

Abstract
Global brain ischemia resulting from cardiac arrest and cardiac surgery can lead to permanent brain damage and mental impairment. A clinical hallmark of global brain ischemia is delayed neurodegeneration, particularly within the CA1 subsector of the hippocampus. Unfortunately, the biochemical mechanisms have not been fully elucidated, hindering optimization of current therapies (i.e., therapeutic hypothermia) or development of new therapies. A major limitation to elucidating the mechanisms that contribute to neurodegeneration and understanding how these are influenced by potential therapies is the inability to relate biochemical markers to alterations in the morphology of individual neurons. Although immunocytochemistry allows imaging of numerous biochemical markers at the sub-cellular level, it is not a direct chemical imaging technique and requires successful “tagging” of the desired analyte. Consequently, important biochemical parameters, particularly those that manifest from oxidative damage to biological molecules, such as aggregated protein levels, have been notoriously difficult to image at the cellular or sub-cellular level. It has been hypothesized that reactive oxygen species (ROS) generated during ischemia and reperfusion facilitate protein aggregation, impairing neuronal protein homeostasis (i.e., decreasing protein synthesis) that in turn promotes neurodegeneration. Despite indirect evidence for this theory, direct measurements of morphology and ROS induced biochemical damage, such as increased protein aggregates and decreased protein synthesis, within the same neuron is lacking, due to the unavailability of a suitable imaging method. Our experimental approach has incorporated routine histology with novel wide-field synchrotron radiation Fourier transform infrared imaging (FTIRI) of the same neurons, ex vivo within brain tissue sections. The results demonstrate for the first time that increased protein aggregation and decreased levels of total protein occur in the same CA1 pyramidal neurons 1 day after global ischemia. Further, analysis of serial tissue sections using X-ray absorption spectroscopy at the sulfur K-edge has revealed that CA1 pyramidal neurons have increased disulfide levels, a direct indicator of oxidative stress, at this time point. These changes at 1 day after ischemia precede a massive increase in aggregated protein and disulfide levels concomitant with loss of neuron integrity 2 days after ischemia. Therefore, this study has provided direct support for a correlative mechanistic link in both spatial and temporal domains between oxidative stress, protein aggregation and altered protein homeostasis prior to irreparable neuron damage following global ischemia.
Keywords: Ischemia, Protein aggregation, FTIR, Hippocampus, Neurodegeneration, Thiol redox, XAS
1. Introduction
Global brain ischemia, with insufficient blood flow, oxygen and energy supply to the entire brain, can occur as a result of cardiac arrest and cardiac surgery, with an important consequence being neurocognitive impairment.[1] Unfortunately, without effective therapeutic intervention, neuron death can continue for days or weeks after global ischemia.[2] Although tragic, this highlights an extended therapeutic window in time over which therapies could be administered to minimize or prevent further delayed neuron death. Numerous rodent models have been used to study the biochemical mechanisms of delayed neurodegeneration in brain regions susceptible to ischemia [1] and specifically that in the pyramidal neurons in the CA1 sector of the hippocampus.[2–5] However, the exact biochemical mechanisms activated by ischemia that drive delayed neurodegeneration remain incompletely elucidated. Expanded understanding of these mechanisms could drive optimization of proven treatment such as therapeutic hypothermia as well as the development of new combination treatment approaches. [1]
It is well established that production of reactive oxygen species (ROS) and oxidative stress occurs during brain ischemia and reperfusion. [6, 7] Specifically, the generation of ROS and oxidative stress result in protein oxidation and altered thiol redox (favouring oxidation of thiols to disulfides), promoting protein aggregation. [8–12] It has been hypothesized that oxidative stress may result in protein aggregation within 24 hours of ischemic insult, which could impair neuron protein synthesis and metabolism resulting in abnormal neuron function and inevitable neuron death. [13–22] Unfortunately, previous studies have not had access to direct biochemical imaging methods that quantify thiol redox, aggregated protein and total protein at the single cell level. Consequently, definitive proof of a cause and effect relationship between protein oxidation and delayed neurodegeneration following global ischemia has remained elusive.[23]
Current imaging methods to study protein aggregation within brain tissue sections are limited to indirect analyses. Electron microscopy and optical microscopy both reveal dense aggregated material in the soma of neurons after global ischemia. Although attributed to aggregated proteins, direct confirmation is not provided by either of these techniques. [4, 13, 24] Whereas the formation of protein aggregates of specific proteins, such as amyloid beta, can be imaged in diseases such as Alzheimer’s disease, using immunofluorescence and a protein specific antibody [25], this is inadequate for brain ischemia, for which many proteins are of interest. Likewise, intra-cellular machinery that “marks” protein aggregates for degradation, (i.e., the ubiquitin pathway) can be stained with immunocytochemistry to indirectly localize aggregated proteins. [13–15] However, this method is indirect, largely non-quantitative, and results are difficult to interpret because the disease itself can modify the ubiquitin pathways that label the aggregated protein. For example, subtle increases in ubiquitin staining with increased protein aggregation are reported in some studies after brain ischemia. [13–15] This is in contrast to studies of more severe ischemic damage in which it has been reported that protein aggregation can prevent normal ubiquitin function and diminish antigenicity such that increased protein aggregation results in decreased ubiquitin staining. [26] Therefore, a technique to directly image aggregated proteins is much needed. In addition to imaging techniques, aggregated proteins may be quantified by chromatographic analysis in homogenates prepared from micro-dissected tissue. However, the spatial resolution is limited to the size of tissue that can be dissected (i.e., hundreds of microns), and the biochemical signature obtained represents the average across the entire piece of dissected tissue. [27] Similarly, quantification of biochemical markers of thiol redox, such as reduced glutathione, oxidized glutathione, protein thiols and protein disulfides is often limited to bulk analysis of dissected tissue. [10] In addition, a large glial response occurs at the same time as delayed neurodegeneration within the CA1 region, [28] which presents a major challenge when studying micro-dissected tissue. In the past, the inability to distinguish between the glial and neuron chemical signature following tissue homogenization has produced serious errors or confounding results. [19, 21]
A promising technique to directly image aggregated proteins ex vivo within tissue sections is Fourier transform infrared (FTIR) spectroscopy.[29–37] Specifically, FTIR is well suited to study alterations in protein secondary structure, for example formation of high molecular weight aggregated protein. It is well established that the position of the amide I band for high molecular weight aggregated proteins occurs at ~1625 cm−1, different from the location of proteins with a β-sheet secondary structure (1640–1630 cm−1), or α-helix secondary structure (1656 cm−1). [31, 36, 38–40] Unfortunately, due to the long wavelengths of light, the diffraction limited spatial resolution associated with FTIR imaging (FTIRI) or FTIR micro-spectroscopy (FTIRM) is an order of magnitude poorer than conventional visible light microscopy. [41] Traditionally, diffraction limited spatial resolution (2–10 μm) has been achieved using FTIRM and a confocal microscopy optical configuration and point by point data collection. [41–44] However, the standard approach used for FTIRM results in a data collection time of many hours or days for an individual image, which limits potential biochemical applications of FTIRM. Fortunately, recent advancements have coupled high magnification and high numerical aperture objectives and focal plane array detectors with synchrotron light sources to perform rapid biochemical imaging within minutes at sub-cellular spatial resolution. [32, 45, 46] This technique, wide-field synchrotron radiation-Fourier transform infrared imaging (SR-FTIRI), has been recently validated and used for direct biochemical imaging of individual neurons within brain tissue sections. [32, 47–49] The method has allowed protein aggregates associated with amyloid-beta plaques in Alzheimer’s disease to be studied at the sub-cellular level. [32, 50, 51] To date, the technique has not been used to image protein aggregates other than those associated with amyloid beta plaques or to image protein aggregates within individual neurons.
In addition to FTIRI, X-ray absorption spectroscopy at the sulfur K-edge is a promising novel technique to directly quantify thiol redox in situ[52, 53] that has been recently validated and applied to the field of neuroscience. [54, 55] Due to the wide energy range of the sulfur K-edge and sensitivity of the technique to both the oxidation state and molecular geometry of sulfur, multiple chemical forms of sulfur can be determined simultaneously. This property was recently used to demonstrate increased disulfide levels as a consequence of complete cerebral ischemia.[54]
In this investigation, we have used wide-field SR-FTIRI to study the levels of total protein and aggregated protein within individual rat hippocampal CA1 pyramidal neurons at 1, 2, and 5 day(s) after global ischemia. Our method incorporated routine haematoxylin and eosin (H&E) histology performed on the same neurons following SR-FTIRI to identify eosinophilic neurons, as a marker of irreparable neuron damage. In addition, serial sections were analysed with micro-X-ray absorption spectroscopy (micro-XAS) at the sulfur K-edge to determine the relative disulfide content of neurons within the CA1 sector. Serial tissue sections were also stained with Fluoro-jade B to assess neuron viability, and ubiquitin immunocytochemistry was performed to correlate biochemical alterations with alterations in proteasome status. The results of this study demonstrate for the first time a direct correlation between increased protein aggregation and decreased total protein in CA1 pyramidal neurons at 1 day after global ischemia, which occurs concomitant with increased disulfide levels, a direct marker of oxidative stress. These changes precede a massive increase in aggregated protein content and loss of neuron integrity 2 days after ischemia. Therefore, this study has provided direct evidence that supports a mechanistic link, in both spatial and temporal domains, between impaired protein homeostasis and protein aggregation, prior to irreparable neuron damage after global ischemia. Further, this study acts as a valuable template for the integration of SR-FTIRI and XAS at the sulfur K-edge into the field of neuroscience, where both techniques are expected to find widespread use to study sub-cellular neuronal chemistry in both the healthy and diseased brain.
2. Materials and methods
2.1. Animal model
Male, Sprague-Dawley rats (8-week old) were acclimatized to the vivarium and subsequently randomized to sham surgery or global brain ischemia (2-vessel occlusion), as previously performed in our laboratory.[56] Brain tissue was obtained from the rats at 1, 2, and 5 days after brain ischemia (n=6 at each time) and from the sham rats at 1 day after surgery (n=6). Prior to, and following surgery, all rats were housed with a 12 h light/12 h dark cycle with ad libitum access to chow and water. This work was approved by the University of Saskatchewan’s Animal Research Ethics Board, and adhered to the Canadian Council on Animal Care guidelines for humane animal use.
2.2. Sample preparation
To avoid introduction of chemical artifacts that can result during sample preparation of biological samples, [47, 55, 57, 58] all rats were anaesthetized with isoflurane, and humanely sacrificed through decapitation, with the head immediately frozen by dropping into liquid nitrogen, as described previously.[54] The frozen brain was chiselled out from the head on dry ice. A 10-μm-thick coronal section of the brain was cut with a cryo-microtome at −18 °C and was melted onto a CaF2 substrate, with three serial sections cut and mounted onto regular glass microscope slides and an additional serial section cut and mounted onto sulfur and metal free Thermanox plastic coverslips for micro-XAS analysis of sulfur speciation. All tissue sections were cut at coordinates between −2.7 and −3.3 mm relative to Bregma. Each sample on CaF2 or glass microscope slides was immediately air-dried at ambient laboratory temperature in a desiccator until the time of SR-FTIRI analysis. The latter was performed within 48 hours of tissue sectioning. The samples on microscope glass slides were immediately processed for H&E, Fluoro-jade B histology or ubiquitin immunocytochemistry. Hippocampal CA1 neuronal damage was assessed in both the left and right hemispheres for each animal, and bilateral damage confirmed with positive Fluoro-jade B staining (days 1, 2 and 5) and the presence of eosinophilic cells (days 2 and 5) (data not shown). SR-FTIRI spectroscopic analysis was performed on tissue sections from the right hemisphere only. Tissue sections prepared for micro-XAS (right hemisphere only) were stored frozen at −80 °C until the day of analysis, when they were air-dried in an ambient helium environment at ambient room temperature (~ 23.5 °C) and analyzed immediately.
2.3. H&E Histology, Fluoro-Jade Histology And Ubiquitin Immunocytochemistry
Three serial tissue sections were analyzed with H&E, Fluoro-jade and FTIRI, respectively. In addition, following FTIRI analysis, the tissue sections mounted on CaF2 were subsequently stained with H&E. In general, histological evaluation of brain sections mounted on CaF2 revealed numerous tissue cracks and artifacts, a product of the high salt content and dehydrating nature of the CaF2 (see supporting information Fig. 1). Nonetheless, sufficient morphological information was obtained from tissue sections mounted on CaF2 to corroborate the presence of the eosinophilic neurons that were also observed on the serial sections prepared exclusively for histological assessment (see supporting information Fig. 1). We observed that degenerating neurons presented with birefringent properties in the unstained tissue sections and subsequent H&E staining revealed the same neurons to be eosinophilic (supporting information Fig. 2). This property allowed degenerating neurons to be selectively targeted for FTIRI imaging in the unstained tissue sections, which was then confirmed by H&E staining following FTIRI analyses. For all sections undergoing histological assessment, tissue sections were vapor-fixed prior to routine H&E or Fluoro-jade B staining performed as previously described. [47, 59] Light microscopic images of the CA1 pyramidal neurons in the H&E stained sections were captured at 400 × magnification with a Nikon Eclipse 50i microscope and Nis Elements F3.0 software. The number of eosinophilic neurons were counted using a microscope grid of 200 μm width placed in the medial sector of the CA1 region in coronal sections obtained at coordinates between −2.7 and −3.3 mm relative to Bregma (see supporting information Fig. 3). Light microscopic images of the CA1 pyramidal neurons that were Fluoro-jade positive were captured at 400 × magnification with an Olympus BX 53 fluorescence microscope and Olympus CellSens software. The average Fluoro-jade optical density per neuron was calculated from tiff image files in Image J V1.36 (http://rsb.info.nih.gov/ij/download.html).
Tissue sections for ubiquitin immunocytochemistry were post-fixed in 10% buffered PFA. Immunocytochemical staining of ubiquitin was performed according to the manufacturer’s instructions using Pierce Peroxidase Detection Kit (Thermo Scientific). Briefly, sections were heated in citrate buffer (pH=6), followed by an overnight incubation at 4°C with a rabbit anti-ubiquitin antibody (1:50; Abcam ab7780). Following a washing step, slides were incubated with a horseradish peroxidase goat anti-rabbit secondary antibody (1:1000; Abcam ab6721) for 2 hours at room temperature and reacted with DAB (3,3′-Diaminobenzidine) for 15 minutes. Sections were counterstained with hematoxylin. Microscope images of ubiquitin positive cells were captured as described for routine H&E histology.
2.4. Single-beam SR-FTIRI
SR-FTIRI images were collected from 5 pyramidal neurons from the medial section of the hippocampal CA1 region for each tissue section, for one tissue section per animal, for 6 animals in each experimental group (6 rats in one experimental group × one tissue section × 5 neurons per tissue section=30 neurons per experimental group). The 5 neurons were selected within a 200 μm wide region of the medial CA1 sector. Single-beam wide-field SR-FTIRI was performed at the Canadian Light Source (CLS) with a Hyperion 3000 microscope fitted with an upper objective of 52 × magnification and a numerical aperture (N.A.) of 0.65, and with a lower condenser of 32 × magnification and a N.A. of 0.65. This arrangement yielded 0.77 μm × 0.77 μm pixel size. The infrared beam was focused and aligned to the center of the array, then defocused such that an array area of approximately 20 μm × 20 μm (26 × 26 pixels) was quasi-homogeneously illuminated.
Spectra were collected with the co-addition of 256 scans, at 4 cm−1 resolution. A background spectrum (256 co-added scans) was collected from blank substrate before each sample measurement, to minimize the effect of beam current decay on data. Normalization to beam current was not performed, as described previously.[47] During data processing individual images were cropped to a region consisting of 26 × 26 pixels.
2.5. Multi-beam wide-field SR-FTIRI
Multi-beam wide-field SR-FTIRI was performed at the Synchrotron Radiation Center (SRC) as previously described. [32, 46, 47] Images were collected from a Hyperion 3000 microscope fitted with an upper objective of 74 × magnification and with a numerical aperture of 0.65, and a lower condenser of 15 × magnification and 0.6 numerical aperture. This arrangement yielded an effective pixel resolution of 0.54 μm × 0.54 μm. The incident infrared beam was focused and aligned to the center of the array, then defocused to cover the complete array (34.6 μm × 34.6 μm).
SR-FTIR-FPA Spectroscopic images were collected with a spectral resolution of 4 cm−1 and the co-addition of 256 scans. A background image was collected from blank substrate using 256 co-added scans. The background was collected immediately prior to each sample. Normalization to beam current was not performed.
2.6. Data processing and data analysis of FTIR spectra
All data processing and image generation was performed using Cytospec software (Cytospec, Version 1.2.04) and Opus software (Version 6.5, Bruker, Ettlingen, Germany). False colour functional group images of total lipid and protein were generated from the integrated band area in un-normalized spectra. Areas were integrated in the range 1755–1715 cm−1 for the lipid ester ν(C=O) band, and 1700–1600 cm−1 for the protein amide I band. A linear baseline was subtracted across the same range as integration. To calculate the average spectrum of each individual neuron, a region of interest was drawn to encompass the neuron soma. Drawing the region of interest around the neuron soma was guided by an overlay of H&E histology and FTIRI images of total lipid ester (sham, day 1 post-ischemic insult) or total protein (days 2 and 5 post-ischemic insult).
For analysis of relative amount and distribution of protein aggregates, spectra were vector normalized to the amide I band (1700–1600 cm−1), and second-derivatives calculated with a 13 smoothing point Savistky-Golay function. Second-derivative intensity at 1625 cm−1 was used as a spectroscopic marker of the relative aggregated protein content. We have previously described the use, advantages and limitations of second-derivatives for biochemical imaging.[47]
To validate the results from second-derivative analysis of relative aggregate protein content, curve fitting of the amide I region (1700–1600 cm−1) was performed. Second-derivative spectra were used to guide the choice of the number and location of underlying bands. The vector normalized spectra were then fitted to 6 mixed Lorentzian and Gaussian curves, located at 1695, 1682, 1665, 1656, 1645, 1638 and 1625 cm−1, across the spectral range 1700–1600 cm−1. The curve centered at 1625 cm−1 was assigned to aggregated protein, and the area underneath this curve was used to assess relative aggregated protein content. The results from curve fitting were in strong agreement with the results from analysis of second-derivative intensity.
2.7. Micro-X-ray absorption spectroscopy at the sulfur K-edge
Micro-X-ray absorption spectra (micro-XAS) at the sulfur K-edge of CA1 pyramidal neurons within brain tissue sections were collected using a Si(111) double crystal monochromator with an upstream collimating mirror set at an angle sufficient to reject the Si(333) and higher harmonics (Beamline 14-3 Stanford Synchrotron Radiation Lightsource). The incident beam was focused to a spot size of approximately 5 μm × 7 μm (horizontal × vertical) using KB optics. The sample was mounted at 45° to the incident beam. Fluorescence emission spectra were recorded with a 4 element vortex detector at 90° to the incident beam. The incident beam intensity was measured with a helium gas filled I0 ion chamber. Prior to spectra collection, the sample chamber was purged with He until the relative O2 content within the chamber was less than 0.5%. A bright field microscope coupled to the X-ray instrumentation was used to align the beam such that spectra could be accurately recorded from individual CA1 pyramidal neurons (See Supporting Information Fig. 5). X-ray absorption spectra were calibrated against the spectrum of an anhydrous Na2S2O3 powder solid standard, with the lowest energy peak assumed to correspond to 2469.2 eV, as described previously [54, 55]. Spectra were collected across the energy range 2450–2515 eV, with a dwell time of 1 second and total collection time of approximately 15 min. For each sample, a spectrum was collected from 5 individual cells.
Bulk sulfur K-edge X-ray absorption spectra of model sulfur were collected using a Si(111) double crystal monochromator with an upstream collimating mirror set at an angle sufficient to reject the Si(333) and higher harmonics (Beamline 4-3 Stanford Synchrotron Radiation Lightsource). The incident beam was reduced to 1 mm × 3 mm by vertical and horizontal slits, and the intensity measured with a helium gas-filled ionization chamber, I0. Samples (model compound solutions) were mounted at 45° to the incident beam, and X-ray fluorescence collected with a Stern-Heald Lytle detector (The EXAFS Company, Pioche, NV) filled with nitrogen gas. Prior to spectra collection, the sample chamber was purged with He as described above. Spectra of calibration standards and model compounds were recorded at room temperature. Standard compounds used for fitting routines were representative of disulfides (oxidized glutathione), thiols (reduced glutathione), thio-ethers (methionine), sulfoxides (methionine sulfoxide), sulfinic acids (hypotaurine), sulfonic acids (taurine), O-linked sulfate esters (N-acetyl D-galactosamine 6-sulfate), N-linked sulfate esters (D-galactosamine 2-sulfate), and inorganic sulfates (Na2SO4) and were analyzed as solutions (to minimize the self-absorption artifacts) made up to 30–100 mM in PBS at pH 7.4. Solutions were analyzed in sulfur-free custom-made polycarbonate cuvettes with a thin polypropylene window.
Spectra were processed using the EXAFSPAK suite of programs.[60] Using the DATFIT program, spectra collected from tissue sections were fitted with a linear combination of reference spectra (see standard compounds discussed above). Standards were excluded from the refinement algorithm if they contributed to <0.5% of the total spectra, at a value less than three times their standard deviation of measurement (calculated from the diagonal elements of the variance-covariance matrix).
2.8. Data analysis and Statistics
All FTIRI and micro-XAS spectroscopic data and Fluoro-jade fluorescence microscopy data were considered as continuous semi-quantitative data. The number of eosinophilic cells was considered as interval data. A one-way ANOVA was performed to determine if there was a significant effect of global ischemia on total protein levels, relative aggregated protein content assessed from curve fitting, relative aggregated protein content assessed from second-derivative spectra, relative disulfide content from micro-XAS analysis, neuron damage assessed by Fluoro-jade fluorescence, and neuron damage assessed by eosinophilic cell counts. When the results from ANOVA indicated a significant effect, a two-tailed Dunnet T3 post hoc test that does not assume equal variance was performed. The 95% confidence limit was used (p < 0.05). All statistical analysis was performed on 6 animals for each group (n=24 total). All statistical analysis was performed with SPSS v. 13.
3. Results
3.1. Histological assessment of neuron damage within the CA1 sector of the rat hippocampus after global brain ischemia
Routine haematoxylin and eosin (H&E) and Fluoro-jade B staining was performed to assess neuron viability following global ischemia. The intensity of Fluoro-jade B staining was used as a marker of the extent of cell damage, [61, 62] while the presence of eosinophilia was used as a marker of irreparable cell damage. Representative examples of the Fluoro-jade B staining are presented in Fig. 1A–D. A one-way ANOVA demonstrated significant differences in Fluoro-jade B staining (F(3,23)=78.900, p < 0.001) (Fig. 1E). Neurons within the CA1 region of the hippocampus from rats at 1, 2 and 5 days after global ischemia displayed a significant increase in the extent of Fluoro-jade staining relative to CA1 neurons from sham animals (p=0.001, < 0.001 and < 0.001, respectively; two-tailed Dunnet T3 post hoc test). CA1 neurons from rats at 2 and 5 days after global ischemia displayed a significant increase in the extent of Fluoro-jade staining relative to CA1 neurons from rats at 1 day after ischemia (p=0.003, and < 0.001, respectively). Neurons within the CA1 sector of the hippocampus at 5 days displayed a significant increase in the extent of Fluoro-jade staining relative to CA1 neurons from rats at 2 days after ischemia (p=0.002).
Fig. 1.

Fluoro-jade staining of degenerating cells demonstrates global ischemia in the rat induces neuron damage in the CA1 sector of the hippocampus at coordinates between −3.3 and −2.7 mm relative to Bregma. Staining was assessed from a 200 μm wide sector, as described in methods Section 2.6 and supporting information Fig. 3. (A) Sham (B) 1 day after global ischemia (C) 2 days after global ischemia (D) 5 days after global ischemia (E) Quantification of mean ± SD fluorescence intensity (n=6 rats for each time point). Scale bar=50 μm. *** p < 0.001 relative to sham. †† p < 0.01 relative to ischemic day 1. ††† p < 0.001 relative to ischemic day 1. ᵻᵻp < 0.01 relative to ischemic day 2. Although subtle relative to day 2 and day 5 animals, positive Fluoro-jade staining was observed 1 day post ischemic insult; see Supporting Information Fig. 4.
Fig. 2 shows the results of the H&E staining. Numerous eosinophilic neurons were observed within the CA1 region of the rat hippocampus at both 2 and 5 days after global ischemia, but no eosinophilic CA1 neurons were observed in brain sections from sham rats or animals at 1 day after global ischemia. A one-way ANOVA demonstrated significant differences in eosinophilic cell counts (F(3,23)=74.861, p < 0.001). The number of eosinophilic cells was significantly increased on days 2 and 5 post-ischemic insult relative to sham animals and animals 1 day post-ischemic insult (p < 0.001 for all cases; two-tailed Dunnet T3 post hoc test). No significant difference in the number of eosinophilic cells within the CA1 sector was observed between rats at day 2 and day 5 after global ischemia (p=0.998). Eosinophilic neurons within the CA1 region displayed irregular and “shrunken” cell morphology at both days 2 and 5 post-ischemia.
Fig. 2.
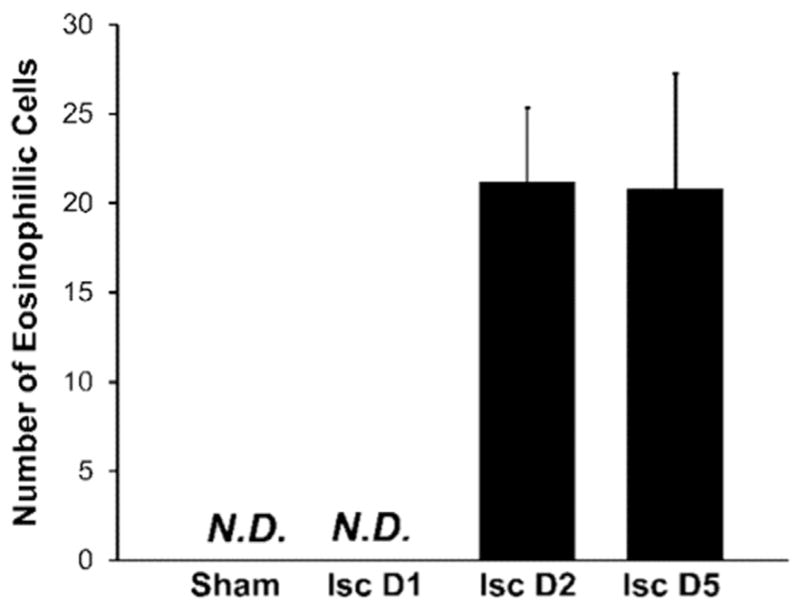
Increased presence of eosinophilic neurons (a marker of dead or dying cells) was observed within H&E stained tissue, and demonstrates that global ischemia in the rat induces neuron damage in the CA1 sector of the hippocampus. Results are expressed as mean ± SD (n=6 rats at each time point). No eosinophilic neurons were present within the hippocampus in sham animals or in animals 1 day after ischemic insult. Large numbers of eosinophilic cells were present within the CA1 sector of the hippocampus on day 2 and day 5 after ischemic insult. No significant difference in the number of eosinophilic neurons was observed between animals on day 2 and day 5 (p=0.998).
3.2. Immunocytochemical analysis of ubiquitination within CA1 pyramidal neurons
Representative images of ubiquitin immunocytochemistry are shown in Fig. 3. Whereas an even distribution of antigenicity to ubiquitin conjugates was observed in CA1 pyramidal neurons from sham animals indicative of background staining, a marked increase in cytoplasmic ubiquitination was observed in CA1 pyramidal neurons 1 day after ischemic insult, consistent with previous reports. [13] A loss of antigenicity was observed in severely damaged or dead CA1 pyramidal neurons (revealed by shrunken triangular nucleus) 2 days and 5 days post-ischemic insult, consistent with previous reports. [13]
Fig. 3.

Representative images of ubiquitin immunocytochemistry. Ubiquitin staining was observed in sham animals, including co-localization with cell nuclei (black arrows). CA1 neurons showed strong staining within the cytoplasm 1 day after global brain ischemia (black arrows). Cells with a normal morphology still displayed ubiquitin antigenicity 2 days after ischemic insult (right black arrow on the panel), whereas damaged cells with a shrunken or distorted nucleus did not display antigenicity (left black arrow). Nearly all cells on day 5 post-ischemic insult displayed a shrunken or distorted nucleus (i.e., dead cells), and ubiquitin antigenicity was not observed (black arrows). Scale bar=50 μm.
3.3. SR-FTIRI analysis of the total protein and relative aggregated protein content of CA1 pyramidal neurons following global ischemia
SR-FTIRI analysis was used to quantify the total protein and relative aggregated protein within individual cells of the CA1 pyramidal neuron layer of the hippocampus in sham animals and animals at 1, 2 and 5 days post-ischemic insult. In order to accurately define the anatomical cell boundary, H&E histology was used as a guide alongside FTIRI molecular images of lipid ester distribution as previously described. [32, 47] Prior to irreparable neuron damage, the distribution of lipid ester was used to define the cell location, as neurons contain less lipid ester than the surrounding neuropil. However, as previously speculated, [47] this study demonstrated that as a consequence of ischemic damage the lipid ester distribution in neurons at 2 days and 5 days following global ischemia did not correlate to the cell morphology observed in the H&E images. Fortuitously, as the soma of neurons is rich in protein, [47] the distribution of total protein provided good correlation with cell morphology at these time points. Therefore, regions of interest were drawn to calculate the average biochemical composition of individual neurons using the lipid ester distribution for sham animals and rats at 1 day post-ischemic insult, whereas the total protein distribution was used for rats at 2 and 5 days post-ischemic insult.
Representative examples of H&E staining and the SR-FTIRI molecular images of the total lipid, total protein and relative aggregated protein content for neurons from all experimental groups are presented in Fig. 4 and Fig. 5. Fig. 4 displays representative images collected with single beam SR-FTIRI. The majority of data was collected with this approach, and statistical analysis was performed on this data set. However, to validate the biochemical images obtained with single beam FTIRI, multi-beam FTIRI, which provides superior signal to noise and improved spatial resolution, was also performed. As can be seen from comparison of Figs. 4 and 5, identical scientific conclusions can be drawn from both methods of image collection.
Fig. 4.

Macromolecule levels in representative single CA1 neurons following global ischemia visualized with single beam FTIRI. Single beam FTIRI analysis of total protein and relative levels of aggregated protein in CA1 pyramidal neurons from (I, M) sham animal, (J, N) animal 1 day after global ischemia, (K, O) animal 2 days after global ischemia, and (L, P) animal 5 days after global ischemia. (A–D) H&E histology of the same neurons analyzed with FTIRI, which highlights that neurons 2 days and 5 days after ischemia are strongly eosinophilic (i.e., bright pink staining). (E–H) The lipid ester distribution (integrated band area 1755–1720 cm−1), which was used to visualize neuron morphology in sham animals and animals 1 day after global ischemia. (I–L) Total protein levels, showing a decrease in total protein 1 day after global ischemia and a large increase in total protein 2 days and 5 days after ischemia. (M–P) Relative levels of aggregated proteins, showing a significant increase for protein aggregation with time following ischemic insult (See Table 1). Scale bar=5 μm. Units for the intensity scale are arbitrary absorbance units.
Fig. 5.

Macromolecule levels in representative single CA1 neurons following global ischemia visualized with multi-beam FTIRI. Multi-beam FTIRI was used to validate the results from single beam FTIRI. (A, E, I, M) sham animal, (B, F, J, N) animal 1 day after global ischemia, (C, G, K, O) animal 2 days after global ischemia (D, H, L, P) animal 5 days after global ischemia. (A–D) Visible light microscopy of the same neurons analyzed with FTIRI. The soma of neurons 2 days and 5 days after global ischemia was substantially darker than in the sham animal or animal 1 day after ischemia. Supporting Information Fig. 2 demonstrates that darkened soma observed on visible light microscopy of unstained tissue correlates with eosinophilic cells in H&E stained tissue. (E–H) The lipid ester distribution (integrated band area 1755–1720 cm−1), which was used to visualize neuron morphology in sham animals and animals 1 day after global ischemia. (I–L) Total protein levels, showing a significant decrease in total protein 1 day after global ischemia and a large increase in total protein 2 days and 5 days after ischemia (See Table 1). (M–P) Relative levels of aggregated proteins, showing an increasing trend for protein aggregation with time following ischemic insult. Scale bar=5 μm. Units for the intensity scale are arbitrary absorbance units.
The SR-FTIRI biochemical images in Fig. 4 and Fig. 5 demonstrate that a subtle increase in aggregated protein content within CA1 pyramidal neurons occurs concomitantly with a decrease in total protein, 1 day after global ischemia. In addition, a large increase in the relative aggregated protein content was observed at post-ischemic days 2 and 5. Interestingly, no decrease in total protein was observed at the later time points and in fact, total protein was increased. In addition to revealing an increase in the relative aggregated protein content at 1, 2 and 5 days after global ischemia, the SR-FTIRI analyses demonstrated that the highest relative concentration of aggregated protein is found at the periphery of the neuron cytoplasm. Although the spatial resolution of SR-FTIRI is not sufficient to resolve the cytoplasm from dendrite and somatic connections, the distribution of aggregated protein at the periphery of the cytoplasm is consistent with a sub-cellular localization to the cytoplasm and soma / dendrite interface. The pattern of distribution of aggregated protein is not consistent with localization within the nucleus of the neuron, or location in nuclear material that spreads from the center of the cell towards the periphery during neuron degeneration.
These visual observations from the representative images reflect statistically significant differences in the neuronal concentration of total protein and aggregated protein (Table 1). The relative aggregated protein content was determined from curve fitting of the amide I band, although analysis of the second-derivative intensity at 1625 cm−1 also produced the same scientific conclusions as curve fitting. Statistically significant differences were determined with one-way ANOVA for total protein levels (F (3,23)=18.770, p < 0.001), relative aggregated protein levels measured by curve-fitting (F(3,23)=71.583, p < 0.001), and relative aggregated protein levels measured by second-derivative intensity (F(3,23)=41.622, p < 0.001). As the results from ANOVA indicated a significant effect, a two-tailed Dunnet T3 post hoc test that does not assume equal variance was performed. Total protein was significantly reduced in neuron soma 1 day after ischemic insult, relative to sham animals (p < 0.05). Total protein was significantly increased in neuron soma on day 2 relative to sham animals and animals 1 day after ischemic insult (p < 0.001 in both cases). Total protein was significantly increased in neuron soma on day 5 relative to sham animals and animals 1 day after ischemic insult (p < 0.01 in both cases). No significant difference in total protein levels was observed in neuron soma between day 2 and day 5 post-ischemic insult. Levels of relative aggregated protein measured via curve fitting were subtly but significantly increased in neuron soma 1 day after ischemic insult, relative to sham animals (p < 0.001). Relative aggregated protein was significantly increased in neuron soma on day 2 relative to sham animals and animals 1 day after ischemic insult (p < 0.001 and p < 0.01, respectively). Relative aggregated protein was significantly increased in neuron soma on day 5 relative to sham animals, and in animals 1 and 2 days after and ischemic insult (p < 0.001, p < 0.01, p < 0.05, respectively). Levels of relative aggregated protein measured via second-derivative intensity were subtly but significantly increased in neuron soma 1 day after ischemic insult, relative to sham animals (p < 0.01). Relative aggregated protein was significantly increased in neuron soma on day 2 relative to sham animals and animals 1 day after ischemic insult (p < 0.01 and p < 0.05, respectively). Relative aggregated protein was significantly increased in neuron soma on day 5 relative to sham animals, and in animals 1 and 2 days after an ischemic insult (p < 0.01, p < 0.01, p < 0.05, respectively).
Table 1.
Total Protein and Relative Aggregated Protein Content in CA1 Pyramidal Neurons After Global Ischemiaa.
| Sham | Ischemic Day 1 | Ischemic Day 2 | Ischemic Day 5 | |
|---|---|---|---|---|
| Total Protein | 23.1 ± 5.2 | 18.8 ± 2.9 * | 28.6 ± 5.5 ***,††† | 32.0 ± 6.1 **,†† |
| Aggregated Protein (Curve Fit) | 4.5 ± 1.6 | 8.2 ± 1.2 *** | 10.1 ± 1.5 ***,†† | 13.4 ± 2.2 ***,††,ᵻ |
| Aggregated Protein (2ndDerivative) | (5.78 ± 0.24) × 10−4 | (4.17 ± 0.58) × 10−4** | (1.12 ± 0.22) × 10−4**,† | (−1.20 ± 0.33) × 10−4**,††,ᵻ |
Data are shown as mean ± SD. n= 6 rats (10 CA1 neurons/rat, averaged to give one data value per rat). Units for total protein are absorbance units. Units for relative aggregated protein content are arbitrary.
p < 0.05 relative to sham.
p < 0.01 relative to sham.
p < 0.001 relative to sham.
p < 0.05 relative to ischemic day 1.
p < 0.01 relative to ischemic day 1.
p < 0.001 relative to ischemic day 1.
p < 0.05 relative to ischemic day 2.
Representative raw spectra and second-derivative spectra from CA1 pyramidal neurons for each experimental group are presented in Fig. 6A and B, respectively. The spectra show the characteristic broadening of the amide I band to lower wave-numbers in the raw spectra, and increased second-derivative intensity at 1625 cm−1 that is associated with increased aggregated protein content. Representative examples of the curve fitting process for each experimental group are presented in Fig. 6C–F.
Fig. 6.

(A) Representative spectra showing the decreased total protein content 1 day after global brain ischemia. (B) Representative second-derivative spectra showing the relative increase in aggregated protein content (intensity at 1625 cm−1) with time after global ischemia. Second-derivative spectra were calculated from vector normalized (1700–1600 cm−1) raw spectra. (C–F) Representative examples of curve fitting spectra to show the increase in aggregated protein content (band centered at 1625 cm−1, in red) with increased time after global ischemia.
3.4. Micro-XAS analysis of thiol redox state within the CA1 pyramidal layer after global ischemia
We and others have previously described the capability of XAS at the sulfur K-edge for direct measurement of sulfur redox in situ within biological samples, [52–55] and we have demonstrated that the technique has immense potential for the field of neuroscience, particularly to study ischemia induced oxidation of thiols to disulfides. [54, 55] The technique differentiates between chemical forms of sulfur based on differences in sulfur oxidation state and molecular geometry. Although the method provides a wealth of information on the sulfur “metabolome” of a sample, the technique was used specifically in this study to quantify the relative disulfide content, as a marker of oxidative stress, within the CA1 pyramidal layer in sham animals and animals 1, 2 and 5 days post ischemic insult. The micro-X-ray beam was focussed to illuminate a 5 μm × 7 μm region of tissue. Accurate and precise X-ray beam alignment was achieved with the aid of a bright field microscope and was used to ensure all spectra were collected from the desired region of interest (i.e., cells within the CA1 pyramidal layer). Due to the small region of tissue interrogated by the synchrotron X-ray beam, 5 adjacent spectra were collected across 5 cells within the pyramidal layer of the CA1 subsector, as demonstrated in Supporting Information Fig. 5. These 5 spectra were averaged together to provide a measurement of the average sulfur speciation for the CA1 pyramidal layer of each sample.
The relative content of thiol, thio-ether, disulfide, sulfoxide, sulfonic acid and sulfate ester, reported as percentage contribution to the total sulfur pool, for the CA1 pyramidal layer of all animals is presented in Table 2. A statistically significant effect of ischemia on disulfide content was determined with a one-way ANOVA (F (3,23)=23.406, p < 0.001). A two-tailed Dunnet T3 post hoc test demonstrated that disulfide levels were subtly but significantly increased 1 day after ischemia, relative to sham animals (p < 0.05). Disulfide was significantly increased in neuron soma on day 2 relative to sham animals and day 1 ischemia rats (p < 0.001 and p < 0.05, respectively). Disulfide was significantly increased in neuron soma on day 5 ischemia rats both relative to sham animals and relative to day 1 ischemia animals (p < 0.001, p < 0.05, respectively). Although the average disulfide content on day 5 post ischemia was greater than on day 2 post-ischemia, the increase was not statistically significant. Representative examples of the micro-XAS spectra and the linear fitting process, which demonstrate the increased contribution of disulfide to the total sulfur pool after global brain ischemia are presented in Fig. 7.
Table 2.
XAS Sulfur Fitting Results of the Relative Contribution (%) of Different Chemical Forms of Sulfur to the Total Sulfur Pool Following Global Brain Ischemiaa.
| Animal | RSH | RSR | RSSR | Sulfoxide | Sulfonic Acid | Sulfate Ester |
|---|---|---|---|---|---|---|
| Sham | 46 (± 2) ± 2 | 27 (± 2) ± 4 | 3.5 (± 0.7) ± 1.4 | 4.0 (± 0.3) ± 0.8 | 18 (± 4) ± 3 | 2.4 (± 0.03) ± 1.4 |
| Day 1 | 43 (± 2) ± 5 | 27 (± 1) ± 4 | 6.6 (± 0.7) ± 1.5 α | 3.7 (± 0.2) ± 1.4 | 17 (± 0.2) ± 1 | 2.6 (± 0.2) ± 1.5 |
| Day 2 | 43 (± 3) ± 1 | 30 (± 2) ± 1 | 8.3 (± 0.1) ± 0.6 α,β | 4.3 (± 0.4) ± 0.8 | 11 (± 0.4) ± 1 α,β | 2.9 (± 0.9) ± 1.7 |
| Day 5 | 48 (± 4) ± 3 | 24 (± 3) ± 5 | 10 (2) ± 3 α,β | 3.4 (± 0.6) ± 1.8 | 12 (± 1) ± 3 α,β | 1.8 (± 0.5) ± 0.9 |
Values given are the mean percentages (± estimated standard deviation from individual fits) ± standard deviation of replicate measurements from 6 animals. The values in parentheses are the average estimated standard deviation from the individual fits obtained from the diagonal elements of the covariance matrices. The estimated standard deviation gives an indication of the precision of determination of the value in the least-squares fit, whereas the standard deviation of replicate measurements, quoted without parentheses, gives an indication of how much variation is present between different samples. As expected for slightly different individual fits, the average percentages do not total to 100%, although the individual fits do total to 100%. Individual values for each animal were calculated from the average of 5 spectra collected from 5 separate cells.
Denotes a significant difference relative to sham.
Denotes a significant difference relative to Day 1.
Fig. 7.

Linear fitting of micro-XAS spectra at the sulfur K-edge to reveal the disulfide contribution (red spectrum) in pyramidal neurons from (A) sham animals, (B) animals 1 day after global brain ischemia, (C) animals 2 days after ischemia, (D) animals 5 days after ischemia. The disulfide contribution of sham animals is also shown as a paler red-dashed line in panels B–D, to demonstrate the increase in disulfide content that occurs following ischemia.
The one-way ANOVA also revealed a significant decrease in sulfonic acid content following brain ischemia (F(3,23)=17.443, p < 0.001). Post-hoc analysis revealed that sulfonic acid content was decreased on day 2 and day 5 post ischemic insult relative to sham animals (Day 2 p < 0.01, Day 5 p < 0.01) and day 1 animals (Day 2 p < 0.05, Day 5 p < 0.05), respectively. No significant difference in sulfonic acid content was observed between animals 1 day after ischemia and sham animals, or between animals 2 and 5 days after ischemia. No significant differences were observed for all other chemical forms of sulfur detected (thiol, thio-ether, sulfoxide, sulfate-ester). In this study, micro-XAS was performed specifically to elucidate the disulfide content of the CA1 pyramidal layer and not to study global changes in sulfur speciation. Therefore, although the decreased sulfonic acid content is of interest, these results will not be discussed further in this manuscript, but will be analyzed further in future experiments.
4. Discussion
4.1. Routine histology and immunocytochemistry reveal subtle damage and increased ubiquitination within CA1 pyramidal neurons 1 day after global brain ischemia and irreparable cell damage by day 2
The results from histological analysis indicate that irreparable cell damage, demonstrated by significantly increased Fluoro-jade staining and eosinophilia, occurs within the CA1 sector of the hippocampus within 2 days of global brain ischemia. These results are in direct accordance with those reports using electron microscopy to investigate the ultrastructure of CA1 pyramidal neurons following global ischemia. The latter demonstrated irreversible structural alterations evident by 2 days following ischemic insult, but not within the first day. [4, 24] Although irreparable ultra-structural alterations are not typically observed within 1 day following an ischemic insult, some studies have identified subtle alterations in neuron morphology within this time frame, including the possible appearance of intracellular molecular aggregates that were proposed but not definitively proven to be protein with an aggregated secondary structure.[4, 24] In addition, Fluoro-jade has been used previously to study neuron degeneration following global ischemia. Similar to investigations of neuron ultrastructure, Fluoro-jade staining indicated substantial neuron damage at 2 days post-ischemic insult, as well as more subtle damage within the first day.[62] These previous studies parallel the results of this current study that revealed a subtle yet significant increase in Fluoro-jade staining within CA1 pyramidal neurons 1 day after brain ischemia, even in the absence of visual alterations to cell morphology in the H&E stained tissue. Therefore, the results of this current study agree well with previous histological investigations and suggest that mild structural damage to CA1 pyramidal neurons occurs within 1 day of ischemic insult, but the damage does not become “irreparable” until 2 days after global ischemia. That is, the neurons are still viable in the first day.
In addition to structural alterations to neuron morphology, previous work by others has revealed increased ubiquitination in the cytoplasm of neurons 1 day after global brain ischemia, concomitant with the appearance of molecular aggregates within the cytoplasm. [13] At later time points during cell death, the previous studies demonstrated a complete loss of ubiquitin immunoreactivity in severely damaged or dead neurons. [13, 14] The results of this current study are in direct accordance with the previous investigations, and show increased cytoplasmic protein ubiquitination 1 day after ischemic insult, with a loss of antigenicity at 2 days and 5 days when severe cell damage has occurred.
4.2. SR-FTIRI and micro-XAS in combination with histological analyses reveals an increase in aggregated protein, a decrease in total protein, and an increase in disulfide levels prior to irreparable CA1 cell damage after global ischemia
This study has provided strong evidence of a spatial and temporal correlation between increased protein aggregation and altered protein homeostasis within CA1 pyramidal neurons prior to irreparable cell damage following global brain ischemia. Previous investigations by others using tissue micro-dissection and biochemical assay have implicated protein oxidation and aggregation in neurodegeneration following global ischemia. [27] Similarly, indirect measurement of protein aggregates (ubiquitin immunoreactivity) has provided evidence of protein aggregate formation in neuron soma following global and also focal ischemia.[13, 14] Further, analysis of amino acid turnover has provided evidence that protein synthesis is decreased after both global and focal ischemia. [16–22] These previous studies helped formulate the hypothesis that oxidative stress within neuron soma promotes protein aggregation that may impair protein synthesis and cause neurodegeneration. [13–15, 63] However, to date, no method has been able to study directly the levels of biochemical markers of oxidative stress (i.e., disulfide levels), total protein, protein aggregates and neuron integrity within the same neuron within brain tissue sections. Consequently, direct evidence for this hypothesis has remained lacking. In this investigation, wide-field SR-FTIRI has provided direct in situ sub-cellular biochemical imaging of total protein and aggregated protein levels in tissue sections from animals 1, 2, and 5 days post-ischemic insult, in combination with routine histology of the same neuron to provide direct biochemical evidence to test this hypothesis. Micro-XAS analysis of disulfide levels in serial tissue sections provided the ability to observe if increased oxidative stress occurred concomitantly within the same tissue region as alterations in protein homeostasis and protein aggregation.
The results of SR-FTIRI in this study correlate well with the results from histological analysis and indicate that substantial levels of protein aggregation are present at the time of irreparable cell damage and cell death (day 2–day 5). Indeed, at these time points some portion of this increase in protein aggregates is likely a secondary effect caused by degeneration of the neuron. However, prior to the large increase in protein aggregates on day 2, a subtle but statistically significant increase in protein aggregates was observed within the soma of neurons 1 day after ischemic insult. This early increase in protein aggregates occurred prior to histological evidence of irreparable neuron damage, since the neurons did not display eosinophilia. To the best of our knowledge, this is the first direct evidence, rather than indirect evidence from labelling with a dye or stain, that conclusively demonstrates protein aggregation prior to neuron death following global ischemia. This provides further evidence that protein aggregation is a key mechanism that occurs prior to neuron death, and thus it could well be a direct contributor and not simply a marker of the end stages of cell death.
One possible mechanism through which protein aggregation could promote cell death is through impaired function of the proteasome. It has been demonstrated that increased levels of protein aggregates can disrupt the normal function of the proteasome resulting in altered protein homeostasis within cells. [64, 65] Indeed, decreased protein synthesis within brain tissue has been demonstrated after both global and focal brain ischemia. [16–22] Our results demonstrated a net decrease in total protein content within CA1 pyramidal neurons 1 day after ischemic insult. Further, the ability of SR-FTIRI to study multiple biochemical parameters within the same neuron revealed that the decrease in total protein was concomitant with increased levels of aggregated protein within the same neuron 24 hours after ischemia. Therefore, this study has provided direct biochemical evidence of a spatial and temporal mechanistic link between impaired protein homeostasis and protein aggregation prior to irreparable cell damage after global brain ischemia.
Interestingly, the total protein content of neurons did not decrease 2 days and 5 days after ischemic insult, but rather increased at these times. This observation is likely due to shrinkage of the cell and concentration of the cell contents into a small volume during the final stages of cell death. This effect would act to enhance the local concentration of chemical components, such as protein.
A homogenous distribution of aggregated protein was not observed within neurons at any time after global ischemia, but rather was highly localized to the neuron periphery. Although not conclusive evidence, this distribution pattern supports a mechanism of protein aggregation occurring from the periphery of the cell inwards. Such an event would be likely to occur during excitotoxicity and increased intracellular Ca2+ concentration, which could accelerate action of the electron transport chain of mitochondria within the dendrites and cytoplasm and drive excessive free radical production, as proposed by others. [6, 66] This pattern of development of protein aggregation is consistent with necrotic cell death within CA1 pyramidal neurons following ischemic insult, [67] and would indicate that oxidative stress holds a key role in the formation of the protein aggregates.
Our results and that of others [13] have shown increased cytoplasmic ubiquitination concomitant with increased protein aggregates 1 day after brain ischemia and a complete absence of ubiquitination in severely damaged or dying cells. These results suggest that the early formation of protein aggregates is not due to an impaired ubiquitin pathway, but rather is driven by another mechanism. Likely, the ubiquitin system initially performs its designed role tagging protein aggregates for degradation, but is either saturated by continued production of protein aggregates or is damaged itself by the same process responsible for aggregated production. Either way, there is a substantial increase in protein aggregation, a loss of ubiquitination and overall destruction of the cell (i.e., the results observed on day 2 and day 5 post ischemic insult). A potential pathway driving protein aggregation within the cytoplasm of pyramidal neurons within the first day after global brain ischemia is oxidative stress and free radical production, as proposed by others. [13] However, the previous work in this field has not been able to correlate increased oxidative stress with protein aggregation, depressed protein synthesis and increased ubiquitination, to prove this hypothesis. The results of this current study, obtained by using a suite of direct bio-spectroscopic techniques in combination with traditional histology and immunocytochemistry, provide the first direct spatial and temporal correlation between increased protein aggregates and cytoplasmic ubiquitination, oxidative stress, and altered protein homeostasis at 1 day following global brain ischemia. The results suggest that free radical induced protein oxidation is a likely cause of the protein aggregation, and therapies that mitigate this process may be of high clinical value. However, it is acknowledged that the results demonstrate a correlation between oxidative stress and increased protein aggregation only, and indeed, these processes could occur concomitantly but independently of each other. Similarly, although these data support the involvement of necrotic mechanisms in cell death after global brain ischemia, it does not rule out apoptotic pathways.
5. Conclusion
Elucidation of the biochemical mechanisms driving delayed degeneration of CA1 pyramidal neurons following global ischemia has long been sought. Accurate knowledge of the mechanisms will help drive development of improved, targeted therapies for patients with stroke, cardiac arrest, and potentially other neurodegenerative conditions for which cerebral ischemia is a component of disease pathogenesis. This investigation has provided a direct biochemical correlation between a biochemical marker of oxidative stress (increased disulfide levels) and protein aggregation, which occurs prior to irreparable neuronal damage. Further, there is a spatial and temporal correlation between increased levels of protein aggregates and decreased total protein content after global brain ischemia. This highlights the need to further understand the relationship between protein aggregation and altered protein homeostasis, which manifest following oxidative stress induced by brain ischemia. Such future insight may identify novel therapeutic strategies for brain ischemia victims. The study results also demonstrate the immense potential for future use of wide-field SR-FTIRI and micro-XAS at the sulfur K-edge, to study protein aggregation and thiol redox alongside many other important biochemical parameters at the sub-cellular level, in a wide range of applications in the field of neuroscience.
Acknowledgments
We thank Dr. Fred Colbourne, University of Alberta, for providing helpful comments on the manuscript. This work was supported by a joint Canadian Institutes of Health Research (CIHR)/ Heart and Stroke Foundation Synchrotron Medical Imaging Team Grant #CIF 99472 awarded to H.N., P.G.P., I.J.P., G.N.G. and others. M.J.H. is a CIHR Postdoctoral Fellow, a Saskatchewan Health Research Foundation postdoctoral fellow. M.J.H and S.C. are SMI postdoctoral fellows and Fellows in the CIHR-Training grant in Health Research Using Synchrotron Techniques (CIHR-THRUST) awarded to I.J.P. and others. G.N.G. and I.J.P. are Canada Research Chairs. This work is based in part upon research conducted at the IRENI beamline at the Synchrotron Radiation Center, which is primarily funded by the University of Wisconsin-Madison with supplemental support from facility Users and the University of Wisconsin-Milwaukee. The IRENI beamline was funded by the NSF (grant 0619759). Research described in this paper was performed in part at the Canadian Light Source, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Di-versification Canada, the National Research Council Canada, and CIHR. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2015.08.029.
References
- 1.Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21. doi: 10.1016/j.ncl.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Durukan A, Tatlisumak T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 3.Corbett D, Nurse S. The problem of assessing effective neuroprotection in experimental cerebral ischemia. Prog Neurobiol. 1998;54:531–548. doi: 10.1016/s0301-0082(97)00078-6. [DOI] [PubMed] [Google Scholar]
- 4.Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984;64:139–147. doi: 10.1007/BF00695577. [DOI] [PubMed] [Google Scholar]
- 5.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 7.Candelario-Jalil E, Mhadu NlH, Al-Dalain SM, Martanez G, Lean OS. Time course of oxidative damage in different brain regions following transient cerebral ischemia in gerbils. Neurosci Res. 2001;41:233–241. doi: 10.1016/s0168-0102(01)00282-6. [DOI] [PubMed] [Google Scholar]
- 8.Mirzaei H, Regnier F. Protein:protein aggregation induced by protein oxidation. J Chromatogr B. 2008;873:8–14. doi: 10.1016/j.jchromb.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 9.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Bio Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 10.Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther. 1995;274:1167–1173. [PubMed] [Google Scholar]
- 11.Liu H, Lightfoot R, Stevens JL. Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J Biol Chem. 1996;271:4805–4812. [PubMed] [Google Scholar]
- 12.Freeman ML, Borrelli MJ, Meredith MJ, Lepock JR. On the path to the heat shock response: destabilization and formation of partially folded protein intermediates, a consequence of protein thiol modification. Free Radical Biol Med. 1999;26:737–745. doi: 10.1016/s0891-5849(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 13.Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neurosci. 2005;134:1273–1284. doi: 10.1016/j.neuroscience.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araki T, Kato H, Inoue T, Kogure K. Regional impairment of protein synthesis following brief cerebral ischemia in the gerbil. Acta Neuropathol. 1990;79:501–505. doi: 10.1007/BF00296109. [DOI] [PubMed] [Google Scholar]
- 17.Cooper HK, Zalewska T, Kawakami S, Hossmann KA, Kleihues P. The effect of ischemia and recirculation on protein synthesis in the rat brian. J Neurochem. 1977;28:929–934. doi: 10.1111/j.1471-4159.1977.tb10652.x. [DOI] [PubMed] [Google Scholar]
- 18.Dienel G, Pulsinelli W, Duffy T. Regional protein synthesis in rat brain following acute hemispheric ischemia. J Neurochem. 1980;35:1216–1226. doi: 10.1111/j.1471-4159.1980.tb07878.x. [DOI] [PubMed] [Google Scholar]
- 19.Frank L, Bruhn T, Diemer NH. The effect of an AMPA antagonist (NBQX) on postischemic neuron loss and protein synthesis in the rat brain. Exp Brain Res. 1993;95:70–76. doi: 10.1007/BF00229655. [DOI] [PubMed] [Google Scholar]
- 20.Nowak TS, Fried RL, Lust WD, Passonneau JV. Changes in brain energy metabolism and protein synthesis following transient bilateral ischemia in the gerbil. J Neurochem. 1985;44:487–494. doi: 10.1111/j.1471-4159.1985.tb05440.x. [DOI] [PubMed] [Google Scholar]
- 21.Raley-Susman KM, Lipton P. In vitro ischemia and protein synthesis in the rat hippocampal slice: the role of calcium and NMDA receptor activation. Brain Res. 1990;515:27–38. doi: 10.1016/0006-8993(90)90572-s. [DOI] [PubMed] [Google Scholar]
- 22.Thilmann R, Xie Y, Kleihues P, Kiessling M. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol. 1986;71:88–93. doi: 10.1007/BF00687967. [DOI] [PubMed] [Google Scholar]
- 23.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deshpande J, Bergstedt K, Lindan T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- 25.Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: Their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer’s disease and cerebral amyloid angiopathy cases. Mol Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita K, Eguchi Y, Kajiwara K, Ito H. Mild hypothermia ameliorates ubiquitin synthesis and prevents delayed neuronal death in the gerbil hippocampus. Stroke. 1991;22:1574–1581. doi: 10.1161/01.str.22.12.1574. [DOI] [PubMed] [Google Scholar]
- 27.Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 29.Kretlow A, Wang Q, Kneipp J, Lasch P, Beekes M, Miller L, Naumann D. FTIR-microspectroscopy of prion-infected nervous tissue. Biochim Biophys Acta. 2006;1758:948–959. doi: 10.1016/j.bbamem.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Kuzyk A, Kastyak M, Agrawal V, Gallant M, Sivakumar G, Rak M, Del Bigio MR, Westaway D, Julian R, Gough KM. Association among amyloid plaque, lipid, and creatine in hippocampus of TgCRND8 mouse model for Alzheimer disease. J Biol Chem. 2010;285:31202–31207. doi: 10.1074/jbc.M110.142174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kneipp J, Miller LM, Joncic M, Kittel M, Lasch P, Beekes M, Naumann D. In situ identification of protein structural changes in prion-infected tissue. Biochim Biophys Acta (BBA)-Mol Basis Dis. 2003;1639:152–158. doi: 10.1016/j.bbadis.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Kastyak-Ibrahim MZ, Nasse MJ, Rak M, Hirschmugl C, Del Bigio MR, Albensi BC, Gough KM. Biochemical label-free tissue imaging with sub-cellular-resolution synchrotron FTIR with focal plane array detector. Neuro-Image. 2012;60:376–383. doi: 10.1016/j.neuroimage.2011.11.069. [DOI] [PubMed] [Google Scholar]
- 33.Chwiej J, Dulinska J, Janeczko K, Dumas P, Eichert D, Dudala J, Setkowicz Z. Synchrotron FTIR micro-spectroscopy study of the rat hippocampal formation after pilocarpine-evoked seizures. J Chem Neuroanat. 2010;40:140–147. doi: 10.1016/j.jchemneu.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol. 2006;155:30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Kretlow A, Wang Q, Kneipp J, Lasch P, Beekes M, Miller L, Naumann D. FTIR-microspectroscopy of prion-infected nervous tissue. Biochim Biophys Acta (BBA)-Biomembranes. 2006;1758:948–959. doi: 10.1016/j.bbamem.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 36.Miller LM, Bourassa MW, Smith RJ. FTIR spectroscopic imaging of protein aggregation in living cells. Biochim Biophys Acta (BBA)-Biomembranes. 2013;1828:2339–2346. doi: 10.1016/j.bbamem.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gough KM, Tzadu L, Kastyak MZ, Kuzyk AC, Julian RL. Theoretical and experimental considerations for interpretation of amide I bands in tissue. Vib Spectrosc. 2010;53:71–76. [Google Scholar]
- 38.Ami D, Natalello A, Gatti-Lafranconi P, Lotti M, Doglia SM. Kinetics of inclusion body formation studied in intact cells by FT-IR spectroscopy. FEBS Lett. 2005;579:3433–3436. doi: 10.1016/j.febslet.2005.04.085. [DOI] [PubMed] [Google Scholar]
- 39.Jackson M, Mantsch HH. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit Rev Biochem Mol Biol. 1995;30:95–120. doi: 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- 40.Troullier A, Reinstadler D, Dupont Y, Naumann D, Forge V. Transient non-native secondary structures during the refolding of [alpha]-lactalbumin detected by infrared spectroscopy. Nat Struct Mol Biol. 2000;7:78–86. doi: 10.1038/71286. [DOI] [PubMed] [Google Scholar]
- 41.Lasch P, Naumann D. Spatial resolution in infrared microspectroscopic imaging of tissues. Biochim Biophys Acta. 2006;1758:814–829. doi: 10.1016/j.bbamem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Miller LM, Dumas P. Chemical imaging of biological tissue with sycnhrotron infrared light. Biochem Biophys Acta. 2006;1758:846–857. doi: 10.1016/j.bbamem.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Dumas P, Jamin N, Teillaud JL, Miller LM, Beccard B. Imaging capabilities of synchrotron infrared microspectroscopy. Faraday Discuss. 2004;126:289–302. doi: 10.1039/b305065c. [DOI] [PubMed] [Google Scholar]
- 44.Carr GL. Resolution limits for infrared microspectroscopy explored with synchrotron radiation. Rev Sci Instrum. 2001;72:1613–1619. [Google Scholar]
- 45.Stavitski E, Smith RJ, Bourassa MW, Acerbo AS, Carr GL, Miller LM. Dynamic full-field infrared imaging with multiple synchrotron beams. Anal Chem. 2013;85:3599–3605. doi: 10.1021/ac3033849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nasse MJ, Walsh MJ, Mattson EC, Reininger R, Kajdacsy-Balla A, Macias V, Bhargava R, Hirschmugl CJ. High-resolution Fourier-transform infrared chemical imaging with multiple synchrotron beams. Nat Meth. 2011;8:413–416. doi: 10.1038/nmeth.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hackett MJ, Borondics F, Brown D, Hirschmugl C, Smith SE, Paterson PG, Nichol H, Pickering IJ, George GN. A sub-cellular biochemical investigation of Purkinje neurons using synchrotron radiation Fourier transform infrared spectroscopic imaging with a focal plane array detector. ACS Chem Neurosci. 2013;4:1071–1080. doi: 10.1021/cn4000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hackett MJ, Caine S, Liu X, May TE, Borondics F. Development of single-beam wide-field infrared imaging to study sub-cellular neuron biochemistry. Vib Spectrosc. 2015 [Google Scholar]
- 49.Barabas ME, Mattson EC, Aboualizadeh E, Hirschmugl CJ, Stucky CL. Chemical structure and morphology of dorsal root ganglion neurons from naive and inflamed mice. J Biol Chem. 2014;289:34241–34249. doi: 10.1074/jbc.M114.570101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Findlay CR, Wiens R, Rak M, Sedlmair J, Hirschmugl CJ, Morrison J, Mundy CJ, Kansiz M, Gough KM. Rapid biodiagnostic ex vivo imaging at 1 μm pixel resolution with thermal source FTIR FPA. Analyst. 2015;140:2493–2503. doi: 10.1039/c4an01982b. [DOI] [PubMed] [Google Scholar]
- 51.Liao CR, Rak M, Lund J, Unger M, Platt E, Albensi BC, Hirschmugl CJ, Gough KM. Synchrotron FTIR reveals lipid around and within amyloid plaques in transgenic mice and Alzheimer’s disease brain. Analyst. 2013;138:3991–3997. doi: 10.1039/c3an00295k. [DOI] [PubMed] [Google Scholar]
- 52.Pickering IJ, Prince RC, Divers T, George GN. Sulfur K-edge X-ray absorption spectroscopy for determining the chemical speciation of sulfur in biological systems. FEBS Lett. 1998;441:11–14. doi: 10.1016/s0014-5793(98)01402-1. [DOI] [PubMed] [Google Scholar]
- 53.Rompel A, Cinco RM, Latimer MJ, McDermott AE, Guiles RD, Quintanilha A, Krauss RM, Sauer K, Yachandra VK, Klein MP. Sulfur K-edge X-ray absorption spectroscopy: a spectroscopic tool to examine the redox state of S-containing metabolites in vivo. Proc Natl Acad Sci. 1998;95:6122–6127. doi: 10.1073/pnas.95.11.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hackett MJ, Britz CJ, Nichol H, Paterson PG, Pickering IJ, George GN. In situ bio-spectroscopic investigation of rapid ischemic and post-mortem induced biochemical alterations in the rat brain. ACS Chem Neurosci. 2014;6:226–238. doi: 10.1021/cn500157j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hackett MJ, Smith SE, Paterson PG, Nichol H, Pickering IJ, George GN. X-ray absorption spectroscopy at the sulfur K-edge: A new tool to investigate the biochemical mechanisms of neurodegeneration. ACS Chem Neurosci. 2012;3:178–185. doi: 10.1021/cn200097s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith SE, Prosser-Loose EJ, Colbourne F, Paterson PG. Protein-energy malnutrition alters thermoregulatory homeostasis and the response to brain ischemia. Curr Neurovasc Res. 2011;8:64–74. doi: 10.2174/156720211794520206. [DOI] [PubMed] [Google Scholar]
- 57.Hackett MJ, McQuillan JA, El-Assaad F, Aitken JB, Levina A, Cohen DD, Siegele R, Carter EA, Grau GE, Hunt NH, Lay PA. Chemical alterations to murine brain tissue induced by formalin fixation: implications for biospectroscopic imaging and mapping studies of disease pathogenesis. Analyst. 2011;136:2941–2952. doi: 10.1039/c0an00269k. [DOI] [PubMed] [Google Scholar]
- 58.Hackett MJ, Lee J, El-Assaad F, McQuillan JA, Carter EA, Grau GE, Hunt NH, Lay PA. FTIR imaging of brain tissue reveals crystalline creatine deposits are an ex vivo marker of localized ischemia during murine cerebral malaria: General implications for disease Neurochemistry. ACS Chem Neurosci. 2012;3:1017–1024. doi: 10.1021/cn300093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 60.George GN. 〈 http://ssrl.slac..stanford.edu/exafspak.html〉.
- 61.Butler TL, Kassed CA, Sanberg PR, Willing AE, Pennypacker KR. Neuro-degeneration in the rat hippocampus and striatum after middle cerebral artery occlusion. Brain Res. 2002;929:252–260. doi: 10.1016/s0006-8993(01)03371-6. [DOI] [PubMed] [Google Scholar]
- 62.Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 63.Grune T, Jung T, Merker K, Davies KJA. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 64.Bence NF, Sampat RM, Kopito RR. Impairment of the Ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 65.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the Ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 66.Frantseva MV, Carlen PL, Perez Velazquez JL. Dynamics of intracellular calcium and free radical production during ischemia in pyramidal neurons. Free Radical Biol Med. 2001;31:1216–1227. doi: 10.1016/s0891-5849(01)00705-5. [DOI] [PubMed] [Google Scholar]
- 67.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]