

Cdc48/p97 provides the energy to retrotranslocate integral membrane ERAD substrates. A series of ERAD substrates with increasingly hydrophobic transmembrane helices is used to show that retrotranslocation efficiency inversely correlates with hydrophobicity.
Abstract
Integral membrane proteins fold inefficiently and are susceptible to turnover via the endoplasmic reticulum–associated degradation (ERAD) pathway. During ERAD, misfolded proteins are recognized by molecular chaperones, polyubiquitinated, and retrotranslocated to the cytoplasm for proteasomal degradation. Although many aspects of this pathway are defined, how transmembrane helices (TMHs) are removed from the membrane and into the cytoplasm before degradation is poorly understood. In this study, we asked whether the hydrophobic character of a TMH acts as an energetic barrier to retrotranslocation. To this end, we designed a dual-pass model ERAD substrate, Chimera A*, which contains the cytoplasmic misfolded domain from a characterized ERAD substrate, Sterile 6* (Ste6p*). We found that the degradation requirements for Chimera A* and Ste6p* are similar, but Chimera A* was retrotranslocated more efficiently than Ste6p* in an in vitro assay in which retrotranslocation can be quantified. We then constructed a series of Chimera A* variants containing synthetic TMHs with a range of ΔG values for membrane insertion. TMH hydrophobicity correlated inversely with retrotranslocation efficiency, and in all cases, retrotranslocation remained Cdc48p dependent. These findings provide insight into the energetic restrictions on the retrotranslocation reaction, as well as a new computational approach to predict retrotranslocation efficiency.
INTRODUCTION
During translation, nearly one-third of all newly synthesized proteins are targeted to the endoplasmic reticulum (ER) where they are cotranslationally inserted. Of these ER-targeted proteins, those with hydrophobic stretches of 19–30 amino acids (Baeza-Delgado et al., 2013) are integrated into the membrane by the Sec61 protein translocation channel (Cymer et al., 2015). Once associated with the ER, nascent polypeptides begin to fold. However, integral membrane proteins are especially difficult to assemble, as they contain regions that encounter the distinct cellular environments of the cytoplasm, the phospholipid bilayer, and the ER lumen. Moreover, given the vast array of conformers any individual protein may adopt while navigating the protein folding landscape (Hartl et al., 2011), it is not surprising that protein misfolding in the ER may be quite common. For example, in some cell types, a substantial amount of clinically relevant proteins are turned over to a high degree, such as the cystic fibrosis transmembrane conductance regulator (CFTR), the epithelial sodium channel, and unassembled T-cell receptor subunits, due to misfolding, failed subunit assembly, or poor membrane insertion (Lippincott-Schwartz et al., 1988; Lukacs et al., 1994; Jensen et al., 1995; Ward et al., 1995; Staub et al., 1997; Valentijn et al., 1998).
Cells have devised an elaborate quality control network to help mitigate the consequences of protein misfolding during both normal and disease states. One element of this network, termed ER-associated degradation (ERAD), is responsible for degrading misfolded secretory proteins that are marked by a polyubiquitin chain (Vembar and Brodsky, 2008; Hampton and Sommer, 2012; Olzmann et al., 2013; Christianson and Ye, 2014; Pisoni and Molinari, 2016). Genetic mutation, errors in transcription or translation, or environmental stress can result in protein misfolding, which gives rise to various ERAD-associated diseases (Guerriero and Brodsky, 2012). During ERAD, misfolded proteins are first recognized by cellular molecular chaperones that monitor protein folding, including heat shock proteins of 40, 70, and 90 kDa, chaperone-like lectins, and protein disulfide isomerases. These proteins play a key role in degradation by maintaining substrate solubility, delivering substrates to the retrotranslocation channel, and enhancing interaction with the ubiquitination machinery (Meacham et al., 1999; Nishikawa et al., 2001; Denic et al., 2006; Nakatsukasa et al., 2008; Hagiwara and Nagata, 2012). In the yeast ER, there are two integral membrane E3 ubiquitin ligases that are required for ERAD, Hrd1p and Doa10p (Hampton et al., 1996; Bays et al., 2001; Swanson et al., 2001). Whereas Hrd1p polyubiquitinates proteins with integral membrane and ER lumenal misfolding lesions, termed ERAD-M and -L for ERAD “membrane” and “lumen,” respectively (Bordallo et al., 1998; Vashist and Ng, 2004; Garza et al., 2009), Doa10p primarily ubiquitinates substrates with misfolding lesions in the cytoplasm (termed ERAD-C, for “cytoplasm”; Vashist and Ng, 2004; Carvalho et al., 2006; Denic et al., 2006).
After or in some cases concomitant with ubiquitination, integral membrane ERAD substrates are retrotranslocated from the ER into the cytoplasm in a process that requires the AAA+ ATPase Cdc48p (p97 in mammals), which provides the driving force to extract substrates (Bagola et al., 2011). Cdc48p forms a complex with the Ufd1-Npl4 heterodimer, which binds to ubiquitinated substrates, recruits cofactors, and is required for degradation of both lumenal and integral membrane ERAD substrates (Meyer et al., 2000, 2002; Ye et al., 2001; Bays and Hampton, 2002; Rabinovich et al., 2002; Schuberth and Buchberger, 2008). During Cdc48p-mediated retrotranslocation, the exact mechanism by which transmembrane helices (TMHs) are removed from the lipid bilayer by Cdc48p has yet to be fully elucidated. There is some evidence that the protein translocation machinery (Plemper et al., 1997; Kalies et al., 2005; Ng et al., 2007; Scott and Schekman, 2008; Schafer and Wolf, 2009) or components of the ERAD machinery, Derlin1 or Hrd1p (Lilley and Ploegh, 2004; Ye et al., 2004; Carvalho et al., 2010; Mehnert et al., 2014; Stein et al., 2014; Baldridge and Rapoport, 2016), form a proteinaceous channel through which membrane proteins pass into the cytoplasm. However, none of these putative retrotranslocons is required for the degradation of some proteins, including the misfolded yeast ATP-binding cassette (ABC) transporter Sterile 6* (Ste6p*; Huyer et al., 2004; Vashist and Ng, 2004). Regardless of whether a proteinaceous channel facilitates retrotranslocation, ERAD substrate TMHs must be removed from the membrane into the aqueous environment of the cytoplasm, which is energetically unfavorable.
Little is known about how the biophysical properties of a TMH in an ERAD substrate influence retrotranslocation. Carlson et al. (2006) examined the degradation of in vitro–translated wild-type and truncated forms of CFTR and found that the truncated species (i.e., those with fewer TMHs) had faster degradation kinetics. Due to limitations of the assay, however, they were unable to distinguish between the retrotranslocation and degradation steps. Therefore, to test directly the role of TMHs in retrotranslocation and predict how biophysical parameters of a TMH influence this energetically unfavorable event, we designed a novel integral membrane ERAD substrate derived from Ste6p*. The wild-type protein, Ste6p, contains 12 TMHs and two cytoplasmic nucleotide-binding domains (NBDs; McGrath and Varshavsky, 1989). Truncating 42 amino acids from the second NBD (NBD2*) in Ste6p* results in ER retention and degradation (Loayza et al., 1998; Huyer et al., 2004). We also showed that a soluble form of NBD2 is stable, whereas the truncated species, NBD2*, is rapidly degraded by the cytoplasmic quality control machinery (Guerriero et al., 2013). Consequently, the NBD2* degron was fused to a dual-pass membrane anchor. We demonstrate here that the resulting protein, Chimera A*, and Ste6p* exhibit nearly identical degradation requirements. We then show that Chimera A*, with two TMHs, is retrotranslocated more efficiently than the 12-membrane-spanning substrate Ste6p* and that there is a direct relationship between the retrotranslocation of Chimera A* variants and TMH hydrophobicity, as determined using a computational model. These findings have implications for the mechanisms underlying the retrotranslocation of integral membrane ERAD substrates and the effects that some disease-causing mutations have on clinically relevant ERAD substrates.
RESULTS
Generation of a model dual-pass integral membrane protein
To define how TMH hydrophobicity influences ERAD substrate retrotranslocation, we sought to examine substrates with the same misfolded domain appended to unique membrane anchors. The domain we chose for this analysis was NBD2*, which derives from the yeast ERAD substrate Ste6p* (see earlier description and Figure 1, A and B). The use of the truncated NBD2* species as a degron has important advantages because the causative lesion is a truncation, so recognition for degradation occurs posttranslationally. Moreover, the cellular factors required for Ste6p* degradation have been extensively characterized. The truncation does not remove any residues critical for ATP binding/hydrolysis (Linton, 2007) but instead disrupts a predicted β-sheet that incorporates β-strand interactions between the N- and C-terminal portions of NBD2 (Supplemental Figure S1A). Moreover, we wanted to avoid complications posed by interactions between the 12 TMHs in Ste6p*. Previous studies established that Ste6p’s TMHs can direct assembly when the protein is expressed as two half-transporters (Berkower and Michaelis, 1991; Berkower et al., 1996). Therefore we designed a minimally anchored substrate containing only the first two TMHs of Ste6p* in order to maintain a native-like ER lumenal loop and also to reduce the possibility of interactions among the 12 TMHs (Figure 1, A and B). Based on the spacing of TMHs in the crystal structure of a homologous ABC transporter, murine p-glycoprotein (3G5U) (Aller et al., 2009), and our homology model (Supplemental Figure S1B), there should be minimal interactions between TMH1 and 2. In fact, as shown in Supplemental Figure 1C, poor packing is observed between the two TMHs in our model. Moreover, based on our homology model of TMH1 and 2 in p-glycoprotein, Ste6p* possesses less than half the number of van der Waals contacts found on a typical, well-defined TMH interface (unpublished data; Walters and DeGrado, 2006). Furthermore, there are no interhelical hydrogen bonds between the two helices, which suggests that at best they interact quite weakly.
FIGURE 1:

Generation of a dual-pass integral membrane ERAD substrate. (A) Linear diagrams of Sterile 6 (Ste6p), Sterile 6p*, and associated substrates. Ste6p is a yeast ABC transporter containing 12 TMHs (black bars) and cytoplasmic NBD1 and NBD2. Truncation of Ste6p’s NBD2 is depicted as an empty rectangle, which results in Ste6p*. Chimera N and Chimera N* are composed of the first two TMHs of Ste6p fused to either full-length or truncated NBD2 (dotted line). Chimera A and Chimera A* contain a nonnative TMH2 (vertical open bar). All constructs contain a triple-HA tag in the ER lumenal loop between TMH1 and 2. (B) Topologies of the constructs in A, with the lumenal HA tag indicated in Ste6p, which was present in all constructs, and the nonnative TMH2 in Chimera A (open rectangle). (C) Sequence of TMH2 for Chimera N* and Chimera A* given next to the predicted ΔG (kcal/mol) for membrane insertion as reported by dgpred.cbr.su.se. (D) S. cerevisiae expressing Chimera N* and Chimera A* were grown to log phase, and cellular protein was extracted by alkaline lysis, precipitated, resuspended, and incubated in the presence or absence of Endo H. Chimeras were detected after SDS–PAGE and immunoblotting. (E) ER-derived microsomes were generated from S. cerevisiae transformed with a Chimera N* or A* expression vector under the control of the PGK promoter. Microsomes were subjected to limited proteolysis with proteinase K on ice for the indicated times. Reactions were quenched and proteins were detected as described in D. Dashed box, Chimera A*-derived proteolytic products. Full-length proteins are denoted by an arrow. Asterisk denotes a small population of Chimera A* that is synthesized with NBD2* in the ER lumen, as observed for the majority of Chimera N*.
To begin to characterize this first chimera, termed Chimera N*, we expressed it in wild-type Saccharomyces cerevisiae, in which it exhibited a lower-than-expected electrophoretic mobility and migrated at ∼55 kDa rather than the predicted mass of ∼45k Da (Figure 1D, Chimera N* (–) lane). Amino acid sequence analysis of Ste6p revealed one ER lumenal, N-linked glycosylation site (NXS/T). However, previous studies examining the glycosylation status of Ste6p* suggested that this site was not used (Vashist and Ng, 2004). Given that the Ste6p* N-glycan site is only 17 amino acids from the predicted end of TMH1, the site may be too close to the membrane to efficiently access the glycosylation machinery (Nilsson and von Heijne, 1993). To examine whether Chimera N* was modified with an N-linked glycan, we treated lysates with endoglycosidase H (Endo H), which resulted in an ∼9-kDa shift, and is consistent with the presence of three N-glycan moieties (Figure 1D, Chimera N* (+) lane). Indeed, there are five potential sites for N-linked glycosylation in the protein—one after TMH1, as described earlier, and four in NBD2* (see supplemental tables for the amino acid sequences). Therefore it appears that TMH1 and 2 from Ste6p*, when expressed in the absence of the other 10 TMHs, are unable to form a hairpin, and so NBD2* is instead deposited in the ER lumen. Consistent with this hypothesis, examination of the predicted amino acid sequence for TMH2 (dgpred.cbr.su.se; Hessa et al., 2007) reveals that Ste6p TMH2 has an unfavorable ΔG for membrane insertion (ΔG = 1.86 kcal/mol; Figure 1C, top).
To correct the topology of Chimera N* so that NBD2* resides instead in the cytoplasm like Ste6p*, we substituted a hydrophobic TMH consisting of alternating alanine and leucine residues for the native TMH2 (Figure 1C, bottom; Hessa et al., 2007). Migration on a polyacrylamide gel and treatment with Endo H confirmed the predicted size (∼45 kDa), with nearly all of the protein lacking N-glycans, as expected (Figure 1D). This new chimeric protein was termed Chimera A* (Figure 1, A and B). To confirm further the different topologies of Chimera N* and A*, we prepared ER-derived microsomes from yeast expressing each substrate and subjected them to limited proteolysis with proteinase K. As anticipated for a cytoplasmic NBD2*, Chimera A* was accessible to the protease, and numerous cleavage fragments were generated (Figure 1E, dashed box). However, for Chimera N*, which contains NBD2* in the ER lumen, substantially fewer proteolytic fragments were evident (Figure 1E). Because our goal was to examine the degradation and retrotranslocation requirements of a simplified, model ERAD substrate whose degradation requirements could be compared with Ste6p*, we completed all further analyses using Chimera A*.
Chimera A* is an ERAD substrate
The mutation that results in ER retention and degradation of Ste6p* is a premature stop codon (Q1249X), which leads to truncation of the C-terminal 42 amino acids of NBD2 (Loayza et al., 1998). Previously we examined the degradation of cytoplasmic, soluble forms of NBD2 and NBD2* to determine how loss of membrane anchorage changes degradation requirements: full-length NBD2 was stable, whereas NBD2* was rapidly degraded in a proteasome-dependent and cytosolic/nuclear E3-dependent manner (Guerriero et al., 2013). Similarly, we set out to characterize whether appending NBD2 and NBD2* to the dual-pass membrane anchor provides a means to determine how ERAD substrates are differentially selected from wild-type proteins. As predicted, more Chimera A was present at steady state than Chimera A* (compare 0-min time points), and Chimera A was significantly more stable (Figure 2). Also note that Chimera A migrated somewhat more slowly (∼4 kDa) than Chimera A*, consistent with the 42–amino acid truncation from the C-terminus. These data show that Chimera A* is destabilized as a result of the truncation, similar to what was shown for Ste6p* and the soluble NBD2* species (Loayza et al., 1998; Guerriero et al., 2013).
FIGURE 2:
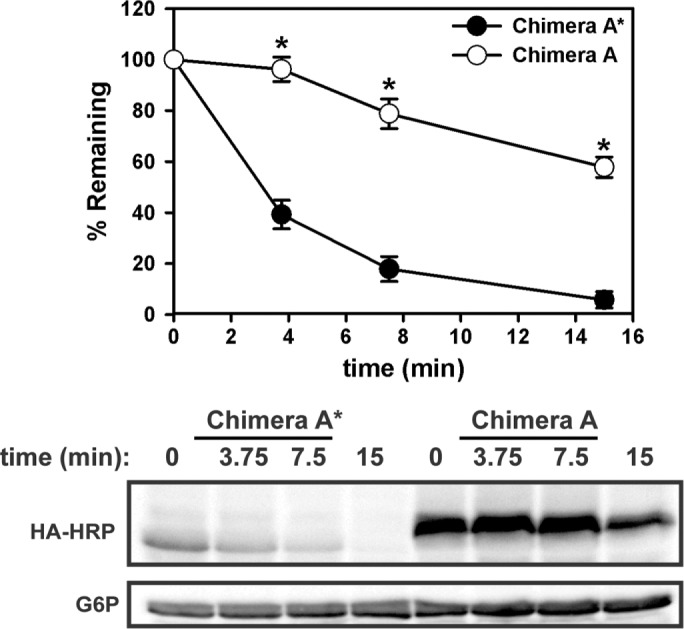
The dual-pass transmembrane-tethered substrate Chimera A* is rapidly degraded. Yeast expressing Chimera A (full-length NBD2; open circles) and Chimera A* (NBD2*; filled circles) under the control of the ADH promoter were grown to log phase and assayed by cycloheximide chase for the indicated times at 26°C. Proteins were extracted and detected as described in Materials and Methods. Representative blots are shown below the graph, and glucose-6-phosphate dehydrogenase (G6P) serves as loading control. Data represent means ± SE from three independent experiments. *p < 0.0000005 as determined by Student’s t test.
Because the truncation in Ste6p*’s NBD2 results in ER retention (Loayza et al., 1998), we next determined Chimera A* residence using indirect immunofluorescence confocal microscopy. As shown in Figure 3A, Chimera A* localization was restricted to the ER as assessed by colocalization with the ER protein Kar2p. To confirm these data, we used sucrose gradient centrifugation to fractionate lysates prepared from wild-type yeast expressing Chimera A*. By using a 10–70% sucrose gradient, we can distinguish the ER and Golgi fractions from the denser plasma membrane fractions (Guerriero et al., 2013; Buck et al., 2016). Similar to what was previously reported for Ste6p* (Loayza et al., 1998), most of Chimera A* overlapped with fractions containing the ER-localized integral membrane protein Sec61p (Figure 3B, lanes 8–13), but only minor overlap was apparent with the plasma membrane marker Pma1p (Figure 3B, lanes 15–23). Chimera A* was also absent from the pellet fraction (Figure 3B, P), which represents aggregated protein (Kruse et al., 2006). Taken together, these results indicate that Chimera A* is an unstable, ER-retained protein.
FIGURE 3:

Chimera A* resides in the ER. (A) Chimera A* localization was determined by indirect immunofluorescence confocal microscopy using mouse anti-HA (Chimera A*), rabbit anti-Kar2p (ER lumen), and DAPI to stain the nuclei. Primary antibodies were labeled with Alexa goat anti-mouse 488 and goat anti-rabbit 568, respectively; scale bar, ∼5 μm. (B) Yeast lysates from Chimera A*–expressing cells were subject to sucrose gradient centrifugation, fractions were collected from the top (low sucrose) to the bottom (high sucrose), and an aliquot of fraction each was analyzed by SDS–PAGE and immunoblot analysis for Sec61p (ER marker), Anp1p (Golgi marker), Pma1p (plasma membrane marker), and Chimera A* (HA-HRP). Lanes containing 0.5% of the total protein loaded (L, on the left) and the pelleted material from the bottom of the gradient tube (P, on the right) were included.
To establish Chimera A* as an ERAD substrate, we next examined dependence on proteasome activity using a cycloheximide chase assay. We used a yeast strain lacking the gene encoding a multidrug pump (pdr5Δ) and treated the cells with either dimethyl sulfoxide (DMSO) or the proteasomal inhibitor MG132 (Lee and Goldberg, 1996; Gaczynska and Osmulski, 2005). As predicted, treatment with MG132 significantly stabilized Chimera A* (Figure 4A). This result was recapitulated when a version of Chimera A* under the control of the stronger phosphoglycerate kinase (PGK) promoter was examined (Supplemental Figure 2A).
FIGURE 4:
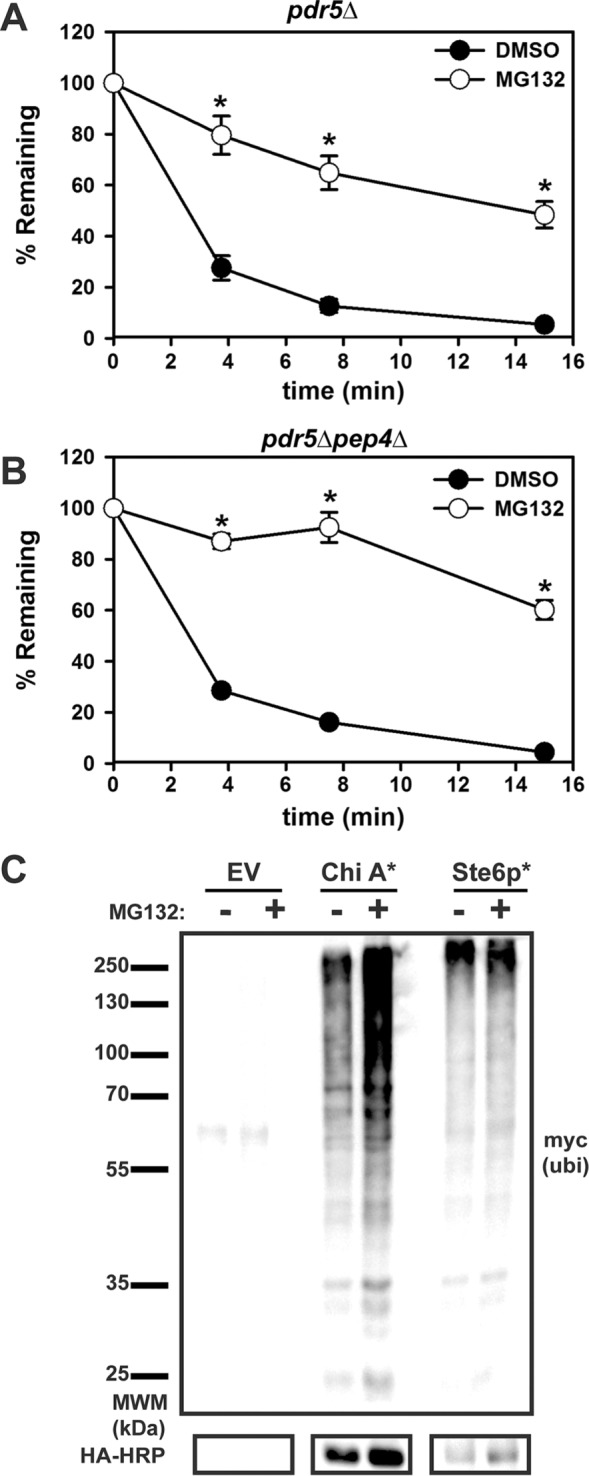
Chimera A* degradation is proteasome-dependent. (A) Chimera A* was expressed under the control of the ADH promoter in (A) pdr5Δ or (B) pdr5Δ pep4Δ yeast. Before the cycloheximide chase analysis, cells were preincubated with DMSO (control; filled circles) or 100 μM MG132 (proteasome inhibitor; open circles) for 20 min and then chased for the indicated times. Graphed data represent the means ± SE from three independent experiments. *p < 0.00003. (C) pdr5Δ cells were transformed with an empty vector or Chimera A* or Ste6p* expression vectors under the control of the PGK promoter, as well as a plasmid for the Cu2+-inducible expression of myc-tagged ubiquitin. Cells were treated for 90 min with DMSO (–) or 50 μM MG132 (+) and then lysed. Total protein was immunoprecipitated with HA-conjugated agarose beads, followed by SDS–PAGE and immunoblot analysis for myc-tagged ubiquitin and the HA tag.
Give that Ste6p normally traffics to the plasma membrane and is internalized to the vacuole, it is possible that a portion of Chimera A* might escape the ER and is instead subject to vacuolar degradation after accessing compartments in the later secretory pathway or being targeted for autophagy (Loayza et al., 1998; Teckman and Perlmutter, 2000; Wang et al., 2011a; Houck et al., 2014). To rule out a contribution of the vacuole in Chimera A* degradation, we measured turnover in a pdr5Δpep4Δ strain. Pep4p acts as an upstream activator of vacuolar proteases, so deleting PEP4 results in a >90% decrease in vacuolar protease activity (Jones, 1984). However, compared with the pdr5Δ strain, there was no significant difference in Chimera A* degradation in the DMSO-treated cells and only minor additional stabilization in the MG132-treated yeast (compare Figure 4, A and B). These data suggest that vacuolar proteases do not play a significant role in Chimera A* proteolysis.
To confirm further that Chimera A* degradation is proteasome-dependent, we immunoprecipitated the protein from pdr5Δ yeast treated with DMSO or MG132 and then immunoblotted it to detect myc-tagged polyubiquitin chains. As shown in Figure 4C, a “smear” of polyubiquitinated species was observed for Chimera A* as well as for Ste6p*, which was used as a control. Treatment with MG132 increased the amount of polyubiquitinated protein (compare – vs. + MG132). Combined with the earlier data, these results establish Chimera A* as a new ERAD substrate.
Chimera A* degradation requires the cytoplasmic ERAD machinery
Next we confirmed that the Chimera A* degradation requirements match what is known for Ste6p*, which contains an identical degron. As noted in the Introduction, ERAD substrates in yeast can be classified based on the site of their misfolded lesion as ERAD-L, ERAD-M, or ERAD-C (Vashist and Ng, 2004). Because Ste6p* uses cytosolic chaperones and other components of the ERAD-C machinery for efficient degradation (Huyer et al., 2004; Nakatsukasa et al., 2008), we first examined Chimera A* turnover in yeast strains harboring temperature-sensitive mutations in genes encoding select cytoplasmic chaperones. Ssa1p is one of the main heat-inducible cytoplasmic Hsp70s in yeast, and a P417L mutation present in ssa1-45 yeast uncouples nucleotide hydrolysis from substrate binding, thus limiting Ssa1p function at the nonpermissive temperature (Becker et al., 1996; Needham et al., 2015). We found that Chimera A* degradation required Ssa1p, as evident from marked stabilization in the ssa1-45 strain (Figure 5A and Supplemental Figure 2B), as well as in strains mutated for the cytoplasmic Hsp40 cochaperones Hlj1p and Ydj1p (Figure 5B). To rule out the acquisition of a lumenal lesion in Chimera A* as a result of the synthetic TMH (i.e., TMH2), we also examined Chimera A* degradation in a strain containing a mutated form of the ER lumenal Hsp70, Kar2p. Degradation was unaffected in the kar2-1 strain, but the canonical ERAD-L substrate CPY* was stabilized in this strain (unpublished data), as previously shown (Kabani et al., 2003).
FIGURE 5:

Chimera A* degradation requires cytosolic chaperones and the E3 ubiquitin ligase, Doa10p. Cycloheximide chase analyses were performed as described in Materials and Methods to measure the turnover of Chimera A* in (A) SSA1 and ssa1-45, (B) HLJ1YDJ1 and hlj1Δydj1-151, (C) KAR2 and kar2-1, and (D) DOA10, doa10Δ, hrd1Δ, and doa10Δhrd1Δ at 37°C (A, B) or at 26°C (C, D). Chimera A* was expressed under the control of the ADH promoter (A, C, D) or the PGK promoter (B). Data represent the means ± SE for at least three independent experiments. *p < 0.0007 for ssa1-45, p < 0.02 for hlj1Δydj1-151, and p < 0.0006 for doa10Δ and doa10Δhrd1Δ compared with the isogenic wild-type strains.
In yeast, Doa10p primarily ubiquitinates ERAD-C substrates, whereas Hrd1p ubiquitinates ERAD-L and -M substrates (Vashist and Ng, 2004; Carvalho et al., 2006; Garza et al., 2009). Ste6p* degradation requires Doa10p, with only a minor contribution from Hrd1p (Huyer et al., 2004; Vashist and Ng, 2004). We found that Chimera A* degradation strictly relied on Doa10p, with no additional stabilization present in the doa10Δhrd1Δ strain (Figure 5D). These data suggest that Chimera A* is a more ideal ERAD-C substrate than Ste6p*. This feature may arise from the fact that Ste6p* has 12 TMHs, increasing the likelihood of a folding lesion in the ER membrane or lumen that may be recognized by Hrd1p (Huyer et al., 2004; Sato et al., 2009).
Before degradation by the 26S proteasome, integral membrane ERAD substrates are retrotranslocated from the ER membrane into the cytoplasm and can even be found in solution before proteasome targeting (Wiertz et al., 1996; Nakatsukasa et al., 2008; Garza et al., 2009; Leichner et al., 2009; Morris et al., 2014; Neal et al., 2017). The AAA+ ATPase Cdc48p (p97 in mammals) provides the mechanical force necessary for the retrotranslocation of ERAD substrates, including Ste6p* (Hitchcock et al., 2001; Ye et al., 2001; Bays and Hampton, 2002; Braun et al., 2002; Jarosch et al., 2002; Rabinovich et al., 2002; Huyer et al., 2004; Nakatsukasa et al., 2008; Nakatsukasa and Kamura, 2016). Cdc48p/p97 functions as a homohexameric ring in a complex together with a heterodimer of Ufd1p and Npl4p. Furthermore, Cdc48p/p97 interacts with a variety of cofactors via its N- and C- termini, which allow it to participate in numerous cellular functions (Wolf and Stolz, 2012; Barthelme and Sauer, 2016). Ste6p* degradation is reduced in yeast harboring a temperature-sensitive mutant of Cdc48p (Huyer et al., 2004), but because Chimera A* contains only two TMHs, it was possible that Chimera A* has a diminished requirement for Cdc48p. However, we found that Chimera A* was also significantly stabilized in a cdc48-2 temperature-sensitive strain (Figure 6A). We then examined whether Chimera A* retrotranslocation in an established in vitro assay (Nakatsukasa et al., 2008) required Cdc48p. In this assay, membranes containing an ERAD substrate (i.e., Chimera A*) are incubated in the presence of 125I-ubiquitin, an ATP-regenerating system, and yeast cytosol. The ERAD substrate is then immunoprecipitated, and the polyubiquitinated species can be detected by autoradiography. However, by introducing a centrifugation step before the immunoprecipitation, we can use this in vitro assay to calculate the percentage of retrotranslocated material in the supernatant (S) relative to the total (S plus pellet [P]). Note that the yeast cytosol, when diluted for this assay, lacks proteasome activity (Nakatsukasa et al., 2008), thereby allowing for measurements of retrotranslocation without the confounding effects arising from concomitant substrate degradation. After immunoprecipitation, we observed that the 125I-ubiquitin signal was equal to or greater than the predicted molecular weight for Chimera A* on the autoradiograph (Figure 6B, arrow), as observed previously for Ste6p* and other substrates (Nakatsukasa et al., 2008; Buck et al., 2016). Furthermore, we found an ∼50% decrease in retrotranslocation efficiency when we performed the reaction using both microsomes and cytosol prepared from temperature-shifted cdc48-2 yeast (compare signals in S, Figure 6, B and C). The incomplete block in retrotranslocation efficiency may result from only partial inactivation of Cdc48p activity in the cdc48-2 strain or the contribution of alternate retrotranslocation factors. Nevertheless, these data demonstrate that Chimera A* retains a robust Cdc48p requirement for both degradation and retrotranslocation, and more generally that Chimera A* is an ERAD-C substrate.
FIGURE 6:
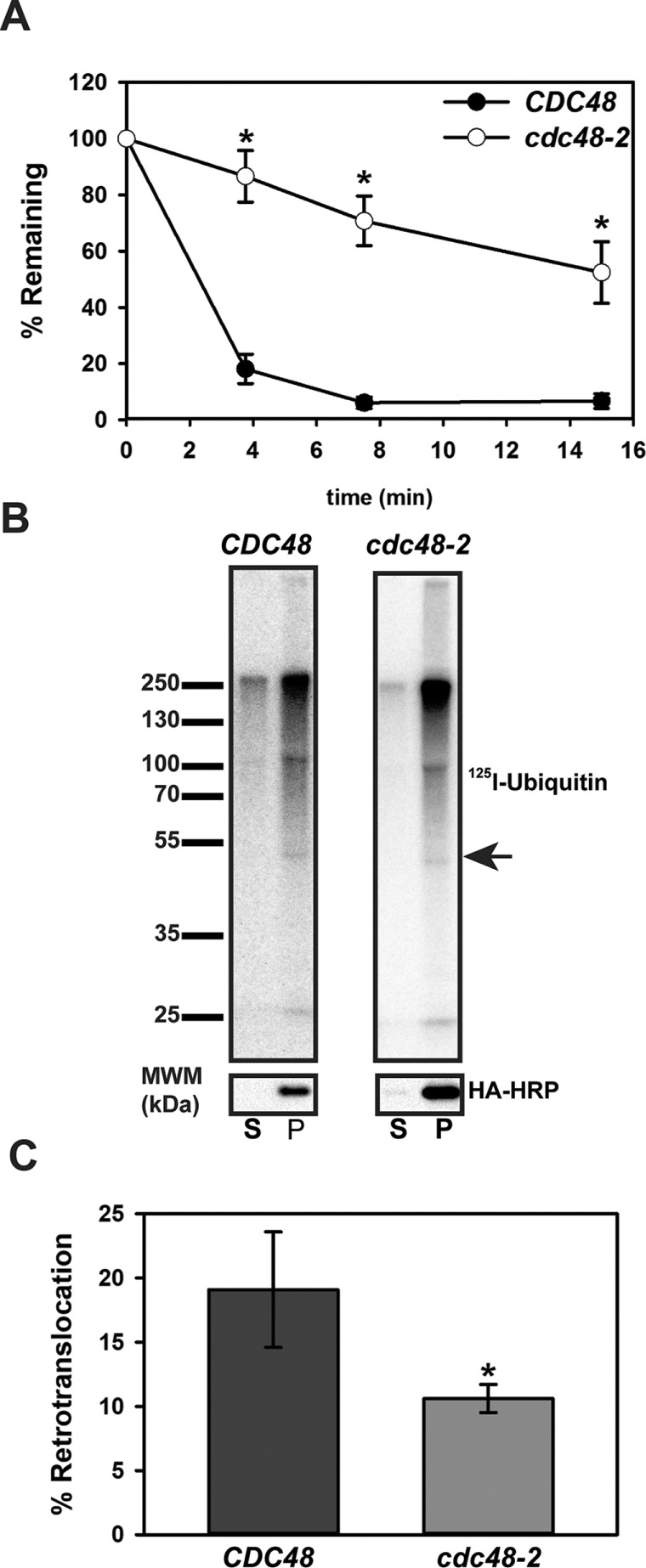
Chimera A* degradation and retrotranslocation require the AAA+ ATPase Cdc48p. (A) Cycloheximide chase analyses were performed as described in Materials and Methods for BY4742 (CDC48, wild type) and cdc48-2 yeast expressing Chimera A* under the control of the ADH promoter. To inactivate cdc48-2, strains were subjected to a 2-h temperature shift to 39°C. Data represent the means ± SE for two independent experiments, *p < 0.04. (B) Microsomes were prepared by glass bead disruption of CDC48 or cdc48-2 yeast expressing Chimera A* under the control of the PGK promoter, which were temperature shifted for 2 h at 39°C. Microsomes were ubiquitinated in vitro with 125-labeled ubiquitin for 40 min using cytosol prepared from CDC48 or cdc48-2 yeast, which was also temperature shifted for 2 h at 39°C. After the reaction, retrotranslocated material (supernatant, S) was separated from membrane-integrated material (pellet, P) by centrifugation before analysis by immunoprecipitation, SDS–PAGE, and autoradiography (top). Half of the material was analyzed separately by immunoblot analysis with anti–HA-HRP (bottom). Arrow indicates the relative migration of Chimera A*. (C) Percent retrotranslocation was determined by comparing the percentage of the radioactive signal in the supernatant (S) divided by the total (S + P) × 100. The data represent the means ± SD from two independent experiments performed in triplicate. *p < 0.0003.
Chimera A* retrotranslocation is more efficient than that of Ste6p*
We next asked how the number of TMHs affects retrotranslocation efficiency, using Chimera A* and Ste6p* as substrates. Given that retrotranslocation is energetically unfavorable, we reasoned that Ste6p* retrotranslocation might be less efficient than that for Chimera A*. When we examined retrotranslocation in vitro after a 40-min ubiquitination reaction, ∼30% of the ubiquitinated Chimera A* had been retrotranslocated, but only ∼20% of the modified Ste6p* species was present in the supernatant (Figure 7, A and B). These data support the prediction that TMHs may serve as a barrier to retrotranslocation and are consistent with data on the retrotranslocation efficiency and partial proteolysis of CFTR and truncated CFTR species (Carlson et al., 2006). Although the difference between the retrotranslocation efficiency of the 12 versus 2 TMH substrates appears modest, one caveat is that we cannot take into account how domain–domain interactions, which are evident in ABC transporters, may influence Ste6p* retrotranslocation (Dawson and Locher, 2006; Li et al., 2014; Zhang and Chen, 2016).
FIGURE 7:
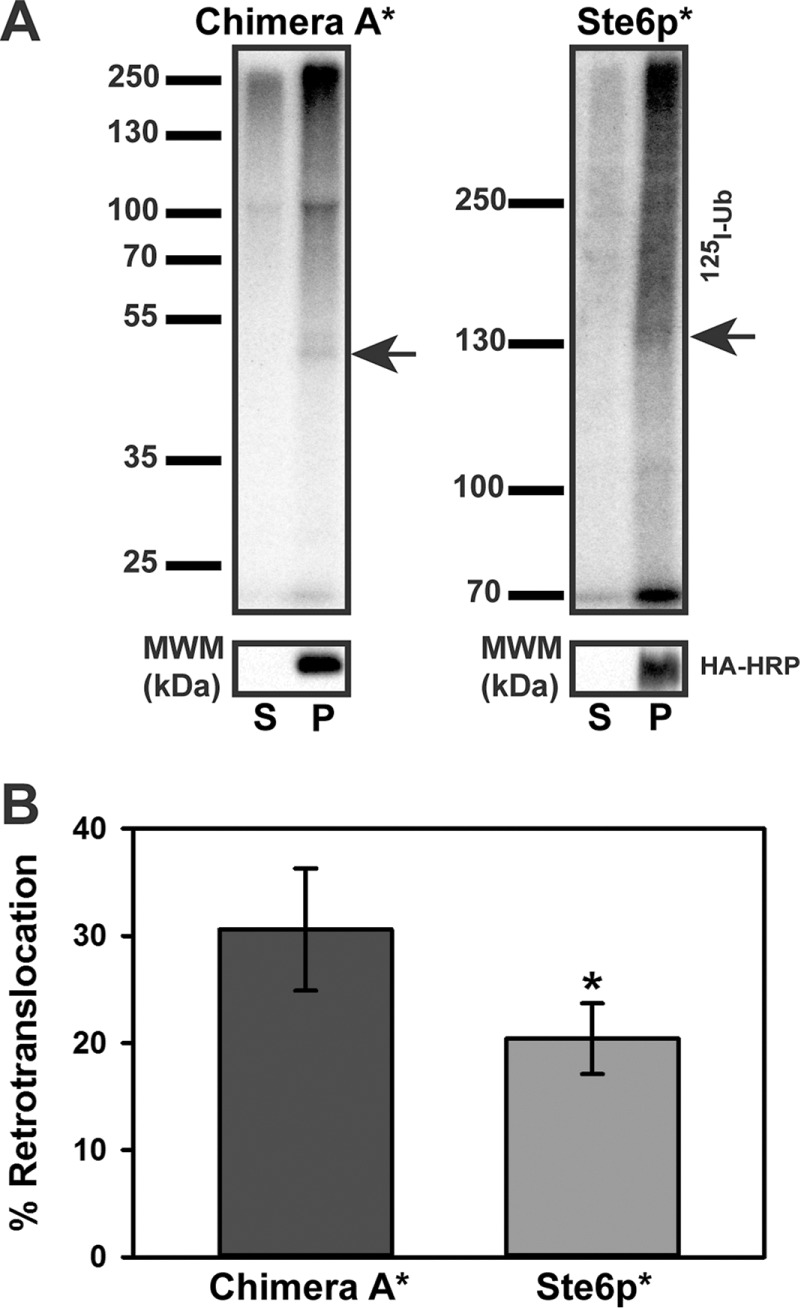
Chimera A* is more efficiently retrotranslocated than Ste6p*. (A) Microsomes prepared from CDC48 yeast expressing Chimera A* or Ste6p* under the control of the PGK promoter grown at 26°C were subjected to an in vitro ubiquitination reaction for 40 min using cytosol prepared from CDC48 yeast grown at 26°C. Samples were processed as described in Figure 6. Representative autoradiographs (top) and corresponding anti–HA-HRP blots (bottom). Arrows indicate the relative migrations of Chimera A* and Ste6p*. (B) The percentage retrotranslocation, calculated as described in the legend to Figure 6, represents the means ± SD from four independent experiments performed in triplicate. *p < 0.03.
Transmembrane helix hydrophobicity reduces retrotranslocation efficiency
To determine more accurately how the biophysical parameters of an ERAD substrate TMH may alter retrotranslocation efficiency, we designed a new series of Chimera A* variants (Figure 8A). Because the native TMH1 for Ste6p* is only marginally stable in the membrane (ΔG = 0.31 kcal/mol; but also see Discussion), we sought to test how increasing the TMH1 hydrophobicity alters retrotranslocation efficiency. In this experiment, TMH2 in Chimera A* was unchanged, and its potential contribution to retrotranslocation efficiency is assumed to remain constant. In contrast, we chose sequences for the TMH1 Chimera A* variants based on those that favor Sec61-mediated insertion of a model integral membrane protein (Hessa et al., 2007). In addition, we used a physics-based continuum model to engineer select mutations into the native TMH1 that increase hydrophobicity (Figure 8A; also see later discussion). The resulting TMH1 sequences provide a broad range of hydrophobic Chimera A* variants (Figure 8B), and in preliminary experiments, none of the Chimera A* variants acquired Hrd1p-dependent degradation, indicating that ERAD-M lesions were not introduced as a result of the TMH1 substitutions (unpublished data).
FIGURE 8:

Retrotranslocation efficiency correlates inversely with transmembrane hydrophobicity. (A) Chimera A* variants were designed to harbor increasingly hydrophobic TMH1 sequences. Predicted ΔG for membrane residence is indicated next to each TMH1 sequence, as determined by dg.pred.cbr.su.se. (B) Calculated TMH1 ΔG (kcal/mol), as described in Materials and Methods. (C) Microsomes were prepared from CDC48 yeast expressing each chimera variant under the control of the PGK promoter, and retrotranslocation was measured in the presence of CDC48 cytosol (open bars) or using temperature-shifted cdc48-2 cytosol (gray bars). Percentage retrotranslocation, calculated as described in Figure 6, is shown. Data represent the means ± SD from least two independent experiments performed in triplicate. *p < 0.03, **p < 0.003, ***p < 0.0004, ****p < 0.00002.
To begin to compare the TMH1 variants, we first maximized reaction efficiency and used a cytosol concentration of 5 instead of 1 mg/ml, which magnified the degree of Chimera A* retrotranslocation (compare Figures 8C and 7B). Next, using cytosol prepared from wild-type (CDC48) yeast, we observed reduced retrotranslocation efficiency for the chimeras, with predicted free energies for insertion of ΔG = −4.38 and −7.15, which represent ∼19 and 36% respective decreases from the chimera with a predicted insertion energy of ΔG = 0.31 (i.e., the chimera containing native TMH1; Figure 8C and Table 1). Of interest, the retrotranslocation efficiencies of variants containing the least stable TMHs (ΔG = 0.31, −0.19, and −1.07) were relatively consistent. We then reasoned that reducing Cdc48p activity by using cytosol prepared from temperature-shifted cdc48-2 yeast may further distinguish substrates with a heightened Cdc48p requirement. On repeating the retrotranslocation analysis with cdc48-2 cytosol, we observed the expected decrease in retrotranslocation efficiency of the Chimera A* protein (Figure 8C; compare ΔG = 0.31, CDC48 vs. cdc48-2), confirming reduced Cdc48p activity in the cytosol. The difference between retrotranslocation efficiency in the presence of CDC48 versus cdc48-2 cytosol in this experiment (Figure 8C) is lower than in Figure 6C primarily because we only inactivated Cdc48p in the cytosol instead of on the microsomes and in the cytosol, as done in Figure 6C. Nevertheless, in all cases, retrotranslocation of each of the chimeras with a modified TMH1 remained Cdc48p-dependent, and the magnitude of the Cdc48p dependence was relatively constant (Figure 8C and Table 1). For example, by using cdc48-2 cytosol, we observed a relative drop in retrotranslocation efficiency for the chimera with ΔG = −1.07 of 12.6% and a further accentuated decrease for ΔG = −4.38 of 37.4% (Figure 8C and Table 1). Taken together, these data demonstrate a complex dependence of retrotranslocation on ERAD substrate TMH hydrophobicity.
TABLE 1.
Retrotranslocation efficiency inversely correlates with TMH1 hydrophobicity.
| ΔG (kcal/mol) | Alias | CDC48 | % decrease vs. Chi A* (construct 1) | cdc48-2 | % decrease vs. Chi A* (construct 1) |
|---|---|---|---|---|---|
| 0.31 | construct 1 | 45.4 | 35.6 | ||
| −0.19 | construct 2 | 47.3 | 36.4 | ||
| −1.07 | construct 3 | 44.8 | 31.1 | 12.6 | |
| −4.38 | construct 4 | 36.9 | 18.7 | 21.9 | 38.5 |
| −7.15 | construct 5 | 29 | 36.1 | 22.3 | 37.4 |
The percentage retrotranslocation (also see Figure 8C) is listed for each condition tested. The percentage reduction in comparison to Chimera A*ΔG = 0.31 (construct 1) harboring the Ste6p* native TMH1 is also included.
A computational model for the relationship between transmembrane helix hydrophobicity and retrotranslocation
We next sought to develop a model for the interdependence of TMH hydrophobicity and retrotranslocation efficiency, one that would allow for predictions of retrotranslocation efficiency for any integral membrane ERAD substrate. Initially, we used a phenomenological energy scale (Hessa et al., 2007) to inform the design of the transmembrane sequences in Figure 8A. In Figure 9A, we replot the percentage retrotranslocation as a function of the theoretical stabilization energies to better explore the relationship between these two quantities (see Supplemental Table S1 for computational values). For brevity, in the following section, we discuss the Chimera A* variants as constructs 1–5, as they are listed in Figure 8A. As stated earlier, retrotranslocation is reduced as the TMH hydrophobicity of the substrate increases, implying that increased membrane stabilization makes it more difficult to extract the TMHs. Of interest, there is little difference in the efficiency of retrotranslocation between constructs 4 and 5, despite the latter being significantly more hydrophobic (Figure 8A). This result suggests a rate-limiting step for retrotranslocation that is independent of transmembrane hydrophobicity for very stable helices in the bilayer. However, we decided instead to turn to a physics-based model of membrane protein stability to reassess the predicted stabilities of each segment (Choe et al., 2008). The phenomenological model that we first used (Hessa et al., 2007) accounts for the membrane insertion depth of each amino acid in the sequence, but it fails to capture other important structural aspects, such as the nonadditivity of amino acid insertion energies (Moon and Fleming, 2011) or helix tilt and orientation in the membrane, which is known to exist in ABC transporters (Li et al., 2014; Zhang and Chen, 2016; Lee and Rosenbaum, 2017).
FIGURE 9:

Retrotranslocation efficiency is more accurately predicted by a physics-based model. Retrotranslocation extent vs. predicted TMH stabilities for the phenomenological insertion energy scale (A) and a physics-based continuum model of protein insertion (B). Energy values in B are reported relative to the wild-type segment (the absolute stability of construct 1 is −48.6 kcal/mol). (C) Optimal orientation of TMH segments in the membrane based on the physics-based model. The upper and lower gray surfaces are the interface of the hydrophobic membrane core with the head-group region. Helices adopt a 20–30° angle with respect to the membrane normal in order to maximize burial of hydrophobic residues in the membrane core.
The structural model that we then used (see Materials and Methods) instead accounts for the nonpolar stabilization of the protein in the greasy interior of the membrane, the electrostatic cost to place the protein in the low-dielectric environment of the lipid tails, and the mechanical distortions incurred in the membrane to accommodate the helix (Argudo et al., 2016). To these ends, we created idealized α-helices for each of the TMH variants using MODELLER, version 9.15 (Sali and Blundell, 1993), and then scanned through hundreds of transmembrane and interfacial configurations to identify the most energetically stable orientation (Figure 9C). For each helix position, we computed the energy in the membrane compared with an extracted state in solution, using bilayer parameters for a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane (see Supplemental Table S2 for all computed values in kcal/mol). We assumed that the designed helix has no interaction with TMH2, which is consistent with structural studies of related ABC transporters that reveal that these two segments share a poorly packed interface, as we noted earlier (Marcoline et al., 2015; Supplemental Figure 1C). Moreover, because the core of each sequence is relatively nonpolar, initial calculations revealed that the membrane distortion term was minimal, and we excluded membrane deformation from the analysis.
The physics-based model also predicts that as the hydrophobic content of the helices increases, the segments become more stable, which correlates with a decrease in the extent of retrotranslocation (Figure 9B). However, the physics-based scale shows a strict monotonic relationship between predicted membrane stability and retrotranslocation efficiency, whereas the phenomenological scale does not. This difference between scales arises because the physics-based scale predicts that construct 2 is less stable than 1 and that construct 5 is less stable than 4 (Supplemental Table S2). The increased stabilities of constructs 1 and 4 relative to constructs 2 and 5, respectively, arise from the four residues at either end of the helices, which partition only partially into the head-group region. These residues are large and bulky for constructs 1 and 4, and therefore they have larger surface areas and increased membrane stability, which correlate with the reduction in retrotranslocation efficiency. Both scales predict a large energetic separation between the first three constructs and the last two, yet the retrotranslocation efficiencies are only modestly reduced for the most stably integrated helices. One explanation for this observation is that there are multiple kinetic or energetic factors involved in retrotranslocation and that helix stability plays only a small part. Thus helix stability may be a smaller component modulating retrotranslocation compared with other steps in the process. Another possibility, which may be related to the first, is that because TMH2 is the same for all five constructs—and it must also be extracted during retrotranslocation—the total membrane stability is changing less for each construct than indicated by Figure 9, A and B.
The most obvious difference between the phenomenological scale and the physics-based model is the predicted stabilization energy of the segment in the membrane, which is much greater for the physics-based model. In fact, the physical model suggests that all of these segments should integrate into the membrane because even the marginally stable wild-type sequence is stabilized by ∼48 kcal/mol (Supplemental Table S2). These differences highlight the importance of the physical state of the retrotranslocated helix, which we know very little about, and which both methods certainly oversimplify. The physical model assumes that the extracted state is a helix in aqueous solution, which is energetically unlikely because it would expose ∼10 hydrophobic residues to water. On the other hand, the translocon-based energies are derived from experimentally measured insertion probabilities (Hessa et al., 2005), in which incorrect topologies are easily identifiable, but the biochemical environment of these improperly inserted segments is unknown. In fact, these segments are likely still associated with the translocon or fold in such a manner as to sequester hydrophobic residues away from water while exposing polar and charged residues to lipid head groups and water. For retrotranslocation, the most appropriate energy difference to calculate is between the membrane-embedded state and the extracted conformation in which the segment is associated with the Cdc48p machinery. As more structural information becomes available, this retrotranslocated reference state can be modeled using the physics-based framework described here.
DISCUSSION
During ERAD, misfolded integral membrane proteins are polyubiquitinated and retrotranslocated from the ER membrane. Although much has been learned about the chaperone-mediated selection of ERAD substrates, the factors that ubiquitinate ERAD substrates, and the retrotranslocation machinery, little attention has been paid to the biophysical features of a TMH in an ERAD substrate that affect retrotranslocation efficiency. In this study, we report on the generation of a new dual-pass chimeric ERAD substrate, Chimera A*, which derives from the misfolded domain from Ste6p*. We generated a series of Chimera A* variants with altered TMH hydrophobicities. We next confirmed that Chimera A* is a simplified version of Ste6p* and requires the same cellular machinery for degradation, although the retrotranslocation of Ste6p* is less efficient. By increasing TMH1 hydrophobicity in Chimera A*, we also discovered that retrotranslocation remains Cdc48p-dependent but becomes less efficient as the ΔG for membrane insertion becomes more favorable. We then used a physics-based model that takes into account the energetics of TMH stability in the lipid bilayer versus the aqueous environment after retrotranslocation to show how retrotranslocation efficiency correlates with TMH hydrophobicity.
Recognition of ERAD substrate transmembrane helices
Significant work has defined the molecular requirements for the insertion of a TMH into a lipid bilayer during translocation (Cymer et al., 2015). Unexpectedly, computational analysis of multispanning membrane proteins with solved structures revealed that up to 25% of TMHs have an unfavorable ΔG for insertion (Hessa et al., 2007; White and von Heijne, 2008). Given this finding, it may not be surprising that the native TMH1 and TMH2 of Ste6p* possess unfavorable energies for membrane partitioning (ΔG = 0.31 and 1.86 kcal/mol, respectively). This explains the inability of TMH2 to become membrane integrated, as observed with Chimera N*. Instead, TMH1-2 integration likely relies on neighboring TMHs in Ste6p for proper membrane partitioning and the acquisition of the correct topology, as described for other proteins (Higy et al., 2004; Pitonzo and Skach, 2006).
Given the fact that many TMHs insert poorly into lipid bilayers, the ERAD machinery has evolved to identify TMHs that may have failed to become membrane integrated. Although it is unclear for Chimera N* whether TMH2 or NBD2* acts as the dominant signal for ERAD, for other proteins, the TMH can be a key determinant in degradation. For example, TCRα must assemble with its partner CD3δ, but loss of charge pairing within their respective TMHs acts as a degradation signal (Bonifacino et al., 1990a,b, 1991). Similarly, lack of expression of the β-subunit causes inefficient membrane partitioning of TMH7 in the Na+/K+ ATPase α-subunit and exposes an ERAD recognition motif (Beggah et al., 1996; Beguin et al., 2000). More recently, Feige and Hendershot (2013) examined the effect of polar residues on the insertion of a single TMH by substituting this region from 24 single-pass cell surface receptors into a reporter protein. Surprisingly, a portion of the proteins with marginally hydrophobic TMHs failed to integrate into the membrane and instead slipped into the ER lumen before retrotranslocation (Feige and Hendershot, 2013). In addition, the degradation kinetics for full-length CFTR, a half-channel containing only the first six TMHs, and a substrate with only TMH1 and 2 was examined (Carlson et al., 2006). It is important to note that these constructs lacked specific degrons, so the degradation requirements likely arose from misfolded lesions in more than one cellular compartment. Regardless, an inverse correlation between the size of each substrate and degradation kinetics was observed through the use of an in vitro degradation assay (Carlson et al., 2006). Although p97 contributed to the degradation of each construct, it was impossible to distinguish between the retrotranslocation and degradation reactions. In contrast, Chimera A* and our series of variants contain a single cytoplasmic degron, and we specifically measured the retrotranslocation reaction. Consistent with these previous data, we observed increased retrotranslocation of Chimera A* compared with Ste6p* (Figure 7). In addition, we showed directly that increasing the hydrophobicity of a TMH decreases retrotranslocation efficiency (Figure 8).
Open questions on Cdc48p/p97 mechanism of action
Based on our work, an area for future investigation relates to the role that Cdc48p plays during the retrotranslocation of substrates with distinct TMH hydrophobicities. The use of Chimera A* instead of Ste6p* will provide several advantages for these future investigations, as the number of potential Cdc48p binding sites and ubiquitination sites are significantly decreased. In turn, several groups recently solved the structure of p97, shedding light on the conformational changes that occur during the ATPase cycle (Davies et al., 2008; Banerjee et al., 2016; Hänzelmann and Schindelin, 2016; Schuller et al., 2016). However, the mechanism by which these conformational changes are translated into force generation during retrotranslocation has not been investigated.
Do multiple Cdc48p/p97 hexamers engage a single misfolded substrate? Although ubiquitin conjugation was initially believed to occur only on lysine side chains, polyubiquitination has now been observed on cysteine, serine, and threonine, as well as at the N-terminus of target proteins (Peng et al., 2003; Shimizu et al., 2010; Wang et al., 2011b; McDowell and Philpott, 2013). Because Cdc48p/p97 interacts with ERAD substrates via polyubiquitin chains, multiple Cdc48p/p97 complexes most likely bind simultaneously to an ERAD substrate. Chimera A* contains 20 lysine residues that reside in the cytoplasm, whereas Ste6p* contains 64 cytoplasmic lysines. Because Chimera A* contains 1/6th of the TMHs present in Ste6p*, we predicted that we would observe a large increase the retrotranslocation efficiency of Chimera A*. Instead, only a 33% increase in efficiency for Chimera A* was observed. One explanation for this result could be that more than one Cdc48p hexamer engages Ste6p* during retrotranslocation. Although there are no studies describing the stoichiometry of Cdc48p/p97 binding to ERAD substrates, it is possible that multipass membrane proteins recruit more than one complex to help extract and/or maintain the solubility of retrotranslocated TMHs. Support for this model emerges from a recent study in which binding of Cdc48p to retrotranslocated ERAD-M substrates was found to be critical to maintain solubility (Neal et al., 2017).
Do Cdc48p/p97 substrates transit through the central pore of the hexamer? Several AAA-ATPases, including ClpA/X (bacteria), PAN (Archaea), and Rpt1-6 (eukaryotes), function as an “unfoldase” by translocating substrates through their central pore (Striebel et al., 2009). For Cdc48p/p97, it is controversial whether it functions as an unfoldase or as a “seggregase,” which separates protein complexes or proteins from membranes (reviewed in (Barthelme and Sauer, 2016; Xia et al., 2016). For ClpX, stable domains, which are likely found in many ERAD substrates, can act as kinetic barriers to unfolding and increase ATP consumption during unfolding (Kenniston et al., 2003). Therefore, if Cdc48p acts as an unfoldase, then we would predict a large difference in retrotranslocation efficiency between Ste6p* and Chimera A*. Moreover, if we invoke the multiple hexamer model, as discussed earlier, it is difficult to envision how several Cdc48p hexamers might bind to different polyubiquitination sites, each independently threading Ste6p* through their central pores. An alternate model of Cdc48p/p97 function suggests that it only engages substrates using conserved residues in the bottom pore (D2) of the hexameric ring (Barthelme and Sauer, 2013). This model is attractive because it allows for Cdc48p/p97 to perform both retrotranslocation and segregation activities using the same mechanism, and central pore unfolding is unnecessary. In either scenario, our data using the Chimera A* variants support the notion that both weakly and well-anchored TMHs require Cdc48p activity during retrotranslocation.
Finally, our results have important implications for design strategies to rescue or accelerate the degradation of disease-related ERAD substrates. For instance, mutations in p97 are the underlying cause of a degenerative disorder that affects multiple organ systems known as inclusion body myopathy, Paget’s disease of the bone, frontotemporal dementia, and amyotrophic lateral sclerosis (IBMPFD/ALS; Meyer and Weihl, 2014). Whereas p97 interacts with many cofactors to function in diverse cellular pathways (Wolf and Stolz, 2012), the neurodegenerative phenotypes and inclusion formation observed in IBMPFD/ALS are highly suggestive of a link to protein quality control, as seen for other neurodegenerative disorders, including Alzheimer, Parkinson, and Huntingtin diseases (Guerriero and Brodsky, 2012). In fact, select mutations associated with IBMPFD/ALS compromise ERAD efficiency (Weihl et al., 2006). This finding strongly suggests that at least some of the pathogenic phenotypes associated with IBMPFD/ALS could arise from impaired retrotranslocation, resulting in prolonged ER stress and neuronal cell death. Alternatively, use of Cdc48p/p97 enhancers could reduce ER stress that has been linked to Cdc48p localization to polyglutamine aggregates (Duennwald and Lindquist, 2008), and much attention has been paid to identifying p97 inhibitors to treat cancer (Chapman et al., 2015; Vekaria et al., 2016). Therefore it is important to understand how these and future cancer therapeutics that target p97 interface with the ERAD pathway and potentially modify retrotranslocation efficiency. With better structural and functional data, as well as an increased understanding of the contribution of Cdc48p/p97 cofactors during retrotranslocation, we envision the development of function-specific modulators for this complex molecular machine.
MATERIALS AND METHODS
Yeast strains, plasmids, and plasmid construction
Yeast were maintained as previously described unless otherwise stated (Adams et al., 1997). Supplemental Table S3 gives a complete list of the S. cerevisiae strains used in this study. The plasmids and primers used are listed in Supplemental Table S4. To generate pKN05, which was designed to express Chimera N*, we amplified the first two TMHs, including the triple hemagglutinin (HA) tag from pSM1911, using oKN57 and oKN58, and we amplified NBD2* using oKN53 and oKN85. The two fragments were blunt-end ligated, digested with XmaI and SacII, and ligated into pSM1911, which had the gene encoding Ste6p* removed using the same enzymes. This plasmid is 2μ (multicopy) and driven by a PGK (high-expression) promoter. To delete TMH2, a two-stage PCR site-directed mutagenesis protocol was used with oCG15 and oCG16 to generate pCG11 (Wang and Malcolm, 1999). To create pCG12, an artificial TMH2 (Hessa et al., 2007) was inserted using the same protocol, with oCG12 and oCG13. To generate full-length NBD2 in pKN05 and pCG12, site-directed mutagenesis was performed using oCG26 and oCG27. To delete TMH1, site-directed mutagenesis was performed using oCG37 and oCG38 to generate pCG32. The same mutagenesis protocol was used to make Chimera A*ΔG = −0.19 (oCG39/40), Chimera A*ΔG = −1.07 (oCG77/78), Chimera A*ΔG = −4.38 (oCG79/80), and Chimera A*ΔG = −7.15 (oCG81/82) using pCG32 as a template. The complete DNA sequence encoding each chimera was confirmed using oKN54, oCG06, and oCG07. To generate Chimera A and Chimera A*, expression vectors under the control of a weaker promoter, pCG12 and 19, were subcloned into a centromeric (single-copy) expression vector under the control of an alcohol dehydrogenase (ADH, low-expression) promoter to generate pCG27 and oCG29, respectively. Full amino acid sequences for each Chimera variant can be found in FASTA format in the Supplemental Material.
Antibodies used in this study
For cycloheximide chase analyses, rat monoclonal anti–HA-horseradish peroxidase (HRP; 3F10; Roche) and rabbit anti–glucose-6-phosphate dehydrogenase (G6PD; A9521; Sigma-Aldrich) were used at a dilution of 1:5000. For sucrose gradient fractionation, polyclonal rabbit anti-Sec61p was used at 1:1000 (Stirling et al., 1992), rabbit anti–Anp1p (a gift from Sean Munro, Cambridge University, Cambridge, United Kingdom) was used at 1:4000, and polyclonal rabbit anti-Pma1p (Abcam) was used at 1:2500. To probe for myc-tagged ubiquitin, a rabbit polyclonal anti-myc (sc987; Santa Cruz Biotechnology) was used at 1:2500. For limited proteolysis studies, rat monoclonal anti–HA-HRP (3F10; Roche) was used at a dilution of 1:5000. In all cases, after overnight incubation with primary antibodies at 4°C, the bound antibodies were decorated with either anti-mouse or anti-rabbit immunoglobulin G (IgG) HRP-conjugated secondary antibodies (Cell Signaling Technology) at 1:5000 for 2 h at room temperature. Proteins were visualized using the SuperSignal Chemiluminescence kit (Thermo Scientific). Blot images were taken using a Bio-Rad ChemiDoc XRS+ with Image Lab software. Images were further analyzed using ImageJ software, version 1.49b (National Institutes of Health). For indirect immunofluorescence microscopy, mouse anti-HA (12CA5; Roche) was used at 1:500 and rabbit anti-Kar2p was used at 1:250 (Brodsky and Schekman, 1993). Primary antibodies were decorated with Alexa Fluor 488 goat anti-mouse or Alexa Fluor 568 goat anti-rabbit at 1:500.
Limited proteolysis assay
Microsomes were prepared from yeast expressing either Chimera A* or Chimera N* under the control of the PGK promoter. In brief, cultures were harvested by centrifugation, washed with ice-cold double-distilled H2O, and flash frozen for storage at −80°C. Cell pellets were thawed on ice and disrupted by glass bead agitation using the medium scale protocol (Nakatsukasa and Brodsky, 2010). Microsomes were mixed on ice with 4 μg/ml proteinase K (Sigma-Aldrich). An aliquot of the reaction was removed at the indicated time points, and proteolysis was halted by precipitation with trichloroacetic acid (TCA). Protein pellets were resuspended in TCA sample buffer (80 mM Tris, pH 8, 8 mM EDTA, 3.5% SDS, 15% glycerol, 0.08% Tris base, 0.01% bromophenol blue) supplemented with freshly added β-mercaptoethanol (to a final concentration of 5%) before analysis by SDS–PAGE and immunoblot analysis.
Assays to monitor protein degradation
To measure protein turnover, we used a cycloheximide chase assay in which yeast cells expressing an ERAD substrate were grown to log phase (OD600 = 0.5–1.5) in synthetic complete (SC) medium lacking uracil (–Ura) and containing glucose. A 1 ml aliquot was removed for the 0-min time point, and then cycloheximide was added to a final concentration of 188 μg/ml. The culture was incubated in a shaking water bath at 26, 37, or 39°C as indicated at 200 rpm. At each time point, 1 ml aliquots were removed to ice-cold tubes containing 35 μl of 0.5 M NaN3 (final concentration of 17.5 mM) and were lysed (Nakatsukasa et al., 2008). TCA-precipitated protein pellets were then disrupted with a mechanical pestle in TCA sample buffer (see earlier description), and the samples were incubated for 30 min at 37°C. An aliquot of each sample was analyzed on a denaturing 10% polyacrylamide gel, and proteins were transferred onto nitrocellulose (BioTrace NT; Pall Corp.) using a Trans-blot turbo transfer system (Bio-Rad).
Sucrose gradient analysis
Sucrose gradients were performed as previously described (Sullivan et al., 2003). Approximately 40 OD600 equivalents of log-phase BY4742 cells expressing HA-tagged Chimera A* were grown in SC –Ura/glucose medium. Cells were pelleted for 3 min at 3000 rpm in a clinical centrifuge at room temperature and resuspended in 400 μl of 10 mM Tris, pH 7.6, 10 mM EDTA, 10% sucrose supplemented with 3 mM phenylmethylsulfonyl fluoride (PMSF), 3 μg/ml leupeptin, and 1.5 μg/ml pepstatin A, and 1 mM dithiothreitol and frozen dropwise into liquid N2, followed by storage at −80°C. Cells were lysed under liquid N2 with a prechilled mortar and pestle by grinding for ∼5 min, with periodic refreshment of the liquid N2. The final yeast powder was thawed, and unbroken cells were removed by two rounds of centrifugation for 2 min at 2000 rpm in a table-top microcentrifuge at 4°C. The cleared lysate was then loaded onto an 11 ml of 20–70% noncontinuous sucrose gradient, and 0.5% of the load was saved. The gradients were centrifuged at 100,000 × g in a Beckman SW41 rotor for 18 h at 4°C. Fractions were collected from the top of the gradient, and pelleted proteins at the bottom of the tube were solubilized in TCA sample buffer. An aliquot from each fraction was mixed with TCA sample buffer and analyzed by immunoblotting using the indicated antibodies.
Assays to monitor protein ubiquitination and retrotranslocation
To measure the amount of substrate ubiquitination in yeast, we grew a 35-ml culture of pdr5Δ yeast transformed with a vector engineered for Cu2+-inducible expression of myc-tagged ubiquitin and either an empty vector or a vector containing the Chimera A* or Ste6p* coding sequence under the control of the PGK promoter to log phase (OD600, ∼ 0.7–0.8) at 26°C. To examine the effect of proteasome inhibition, cultures were treated with DMSO or 20 μM MG132 for 1 h with simultaneous induction of myc-tagged ubiquitin production with 100 μM copper sulfate. Cells were harvested by centrifugation for 3 min at 3000 rpm in a table-top clinical centrifuge, and pellets were washed once with ice-cold water, recentrifuged, and stored at −80°C. The cell pellets were then thawed on ice and resuspended in 1 ml of radioimmunoprecipitation assay buffer (25 mM Tris, pH 7, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% deoxycholate) supplemented with 3 mM PMSF, 3 μg/ml leupeptin, 1.5 μg/ml pepstatin A, and 10 μM N-ethylmaleimide (NEM) and lysed by glass bead disruption three times for 1 min. The supernatant was removed and combined with a 500 μl wash of the beads with buffer, and the unbroken cells were removed by centrifugation in a table-top microcentrifuge for 10 min at 13,000 rpm at 4°C. The protein concentration was determined by measuring the A280 of each sample, and equal amounts of lysate were immunoprecipitated overnight at 4°C using 30 μl of anti-HA-conjugated agarose beads (Roche Applied Science). The beads were washed four times with RIPA buffer supplemented with protease inhibitors and NEM. Protein was eluted from the beads with TCA sample buffer supplemented with fresh β-mercaptoethanol at a final concentration of 5%. The liberated proteins were run on 10% denaturing polyacrylamide gels and transferred to nitrocellulose as described above. Before blocking of the nitrocellulose membrane, the membrane was incubated in boiling water for 1 h to expose antibody epitopes on polyubiquitin chains. The polyubiquitin profile was visualized using an anti-myc antibody.
An in vitro retrotranslocation assay using 125I-ubiquitin was performed essentially as described (Nakatsukasa et al., 2008; Nakatsukasa and Brodsky, 2010). In brief, microsomes were prepared from yeast expressing the indicated chimera variant or Ste6p* under the control of the PGK promoter. Cytosol was prepared from CDC48 or cdc48-2 yeast grown at either 26°C or 2 h at 39°C, as indicated, by liquid nitrogen lysis, and the supernatant (cytosol) was collected after a 1 h spin at 300,000 × g in a Beckman L8-70M ultracentrifuge using the SW 55 Ti rotor at 4°C. The in vitro reactions were mixed on ice and contained either 1 or 5 mg/ml yeast cytosol as indicated, as well as 1 mg/ml ER-derived microsomes and an ATP-regenerating system (1 mM ATP, 40 M creative phosphate, and 0.2 mg/ml creatine phosphokinase in Buffer 88 [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 6.8, 150 mM KOAc, 250 mM sorbitol, 5 mM MgOAc]). The reactions were assembled on ice and shifted to room temperature for 10 min before the addition of 125I-ubiquitin to a final concentration of ∼2 μg/ml. The reactions were then allowed to proceed for 40 min (CDC48 cytosol) or 60 min (cdc48-2 cytosol) at room temperature. The reactions were stopped by the addition of 125 μl of 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1.25% SDS plus protease inhibitors, and 10 mM NEM. After incubation at 37°C for 30 min, the samples were mixed with 400 μl of 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 2% Triton X-100 plus protease inhibitors, and 10 mM NEM. Ste6p* and Chimera A* were immunoprecipitated with anti-HA antibody and protein A–Sepharose beads and resolved by SDS–PAGE. Each sample was split so that half was used for phosphorimager analysis and the other half for immunoblot analysis. To quantitate the amount of 125I-ubiquitin–tagged substrate present in each lane, the signal was quantified using the measure tool contained in the ImageJ software, version 1.49b. A rectangular box was drawn around the ubiquitin smear starting at the predicted molecular weight for the substrate being examined to the top of the resolving gel for the supernatant lane (S), pellet lane (P), and an empty lane (used for background subtraction). We calculated percentage retrotranslocation = [S/(S + P)] × 100.
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy was performed as described previously (Amberg et al., 2005). Briefly, cells were grown in SC–Ura/glucose medium to an OD600 of ∼0.5 and fixed for 1 h in 4% formaldehyde. The cells were spheroplasted by incubation with Zymolyase 20T (U.S. Biological) at 37°C for 15–20 min and spotted onto slides treated with 1% polylysine and permeabilized with methanol/acetone treatment. The fixed cells were blocked and further permeabilized using 0.5% bovine serum albumin, 0.5% ovalbumin, and 0.6% fish-skin gelatin supplemented with 0.1% Triton X-100. Cells were incubated with primary and secondary antibodies as described earlier, and 4′,6-diamidino-2-phenylindole (DAPI) was included at 1:250 to stain the nuclei. The slides were mounted using Prolong Antifade Gold (Invitrogen) and imaged with an Olympus FV1000, 100× UPlanSApo oil immersion objective, numerical aperture 1.40. Images were analyzed using ImageJ software, version 1.49b.
Physics-based insertion model
Idealized helices were generated with MODELLER, version 9.15 (Sali and Blundell, 1993), and then APBSmem was used to center the helices and orient them in the membrane (Marcoline et al., 2015). We scanned tilts from 0 to 60° in increments of 10°, rotations from 0 to 360° in increments of 30°, and z offsets from −5 to 5 Å in increments of 1 Å. At every orientation, the electrostatic and nonpolar energies were calculated using the same procedures as described (Marcoline et al., 2015). Briefly, charges and atomic radii were assigned using PDB2PQR (Dolinsky et al., 2004) and the PARSE parameter set. The electrostatic energies were calculated using the Adaptive Poisson–Boltzmann Solver (Baker et al., 2001), where the dielectric influence of the membrane was added using APBSmem. Nonpolar stabilization energies were calculated as proportional to the solvent-accessible surface area embedded in the membrane. The surface tension used in the nonpolar calculation was constant in the membrane core (23 cal/mol/Å2) and decayed linearly to zero across the head-group regions. The total energy was calculated for every orientation, and the minimum energy for each helix is reported.
Homology modeling
The closest crystalized homologue for NBD2 was determined by querying the Research Collaboratory for Structural Bioinformatics Protein Data Bank (www.rcsb.org; Berman et al., 2002). A solved structure for mouse P-glycoprotein, Protein Data bank ID 4M1M (Li et al., 2014), was chosen based on 35% sequence identity to the NBD2 sequence. Homology models were constructed with MODELLER, version 9.15 (Sali and Blundell, 1993). The homology model of Ste6p TMH1 and 2 was based on another structure of murine p-glycoprotein, 3G5U (Li et al., 2014). A magnified depiction of TMH1 and 2 (Supplemental Figure S1C) was made using visual molecular dynamics (Humphrey et al., 1996; www.ks.uiuc.edu/Research/vmd/).
Supplementary Material
Acknowledgments
We thank the members of the Brodsky laboratory for their thoughtful insights into experimental design and for feedback during the preparation of the manuscript, and especially Olivia Hilal for technical assistance. This work was supported by National Institutes of Health Grants DK101584 to C.J.G. and DK061296 and DK72506 to G.M.P., a Cystic Fibrosis Foundation Student Traineeship (PRESTO15H0) to G.M.P., a Beckman Scholars Award to K.F.W., an American Heart Association Predoctoral Fellowship (7028397) to N.B.P., and National Institutes of Health Grants GM117593 to M.G. and GM75061 and DK079307 to J.L.B.
Abbreviations used:
- ABC
ATP-binding cassette
- ADH
alcohol dehydrogenase
- CFTR
cystic fibrosis transmembrane conductance regulator
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum–associated degradation
- NBD
nucleotide-binding domain
- PGK
phosphoglycerate kinase
- Ste6p*
Sterile 6*
- TMH
transmembrane helix
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-03-0184) on May 24, 2017.
REFERENCES
- Adams A, Gottschling DE, Kaiser CA, Stearns T. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. [Google Scholar]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg DC, Burke D, Strathern JN, Cold Spring Harbor Laboratory . Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Argudo D, Bethel NP, Marcoline FV, Grabe M. Continuum descriptions of membranes and their interaction with proteins: towards chemically accurate models. Biochim Biophys Acta. 2016;1858:1619–1634. doi: 10.1016/j.bbamem.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Delgado C, Marti-Renom MA, Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur Biophys J. 2013;42:199–207. doi: 10.1007/s00249-012-0813-9. [DOI] [PubMed] [Google Scholar]
- Bagola K, Mehnert M, Jarosch E, Sommer T. Protein dislocation from the ER. Biochim Biophys Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge RD, Rapoport TA. Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell. 2016;166:394–407. doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelme D, Sauer RT. Bipartite determinants mediate an evolutionarily conserved interaction between Cdc48 and the 20S peptidase. Proc Natl Acad Sci USA. 2013;110:3327–3332. doi: 10.1073/pnas.1300408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelme D, Sauer RT. Origin and functional evolution of the Cdc48/p97/VCP AAA+ protein unfolding and remodeling machine. J Mol Biol. 2016;428:1861–1869. doi: 10.1016/j.jmb.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Bays NW, Hampton RY. Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr Biol. 2002;12:R366–R371. doi: 10.1016/s0960-9822(02)00862-x. [DOI] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggah A, Mathews P, Beguin P, Geering K. Degradation and endoplasmic reticulum retention of unassembled alpha- and beta-subunits of Na,K-ATPase correlate with interaction of BiP. J Biol Chem. 1996;271:20895–20902. doi: 10.1074/jbc.271.34.20895. [DOI] [PubMed] [Google Scholar]
- Beguin P, Hasler U, Staub O, Geering K. Endoplasmic reticulum quality control of oligomeric membrane proteins: topogenic determinants involved in the degradation of the unassembled Na,K-ATPase alpha subunit and in its stabilization by beta subunit assembly. Mol Biol Cell. 2000;11:1657–1672. doi: 10.1091/mbc.11.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkower C, Michaelis S. Mutational analysis of the yeast a-factor transporter STE6, a member of the ATP binding cassette (ABC) protein superfamily. EMBO J. 1991;10:3777–3785. doi: 10.1002/j.1460-2075.1991.tb04947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkower C, Taglicht D, Michaelis S. Functional and physical interactions between partial molecules of STE6, a yeast ATP-binding cassette protein. J Biol Chem. 1996;271:22983–22989. doi: 10.1074/jbc.271.38.22983. [DOI] [PubMed] [Google Scholar]
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Klausner RD. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell. 1990a;63:503–513. doi: 10.1016/0092-8674(90)90447-m. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Shah N, Klausner RD. Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J. 1991;10:2783–2793. doi: 10.1002/j.1460-2075.1991.tb07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Klausner RD. A peptide sequence confers retention and rapid degradation in the endoplasmic reticulum. Science. 1990b;247:79–82. doi: 10.1126/science.2294595. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Schekman R. A Sec 63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J Cell Biol. 1993;123:1355–1363. doi: 10.1083/jcb.123.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Jordahl AS, Yates ME, Preston M, Cook E, Kleyman TR, Brodsky JL. Interactions between intersubunit transmembrane domains regulate the chaperone dependent degradation of an oligomeric membrane protein. Biochem J. 2016;474:357–376. doi: 10.1042/BCJ20160760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E, Maksim N, de la Cruz F, La Clair JJ. Inhibitors of the AAA+ chaperone p97. Molecules. 2015;20:3027–3049. doi: 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S, Hecht KA, Grabe M. A continuum method for determining membrane protein insertion energies and the problem of charged residues. J Gen Physiol. 2008;131:563–573. doi: 10.1085/jgp.200809959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymer F, von Heijne G, White SH. Mechanisms of integral membrane protein insertion and folding. J Mol Biol. 2015;427:999–1022. doi: 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16:715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MJ, Hendershot LM. Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol Cell. 2013;51:297–309. doi: 10.1016/j.molcel.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaczynska M, Osmulski PA. Small-molecule inhibitors of proteasome activity. Methods Mol Biol. 2005;301:3–22. doi: 10.1385/1-59259-895-1:003. [DOI] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY. In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem. 2009;284:14710–14722. doi: 10.1074/jbc.M809607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J Biol Chem. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara M, Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxid Redox Signal. 2012;16:1119–1128. doi: 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24:460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Hänzelmann P, Schindelin H. Structural basis of ATP hydrolysis and intersubunit signaling in the AAA+ ATPase p97. Structure. 2016;24:127–139. doi: 10.1016/j.str.2015.10.026. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Molecular code for transmembrane-helix recognition by the Sec 61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- Higy M, Junne T, Spiess M. Topogenesis of membrane proteins at the endoplasmic reticulum. Biochemistry. 2004;43:12716–12722. doi: 10.1021/bi048368m. [DOI] [PubMed] [Google Scholar]
- Hitchcock AL, Krebber H, Frietze S, Lin A, Latterich M, Silver PA. The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol Biol Cell. 2001;12:3226–3241. doi: 10.1091/mbc.12.10.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck SA, Ren HY, Madden VJ, Bonner JN, Conlin MP, Janovick JA, Conn PM, Cyr DM. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol Cell. 2014;54:166–179. doi: 10.1016/j.molcel.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38, 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Jones EW. The synthesis and function of proteases in Saccharomyces: genetic approaches. Annu Rev Genet. 1984;18:233–270. doi: 10.1146/annurev.ge.18.120184.001313. [DOI] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol Biol Cell. 2003;14:3437–3448. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- Lee J-Y, Rosenbaum DM. Transporters revealed. Cell. 2017;168:951–953. doi: 10.1016/j.cell.2017.02.033. [DOI] [PubMed] [Google Scholar]
- Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell. 2009;20:3330–3341. doi: 10.1091/mbc.E08-09-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- Linton KJ. Structure and function of ABC transporters. Physiology. 2007;22:122–130. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- Loayza D, Tam A, Schmidt WK, Michaelis S. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2767–2784. doi: 10.1091/mbc.9.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoline FV, Bethel N, Guerriero CJ, Brodsky JL, Grabe M. Membrane protein properties revealed through data-rich electrostatics calculations. Structure. 2015;23:1526–1537. doi: 10.1016/j.str.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell GS, Philpott A. Non-canonical ubiquitylation: mechanisms and consequences. Int J Biochem Cell Biol. 2013;45:1833–1842. doi: 10.1016/j.biocel.2013.05.026. [DOI] [PubMed] [Google Scholar]
- McGrath JP, Varshavsky A. The yeast STE6 gene encodes a homologue of the mammalian multidrug resistance P-glycoprotein. Nature. 1989;340:400–404. doi: 10.1038/340400a0. [DOI] [PubMed] [Google Scholar]
- Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M, Sommer T, Jarosch E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat Cell Biol. 2014;16:77–86. doi: 10.1038/ncb2882. [DOI] [PubMed] [Google Scholar]
- Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. 2014;127:3877–3883. doi: 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Wang Y, Warren G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002;21:5645–5652. doi: 10.1093/emboj/cdf579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci USA. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris LL, Hartman IZ, Jun DJ, Seemann J, DeBose-Boyd RA. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2014;289:19053–19066. doi: 10.1074/jbc.M114.576652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL. in vitro reconstitution of the selection, ubiquitination, and membrane extraction of a polytopic ERAD substrate. Methods Mol Biol. 2010;619:365–376. doi: 10.1007/978-1-60327-412-8_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Kamura T. Subcellular fractionation analysis of the extraction of ubiquitinated polytopic membrane substrate during ER-associated degradation. PLoS One. 2016;11:e0148327. doi: 10.1371/journal.pone.0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal SE, Mak R, Bennett EJ, Hampton R. A “retrochaperone” function for Cdc48: the Cdc48 complex is required for retrotranslocated ERAD-M substrate solubility. J Biol Chem. 2017;292:3112–3128. doi: 10.1074/jbc.M116.770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham PG, Patel HJ, Chiosis G, Thibodeau PH, Brodsky JL. Mutations in the yeast Hsp70, Ssa1, at P417 alter ATP cycling, interdomain coupling, and specific chaperone functions. J Mol Biol. 2015;427:2948–2965. doi: 10.1016/j.jmb.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng W, Sergeyenko T, Zeng N, Brown JD, Romisch K. Characterization of the proteasome interaction with the Sec 61 channel in the endoplasmic reticulum. J Cell Sci. 2007;120:682–691. doi: 10.1242/jcs.03351. [DOI] [PubMed] [Google Scholar]
- Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem. 1993;268:5798–5801. [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013;5:a013185. doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Pisoni GB, Molinari M. Five questions (with their answers) on ER-associated degradation. Traffic. 2016;17:341–350. doi: 10.1111/tra.12373. [DOI] [PubMed] [Google Scholar]
- Pitonzo D, Skach WR. Molecular mechanisms of aquaporin biogenesis by the endoplasmic reticulum Sec 61 translocon. Biochim Biophys Acta. 2006;1758:976–988. doi: 10.1016/j.bbamem.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer A, Wolf DH. Sec 61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009;28:2874–2884. doi: 10.1038/emboj.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell Mol Life Sci. 2008;65:2360–2371. doi: 10.1007/s00018-008-8072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller JM, Beck F, Lossl P, Heck AJ, Forster F. Nucleotide-dependent conformational changes of the AAA+ ATPase p97 revisited. FEBS Lett. 2016;590:595–604. doi: 10.1002/1873-3468.12091. [DOI] [PubMed] [Google Scholar]
- Scott DC, Schekman R. Role of Sec 61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Okuda-Shimizu Y, Hendershot LM. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol Cell. 2010;40:917–926. doi: 10.1016/j.molcel.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling CJ, Rothblatt J, Hosobuchi M, Deshaies R, Schekman R. Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol Biol Cell. 1992;3:129–142. doi: 10.1091/mbc.3.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 2009;19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Sullivan ML, Youker RT, Watkins SC, Brodsky JL. Localization of the BiP molecular chaperone with respect to endoplasmic reticulum foci containing the cystic fibrosis transmembrane conductance regulator in yeast. J Histochem Cytochem. 2003;51:545–548. doi: 10.1177/002215540305100417. [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Deve. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH. Retention of mutant α1-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G961–G974. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- Valentijn JA, Fyfe GK, Canessa CM. Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J Biol Chem. 1998;273:30344–30351. doi: 10.1074/jbc.273.46.30344. [DOI] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekaria PH, Home T, Weir S, Schoenen FJ, Rao R. Targeting p97 to disrupt protein homeostasis in cancer. Frontiers Oncol. 2016;6:181. doi: 10.3389/fonc.2016.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RF, DeGrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci USA. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Thibault G, Ng DT. Routing misfolded proteins through the multivesicular body (MVB) pathway protects against proteotoxicity. J Biol Chem. 2011a;286:29376–29387. doi: 10.1074/jbc.M111.233346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site-directed mutagenesis. Biotechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guan S, Acharya P, Koop DR, Liu Y, Liao M, Burlingame AL, Correia MA. Ubiquitin-dependent proteasomal degradation of human liver cytochrome P450 2E1: identification of sites targeted for phosphorylation and ubiquitination. J Biol Chem. 2011b;286:9443–9456. doi: 10.1074/jbc.M110.176685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, Hanson PI. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet. 2006;15:189–199. doi: 10.1093/hmg/ddi426. [DOI] [PubMed] [Google Scholar]
- White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec 61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- Wolf DH, Stolz A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta. 2012;1823:117–124. doi: 10.1016/j.bbamcr.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Xia D, Tang WK, Ye Y. Structure and function of the AAA + ATPase p97/Cdc48p. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell. 2016;167:1586–1597.e1589. doi: 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.