

Abstract
Mitochondrial dynamics is a conserved process by which mitochondria undergo repeated cycles of fusion and fission, leading to exchange of mitochondrial genetic content, ions, metabolites, and proteins. Here, we examine the role of the mitochondrial fusion protein optic atrophy 1 (OPA1) in differentiated skeletal muscle by reducing OPA1 gene expression in an inducible manner. OPA1 deficiency in young mice results in non‐lethal progressive mitochondrial dysfunction and loss of muscle mass. Mutant mice are resistant to age‐ and diet‐induced weight gain and insulin resistance, by mechanisms that involve activation of ER stress and secretion of fibroblast growth factor 21 (FGF21) from skeletal muscle, resulting in increased metabolic rates and improved whole‐body insulin sensitivity. OPA1‐elicited mitochondrial dysfunction activates an integrated stress response that locally induces muscle atrophy, but via secretion of FGF21 acts distally to modulate whole‐body metabolism.
Keywords: ER stress, FGF21, mitochondrial dysfunction, OPA1, skeletal muscle
Subject Categories: Ageing, Metabolism, Molecular Biology of Disease
Introduction
Mitochondria are highly dynamic organelles, which experience cycles of fusion and fission, a process known as mitochondrial dynamics. Mitochondrial dynamics regulate various cellular processes, including organelle distribution during cell proliferation, bioenergetics, mitochondrial calcium flux, and autophagy (Dorn & Kitsis, 2015). Fission is regulated by dynamin‐related protein 1 (DRP1) and by its partners fission 1 (FIS1), mitochondrial fission factor (MFF), and mitochondrial division (MID). Mitochondrial fusion is regulated by a family of outer mitochondrial membrane GTPases mitofusin 1 and mitofusin 2 (Mfn1 and Mfn2) and optic atrophy 1 (OPA1), which resides on the inner mitochondrial membrane (Kasahara & Scorrano, 2014). In addition to, and independently from its role in mitochondrial fusion, OPA1 regulates apoptotic cristae remodeling and maintenance (Frezza et al, 2006). Moreover, OPA1, by maintaining mitochondrial cristae morphology, has a direct metabolic effect, stabilizing respiratory chain supercomplexes. Indeed, mild Opa1 overexpression corrects a model of Cox15 skeletal muscle‐specific knockout (Civiletto et al, 2015) and protects from acute muscle atrophy induced by denervation, by blunting mitochondrial dysfunction and expression of MuRF1 (Varanita et al, 2015).
Insulin resistance is oftentimes associated with mitochondrial dysfunction. Mechanisms include transcriptional repression of mitochondrial genes, lipotoxicity, and direct effects of insulin resistance (Montgomery & Turner, 2015). However, in skeletal muscle, mitochondrial dysfunction can occur without insulin resistance and conversely, insulin resistance has been demonstrated in the absence of mitochondrial dysfunction (Pagel‐Langenickel et al, 2010). An additional paradox is the observation that mutations that limit mitochondrial function in skeletal muscle may lead to a striking improvement in insulin sensitivity and protection against diet‐induced obesity and insulin resistance (Pospisilik et al, 2007; Kim et al, 2013). These adaptations that develop in response to mitochondrial stress exemplify the phenomenon of hormesis (Pulliam et al, 2013). Pathways implicated in this process include reactive oxygen species (ROS; Ristow & Schmeisser, 2014), AMPK activation (Sharma, 2015), and activation of ER stress pathways (Kim et al, 2013). Studies also suggest that such adaptations may involve secretion of myokines, including IL‐6 (Munoz‐Canoves et al, 2013), Fndc5/Irisin (Bostrom et al, 2012), and, more recently, fibroblast growth factor 21 (FGF21) (Kim et al, 2013), a hormone that is expressed in multiple tissues and functions physiologically to maintain energy homeostasis, by increasing energy expenditure and insulin sensitivity (Potthoff & Finck, 2014).
Recent studies have suggested that perturbations in mitochondrial dynamics may contribute to the pathophysiology of insulin resistance (Jheng et al, 2012; Quiros et al, 2012). Reduced levels of Opa1 and Mfn2 have been described in humans with insulin resistance (elderly subjects and patients with type 2 diabetes) and in obese primates (Liesa et al, 2009). Moreover, insulin resistance might be exacerbated by mutations in or altered expression of mitochondrial dynamics proteins that impair fusion or promote mitochondrial fission (Civitarese et al, 2010; Zorzano et al, 2010; Sebastian et al, 2012). We have previously reported that exposure of cardiac and skeletal muscle cells (in vivo or in vitro) to physiological hyperinsulinemia increases OPA1 mRNA and protein, promoting mitochondrial fusion in concert with increased mitochondrial oxygen consumption and ATP generation (Parra et al, 2014). These studies underscore the complex interactions between mitochondrial dynamics, metabolic homeostasis, and insulin resistance, but leave unresolved a precise understanding of the tissue‐specific role of OPA1 in muscle, to regulate insulin sensitivity in vivo.
In the present study, we fill an important gap in knowledge regarding the interaction of insulin sensitivity, OPA1 function in skeletal muscle, mitochondrial bioenergetics, and in vivo metabolic homeostasis. Our data reveal that OPA1 deficiency in muscle results in progressive non‐lethal mitochondrial dysfunction and reduced ATP synthesis. Locally, OPA1‐elicited mitochondrial dysfunction was accompanied by increased MuRF1 protein levels and modest muscle atrophy; however, via mechanisms that include ER stress activation, OPA1‐deficient muscle induces Fgf21 expression and secretion, thereby counteracting age‐ and diet‐induced obesity and insulin resistance.
Results
OPA1 deficiency disrupts mitochondrial cristae structure and progressively impairs mitochondrial respiratory capacity
To understand the role of OPA1 in skeletal muscle fibers, we generated mice that were homozygous for a floxed OPA1 allele and also expressed a tamoxifen‐inducible Cre transgene (Cre‐ERT2) under the control of the human alpha‐skeletal actin (HSA) promoter. To excise the floxed OPA1 alleles, we treated the mice with tamoxifen at 4 weeks of age, thereby generating muscle‐specific OPA1 hypomorph mice (mOPA1 KO mice). Relative to WT mice (mice lacking the Cre‐ERT2), mOPA1 KO mice have a 70% lower level of OPA1 protein in gastrocnemius muscle (Fig 1A) and about 50% lower level in soleus muscle (Fig EV1A). OPA1 deficiency resulted in impaired mitochondria cristae structure in soleus muscle, characterized by loss of cristae content and abnormal vacuolated ultrastructure (Fig 1B). Various indices of mitochondrial capacity such as BN‐PAGE gel quantification of mitochondrial OXPHOS proteins (Fig 1C), citrate synthase activity (Fig 1D), and mtDNA copy number (Fig 1E) were unchanged between WT and KO mice. Additionally, mRNA expression of genes involved in mitochondrial biogenesis, namely Pgc‐1α and Tfam, was equivalent between genotypes at 12 weeks of age (Fig 1F), suggesting maintenance of mitochondrial number, despite morphological changes. Interestingly, OPA1 deficiency induced protein levels of the outer mitochondrial membrane fusion proteins Mfn1 and Mfn2 (Fig EV1B). To investigate the effects of reduced OPA1 levels on mitochondrial respiratory capacity, we measured ADP‐driven mitochondrial oxygen consumption (v‐ADP) and ATP synthesis rates in the presence of succinate and rotenone in permeabilized soleus fibers at three different time points (12, 20 and 40 weeks of age). Oxygen consumption was impaired starting at 20 weeks of age (Fig 1G) and was further reduced at 40 weeks of age (Fig EV1C) in mOPA1 KO mice relative to WT mice. ATP synthesis rates were reduced as early as 12 weeks and persisted at later time points (Figs 1H and EV1D). ATP/O ratios were decreased at the age of 12 weeks (Data not shown), which could reflect mitochondrial uncoupling, but differences in ATP/O were not seen in older animals. Consistently, oxygen consumption was also mildly but significantly reduced in the presence of various substrates in permeabilized white and red gastrocnemius muscle fibers obtained from 20‐week‐old OPA1 KO mice relative to WT mice. Palmitoyl‐carnitine/malate‐supported respirations were unchanged in red gastrocnemius, but were reduced in white gastrocnemius of KO mice. Conversely, succinate‐supported respirations were reduced in red gastrocnemius muscle but were unchanged in white gastrocnemius of KO mice. Pyruvate/malate and glutamate/malate‐supported respirations were reduced in both muscle types (Fig 1I and J). These data underscore a critical role for OPA1 in mitochondrial energetics.
Figure 1. OPA1 deficiency disrupts mitochondrial cristae structure and progressively impairs mitochondrial respiratory capacity.
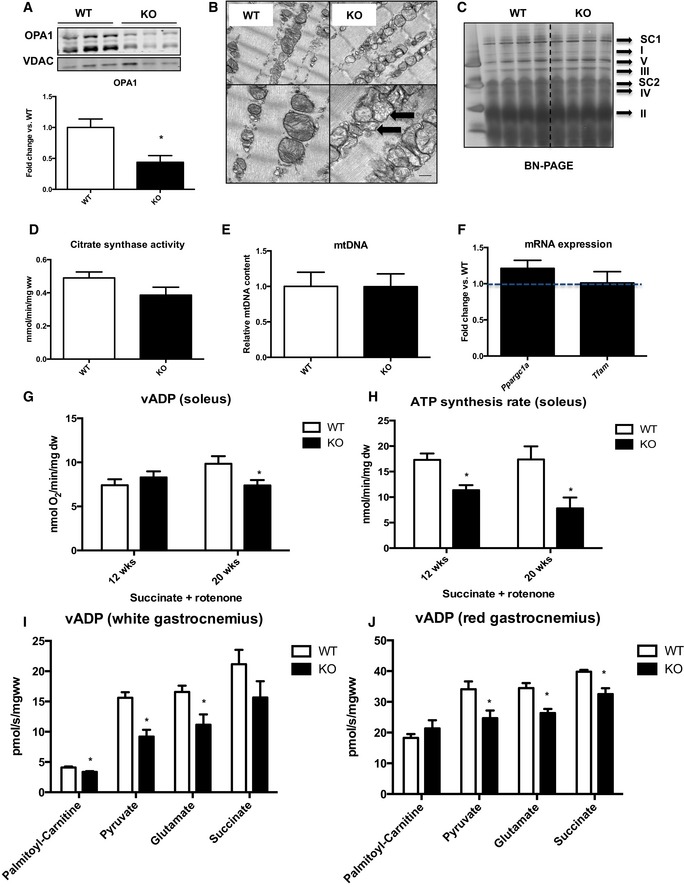
- Representative immunoblot of OPA1 in gastrocnemius muscle from 12‐week‐old WT and mOPA1 KO (KO) mice and densitometric analysis of OPA1 normalized by VDAC (n = 6).
- Electron micrographs from soleus muscle of 12‐week‐old mice (arrows highlight mitochondria with abnormal cristae structure) (n = 4). Scale bar = 0.32 μm.
- BN‐PAGE in gastrocnemius muscle of 12‐week‐old mice (n = 4). I‐V, mitochondrial OXPHOS complexes I‐V; SC1, super complex 1; SC2 super complex 2.
- Citrate synthase activity in gastrocnemius muscle of 12‐week‐old mice (n = 6).
- mtDNA copy number in gastrocnemius muscle of 12‐week‐old mice (n = 6).
- mRNA expression of mitochondrial biogenesis genes (n = 4–6).
- Maximally stimulated succinate‐supported mitochondrial respirations in soleus from 12‐ and 20‐week‐old mice (n = 4–7).
- Succinate‐supported ATP synthesis rates in soleus from 12‐ and 20‐week‐old mice (n = 3–6).
- Maximally stimulated mitochondrial respirations in white gastrocnemius muscle from 20‐week‐old mice (n = 3–4).
- Maximally stimulated mitochondrial respirations in red gastrocnemius muscle from 20‐week‐old mice (n = 3–4).
Figure EV1. Mitochondrial dynamics proteins and mitochondrial oxygen consumption.
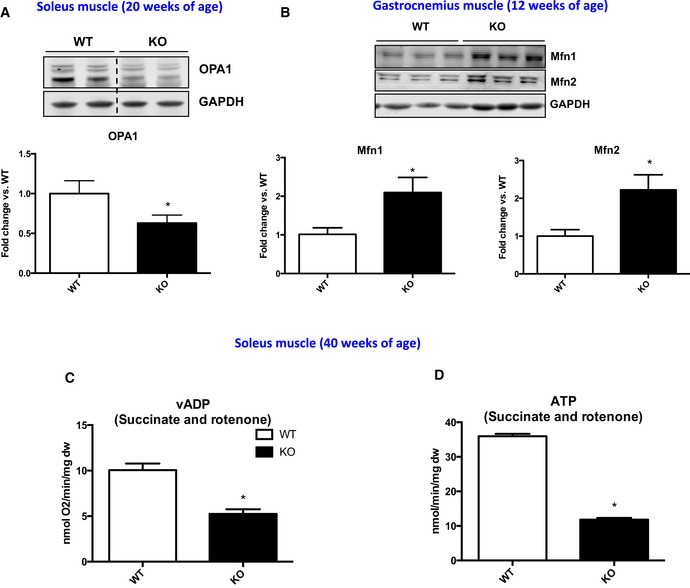
-
A, BRepresentative immunoblots and densitometric quantification in muscle homogenates from WT and mOPA1 KO mice. (A) OPA1 levels in soleus muscle normalized to GAPDH (n = 3). (B) Mfn1 and Mfn2 normalized to GAPDH (n = 4–5).
-
CMaximally stimulated succinate‐supported mitochondrial respirations in soleus from 40‐week‐old WT and mOPA1 KO (KO) mice (n = 5).
-
DSuccinate‐supported ATP synthesis rates in soleus from 40‐week‐old WT and mOPA1 KO (KO) mice (n = 5).
tPData information: Data are expressed as means ± SEM. Significant differences were determined by Student's t‐test, using a significance level of P < 0.05. (*) Significantly different vs. WT mice. Source data are available online for this figure.
Reduced OPA1 levels in muscle attenuate age‐induced weight gain and glucose intolerance and induce muscle atrophy
As early as 12 weeks of age, a small but consistent reduction in body weight was observed in mOPA1 KO mice (Fig 2A). To determine whether this decrease in body weight was attributable to loss of fat or lean mass, nuclear magnetic resonance (NMR) was performed. Interestingly, at 12 weeks, fat mass was unchanged between WT and mOPA1 KO mice (Fig 2B), however, total lean mass (Fig 2C) was significantly reduced in mOPA1 KO mice. Weight gain was dramatically attenuated over time (Fig 2A), which was accompanied by reduced fat mass starting at 20 weeks of age (Fig 2B), and reduced lean mass (Fig 2C). To determine whether the reduction in muscle mass was due to reduced fiber diameter, cross sections of gastrocnemius and soleus muscles were analyzed after wheat‐germ agglutinin staining. This analysis revealed reduced muscle fiber diameter at 20 weeks of age in mOPA1 KO mice relative to their WT littermate controls (Figs 2D and E, and EV2A and B), and the number of myocytes per area was increased in KO mice relative to WT (Fig 2F). Protein levels of the E3 ubiquitin ligase MuRF1 were significantly increased in mOPA1 KO mice relative to WT mice (Fig 2G and H), and mRNA expression of the E3 ubiquitin ligase MAF‐bx/Atrogin‐1 was also elevated in gastrocnemius muscle of mOPA1 KO mice, consistent with muscle atrophy (Fig 2I). Because muscle mass was reduced in mOPA1 KO mice, overall muscle health was assessed. Surprisingly, exhaustion tests revealed that 20‐week‐old mOPA1 KO mice reached comparable distances as WT mice; however, at 40 weeks, KO mice ran significantly more than WT mice before reaching exhaustion (Fig EV2C). Blood lactate levels under random‐fed conditions were elevated to the same extent at the end of the test, indicating equivalent exhaustion levels were achieved (Fig EV2D). To account for body weight differences between WT and mOPA1 KO mice, especially at 40 weeks of age, vertical work was calculated. Although 40‐week‐old mOPA1 KO mice ran longer, the work performed was the same between WT and mOPA1 KO mice at both ages (Fig EV2E). Additionally, grip strength was significantly reduced in mOPA1 KO mice relative to WT mice at 40 weeks of age (Fig EV2F).
Figure 2. Reduced OPA1 levels in muscle attenuates age‐induced weight gain and glucose intolerance and induces muscle atrophy.
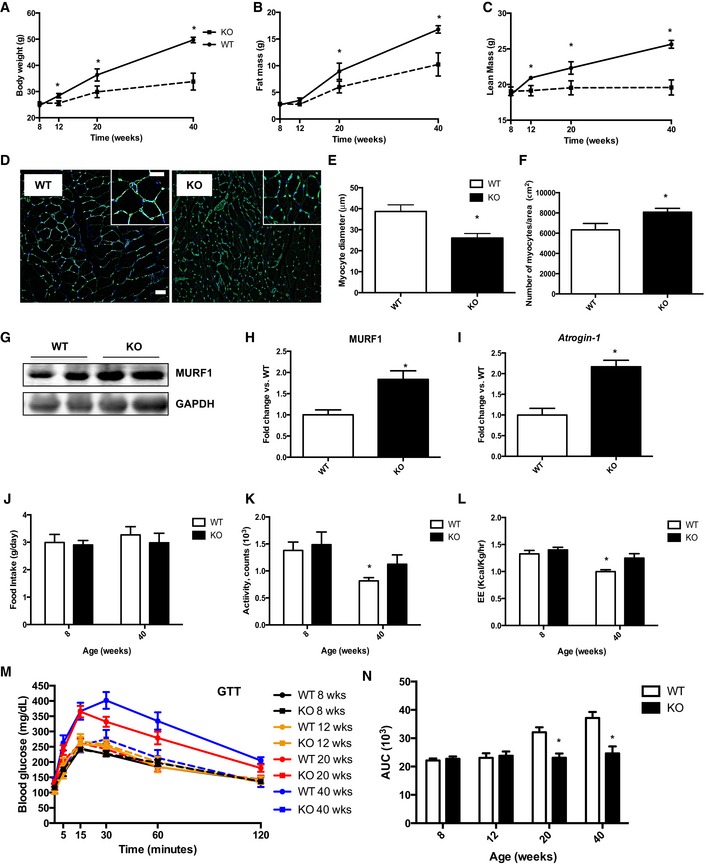
-
ABody weight over time (n = 8–13).
-
BTotal fat mass over time (n = 8–13).
-
CTotal lean mass over time (n = 8–13).
-
DCross sections of gastrocnemius muscle from 20‐week‐old mice stained with wheat‐germ agglutinin (WGA). Scale bar = 20 μm (main images) and 40 μm (magnification inserts).
-
EMeasurements of muscle fiber diameter in gastrocnemius muscle from 20‐week‐old mice (n = 3–5).
-
FMeasurements of the number of myocyte/area (n = 3–5).
-
G, H(G) Western blot analysis and (H) densitometric quantification of MuRF1 and GAPDH in gastrocnemius muscle from 40‐week‐old mice. Densitometric quantification is relative to GAPDH (n = 5).
-
ImRNA expression of Atrogin‐1 in gastrocnemius muscle of 20‐week‐old mice (n = 5). Data are represented as fold change vs. WT mice.
-
J–LCLAMS data in 8‐ and 40‐week‐old WT and mOPA1 KO (KO) mice. (J) Daily food intake (n = 5–9). (K) Activity levels (n = 6–9). (L) Energy expenditure (n = 6–9).
-
MGlucose tolerance tests (n = 7–11).
-
NArea under the curve (AUC) for glucose tolerance tests (n = 7–11).
Figure EV2. Assessment of overall muscle health of mOPA1 KO mice.
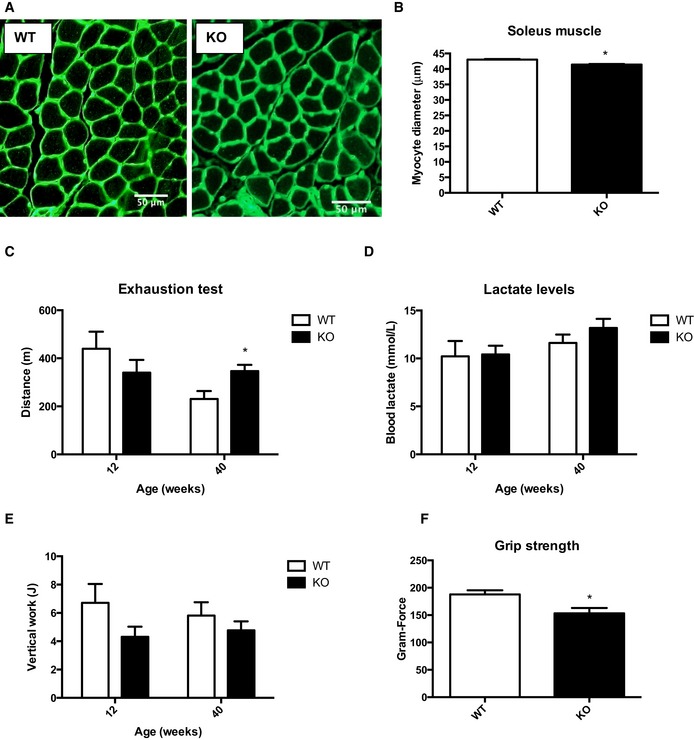
- Cross sections of soleus muscle from 20‐week‐old mice stained with wheat‐germ agglutinin (WGA). Scale bar = 50 μm.
- Measurements of muscle fiber diameter in soleus muscle from 20‐week‐old mice (n = 4).
- Exhaustion test in 20‐ and 40‐week‐old WT and mOPA1 KO mice (n = 4–9).
- Blood lactate after the exhaustion test (n = 4–9).
- Measurements of vertical work performed by 20‐ and 40‐week‐old mice during the exhaustion test (n = 4–9).
- Measurements of grip strength in 40‐week‐old mice (n = 5).
tPData information: Data are expressed as means ± SEM. Significant differences were determined by Student's t‐test, using a significance level of P < 0.05. (*) Significantly different vs. WT mice. Source data are available online for this figure.
To investigate the mechanisms for reduced body weight and fat mass, animals were placed in metabolic chambers, before body weight differences were observed (8 weeks of age) and at 40 weeks of age. No differences in food consumption or activity levels were observed between WT and mOPA1 KO mice at 8 or 40 weeks of age (Fig 2J and K). Activity levels and energy expenditure declined with age in WT mice, but were preserved in mOPA1 KO mice (Fig 2K and L), which could have contributed to the slower weight gain rate observed in these mice. Reduced body weight in mOPA1 KO mice was accompanied by improved whole‐body glucose homeostasis. Glucose tolerance tests demonstrated that the age‐induced impairment in glucose tolerance observed in WT mice at 20 and 40 weeks of age was completely prevented in mOPA1 KO mice (Fig 2M and N).
Muscle‐specific OPA1 deficiency prevents diet‐induced obesity and insulin resistance and increases circulating FGF21
To test the response of mOPA1 KO mice to a metabolic challenge, mice were fed a high‐fat diet (HFD) for 12 weeks. Consistent with findings on normal chow, weight gain was prevented in high‐fat‐fed mOPA1 KO mice (Fig 3A). The increase in fat mass observed in WT mice after 12 weeks of HFD was completely blunted in mOPA1 KO mice (Fig 3B). Total lean mass was equivalently reduced in mOPA1 KO mice regardless of the diet (Fig 3C). Therefore, reduced body weight in high‐fat‐fed mOPA1 KO mice can be attributed to both reduced fat mass and lean mass. To investigate the basis for this reduction in body weight, HF‐fed WT and mOPA1 KO mice were placed in metabolic chambers. Although food consumption (Fig 3D) and activity levels (Fig 3E) were unchanged between WT and mOPA1 KO mice, energy expenditure was significantly increased in mOPA1 KO mice (Fig 3F). In addition, HFD fed mOPA1 KO mice showed improved glucose tolerance (Fig 3G and H), decreased fasting insulin levels (Fig 3I), and improved insulin sensitivity compared to WT controls (Fig 3J and K). Taken together, these data indicate that mOPA1 KO mice have significantly increased insulin sensitivity and improved glucose homeostasis in response to a HFD challenge. Diet‐induced hepatic steatosis and increased serum concentrations of triglycerides were also prevented in mOPA1 KO mice (Fig EV3A and B). A brief survey for myokines in gastrocnemius muscle revealed that mRNA expression of IL‐6 and IL‐15, which have been reported to have distal effects to improve whole‐body metabolism (Ahima & Park, 2015), was unchanged between WT and mOPA1 KO mice (Fig EV3C). However, Fgf21 mRNA expression was dramatically induced in muscle of mOPA1 KO mice (Fig 3L), but not in liver (Fig 3M) or white adipose tissue (WAT; Fig 3N), organs that have been previously shown to produce FGF21 (Markan et al, 2014; Fasshauer & Bluher, 2015). Increased Fgf21 mRNA levels in gastrocnemius muscle correlated with an approximate eightfold increase in circulating levels of FGF21 in mOPA1 KO mice relative to WT controls (Fig 3O). FGF21 protein levels were also elevated in soleus muscle of 20‐week‐old mOPA1 KO mice (Fig EV3D). To explore potential mechanisms for FGF21‐mediated increase in energy expenditure, we examined markers of brown adipose tissue activation and beiging of white adipose tissue (WAT). UCP1 and PGC‐1α protein levels were unchanged in BAT of 20‐week‐old mOPA1 KO mice (Fig EV3E); however, UCP1 levels were elevated in inguinal fat (scWAT) (Fig EV3F), indicative of browning of white adipose tissue.
Figure 3. Muscle‐specific OPA1 deficiency prevents diet‐induced obesity and insulin resistance and increases circulating FGF21.
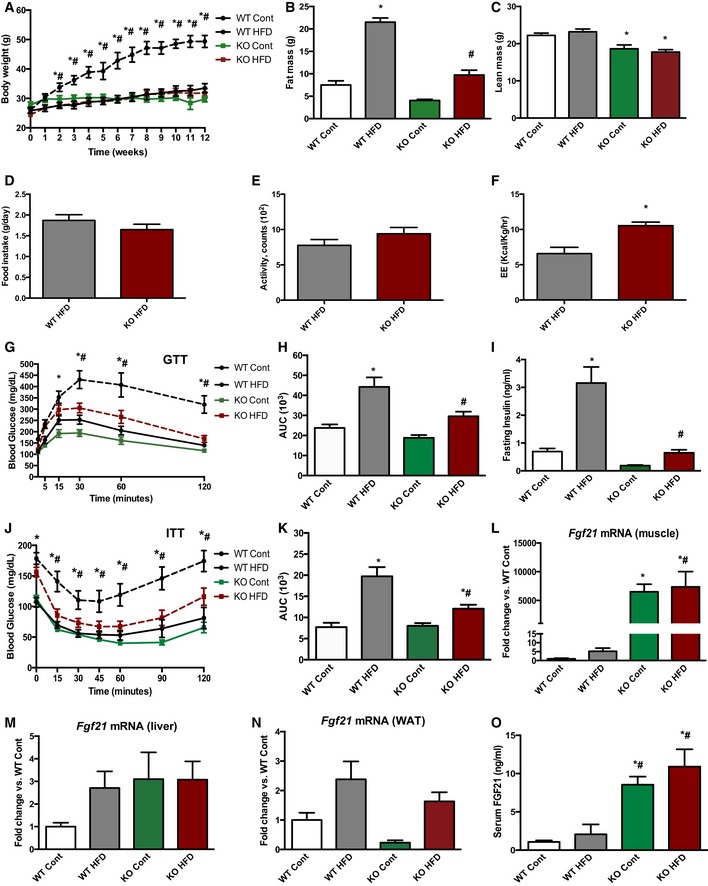
-
AWeekly body weight during 12 weeks of control or HFD feeding (n = 5–9).
-
BTotal fat mass after 12 weeks on control or HFD (n = 9–11).
-
CTotal lean mass after 12 weeks on control or HFD (n = 9–11).
-
D–FCLAMS data in high‐fat‐fed WT and mOPA1 KO (KO) mice at the end of 12 weeks of feeding. (D) Daily food intake (n = 5–7). (E) Activity level (n = 5–7). (F) Energy expenditure (n = 6–7).
-
GGlucose tolerance tests in control and high‐fat‐fed WT and mOPA1 KO (KO) mice at the end of 12 weeks of feeding (n = 5–9).
-
HArea under the curve (AUC) for glucose tolerance tests (n = 5–9).
-
IFasting insulin levels (n = 5–9).
-
JInsulin tolerance test in control and high‐fat‐fed WT and KO mice at the end of 12 weeks of feeding (n = 5–8).
-
KArea under the curve (AUC) for insulin tolerance tests (n = 5–8).
-
L–NFGF21 mRNA expression in 20‐week‐old mice at the end of 12 weeks of feeding. (L) Skeletal muscle (n = 3–5). (M) Liver (n = 4–5). (N) White adipose tissue (WAT) (n = 3–7).
-
OFGF21 plasma levels in control and high‐fat‐fed WT and KO mice at the end of 12 weeks of feeding (n = 3–6).
Figure EV3. Metabolic parameters in mOPA1 KO mice.
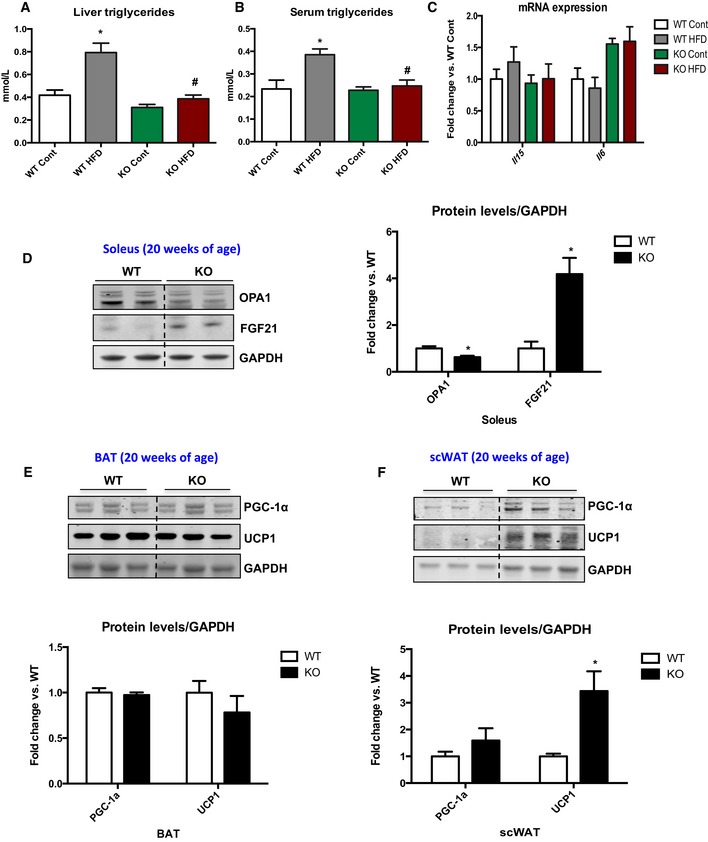
- Triglycerides levels in mice fed either a control or a HFD for 12 weeks (n = 5–6).
- Fasting triglyceride levels in the serum of mice fed either a control or a HFD for 12 weeks (n = 5–8).
- IL‐15 and IL‐6 mRNA expression in gastrocnemius muscle of 20‐week‐old WT and mOPA1 KO mice (n = 4–5).
- Representative immunoblot and densitometric quantification of OPA1 and FGF21 normalized to GAPDH in soleus muscle of 20‐week‐old mice (n = 3).
- Representative immunoblot and densitometric quantification of PGC‐1α and UCP1 normalized to GAPDH in BAT of 20‐week‐old mice (n = 4).
- Representative immunoblot and densitometric quantification of PGC‐1α and UCP1 normalized to GAPDH in sc‐WAT of 20‐week‐old mice (n = 3–4). Data are represented as fold change vs. WT mice.
tPPData information: Data are expressed as means ± SEM. Significant differences were determined by Student's t‐test (D, F), using a significance level of P < 0.05 or by ANOVA followed by Tukey multiple comparison test (A, B), using a significance level of P < 0.05. (*) Significantly different vs. WT mice. (*) vs. WT Cont (#) vs. WT HFD. Source data are available online for this figure.
Established diet‐induced obesity and insulin resistance are reversed by reducing OPA1 levels in muscle in parallel with increased FGF21
To test whether OPA1 deficiency could also ameliorate HFD‐induced weight gain and insulin resistance, WT and mOPA1 KO mice were fed either a control or a HFD for 8 weeks, prior to inducing OPA1 gene recombination. After 8 weeks of HF feeding, body weight was significantly increased (Fig 4A) and glucose tolerance was equivalently impaired (Fig 4C and D) in WT and mOPA1 KO mice. Mice were then administered tamoxifen and metabolically phenotyped. Five weeks after administration of tamoxifen, body weight progressively declined in mOPA1 KO mice (Fig 4A). At the end of 20 weeks of HF feeding, body weight was completely normalized in mOPA1 KO mice fed a HFD compared to WT mice fed the same diet (Fig 4B). Body weight was also reduced in mOPA1 KO mice fed a control diet relative to WT mice (Fig 4B). Glucose intolerance was completely reversed at the end of 20 weeks of HF feeding in mOPA1 KO mice fed a HFD, relative to WT mice (Fig 4E and F). These metabolic improvements correlated with elevated circulating levels of FGF21 (Fig 4G).
Figure 4. Reducing skeletal muscle OPA1 in mice with diet‐induced obesity and insulin resistance promotes weight loss and normalizes glucose tolerance in parallel with increased FGF21.
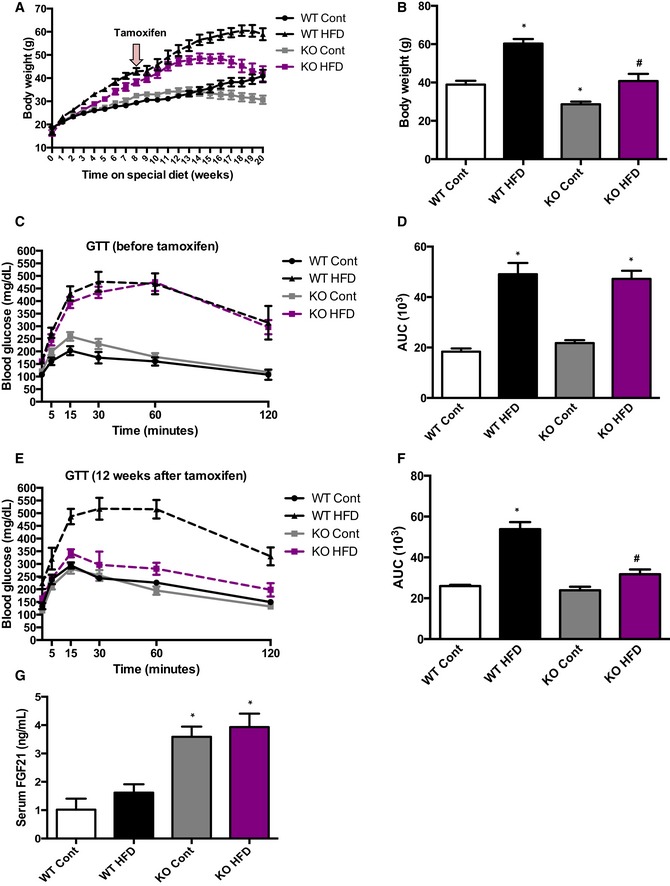
- Weekly body weight before and after tamoxifen injections (total of 20 weeks of control or HFD) (n = 6).
- Final body weight after 20 weeks of control or HFD (n = 6).
- Glucose tolerance tests in control and high‐fat‐fed WT and mOPA1 KO (KO) before tamoxifen injection (8 weeks of control or HFD) (n = 6–9).
- Area under the curve (AUC) for glucose tolerance tests (n = 6–9).
- Glucose tolerance tests in control and high‐fat‐fed WT and mOPA1 KO (KO) 12 weeks after tamoxifen injection (20 weeks total of control or HFD) (n = 5–8).
- Area under the curve (AUC) for glucose tolerance tests (n = 5–8).
- FGF21 plasma levels after 20 weeks of control or HFD (12 weeks after tamoxifen injections) (n = 6–8).
Muscle‐derived FGF21 plays an essential role in the favorable metabolic phenotype observed in OPA1‐deficient mice
Based on these data, we hypothesized that muscle‐derived FGF21 mediates the metabolic improvements observed in mOPA1 KO mice. To test this hypothesis, we generated muscle‐specific OPA1/FGF21 double knockout (DKO) mice and phenotyped these mice on a HFD. Consistent with our gene expression data, FGF21 protein levels were elevated in mOPA1 KO gastrocnemius muscle, but not in DKO mice (Fig 5A). Plasma FGF21 levels were also elevated in mice with reduced OPA1 expression in muscle, but were completely blunted in mOPA1/FGF21 DKO mice (Fig 5B), confirming that mice lacking the Fgf21 gene in muscle are unable to increase FGF21 circulating levels in response to OPA1‐elicited mitochondrial stress. OPA1 levels were reduced by approximately 50% in DKO mice relative to WT mice at 12 weeks of age (Fig EV4A). Similar to what was observed in mOPA1 KO mice, no differences were found in mitochondrial OXPHOS proteins by BN‐PAGE (Fig EV4B) or mtDNA copy number (Fig EV4C) in DKO mice relative to their WT littermate controls. Nonetheless, mitochondrial oxygen consumption (Fig EV4D) and ATP synthesis rates (Fig EV4E) in permeabilized soleus fibers were reduced in 20‐week‐old DKO mice. ADP‐supported mitochondrial respiration (Fig EV4F) and grip strength (Fig EV4G) were also significantly reduced in 40‐week‐old DKO mice. Although no significant differences were observed in body weight, total fat mass, and total lean mass in 12‐week‐old DKO mice compared to WT (Fig EV4H), myocyte diameter was significantly reduced in DKO mice, suggesting mild muscle atrophy (Fig EV4I and J). After DIO, NMR revealed that DKO and their WT control mice gained equivalent amounts of weight when fed a HFD (Fig 5C) and exhibited equivalent fat mass expansion (Fig 5D). Total lean mass was unchanged between WT and DKO regardless of the diet (Fig 5E). Food intake (Fig 5F), activity level (Fig 5G), and energy expenditure (Fig 5H) were all unchanged between WT and DKO mice. Glucose tolerance was equivalently impaired (Fig 5I and J), and fasting insulin levels were similarly increased (Fig 5K) in WT and DKO mice fed a HFD relative to mice fed a control diet. Insulin tolerance tests revealed that upon high‐fat feeding, both WT and DKO mice became equivalently insulin resistant relative to mice fed a control diet (Fig 5L and M). As expected, the rise in FGF21 circulating levels observed in mOPA1 KO mice was entirely prevented in DKO mice (Fig 5N). These data confirm that muscle is the source for increased circulating FGF21 levels in mOPA1 KO mice and that muscle‐derived FGF21 prevents the diet‐induced obesity and insulin resistance observed in these mice.
Figure 5. Muscle‐derived FGF21 mediates the metabolic phenotype observed in OPA1‐deficient mice.
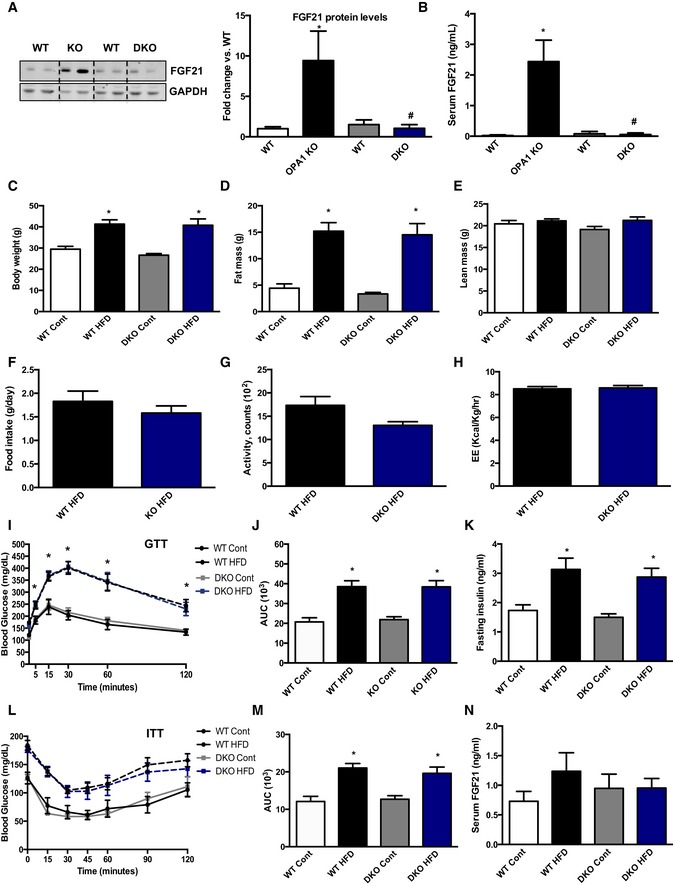
-
ARepresentative immunoblot of FGF21 in gastrocnemius muscle from 12‐week‐old mice and densitometric analysis of FGF21 normalized by GAPDH (n = 4).
-
BFGF21 serum levels in 12‐week‐old mOPA1 KO (KO), DKO mice, and their respective WT controls (WT) (n = 5).
-
C–NMetabolic data in DKO mice and their respective WT controls after 10 weeks of control or HFD. (C) Final body weight (n = 5–9). (D) Total fat mass (n = 5–9). (E) Total lean mass (n = 5–9). (F) Daily food intake (n = 4). (G) Activity level (n = 4). (H) Energy expenditure (n = 4). (I) Glucose tolerance tests (n = 9–16). (J) Area under the curve (AUC) for the GTT (n = 9–16). (K) Fasting insulin levels (n = 5–11). (L) Insulin tolerance test (n = 7–10). (M) Area under the curve (AUC) for the ITT (n = 7–10). (N) FGF21 plasma levels (n = 5–10).
Figure EV4. Assessment of overall mitochondrial function and muscle health in mOPA1/FGF21 DKO mice.
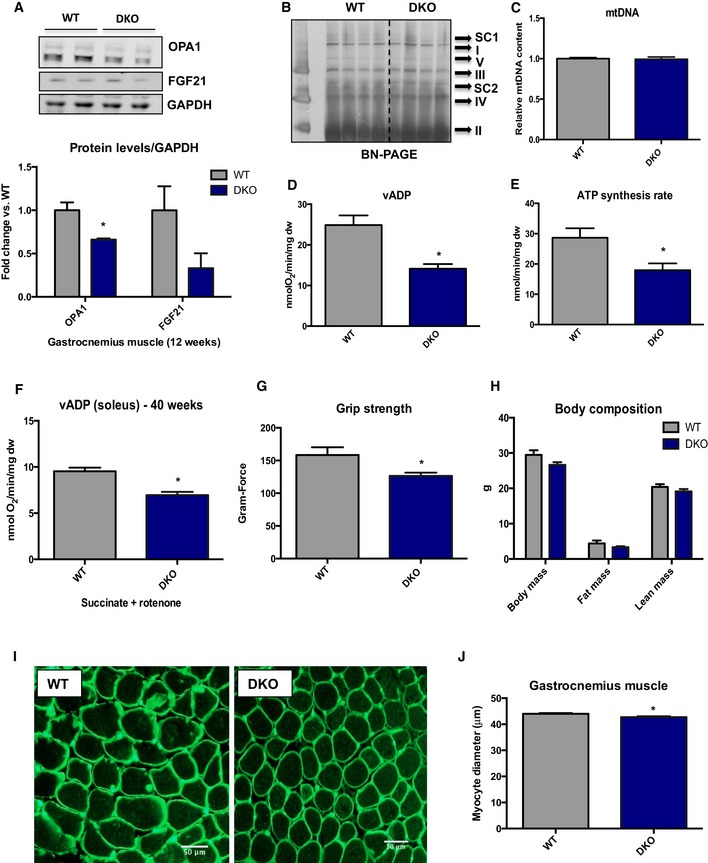
- Representative immunoblots and densitometric quantification of OPA1 normalized to GAPDH in gastrocnemius muscle from 12‐week‐old WT and mOPA1/FGF21 KO mice (n = 3).
- BN‐PAGE in gastrocnemius muscle of 12‐week‐old mice (n = 4).
- mtDNA copy number in gastrocnemius muscle of 12‐week‐old mice (n = 3–4).
- Maximally stimulated succinate‐supported mitochondrial respirations in soleus from 20‐week‐old mice (n = 4–6).
- Succinate‐supported ATP synthesis rates in soleus from 20‐week‐old mice (n = 4–5).
- Maximally stimulated succinate‐supported mitochondrial respirations in soleus from 40‐week‐old mice (n = 5).
- Measurements of grip strength in 40‐week‐old mice (n = 3).
- Body weight and body composition in 20‐week‐old WT and DKO mice (n = 5–9).
- Cross sections of gastrocnemius muscle from 20‐week‐old mice stained with wheat‐germ agglutinin (WGA). Scale bar = 50 μm.
- Measurements of muscle fiber diameter in gastrocnemius muscle from 20‐week‐old mice (n = 3).
tPData information: Data are expressed as means ± SEM. Significant differences were determined by Student's t‐test, using a significance level of P < 0.05. (*) Significantly different vs. WT mice. Source data are available online for this figure.
Activation of ER stress plays a significant role on FGF21 induction in OPA1‐deficient muscle cells
To identify potential mechanisms involved in FGF21 induction, pathways previously implicated in Fgf21 mRNA induction in muscle were evaluated. Measurements of 4‐hydroxynonenal, nitrotyrosine, and MnSOD, markers of oxidative stress, were unchanged between WT and mOPA1 KO mice at 12 weeks of age (Fig 6A and B). Conversely, AMP‐dependent kinase (AMPK) activation, which is normally induced during bioenergetic stress, was significantly increased in mOPA1 KO mice (Fig 6A and B) compared to WT mice. Markers of ER stress activation, such as increased phosphorylation of eIF2α, augmented BiP protein levels (Fig 6A and B), and increased mRNA expression of Atf4, Chop, BiP, and spliced Xbp1 (Fig 6A–C) were all observed in mOPA1 KO mice relative to WT mice. Insulin‐mediated activation of AKT was equivalent in gastrocnemius muscle of WT and mOPA1 KO mice at 12 weeks of age (Fig 6D and E), whereas insulin‐stimulated phosphorylation of S6, downstream of mTORC1, was significantly increased in KO mice relative to WT (Fig 6D and E). Because ER stress markers were robustly induced in our model, we sought to determine whether attenuation of ER stress could prevent the rise in FGF21 in mOPA1 KO mice. Four weeks after tamoxifen injections, mice were treated either with PBS or with tauroursodeoxycholic acid (TUDCA), a known ER chaperone, for 4 weeks (12 weeks of age). TUDCA treatment attenuated the induction of FGF21 in muscle of mOPA1 KO mice, concurrent with reduced activation of ER stress, as demonstrated by decreased BiP protein levels (Fig 6F and G). The rise in FGF21 circulating levels in mOPA1 KO mice was also prevented after TUDCA treatment (Fig 6H). Similar to earlier observations (Fig 2M), 4 weeks of OPA1 deletion was not associated with a change in glucose tolerance or significant changes in body composition (Fig 6I). At the end of TUDCA treatment, no changes in body weight, body composition (Fig 6I), or glucose tolerance (Fig 6J) were observed between WT and mOPA1 KO mice.
Figure 6. ER stress activation is necessary for FGF21 induction in OPA1‐deficient muscle.
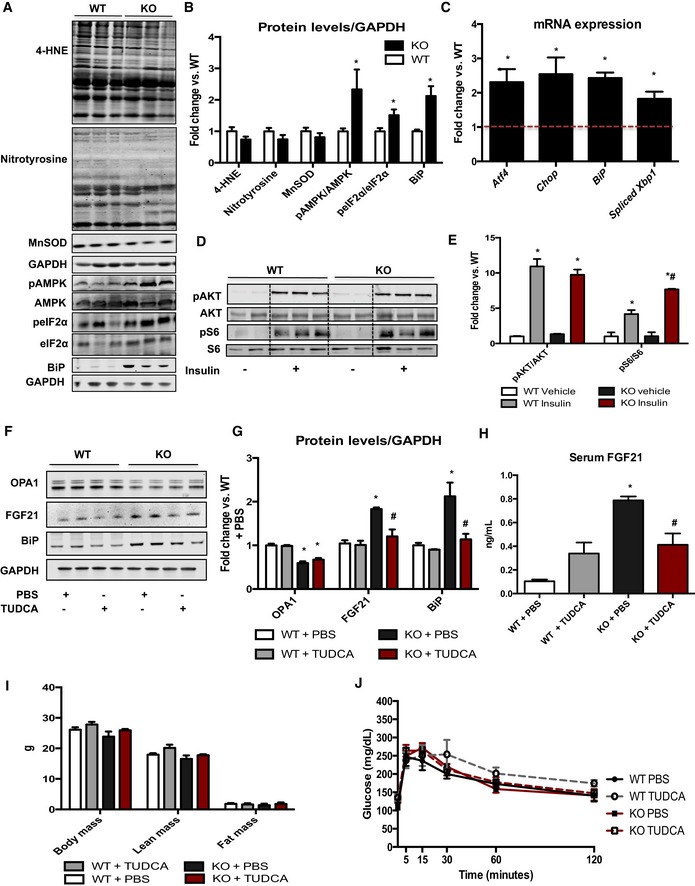
- Representative immunoblots in gastrocnemius muscle from 12‐week‐old WT or mOPA1 KO (KO) mice.
- Densitometric analysis of immunoblots of 4‐hydroxynonenal (4‐HNE), nitrotyrosine, MnSOD (n = 5), and BiP (n = 3) normalized to GAPDH protein levels (n = 5) and of pAMPK/AMPK (n = 5) and peIF2α/eIF2α ratios (n = 5).
- mRNA expression of ER stress genes in gastrocnemius muscle from 20‐week‐old WT or mOPA1 KO (KO) mice (data are expressed as fold change vs. WT mice, represented as the dashed line) (n = 3–5).
- AKT phosphorylation at Ser 473 and S6 phosphorylation at Ser235/236, 30 min after an i.p. injection of either saline or insulin, normalized by total AKT and total S6 levels, respectively.
- Densitometric analysis of pAKT/AKT and pS6/S6 immunoblots (n = 3).
- Representative immunoblots in soleus muscle from 12‐week‐old WT or mOPA1 KO (KO) mice treated with TUDCA for 4 weeks.
- Densitometric analysis of immunoblots of OPA1, FGF21, and BiP normalized to GAPDH protein levels (n = 4).
- FGF21 serum levels in 12‐week‐old mOPA1 KO mice and their respective WT controls 4 weeks after TUDCA treatment (n = 3–4).
- Body weight and body composition in 12‐week‐old mice 4 weeks after TUDCA treatment (n = 3–5).
- Glucose tolerance test in 12‐week‐old mice 4 weeks after TUDCA treatment (n = 4–6).
To further investigate the role of ER stress activation on FGF21 induction, we utilized an in vitro system. Primary satellite cells were obtained from OPA1 floxed mice. After differentiation into primary myotubes, cells were infected either with a GFP‐expressing adenovirus (WT) or with an adenovirus expressing a GFP‐tagged Cre recombinase (KO). Similar to in vivo observations, OPA1 deletion in vitro activated ER stress pathways and induced FGF21 protein levels in cells lysates (Fig 7A), and in the culture media (Fig 7B). Upon attenuation of ER stress with 4‐phenylbutyrate (PBA), a molecular chaperone, FGF21 levels were significantly reduced in OPA1 KO myotubes, and in the media (Fig 7A and B). Short‐term deletion of OPA1 in vitro also induced activation of AMPK, indicating energetic stress, which was not prevented with PBA treatment (Fig 7A). Basal and ATP‐linked mitochondrial respirations were reduced in OPA1 KO myotubes. Proton leak was also reduced in these cells, while maximally stimulated respirations and reserve capacity were unchanged between OPA1 KO and WT myotubes (Fig 7C). Conversely, glycolysis and glycolytic capacity were both increased in OPA1 KO myotubes upon a glycolysis stress test (Fig 7D). Mitochondrial membrane potential, as measured by TMRM staining, was more rapidly reduced in OPA1 KO myotubes in response to H2O2 treatment (Fig 7E and F). Studies in C2C12 myotubes corroborate the role of bioenergetic stress and ER stress on FGF21 induction. Myotubes were treated for 8 h with either oligomycin, an inhibitor of mitochondrial respiratory complex V, or tunicamycin, an ER stress inducer. Both treatments increased FGF21 levels in cell lysates as well in the media (Fig EV5A and B). Oligomycin treatment also induced the expression of Fgf21 and ER stress genes (Fig EV5C). Similar to findings in OPA1 KO myotubes, co‐treatment of C2C12 myotubes with PBA significantly reduced oligomycin‐ and tunicamycin‐mediated induction of FGF21 (Fig EV5A and B). Because AMPK activation was induced in OPA1 KO muscles in vivo and OPA1 KO myotubes, we tested whether agonist‐mediated activation of AMPK would be sufficient to induce Fgf21 mRNA expression. C2C12 myotubes were treated with 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) to induce phosphorylation and activation of AMPK (Fig EV5D); however, direct activation of AMPK failed to induce Fgf21 mRNA levels (Fig EV5E).
Figure 7. OPA1 deletion in primary myotubes confirms a role for OPA1‐induced mitochondrial dysfunction and ER stress on FGF21 secretion.
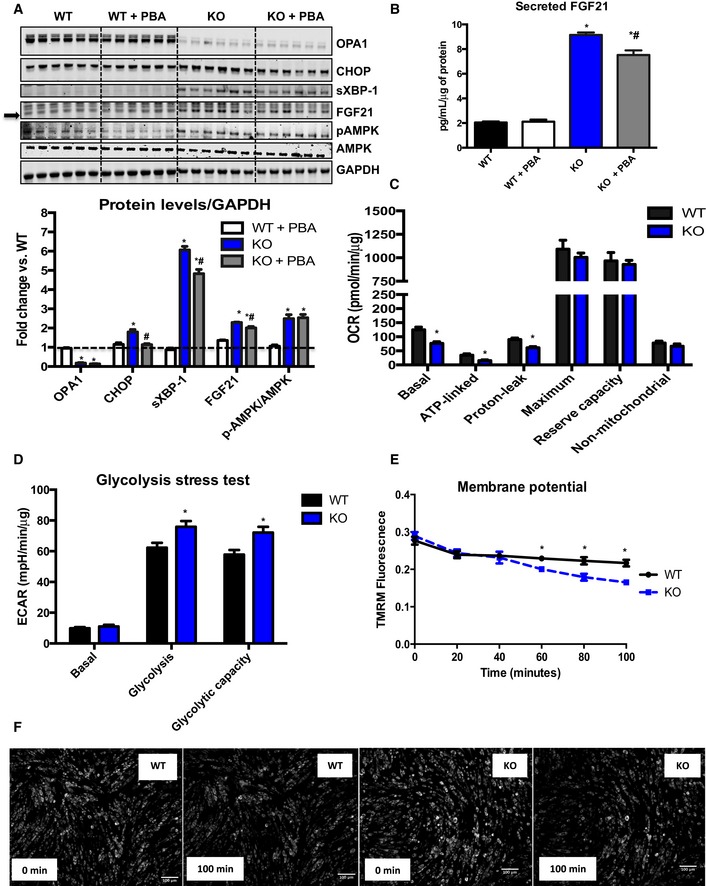
- Representative immunoblots in primary myotubes obtained from OPA1fl/fl mice and infected with Ad‐GFP or Ad‐Cre. Cells were treated with either vehicle (PBS) or PBA for 3 days. Densitometric analysis of immunoblots of OPA1, CHOP, XBP‐1, and FGF21 normalized to GAPDH protein levels and of pAMPK/AMPK ratios (n = 6).
- Secreted FGF21 levels measured in the media (n = 6).
- Measurement of oxygen consumption rates (OCR) in primary myotubes 3 days after adenoviral infection (n = 8–10 technical replicates).
- Measurements of extracellular acidification rates during a glycolysis stress test in primary myotubes 3 days after adenoviral infection (n = 8–10 technical replicates).
- TMRM fluorescence over time after treatment with H2O2 (n = 3 technical replicates).
- Representative pictures of primary myotubes stained with TMRM at times 0 (immediately after addition of H2O2) and 100 min after H2O2 treatment. Scale bar = 100 μm.
Figure EV5. Mitochondrial stress and ER stress in C2C12 myotubes.
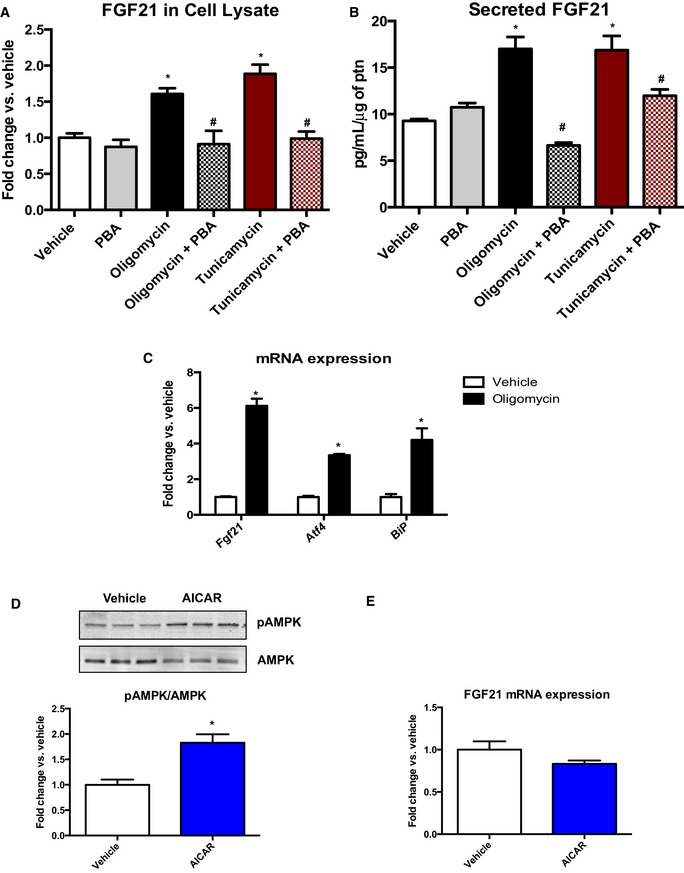
- FGF21 protein levels measured by immunoblot, in cell lysates from C2C12 myotubes treated either with vehicle, oligomycin, or tunicamycin ± PBA for 8 h. Data are represented as fold change vs. vehicle normalized to GAPDH (n = 4–8).
- FGF21 in the media secreted from C2C12 myotubes treated either with vehicle, oligomycin, or tunicamycin ± PBA for 8 h (n = 3).
- mRNA expression of Fgf21, Atf4, and BiP in C2C12 myotubes treated with vehicle or oligomycin for 8 h (data are expressed as fold change vs. vehicle) (n = 4–6).
- Western blot analysis and densitometric quantification of phosphorylated AMPK normalized to total AMPK in C2C12 cells treated with vehicle or AICAR for 12 h (n = 4).
- FGF21 mRNA expression in C2C12 cells treated with vehicle or AICAR for 12 h (n = 3–4). Data are represented as fold change vs. vehicle.
tPPData information: Data are expressed as means ± SEM. Significant differences were determined by Student's t‐test (C, D), using a significance level of P < 0.05 or by ANOVA followed by Tukey multiple comparison test (A, B), using a significance level of P < 0.05. (*) Significantly different vs. vehicle. (*) and (#) vs. oligomycin or tunicamycin. Source data are available online for this figure.
Discussion
Mitochondrial dynamics plays a crucial role in various cellular processes and is an essential mechanism for mitochondrial quality control (Dorn & Kitsis, 2015). Mild overexpression of the fusion protein OPA1 has recently been shown to ameliorate two models of mitochondrial disease (Civiletto et al, 2015) and to protect from acute muscle atrophy induced by denervation (Varanita et al, 2015). In addition, reduced OPA1 levels have been described in humans with insulin resistance and in obese primates (Liesa et al, 2009). Taking together, these data suggest maintaining OPA1 levels in muscle may be protective in these pathologies. Here, we investigated the role of OPA1 in skeletal muscle fibers under normo‐caloric conditions and in response to a high‐fat diet challenge, by reducing OPA1 levels specifically in skeletal muscle of young adult mice. We demonstrate that OPA1 is critical for normal mitochondrial morphology and function, with its deficiency precipitating progressive mitochondrial dysfunction in muscle in vivo and in vitro. OPA1‐deficient muscle cells also display glycolytic compensation and impaired ability to maintain mitochondrial membrane potential in vitro. Although, locally, OPA1 deficiency activates atrophy signaling pathways resulting in mild muscle loss as early as 8 weeks after recombination, OPA1‐elicited mitochondrial dysfunction induces an integrated stress response, leading to increased expression and secretion of FGF21 from muscle, which acts distally to improve whole‐body energy expenditure and insulin sensitivity.
Our findings underscore the importance of OPA1 for maintaining mitochondrial structure and function, and how its disruption was detrimental to overall muscle health. Muscle atrophy has been observed in other mouse models in which mitochondrial dynamics and function are disrupted, such as in the Mfn1/2 muscle‐specific double knockout mice (Chen et al, 2010) and in Atg7 muscle‐specific knockout mice (Kim et al, 2013). The severity of muscle atrophy in these models seems to correlate with the degree of mitochondrial dysfunction. Mitochondrial defects that have been associated with muscle wasting include decreased biogenesis, impaired protein folding, reduced mitophagy, increased fission (Russell et al, 2014), accumulation of mtDNA point mutations, and severe mtDNA depletion (Chen et al, 2010). Here, we report that mild mitochondrial dysfunction due to reduced levels of OPA1 elicits modest muscle atrophy, resulting in loss of total lean mass, and reduced muscle strength. Although the precise mechanisms for the decline in muscle mass were not investigated in the present study, we have observed that muscle atrophy in mOPA1 KO mice is accompanied by increased levels of MuRF1, an E3 ubiquitin ligase that promotes skeletal muscle atrophy during muscle denervation and certain other stress conditions (Bodine & Baehr, 2014). This finding, coupled with the previous finding that OPA1 overexpression reduces MuRF1 expression in denervated muscle (Varanita et al, 2015), suggests that OPA1 is necessary and sufficient to repress MuRF1 expression in skeletal muscle and supports the notion that MuRF1 may be a crucial mediator of skeletal muscle atrophy in mOPA1 KO mice. It is also unlikely that FGF21 induction drives the muscle atrophy observed, as muscle atrophy persisted when FGF21 expression was deleted in muscle of OPA1 KO mice. Another potential mediator of muscle atrophy in mOPA1 KO mice is ATF4, a bZIP transcription factor subunit that is sufficient to induce skeletal muscle fiber atrophy and is required for skeletal muscle atrophy during fasting, immobilization, and aging (Ebert et al, 2010, 2012, 2015; Bongers et al, 2013; Fox et al, 2014). Our data indicate that skeletal muscle atrophy in mOPA1 KO mice is associated with an increased level of Atf4 mRNA, as a result of ER stress, which could potentially promote muscle loss by elevating the level of ATF4 protein in skeletal muscle fibers. Interestingly, growth‐promoting pathways, such as insulin signaling through AKT leading to mTORC1 activation, were somewhat augmented in mOPA1 KO mice, which could be counteracting the effects secondary to activation of the canonical atrophy signaling pathways.
Surprisingly, mOPA1 KO mice were leaner and exhibited improved insulin sensitivity and glucose homeostasis in response to aging and DIO. Our data demonstrate that these effects are mediated by muscle‐derived FGF21, which is locally induced in response to OPA1 deficiency. Several myokines have been suggested to be secreted by skeletal muscle, which exert distal effects to improve whole‐body metabolism, those include IL‐15, IL‐6, and BNDF (Ahima & Park, 2015). Studies in animal models (Keipert et al, 2014; Kim et al, 2013; Tyynismaa et al, 2010) and in humans (Suomalainen et al, 2011) suggest that, specifically in the context of mitochondrial stress, FGF21 mRNA expression is induced in skeletal muscle, which also correlates with increased FGF21 circulating levels. Although skeletal muscle may be able to induce expression of FGF21 (Keipert et al, 2014; Guridi et al, 2015; Harris et al, 2015), to date, definitive data confirming that skeletal muscle is the specific origin of circulating FGF21 in mouse models of muscle mitochondrial stress were lacking. Here, we provide compelling evidence that skeletal muscle can secrete FGF21 in a cell autonomous manner, and this capacity is increased in response to OPA1 deficiency. These findings open numerous possibilities to study the role of muscle‐derived FGF21 in physiology and therapeutically. Of note, our data challenge a recent study, suggesting that FGF21 plays a negligible role in the whole body metabolic adaptations reported in mice with decreased muscle mitochondrial efficiency due to increased respiratory uncoupling (Ost et al, 2016). In contrast, we were able to completely reverse the whole body metabolic adaptations observed in OPA1‐deficient mice, by simultaneously deleting FGF21 from skeletal muscle, suggesting a clear role for muscle‐derived FGF21 in the metabolic improvements observed in our model. Differences in the source of mitochondrial stress, the temporal nature of our inducible OPA1 deletion, and the tissue specificity of FGF21 deletion in our model could underlie the differences between our data and that of the aforementioned study (Ost et al, 2016). Decreased ATP/O ratios in young mice suggest that mitochondrial uncoupling could exist in the OPA1‐deficient mice examined in this study, and we did not directly determine whether increased complex III‐generated superoxide could contribute to this. However, our in vivo and in vitro models suggest that additional mechanisms contribute to the bioenergetic failure following OPA1 reduction. These include impaired electron transport chain function, decreased ATP generation, and potentially impaired mitochondrial membrane potential, likely related to disrupted crista structure. These defects in aggregate may activate mechanisms that lead to induction of FGF21, in contrast to the scenario when mitochondrial uncoupling is the primary basis for mitochondrial energetic compromise.
The molecular mechanisms governing FGF21 regulation in skeletal muscle are incompletely understood. Studies have linked diverse signaling pathways with FGF21 induction in muscle; those include activation of AKT (Izumiya et al, 2008), ER stress, activation of ATF4 (Kim et al, 2013), oxidative stress (Ribas et al, 2014), and more recently activation of mTORC1 (Guridi et al, 2015). Our data indicate that AKT activation and induction of oxidative stress are probably unlikely factors contributing to FGF21 induction in our model. However, our findings reinforce the idea that activation of ER stress may be necessary and sufficient to induce FGF21 in muscle cells. OPA1‐induced mitochondrial dysfunction in mice lead to activation of eIF2α and induced several genes involved in the ER stress response. Activation of ER stress and induction of FGF21 were also observed in primary myotubes deficient for OPA1 and in C2C12 myotubes in which mitochondrial stress or ER stress was induced pharmacologically. Interestingly, pharmacological inhibition of ER stress with PBA in vitro and TUDCA in vivo partially prevented FGF21 induction, suggesting that ER stress is at least one of the mechanisms downstream of bioenergetic stress to induce FGF21 in muscle. We also examined the role of AMPK activation on FGF21 induction. Although AMPK is activated in both in vivo and in vitro in response to OPA1 deficiency, activation of AMPK with AICAR was insufficient to induce Fgf21 mRNA expression in C2C12 myotubes. Indeed, a recent publication excludes a role for AMPK on FGF21 induction in UCP1‐expressing muscle (Ost et al, 2014). Thus, our data suggest that OPA1‐elicited mitochondrial dysfunction in skeletal muscle activates an integrated stress response pathway, resulting in increased FGF21 expression and secretion, and reveals that activation of ER stress is sufficient and necessary to induce FGF21 expression in and secretion from muscle cells.
Together, these observations underscore a conserved mitochondrial stress pathway by which mitochondrial stress in skeletal muscle is communicated to the entire organism via a stress‐related endocrine pathway that is mediated via FGF21. It remains to be determined whether modest activation of this pathway, as seen in our model of hypomorphic OPA1 insufficiency in skeletal muscle, could be harnessed to combat the metabolic sequelae of obesity and insulin resistance. The specific mechanisms by which mitochondrial dysfunction activates ER stress and the molecular mediators linking ER stress and induction of FGF21 remain to be elucidated.
In conclusion, these studies support a conserved role for Opa1 in maintaining mitochondrial structure and function in differentiated skeletal muscle, and that mitochondrial dynamics may represent an important homeostatic mechanism to preserve mitochondrial bioenergetics in adult skeletal muscle. Moreover, although locally a modest reduction in OPA1 protein levels mediates mild muscle atrophy, mOPA1 KO mice have normal survival rates and are resistant to age‐ and diet‐induced obesity and insulin resistance via mechanisms that include local activation of ER stress pathways and secretion of FGF21 as a myokine. Therefore, mitochondrial dynamics may represent a critical mechanism by which skeletal muscle may regulate systemic metabolism via an FGF21‐mediated endocrine mechanism.
Materials and Methods
Animals and animal care
Animal work was performed in accordance with protocols approved by the University of Iowa Animal Care and Use Committee (IACUC). Unless otherwise stated, all experiments were performed in male mice on a C57Bl/6J background or that were backcrossed into the C57Bl/6J background for more than five generations. OPA1fl/fl mice were generated as previously described (Zhang et al, 2011) and were kindly provided by Dr. Sesaki. FGF21fl/fl mice were generated as previously described (Potthoff et al, 2009). Tamoxifen‐inducible HSA‐CreERT2 mice were a kind gift from Dr. Pierre Chambon, University of Strasbourg. For generation of mOPA1 KO mice and OPA1/FGF21 DKO mice, recombination was induced in 4‐week‐old mice via i.p. injections of tamoxifen (Sigma, St. Louis, MO, USA, T5648; 20 mg/kg) for 5 days. Mice were allowed 4 weeks to recover from tamoxifen injections, after which they were either kept on standard chow (2029X Harlan Teklad, Indianapolis, IN, USA) or were fed special diets. Mice were divided into a control‐diet group (Cont; 10% Kcal from fat—Research Diets, New Brunswick, NJ, USA, D12450J) or a high‐fat diet group (HFD; 60% Kcal from fat—Research Diets D12492) and were kept on these respective diets for 10–12 weeks (special diet was initiated when mice were 8 weeks of age). A subset of animals was fed the aforementioned special diets for 8 weeks, after which they were then injected with tamoxifen (20 mg/kg) for 5 days. To inhibit ER stress, a different cohort of mice was injected intraperitoneally with TUDCA (Millipore, Billerica, MA, USA; 250 mg/kg) once a day, five times a week for a total a 4 weeks. Controls for TUDCA received the same volume of vehicle (PBS) by intraperitoneal injection. Animals were housed at 22°C with a 12‐h light, 12‐h dark cycle with free access to water and standard chow.
Measurements of mitochondrial function
Soleus fibers were freshly excised and permeabilized with saponin. Respiration and ATP synthesis were measured using succinate (5 mM) with rotenone (10 mM). Soleus muscles were permeabilized for 30 min at 4°C in buffer A containing 50 μg/ml saponin and (in mmol/l) 7.23 K2EGTA, 2.77 K2CaEGTA, 6.56 MgCl2, 20 imidazole, 0.5 dithiothreitol, 53.3 K‐methans, 20 taurine, 5.3 Na2ATP, 15 PCr, and 3 KH2PO4, (pH 7.1 adjusted at 25°C). Next, fibers were washed twice for 10 min in buffer B containing (in mmol/l) 1.38 MgCl2, 20 imidazole, 0.5 dithiothreitol, 100 K‐methans, 20 taurine, 3 KH2PO4, 2 mg/ml BSA, 5 succinate, and 10 rotenone as substrate, (pH 7.1 adjusted at 25°C).
Mitochondrial oxygen consumption
The respiratory rates of soleus fibers were measured using an oxygen sensor probe (Ocean Optics, Dunedin, FL, USA) in 1 ml of KCl buffer at 25°C containing (in mmol/l) 125 KCl, 20 HEPES, 3 Mg‐acetate, 0.4 EGTA, 2 mg/ml BSA, 5 KH2PO4 and 0.3 dithiothreitol, 5 succinate, and 10 rotenone as substrate, (pH 7.1 adjusted at 25°C). Oxygen consumption was determined following ADP stimulation (1 mM; VADP). The solubility of oxygen in KCl buffer was 246.87 nmol of O2/ml. Oxygen consumption rates were expressed as nmol of O2 × min−1 × mg dry fiber weight−1.
Mitochondrial ATP production
For measurement of ATP production, ADP was added to 1 ml of buffer B to a final concentration of 1 mmol/l. Next, 10 μl buffer B from the respiration chamber was added to 190 μl DMSO on ice, every 10 s for a 1‐min time period. ATP production was determined by a bioluminescence assay based on the luciferin/luciferase reaction with the ATP assay kit (Promega Corporation, Madison, WI, USA; Boudina et al, 2012).
Mitochondrial respiration in permeabilized fibers from red and white gastrocnemius muscle was measured using the Oroboros O2K Oxygraph system (Oroboros Instruments, Innsbruck, Austria). Following dissection, muscle samples were placed in ice‐cold (4°C) Buffer X containing (mM) 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 20 taurine, 5.7 ATP, 14.3 phosphocreatine, 6.56 MgCl2.6H2O, and 50 MES (pH 7.1, 295 mosmol/l). Under a dissecting microscope, small bundles of fibers were prepared (1–3 mg wet weight per fiber bundle). Fiber bundles were treated with 50 μg/ml saponin for 30 min. Following permeabilization, myofiber bundles were washed in ice‐cold buffer Z containing (mM) 110 K‐MES, 35 KCl, 1 EGTA, 5 K2HPO2, 3 MgCl2.6H2O, and 5 mg/ml bovine serum albumin (BSA; pH 7.4, 295 mosmol/l) and remained in buffer Z on a rotator at 4°C until analysis. To prevent contraction, 20 μM blebbistatin was added to the wash buffer, in addition to the respiration medium during experiments (Fisher‐Wellman et al, 2013). The following substrates and nucleotides were utilized: palmitoyl‐carnitine (50 mM) + malate (2 mM), pyruvate (5 mM) + malate (2 mM), glutamate (5 mM), and succinate (5 mM), followed by addition of ADP (5 mM).
Electron microscopy
Soleus muscle was freshly excised and immediately washed in ice‐cold saline. Samples were processed as previously described (Boudina et al, 2007) at the University of Iowa Microscopy Core Facility.
Blue native PAGE
BN‐PAGE was performed as described previously (Wittig et al, 2006). Gastrocnemius muscle was homogenized in 5% digitonin. The homogenate was then solubilized in lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing 1% digitonin and 0.2% G‐250 sample additive (Thermo Fisher Scientific) and resolved on a 4–16% gradient native gel (Thermo Fisher Scientific).
Mitochondrial DNA content
Mitochondrial DNA content was quantified by real‐time polymerase chain reaction (RT–PCR). Briefly, total DNA was extracted and purified from gastrocnemius muscle with the DNeasy Kit (Qiagen Inc., Valencia, CA, USA). Five ng of DNA was used to quantify mitochondrial and nuclear DNA markers. RT–PCR was performed using an ABI Prism 7900HT instrument (Applied Biosystems, Foster City, CA, USA) in 384‐well plate format with SYBR Green I chemistry and ROX internal reference (Invitrogen). Analysis of results was automated using scripting with SDS 2.1 (Applied Biosystems), Microsoft Access, and Microsoft Excel. β‐actin was used as a nuclear DNA marker. Mitochondrial DNA content (Cox1) was expressed relative to genomic Rpl13a gene. The following primers were utilized:
Primers (5′ to 3′):
Cox1‐fwd: gcc cca gat ata gca ttc cc
Cox1‐rev: gtt cat cct gtt cct gct cc
Rpl13a‐fwd: gag gcc cct acc att tcc ga
Rpl13a‐rev: ggc ttc agc cga aca acc tt
Citrate synthase activity
Citrate synthase enzyme activity was determined as previously described (Boudina et al, 2005). Briefly, muscle fragments were homogenized on ice in 20% (wt/vol) homogenization buffer containing (in mmol/l) HEPES 20, EDTA 10, pH 7.4. The homogenates were then frozen for 1 h to liberate CS from mitochondrial matrix and were then diluted 1:10. The reaction was performed in 200 μl of reaction buffer containing (in mmol/l) HEPES 20, EGTA 1, sucrose 220, KCl 40, DTNB 0.1, and acetyl‐CoA 0.1, pH 7.4 at 25°C, and was started by the addition of 0.05 mmol/l oxaloacetate and finally monitored at 412 nm for 3 min with a Synergy HT Multi‐Detection Reader (BioTek Instruments).
WGA staining and stereological quantification
Fragments of gastrocnemius muscle were embedded in paraffin, portioned into 5‐μm‐thick sections, stained with wheat‐germ agglutinin (WGA)–Alexa Fluor 488 conjugate (Invitrogen Corporation, Carlsbad, CA, USA), and covered with SlowFade Gold antifade reagent with 4′,6‐diamidino‐2‐phenylindole (DAPI; Invitrogen). Images were obtained and processed at the University of Utah Fluorescence Microscopy Core Facility. From each sample, 20 microscopic fields were analyzed at random, the stage of the microscope being moved blindly. For stereological analysis, cross‐sectioned myocytes were selected. Myocyte diameter and number were determined using the Image‐Pro Plus software package (Media Cybernetics, Bethesda, MD, USA; Riehle et al, 2014). WGA‐stained images from soleus and gastrocnemius muscle from DKO mice were analyzed using the CellProfiler software (Broad Institute, Cambridge, MA, USA).
Glucose and insulin tolerance tests, CLAMS, nuclear magnetic resonance, and serum analysis
Glucose tolerance tests (GTT) were performed after a 6‐h fast, and mice were administered glucose (2 g/kg body weight), as described (Tabbi‐Anneni et al, 2008). Insulin tolerance tests (ITT) were performed after a 2‐h fast by injecting insulin (0.75 U/kg body weight; Humulin, Eli Lilly, Indianapolis, IN, USA). Blood glucose was determined using a glucometer (Glucometer Elite; Bayer, Tarrytown, NY, USA). Dosages were based on body weight, prepared in sterile 0.9% saline, and administered intraperitoneally. Plasma insulin (Ultra Sensitive Mouse Insulin ELISA Kit, Chrystal Chem, Downers Grove, IL, USA) and FGF21 (BioVendor ELISA kit, Asheville, NC, USA) were measured using commercially available kits according to the manufacturers’ directions. Whole body composition was measured using the Bruker Minispec NF‐90 instrument (Bruker, Billerica, MA, USA). For analysis of whole animal energy expenditure, animals were placed in the CLAMS (Comprehensive Lab Animal Monitoring System, Columbus Instruments) instruments (Columbus, OH, USA), an open circuit system that directly measures various parameters over a 72‐h period, such as heat production, food intake, and movement. NMR and CLAMS were performed at the University of Iowa Fraternal Order Of Eagles Diabetes Research Center Metabolic Phenotyping Core. A subset of 12‐week‐old mice fed regular chow was given intraperitoneal injections of either vehicle (PBS) or 5 mU/g body weight insulin (Novo Nordisk, Plainsboro, NJ, USA) after a 6‐h fast. Ten minutes after insulin injection, mice were sacrificed and gastrocnemius muscles were collected for analysis of insulin signaling (Hinchee‐Rodriguez et al, 2013).
Analysis of triglyceride levels
Triglycerides levels were measured in liver and in serum collected after a 6‐h fast using the EnzyChrom™ Triglyceride Assay Kit (BioAssay Systems, Hayward, CA, USA). Liver triglycerides were extracted using a solution of isopropanol and Triton X‐100, as recommended by the manufacturers (Tam et al, 2010).
RNA extraction and quantitative RT–PCR
Total RNA was extracted from tissues with TRIzol reagent (Invitrogen) and purified with the RNeasy kit (Qiagen Inc). RNA concentration was determined by measuring the absorbance at 260 and 280 nm using a spectrophotometer (NanoDrop 1000, NanoDrop products, Wilmington, DE, USA). Total RNA (~3 μg) was reverse‐transcribed using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA), followed by qPCR reactions using SYBR Green (Life Technologies, Carlsbad, CA) (Boudina et al, 2007). Samples were loaded in a 384‐well plate in triplicate, and real‐time polymerase chain reaction was performed with an ABI Prism 7900HT instrument (Applied Biosystems). The following cycle profile was used: 1 cycle at 95°C for 10 min; 40 cycles of 95°C for 15 s; 59°C for 15 s, 72°C for 30 s, and 78°C for 10 s; 1 cycle of 95°C for 15 s; 1 cycle of 60°C for 15 s; and 1 cycle of 95°C for 15 s. Data were normalized to GAPDH, and results are shown as fold change vs. WT mice. qPCR primers were designed using Primer‐Blast or previously published sequences (Kim et al, 2013; Baehr et al, 2014; Cubillos‐Ruiz et al, 2015). Utilized primers are listed in Table 1.
Table 1.
Primers
| Gene name | Forward | Reverse |
|---|---|---|
| Fgf21 | TGACGACCAAGACACTGAAGC | TTTGAGCTCCAGGAGACTTTCTG |
| Atf4 | AGCAAAACAAGACAGCAGCC | ACTCTCTTCTTCCCCCTTGC |
| Chop | GTCCCTAGCTTGGCTGACAGA | TGGAGAGCGAGGGCTTTG |
| BiP | TCATCGGACGCACTTGGAA | CAACCACCTTGAATGGCAAGA |
| Xbp‐1 splicing | ACACGTTTGGGAATGGACAC | CCATGGGAAGATGTTCTGGG |
| Atrogin‐1 | CTTTCAACAGACTGGACTTCTCGA | CAGCTCCAACAGCCTTACTACGT |
| Gapdh | ACTCTCTTCTTCCCCCTTGC | TCCACGACATACTCAGCAC |
| Ppargc1a | GTAAATCTGCGGGATGATGG | AGCAGGGTCAAAATCGTCTG |
| Tfam | CAAAAAGACCTCGTTCAGCA | CTTCAGCCATCTGCTCTTCC |
Western blot analysis
Immunoblotting analysis was performed as previously described (Pereira et al, 2013). Approximately, 50 mg of frozen tissue was homogenized in 200 μl lysis buffer containing (in mmol/l) 50 HEPES, 150 NaCl, 10% glycerol, 1% Triton X‐100, 1.5 MgCl2, 1 EGTA, 10 sodium pyrophosphate, 100 sodium fluoride, and 100 μmol/l sodium vanadate. Right before use, HALT protease/phosphatase inhibitors (Thermo Fisher Scientific) were added to the lysis buffer and samples were processed using the TissueLyser II (Qiagen Inc.). Tissue lysates were resolved on SDS–PAGE and transferred to PVDF membranes (Millipore Corp). Membranes were incubated with primary antibodies overnight and with secondary antibodies for 1 h.
Antibodies
Primary Antibodies: OPA1 (1:1,000, BD Biosciences, San Jose, CA, USA, #612606), FGF21 (1:1,000, Abcam, Cambridge, UK #ab171941), VDAC (1:1,000, Thermo Scientific, #PA1‐954A), GAPDH (1:1,000, Cell Signaling Technology, Danvers, MA, USA, #2118), phospho‐Akt Ser 473 (1:1,000, Cell Signaling Technology #9271), Akt (1:1,000, Cell Signaling Technology #2920), 4‐HNE (1:1,000, Abcam #ab46545), nitrotyrosine (1:1,000, Cell Signaling Technology #9691), phospho‐eIF2α (1:1,000, Cell Signaling Technology #3597), eIF2α (1:1,000, Santa Cruz Biotechnology, Dallas, TX, USA, #SC81261), AMPK (1:1,000, Cell Signaling Technology #2793), phospho‐AMPK (1:1,000, Cell Signaling Technology #4181), PGC1‐α (1:500, Santa Cruz Biotechnology #SC13067), Mfn1 (1:1,000, Abcam #ab57602), Mfn2 (1:1,000, Abcam # ab101055), MuRF1 (1:1,000, Abcam #ab172479), pS6 (1:1,000, Cell Signaling Technology #2211), S6 (1:1,000, Cell Signaling Technology #2317), MnSOD (1:1,000, Cell Signaling Technology #13141), BiP (BD Biosciences, 1:1,000, #610978), XBP‐1 (1:500, Santa Cruz Biotechnology #SC7160), CHOP (1:500, Santa Cruz Biotechnology #SC7351 H1908). Secondary antibodies: IRDye 800CW anti‐mouse (1:10,000, LI‐COR, Lincoln, NE, USA, #925‐32212) and Alexa Fluor anti‐rabbit 680 (1:10,000, Invitrogen #A27042). Fluorescence was quantified using the LiCor Odyssey imager.
Cell culture and treatments
Satellite cell isolation was performed as previously described (Groh et al, 2009). Satellite cells from OPA1fl/fl were plated on BD Matrigel‐coated dishes and activated to differentiate into myoblasts in DMEM‐F12, 20% fetal bovine serum (FBS), 40 ng/ml basic fibroblast growth factor (R&D Systems, 233‐FB/CF), 1× non‐essential amino acids, 0.14 mM β‐mercaptoethanol, 1× penicillin/streptomycin, and Fungizone. Myoblasts were maintained with 10 ng/ml basic fibroblast growth factor and differentiated in DMEM‐F12, 2% FBS, 1× insulin–transferrin–selenium, when 90% confluency was reached. Three days after differentiation, myotubes were infected with adenovirus for ntGFP or GFP‐Cre for OPA1 deletion. Adenoviruses were obtained from the University of Iowa Viral Vector Core facility. Experiments were performed between 3 and 7 days after infection. To inhibit ER stress, myotubes were treated with either vehicle or 500 μM PBA for 3 days (4 days after adenoviral infection). On the third day, the media was switched to 2% FBS phenol‐red‐free DMEM/F12 (differentiation media), and fresh PBA was added at 8 am. Eight hours later, the supernatant was collected for FGF21 measurements and cells were processed for protein analysis by Western blot. This experiment was performed three separate times. Each time four to six replicates/condition were obtained. Representative data from six replicates are shown. Additional sets of cells were used for cellular respiration with the XF24 extracellular flux (XF) bioanalyzer (Agilent Technologies/Seahorse Bioscience, North Billerica, MA, USA). Cells were plated at a density of 20 × 103 per well and differentiated. After 3 days of differentiation, adenoviruses for ntGFP or GFP‐Cre were added, as described above, at a multiplicity of infection sufficient to infect > 95% of the cells based on the GFP fluorescence with minimal cell death. Three days after infection, medium was changed to XF‐DMEM and cells were kept in a non‐CO2 incubator for 60 min. Basal oxygen consumption rate (OCR) was measured in XF‐DMEM. Oxygen consumption was then measured after the addition of each of the following compounds: oligomycin (1 μg/ml), carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP; 1 μM), rotenone (1 μM), and antimycin A (10 μM; Wende et al, 2015). For the glycolysis stress test, prior to analysis cells were switched to glucose‐free XF‐DMEM and were kept in a non‐CO2 incubator for 60 min. Extracellular acidification rate (ECAR) was measured in XF‐DMEM followed by these additional conditions: glucose (10 mM), oligomycin (1 μM), and 2‐DG (100 mM). Data shown for Seahorse experiments reflect the results of one Seahorse run/condition in which eight replicates were utilized. For analysis of mitochondrial membrane potential (Δψ) in primary myotubes, primary myoblasts were seeded on 24‐well glass‐bottom plates (Mattek, Thermo Fisher Scientific) coated with BD Matrigel. Cells were differentiated and infected, as described above. Three days after infection, media were switched to FluoroBrite DMEM Media (Thermo Fisher Scientific) supplemented with 2% FBS and 2 mM GlutaMax. Cells were then incubated with 50 nM tetramethylrhodamine methyl ester (TMRM) for 30 min. Media were refreshed, and 100 μM H2O2 was added. TMRM fluorescence was then continuously measured across identical fields at 5‐min intervals in live cells using RFP/TRITC filter set on a EVOS FL Auto 2 microscope (Thermo Fisher Scientific) for a total of 100 min. Quantitative analysis of the decline of TMRM fluorescence in the same field over time was measured using ImageJ (National Institutes of Health, Bethesda, MA, USA). Data represent the mean ± standard error of the mean (SEM), with n of three replicates per group. A total of four fields/well/time point were analyzed. C2C12 myoblasts were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/l glucose supplemented with 10% fetal bovine serum. When cells were confluent, medium was changed to DMEM supplemented with 2% horse serum for myotube differentiation. Five days after differentiation, myotubes were treated with either vehicle (ethanol), 0.1 μM oligomycin, or tunicamycin (1 μg/ml) ± 500 μM PBA for 8 h, after which cells were harvested and processed for Western blot or qPCR analysis as described above. For experiments in which secreted FGF21 was measured in the media, cells were cultured in phenol‐red‐free DMEM during the 8‐h treatment. Media were collected and processed, and FGF21 was then measured in the supernatant and in cell lysates using a BioVendor ELISA kit (Asheville, NC, USA), according to the manufacturers’ directions. These studies were repeated independently three times.
Exhaustion test
Exercise tolerance was measured on a motor‐driven treadmill as previously described (Arany et al, 2007; Hakimi et al, 2007). To encourage the mice to run, the treadmill is equipped with an electrical shock grid at the rear of the treadmill. The shock grid was set to deliver 0.2 mA, an uncomfortable shock that does not cause harm or injury. On days 0–2, mice were acclimated by being placed on the treadmill (set at 14 m/min at 0% grade) for 5 min/day. On day 3, exercise capacity was determined at a constant treadmill incline of 10% using the following protocol: First, mice underwent a 5‐min run‐in period at 10 m/min. Next, treadmill speed was increased by 2 m/min every 2 min. Mice were then forced to run to exhaustion, which was defined as sitting on the shock pad for 10 s without attempting to restart running. Blood lactate was measured under random‐fed conditions before the test and after exhaustion was reached using a Fisher Lactate Scout Analyzer (San Antonio, TX, USA).
Grip strength
Forty‐week‐old WT and mOPA1 KO mice were allowed to grab the pull bar attached to the force meter with their forelimbs and were pulled backward by the tail. The peak tension was recorded by the meter when their grasp failed. This procedure was repeated five times total per session, with at least 7 days between sessions (Meyer et al, 1979; Costa et al, 1999).
Calculations of vertical work
Vertical work was calculated using the following formula:
| (1) |
where F = body weight (kg) × 9.8 m/s2.
Data analysis
Unless otherwise noted, all data are reported as mean ± SEM. Student's t‐test was performed for comparison of two groups, and ANOVA followed by Tukey multiple comparison test was utilized when more than three groups were compared. A probability value of P ≤ 0.05 was considered significantly different. Statistical calculations were performed using the GraphPad Prism software (La Jolla, CA, USA).
Author contributions
ROP designed the study, researched data, and wrote the manuscript. SMT, FMZ, KJO, KMPP, AO, JJ, RS, RM, AS, and TF researched data and helped design experiments. HS, MJP, CMA, EJA helped design experiments and provided critical materials. EDA conceived and supervised the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This work was supported by NIH grants R01 R01HL108379 and R01DK092065 to E.D.A., who is an established investigator of the American Heart Association (AHA), and AHA Scientist Development Grant 15SDG25710438 (to R.O.P.). We thank Andrew Gulden and Conor Bryant for their technical support. Metabolic phenotyping was performed in the Metabolic Phenotyping Core at the Fraternal Order of Eagles Diabetes Research Center.
The EMBO Journal (2017) 36: 2126–2145
References
- Ahima RS, Park HK (2015) Connecting myokines and metabolism. Endocrinol Metab (Seoul) 30: 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S, Spiegelman BM (2007) The transcriptional coactivator PGC‐1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab 5: 35–46 [DOI] [PubMed] [Google Scholar]
- Baehr LM, Tunzi M, Bodine SC (2014) Muscle hypertrophy is associated with increases in proteasome activity that is independent of MuRF1 and MAFbx expression. Front Physiol 5: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Baehr LM (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab 307: E469–E484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, Dierdorff JM, Adams CM (2013) Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J Physiol Endocrinol Metab 305: E907–E915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM (2012) A PGC1‐alpha‐dependent myokine that drives brown‐fat‐like development of white fat and thermogenesis. Nature 481: 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112: 2686–2695 [DOI] [PubMed] [Google Scholar]
- Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED (2007) Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466 [DOI] [PubMed] [Google Scholar]
- Boudina S, Sena S, Sloan C, Tebbi A, Han YH, O'Neill BT, Cooksey RC, Jones D, Holland WL, McClain DA, Abel ED (2012) Early mitochondrial adaptations in skeletal muscle to diet‐induced obesity are strain dependent and determine oxidative stress and energy expenditure but not insulin sensitivity. Endocrinology 153: 2677–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M (2015) Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21: 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civitarese AE, MacLean PS, Carling S, Kerr‐Bayles L, McMillan RP, Pierce A, Becker TC, Moro C, Finlayson J, Lefort N, Newgard CB, Mandarino L, Cefalu W, Walder K, Collier GR, Hulver MW, Smith SR, Ravussin E (2010) Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metab 11: 412–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Walsh K, Davisson MT (1999) Motor dysfunction in a mouse model for Down syndrome. Physiol Behav 68: 211–220 [DOI] [PubMed] [Google Scholar]
- Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales‐Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo‐Garcia JR, Glimcher LH (2015) ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell 161: 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW 2nd, Kitsis RN (2015) The mitochondrial dynamism‐mitophagy‐cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res 116: 167–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert SM, Monteys AM, Fox DK, Bongers KS, Shields BE, Malmberg SE, Davidson BL, Suneja M, Adams CM (2010) The transcription factor ATF4 promotes skeletal myofiber atrophy during fasting. Mol Endocrinol 24: 790–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM (2012) Stress‐induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 287: 27290–27301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, Bongers KS, Lira VA, Meyerholz DK, Talley JJ, Adams CM (2015) Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)‐dependent pathway to age‐related skeletal muscle weakness and atrophy. J Biol Chem 290: 25497–25511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer M, Bluher M (2015) Adipokines in health and disease. Trends Pharmacol Sci 36: 461–470 [DOI] [PubMed] [Google Scholar]
- Fisher‐Wellman KH, Mattox TA, Thayne K, Katunga LA, La Favor JD, Neufer PD, Hickner RC, Wingard CJ, Anderson EJ (2013) Novel role for thioredoxin reductase‐2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J Physiol 591: 3471–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DK, Ebert SM, Bongers KS, Dyle MC, Bullard SA, Dierdorff JM, Kunkel SD, Adams CM (2014) p53 and ATF4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am J Physiol Endocrinol Metab 307: E245–E261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189 [DOI] [PubMed] [Google Scholar]
- Groh S, Zong H, Goddeeris MM, Lebakken CS, Venzke D, Pessin JE, Campbell KP (2009) Sarcoglycan complex: implications for metabolic defects in muscular dystrophies. J Biol Chem 284: 19178–19182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guridi M, Tintignac LA, Lin S, Kupr B, Castets P, Ruegg MA (2015) Activation of mTORC1 in skeletal muscle regulates whole‐body metabolism through FGF21. Sci Signal 8: ra113 [DOI] [PubMed] [Google Scholar]
- Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino‐Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC, Hanson RW (2007) Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem 282: 32844–32855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LA, Skinner JR, Shew TM, Pietka TA, Abumrad NA, Wolins NE (2015) Perilipin 5‐driven lipid droplet accumulation in skeletal muscle stimulates the expression of fibroblast growth factor 21. Diabetes 64: 2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchee‐Rodriguez K, Garg N, Venkatakrishnan P, Roman MG, Adamo ML, Masters BS, Roman LJ (2013) Neuronal nitric oxide synthase is phosphorylated in response to insulin stimulation in skeletal muscle. Biochem Biophys Res Comm 435: 501–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K (2008) FGF21 is an Akt‐regulated myokine. FEBS Lett 582: 3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS (2012) Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32: 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A, Scorrano L (2014) Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol 24: 761–770 [DOI] [PubMed] [Google Scholar]
- Keipert S, Ost M, Johann K, Imber F, Jastroch M, van Schothorst EM, Keijer J, Klaus S (2014) Skeletal muscle mitochondrial uncoupling drives endocrine cross‐talk through the induction of FGF21 as a myokine. Am J Physiol Endocrinol Metab 306: E469–E482 [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N et al (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19: 83–92 [DOI] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845 [DOI] [PubMed] [Google Scholar]
- Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, Mohammadi M, Potthoff MJ (2014) Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63: 4057–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer OA, Tilson HA, Byrd WC, Riley MT (1979) A method for the routine assessment of fore‐ and hindlimb grip strength of rats and mice. Neurobehav Toxicol 1: 233–236 [PubMed] [Google Scholar]
- Montgomery MK, Turner N (2015) Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 4: R1–R15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Canoves P, Scheele C, Pedersen BK, Serrano AL (2013) Interleukin‐6 myokine signaling in skeletal muscle: a double‐edged sword? FEBS J 280: 4131–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ost M, Werner F, Dokas J, Klaus S, Voigt A (2014) Activation of AMPKalpha2 is not crucial for mitochondrial uncoupling‐induced metabolic effects but required to maintain skeletal muscle integrity. PLoS One 9: e94689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ost M, Coleman V, Voigt A, van Schothorst EM, Keipert S, van der Stelt I, Ringel S, Graja A, Ambrosi T, Kipp AP, Jastroch M, Schulz TJ, Keijer J, Klaus S (2016) Muscle mitochondrial stress adaptation operates independently of endogenous FGF21 action. Mol Metab 5: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel‐Langenickel I, Bao J, Pang L, Sack MN (2010) The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev 31: 25–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez‐Crisosto C, Jana F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A et al (2014) Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt‐mTOR‐NFkappaB‐Opa‐1 signaling pathway. Diabetes 63: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED (2013) Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc 2: e000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM (2007) Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: 476–491 [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, Burgess SC (2009) FGF21 induces PGC‐1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA 106: 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Finck BN (2014) Head over hepatocytes for FGF21. Diabetes 63: 4013–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam DA, Bhattacharya A, Van Remmen H (2013) Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid Redox Signal 19: 1373–1387 [DOI] [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez‐Vizarra E, Rodriguez F, Peinado JR, Fernandez‐Garcia MS, Vega JA, Enriquez JA, Zorzano A, Lopez‐Otin C (2012) Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J 31: 2117–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas F, Villarroya J, Hondares E, Giralt M, Villarroya F (2014) FGF21 expression and release in muscle cells: involvement of MyoD and regulation by mitochondria‐driven signalling. Biochem J 463: 191–199 [DOI] [PubMed] [Google Scholar]
- Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, Abel ED (2014) Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol 34: 3450–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Schmeisser K (2014) Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12: 288–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Foletta VC, Snow RJ, Wadley GD (2014) Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochem Biophys Acta 1840: 1276–1284 [DOI] [PubMed] [Google Scholar]
- Sebastian D, Hernandez‐Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K (2015) Mitochondrial hormesis and diabetic complications. Diabetes 64: 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietilainen KH, Hakonen AH, Sevastianova K, Korpela M, Isohanni P, Marjavaara SK, Tyni T, Kiuru‐Enari S, Pihko H, Darin N, Ounap K, Kluijtmans LA, Paetau A, Buzkova J, Bindoff LA, Annunen‐Rasila J, Uusimaa J, Rissanen A et al (2011) FGF‐21 as a biomarker for muscle‐manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 10: 806–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabbi‐Anneni I, Buchanan J, Cooksey RC, Abel ED (2008) Captopril normalizes insulin signaling and insulin‐regulated substrate metabolism in obese (ob/ob) mouse hearts. Endocrinology 149: 4043–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, Osei‐Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G (2010) Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Investig 120: 2953–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Carroll CJ, Raimundo N, Ahola‐Erkkila S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola‐Mjosund KE, Tulkki V, Oresic M, Moraes CT, Pietilainen K, Hovatta I, Suomalainen A (2010) Mitochondrial myopathy induces a starvation‐like response. Hum Mol Genet 19: 3948–3958 [DOI] [PubMed] [Google Scholar]
- Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana‐Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Di Lisa F, Mongillo M, Sandri M, Scorrano L (2015) The OPA1‐dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21: 834–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, O'Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, Sloan C, Kim BJ, Wang X, Pereira RO, McCrory MA, Nye BG, Benavides GA, Darley‐Usmar VM, Shioi T, Weimer BC et al (2015) Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion‐targeted nuclear genes. Mol Cell Biol 35: 831–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H (2006) Blue native PAGE. Nat Protoc 1: 418–428 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S, Clerc P, Polster BM, Aja SM, Pletnikov MV, Kensler TW, Shirihai OS, Iijima M, Hussain MA, Sesaki H (2011) The dynamin‐related GTPase Opa1 is required for glucose‐stimulated ATP production in pancreatic beta cells. Mol Biol Cell 22: 2235–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorzano A, Hernandez‐Alvarez MI, Palacin M, Mingrone G (2010) Alterations in the mitochondrial regulatory pathways constituted by the nuclear co‐factors PGC‐1alpha or PGC‐1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochem Biophys Acta 1797: 1028–1033 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7