

Abstract
Background
Patients with temporomandibular joint disorders (TMD), reactive arthritis, and rheumatoid arthritis often have combined etiology of hereditary and microenvironmental factors contributing to joint pain. Multiple clinical and animal studies indicate “double-hit” inflammatory insults can cause chronic inflammation. The first inflammatory insult primes the immune system and subsequent insults elicit amplified responses. The present “double hit” study produced a chronic orofacial pain model in mice with genetic deletion of both TNFα receptors (TNFR1/R2−/−), investigating the main nociceptive signaling pathways in comparisons to wild type mice.
Methods
An initial inflammatory insult was given unilaterally into the temporomandibular joint (TMJ). Secondary hypersensitivity was tested on the skin over the TMJ throughout the experiment. Three weeks later after complete reversal of hypersensitivity, a second inflammatory insult was imposed on the colon. Pharmacological interventions were tested for efficacy after week 10 when hypersensitivity was chronic in TNFR1/R2−/− mice. Serum cytokines were analyzed Days 1, 14, and Week 18.
Results
The double hit insult produced chronic hypersensitivity continuing through the four month experimental timeline in the absence of TNFα signaling. P2X7 and NMDA receptor antagonists temporarily attenuated chronic hypersensitivity. Serum cytokine/chemokine analysis on Day 14 when CFA induced hypersensitivity was resolved identified increased levels of pro-inflammatory cytokines CCL2, CXCL9, CXCL10, RANTES and decreased levels of anti-inflammatory cytokines IL-1ra and IL-4 in TNFR1/R2−/− compared to WT mice.
Conclusions
These data suggest a causal feed-forward signaling cascade of these little studied cytokines have the potential to cause recrudescence in this orofacial inflammatory pain model in the absence of TNFα signaling.
Keywords: orofacial pain, proteome, cytokine, mechanical allodynia, P2X7 receptor, NMDA receptor
1. INTRODUCTION
Similar to rheumatoid arthritis (RA) in other joints, temporomandibular joint disorders (TMD) are characterized by severe pain, but can also induce earaches, toothaches and chronic headaches (Buescher, 2007; Lupoli and Lockey, 2007; Schiffman et al., 2014). As a major early onset pro-inflammatory cytokine, tumor necrosis factor alpha (TNFα) is released not only by immune cells but also by glia and neurons of the peripheral and central nervous system (Wagner and Myers, 1996; Schafers et al., 2003; Ohtori et al., 2004; Gao and Ji, 2010). Similar to patients with RA, patients with inflammatory TMD also have elevated levels of TNFα in their temporomandibular joint (TMJ) synovial fluid concurrent with pain (Takahashi et al., 1998; Kaneyama et al., 2002; Kopp et al., 2005; Ahmed et al., 2015; Kellesarian et al., 2016). A critical role is implicated for dysregulated TNFα and malfunctioning TNFα receptors, TNFR1 and TNFR2, in the pathogenesis of chronic pain in multifactorial diseases such as RA (Maini et al., 1993). Many patients with RA have gene polymorphisms in TNFα and the TNFα receptor genes (Hughes et al., 2004; Glossop et al., 2005; Korczowska, 2014). Patients with reactive arthritis and RA often have a combined etiology of hereditary and microenvironmental factors such as altered microbial flora in the gastrointestinal system (Toivanen, 2003). Bacterial degradation products, their cell walls and DNA, have been identified in synovial fluids from patients with TMD, RA, and reactive arthritis (Klineberg et al., 1998; Toivanen, 2001; Chen et al., 2002; Olsen-Bergem et al., 2016). Multiple experimental studies have demonstrated dysregulated TNFα and malfunctioning TNFR1 and TNFR2 caused by inflammation (Peschon et al., 1998; Kagari et al., 2002; Choi et al., 2010), or neuropathy (Sommer and Kress, 2004), as well as by “double-hit” inflammatory insults that can cause chronic inflammation not only in joints (Hains et al., 2010; Westlund et al., 2012; Traub et al., 2014) but also in other organs (Mariotti et al., 2002; Ilievski and Hirsch, 2010). The first inflammatory insult primes the immune system so that subsequent insults elicit amplified responses (Rubin and Coons, 1972).
In the present study, the role of TNFα in models of inflammatory orofacial pain was investigated using both wild type mice (WT) and mice with genetic deletion of both TNFα receptors (TNFR1/R2−/−). Injection of formalin into the lip identified increased nocifensive responses in TNFR1/R2−/− compared to WT mice. A chronic inflammatory TMD model was induced by an initial inflammatory insult given unilaterally into the TMJ. After hypersensitivity completely resolved, a second inflammatory insult was imposed on the colon three weeks later that evoked recrudescence of TMJ inflammatory hypersensitivity only in the TNFR1/R2−/− mice. The chronic hypersensitivity continued through the four month experimental timeline. Analysis of serum cytokines and chemokines at multiple time points (Baseline, Days 1, 14, and Week 18) identified both increased and decreased levels of several pro- and anti-inflammatory cytokines in TNFR1/R2−/− compared to wild type mice. Several drugs inhibiting the main inflammatory nociceptive signaling pathways during chronic pain were administered to begin characterization of factors contributing to the chronic inflammatory hypersensitivity many months after the initial TMJ inflammatory flare (Sessle, 2011). Our data suggest an alternate causal feed-forward signaling cascade causes recrudescence in this orofacial inflammatory pain model in the absence of TNFα signaling.
2. MATERIALS AND METHODS
2.1 Animals
All procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee and followed Guidelines of the National Institutes of Health, the International and the American Pain Associations. Adult male and female mice were housed on a reverse light cycle and given water and food ad libitum. TNFR1/R2−/− animals (B6.129S-Tnfrsf1atm1/mx Tnfrsf1btm1/mx/J) were bred in house after purchase of 2 breeding pairs (Jackson Laboratory, Bar Harbor, ME). WT control animals (B6129SF2/J) were ordered directly (Jackson Laboratory). The ovarian hormone cycle of female mice was not tested in this study. A total of 60 mice were tested.
2.2 Induction of Chronic Inflammation: Inflammatory “Double Hit” Model
The inflammatory double hit model (Fig. 1A) is induced with two consecutive inflammatory insults spaced three weeks apart. Six female and 8 male TNFR1/R2−/− and 6 female and 7 male WT mice were tested. The first insult is to the TMJ and the second to the colon, to model clinical reports that rheumatoid arthritis is associated with the presence and changes in colonic bacteria (Klineberg et al., 1998; Chen et al., 2002; Toivanen, 2003). Recrudescence of hypersensitivity and unilateral swelling occurred three weeks after mice were given the “second hit” and persisted through the 18 week experimental time course.
Figure 1. Induction of the “double hit” inflammation model.

(A) Experimental timeline depicting behavioral testing at baseline and continuing weekly throughout the 18 week study. Stars indicate time points when cytokines were analyzed. (B) Unilateral injection of CFA into the TMJ induced acute ipsilateral mechanical hypersensitivity and (C) referred heat hypersensitivity that were not different in WT and TNFR1/R2−/− mice. (D) After resolution of CFA induced hypersensitivity infusion of mustard oil (MO) into the colon induced recrudescence of mechanical and (E) heat hypersensitivity in TNFR1/R2−/− mice, only, that persisted for at least 18 weeks post MO infusion. WT mice did not develop hypersensitivity after the “second hit” inflammatory insult (* P < 0.05 in B-E).
First Hit
Animals were lightly anesthetized with 2% isoflurane inhalation. The right TMJ was injected with 10 μl complete Freund’s adjuvant (CFA) emulsion (1 mg/ml heat killed, desiccated Mycobacterium tuberculosis H37 (Sigma) dissolved in sterile saline mixed 1:1 with peanut oil). Similar to our previous study (Westlund et al., 2012), CFA produced temporary inflammation and pain-like behaviors in both WT and TNFR1/R2−/− mice with recovery within 2 weeks.
Second Hit
Three weeks after the first insult, animals were again lightly anesthetized with 2% isoflurane inhalation. Petroleum jelly was applied to the perianal area and a soft, perforated polyethylene tube (PE 90, 1.27 mm diameter) with a blunt, bulbous tip was inserted into the colon (4 cm length). Colonic mustard oil (MO) infusion (50 μl, 0.5% in peanut oil, Sigma-Aldrich, St. Louis, MO) was administered using a syringe connected to the tubing (Lu and Westlund, 2001). The tube was removed after 5 min and animals returned to their home cage.
2.3 Behavioral assays
For all behavioral experiments, animals were acclimated to the testing room in their home cage for 1 hour. Baseline testing for mechanical threshold occurred daily for 1 week prior to experimental start and one day prior to drug tests. All animals were tested at least once a week throughout the experiment, and were tested on days 1, 3, 5, 7, 10, and 14 after the “First Hit”. Three weeks after the “Second Hit” when a spontaneous recrudescence of mechanical hypersensitivity occurred, the animals were tested once 24 h prior to drug treatments and at multiple time points afterwards to determine drug efficacy. The short-lived effect of the drugs allowed testing of a different drug each week since behavioral tests 24 h prior to a drug treatment evidenced return to the reduced threshold indicating that the drugs did not have additive effects. The experiment was conducted in 2 cohorts using the Latin squares design alternating the sequence of drugs randomly. Results were pooled since no differences were noted between the cohorts.
2.3.1 Head Withdrawal Mechanical Threshold
Mechanical sensitivity was determined by probing the area of the face directly overlaying the TMJ with von Frey filaments to determine the withdrawal threshold using a modified up-down method (Chaplan et al., 1994). The animal was gently restrained in a tapered plastic bag so that only its head protruded for testing. Head withdrawal threshold was determined using a series of 8 von Frey filaments with logarithmically incremental bending force or stiffness (1.0, 2.0, 4.0, 6.0, 8.0, 10.0, 15.0, 26.0 g) (Stoelting Co., Wood Dale, IL). A monofilament was applied 3 times perpendicular to skin over the TMJ. If the animal responded (head withdrawal) then a filament of lesser force was used. In case of a negative response, a filament exerting higher force was tested until the mechanical sensitivity threshold was determined.
2.3.2 Heat Response Latency
Referred heat sensitivity was tested using a hot plate analgesia meter (Columbus Instruments, Columbus, OH). The heated surface (25.4 × 25.4 cm) was held constant at 52°C in a clear acrylic chamber (height: 28 cm) (Zimmer et al., 1999). The mouse was placed on the surface and the time until a response (hindpaw shaking, licking, jumping, etc.) was measured. Upon response, the animal was immediately removed and returned to its home cage. If an animal did not respond within 30 s (cut-off time), it was removed to prevent tissue damage. This was never noted. Each measurement consisted of 3 trials at 20 min intervals, and the averaged response latency is reported.
2.3.3 Orofacial Formalin Test
Animal behaviors were recorded with a computer controlled video camera. Baseline behavior was recorded for 10 min. Animals were gently restrained while 20 μl formalin (5% in 0.9% saline) was injected into the upper right lip with a syringe and 27-gauge needle (Gonzales et al., 2011). Mice were immediately returned to the clear cage (29.5 × 18.8 × 13 cm) and behaviors videotaped for 45 min in the absence of a human observer. The data was analyzed offline by an investigator blinded to experimental groups. By literature consensus, the formalin response is characterized in distinct phases. Phase 1 lasts from 0 until 6 minutes after formalin injection. The quiescent interphase during which animals showed no pain related behaviors lasts from minute 6 until 15 post injection. Phase 2 extends from minute 15 until 45 minutes post formalin injection. Statistical comparison of the responses during the two phases was done using a non-parametric Kruskal-Wallis with Dunn’s post hoc test.
2.4 Chemokine/Cytokine Analysis
Analysis of serum cytokines and chemokines at multiple time points was done in male mice to identify both increased and decreased levels of several pro- and anti-inflammatory cytokines in TNFR1/R2−/− for comparison to wild type mice. On Days 1, 14, and Week 18 (n = 3/group), groups of mice were deeply anesthetized, and blood was collected transcardially into microtainer tubes (BD Diagnostics, Franklin Lakes, NJ, USA). Samples were kept on ice for 60 min to allow clotting, centrifuged at 5000 × g for 5 min, and the serum stored at −80°C for analysis. The Proteome Profiler mouse cytokine array Panel A Kit (#ARY006, R&D Systems, Minneapolis, MN, USA) was used to quantify 40 different cyto- and chemokines simultaneously by membrane-based dot blot following manufacturer’s instructions. Digitalized radiographic films were analyzed using NIH ImageJ software and values expressed as relative pixel density.
2.5 Drugs
The efficacy of several different compounds to decrease the recrudesced hypersensitivity was determined in weeks 7–18 after the second insult. The compounds antagonizing cytokines, radical oxygen species, or ion channels were purchased from Tocris (Bristol, UK) unless otherwise stated. For injections, the drugs were dissolved in sterile 0.9% saline. The TNFα neutralizing fusion protein etanercept (Enbrel®) consisting of the human TNFR2 receptor fused to the Fc (constant) end of the immunoglobulin G1 antibody (Peppel et al., 1991) was injected intraperitoneally (i.p.; 5 mg/kg bodyweight) (Pierno et al., 2007; Malleo et al., 2008). The unspecific reactive oxygen species (ROS) scavenger alpha-phenyl-N-tert-butyl nitrone (PBN; Sigma, St. Louis, MO, USA) was administered intraperitoneally (i.p.) (100 mg/kg) (Schwartz et al., 2008) or in a separate experiment directly into the TMJ (100 mg/kg in 10 μl). The TRPV1 antagonist capsazepine was injected into the TMJ (10 mg/kg in 5 μl) (Liang et al., 2007). The selective non-competitive NMDA receptor antagonist MK-801 maleate was injected into the TMJ (10 μg/kg in 10 μl) (Du et al., 2003). The P2X7 antagonist A438079 was injected directly into the TMJ (10 mg/kg in 10 μl).
2.6 Statistical analysis
All data are reported as mean ± standard error of the mean (SEM) and compared using non-parametric tests (Friedman or Kruskal-Wallis tests) followed by Dunn’s Multiple Comparison Test or Mann-Whitney test using GraphPad Prism Software (San Diego, CA, USA). A P value of less than 0.05 was considered significant.
3. RESULTS
3.1 Inflammatory “Double Hit” Model
Baseline mechanical withdrawal thresholds were similar in animals of both genetic strains (Fig. 1B; WT: 22.6 ± 0.9 g; TNFR1/R2−/−: 22.6 ± 0.9 g). Unilateral injection of CFA into the TMJ significantly reduced the ipsilateral head withdrawal threshold from baseline in both animal strains to 6.8 ± 0.3 g in wild type and 6.1 ± 0.1 g in TNFR1/R2−/− animals (Fig. 1B, baseline vs d1, P<0.005, Kruskal-Wallis test with Dunn’s multiple comparison post hoc test). Mechanical sensitivity on the contralateral side was not different from baseline (WT: 21.6 ± 0.9 g; TNFR1/R2−/−: 23.5 ± 0.1 g). Reduction of head withdrawal threshold was maximal on day 1 post injection and returned to baseline levels 5 days after injection. Incomplete Freud’s Adjuvant did not induce any behavioral changes.
Heat sensitivity testing determined with a hotplate analgesia meter setting of 52°C was not different between wild type (18.3 ± 0.1 s) and TNFR1/R2−/− (18.6 ± 0.2 s) at baseline. One day after unilateral CFA injection response latencies were significantly reduced compared to baseline in both strains (WT: 11.0 ± 0.3; TNFR1/R2−/−: 11.7 ± 0.6 s; baseline vs d1, P<0.005, Kruskal-Wallis test with Dunn’s multiple comparison post hoc test) and returned to baseline on day 5 (Fig. 1C).
On day 21 after the CFA injection when mechanical and heat hypersensitivity were resolved, the “second hit” inflammatory colon insult was induced by infusing a 0.5% mustard oil (MO) solution into the colon for 5 min. Recrudescence of both mechanical and heat hypersensitivity occurred only in the TNFR1/R2−/− after mice given the “second hit”. Three weeks after MO infusion recrudescence included a statistically significant decrease in mechanical withdrawal thresholds and response latencies in the hotplate analgesia meter, only in the TNFR1/R2−/− mice. The recrudesced hypersensitivity was maintained chronically (at least through 18 weeks) while thresholds were unchanged from baseline in wild type animals (Fig. 1D & E). Several different compounds were tested between weeks 7 and 18 to identify contributing cellular and molecular mechanisms for recrudescence in the TNFR1/R2−/− mice.
3.2 Orofacial Formalin Test
During both Phase 1 and 2 of the formalin test, TNFR1/R2−/− mice displayed significantly increased nocifensive behavior (wiping and scratching the face, jumping, freezing) compared to wild types (Fig. 2), while the overall response curve time course was not different between the two strains.
Figure 2. Orofacial formalin test.
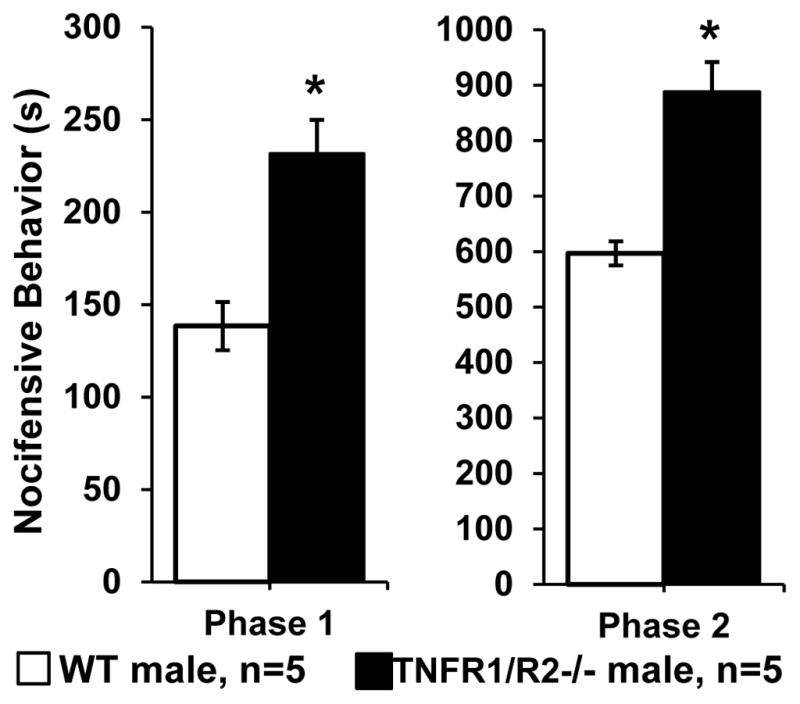
Nocifensive behavior in response to lip injection of formalin was significantly increased during phase 1 (0–10 minutes) and phase 2 (20–45 minutes) in TNFR1/R2−/− mice compared to WT animals (* P < 0.01).
The Phase 1 nocifensive response (0 – 6 minutes) peaked during the first 3 min after injection. TNFR1/R2−/− mice spent 64.4% of this time (231 ± 18.2 s) in nocifensive behaviors while wild type mice spent only 38.3% (138 ± 16.9 s, P<0.05, Mann-Whitney test). Phase 2 followed after a 9 min quiescent interphase during which animal behaviors were similar to baseline. Phase 2 (15 – 45 minutes) lasted through the end of the 45 min experiment and did not return to the original baseline. During Phase 2 TNFR1/R2−/− mice spent 46.3% of this time (888 ± 97.2 s) in nocifensive behavior while wild types spent only 30.5% (585 ± 36.8 s, P<0.05, Mann-Whitney test). The TNFR1/R2−/− mice had freezing behavior and jumped during both phases trying to escape the experimental cage.
3.3 Cytokine/chemokine proteome profiles
Serum cytokine and chemokine profiles were determined for naïve male WT and TNFR1/R2−/− mice, as well as for both genotypes 1 and 14 days post CFA injection and at experimental end, 18 weeks post colonic MO instillation (Fig. 3; figure S1).
Figure 3. Time course of increased blood serum cytokine levels.

Blood serum cytokine/chemokine levels were measured prior to acute CFA inflammation, day 1 post when behavioral hypersensitivity peaked, day 14 post CFA injection when hypersensitivity was resolved, and at experimental end 18 weeks after the “second hit” gastrointestinal inflammation. (A) Increased cytokines/chemokines. Five pro-inflammatory cytokines were elevated in TNFR1/R2−/− mice particularly on day 14 post CFA inflammation. (B) and (C) Decreased cytokines/chemokines. Twelve, anti-inflammatory and pro-inflammatory, of the tested 40 cytokines were decreased in TNFR1/R2−/− mice compared to WT animals in response to acute CFA inflammation (* P < 0.05).
3.3.1 Initial naïve levels
In naïve TNFR1/R2−/− animals, serum levels of TNFα were twice as high compared to naïve wildtype animals. Also in naïve TNFR1/R2−/− animals, chemokine (C-X-C motif) ligand 12 (CXCL12 [SDF-1]), interleukin-1α (IL-1α), IL-7, and IL-17 were elevated while chemokine (C-C motif) ligand 2 (CCL2 [MCP-1, JE]), CXCL1 (KC, NAP-3), and IL-13 were decreased compared to naïve wild type mice. All other inflammatory mediators tested were not different.
3.3.2 Cytokine/chemokine levels increased more than in naïve mice
Day 1 levels
Of note, CCL2 was elevated in TNFR1/R2−/− animals compared to wild types.
Recovery Day 14 levels
At day 14 post CFA injection when no behavioral hypersensitivity was detected, high levels of several proinflammatory mediators were detected the TNFR1/R2−/− animals including TNFα, CCL-2, CCL5 (RANTES), CXCL9 (MIG), and CXCL10 (IP-10) compared to WT mice (Fig. 3A).
Week 18 levels
While CXCL9 remained elevated in TNFR1/R2−/− animals at experiment end, the other cytokines returned to basal levels, similar to those in wildtype animals.
3.3.3 Cytokine/chemokine levels increased less than in naïve mice
Day 1 levels
Acute inflammation on Day 1 induced less increase of 12 of the 40 cytokines in serum from TNFR1/R2−/− animals than in WT mice. One third of these inflammatory mediators were anti-inflammatory such as the hematopoetic factor granulocyte colony-stimulating factor (G-CSF), IL-1ra, IL-4, and tissue inhibitor of matrix metalloproteases-1 (TIMP-1). Other decreased pro-inflammatory mediators included IL-1α, IL-1β, IL-3, CXCL1 (KC, NAP-3), CXCL12 (SDF-1), and triggering receptor expressed on myeloid cells-1 (TREM-1) compared to WT mice (Fig. 3B & C). At experiment end, expression of most of these cytokines/chemokines had returned to basal levels in animals of both genotypes.
Recovery Day 14 levels
CXCL12 in TNFR1/R2−/− animals remained lower than levels in WT mice at all post inflammation time points.
Week 18 levels
Several cytokine/chemokine levels were much lower in TNFR1/R2−/− animals than in WT mice, including anti-inflammatory IL-1ra, G-CSF, IL-1β, IL-4.
3.3.4 Cytokine/chemokine levels increased similarly in both genotypes
Expression of several proinflammatory cytokines, however, was not different between genotypes at all post inflammatory time points and is shown in Supplemental figureS1. Serum levels of complement component 5a (C5a), cluster of differentiation 54 (CD54 [ICAM-1]), granulocyte-macrophage colony-stimulating factor (GM-CSF [CSF2]), IL-7, IL-13, IL-16, interferon gamma (IFNγ), macrophage colony-stimulating factor (M-CSF [CSF1]), CCL12 (MCP-5), and CCL17 (TARC) were increased similarly on days 1 and 14 post CFA injection and were not different between genotypes (figure S1). Expression of CCL1 (I-309), CCL11 (eotaxin), IL-2, IL-10, IL-23, IL-27, CCL3 (MIP-1α), CCL4 (MIP-1β), and CXCL2 (MIP-2) in serum did not change during the experiment and was not different in WT and TNFR1/R2−/− animals (data not shown). Ten of the 40 inflammatory mediators tested had similar time course profiles in both genotypes. IL-17 was the only cytokine identified with initially similar concentrations in both genotypes that had decreased expression during the experiment in both genotypes (data not shown). The expression of IL-5, IL-6, and IL-12p was not detected in our samples in either genotype.
3.4 Systemic TNFα antagonist reverses mechanical but not heat hypersensitivity
To determine if increased levels of circulating TNFα contribute to recrudesced hypersensitivity in the TNFR1/R2−/− mice, we administered a single dose of etanercept. One day prior to administration of the TNFα neutralizing antibody both male and female TNFR1/R2−/− mice had significantly reduced mechanical (female: 8.6 ± 0.7 g; male: 11.4 ± 1.8 g; Fig. 4A) compared to wild type mice (female: 22.9 ± 0.6 g; male: 23.0 ± 0.5 g) ipsilateral to the “first hit” CFA injection. Thirty minutes after etanercept injection the mechanical thresholds significantly increased to baseline levels in both male and female TNFR1/R2−/− mice (female: 20.1 ± 2.2 s; male: 21.4 ± 1.5 s, P<0.05, Kruskal-Wallis tests with Dunn’s post hoc test) and remained elevated for 3 h (female: 19.0 ± 2.3 g; male: 18.7 ± 2.1 g, P<0.05, Kruskal-Wallis tests with Dunn’s post hoc test). One day after the single dose etanercept treatment mechanical head withdrawal thresholds returned to decreased hypersensitive levels (female: 8.9 ± 1.0 g; male: 9.1 ± 1.3 g). Mechanical withdrawal thresholds of wild type mice were not altered by etanercept and remained at baseline levels. Head withdrawal thresholds on the contralateral side were not decreased by the low dose CFA injection and treatment with etanercept did not alter them (figure S2). Response latencies to 52°C heat were not significantly improved by treatment with etanercept (TNFR1/R2−/− female: 10.7 ± 0.3 s; male: 10.7 ± 0.4 s 24 h prior to treatment; female: 11.9 ± 0.4 s; male: 12.8 ± 0.7 s 30 min post treatment; Fig. 4B). No differences between male and female animals were determined.
Figure 4. Acute administration of the TNFα neutralizing fusion protein etanercept and ROS scavenger PBN.

(A) Systemic etanercept significantly increased the mechanical withdrawal threshold ipsilaterally in TNFR1/R2−/− mice to levels observed in the recovered WT mice (#, P < 0.05). (B) Response latency in the hotplate test was not attenuated by etanercept (*, P < 0.05). (C) Systemic administration of PBN acutely resolved hypersensitive mechanical withdrawal threshold in TNFR1/R2−/− mice. (D) No change of the response latency in the hotplate test was determined. (E) Local injection of PBN into the TMJ also increased mechanical withdrawal thresholds of TNFR1/R2−/− mice to levels observed in WT mice while (F) heat responses remained hypersensitive (# P < 0.05 between timepoints; * P < 0.05 between groups).
3.5 Systemic and local treatment with radical oxygen species scavenger reduces mechanical hypersensitivity
Numerous studies using acute and persistent models inflammatory nociception have reported increased concentrations of free. Here the unspecific ROS scavenger PBN was used to determine their contribution to hypersensitivity in the inflammatory double hit model. Systemic injection of PBN resulted in temporarily significantly increased mechanical withdrawal thresholds in both male and female TNFR1/R2−/− mice (P<0.001, two-way ANOVA with Bonferroni post hoc test). Within 30 min post injection mechanical thresholds significantly increased from 9.1± 0.4 g in female and 11.1 ± 2.3 g in male TNFR1/R2−/− mice to 21.9 ± 1.6 g and 21.9 ± 1.5 g, respectively (P<0.01, Kruskal-Wallis test with Dunn’s post hoc test) and returned to the hypersensitive state within 3 h post injection (Fig. 4C). Single local injections of PBN directly into the ipsilateral TMJ were similarly effective in acutely reducing mechanical hypersensitivity 30 min post injection and did not persist (Fig. 4E). Unlike the systemic administration, injections into the TMJ showed a better trend to improved mechanical sensitivity in male than in female mice (male: 23.5 ± 0.1 g; female: 19.4 ± 2.3 g; P<0.05, Kruskal-Wallis test with Dunn’s post hoc test). PBN did not alter mechanical sensitivity on the contralateral side (data not shown). Response latencies on the hotplate test was improved neither after systemic PBN treatment (TNFR1/R2−/− female: 14.2 ± 0.8 s; male: 14.9 ± 0.5 s; Fig. 4D) nor after local TMJ injections (TNFR1/R2−/− female: 10.8 ± 0.7 s; male: 13.7 ± 0.6 s; Fig. 4F).
3.6 P2X7 and NMDA receptor antagonists improve mechanical and heat sensitivity while TRPV1 antagonists improves neither
Several different ion channels were investigated that have been shown to be localized either on peripheral immune cells and/or axonal terminals of nociceptive sensory neurons. Local injection of the selective P2X7 antagonist A438079 into the ipsilateral TMJ briefly reversed mechanical hypersensitivity (TNFR1/R2−/− female: 19.9. ± 1.8 g; male: 20.7 ± 1.0 g; P<0.01, Kruskal-Wallis test with Dunn’s post hoc test; Fig. 5A) as well as improved heat hypersensitivity 30 min post injection (TNFR1/R2−/− female: 17.8 ± 0.4 s; male: 16.6 ± 0.5 s; P<0.01, Kruskal-Wallis test with Dunn’s post hoc test; Fig. 5B), but did not persist. Injection of the NMDA specific antagonist MK801 into the TMJ significantly improved mechanical hypersensitivity within 30 min (TNFR1/R2−/− female: 19.7 ± 0.6 g; male: 20.4 ± 1.6 s; P<0.01, Kruskal-Wallis test with Dunn’s post hoc test; Fig. 5C) and persisted for 3 h (TNFR1/R2−/− female: 19.0 ± 0.9 g; male: 16.9 ± 2.2 g). Heat sensitivity was only alleviated 30 min post injection (TNFR1/R2−/− female: 14.8 ± 0.3 s; male: 14.6 ± 0.6 s; 3 h post injection; P<0.01, Kruskal-Wallis test with Dunn’s post hoc test; Fig. 5D). Use of the TRPV1 antagonist capsazepine did not reduce either mechanical (Fig. 5E) or heat sensitivity (Fig. 5F).
Figure 5. Acute administration of ion channel antagonists.

(A) Local injection of the P2X7 receptor antagonist A438079 resolved mechanical hypersensitivity in TNFR1/R2−/− mice only at the 30 min time point post injection. Mechanical withdrawal thresholds and (B) response latencies in the hotplate test were significantly increased and then returned to their hypersensitive levels. (C) Local injection of the NMDA receptor antagonist MK801 in TNFR1/R2−/− mice significantly increased mechanical withdrawal thresholds for up to 3 h post injection (P < 0.05). (D) Response latencies of TNFR1/R2−/− mice in the hotplate test were significantly attenuated only at 30 min post injection. (E) Local administration of the TRPV1 antagonist capsazepine had no effect on mechanical withdrawal threshold or (F) heat response latencies. None of the compounds tested altered the responses of WT animals (# P < 0.05 between timepoints; * P < 0.05 between groups).
4. DISCUSSION
The data presented demonstrate that unilateral chronic TMJ inflammation could be initiated in TNFR1/R2−/− mice when a “first hit” TMJ inflammatory insult with CFA was followed later by a mild gastrointestinal inflammation “second hit” using MO. The “double hit” model in wild type mice did not induce recrudescence of TMJ inflammation and associated hypersensitivity. In TNFR1/R2−/− mice, the chronic hypersensitivity continued through the four month experimental timeline. Cytokine levels of TNFα and several other pro-inflammatory cytokines were increased while several anti-inflammatory cytokines were decreased in blood serum samples of TNFR1/R2−/− mice compared to wild type mice. This was particularly striking on day 14 after CFA injection when behavioral hypersensitivity in animals of both genotypes was recovered. ROS scavenger PBN and TNFα neutralizing fusion protein etanercept, as well as specific antagonists for P2X7 and NMDA receptors were able to attenuate behavioral hypersensitivity while TRPV1 antagonist capsazepine had no effect. These data demonstrate the complexity of molecular signaling pathways involved in the chronic TMJ inflammation. We also determined that TNFR1/R2−/− mice respond to acute inflammation induced by formalin injection into the lip with increased nocifensive behavior during both phases compared to WT animals. TNFR1/R2−/− mice not only showed wiping and scratching of the injected side of the face, but also responded by freezing and unlike WT mice, jumped to escape the cage. This was unexpected as previous studies reported the absence of functional TNFR1/R2 by use of antibodies or knockout mice to be protective in the formalin test (Seadi Pereira et al., 2009; Zhang et al., 2011a; Zhang et al., 2011b). A possible explanation is differences in experienced environmental stress that reportedly alters susceptibility to arthritis model induction (Ji et al., 2002). Most recently, a feature in Science magazine attributed differences in gut microbiota to reproducibility differences in published experimental results even when using the identical mouse strain purchased from the same vendor (Servick, 2016).
The “double hit” inflammatory insult model produced recrudescence of mechanical hypersensitivity only in the ipsilateral TMJ area injected with the CFA similar to our study using the “double hit” inflammatory model to model knee joint arthritis (Westlund et al., 2012). Behavioral hypersensitivity re-occurred weeks after gastrointestinal inflammation only in TNFR1/R2−/− mice while WT animals fully recovered from both insults. The use of CFA to model RA as well as persistent TMJ inflammation is well established, inducing edema in the joint and behavioral hypersensitivity (Spears et al., 2003), symptoms similar to clinical patients (Arthritis Foundation, http://www.arthritis.org). On a technical note, similar to a previous study, we tested the area over the TMJ with von Frey filaments (do Val et al., 2014). In our experiments, the stimulated skin and underlying tissue was relatively insensitive, requiring higher strength von Frey filaments to elicit a response compared to the whiskerpad and feet. We tested heat sensitivity using the hotplate, demonstrating whole body heat hypersensitivity. A recent publication demonstrated that that orofacial pain induces widespread pain sensitization in rodent models (Zhang et al., 2016). That study also reported paradoxical mechanical allodynia in the hindpaw with concurrent heat hyperalgesia.
Several clinical studies on patients with RA have reported finding gut residing bacteria, cell walls, and their DNA within inflamed joints and to actively drive disease progression (Klineberg et al., 1998; Toivanen, 2001; Chen et al., 2002). Experimental animal studies have been able to model this for more detailed investigation (Wu et al., 2010). To model this, the present study used a mild colonic insult with MO as the “second hit”. We previously demonstrated resolution within 4–8 hours of the induced colonic ulceration and cFOS activation in the spinal cord using a higher dose of mustard oil in rats (Lu and Westlund, 2001). The colonic injection of MO may have interrupted the neuroimmune interactions in the gut and altered circulating microbial metabolites (Holzer et al., 2015). The combination of the “double hit” inflammatory insults with hereditary dysregulation of TNFα in TNFR1/R2−/− mice produced a model of chronic TMJ hypersensitivity and altered serum cytokine profiles that characterize a suitable model of TMD. However, future studies are needed to determine if the “double hit” inflammatory model can also be induced in single TNFR knockout mice and to further characterize the role of the two TNFRs in chronic inflammatory pain.
Time course analysis of 40 mixed anti- and pro-inflammatory cyto-/chemokines identified differential expression in animals of both genotypes. Approximately ¼ of the analyzed pro-inflammatory cytokines increased after acute inflammation with CFA and then decreased to basal levels in TNFR1/R2−/− and WT mice. Cyto-/chemokine levels that returned to basal conditions in the WT mice prevented the development of chronic inflammation. In TNFR1/R2−/− mice several pro-inflammatory cytokines, TNFα, CCL2, CXCL9, CXCL10, and RANTES were elevated compared to WT animals. Levels of TNFα were increased 3-fold compared to WTs, similar to previous studies (Peschon et al., 1998; Westlund et al., 2012). Typically, TNFα signaling is pro-inflammatory and its inhibition decreases inflammation (Kopp et al., 2005). Yet, several reports have shown that it also initiates the release of anti-inflammatory cytokines, those decreasing inflammation. TNFα signaling has been shown to inhibit IL-12 and IL-23 in macrophages and dendritic cells (Zakharova and Ziegler, 2005). Tolerance and secretion of IL-12 of transforming growth factor beta (TGF-β) activated antigen-presenting cells in the eye is negatively regulated by TNFα, resulting in a decreased immune response (Masli and Turpie, 2009).
Elevated concentrations of CCL2, CXCL9, CXCL10, and RANTES in the synovial tissue and blood serum of patients diagnosed with RA and chronic TMJ inflammation have all been extensively described in the literature. Patient serum levels of CCL2, CXCL9, and CXCL10 correlate with severity of RA progression, and have been used to monitor efficacy of novel RA treatments (König et al., 2000; Xia et al., 2011; Shah et al., 2011). Their increased levels in our “double hit” inflammatory model in TNFR1/R2−/− mice only indicates that in the absence of TNFα their levels are dysregulated, though their upregulation is supportive of the relevance of the “double-hit” model described here. Similar findings and correlation of pain scores with selected cytokines are reported in collagen induced hindpaw arthritis (Kagari et al., 2002). Concurrent decreased levels of anti-inflammatory IL-1ra and IL-4 were measured in TNFR1/R2−/− mice that remained lower than in WT animals until experimental end. Elevated levels of IL-1ra in clinical patients have been measured in the TMJ of patients with polyarthritis who reported minimal amounts of pain (Alstergren et al., 2003; Kopp et al., 2005). Treatment with recombinant human IL-1ra was shown to not only decrease cartilage but to facilitate cartilage repair degradation in experimentally induced arthritis in the TMJ (Zhang et al., 2011a; Zhang et al., 2011b). These data suggest that the combination of elevated levels of CCL2, CXCL9, CXCL10, and RANTES combined with decreases of anti-inflammatory IL-1ra and IL-4 may have contributed to chronic inflammation in the TMJ and behavioral hypersensitivity in the TNFR1/R2−/− mice.
Surprisingly, use of etanercept reduced mechanical hypersensitivity in TNFR1/R2−/− mice. It has been repeatedly demonstrated in the last 20 years that TNFα specifically binds the TNFR1 and TNFR2 receptors (Beutler and Van, 1994; Locksley et al., 2001). No other receptor bindings have so far been demonstrated for TNFα. In the absence of both TNFα receptors, even 3-fold increased levels over WT animals are unable to initiate any signaling cascades. Etanercept is a unique biological in that it is a human fusion protein consisting of the TNFR2 receptor and the IgG heavy chain (Peppel et al., 1991). It is unclear if the ability of etanercept to reduce hypersensitivity briefly in TNFR1/R2−/− mice during chronic inflammation was due to neutralization of LTα since etanercept not only binds TNFα but also LTα (Beutler and Van Huffel, 1994; Locksley et al., 2001). In any event, a short efficacy is noted in mice with this human protein in the present study and in others (Christianson et al., 2010; Park et al., 2013).
Several limitations can be noted in this study. Limitations imposed by the breeding colony did not allow completion of all testing in both male and female mice. Formalin and cytokine analysis was only performed in male mice. While ovarian hormone cycling information was not collected in the female mice, we did not find sex differences in mechanical and heat sensitivity in the “double hit” TMJ chronic inflammation model. Gender differences in orofacial pain signaling and incidence in clinical patients has been reported (Warren and Fried, 2001; Gazerani et al., 2010). We utilized von Frey filaments, a well-established method, that required of the animal’s movements in order to determine mechanical withdrawal thresholds by stimulation the area of the face overlaying the TMJ without touching the highly sensitive sinus hairs located in close proximity. It is therefore possible that stress caused by gentle restraint differed between male and female animals, overshadowing gender differences of hypersensitivity.
We also investigated peripheral tissue mechanisms for their contribution to maintenance of chronic pain. Clinical studies have identified several genes encoding ion channels and receptors with single nucleotide polymorphisms (SNPs) expressed by local immune cells, neurons and glia that alter the susceptibility for chronic pain (Lacroix-Fralish and Mogil, 2009). Acute local inhibition of P2X7 and NMDA receptors was able to normalize both mechanical and heat sensitivity. Both compounds are able to decrease peripheral sensory neuron activity either indirectly or directly, thus reducing mechanical hypersensitivity tested by applying force directly over the TMJ (Gazerani et al., 2010; Arandjelovic et al., 2012).
The P2X7 purinergic receptor is localized on immune and trigeminal ganglia satellite glial cells, suggesting that its inhibition may have decreased the release of excitatory molecules onto nociceptors, thus reducing behavioral hypersensitivity to noxious stimuli (Donnelly-Roberts and Jarvis, 2007; Kushnir et al., 2011). The P2X7 ion channel is expressed on mast cells and use of the antagonist may have decreased inflammatory signaling cascades initiated by ATP that result in mast cell activation (Arandjelovic et al., 2012). Treatment with A438079 may have also decreased local levels of pro-inflammatory cytokines IL-1β and IL-18, thus decreasing immune-neural interactions resulting in decreased hypersensitivity (McGaraughty et al., 2007).
Glutamate is released by damaged cells as well as by primary sensory neurons innervating the tissue (deGroot et al., 2000) and activates neurons by binding to both ionotropic and metabotropic receptors. The NMDA receptor is localized on primary afferents innervating the TMJ as well as peripheral immune cells (Gazerani et al., 2010). After peripheral nerve injury, increased expression of the NMDA receptor in DRG neurons has been correlated with an influx of immune cells into the injury site, behavioral hypersensitivity, and an increase of activated microglia in the spinal cord, suggesting a role for glutamate and the NMDA receptor in peripheral and central neuroimmune responses (Gazerani et al., 2010). Our data demonstrate that MK801 efficaciously decreased mechanical and heat hypersensitivity. While the exact mechanisms are still being elucidated, it is known that both antagonists initiate different second messenger signaling cascades and have different efficacy in reducing nociceptive responses after injury (Suzuki et al., 2001). The data presented suggest involvement of the NMDA receptor maintaining hypersensitivity produced by the “double hit” inflammatory insult.
Afferents expressing TRPV1 have been reported to have an essential role in establishing inflammatory heat hypersensitivity, yet, our data similar to others suggests that it is not essential to transduce noxious heat stimuli chronically (Woodbury et al., 2004). In the present study only the effect of local capsazepine application was tested. It is therefore possible that its systemic use might reduce chronic inflammation induced hypersensitivity.
Cellular stress and damage cause the production and release of ROS, which combined with an imbalance of pro- and anti-inflammatory cytokines are universal biomarkers of inflammatory diseases including RA and chronic inflammation of the TMJ (Thacker et al., 2007). We have previously shown ROS increase in the spinal cord in a knee joint arthritis model, as well as secondary mechanical and heat sensitization that is ameliorated with the ROS scavenger PBN (Westlund et al., 2010). A recent study demonstrated a positive correlation between levels of ROS and pro-inflammatory cytokines CCL2, CXCL10, and RANTES (Shah et al., 2011), speculating that these molecules act in a positive feed-forward loop. Our data identified a contribution of an altered redox status in TNFR1/R2−/− mice with chronic inflammation as use of the ROS scavenger PBN was able to normalize mechanical hypersensitivity acutely.
The data presented here identify several different pro-inflammatory systems that act in concert in a feed-forward mechanism in the “double hit” inflammatory model. The chronic inflammation and hypersensitivity produced long term in these animals with hereditary dysregulated TNFα signaling identifying them as novel targets for pharmacological interventions.
Supplementary Material
Ten of the tested 40 cytokines showed similar response profiles in both TNFR1/R2−/− and WT mice. Cytokines were elevated on days 1 and 14 post CFA injection and returned to baseline levels at the 18 week experimental end time point.
Mechanical withdrawal threshold of TNFR1/R2−/− mice on the contralateral side was not affected by etanercept and remained similar to baseline and levels observed in the WT mice.
What does this study add?
Using a mouse model of chronic inflammatory temporomandibular joint disorder, we determined that absence of functional TNFR1/R2 induces aberrant inflammatory signaling caused by other increased pro-inflammatory and decreased anti-inflammatory cytokines that could serve as blood biomarkers and may predict disease progression.
Acknowledgments
Funding Sources: This research was further supported by the National Institute of Health award R01 NS039041 (KNW) and the University of Kentucky President’s Research Fund (KNW).
ABBREVIATIONS
- C5a
complement component 5a
- CCL
C-C motif ligand
- CD54
cluster of differentiation 54
- CFA
complete Freund’s adjuvant
- CXCL
C-X-C motif ligand
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- i.p
intraperitoneally
- IFNγ
interferon γ
- IL-1ra
interleukin-1 receptor antagonist
- IL
interleukin
- M-CSF
macrophage colony-stimulating factor
- MO
mustard oil
- NMDA
N-methyl-D-aspartate
- PBN
alpha-phenyl-N-tert-butyl nitrone
- RA
rheumatoid arthritis
- ROS
reactive oxygen species
- SEM
standard error of the mean
- SNPs
single nucleotide polymorphisms
- TGF-β
transforming growth factor beta
- TIMP-1
tissue inhibitor of matrix metalloproteases-1
- TMD
temporomandibular disorder
- TMJ
temporomandibular joint
- TNFR, TNFα
tumor necrosis factor α; TNFα receptor
- TNFR1/R2−/−
mice with genetic deletion of both TNFα receptors
- TREM -1
triggering receptor expressed on myeloid cells-1
- TRPV1
transient receptor potential vanilloid receptor 1
- WT
wild type
Footnotes
Conflicts of Interest: The Authors declare no conflicts of interest.
Author contribution: S.L.M. performed all behavioral experiments, data analysis and graphing, manuscript preparation and revision. R.N. performed behavioral experiments. F.M. performed serum cytokine analysis and tissue dissections. H.S.O. performed serum cytokine assays. L.Z. performed tissue dissections. K.N.W. designed the experiments and revised manuscript. All authors discussed the results and commented on the manuscript.
References
- Ahmed N, Catrina AI, Alyamani AO, Mustafa H, Alstergren P. Deficient cytokine control modulates temporomandibular joint pain in rheumatoid arthritis. Eur J Oral Sci. 2015;123:235–241. doi: 10.1111/eos.12193. [DOI] [PubMed] [Google Scholar]
- Alstergren P, Benavente C, Kopp S. Interleukin-1beta, interleukin-1 receptor antagonist, and interleukin-1 soluble receptor II in temporomandibular joint synovial fluid from patients with chronic polyarthritides. J Oral Maxillofac Surg. 2003;61:1171–1178. doi: 10.1016/s0278-2391(03)00678-5. [DOI] [PubMed] [Google Scholar]
- Arandjelovic S, McKenney KR, Leming SS, Mowen KA. ATP induces protein arginine deiminase 2-dependent citrullination in mast cells through the P2X7 purinergic receptor. J Immunol. 2012;189:4112–4122. doi: 10.4049/jimmunol.1201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, van Huffel C. Unraveling function in the TNF ligand and receptor families. Science. 1994;264:667–668. doi: 10.1126/science.8171316. [DOI] [PubMed] [Google Scholar]
- Buescher JJ. Temporomandibular joint disorders. Am Fam Physician. 2007;76:1477–1482. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen T, Rimpilainen M, Luukkainen R, Mottonen T, Yli-Kerttula U, Saario R, Toivanen P. Mononuclear cell response to enterobacteria and Gram-positive cell walls of normal intestinal microbiota in early rheumatoid arthritis and other inflammatory arthritides. Clin Exp Rheumatol. 2002;20:193–200. [PubMed] [Google Scholar]
- Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain. 2010;149:243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson CA, Corr M, Firestein GS, Mobargha A, Yaksh TL, Svensson CI. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain. 2010;151:394–403. doi: 10.1016/j.pain.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deGroot J, Zhou S, Carlton SM. Peripheral glutamate release in the hindpaw following low and high intensity sciatic stimulation. Neuroreport. 2000;11:497–502. doi: 10.1097/00001756-200002280-00014. [DOI] [PubMed] [Google Scholar]
- do Val DR, Bezerra MM, Silva AA, Pereira KM, Rios LC, Lemos JC, Arriaga NC, Vasconcelos JN, Benevides NM, Pinto VP, Cristino-Filho G, Brito GA, Silva FR, Santiago GM, Arriaga AM, Chaves HV. Tephrosia toxicaria Pers. reduces temporomandibular joint inflammatory hypernociception: the involvement of the HO-1 pathway. Eur J Pain. 2014;18:1280–1289. doi: 10.1002/j.1532-2149.2014.488.x. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Jarvis MF. Discovery of P2X7 receptor-selective antagonists offers new insights into P2X7 receptor function and indicates a role in chronic pain states. Br J Pharmacol. 2007;151:571–579. doi: 10.1038/sj.bjp.0707265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou S, Coggeshall RE, Carlton SM. N-methyl-D-aspartate-induced excitation and sensitization of normal and inflamed nociceptors. Neuroscience. 2003;118:547–562. doi: 10.1016/s0306-4522(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazerani P, Dong X, Wang M, Kumar U, Cairns BE. Sensitization of rat facial cutaneous mechanoreceptors by activation of peripheral N-methyl-d-aspartate receptors. Brain Res. 2010;1319:70–82. doi: 10.1016/j.brainres.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Glossop JR, Dawes PT, Nixon NB, Mattey DL. Polymorphism in the tumour necrosis factor receptor II gene is associated with circulating levels of soluble tumour necrosis factor receptors in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1227–1234. doi: 10.1186/ar1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Zegpi C, Noriega V, Prieto JC, Miranda HF. Synergism between dexketoprofen and meloxicam in an orofacial formalin test was not modified by opioid antagonists. Pharmacol Rep. 2011;63:433–440. doi: 10.1016/s1734-1140(11)70509-6. [DOI] [PubMed] [Google Scholar]
- Hains LE, Loram LC, Weiseler JL, Frank MG, Bloss EB, Sholar P, Taylor FR, Harrison JA, Martin TJ, Eisenach JC, Maier SF, Watkins LR. Pain intensity and duration can be enhanced by prior challenge: initial evidence suggestive of a role of microglial priming. J Pain. 2010;11:1004–1014. doi: 10.1016/j.jpain.2010.01.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, Hassan A, Jain P, Reichmann F, Farzi A. Neuroimmune pharmacological approaches. Curr Opin Pharmacol. 2015;25:13–22. doi: 10.1016/j.coph.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LB, Criswell LA, Beasley TM, Edberg JC, Kimberly RP, Moreland LW, Seldin MF, Bridges SL. Genetic risk factors for infection in patients with early rheumatoid arthritis. Genes Immun. 2004;5:641–647. doi: 10.1038/sj.gene.6364137. [DOI] [PubMed] [Google Scholar]
- Ilievski V, Hirsch E. Synergy between viral and bacterial toll-like receptors leads to amplification of inflammatory responses and preterm labor in the mouse. Biol Reprod. 2010;83:767–773. doi: 10.1095/biolreprod.110.085464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagari T, Doi H, Shimozato T. The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J Immunol. 2002;169:1459–1466. doi: 10.4049/jimmunol.169.3.1459. [DOI] [PubMed] [Google Scholar]
- Kaneyama K, Segami N, Nishimura M, Suzuki T, Sato J. Importance of proinflammatory cytokines in synovial fluid from 121 joints with temporomandibular disorders. Br J Oral Maxillofac Surg. 2002;40:418–423. [PubMed] [Google Scholar]
- Kellesarian SV, Al-Kheraif AA, Vohra F, Ghanem A, Malmstrom H, Romanos GE, Javed F. Cytokine profile in the synovial fluid of patients with temporomandibular joint disorders: A systematic review. Cytokine. 2016;77:98–106. doi: 10.1016/j.cyto.2015.11.005. [DOI] [PubMed] [Google Scholar]
- Klineberg I, McGregor N, Butt H, Dunstan H, Roberts T, Zerbes M. Chronic orofacial muscle pain: a new approach to diagnosis and management. Alpha Omegan. 1998;91:25–28. [PubMed] [Google Scholar]
- Konig A, Krenn V, Toksoy A, Gerhard N, Gillitzer R. Mig, GRO alpha and RANTES messenger RNA expression in lining layer, infiltrates and different leucocyte populations of synovial tissue from patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Virchows Arch. 2000;436:449–458. doi: 10.1007/s004280050472. [DOI] [PubMed] [Google Scholar]
- Kopp S, Alstergren P, Ernestam S, Nordahl S, Morin P, Bratt J. Reduction of temporomandibular joint pain after treatment with a combination of methotrexate and infliximab is associated with changes in synovial fluid and plasma cytokines in rheumatoid arthritis. Cells Tissues Organs. 2005;180:22–30. doi: 10.1159/000086195. [DOI] [PubMed] [Google Scholar]
- Korczowska I. Rheumatoid arthritis susceptibility genes: An overview. World J Orthop. 2014;5:544–549. doi: 10.5312/wjo.v5.i4.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir R, Cherkas PS, Hanani M. Peripheral inflammation upregulates P2X receptor expression in satellite glial cells of mouse trigeminal ganglia: a calcium imaging study. Neuropharmacology. 2011;61:739–746. doi: 10.1016/j.neuropharm.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Lacroix-Fralish ML, Mogil JS. Progress in genetic studies of pain and analgesia. Annu Rev Pharmacol Toxicol. 2009;49:97–121. doi: 10.1146/annurev-pharmtox-061008-103222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS. The synthetic cannabinoids attenuate allodynia and hyperalgesia in a rat model of trigeminal neuropathic pain. Neuropharmacology. 2007;53:169–177. doi: 10.1016/j.neuropharm.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Lu Y, Westlund KN. Effects of baclofen on colon inflammation-induced Fos, CGRP and SP expression in spinal cord and brainstem. Brain Res. 2001;889:118–130. doi: 10.1016/s0006-8993(00)03124-3. [DOI] [PubMed] [Google Scholar]
- Lupoli TA, Lockey RF. Temporomandibular dysfunction: an often overlooked cause of chronic headaches. Ann Allergy Asthma Immunol. 2007;99:314–318. doi: 10.1016/S1081-1206(10)60546-7. [DOI] [PubMed] [Google Scholar]
- Maini RN, Brennan FM, Williams R, Chu CQ, Cope AP, Gibbons D, Elliott M, Feldmann M. TNF-alpha in rheumatoid arthritis and prospects of anti-TNF therapy. Clin Exp Rheumatol. 1993;11(Suppl 8):S173–175. [PubMed] [Google Scholar]
- Malleo G, Mazzon E, Genovese T, Di Paola R, Muia C, Caminiti R, Esposito E, Di Bella P, Cuzzocrea S. Etanercept reduces acute tissue injury and mortality associated to zymosan-induced multiple organ dysfunction syndrome. Shock. 2008;29:560–571. doi: 10.1097/shk.0b013e3181507234. [DOI] [PubMed] [Google Scholar]
- Mariotti R, Tongiorgi E, Bressan C, Kristensson K, Bentivoglio M. Priming by muscle inflammation alters the response and vulnerability to axotomy-induced damage of the rat facial motor nucleus. Exp Neurol. 2002;176:133–142. doi: 10.1006/exnr.2002.7908. [DOI] [PubMed] [Google Scholar]
- Masli S, Turpie B. Anti-inflammatory effects of tumour necrosis factor (TNF)-alpha are mediated via TNF-R2 (p75) in tolerogenic transforming growth factor-beta-treated antigen-presenting cells. Immunology. 2009;127:62–72. doi: 10.1111/j.1365-2567.2008.02933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Namovic MT, Donnelly-Roberts DL, Harris RR, Zhang XF, Shieh CC, Wismer CT, Zhu CZ, Gauvin DM, Fabiyi AC, Honore P, Gregg RJ, Kort ME, Nelson DW, Carroll WA, Marsh K, Faltynek CR, Jarvis MF. P2X7-related modulation of pathological nociception in rats. Neuroscience. 2007;146:1817–1828. doi: 10.1016/j.neuroscience.2007.03.035. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- Olsen-Bergem H, Kristoffersen AK, Bjornland T, Reseland JE, Aas JA. Juvenile idiopathic arthritis and rheumatoid arthritis: bacterial diversity in temporomandibular joint synovial fluid in comparison with immunological and clinical findings. Int J Oral Maxillofac Surg. 2016;45:318–322. doi: 10.1016/j.ijom.2015.08.986. [DOI] [PubMed] [Google Scholar]
- Park HJ, Stokes JA, Pirie E, Skahen J, Shtaerman Y, Yaksh TL. Persistent hyperalgesia in the cisplatin-treated mouse as defined by threshold measures, the conditioned place preference paradigm, and changes in dorsal root ganglia activated transcription factor 3: the effects of gabapentin, ketorolac, and etanercept. Anesth Analg. 2013;116:224–231. doi: 10.1213/ANE.0b013e31826e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–1489. doi: 10.1084/jem.174.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- Pierno S, Nico B, Burdi R, Liantonio A, Didonna MP, Cippone V, Fraysse B, Rolland JF, Mangieri D, Andreetta F, Ferro P, Camerino C, Zallone A, Confalonieri P, De Luca A. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol. 2007;33:344–359. doi: 10.1111/j.1365-2990.2007.00798.x. [DOI] [PubMed] [Google Scholar]
- Rubin AS, Coons AH. Specific heterologous enhancement of immune responses. IV. Specific generation of a thymus-derived enhancing factor. J Exp Med. 1972;136:1501–1517. doi: 10.1084/jem.136.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17:791–804. doi: 10.1046/j.1460-9568.2003.02504.x. [DOI] [PubMed] [Google Scholar]
- Schiffman E, Ohrbach R, Truelove E, Look J, Anderson G, Goulet JP, List T, Svensson P, Gonzalez Y, Lobbezoo F, Michelotti A, Brooks SL, Ceusters W, Drangsholt M, Ettlin D, Gaul C, Goldberg LJ, Haythornthwaite JA, Hollender L, Jensen R, John MT, De Laat A, de Leeuw R, Maixner W, van der Meulen M, Murray GM, Nixdorf DR, Palla S, Petersson A, Pionchon P, Smith B, Visscher CM, Zakrzewska J, Dworkin SF International RDC/TMD Consortium Network, International association for Dental Research; Orofacial Pain Special Interest Group, International Association for the Study of Pain. Diagnostic Criteria for Temporomandibular Disorders (DC/TMD) for Clinical and Research Applications: recommendations of the International RDC/TMD Consortium Network* and Orofacial Pain Special Interest Group†. J Oral Facial Pain Headache. 2014;28:6–27. doi: 10.11607/jop.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ES, Lee I, Chung K, Chung JM. Oxidative stress in the spinal cord is an important contributor in capsaicin-induced mechanical secondary hyperalgesia in mice. Pain. 2008;138:514–524. doi: 10.1016/j.pain.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seadi Pereira PJ, Noronha Dornelles F, Santiago Santos D, Batista Calixto J, Bueno Morrone F, Campos MM. Nociceptive and inflammatory responses induced by formalin in the orofacial region of rats: effect of anti-TNFalpha strategies. Int Immunopharmacol. 2009;9:80–85. doi: 10.1016/j.intimp.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Servick K. Of mice and microbes. Science. 2016;353:741–743. doi: 10.1126/science.353.6301.741. [DOI] [PubMed] [Google Scholar]
- Sessle BJ. Peripheral and central mechanisms of orofacial inflammatory pain. Int Rev Neurobiol. 2011;97:179–206. doi: 10.1016/B978-0-12-385198-7.00007-2. [DOI] [PubMed] [Google Scholar]
- Shah D, Wanchu A, Bhatnagar A. Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology. 2011;216:1010–1017. doi: 10.1016/j.imbio.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Spears R, Oakes R, Bellinger LL, Hutchins B. Tumour necrosis factor-alpha and apoptosis in the rat temporomandibular joint. Arch Oral Biol. 2003;48:825–834. doi: 10.1016/s0003-9969(03)00175-4. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Matthews EA, Dickenson AH. Comparison of the effects of MK-801, ketamine and memantine on responses of spinal dorsal horn neurones in a rat model of mononeuropathy. Pain. 2001;91:101–109. doi: 10.1016/s0304-3959(00)00423-1. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kondoh T, Fukuda M, Yamazaki Y, Toyosaki T, Suzuki R. Proinflammatory cytokines detectable in synovial fluids from patients with temporomandibular disorders. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:135–141. doi: 10.1016/s1079-2104(98)90415-2. [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg. 2007;105:838–847. doi: 10.1213/01.ane.0000275190.42912.37. [DOI] [PubMed] [Google Scholar]
- Toivanen P. From reactive arthritis to rheumatoid arthritis. J Autoimmun. 2001;16:369–371. doi: 10.1006/jaut.2000.0496. [DOI] [PubMed] [Google Scholar]
- Toivanen P. Normal intestinal microbiota in the aetiopathogenesis of rheumatoid arthritis. Ann Rheum Dis. 2003;62:807–811. doi: 10.1136/ard.62.9.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RJ, Cao DY, Karpowicz J, Pandya S, Ji Y, Dorsey SG, Dessem D. A clinically relevant animal model of temporomandibular disorder and irritable bowel syndrome comorbidity. J Pain. 2014;15:956–966. doi: 10.1016/j.jpain.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Myers RR. Schwann cells produce tumor necrosis factor alpha: expression in injured and non-injured nerves. Neuroscience. 1996;73:625–629. doi: 10.1016/0306-4522(96)00127-3. [DOI] [PubMed] [Google Scholar]
- Warren MP, Fried JL. Temporomandibular disorders and hormones in women. Cells Tissues Organs. 2001;169:187–192. doi: 10.1159/000047881. [DOI] [PubMed] [Google Scholar]
- Westlund KN, Kochukov MY, Lu Y, McNearney TA. Impact of central and peripheral TRPV1 and ROS levels on proinflammatory mediators and nociceptive behavior. Mol Pain. 2010;6:46. doi: 10.1186/1744-8069-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlund KN, Zhang L, Ma F, Oz HS. Chronic inflammation and pain in a tumor necrosis factor receptor (TNFR) (p55/p75−/−) dual deficient murine model. Transl Res. 2012;160:84–94. doi: 10.1016/j.trsl.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM. Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci. 2004;24:6410–6415. doi: 10.1523/JNEUROSCI.1421-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Lu J, Xiao W. Blockage of TNF-alpha by infliximab reduces CCL2 and CCR2 levels in patients with rheumatoid arthritis. J Investig Med. 2011;59:961–963. doi: 10.2310/JIM.0b013e31821c0242. [DOI] [PubMed] [Google Scholar]
- Zakharova M, Ziegler HK. Paradoxical anti-inflammatory actions of TNF-alpha: inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J Immunol. 2005;175:5024–5033. doi: 10.4049/jimmunol.175.8.5024. [DOI] [PubMed] [Google Scholar]
- Zhang B, Hu J, Man C, Zhu S. Effect of intra-articular administration of interleukin 1 receptor antagonist on cartilage repair in temporomandibular joint. J Craniofac Surg. 2011a;22:711–714. doi: 10.1097/SCS.0b013e31820873c6. [DOI] [PubMed] [Google Scholar]
- Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain. 2011b;152:419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Yu J, Lou GD, Tang YY, Wang RR, Hou WW, Chen Z. Widespread pain sensitization after partial infraorbital nerve transection in MRL/MPJ mice. Pain. 2016;157:740–749. doi: 10.1097/j.pain.0000000000000432. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ten of the tested 40 cytokines showed similar response profiles in both TNFR1/R2−/− and WT mice. Cytokines were elevated on days 1 and 14 post CFA injection and returned to baseline levels at the 18 week experimental end time point.
Mechanical withdrawal threshold of TNFR1/R2−/− mice on the contralateral side was not affected by etanercept and remained similar to baseline and levels observed in the WT mice.