

Abstract
XPF‐ERCC1 is a structure‐specific endonuclease pivotal for several DNA repair pathways and, when mutated, can cause multiple diseases. Although the disease‐specific mutations are thought to affect different DNA repair pathways, the molecular basis for this is unknown. Here we examine the function of XPF‐ERCC1 in DNA interstrand crosslink (ICL) repair. We used Xenopus egg extracts to measure both ICL and nucleotide excision repair, and we identified mutations that are specifically defective in ICL repair. One of these separation‐of‐function mutations resides in the helicase‐like domain of XPF and disrupts binding to SLX4 and recruitment to the ICL. A small deletion in the same domain supports recruitment of XPF to the ICL, but inhibited the unhooking incisions most likely by disrupting a second, transient interaction with SLX4. Finally, mutation of residues in the nuclease domain did not affect localization of XPF‐ERCC1 to the ICL but did prevent incisions on the ICL substrate. Our data support a model in which the ICL repair‐specific function of XPF‐ERCC1 is dependent on recruitment, positioning and substrate recognition.
Keywords: Fanconi anemia, interstrand crosslink repair, nucleotide excision repair, Xenopus egg extract, XPF‐ERCC1
Subject Categories: DNA Replication, Repair & Recombination; Molecular Biology of Disease
Introduction
The structure‐specific endonuclease XPF‐ERCC1 participates in multiple genome maintenance pathways, including nucleotide excision repair (NER), DNA interstrand crosslink (ICL) repair, certain branches of double‐stranded break (DSB) repair, and telomere maintenance. Mutations in XPF‐ERCC1 have been associated with the genetic disorders Xeroderma pigmentosum (XP), Cockayne syndrome (CS), cerebro‐oculo‐facio‐skeletal syndrome (COFS), Fanconi anemia (FA), and premature aging. These phenotypes are believed to be caused by a defect in one, or several, of the genome maintenance pathways XPF‐ERCC1 is involved in, but the molecular basis for this is unknown.
XPF is a structure‐specific endonuclease that contains an N‐terminal helicase‐like domain, a central ERCC4‐type nuclease domain, and a C‐terminal helix‐hairpin‐helix (HhH) domain with which it interacts with its cofactor ERCC1 (Fig 1A). Very little is known about the role of the helicase‐like domain, but it is important for nuclease activity (Bowles et al, 2012). The function of XPF‐ERCC1 in NER, a pathway that removes helix distorting lesions, has been extensively studied (Gillet & Schärer, 2006; Friedberg, 2011). After damage recognition by upstream NER factors, XPF‐ERCC1 is recruited to the lesion by XPA and excises a short oligo containing the damage in collaboration with another nuclease XPG (Huang et al, 1992; Li et al, 1995; Spivak, 2015). Defects in NER factors are associated with the genetic disease Xeroderma pigmentosum (XP), which is characterized by sunlight sensitivity and skin cancer predisposition. Uniquely among NER factors, deficiency in XPF‐ERCC1 not only causes UV sensitivity, but also results in hypersensitivity to ICL‐inducing agents, indicating an additional role for this protein in the repair of interstrand crosslinks (De Silva et al, 2000; Kuraoka et al, 2000; Niedernhofer et al, 2004).
Figure 1. Characterization of mutant XPF‐ERCC1 complexes.
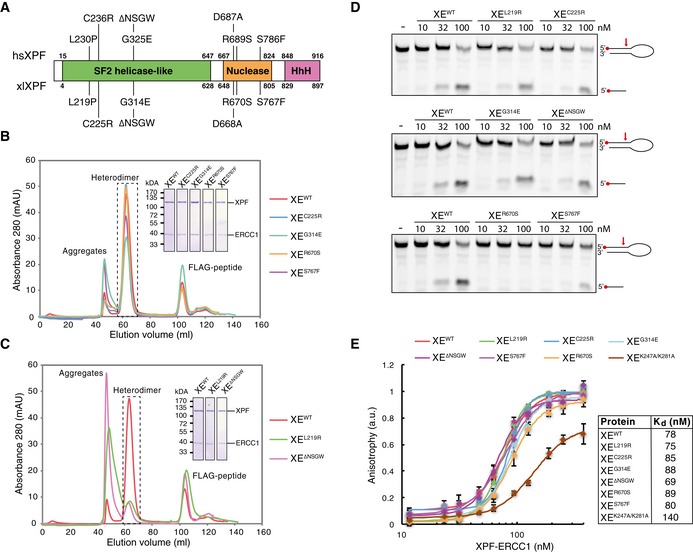
- Schematic representation of the domain organization of the XPF protein. Domain boundaries of human and Xenopus laevis XPF are indicated. Relevant mutations of the human protein, and the Xenopus laevis equivalents, are indicated on top and bottom, respectively.
- Superdex 200 gel filtration column elution profile of wild‐type XPF‐ERCC1 and indicated mutant complexes. Aggregates eluted in the void volume of the column (˜45 ml) while the active XPF‐ERCC1 heterodimer eluted at ˜65 ml. The peak eluting at ˜105 ml contains the FLAG peptide used to elute the protein from the FLAG affinity resin. The heterodimer peak was isolated, and proteins were separated on SDS–PAGE and stained with Coomassie blue (inset).
- As in (B) but for different mutant complexes that showed more aggregation.
- Wild‐type and indicated mutant XPF‐ERCC1 complexes were incubated with a 5′‐FAM‐labeled stem‐loop DNA substrate (10 nM) at room temperature for 30 min. Reaction products were separated on a 12% urea–PAGE gel and visualized using a fluorescence imaging system. Red arrow indicates position of incision by XPF‐ERCC1.
- Wild‐type and mutant XPF‐ERCC1 complexes at various concentrations were incubated with a 5′‐FAM‐labeled 3′ flap DNA substrate (10 nM) and fluorescent anisotropy was measured. Graphs were fitted to calculate dissociation constants (K ds) as described in the Materials and Methods section. The error bars represent s.d. from three measurements. Experimental replicates are shown in Fig EV2.
Source data are available online for this figure.
ICLs are toxic DNA lesions that covalently link both strands of the DNA together, thereby blocking DNA replication and transcription. ICLs are formed endogenously by products of cellular metabolism, but are also induced at high doses by certain chemotherapeutic drugs. The major pathway of ICL repair is coupled to DNA replication and involves the coordinated action of many DNA repair proteins including the Fanconi anemia (FA) pathway proteins. Mutations in any of the 21 currently known FA genes give rise to Fanconi anemia (FA), a cancer susceptibility disorder characterized by cellular sensitivity to ICL‐inducing agents (Kottemann & Smogorzewska, 2013; Dong et al, 2015). Using a Xenopus egg extract‐based assay, we and others have recently elucidated a molecular mechanism of replication‐coupled ICL repair (Fig EV1; Räschle et al, 2008). This mechanism requires the convergence of two replication forks at an ICL during DNA replication (Zhang et al, 2015). Both forks initially stall 20–40 nucleotides from the crosslink followed by CMG helicase unloading allowing one fork to approach to within 1 nucleotide of the crosslink (Räschle et al, 2008; Fu et al, 2011; Long et al, 2011). Dual incisions on either side of the ICL then unhook the lesion from one of the strands. This critical repair step requires the endonuclease XPF (FANCQ)‐ERCC1, which is recruited to the ICL by the large scaffold protein SLX4 (FANCP), and depends on the activation of the Fanconi anemia pathway by ubiquitylation of the FANCI‐FANCD2 complex (Knipscheer et al, 2009; Klein Douwel et al, 2014). After unhooking, a nucleotide is inserted across from the adducted base, followed by strand extension by REV1 and polymerase ζ, consisting of REV7 (FANCV) and REV3 (Räschle et al, 2008; Budzowska et al, 2015; Mamrak et al, 2016). This strand now acts as a template for repair of the opposite strand by HR (Long et al, 2011), leading to fully repaired products.
Figure EV1. Schematic representation of replication‐dependent ICL repair in Xenopus egg extract.
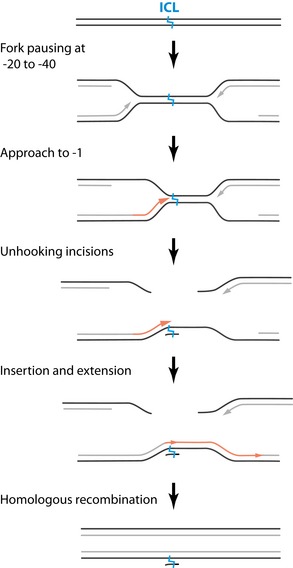
Arrow heads represent 3' ends of leading strands.
Patient phenotypes linked to specific XPF mutations can be extremely valuable in determining pathway‐specific functions. Most patients with a mutation in XPF suffer from a mild form of XP and are deficient in NER. These patients express residual protein and are likely proficient in ICL repair, because they do not show features of FA (Ahmad et al, 2010). In some cases, XPF mutations can lead to much more severe phenotypes. An extreme progeroid syndrome was caused by a mutation in the helicase‐like domain of XPF (R153P). This patient suffered from neurological and hematological defects and a cellular sensitivity to UV and ICLs indicating both NER and ICL repair were defective (Niedernhofer et al, 2006). Another patient, with a mutation in the same XPF domain (C236R), presented with phenotypes of XP, but also of CS, such as developmental and neurological abnormalities (Kashiyama et al, 2013). This patient also showed FA‐like features and ICL sensitivity suggestive of a defect in ICL repair. In addition, some patients with specific mutations in XPF were diagnosed with Fanconi anemia and showed no signs of XP (Bogliolo et al, 2013). These mutations were mapped to the helicase‐like domain (L230P), and the nuclease domain (R689S) of XPF.
To examine what features of XPF‐ERCC1 determine its specificity in ICL repair, we employed the Xenopus egg extract system. We monitored both replication‐coupled ICL repair and nucleotide excision repair and identified five XPF mutants that are deficient in ICL repair and proficient in NER. Although all of these mutants showed a defect in ICL unhooking, the majority was still efficiently recruited to the ICL. In contrast, mutation of xlXPF leucine 219, equivalent to the human leucine 230 mutated in Fanconi anemia, abrogated this ICL localization. This was caused by a defect in interaction with SLX4. We propose there are two interaction sites between XPF and SLX4, one ensuring recruitment of XPF to the ICL, and another to promote its nuclease activity. This dual interaction site, in combination with residues in the nuclease domain ensuring substrate specificity, dictates the ICL repair‐specific function of XPF‐ERCC1.
Results
XPF mutants form functional complexes with ERCC1
To study the role of XPF‐ERCC1 in ICL repair, we selected a set of XPF mutations that we predicted to specifically affect this process. In the helicase‐like domain, two point mutations were found in patients with FA and FA‐like symptoms, L230P and C236R (Bogliolo et al, 2013; Kashiyama et al, 2013). These mutated residues correspond to residues L219 and C225 in Xenopus laevis XPF that is 75% identical to human XPF (Fig 1A). Another mutation in XPF's helicase‐like domain, hsG325E (xlG314E), was reported to disrupt the interaction of XPF with the BTB domain of SLX4 (Andersen et al, 2009). This interaction is likely specific to ICL repair and not NER because SLX4‐deficient cells are not UV sensitive (Crossan et al, 2011). With the aim to further disrupt this interaction, we generated a deletion mutant lacking G314 and three surrounding residues that were predicted to form a loop (XPFΔNSGW). Finally, in the nuclease domain, we analyzed two mutations, R689S and S786F (xlR670S and xlS676F). The R689S mutation was associated with Fanconi anemia (Bogliolo et al, 2013) and the S786F mutation sensitizes cells to MMC, but not UV radiation (Osorio et al, 2013).
We co‐expressed FLAG‐tagged xlXPF wild type and mutants with His‐tagged hsERCC1 in Sf9 insect cells and purified the complex by affinity purification. We previously showed that the xlXPF‐hsERCC1 complex (referred to as XPF‐ERCC1 from here on) supports ICL repair in Xenopus egg extracts (Klein Douwel et al, 2014). Expression levels of all mutant complexes were similar to wild‐type levels except for the XPFL219P‐ERCC1 (XEL219P) complex that was previously reported to be unstable (Bogliolo et al, 2013; Hashimoto et al, 2015). To examine the function of leucine 219, we instead mutated it to an arginine and found that XEL219R was expressed at normal levels. When wild‐type XPF‐ERCC1 was subjected to gel filtration chromatography, the majority of the protein eluted at the expected range for a heterodimer while a small fraction of inactive aggregates eluted in the void volume of the column, similar to what was previously described (Fig 1B; Enzlin & Schärer, 2002). The XEC225R, XEG314E, XER670S, and XES767F mutant complexes behaved similarly to the wild type on gel filtration (Fig 1B) and the peak containing the heterodimer was isolated and used for further experiments. However, the XEL219R and XEΔNSGW mutant complexes showed an increased aggregate peak and lower heterodimer peak (Fig 1C). Nevertheless, when this heterodimer peak was isolated and rerun on a gel filtration column, it did not aggregate (Fig EV2A).
Figure EV2. Characterization of mutant XPF‐ERCC1 complexes.
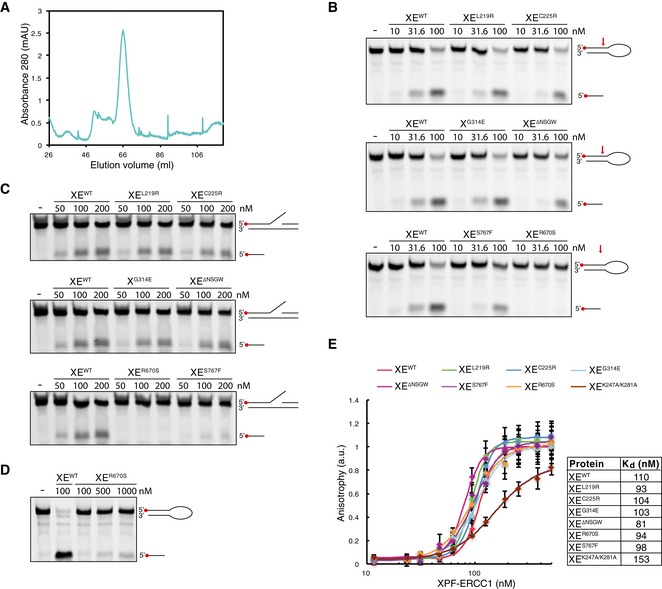
- Superdex 200 gel filtration column elution profile of XPFL219R‐ERCC1. The heterodimeric fraction depicted in Fig 1A was collected, concentrated, and rerun on the same column. The protein did not aggregate and eluted as a heterodimer at ˜65 ml.
- Replicate of Fig 1D. Wild‐type and indicated mutant XPF‐ERCC1 complexes were incubated with a 5′‐FAM‐labeled stem‐loop DNA substrate (10 nM) at room temperature for 30 min. Reaction products were separated on a 12% urea–PAGE gel and visualized using a fluorescence imaging system. Red arrow indicates position of incision by XPF‐ERCC1.
- As in (B) but using a 5′‐FAM‐labeled 3′ flap DNA substrate.
- As in (B) but using higher concentrations of the XER670S mutant.
- Replicate of Fig 1E. Wild‐type and mutant XPF‐ERCC1 complexes at various concentrations were incubated with a 5′‐FAM‐labeled 3′ flap DNA substrate (10 nM) and fluorescent anisotropy was measured. Graphs were fitted to calculate dissociation constants (K ds) as described in the Materials and Methods section. The error bars represent s.d. from three measurements.
Source data are available online for this figure.
We next examined the endonuclease activity of the mutant XPF‐ERCC1 complexes. To this end, a fluorescently labeled stem‐loop substrate was incubated with increasing concentrations of protein, and reaction products were separated by denaturing urea–PAGE. All XPF‐ERCC1 complexes with mutations in the helicase‐like domain showed nuclease activity similar to wild‐type protein, as seen by the appearance of the incision product (Fig 1D, top two panels, and Fig EV2B). Importantly, this demonstrates that the XEL219R and XEΔNSGW complexes, which showed increased aggregation upon expression, are fully active after isolation of the heterodimer peak. The XES767F complex was slightly reduced in nuclease activity while the XER670S complex showed a more dramatic reduction and was only capable of cutting the substrate at high concentrations (Figs 1D and EV2D). This was not surprising as both mutations are located in the nuclease domain of XPF, and the human equivalent of the R670S mutant has decreased nuclease activity (Enzlin & Schärer, 2002; Su et al, 2012). We also analyzed the endonuclease activity on a 3′ flap substrate and obtained similar results (Fig EV2C). In conclusion, all mutants except R670S have nuclease activity similar to wild‐type protein.
Finally, we analyzed the DNA binding of the mutant XPF‐ERCC1 complexes. For this, a fluorescently labeled 3′ flap substrate was incubated with increasing concentrations of XPF‐ERCC1 and fluorescence anisotropy was measured to assess binding. All our mutants showed very similar binding curves and the K d values derived from these curves were comparable to each other and to wild‐type XPF‐ERCC1 (Figs 1E and EV2E). This indicates that the mutations do not affect DNA binding affinity. To validate our results, we measured fluorescence anisotropy of a mutant XPF‐ERCC1 carrying two point mutations in ERCC1 (K247A and K281A) that were previously shown to affect DNA binding (Su et al, 2012). Consistent with this, we found that this XEKAKA mutant had reduced affinity for DNA (Figs 1E and EV2E).
In summary, we purified six mutant XPF‐ERCC1 complexes that are predicted to affect ICL repair. All mutant complexes form stable heterodimers and interact normally with DNA. The nuclease activity of the mutants is similar to wild type, with the exception of one mutation in the nuclease domain.
Mutations in helicase‐like and nuclease domains abrogate ICL repair
To investigate the effect of the XPF mutations on ICL repair, we used Xenopus egg extracts. This system recapitulates DNA replication‐coupled repair of a sequence‐specific cisplatin ICL situated on a plasmid template (pICL; Räschle et al, 2008). Moreover, it enables the quantification of repair by the regeneration of a SapI restriction site that is blocked by the crosslink (Fig 2A). We immunodepleted ERCC1 from egg extract and complemented the repair reaction with wild‐type or mutant XPF‐ERCC1. Since depletion of ERCC1 leads to equal depletion of XPF (Klein Douwel et al, 2014), we refer to this depletion as an XPF‐ERCC1 depletion. Because depletion of XPF‐ERCC1 leads to co‐depletion of SLX4, we also complemented all the depleted reactions with purified xlSLX4 protein unless stated otherwise (Fig EV3A; Klein Douwel et al, 2014). Reactions were stopped at various time points, and DNA repair intermediates were isolated and digested with SapI to quantify ICL repair. A small fraction of non‐crosslinked plasmids is present in pICL preparations leading to a constant background of SapI digestible replication products. XPF‐ERCC1‐depleted extracts did not support ICL repair above this background while addition of recombinant wild‐type XPF‐ERCC1 (XEwt) restored ICL repair (Fig 2B–D; Klein Douwel et al, 2014). XEL219R and XEC225R, which carry mutations in the helicase‐like domain of XPF, did not efficiently rescue ICL repair (Figs 2B and EV3B). We then tested the other two helicase‐like domain mutants that were expected to affect the interaction with the BTB domain of SLX4. While addition of XEG314E to XPF‐ERCC1‐depleted extract supported ICL repair, the deletion mutant XEΔNSGW was defective in ICL repair (Figs 2C and EV3C). This suggests that this region is important for ICL repair, likely through mediating interaction with SLX4. Finally, XPF‐ERCC1‐depleted extracts were supplemented with the nuclease domain mutants XER670S and XES767F. Both mutants were unable to restore ICL repair (Figs 2D and EV3B). These results show that specific residues in the helicase‐like domain and the nuclease domain of XPF‐ERCC1 are required for ICL repair.
Figure 2. Effect of mutations in XPF‐ERCC1 on ICL repair in Xenopus egg extract.
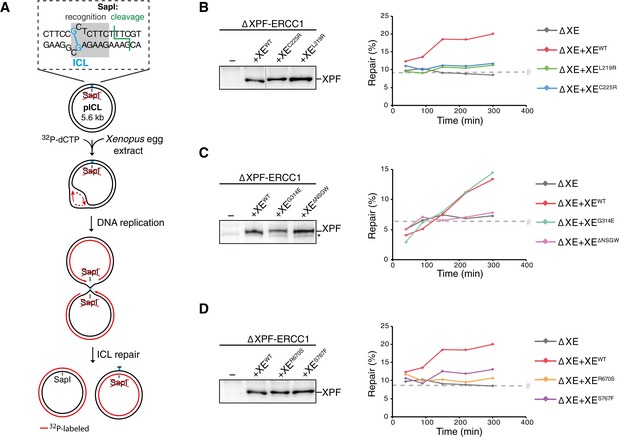
-
ASchematic representation of repair of a plasmid containing a site‐specific cisplatin ICL (pICL) in Xenopus egg extract. The SapI site that is blocked by the ICL becomes available on one of the replicated molecules after full repair via HR using the sister molecule (Fig EV1). The sister molecule is repaired by lesion bypass, but retains the unhooked ICL that is not removed efficiently in Xenopus egg extract (Räschle et al, 2008).
-
BXPF‐ERCC1‐depleted (ΔXE) and XPF‐ERCC1‐depleted extracts complemented with wild‐type (XEWT) or indicated mutant XPF‐ERCC1 (XEMUT) were analyzed by Western blot using α‐XPF antibodies (left panel). Line within blot indicates position where irrelevant lanes were removed. These extracts were used to replicate pICL. Replication intermediates were isolated and digested with HincII, or HincII and SapI, and separated on agarose gel. Repair efficiency, represented by SapI regeneration, was calculated as described (Räschle et al, 2008) and plotted (right panel).
-
C, DAs in (B) but analyzing different XPF‐ERCC1 mutant complexes. Experimental replicates are shown in Fig EV3. Note: repair levels can differ per batch of individually prepared extract or per depletion experiment and can only be compared within an experiment. *, background band.
Figure EV3. Effect of mutations in XPF‐ERCC1 on ICL repair in Xenopus egg extract.
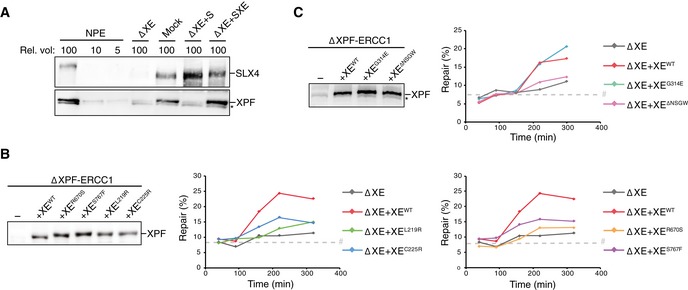
- Mock‐depleted, XPF‐ERCC1‐depleted (ΔXE), and XPF‐ERCC1‐depleted NPE complemented with SLX4 (ΔXE+S) or XPF‐ERCC1 and SLX4 (ΔXE+SXE) were analyzed by Western blot using α‐XPF or α‐SLX4 antibodies. A dilution series of undepleted NPE was loaded on the same blot to determine the degree of depletion. A relative volume of 100 corresponds to 0.2 μl of NPE.
- Replicates of Fig 2B. XPF‐ERCC1‐depleted (ΔXE) and XPF‐ERCC1‐depleted extracts complemented with wild‐type (XEWT) or indicated mutant XPF‐ERCC1 (XEMUT) were analyzed by Western blot using α‐XPF antibodies (left panel). These extracts were used to replicate pICL. Replication intermediates were isolated and digested with HincII, or HincII and SapI, and separated on an agarose gel. Repair efficiency was calculated and plotted (right panels).
- As in (B) but analyzing different mutant complexes. Note: repair levels can differ per batch of individually prepared extract or per depletion experiment and can only be compared within an experiment.
ICL repair‐deficient XPF mutants are proficient in NER
To determine whether the XPF mutations specifically affect ICL repair, we investigated their activity in nucleotide excision repair (NER). During NER, the endonucleases XPF‐ERCC1 and XPG make incisions on either side of a lesion, creating a gap that is subsequently filled in. This gap‐filling DNA synthesis is called unscheduled DNA synthesis (UDS) to differentiate it from the semi‐conservative DNA synthesis that takes place during replication. UDS can be measured on UV‐damaged plasmids incubated in a high‐speed supernatant Xenopus egg extract and used as a readout for NER activity (Fig 3A; Shivji et al, 1994; Gaillard et al, 1996). To this end, we incubated non‐damaged or UV‐damaged plasmid in a non‐replicating Xenopus egg extract in the presence of 32P‐dCTP. The DNA was subsequently isolated and linearized, and the products were separated on an agarose gel (Fig 3B). While a UV‐damaged plasmid showed clear incorporation of 32P‐dCTP indicative of UDS, a non‐damaged plasmid only showed some background incorporation, probably due to nicks created during plasmid preparation (Fig 3B, lanes 1 and 2). To confirm that the UDS on the UV‐damaged template is a result of the repair, we directly monitored cyclobutane pyrimidine dimers and showed that Xenopus egg extract is capable of removing these lesions (Fig EV4C). To further validate that the unscheduled DNA synthesis is caused by NER, we showed that UDS is strongly reduced after depletion of NER factors PCNA as well as XPA (Fig EV4D and E). In addition, we found that immunodepletion of XPF (Fig EV4A) strongly reduced UV‐dependent UDS (Fig 3B, compare lanes 2 and 4, and Fig 3C). The slight increase in UDS compared to the non‐damaged plasmid is likely caused by an incomplete depletion of XPF‐ERCC1 or other repair mechanisms present in the extract. Addition of wild‐type XPF‐ERCC1 (XEWT) to XPF‐depleted extracts fully rescued UDS, while addition of a catalytically inactive XED668A mutant did not support UDS (Fig 3B, compare lanes 5 and 10, Figs 3C and EV4B). This shows that XPF‐ERCC1 is required for UDS in Xenopus egg extract. We then complemented an XPF‐ERCC1‐depleted extract with the XPF‐ERCC1 mutants and found that all mutants were able to rescue the NER defect (Fig 3B and C). This observation was especially striking for the XER670S mutant whose nuclease activity on model DNA templates was strongly reduced (Fig 1D). This finding is consistent with a previous report in which the human equivalent of this mutant was able to make NER incisions, although there was a difference in the position of the incisions compared to the wild‐type protein (Su et al, 2012).
Figure 3. XPF‐ERCC1 mutant complexes are active in nucleotide excision repair (NER).

- Schematic representation of unscheduled DNA synthesis (UDS) during NER on a UV‐treated template in high‐speed supernatant (HSS) egg extract.
- Mock‐depleted, XPF‐ERCC1‐depleted (ΔXE), and XPF‐ERCC1‐depleted extracts complemented with wild‐type (XEWT) or mutant XPF‐ERCC1 (XEMUT) were incubated with untreated or UV‐treated plasmids for 2 h at room temperature in the presence of 32P‐α‐dCTP. Reaction products were isolated, linearized with HincII, and separated on a 0.8% agarose gel. The DNA was visualized by autoradiography to show incorporation of 32P‐α‐dCTP during UDS (upper panel) and stained with SYBR gold for total DNA (lower panel).
- The incorporation of 32P‐α‐dCTP was quantified, the background signal from non‐damaged plasmid was subtracted, and the signal for the mock depletion condition was set to 100% to normalize the data. Error bars represent s.e.m. of three independent experiments. **P = 0.003, ***P = 0.0004, paired t‐test comparing all conditions to the mock. All non‐marked conditions did not show a statistical difference from the mock condition.
Source data are available online for this figure.
Figure EV4. XPF‐ERCC1 mutant complexes are active in NER.
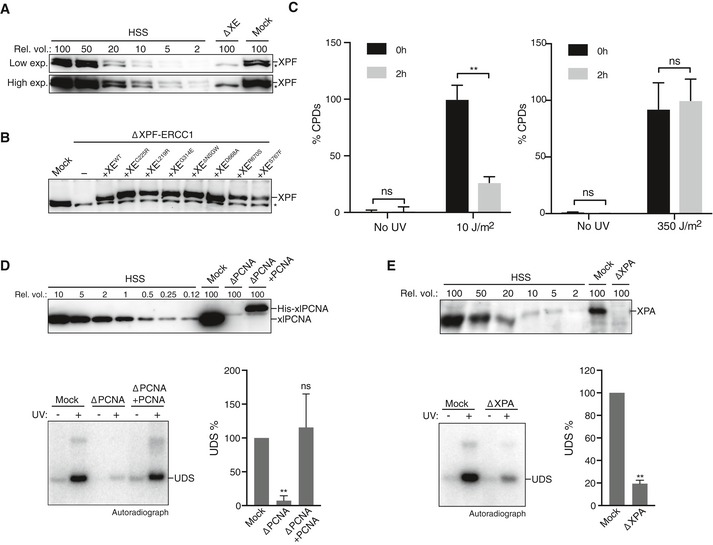
- Mock‐depleted and XPF‐ERCC1‐depleted (ΔXE) high‐speed supernatant (HSS) egg extracts used in Fig 3B were analyzed by Western blot using α‐XPF antibodies. A dilution series of undepleted NPE was loaded on the same blot to determine the degree of depletion. A relative volume of 100 corresponds to 0.2 μl of NPE. *, background band.
- Mock‐depleted, XPF‐ERCC1‐depleted (ΔXE), and XPF‐ERCC1‐depleted HSS complemented with wild‐type (XEWT) or mutant XPF‐ERCC1 (XEMUT) were analyzed by Western blot using α‐XPF antibodies. *, background band.
- Untreated or UV‐treated (10 J/m2 , left panel, 350 J/m2 right panel) plasmid DNA was incubated in HSS for 2 h. Samples were taken at time 0 and 2 h, and DNA was extracted and analyzed by an enzyme‐linked immunosorbent assay (ELISA) for the presence of CPDs. The highest value within one experiment was set to 100%. Error bars represent s.e.m. of three independent experiments. **P = 0.0061, paired t‐test. ns, not significant.
- Mock‐depleted, PCNA‐depleted (ΔPCNA), and PCNA‐depleted HSS complemented with recombinant His‐xlPCNA (ΔPCNA + PCNA) were analyzed by Western blot using α‐PCNA antibodies (Kochaniak et al, 2009). A dilution series of undepleted NPE was loaded on the same blot to determine the degree of depletion. A relative volume of 100 corresponds to 0.2 μl of NPE (top panel). These extracts were incubated with untreated or UV‐treated (350 J/m2) plasmids for 0 or 2 h at room temperature in the presence of 32P‐α‐dCTP. Reaction products were isolated, linearized with HincII, and separated on a 0.8% agarose gel. The DNA was visualized by autoradiography to show incorporation of 32P‐α‐dCTP during UDS (bottom left panel). The signal was quantified, the background signal from non‐damaged plasmid was subtracted, and the signal for the mock depletion condition was set to 100% to normalize the data. Error bars represent s.e.m. of three independent experiments. **P = 0.0059, paired t‐test compared to the mock condition. ns, not significant.
- As in (D) but using mock‐depleted or XPA‐depleted HSS. **P = 0.0014.
Source data are available online for this figure.
In summary, we identified three mutations in the helicase‐like domain (XEL219R, XEC225R and XEΔNSGW) and two in the nuclease domain (XER670S and XES767F) that are defective in ICL repair, while supporting functional NER. Strikingly, this separation of function is achieved by mutations in two different domains of XPF‐ERCC1.
Separation‐of‐function mutations in XPF specifically affect ICL incisions
To determine the mechanism underlying the specific inhibition of ICL repair in these mutants, we examined which step in ICL repair was affected. We previously showed that XPF is required for ICL unhooking (Klein Douwel et al, 2014). However, XPF could also have additional roles downstream, for example, in HR, that might be affected by our mutations (Bergstralh & Sekelsky, 2008). To directly monitor unhooking incisions that take place on the parental strand, we pre‐labeled pICL with 32P‐dCTP using nick translation and replicated it in Xenopus egg extract. Replication intermediates were linearized and separated on a denaturing agarose gel. At early times, the parental strand migrates as a large X‐structure, while after crosslink unhooking during repair, it is converted to a linear molecule and arms (Fig 4A; Knipscheer et al, 2009). The decline of the X‐shaped structures and the accumulation of the linears are a direct readout of unhooking incisions. In XPF‐ERCC1‐depleted extract (Appendix Fig S1), the X‐structures persist and the appearance of linear structures is greatly reduced (Fig 4B–D and Appendix Fig S1B–D), indicating the unhooking incisions are blocked (see also Klein Douwel et al, 2014). Addition of wild‐type XPF‐ERCC1 (XEWT) rescues this incision defect (Fig 4B–D and Appendix Fig S1B–D) whereas the helicase‐like domain mutants XEL219R or XEC225R did not (Fig 4B and Appendix Fig S1B). The XEG314E mutant did not inhibit incisions, while the XEΔNSGW mutant caused a strong reduction in ICL unhooking (Fig 4C and Appendix Fig S1C). This is consistent with our earlier observation that the point mutant is functional in ICL repair while the deletion mutant is not. Finally, we found that the nuclease domain mutants XER670S and XES767F were unable to support efficient incisions (Fig 4D and Appendix Fig S1D). These results show that all our separation‐of‐function mutants are defective in ICL unhooking, which explains their inability to support ICL repair.
Figure 4. XPF‐ERCC1 separation‐of‐function mutants are all defective in ICL unhooking.

-
ASchematic representation of the assay used to directly measure unhooking incisions. 32P‐labeled parental stands are indicated in red. Products before and after ICL unhooking during repair are indicated. HincII digestion of repair intermediates yields X‐structures, arms, and linears under denaturing conditions.
-
BXPF‐ERCC1‐depleted (ΔXE) or XPF‐ERCC1‐depleted egg extract complemented with wild‐type (XEWT) or mutant XPF‐ERCC1 (XEMUT) were incubated with pre‐labeled pICL. Repair products were isolated at indicated times, linearized with HincII, separated on a denaturing agarose gel, and visualized by autoradiography. The X‐structures and linear products were quantified and plotted.
-
C, DAs in (B) but using different XPF‐ERCC1 mutant complexes. Experimental replicates are shown in Appendix Fig S1.
The XPFL219R‐ERCC1 mutant complex is not recruited to the ICL
A possible explanation for why our mutants were not able to support ICL unhooking is that they are not recruited to the site of damage. To test this, we examined XPF recruitment by chromatin immunoprecipitation (ChIP). We replicated pICL in extract depleted of XPF‐ERCC1 and supplemented with wild‐type or mutant XPF‐ERCC1 (Appendix Fig S2A), and performed chromatin immunoprecipitation with XPF antibodies at various time points. An unrelated plasmid (pQuant) was added to the reaction to determine background protein recruitment to undamaged DNA. The co‐precipitated DNA was recovered and amplified by quantitative PCR with primers specific to the ICL region or to pQuant (Fig 5A). Using this assay, we recently showed that XPF is specifically recruited to the ICL at the time of unhooking incisions (Klein Douwel et al, 2014). The exact timing of recruitment can vary as a result of the immunodepletion procedure. We first examined the most N‐terminal mutant XEL219R and found that, in contrast to the wild‐type protein, recruitment of this mutant to the ICL was completely blocked (Fig 5B). In contrast, the XEC225R complex, containing a mutation just six residues further downstream, was recruited to the ICL as efficiently as the wild‐type protein (Fig 5C and Appendix Fig S2B). We then examined the XEΔNSGW mutant and found that it was recruited normally to the ICL (Fig 5D and Appendix Fig S2C). This is striking, because this region was shown to be important for the interaction between XPF and SLX4 (Andersen et al, 2009) and we have previously shown that SLX4 is important for the recruitment of XPF to the ICL. Lastly, we examined the recruitment of the nuclease domain mutants XER670S and XES767F. Both mutants were recruited to the ICL as efficiently as the wild‐type protein (Fig 5E and F and Appendix Fig S6D).
Figure 5. Recruitment of XPF‐ERCC1 mutants to the ICL during repair.
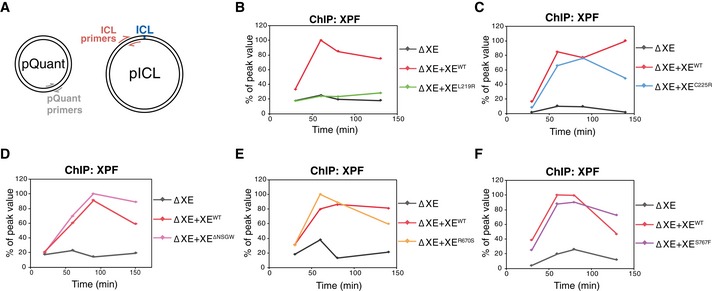
-
ASchematic representation showing the primer locations on pICL and pQuant.
-
BpICL was replicated in XPF‐ERCC1‐depleted (ΔXE) or XPF‐ERCC1‐depleted egg extract supplemented with wild‐type (XEWT) or mutant XPF‐ERCC1 (XEMUT; see Appendix Fig S2). Samples were taken at various times and immunoprecipitated with α‐XPF antibodies. Co‐precipitated DNA was isolated and analyzed by quantitative PCR using the primers depicted in (A). The qPCR data were plotted as the percentage of peak value with the highest value within one experiment set to 100%.
-
C–FAs in (B) but using the indicated XPF‐ERCC1 mutant complexes. Experimental replicates are shown in Appendix Fig S2.
We conclude that all mutants, except for XEL219R, are recruited normally to the site of damage, suggesting the defect in incisions observed for these mutants is due to a defect in proper positioning of the nuclease within the repair complex.
XPF leucine 219 is part of the major binding site between XPF and SLX4
To determine why XEL219R is not recruited to the ICL, we examined recruitment of both SLX4 and XPF to the ICL by ChIP. When we supplemented an XPF‐ERCC1‐depleted reaction with XPF‐ERCC1 only, and not SLX4, XPF was not recruited to the ICL (Figs 6A and EV5A–C; and Klein Douwel et al, 2014). Supplemented SLX4 bound to the ICL and rescued the recruitment of wild‐type XPF‐ERCC1 (Figs 6A and EV5A–C), but not of the XEL219R mutant complex (Figs 6A and EV5A–C). These results show that a single point mutations can abrogate XPF recruitment and strongly suggest that this is caused by a defect in the direct interaction with SLX4.
Figure 6. XPF leucine 219 is part of the major interaction site between XPF and SLX4.
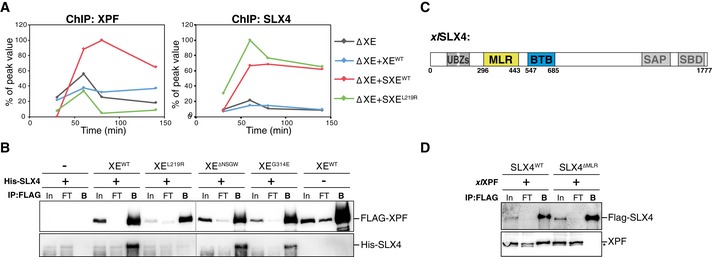
- pICL was replicated in XPF‐ERCC1‐depleted (ΔXE) extract or in XPF‐ERCC1‐depleted extract supplemented with wild‐type XPF‐ERCC1 only (+XEWT), wild‐type XPF‐ERCC1 and SLX4 (+SXEWT), or XPFL219R‐ERCC1 and SLX4 (+SXEL219R; see Fig EV5A). Samples were taken at the indicated times and immunoprecipitated with α‐XPF (left panel) or α‐SLX4 antibodies (right panel). Co‐precipitated DNA was isolated and analyzed by quantitative PCR using ICL or pQuant primers. The qPCR data were plotted as the percentage of peak value with the highest value set to 100%.
- Wild‐type and mutant FLAG‐XPF‐ERCC1 were co‐expressed with His‐SLX4 in Sf9 insect cells. Cells were lysed and XPF was immunoprecipitated via the FLAG‐tag. Samples were analyzed by Western blot using α‐FLAG and α‐His antibodies. In, input; FT, flow‐through fraction; B, fraction bound to beads.
- Schematic representation of xlSLX4 proteins, with the MLR and BTB domains indicated. Experimental replicates are shown in Fig EV5.
- Purified wild‐type FLAG‐SLX4 and FLAG‐SLX4∆MLR were added to Xenopus egg extract. SLX4 was immunoprecipitated via the FLAG‐tag. Samples were analyzed by Western blot using α‐FLAG and α‐XPF antibodies. Line within blot indicates position where irrelevant lanes were removed. *, background band.
Source data are available online for this figure.
Figure EV5. XPF leucine 219 is part of the major interaction site between XPF and SLX4.
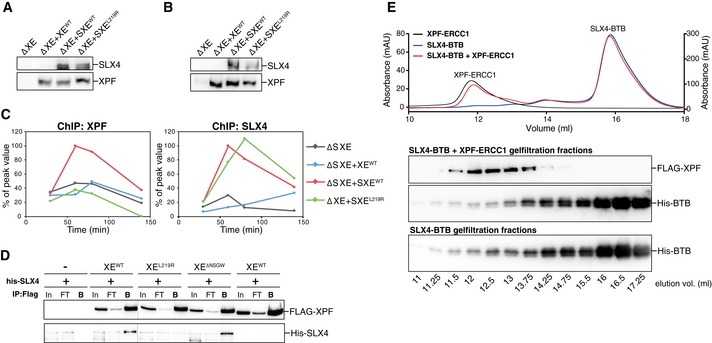
- XPF‐ERCC1‐depleted (ΔXE) and XPF‐ERCC1‐depleted NPE supplemented with XPF‐ERCC1 (+XEWT), XPF‐ERCC1 and SLX4 (+SXEWT), or XPFL219R‐ERCC1 and SLX4 (+SXEL219R) were analyzed by Western blot using α‐XPF and α‐SLX4 antibodies. Extracts were used for Fig 6A.
- As in (A).
- Replicate of Fig 6A. The extracts from (B), with similarly treated HSS, were used to replicate pICL. Samples were taken at the indicated times and analyzed by XPF (left) and SLX4 (right) ChIP using pICL and pQuant primers. The qPCR data were plotted as the percentage of peak value with the highest value set to 100%.
- Replicate of Fig 6C. Wild‐type and mutant FLAG‐XPF‐ERCC1 were co‐expressed with His‐SLX4 in Sf9 insect cells. Cells were lysed and XPF was immunoprecipitated via the FLAG‐tag. Samples were analyzed by Western blot using α‐FLAG and α‐His antibodies. In, input; FT, flow‐through fraction; B, fraction bound to beads.
- Size exclusion chromatography of recombinant XPF‐ERCC1 and BTB domain of SLX4. Superdex 200 gel filtration column elution profile of FLAG‐XPF‐ERCC1, His‐tagged BTB domain, and both proteins combined (top panel). The XPF‐ERCC1 heterodimer eluted at ˜12 ml, while His‐BTB eluted around ˜16 ml. Collected fractions during elution were analyzed by Western blot using α‐XPF and α‐His antibodies (bottom panel). The BTB domain protein does not shift to a higher elution volume when incubated with XPF‐ERCC1 indicating the affinity is not high enough to show binding between the two proteins.
Source data are available online for this figure.
To confirm this, we co‐expressed FLAG‐tagged XEWT and XEL219R with His‐tagged SLX4 in Sf9 insect cells, immunoprecipitated XPF, and examined co‐precipitation of SLX4. His‐SLX4 was enriched after immunoprecipitation of wild‐type XPF‐ERCC1, but not XPFL219R‐ERCC1, indicating this mutant does not bind SLX4 (Figs 6B and EV5D). These findings indicate that XPF's leucine 219 is essential for the interaction between XPF and SLX4 and therefore required for the recruitment of XPF to the site of damage.
Two domains in SLX4 have been implicated in the interaction between SLX4 and XPF. The previously mentioned BTB domain and the MUS312/MEI9 interaction‐like, or MLR, domain (Fig 6C; Fekairi et al, 2009; Kim et al, 2013). To further investigate the importance of the MLR domain for the interaction with XPF, we purified xlSLX4WT and xlSLX4ΔMLR. In contrast to wild‐type SLX4, the ΔMLR mutant was not able to bind XPF from Xenopus egg extract (Fig 6D). This shows that the MLR domain of SLX4 acts as the major interaction site with XPF, which is in line with previous reports in human cells (Kim et al, 2013). Based on our data, this domain most likely interacts with leucine 219 of XPF.
Finally, we set out to examine the role of the interaction between the SLX4 BTB domain and XPF. The hsG325E mutation in XPF abrogates the interaction between XPF and the BTB domain of SLX4 (Andersen et al, 2009). We found that the XEG314E and XEΔNSGW mutants were both able to interact normally with full‐length SLX4 (Figs 6B and EV5D). This is consistent with previous reports showing that the BTB domain is not essential for SLX4 and XPF interaction (Kim et al, 2013; Guervilly et al, 2015). One explanation for these observations is that this interaction is transient and can only be observed in the absence of the major interaction site involving the MLR domain. To be able to study this, we cloned and purified the SLX4 BTB domain alone and examined the interaction with XPF using size exclusion chromatography. However, even using an excess of BTB domain protein, we did not observe an interaction with XPF‐ERCC1 (Fig EV5E). This indicates that this is not a high‐affinity interaction site.
These findings, together with previous reports, support a model in which XPF and SLX4 interact through two binding sites (Fig 7). The first consists of the MLR domain of SLX4 and XPF leucine 219, and possibly a region around this residue. This is a high‐affinity binding site that is responsible for the recruitment of XPF via SLX4 to ICLs. The second comprises the BTB domain of SLX4 and residues 312–315 of XPF. This interaction is transient, but important to promote nuclease activity of XPF possibly by orienting it properly.
Figure 7. Model for ICL repair‐specific features of XPF.
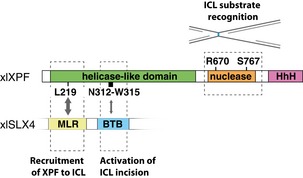
Leucine 219 in the helicase‐like domain of XPF is essential for the interaction of XPF with the MLR domain of SLX4. This interaction mediates the recruitment of XPF to an ICL. Residues 312–315 transiently interact with the BTB domain of SLX4 and are required for the incisions of an ICL by XPF. Arginine 670 and serine 767 in the nuclease domain of XPF are crucial for the recognition of the ICL substrate.
Discussion
Mutations in XPF‐ERCC1 affect several DNA repair pathways and can cause multiple diseases likely due to differential inhibition of these pathways. Using Xenopus egg extracts, we have examined how certain mutations in XPF inhibit ICL repair, while maintaining proficient nucleotide excision repair. We have characterized five separation‐of‐function mutations that reside in the helicase‐like and nuclease domains of XPF. While all these mutants are defective in ICL unhooking, this is caused by different mechanisms. The nuclease domain mutants are normally recruited to the ICL and most likely affect interactions with the DNA template or specific protein–protein interactions important for substrate recognition (Fig 7). The helicase‐like domain mutants are part of a dual interaction site with SLX4. XPF's leucine 219 is part of a high‐affinity interaction site that interacts with the MLR domain of SLX4, while deletion of residues 312–315 of XPF disrupts a transient second interaction site with the BTB domain of SLX4 (Fig 7 and Table 1).
Table 1.
Summary of features of different XPF mutations
| xl | hs | Patient mutation | Clinical features | Nuclease activity | DNA binding | NER | ICL repair | Unhooking | Recruitment to ICL | SLX4 binding |
|---|---|---|---|---|---|---|---|---|---|---|
| XPFL219R | XPFL230R | L230P | FA | + | + | + | − | − | − | − |
| XPFC225R | XPFC236R | C236R | CS v CS/XP/FA | + | + | + | − | − | + | N.D |
| XPFG314E | XPFG325E | N.A. | N.A. | + | + | + | + | + | N.D. | N.D. |
| XPFΔNSGW | XPFΔNSGW | N.A. | N.A. | + | + | + | − | − | + | + |
| XPFR670S | XPFR689S | R689S | FA | − | + | + | − | − | + | N.D. |
| XPFS767F | XPFS786F | N.A. | N.A. | + | + | + | − | − | + | N.D. |
Abbreviations are as follows: +, normal; − absent or defective; NA, not applicable; ND, not determined.
We have previously shown that XPF‐ERCC1 is recruited to the site of damage by SLX4 (Klein Douwel et al, 2014). A specific residue in the helicase‐like domain of XPF has been implicated in the interaction with SLX4 (Yildiz et al, 2002; Andersen et al, 2009). A glycine to glutamic acid mutation at residue 325 of human XPF abrogated the interaction with SLX4 in a yeast two‐hybrid assay (Andersen et al, 2009). This yeast two‐hybrid assay was performed with C‐terminal deletion mutants of SLX4 and the interaction with XPF was pinpointed to the BTB domain. However, these mutants lacked the MLR domain which has also been implicated in the interaction with XPF (Kim et al, 2013). In our hands, the equivalent mutation in xlXPF, G314E, did not abrogate ICL repair or recruitment. Moreover, a deletion mutant in which this glycine and three additional residues around it were removed (XPFΔNSGW) interacted normally with SLX4. Based on these observations, we suggest that the interaction site between the BTB domain of SLX4 and residues 312–315 of XPF is a minor interaction site. This is consistent with our data showing that the isolated BTB domain does not interact strongly with XPF and with results reported by Guervilly et al that show only a slight decrease in XPF binding after mutation of the SLX4 BTB domain (Guervilly et al, 2015). However, this interaction is important because the XPFΔNSGW‐ERCC1 mutant is deficient in ICL unhooking and repair (Figs 2 and 4). Therefore, we propose that this transient interaction site is important for activation of XPF‐ERCC1 by ensuring correct positioning onto its substrate. Interestingly, SLX4 has been shown to stimulate XPF‐ERCC1 activity on model substrates (Hodskinson et al, 2014) which could be mediated through this interaction. Although we cannot exclude that this ICL repair defect of the XPFΔNSGW‐ERCC1 mutant is caused by a different mechanism, the fact that this mutant completely overlaps with a previously identified interaction site strongly supports this explanation.
We and others have shown that the MLR domain of SLX4 is essential for the interaction with XPF (Fig 6 and Kim et al, 2013), but it was not known which site on XPF was involved in this interaction. We now show that the XPF leucine 219 to arginine mutant is defective in binding to SLX4 indicating this is the site that interacts with the MLR domain. Further examination of the residues surrounding leucine 219 is required to better characterize this interaction site. Notably, our ChIP results indicate that the cysteine at position 225 is not required for the interaction. We further show that the XPFL219R‐ERCC1 mutant complex is defective in ICL repair but not in NER. This shows that the interaction with SLX4 is specific to the role of XPF‐ERCC1 in ICL repair. This is consistent with the fact that patients with an L230P mutation suffer from FA and not XP. While previously it was assumed that poor stability of the XEL230P protein was causing the FA phenotype, these data suggest that a functional defect, namely impaired interaction with SLX4, may cause, or contribute to, the disease.
Interestingly, the XPFC225R‐ERCC1 mutant does affect ICL recruitment but is still defective in ICL repair. Possibly this mutant does not prevent binding to SLX4, but does affect the interaction site in a way that it cannot properly position XPF for incisions. Two patients have been identified carrying the C236R (xlC225R) mutation, both show Cockayne syndrome (CS) phenotypes, while only one patient additionally shows a Fanconi anemia phenotype. The Cockayne syndrome phenotype is thought to be caused by a specific defect in transcription‐coupled nucleotide excision repair (TC‐NER). While mutations in XPF are not expected to specifically affect TC‐NER, because it acts downstream in the NER pathway where the transcription‐coupled and global NER pathways have come together, a CS phenotype has been observed previously in patients with mutations in XPF (Kashiyama et al, 2013). Our data show that the xlC225R mutation prevents ICL repair, but does not affect NER. However, Xenopus egg extracts are transcription incompetent and we may therefore not identify a defect in TC‐NER. Why only one of the patients carrying the C236R mutation presents with a clear FA phenotype is currently unclear. Possibly the other patient has an additional mutation or specific genetic background that neutralizes the ICL repair defect.
In addition to the helicase‐like domain mutants, we found two separation‐of‐function mutants in the nuclease domain of XPF. Arginine 670 is located within the active site of xlXPF and mutating it to a serine severely reduces nuclease activity as was shown by us and others (Bogliolo et al, 2013). Nevertheless, our data indicate that the XER670S mutant can still support NER to wild‐type levels. This is in line with previous data showing the human equivalent, hsXER689A, can incise an NER substrate (Enzlin & Schärer, 2002; Staresincic et al, 2009; Su et al, 2012). Interestingly, the human mutant protein did show a shift in incision position, suggesting the residue is not directly involved in catalysis, but contributes to the proper orientation of the active site onto the DNA substrate (Su et al, 2012). This aberrant positioning is apparently not detrimental for NER but does prevent ICL repair in our assays. This is a likely explanation as the DNA template for incision differs in both repair pathways. Moreover, it is supported by the identification of a patient with the hsR689S mutation that suffers from FA, but not XP (Bogliolo et al, 2013).
The S767F mutation is also located in the nuclease domain and structure predictions based on the crystal structure of the nuclease domain of archaeal XPF in complex with DNA, indicating that it could be involved in protein–DNA interaction (Newman et al, 2005). In our experiments, this mutant shows a mild reduction in nuclease activity, is proficient in NER but largely deficient in ICL repair. We propose that, like the arginine 670, this residue is important in positioning the active site specifically on an ICL template likely by direct contact with the DNA. We did not observe reduced DNA binding affinity for these mutants most likely because XPF‐ERCC1 contains multiple DNA interacting domains and it was shown that mutation of at least two of those is required to reduce this affinity (Su et al, 2012).
XPF‐ERCC1 is essential for the repair of ICLs induced by chemotherapy agents, such as derivatives of cisplatin and nitrogen mustards (Kirschner & Melton, 2010). Moreover, high expression of ERCC1 has been associated with poor response to chemotherapy in many cancers and could be a potential target to overcome resistance (McNeil & Melton, 2012). A better understanding of the ICL repair function of XPF‐ERCC1 could potentially lead to the design of ICL‐specific inhibitors that could be beneficial in cancer treatment.
Materials and Methods
Protein expression and purification
His‐tagged hsERCC1 was cloned into pDONR201 (Life Technologies). FLAG‐tagged xlXPF was cloned into pFastBac1 (Life Technologies) and in pDONR201. The XPF mutations (L219R, C225R, G314E, ΔNSGW, R670S, D668A, and S767F) and ERCC1 mutation (K247A/K281A) were introduced in pDONR‐XPF using QuikChange site‐directed mutagenesis protocol. Baculoviruses were produced using the BAC‐to‐BAC system (xlXPFWT), or the BaculoDirect system (hsERCC1 and xlXPFMUTs) following manufacturer's protocol (Life Technologies). Proteins were expressed in suspension cultures of Sf9 insect cells by co‐infection with His‐hsERCC1 (or His‐hsERCC1K247A/K281A) and FLAG‐xlXPF (or FLAG‐xlXPF mutants) viruses for 72 h. Cells from 750 ml culture were collected by centrifugation, resuspended in 30 ml of lysis buffer (50 mM K2HPO4 pH 8.0, 500 mM NaCl, 0.1% NP‐40, 10% glycerol, 0.4 mM PMSF, 1 tablet/50 ml Complete Mini EDTA‐free Protease Inhibitor Cocktail (Roche), 10 mM imidazole), and lysed by sonication. The soluble fraction obtained after centrifugation (40,000 × g for 40 min at 4°C) was incubated for 1 h at 4°C with 1 ml of Ni‐NTA‐agarose (Qiagen) that was pre‐washed with lysis buffer. After incubation, the beads were washed using 50 ml of wash buffer (50 mM K2HPO4 pH 8.0, 300 mM NaCl, 0.1% NP‐40, 10% glycerol, 0.1 mM PMSF, 10 μg/ml apropotin/leupeptin, 20 mM imidazole). The xlXPF‐hsERCC1 complex was eluted in elution buffer (50 mM K2HPO4 pH 8.0, 300 mM NaCl, 0.1% NP‐40, 10% glycerol, 0.1 mM PMSF, 10 μg/ml apropotin/leupeptin, 250 mM imidazole). The eluate was diluted with FLAG‐wash buffer I (20 mM K2HPO4 pH 8.0, 200 mM NaCl, 0.1% NP‐40, 10% glycerol, 0.4 mM PMSF) and incubated for 1 h at 4°C with 500 μl of anti‐FLAG M2 affinity gel (Sigma) that was pre‐washed with FLAG‐wash buffer I. After incubation, the beads were washed with 30 ml of FLAG‐wash buffer I, and subsequently with 30 ml of GF buffer (25 mM Hepes pH 8.0, 200 mM NaCl, 10% glycerol, 5 mM β‐mercaptoethanol). The xlXPF‐hsERCC1 complex was eluted in 3 ml of GF buffer containing 100 μg/ml 3× FLAG peptide (Sigma). The protein was then loaded onto a HiLoad 16/600 Superdex 200 pg gel filtration column (GE Healthcare) equilibrated in GF buffer. Fractions containing the xlXPF‐hsERCC1 heterodimer were eluted between 60 and 70 ml (Enzlin & Schärer, 2002), pooled, and concentrated with an Amicon Ultra‐4 centrifuge filter unit, 30 kDa (Merck Millipore). Protein was aliquoted, flash‐frozen, and stored at −80°C. FLAG‐tagged xlSLX4 was purified as previously described (Klein Douwel et al, 2014). His‐tagged xlSLX4 was cloned into pDONR201 (Life Technologies) and baculoviruses were produced using the BaculoDirect system following manufacturer's protocol (Life Technologies). His‐tagged xlSLX4 was expressed in 150 ml suspension cultures of Sf9 insect cells for 72 h. Cells were collected by centrifugation, resuspended in lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 0.1% NP‐40, 10% glycerol, 0.4 mM PMSF, 10 mM imidazole, 1 tablet/10 ml Complete Mini EDTA‐free Protease Inhibitor Cocktail (Roche)), and lysed by sonication. The soluble fraction obtained after centrifugation (40,000 × g for 40 min at 4°C) was incubated for 1 h at 4°C with 750 μl of Ni‐NTA‐agarose (Qiagen) that were pre‐washed with lysis buffer. After incubation, the beads were washed with wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 0.1% NP‐40, 5% glycerol, 0.1 mM PMSF, 20 mM imidazole, 10 μg/ml apropotin/leupeptin). His‐tagged xlSLX4 protein was eluted in wash buffer containing 250 mM imidazole. The protein was aliquoted and stored at −80°C. The pDONR201 construct for FLAG‐tagged xlSLX4 was used to create the xlSLX4ΔMLR mutant. PCR amplification was used to replace the MLR domain with a short linker containing a KpnI restriction site. Expression and purification of FLAG‐tagged xlSLX4ΔMLR were identical to the wild‐type protein.
Nuclease assay
Nuclease assay was performed as previously described (De Laat et al, 1998). The following primers were obtained (Integrated DNA technologies): SL: 5′‐FAM‐CGCCAG CGC TCGGTTTTTTTTTTTTTTTTTTTTTTCCGAGCGCTGGC‐'3; F1: 5′‐FAM‐ CGCGATGCGG ATCCAA‐3′; F2: 5′‐CCTAGACTTAAGAGGCCAGACTTGGATCCGCATCGC‐3′; F3: 5′‐GGCCTCTTAAGTCTAGG‐3′. For the stem‐loop structure, primer SL was heated for 3 min at 95°C, followed by stepwise cooling to allow annealing (30 min at 60°C, 30 min at 37°C, 30 min at 25°C, 30 min on ice). To assemble the 3′ flap substrate, primer F1 was annealed to primer F2 and F3 in a 1:1:1 ratio and annealed similar to the stem‐loop substrate. Nuclease reactions (15 μl) were carried out in nuclease buffer (50 mM Tris pH 8.0, 0.2 mM MnCl2, 0.1 mg/ml bovine serum albumin, and 0.5 mM β‐mercaptoethanol) containing 100 nmol of substrate DNA and 10–100 nM of recombinant XPF‐ERCC1 wild‐type or mutant protein complex. Reactions were incubated for 30 min at room temperature and stopped by addition of 15 μl denaturing PAGE Gel Loading Buffer II (Life Technologies, Inc.). Samples were heated to 72°C for 3 min, snap‐cooled, and loaded onto a 12% denaturing urea–PAGE gel. Gels were directly measured on a Typhoon phosphor imager (GE Healthcare) on the blue 488 channel.
Fluorescent anisotropy binding assay
Increasing concentrations of protein were incubated with 10 nM of a 3′ flap DNA substrate containing a 5′‐fluorescent FAM label (see Nuclease assay). The reaction was incubated with annealing buffer (25 mM Hepes pH 8.0, 15% glycerol, 0.1 mg/ml BSA, 2 mM CaCl2) in a 384‐well plate (kBioscience) for 1 h at room temperature, and fluorescent anisotropy was measured on a Spectramax I3 (Molecular Devices). The data were fitted using Origin 8.5 to the equation y = a + b × x n/(k n + x n), where x is the protein concentration, y is the fluorescence anisotropy, and k is the K d value.
DNA replication and repair assay in Xenopus egg extracts
DNA replication and preparation of Xenopus egg extracts (HSS and NPE) were performed as described previously (Walter et al, 1998; Tutter & Walter, 2006). Preparation of plasmid with a site‐specific cisplatin ICL (pICL), and ICL repair assays were performed as described (Räschle et al, 2008; Enoiu et al, 2012). Briefly pICL was incubated with HSS for 20 min, following addition of two volumes of NPE (t = 0) containing 32P‐α‐dCTP. Aliquots of replication reaction (4–10 μl) were stopped at various times with ten volumes of Stop Solution II (0.5% SDS, 10 mM EDTA, 50 mM Tris pH 7.5). Samples were incubated with RNase (0.13 μg/μl) followed by proteinase K (0.5 μg/μl) for 30 min at 37°C each. DNA was extracted using phenol/chloroform, ethanol‐precipitated in the presence of glycogen (30 mg/ml), and resuspended in 5–10 μl of 10 mM Tris pH 7.5. ICL repair was analyzed by digesting 1 μl of extracted DNA with HincII, or HincII and SapI, separation on a 0.8% native agarose gel, and quantification using autoradiography. Repair efficiency was calculated as described (Knipscheer et al, 2012).
Unscheduled DNA synthesis
The assay to monitor unscheduled DNA synthesis (UDS) in Xenopus egg extract was adapted from Gaillard et al (1996). A 6.25‐μl reaction containing 2.5 μl HSS and 6 ng/μl non‐treated or UV‐C‐irradiated (350 μJ/m2) pControl was supplemented with 5 mM MgCl2, 0.5 mM DTT, 4 mM ATP, 40 mM phosphocreatine, 0.5 μg creatine phosphokinase, and 80 μCi/ml 32P‐α‐dCTP (3,000 Ci/mmol). Reactions were incubated at room temperature for 2 h and stopped by addition of ten volumes of Stop Solution II (0.5% SDS, 10 mM EDTA, 50 mM Tris pH 7.5). Samples were incubated with proteinase K (0.5 μg/μl) for 30 min at 37°C. DNA was extracted using phenol/chloroform, ethanol‐precipitated in the presence of glycogen (30 mg/ml), and resuspended in 5–10 μl of 10 mM Tris pH 7.5. Extracted DNA (2 μl) was digested with HincII and separated on a 0.8% native agarose gel. The gel was stained with SYBR GOLD (Fisher) and subsequently dried and quantified using autoradiography. The background signal from non‐treated plasmid was subtracted. To compare the levels of UDS between experiments, the signal for wild‐type XPF‐ERCC1 add back was set to 100%. This is because the efficiency of UDS can differ between extract preparations and depletions.
Antibodies and immunodepletions
Antibodies were raised against residues 444–797 of xlXPF, full‐length xlERCC1, and residues 825–1,052 of xlSLX4. Specificity was confirmed using Western blot (Klein Douwel et al, 2014). XPF‐ERCC1 was removed from extract using three rounds of depletion with the α‐ERCC1 serum (HSS and NPE). ERCC1 depletion was described previously (Klein Douwel et al, 2014). For the unscheduled DNA synthesis assay, HSS was depleted using three rounds of an ERCC1 depletion (one volume of PAS was bound to one volumes of anti‐serum or pre‐immune serum, and added to four volumes of HSS) followed by three rounds of depletion with the α‐XPF serum (one volume of PAS was bound to three volumes of anti‐serum or pre‐immune serum, and added to five volumes of HSS). Anti‐FLAG M2 antibody was purchased from Sigma and His‐antibody from Westburg.
Incision assay
Incision assay was performed as described in Klein Douwel et al (2014). Briefly, pICL and pQuant were labeled via nick translation. pQuant was added as an internal control to allow accurate calculation of incision efficiency. pICL (225 ng) and pQuant (11.25 ng) were incubated in 1.5 units of NB‐BSR DI enzyme (NEB) and 1× NEBuffer 2 for 30 min at room temperature. Subsequently, 11 μl of DNA Polymerase I mix (5 units of DNA polymerase I (NEB), dATP, dGTP, dTTP (0.5 mM each), dCTP (0.4 μM), 32P‐α‐dCTP (3.3 μM) in 1× NEBuffer 2) was added and this was incubated for 3 min at 16°C. The reaction was stopped with 180 μl of Stop Solution II, treated with proteinase K, and phenol/chloroform‐extracted. Excess label was removed using a Micro Bio‐Spin 6 Column (Bio‐Rad). After ethanol precipitation, the pellet was resuspended in 5 μl of ELB (10 mM HEPES‐KOH pH 7.7, 50 mM KCl, 2.5 mM MgCl2, and 250 mM sucrose). The labeled plasmid (pICL*) was used in a replication reaction and samples at various times were extracted and digested with HincII. Fragments were separated on a 0.8% alkaline denaturing agarose gel for 18 h at 0.85 Volts/cm, after which the gel was dried and exposed to a phosphor screen. Quantification was performed using ImageQuant software (GE healthcare). The highest value was set at 100% for the X‐shape and the linear products.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described (Pacek et al, 2006). Briefly, reaction samples were crosslinked with formaldehyde, sonicated to yield DNA fragments of roughly 100–500 bp, and immunoprecipitated with the indicated antibodies. Protein–DNA crosslinks were reversed and DNA was phenol/chloroform‐extracted for analysis by quantitative real‐time PCR with the following primers: ICL (5′‐AGCCAGATTTTTCCTCCTCTC‐3′ and 5′‐CATGCATTGGTTCTGCACTT‐3′) and pQuant (5′‐TACAAATGTACGGCCAGCAA‐3′ and 5′‐GAGTATGAGGGAAGCGGTGA‐3′). The values from pQuant primers were subtracted from the values for pICL primers.
Immunoprecipitations
Proteins were expressed in adherent cultures of Sf9 insect cells in 6‐well plates by co‐infection with His‐hsERCC1, His‐xlSLX4, and FLAG‐xlXPF (or FLAG‐xlXP mutants) viruses for 72 h. Cells were resuspended in medium and collected by centrifugation, resuspended in 250 μl of lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1% Triton, 4 mM EDTA, 10 μg/ml apropotin/leupeptin), and lysed by sonication. After centrifugation (20,000 × g for 20 min at 4°C), 200 μl of soluble fraction was incubated for 30 min at 4°C with 8 μl of FLAG M2 beads (Sigma‐Aldrich) that were pre‐washed with lysis buffer. After incubation, the beads were washed using 2 ml lysis buffer. Beads were taken up in 50 μl of 2× SDS sample buffer and incubated for 5 min at 95°C. Proteins were loaded on SDS–PAGE and visualized by Western blot using respective antibodies.
For immunoprecipitations from Xenopus egg extract, FLAG‐tagged xlSLX4WT or xlSLX4ΔMLR protein was added to NPE/HSS at a concentration of 5 ng/μl. To each 20 μl extract, 45.5 μl of IP buffer (1×ELB salts, 0.25 M sucrose, 75 mM NaCl, 2 mM EDTA, 10 μg/ml apropotin/leupeptin, 0.1% NP‐40) and 10 μl pre‐washed FLAG M2 beads (Sigma‐Aldrich) were added. Beads were incubated for 90 min at 4°C and subsequently washed using 2.5 ml IP buffer. Beads were taken up in 30 μl of 2× SDS sample buffer and incubated for 5 min at 95°C. Proteins were loaded on SDS–PAGE and visualized by Western blot using respective antibodies.
Author contributions
DKD and PK designed the experiments. DKD performed the majority of the experiments. WSH performed IP assays from extract and insect cells. RACMB performed some of the ChIP assays. DKD analyzed the data, and DKD and PK discussed the data with the help of the other authors. DKD and PK wrote the manuscript and WSH and RACMB commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 6
Acknowledgements
This work was supported by the Netherlands organization for Scientific Research (VIDI 700.10.421 to P.K.) and a project grant from the Dutch Cancer Society (KWF HUBR 2015‐7736 to P.K.). We thank N. Martin‐Pintado for assistance with the anisotropy assays and suggestions on the manuscript. We also thank J.C. Walter for the xlPCNA antibody and K.A. Cimprich for the xlXPA antibody. In addition, we thank J.C. Walter and J.A.F. Marteijn for suggestions on the manuscript, the Hubrecht animal caretakers for animal support, and the other members of the Knipscheer laboratory for feedback.
The EMBO Journal (2017) 36: 2034–2046
See also: UB Abdullah et al (July 2017) and OD Schärer (July 2017)
References
- Ahmad A, Enzlin JH, Bhagwat NR, Wijgers N, Raams A, Appledoorn E, Theil AF, Hoeijmakers JH, Vermeulen W, Jaspers NG, Schärer OD, Niedernhofer LJ (2010) Mislocalization of XPF‐ERCC1 nuclease contributes to reduced DNA repair in XP‐F patients. PLoS Genet 6: e1000871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SL, Bergstralh DT, Kohl KP, LaRocque JR, Moore CB, Sekelsky J (2009) Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure‐specific endonucleases in DNA repair and recombination. Mol Cell 35: 128–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh DT, Sekelsky J (2008) Interstrand crosslink repair: can XPF‐ERCC1 be let off the hook? Trends Genet 24: 70–76 [DOI] [PubMed] [Google Scholar]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, Casado JA, Baños R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NGJ, Schärer OD, de Winter JP et al (2013) Mutations in ERCC4, encoding the DNA‐repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92: 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles M, Lally J, Fadden AJ, Mouilleron S, Hammonds T, McDonald NQ (2012) Fluorescence‐based incision assay for human XPF‐ERCC1 activity identifies important elements of DNA junction recognition. Nucleic Acids Res 40: e101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzowska M, Graham TGW, Sobeck A, Waga S, Walter JC (2015) Regulation of the Rev1‐pol ζ complex during bypass of a DNA interstrand cross‐link. EMBO J 34: 1971–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard P‐HL, McIntyre RE, Sanger Mouse Genetics Project , Gallagher F, Kettunen MI, Lewis DY, Brindle K, Arends MJ, Adams DJ, Patel KJ (2011) Disruption of mouse Slx4, a regulator of structure‐specific nucleases, phenocopies Fanconi anemia. Nat Genet 43: 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laat WL, Appeldoorn E, Jaspers NGJ, Hoeijmakers JHJ (1998) DNA structural elements required for ERCC1‐XPF endonuclease activity. J Biol Chem 273: 7835–7842 [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross‐links in mammalian cells. Mol Cell Biol 20: 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V (2015) Update of the human and mouse Fanconi anemia genes. Hum Genomics 9: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoiu M, Jiricny J, Schärer OD (2012) Repair of cisplatin‐induced DNA interstrand crosslinks by a replication‐independent pathway involving transcription‐coupled repair and translesion synthesis. Nucleic Acids Res 40: 8953–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enzlin JH, Schärer OD (2002) The active site of the DNA repair endonuclease XPF‐ERCC1 forms a highly conserved nuclease motif. EMBO J 21: 2045–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR, Russell P, Fuchs RP, McGowan CH, Gaillard PHL (2009) Human SLX4 is a holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138: 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC (2011) Nucleotide excision repair of DNA: the very early history. DNA Repair 10: 668–672 [DOI] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Schärer OD, Walter JC (2011) Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146: 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard PH, Martini EM, Kaufman PD, Stillman B, Moustacchi E, Almouzni G (1996) Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell 86: 887–896 [DOI] [PubMed] [Google Scholar]
- Gillet LCJ, Schärer OD (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 106: 253–276 [DOI] [PubMed] [Google Scholar]
- Guervilly J, Takedachi A, Naim V, Scaglione S, Chawhan C, Lovera Y, Despras E, Kuraoka I, Kannouche P, Rosselli F, Gaillard P‐HL (2015) The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol Cell 57: 123–137 [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Wada K, Matsumoto K, Moriya M (2015) Physical interaction between SLX4 (FANCP) and XPF (FANCQ) proteins and biological consequences of interaction‐defective missense mutations. DNA Repair 35: 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodskinson MRG, Silhan J, Crossan GP, Garaycoechea JI, Mukherjee S, Johnson CM, Schärer OD, Patel KJ (2014) Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF‐ERCC1 in DNA crosslink repair. Mol Cell 54: 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JC, Svoboda DL, Reardon JT, Sancar A (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc Natl Acad Sci USA 89: 3664–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li T, Yoshiura K, Utani A, Hirano A (2013) Malfunction of nuclease ERCC1‐XPF results in diverse clinical manifestations and causes cockayne syndrome, Xeroderma Pigmentosum, and Fanconi Anemia. Am J Hum Genet 92: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Spitz GS, Veturi U, Lach FP, Auerbach AD, Smogorzewska A (2013) Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121: 54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K, Melton DW (2010) Multiple roles of the ERCC1‐XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res 30: 3223–3232 [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RACM, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P (2014) XPF‐ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54: 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication‐dependent DNA interstrand cross‐link repair. Science 326: 1698–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Schärer OD, Walter JC (2012) Replication‐coupled DNA interstrand cross‐link repair in Xenopus egg extracts. Methods Mol Biol 920: 221–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, van Oijen AM (2009) Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem 284: 17700–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD (2000) Repair of an interstrand DNA cross‐link initiated by ERCC1‐XPF repair/recombination nuclease. J Biol Chem 275: 26632–26636 [DOI] [PubMed] [Google Scholar]
- Li L, Peterson CA, Lu X, Legerski RJ (1995) Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol Cell Biol 15: 1993–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Räschle M, Joukov V, Walter JC (2011) Mechanism of RAD51‐dependent DNA interstrand cross‐link repair. Science 333: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamrak NE, Shimamura A, Howlett NG (2016) Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev doi: 10.1016/j.blre.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil EM, Melton DW (2012) DNA repair endonuclease ERCC1‐XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res 40: 9990–10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M, Murray‐Rust J, Lally J, Rudolf J, Fadden A, Knowles PP, White MF, McDonald NQ (2005) Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J 24: 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo HB, Hoeijmakers JHJ, Kanaar R (2004) The structure‐specific endonuclease Ercc1‐Xpf is required to resolve DNA interstrand cross‐link‐induced double‐strand breaks. Mol Cell Biol 24: 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GTJ, Meinecke P, Kleijer WJ, Vijg J, Jaspers NGJ, Hoeijmakers JHJ (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444: 1038–1043 [DOI] [PubMed] [Google Scholar]
- Osorio A, Bogliolo M, Fernández V, Barroso A, de la Hoya M, Caldés T, Lasa A, Ramón y Cajal T, Santamariña M, Vega A, Quiles F, Lázaro C, Díez O, Fernández D, González‐Sarmiento R, Durán M, Piqueras JF, Marín M, Pujol R, Surrallés J et al (2013) Evaluation of rare variants in the new Fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum Mutat 34: 1615–1618 [DOI] [PubMed] [Google Scholar]
- Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC (2006) Localization of MCM2‐7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell 21: 581–587 [DOI] [PubMed] [Google Scholar]
- Räschle M, Knipscheer P, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC (2008) Mechanism of replication‐coupled DNA interstrand crosslink repair. Cell 134: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MK, Grey SJ, Strausfeld UP, Wood RD, Blow JJ (1994) Cip1 inhibits DNA replication but not PCNA‐dependent nucleotide excision‐repair. Curr Biol 4: 1062–1068 [DOI] [PubMed] [Google Scholar]
- Spivak G (2015) Nucleotide excision repair in humans. DNA Repair 36: 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand‐Sauthier I, Giglia‐Mari G, Clarkson SG, Vermeulen W, Schärer OD (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 28: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Orelli B, Madireddy A, Niedernhofer LJ, Schärer OD (2012) Multiple DNA binding domains mediate the function of the ERCC1‐XPF protein in nucleotide excision repair. J Biol Chem 287: 21846–21855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutter AV, Walter JC (2006) Chromosomal DNA replication in a soluble cell‐free system derived from Xenopus eggs. Methods Mol Biol 322: 121–137 [DOI] [PubMed] [Google Scholar]
- Walter J, Sun L, Newport J (1998) Regulated chromosomal DNA replication in the absence of a nucleus. Mol Cell 1: 519–529 [DOI] [PubMed] [Google Scholar]
- Yildiz Ö, Majumder S, Kramer B, Sekelsky JJ (2002) Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI‐9 to generate meiotic crossovers. Mol Cell 10: 1503–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC (2015) DNA interstrand cross‐link repair requires replication‐fork convergence. Nat Struct Mol Biol 22: 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 6