Zou J, Cheng C. Synthesis and biomedical applications of functional poly(α-hydroxyl acid)s. Polym Chem. 2014;5:5854–5872. [Google Scholar]
- 416.Guo X, Li D, Yang G, Shi C, Tang Z, Wang J, Zhou S. Thermo-triggered drug release from actively targeting polymer micelles. ACS Appl Mater Interfaces. 2014;6:8549–8559. doi: 10.1021/am501422r. [DOI] [PubMed] [Google Scholar]
- 417.Zhang Z, Wang L, Wang J, Jiang X, Li X, Hu Z, Ji Y, Wu X, Chen C. Mesoporous silica-coated gold nanorods as a light-mediated multifunctional theranostic platform for cancer treatment. Adv Mater. 2012;24:1418–1423. doi: 10.1002/adma.201104714. [DOI] [PubMed] [Google Scholar]
- 418.Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–1137. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- 419.Tong R, Hemmati HD, Langer R, Kohane DS. Photoswitchable nanoparticles for triggered tissue penetration and drug delivery. J Am Chem Soc. 2012;134:8848–8855. doi: 10.1021/ja211888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.del Barrio Js, Oriol L, Alcalá R, Sánchez C. Azobenzene-Containing Linear–Dendritic Diblock Copolymers by Click Chemistry: Synthesis, Characterization, Morphological Study, and Photoinduction of Optical Anisotropy. Macromolecules. 2009;42:5752–5760. [Google Scholar]
- 421.Blasco E, Barrio Jd, Sánchez-Somolinos C, Piñol M, Oriol L. Light induced molecular release from vesicles based on amphiphilic linear-dendritic block copolymers. Polym Chem. 2013;4:2246. [Google Scholar]
- 422.Blasco E, Serrano JL, Piñol M, Oriol L. Light Responsive Vesicles Based on Linear–Dendritic Block Copolymers Using Azobenzene–Aliphatic Codendrons. Macromolecules. 2013;46:5951–5960. [Google Scholar]
- 423.Buback J, Kullmann M, Langhojer F, Nuernberger P, Schmidt R, Wurthner F, Brixner T. Ultrafast Bidirectional Photoswitching of a Spiropyran. J Am Chem Soc. 2010;132:16510–16519. doi: 10.1021/ja1062746. [DOI] [PubMed] [Google Scholar]
- 424.Rini M, Holm A-K, Nibbering ETJ, Fidder H. Ultrafast UV-mid-IR Investigation of the Ring Opening Reaction of a Photochromic Spiropyran. J Am Chem Soc. 2003;125:3028–3034. doi: 10.1021/ja028262j. [DOI] [PubMed] [Google Scholar]
- 425.Klajn R. Spiropyran-based dynamic materials. Chem Soc Rev. 2014;43:148–184. doi: 10.1039/c3cs60181a. [DOI] [PubMed] [Google Scholar]
- 426.Minkin VI. Photo-, Thermo-, Solvato-, and Electrochromic Spiroheterocyclic Compounds. Chem Rev. 2004;104:2751–2776. doi: 10.1021/cr020088u. [DOI] [PubMed] [Google Scholar]
- 427.Chen C-J, Jin Q, Liu G-Y, Li D-D, Wang J-L, Ji J. Reversibly light-responsive micelles constructed via a simple modification of hyperbranched polymers with chromophores. Polymer. 2012;53:3695–3703. [Google Scholar]
- 428.Son S, Shin E, Kim BS. Light-responsive micelles of spiropyran initiated hyperbranched polyglycerol for smart drug delivery. Biomacromolecules. 2014;15:628–634. doi: 10.1021/bm401670t. [DOI] [PubMed] [Google Scholar]
- 429.Yang H, Jia L, Wang Z, Di-Cicco Al, Lévy D, Keller P. Novel Photolabile Diblock Copolymers Bearing Truxillic Acid Derivative Junctions. Macromolecules. 2011;44:159–165. [Google Scholar]
- 430.Lendlein A, Jiang H, Jünger O, Langer R. Light-induced shape-memory polymers. Nature. 2005;434:879–882. doi: 10.1038/nature03496. [DOI] [PubMed] [Google Scholar]
- 431.Klinger M, Tolbod LP, Gothelf KV, Ogilby PR. Effect of polymer cross-links on oxygen diffusion in glassy PMMA films. ACS Appl Mater Interfaces. 2009;1:661–667. doi: 10.1021/am800197j. [DOI] [PubMed] [Google Scholar]
- 432.Jayakumar MK, Idris NM, Zhang Y. Remote activation of biomolecules in deep tissues using near-infrared-to-UV upconversion nanotransducers. Proc Natl Acad Sci U S A. 2012;109:8483–8488. doi: 10.1073/pnas.1114551109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Smith AM, Mancini MC, Nie S. Bioimaging: second window for in vivo imaging. Nat Nanotechnol. 2009;4:710–711. doi: 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Fodor L, Ullmann Y, Elman M. Aesthetic Applications of Intense Pulsed Light. Springer; New York: 2011. pp. 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 436.Liu G-Y, Chen C-J, Li D-D, Wang S-S, Ji J. Near-infrared light-sensitive micelles for enhanced intracellular drug delivery. J Mater Chem. 2012;22:16865. [Google Scholar]
- 437.de Gracia Lux C, Joshi-Barr S, Sankaranarayanan J, Fomina N, Almutairi A, McFearin CL. Single UV or Near IR Triggering Event Leads to Polymer Degradation into Small Molecules. ACS Macro Lett. 2012;1:922–926. doi: 10.1021/mz3002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Haase M, Schafer H. Upconverting nanoparticles. Angew Chem Int Ed. 2011;50:5808–5829. doi: 10.1002/anie.201005159. [DOI] [PubMed] [Google Scholar]
- 439.Fomina N, Sermsakdi M, Edigin O, Almutairi A, McFearin C. UV and Near-IR Triggered Release from Polymeric Nanoparticles. J Am Chem Soc. 2010;132:9540–9542. doi: 10.1021/ja102595j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Viger ML, Grossman M, Fomina N, Almutairi A. Low Power Upconverted Near-IR Light for Efficient Polymeric Nanoparticle Degradation and Cargo Release. Adv Mater. 2013;25:3733–3738. doi: 10.1002/adma.201300902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Leighton TG. What is ultrasound? Prog Biophys Mol Biol. 2007;93:3–83. doi: 10.1016/j.pbiomolbio.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 442.Ashihara K, Kurakata K, Mizunami T, Matsushita K. Hearing threshold for pure tones above 20 kHz. Acoust Sci Technol. 2006;27:12–19. [Google Scholar]
- 443.Husseini GA, Pitt WG. Ultrasonic-activated micellar drug delivery for cancer treatment. J Pharm Sci. 2009;98:795–811. doi: 10.1002/jps.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Pitt WG, Husseini GA, Kherbeck LN. Smart Materials for Drug Delivery. Royal Society of Chemistry; Letchworth, UK: 2013. pp. 148–178. [Google Scholar]
- 445.Kennedy JE. High-intensity focused ultrasound in the treatment of solid tumours. Nat Rev Cancer. 2005;5:321–327. doi: 10.1038/nrc1591. [DOI] [PubMed] [Google Scholar]
- 446.Geers B, Dewitte H, De Smedt SC, Lentacker I. Crucial factors and emerging concepts in ultrasound-triggered drug delivery. J Controlled Release. 2012;164:248–255. doi: 10.1016/j.jconrel.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 447.Ahmed SE, Martins AM, Husseini GA. The use of ultrasound to release chemotherapeutic drugs from micelles and liposomes. J Drug Target. 2015;23:16–42. doi: 10.3109/1061186X.2014.954119. [DOI] [PubMed] [Google Scholar]
- 448.Marmottant P, Hilgenfeldt S. Controlled vesicle deformation and lysis by single oscillating bubbles. Nature. 2003;423:153–156. doi: 10.1038/nature01613. [DOI] [PubMed] [Google Scholar]
- 449.Husseini GA, Pitt WG. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv Drug Delivery Rev. 2008;60:1137–1152. doi: 10.1016/j.addr.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Husseini GA, Pitt WG, Martins AM. Ultrasonically triggered drug delivery: breaking the barrier. Colloids Surf B. 2014;123:364–386. doi: 10.1016/j.colsurfb.2014.07.051. [DOI] [PubMed] [Google Scholar]
- 451.Rapoport N. Phase-shift, stimuli-responsive perfluorocarbon nanodroplets for drug delivery to cancer. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4:492–510. doi: 10.1002/wnan.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Rapoport N, Nam KH, Gupta R, Gao Z, Mohan P, Payne A, Todd N, Liu X, Kim T, Shea J, Scaife C, Parker DL, Jeong EK, Kennedy AM. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. J Controlled Release. 2011;153:4–15. doi: 10.1016/j.jconrel.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Gao Z, Fain HD, Rapoport N. Ultrasound-Enhanced Tumor Targeting of Polymeric Micellar Drug Carriers. Mol Pharmaceutics. 2004;1:317–330. doi: 10.1021/mp049958h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Husseini GA, Myrup GDM, Pitt WG, Christensen DA, Rapoport NY. Factors affecting acoustically triggered release of drugs from polymeric micelles. J Controlled Release. 2000;69:43–52. doi: 10.1016/s0168-3659(00)00278-9. [DOI] [PubMed] [Google Scholar]
- 455.Zhang H, Xia H, Wang J, Li Y. High intensity focused ultrasound-responsive release behavior of PLA-b-PEG copolymer micelles. J Controlled Release. 2009;139:31–39. doi: 10.1016/j.jconrel.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 456.Davis DA, Hamilton A, Yang J, Cremar LD, Van Gough D, Potisek SL, Ong MT, Braun PV, Martinez TJ, White SR, Moore JS, Sottos NR. Force-induced activation of covalent bonds in mechanoresponsive polymeric materials. Nature. 2009;459:68–72. doi: 10.1038/nature07970. [DOI] [PubMed] [Google Scholar]
- 457.Potisek SL, Davis DA, Sottos NR, White SR, Moore JS. Mechanophore-Linked Addition Polymers. J Am Chem Soc. 2007;129:13808–13809. doi: 10.1021/ja076189x. [DOI] [PubMed] [Google Scholar]
- 458.Caruso MM, Davis DA, Shen Q, Odom SA, Sottos NR, White SR, Moore JS. Mechanically-Induced Chemical Changes in Polymeric Materials. Chem Rev. 2009;109:5755–5798. doi: 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]
- 459.Berkowski K, Potisek SL, Hickenboth CR, Moore JS. Ultrasound-Induced Site-Specific Cleavage of Azo-Functionalized Poly(ethylene glycol) Macromolecules. 2005;38:8975–8978. [Google Scholar]
- 460.Hickenboth CR, Moore JS, White SR, Sottos NR, Baudry J, Wilson SR. Biasing reaction pathways with mechanical force. Nature. 2007;446:423–427. doi: 10.1038/nature05681. [DOI] [PubMed] [Google Scholar]
- 461.Black Ramirez AL, Ogle JW, Schmitt AL, Lenhardt JM, Cashion MP, Mahanthappa MK, Craig SL. Microstructure of Copolymers Formed by the Reagentless, Mechanochemical Remodeling of Homopolymers via Pulsed Ultrasound. ACS Macro Lett. 2012;1:23–27. doi: 10.1021/mz200005u. [DOI] [PubMed] [Google Scholar]
- 462.Wang J, Pelletier M, Zhang H, Xia H, Zhao Y. High-frequency ultrasound-responsive block copolymer micelle. Langmuir. 2009;25:13201–13205. doi: 10.1021/la9018794. [DOI] [PubMed] [Google Scholar]
- 463.Xuan J, Boissiere O, Zhao Y, Yan B, Tremblay L, Lacelle S, Xia H, Zhao Y. Ultrasound-responsive block copolymer micelles based on a new amplification mechanism. Langmuir. 2012;28:16463–16468. doi: 10.1021/la303946b. [DOI] [PubMed] [Google Scholar]
- 464.Li Y, Tong R, Xia H, Zhang H, Xuan J. High intensity focused ultrasound and redox dual responsive polymer micelles. Chem Commun. 2010;46:7739–7741. doi: 10.1039/c0cc02628j. [DOI] [PubMed] [Google Scholar]
- 465.Tong R, Lu X, Xia H. A facile mechanophore functionalization of an amphiphilic block copolymer towards remote ultrasound and redox dual stimulus responsiveness. Chem Commun. 2014;50:3575–3578. doi: 10.1039/c4cc00103f. [DOI] [PubMed] [Google Scholar]
- 466.Skinner HA. A Revision of Some Bond-Energy Values and the Variation of Bond-Energy with Bond-Length. Trans Faraday Soc. 1945;41:645–662. [Google Scholar]
- 467.Schafer FQ, Buettner GR. Redox environment of the as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 468.Saito G, Lee K-D, Swanson JA. Drug delivery strategy utilizing conjugation via reversible disulfide linkages- role and site of cellular reducing activities. Adv Drug Delivery Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 469.Huo M, Yuan J, Tao L, Wei Y. Redox-responsive polymers for drug delivery: from molecular design to applications. Polym Chem. 2014;5:1519–1528. [Google Scholar]
- 470.Manickam DS, Li J, Putt DA, Zhou QH, Wu C, Lash LH, Oupicky D. Effect of innate glutathione levels on activity of redox-responsive gene delivery vectors. J Controlled Release. 2010;141:77–84. doi: 10.1016/j.jconrel.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Kuppusamy P, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB, Li H. Noninvasive Imaging of Tumor Redox Status and Its Modification by Tissue Glutathione Levels. J Cancer Res. 2002;62:307–312. [PubMed] [Google Scholar]
- 472.Wu G, Yang S, Turner ND, Lupton ND, Fang YZ. Glutathione Metabolism and Its Implications for Health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 473.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta, Gen Subj. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Arunachalam B, Geuze HJ, Cresswell P, Phan UT. Enzymatic reduction of disulfide bonds in lysosomes- Characterization of a Gamma-interferon-inducible lysosomal thiol reductase (GILT) Proc Natl Acad Sci U S A. 2000;97:745–750. doi: 10.1073/pnas.97.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Kurz T, Eaton JW, Brunk UT. Redox Activity Within the Lysosomal Compartment: Implications for Aging and Apoptosis. Antioxid Redox Signaling. 2010;13:511–523. doi: 10.1089/ars.2009.3005. [DOI] [PubMed] [Google Scholar]
- 476.Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Controlled Release. 2011;152:2–12. doi: 10.1016/j.jconrel.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 477.Son S, Kim J, Singha K, Kim WJ, Namgung R. Bioreducible Polymers for Gene Silencing and Delivery. Acc Chem Res. 2012;45:1100–1112. doi: 10.1021/ar200248u. [DOI] [PubMed] [Google Scholar]
- 478.Bauhuber S, Hozsa C, Breunig M, Gopferich A. Delivery of nucleic acids via disulfide-based carrier systems. Adv Mater. 2009;21:3286–3306. doi: 10.1002/adma.200802453. [DOI] [PubMed] [Google Scholar]
- 479.Xu H, Cao W, Zhang X. Selenium-Containing Polymers-Promising Biomaterials for Controlled Release and Enzyme Mimics. Acc Chem Res. 2013;46:1647–1658. doi: 10.1021/ar4000339. [DOI] [PubMed] [Google Scholar]
- 480.Fleige E, Quadir MA, Haag R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv Drug Delivery Rev. 2012;64:866–884. doi: 10.1016/j.addr.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 481.Li J, Huo M, Wang J, Zhou J, Mohammad JM, Zhang Y, Zhu Q, Waddad AY, Zhang Q. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33:2310–2320. doi: 10.1016/j.biomaterials.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 482.Xu M, Qian J, Suo A, Wang H, Yong X, Liu X, Liu R. Reduction/pH dual-sensitive PEGylated hyaluronan nanoparticles for targeted doxorubicin delivery. Carbohydr Polym. 2013;98:181–188. doi: 10.1016/j.carbpol.2013.05.077. [DOI] [PubMed] [Google Scholar]
- 483.Jia L, Li Z, Zhang D, Zhang Q, Shen J, Guo H, Tian X, Liu G, Zheng D, Qi L. Redox-responsive catiomer based on PEG-ss-chitosan oligosaccharide-ss-polyethylenimine copolymer for effective gene delivery. Polym Chem. 2013;4:156–165. [Google Scholar]
- 484.Thambi T, You DG, Han HS, Deepagan VG, Jeon SM, Suh YD, Choi KY, Kim K, Kwon IC, Yi G-R, Lee JY, Lee DS, Park JH. Bioreducible Carboxymethyl Dextran Nanoparticles for Tumor-Targeted Drug Delivery. Adv Healthcare Mater. 2014;3:1829–1838. doi: 10.1002/adhm.201300691. [DOI] [PubMed] [Google Scholar]
- 485.Kim SH, Jeong JH, Kim TI, Kim SW, Bull DA. VEGF siRNA delivery system using arginine-grafted bioreducible poly(disulfide amine) Mol Pharmaceutics. 2009;6:718–726. doi: 10.1021/mp800161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Vader P, van der Aa LJ, Engbersen JF, Storm G, Schiffelers RM. Disulfide-based poly(amido amine)s for siRNA delivery: effects of structure on siRNA complexation, cellular uptake gene silencing and toxicity. Pharm Res. 2011;28:1013–1022. doi: 10.1007/s11095-010-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Stevenson M, Ramos-Perez V, Singh S, Soliman M, Preece JA, Briggs SS, Read ML, Seymour LW. Delivery of siRNA mediated by histidine-containing reducible polycations. J Controlled Release. 2008;130:46–56. doi: 10.1016/j.jconrel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 488.Takemoto H, Ishii A, Miyata K, Nakanishi M, Oba M, Ishii T, Yamasaki Y, Nishiyama N, Kataoka K. Polyion complex stability and gene silencing efficiency with a siRNA-grafted polymer delivery system. Biomaterials. 2010;31:8097–8105. doi: 10.1016/j.biomaterials.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 489.Romberg B, Hennink WE, Storm G. Sheddable coatings for long-circulating nanoparticles. Pharm Res. 2008;25:55–71. doi: 10.1007/s11095-007-9348-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Sun H, Cheng R, Meng F, Liu H, Zhong Z, Guo B. Biodegradable micelles with sheddable poly(ethylene glycol) shells for triggered intracellular release of doxorubicin. Biomaterials. 2009;30:6358–6366. doi: 10.1016/j.biomaterials.2009.07.051. [DOI] [PubMed] [Google Scholar]
- 491.Wang W, Sun H, Meng F, Ma S, Liu H, Zhong Z. Precise control of intracellular drug release and anti-tumor activity of biodegradable micellar drugs via reduction-sensitive shell-shedding. Soft Matter. 2012;8:3949. [Google Scholar]
- 492.Zhong Y, Yang W, Sun H, Cheng R, Meng F, Deng C, Zhong Z. Ligand-directed reduction-sensitive shell-sheddable biodegradable micelles actively deliver doxorubicin into the nuclei of target cancer cells. Biomacromolecules. 2013;14:3723–3730. doi: 10.1021/bm401098w. [DOI] [PubMed] [Google Scholar]
- 493.Chen W, Zhong P, Meng F, Cheng R, Deng C, Feijen J, Zhong Z. Redox and pH-responsive degradable micelles for dually activated intracellular anticancer drug release. J Controlled Release. 2013;169:171–179. doi: 10.1016/j.jconrel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 494.Ren T-B, Feng Y, Zhang Z-H, Li L, Li Y-Y. Shell-sheddable micelles based on star-shaped poly(ε-caprolactone)-SS-poly(ethyl glycol) copolymer for intracellular drug release. Soft Matter. 2011;7:2329. [Google Scholar]
- 495.Shi C, Guo X, Qu Q, Tang Z, Wang Y, Zhou S. Actively targeted delivery of anticancer drug to tumor cells by redox-responsive star-shaped micelles. Biomaterials. 2014;35:8711–8722. doi: 10.1016/j.biomaterials.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 496.Song N, Liu W, Tu Q, Liu R, Zhang Y, Wang J. Preparation and in vitro properties of redox-responsive polymeric nanoparticles for paclitaxel delivery. Colloids Surf B. 2011;87:454–463. doi: 10.1016/j.colsurfb.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 497.Wen HY, Dong HQ, Xie WJ, Li YY, Wang K, Pauletti GM, Shi DL. Rapidly disassembling nanomicelles with disulfide-linked PEG shells for glutathione-mediated intracellular drug delivery. Chem Commun. 2011;47:3550–3552. doi: 10.1039/c0cc04983b. [DOI] [PubMed] [Google Scholar]
- 498.Thambi T, Yoon HY, Kim K, Kwon IC, Yoo CK, Park JH. Bioreducible block copolymers based on poly(ethylene glycol) and poly(gamma-benzyl L-glutamate) for intracellular delivery of camptothecin. Bioconjugate Chem. 2011;22:1924–1931. doi: 10.1021/bc2000963. [DOI] [PubMed] [Google Scholar]
- 499.Liu J, Pang Y, Huang W, Huang X, Meng L, Zhu X, Zhou Y, Yan D. Bioreducible micelles self-assembled from amphiphilic hyperbranched multiarm copolymer for glutathione-mediated intracellular drug delivery. Biomacromolecules. 2011;12:1567–1577. doi: 10.1021/bm200275j. [DOI] [PubMed] [Google Scholar]
- 500.Wang YC, Wang F, Sun TM, Wang J. Redox-responsive nanoparticles from the single disulfide bond-bridged block copolymer as drug carriers for overcoming multidrug resistance in cancer cells. Bioconjugate Chem. 2011;22:1939–1945. doi: 10.1021/bc200139n. [DOI] [PubMed] [Google Scholar]
- 501.Tang L-Y, Li Y, Du J, Wang J, Wang Y-C. Shell-Detachable Micelles Based on Disulfide-Linked Block Copolymer As Potential Carrier for Intracellular Drug Delivery. Bioconjugate Chem. 2009;20:1095–1099. doi: 10.1021/bc900144m. [DOI] [PubMed] [Google Scholar]
- 502.Sun H, Li X, Cheng R, Meng F, Liu H, Zhong Z, Guo B. Shell-Sheddable Micelles Based on Dextran-SS-Poly(ε-caprolactone) Diblock Copolymer for Efficient Intracellular Release of Doxorubicin. Biomacromolecules. 2010;11:848–854. doi: 10.1021/bm1001069. [DOI] [PubMed] [Google Scholar]
- 503.Li W, Zhang P, Zheng K, Hu Q, Wang Y. Redox-triggered intracellular dePEGylation based on diselenide-linked polycations for DNA delivery. J Mater Chem B. 2013;1:6418. doi: 10.1039/c3tb21241f. [DOI] [PubMed] [Google Scholar]
- 504.Lin C, Zhong Z, Lok MC, Jiang X, Hennink WE, Feijen J, Engbersen JF. Linear poly(amido amine)s with secondary and tertiary amino groups and variable amounts of disulfide linkages: synthesis and in vitro gene transfer properties. J Controlled Release. 2006;116:130–137. doi: 10.1016/j.jconrel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 505.Martello F, Piest M, Engbersen JF, Ferruti P. Effects of branched or linear architecture of bioreducible poly(amido amine)s on their in vitro gene delivery properties. J Controlled Release. 2012;164:372–379. doi: 10.1016/j.jconrel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 506.van der Aa LJ, Vader P, Storm G, Schiffelers RM, Engbersen JF. Intercalating quaternary nicotinamide-based poly(amido amine)s for gene delivery. J Controlled Release. 2014;195:11–20. doi: 10.1016/j.jconrel.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 507.Chen J, Qiu X, Ouyang J, Kong J, Zhong W, Xing MM. pH and reduction dual-sensitive copolymeric micelles for intracellular doxorubicin delivery. Biomacromolecules. 2011;12:3601–3611. doi: 10.1021/bm200804j. [DOI] [PubMed] [Google Scholar]
- 508.Chen J, Zehtabi F, Ouyang J, Kong J, Zhong W, Xing MMQ. Reducible self-assembled micelles for enhanced intracellular delivery of doxorubicin. J Mater Chem. 2012;22:7121. [Google Scholar]
- 509.Kozielski KL, Tzeng SY, Hurtado De Mendoza BA, Green JJ. Bioreducible Cationic Polymer-Based Nanoparticles for Efficient and Environmentally Triggered Cytoplasmic siRNA Delivery to Primary Human Brain Cancer Cells. ACS Nano. 2014;8:3232–3241. doi: 10.1021/nn500704t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Kozielski KL, Tzeng SY, Green JJ. A bioreducible linear poly(beta-amino ester) for siRNA delivery. Chem Commun. 2013;49:5319–5321. doi: 10.1039/c3cc40718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Zhu W, Wang Y, Cai X, Zha G, Luo Q, Sun R, Li X, Shen Z. Reduction-Triggered Release of Paclitaxel From In Situ Formed Biodegradable Core-Crosslinked Micelles. J Mater Chem B. 2015;3:3024–3031. doi: 10.1039/c4tb01834f. [DOI] [PubMed] [Google Scholar]
- 512.Yan L, Wu W, Zhao W, Qi R, Cui D, Xie Z, Huang Y, Tong T, Jing X. Reduction-sensitive core-cross-linked mPEG–poly(ester-carbonate) micelles for glutathione-triggered intracellular drug release. Polym Chem. 2012;3:2403. [Google Scholar]
- 513.Wu L, Zou Y, Deng C, Cheng R, Meng F, Zhong Z. Intracellular release of doxorubicin from core-crosslinked polypeptide micelles triggered by both pH and reduction conditions. Biomaterials. 2013;34:5262–5272. doi: 10.1016/j.biomaterials.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 514.Zhong Y, Zhang J, Cheng R, Deng C, Meng F, Xie F, Zhong Z. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Controlled Release. 2015;205:144–154. doi: 10.1016/j.jconrel.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 515.Yu S, Ding J, He C, Cao Y, Xu W, Chen X. Disulfide Cross-Linked Polyurethane Micelles as a Reduction-Triggered Drug Delivery System for Cancer Therapy. Adv Healthcare Mater. 2014;3:752–760. doi: 10.1002/adhm.201300308. [DOI] [PubMed] [Google Scholar]
- 516.Li Y, Wang S, Zhu D, Shen Y, Du B, Liu X, Zheng Y. Reversibly cross-linked poly(ethylene glycol) –poly(amino acid)s copolymer micelles: a promising approach to overcome the extracellular stability versus intracellular drug release challenge. RSC Adv. 2015;5:20025–20034. [Google Scholar]
- 517.Yue J, Wang R, Liu S, Wu S, Xie Z, Huang Y, Jing X. Reduction-responsive shell-crosslinked micelles prepared from Y-shaped amphiphilic block copolymers as a drug carrier. Soft Matter. 2012;8:7426. [Google Scholar]
- 518.Kietzmann T. Intracellular Redox Compartments: Mechanisms and Significances. Antioxid Redox Signaling. 2010;13:395–398. doi: 10.1089/ars.2009.3001. [DOI] [PubMed] [Google Scholar]
- 519.Bystrom LM, Guzman ML, Rivella S. Iron and Reactive Oxygen Species: Friends or Foes of Cancer Cells? Antioxid Redox Signaling. 2014;20:1917–1924. doi: 10.1089/ars.2012.5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Phillips D, Gibson MI. Redox-Sensitive Materials for Drug Delivery: Targeting the Correct Intracellular Environment, Tuning Release Rates and Appropriate Predictive Systems. Antioxid Redox Signaling. 2014;21:786–803. doi: 10.1089/ars.2013.5728. [DOI] [PubMed] [Google Scholar]
- 521.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122:339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 522.Lee SH, Boire TC, Lee JB, Gupta MK, Zachman AL, Rath R, Sung HJ. ROS-cleavable proline oligomer crosslinking of polycaprolactone for pro-angiogenic host response. J Mater Chem B. 2014;2:7109–7113. doi: 10.1039/C4TB01094A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Colson YL, Grinstaff MW. Biologically Responsive Polymeric Nanoparticles for Drug Delivery. Adv Mater. 2012;24:3878–3886. doi: 10.1002/adma.201200420. [DOI] [PubMed] [Google Scholar]
- 524.Sugamura K, Keaney JF., Jr Reactive oxygen species in cardiovascular disease. Free Radical Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 526.Rastew E, Vicente JB, Singh U. Oxidative stress resistance genes contribute to the pathogenic potential of the anaerobic protozoan parasite, Entamoeba histolytica. Int J Parasitol. 2012;42:1007–1015. doi: 10.1016/j.ijpara.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Rahman I. Oxidative Stress, Chromatin Remodeling and Gene Transcription in Inflammation and Chronic Lung Diseases. J Biochem Mol Biol. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- 528.Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, Alcaide P, Grabie N, Luscinskas FW, Croce KJ, Lichtman AH. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. 2012;188:6287–6299. doi: 10.4049/jimmunol.1200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 530.Yu BP. Cellular Defenses Against Damage From Reactive Oxygen Species. Physiol Rev. 1994;74:139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 531.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signaling. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Ibanez IL, Notcovich C, Catalano PN, Bellino MG, Duran H. The redox-active nanomaterial toolbox for cancer therapy. Cancer Lett. 2015;359:9–19. doi: 10.1016/j.canlet.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 533.Napoli A, Valentini M, Tirelli N, Muller M, Hubbell JA. Oxidation-responsive polymeric vesicles. Nat Mater. 2004;3:183–189. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- 534.Poole KM, Nelson CE, Joshi RV, Martin JR, Gupta MK, Haws SC, Kavanaugh TE, Skala MC, Duvall CL. ROS-responsive microspheres for on demand antioxidant therapy in a model of diabetic peripheral arterial disease. Biomaterials. 2015;41:166–175. doi: 10.1016/j.biomaterials.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Ma N, Xu H, Wang Z, Zhang Xi, Li Y. Dual Redox Responsive Assemblies Formed from Diselenide Block Copolymers. J Am Chem Soc. 2010;132:442–443. doi: 10.1021/ja908124g. [DOI] [PubMed] [Google Scholar]
- 536.Su Z, Chen M, Xiao Y, Sun M, Zong L, Asghar S, Dong M, Li H, Ping Q, Zhang C. ROS-triggered and regenerating anticancer nanosystem: an effective strategy to subdue tumor’s multidrug resistance. J Controlled Release. 2014;196:370–383. doi: 10.1016/j.jconrel.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 537.Zhang D, Wei Y, Chen K, Zhang X, Xu X, Shi Q, Han S, Chen X, Gong H, Li X, Zhang J. Biocompatible reactive oxygen species (ROS)-responsive nanoparticles as superior drug delivery vehicles. Adv Healthcare Mater. 2015;4:69–76. doi: 10.1002/adhm.201400299. [DOI] [PubMed] [Google Scholar]
- 538.Broaders KE, Grandhe S, Frechet JM. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J Am Chem Soc. 2011;133:756–758. doi: 10.1021/ja110468v. [DOI] [PubMed] [Google Scholar]
- 539.Aguirre-Chagala YE, Aguilar-Castillo BA, Herrera-Alonso M, Santos JL. Synthesis of Copolymers from Phenylboronic Acid-Installed Cyclic Carbonates. ACS Macro Lett. 2014;3:353–358. doi: 10.1021/mz500047p. [DOI] [PubMed] [Google Scholar]
- 540.Song C-C, Du F-S, Li Z-C. Oxidation-responsive polymers for biomedical applications. J Mater Chem B. 2014;2:3413. doi: 10.1039/c3tb21725f. [DOI] [PubMed] [Google Scholar]
- 541.Lee SH, Gupta MK, Bang JB, Bae H, Sung H-J. Current Progress in Reactive Oxygen Species (ROS)-Responsive Materials for Biomedical Applications. Adv Healthcare Mater. 2013;2:908–915. doi: 10.1002/adhm.201200423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Xiao C, Ding J, Ma L, Yang C, Zhuang X, Chen X. Synthesis of thermal and oxidation dual responsive polymers for reactive oxygen species (ROS)-triggered drug release. Polym Chem. 2015;6:738–747. [Google Scholar]
- 543.Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9:923–928. doi: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- 545.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 546.Vaupel P, Kallinowski F, Okunieff P. Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors—A Review. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 547.Gao W, Chan JM, Farokhzad OC. pH-Responsive Nanoparticles for Drug Delivery. Mol Pharmaceutics. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Controlled Release. 2014;190:485–499. doi: 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Quadir MA, Morton SW, Deng ZJ, Shopsowitz KE, Murphy RP, Epps TH, 3rd, Hammond PT. PEG-polypeptide block copolymers as pH-responsive endosome-solubilizing drug nanocarriers. Mol Pharmaceutics. 2014;11:2420–2430. doi: 10.1021/mp500162w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Boussif O, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr J-P, Lezoualc’h F. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo- Polyethylenimine. Proc Natl Acad Sci U S A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Guidry EN, Farand J, Soheili A, Parish CA, Kevin NJ, Pipik B, Calati KB, Ikemoto N, Waldman JH, Latham AH, Howell BJ, Leone A, Garbaccio RM, Barrett SE, Parmar RG, Truong QT, Mao B, Davies IW, Colletti SL, Sepp-Lorenzino L. Improving the in vivo therapeutic index of siRNA polymer conjugates through increasing pH responsiveness. Bioconjugate Chem. 2014;25:296–307. doi: 10.1021/bc400442p. [DOI] [PubMed] [Google Scholar]
- 552.Deng H, Liu J, Zhao X, Zhang Y, Liu J, Xu S, Deng L, Dong A, Zhang J. PEG-b-PCL copolymer micelles with the ability of pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. Biomacromolecules. 2014;15:4281–4292. doi: 10.1021/bm501290t. [DOI] [PubMed] [Google Scholar]
- 553.Kang Z, Zhang Z, Zhang M, Wong N-b, Zapien YS, Lee S-T, Tsang CHA, Shan Y. A Polyoxometalate-Assisted Electrochemical Method for Silicon Nanostructures Preparation- From Quantum Dots to Nanowires. J Am Chem Soc. 2007;129:5326–5327. doi: 10.1021/ja068894w. [DOI] [PubMed] [Google Scholar]
- 554.Lee Y, Miyata K, Oba M, Ishii T, Fukushima S, Han M, Koyama H, Nishiyama N, Kataoka K. Charge-conversion ternary polyplex with endosome disruption moiety: a technique for efficient and safe gene delivery. Angew Chem Int Ed. 2008;47:5163–5166. doi: 10.1002/anie.200800963. [DOI] [PubMed] [Google Scholar]
- 555.Jiang H, Taranekar P, Reynolds JR, Schanze KS. Conjugated polyelectrolytes: synthesis photophysics, and applications. Angew Chem Int Ed. 2009;48:4300–4316. doi: 10.1002/anie.200805456. [DOI] [PubMed] [Google Scholar]
- 556.Chen J, Ding J, Zhang Y, Xiao C, Zhuang X, Chen X. Polyion complex micelles with gradient pH-sensitivity for adjustable intracellular drug delivery. Polym Chem. 2015;6:397–405. [Google Scholar]
- 557.Binauld S, Stenzel MH. Acid-degradable polymers for drug delivery: a decade of innovation. Chem Commun. 2013;49:2082–2102. doi: 10.1039/c2cc36589h. [DOI] [PubMed] [Google Scholar]
- 558.Wei H, Zhuo R-X, Zhang X-Z. Design and development of polymeric micelles with cleavable links for intracellular drug delivery. Prog Polym Sci. 2013;38:503–535. [Google Scholar]
- 559.Petrova S, Jäger E, Konefał R, Jäger A, Venturini CG, Spěváček J, Pavlova E, Štěpánek P. Novel poly(ethylene oxide monomethyl ether)-b-poly(ε-caprolactone) diblock copolymers containing a pH-acid labile ketal group as a block linkage. Polym Chem. 2014;5:3884. [Google Scholar]
- 560.Tomlinson R, Klee M, Garrett S, Heller J, Duncan R, Brocchini S. Pendent Chain Functionalized Polyacetals That Display pH-Dependent Degradation- A Platform for the Development of Novel Polymer Therapeutics. Macromolecules. 2002;35:473–480. [Google Scholar]
- 561.Paramonov P, Bachelder EM, Beaudette TT, Standley SM, Lee CC, Dashe J, Fréchet JMJ. Fully Acid-Degradable Biocompatible Polyacetal Microparticles for Drug Delivery. Bioconjugate Chem. 2008;19:911–919. doi: 10.1021/bc7004472. [DOI] [PubMed] [Google Scholar]
- 562.Shenoi RA, Lai BF, Imran ul-haq M, Brooks DE, Kizhakkedathu JN. Biodegradable polyglycerols with randomly distributed ketal groups as multi-functional drug delivery systems. Biomaterials. 2013;34:6068–6081. doi: 10.1016/j.biomaterials.2013.04.043. [DOI] [PubMed] [Google Scholar]
- 563.Tonhauser C, Dingels C, Frey H, Schull C. Branched Acid-Degradable, Biocompatible Polyether Copolymers via Anionic Ring-Opening Polymerization Using an Epoxide Inimer. ACS Macro Lett. 2012;1:1094–1097. doi: 10.1021/mz300265z. [DOI] [PubMed] [Google Scholar]
- 564.Shenoi RA, Narayanannair JK, Hamilton JL, Lai BF, Horte S, Kainthan RK, Varghese JP, Rajeev KG, Manoharan M, Kizhakkedathu JN. Branched multifunctional polyether polyketals: variation of ketal group structure enables unprecedented control over polymer degradation in solution and within cells. J Am Chem Soc. 2012;134:14945–14957. doi: 10.1021/ja305080f. [DOI] [PubMed] [Google Scholar]
- 565.Schopf E, Sankaranarayanan J, Chan M, Mattrey R, Almutairi A. An extracellular MRI polymeric contrast agent that degrades at physiological pH. Mol Pharmaceutics. 2012;9:1911–1918. doi: 10.1021/mp2005998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Li L, Xu Y, Milligan I, Fu L, Franckowiak EA, Du W. Synthesis of highly pH-responsive glucose poly(orthoester) Angew Chem, Int Ed. 2013;52:13699–13702. doi: 10.1002/anie.201306391. [DOI] [PubMed] [Google Scholar]
- 567.Jin Y, Song L, Su Y, Zhu L, Pang Y, Qiu F, Tong G, Yan D, Zhu B, Zhu X. Oxime linkage: a robust tool for the design of pH-sensitive polymeric drug carriers. Biomacromolecules. 2011;12:3460–3468. doi: 10.1021/bm200956u. [DOI] [PubMed] [Google Scholar]
- 568.Ganivada MN, Rao NV, Dinda H, Kumar P, Das Sarma J, Shunmugam R. Biodegradable Magnetic Nanocarrier for Stimuli Responsive Drug Release. Macromolecules. 2014;47:2703–2711. [Google Scholar]
- 569.Wang H, Wang Y, Chen Y, Jin Q, Ji J. A biomimic pH-sensitive polymeric prodrug based on polycarbonate for intracellular drug delivery. Polym Chem. 2014;5:854–861. [Google Scholar]
- 570.Du JZ, Du XJ, Mao CQ, Wang J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J Am Chem Soc. 2011;133:17560–17563. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 571.Yang B, Lv Y, Zhu JY, Han YT, Jia HZ, Chen WH, Feng J, Zhang XZ, Zhuo RX. A pH-responsive drug nanovehicle constructed by reversible attachment of cholesterol to PEGylated poly(l-lysine) via catechol-boronic acid ester formation. Acta Biomater. 2014;10:3686–3695. doi: 10.1016/j.actbio.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 572.Aguirre-Chagala YE, Santos JL, Huang Y, Herrera-Alonso M. Thermoresponsive nanostructured polycarbonate block copolymers as biodegradable therapeutic delivery carriers. ACS Macro Lett. 2014;3:1249–1253. doi: 10.1021/mz500594m. [DOI] [PubMed] [Google Scholar]
- 573.Yu Y, Chen C-K, Law W-C, Sun H, Prasad PN, Cheng C. A degradable brush polymer–drug conjugate for pH-responsive release of doxorubicin. Polym Chem. 2015;6:953–961. [Google Scholar]
- 574.Ling D, Park W, Park SJ, Lu Y, Kim KS, Hackett MJ, Kim BH, Yim H, Jeon YS, Na K, Hyeon T. Multifunctional tumor pH-sensitive self-assembled nanoparticles for bimodal imaging and treatment of resistant heterogeneous tumors. J Am Chem Soc. 2014;136:5647–5655. doi: 10.1021/ja4108287. [DOI] [PubMed] [Google Scholar]
- 575.Liu Q, Chen X, Jia J, Zhang W, Yang T, Wang L, Ma G. pH-Responsive Poly(d,l-lactic-co-glycolic acid) Nanoparticles with Rapid Antigen Release Behavior Promote Immune Response. ACS Nano. 2015;9:4925–4938. doi: 10.1021/nn5066793. [DOI] [PubMed] [Google Scholar]
- 576.Dreaden EC, Morton SW, Shopsowitz KE, Choi JH, Deng ZJ, Cho NJ, Hammond PT. Bimodal tumor-targeting from microenvironment responsive hyaluronan layer-by-layer (LbL) nanoparticles. ACS Nano. 2014;8:8374–8382. doi: 10.1021/nn502861t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Morton SW, Poon Z, Hammond PT. The architecture and biological performance of drug-loaded LbL nanoparticles. Biomaterials. 2013;34:5328–5335. doi: 10.1016/j.biomaterials.2013.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Costa RR, Alatorre-Meda M, Mano JF. Drug nano-reservoirs synthesized using layer-by-layer technologies. Biotechnol Adv. 2015;33:1310–1326. doi: 10.1016/j.biotechadv.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 579.Radovic-Moreno AF, Lu TK, Puscasu VA, Yoon CJ, Langer R, Farokhzad OC. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano. 2012;6:4279–4287. doi: 10.1021/nn3008383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Yoshida T, Lai TC, Kwon GS, Sako K. pH- and ion-sensitive polymers for drug delivery. Expert Opin Drug Delivery. 2013;10:1497–1513. doi: 10.1517/17425247.2013.821978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Gao GH, Li Y, Lee DS. Environmental pH-sensitive polymeric micelles for cancer diagnosis and targeted therapy. J Controlled Release. 2013;169:180–184. doi: 10.1016/j.jconrel.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 582.Colson YL, Grinstaff MW. Biologically responsive polymeric nanoparticles for drug delivery. Adv Mater. 2012;24:3878–3886. doi: 10.1002/adma.201200420. [DOI] [PubMed] [Google Scholar]
- 583.Sung HW, Sonaje K, Liao ZX, Hsu LW, Chuang EY. pH-responsive nanoparticles shelled with chitosan for oral delivery of insulin: from mechanism to therapeutic applications. Acc Chem Res. 2012;45:619–629. doi: 10.1021/ar200234q. [DOI] [PubMed] [Google Scholar]
- 584.Manchun S, Dass CR, Sriamornsak P. Targeted therapy for cancer using pH-responsive nanocarrier systems. Life Sci. 2012;90:381–387. doi: 10.1016/j.lfs.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 585.Delcea M, Mohwald H, Skirtach AG. Stimuli-responsive LbL capsules and nanoshells for drug delivery. Adv Drug Delivery Rev. 2011;63:730–747. doi: 10.1016/j.addr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 586.Gao W, Chan JM, Farokhzad OC. pH-Responsive nanoparticles for drug delivery. Mol Pharmaceutics. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Behrendt N. Matrix Proteases in Health and Disease. Wiley-VCH; Weinheim, Germany: 2012. [Google Scholar]
- 588.Ding Y, Kang Y, Zhang X. Enzyme-responsive polymer assemblies constructed through covalent synthesis and supramolecular strategy. Chem Commun. 2015;51:996–1003. doi: 10.1039/c4cc05878j. [DOI] [PubMed] [Google Scholar]
- 589.de la Rica R, Aili D, Stevens MM. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv Drug Delivery Rev. 2012;64:967–978. doi: 10.1016/j.addr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 590.Hu Q, Katti PS, Gu Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale. 2014;6:12273–12286. doi: 10.1039/c4nr04249b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Lv C, Wang Z, Wang P, Tang X. Photodegradable polyesters for triggered release. Int J Mol Sci. 2012;13:16387–16399. doi: 10.3390/ijms131216387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Bawa P, Pillay V, Choonara YE, du Toit LC. Stimuliresponsive polymers and their applications in drug delivery. Biomed Mater. 2009;4:022001. doi: 10.1088/1748-6041/4/2/022001. [DOI] [PubMed] [Google Scholar]
- 593.Kost J, Langer R. Responsive polymer systems for controlled delivery of therapeutics. Trends Biotechnol. 1992;10:127–131. doi: 10.1016/0167-7799(92)90194-z. [DOI] [PubMed] [Google Scholar]
- 594.Hu J, Zhang G, Liu S. Enzyme-responsive polymeric assemblies, nanoparticles and hydrogels. Chem Soc Rev. 2012;41:5933–5949. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 595.Samarajeewa S, Shrestha R, Li Y, Wooley KL. Degradability of poly(lactic acid)-containing nanoparticles: enzymatic access through a cross-linked shell barrier. J Am Chem Soc. 2012;134:1235–1242. doi: 10.1021/ja2095602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets. 2013;17:281–291. doi: 10.1517/14728222.2013.740461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- 598.Duncan R. Development of HPMA copolymer-anticancer conjugates: clinical experience and lessons learnt. Adv Drug Delivery Rev. 2009;61:1131–1148. doi: 10.1016/j.addr.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 599.Gianasi E, Wasil M, Evagorou EG, Keddle A, Wilson G, Duncan R. HPMA copolymer platinates as novel antitumour agents: in vitro properties, pharmacokinetics and antitumour activity in vivo. Eur J Cancer. 1999;35:994–1002. doi: 10.1016/s0959-8049(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 600.Dong L, Xia S, Wu K, Huang Z, Chen H, Chen J, Zhang J. A pH/enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials. 2010;31:6309–6316. doi: 10.1016/j.biomaterials.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 601.Khokha R, Denhardt DT. Matrix metalloproteinases and tissue inhibitor of metalloproteinases: A review of their role in tumorigenesis and tissue invasion. Invasion Metastasis. 1989;9:391–405. [PubMed] [Google Scholar]
- 602.Schultz RM. The role of metalloproteinases in tumor cell metastasis. Cancer Treat Res. 1991;54:119–133. doi: 10.1007/978-1-4615-3938-4_7. [DOI] [PubMed] [Google Scholar]
- 603.Shay G, Lynch CC, Fingleton B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015;44–46:200–206. doi: 10.1016/j.matbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Wieczorek E, Jablonska E, Wasowicz W, Reszka E. Matrix metalloproteinases and genetic mouse models in cancer research: a mini-review. Tumor Biol. 2015;36:163–175. doi: 10.1007/s13277-014-2747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Liu P, Sun M, Sader S. Matrix metalloproteinases in cardiovascular disease. Can J Cardiol. 2006;22:25B–30B. doi: 10.1016/s0828-282x(06)70983-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci Landmark Ed. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- 607.Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015:44–46. doi: 10.1016/j.matbio.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Rao J, Khan A. Enzyme sensitive synthetic polymer micelles based on the azobenzene motif. J Am Chem Soc. 2013;135:14056–14059. doi: 10.1021/ja407514z. [DOI] [PubMed] [Google Scholar]
- 609.Elani Y, Law RV, Ces O. Vesicle-based artificial cells as chemical microreactors with spatially segregated reaction pathways. Nat Commun. 2014;5:5305. doi: 10.1038/ncomms6305. [DOI] [PubMed] [Google Scholar]
- 610.Kretlow JD, Klouda L, Mikos AG. Injectable matrices and scaffolds for drug delivery in tissue engineering. Adv Drug Delivery Rev. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 611.Li Y, Rodrigues J, Tomas H. Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications. Chem Soc Rev. 2012;41:2193–2221. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 612.Thornton PD, McConnell G, Ulijn RV. Enzyme responsive polymer hydrogel beads. Chem Commun. 2005:5913–5915. doi: 10.1039/b511005j. [DOI] [PubMed] [Google Scholar]