

Abstract
Early postnatal mammals, including human babies, can perform only basic motor tasks. The acquisition of skilled behaviors occurs later, requiring anatomical changes in neural circuitry to support the development of coordinated activation or suppression of functionally related muscle groups. How this circuit reorganization occurs during postnatal development remains poorly understood. Here we explore the connectivity between corticospinal (CS) neurons in the motor cortex and muscles in mice. Using trans-synaptic viral and electrophysiological assays, we identify the early postnatal reorganization of CS circuitry for antagonistic muscle pairs. We further show that this synaptic rearrangement requires the activity-dependent, non-apoptotic Bax/Bak-caspase signaling cascade. Adult Bax/Bak mutant mice exhibit aberrant co-activation of antagonistic muscle pairs and skilled grasping deficits but normal reaching and retrieval behaviors. Our findings reveal key cellular and molecular mechanisms driving postnatal motor circuit reorganization and the resulting impacts on muscle activation patterns and the execution of skilled movements.
Introduction
The acquisition of appropriate muscle activation patterns for skilled movements requires motor circuit reorganization during postnatal development (Dominici et al., 2011; Gerber et al., 2010; Martin, 2005). Initially, human infants exhibit a high prevalence of co-activation of antagonistic muscles, which diminishes as infants grow (Myklebust et al., 1986; Pang and Yang, 2001; Teulier et al., 2012). Gradually, a permanent shift occurs to mainly reciprocal activation of antagonistic muscles, as typically seen in adults (Myklebust and Gottlieb, 1993). This conversion from co-activation to reciprocal activation of antagonistic muscles is an integral part of motor development, but how these motor circuits are reorganized during postnatal stages to generate the precise muscle activation patterns required for skilled movements remains unclear.
Extensive studies over the last two decades reveal how initial motor circuits are genetically determined by transcription factors as well as axon guidance molecules (Arber, 2012; Bonanomi and Pfaff, 2010; Goulding, 2009; Jessell, 2000; Kiehn, 2011). In contrast, our knowledge is quite limited about how initial motor circuits become reorganized to form more sophisticated adult circuits by neuronal activity-driven mechanisms. In other regions of the central nervous system, different molecular mechanisms have been reported to underlie activity-dependent circuit modification. For example, neuronal activity can induce various changes in gene expression through transcription factors such as CREB and MEF2, which regulate circuit remodeling and function (Flavell and Greenberg, 2008). In addition, the non-apoptotic Bax-caspase signaling pathway, triggered by neuronal activity, has been shown to control some late aspects of neural circuit formation and synaptic plasticity (Jiao and Li, 2011; Li et al., 2010; Miura, 2012; Ohsawa et al., 2010; Simon et al., 2012; Williams et al., 2006).
Corticospinal (CS) circuits are essential for complex voluntary motor skills by coordinating the activation of multiple limb muscles (Porter and Lemon, 1993). In all mammals, CS axons descend from the motor cortex to connect with interneurons (INs) in the spinal cord to regulate motor neuron activity (Kalaska, 2009; Lemon, 2008). Disruption of these disynaptic CS connections results in varying degrees of skilled movement deficits in rodents, cats, and primates (Alstermark and Isa, 2012). The emergence of skilled behaviors during development is correlated with CS axon remodeling, and it has been postulated that this circuit reorganization occurs in an activity-dependent manner after initial CS circuits have been formed (Martin, 2005). Previous studies in cats reveal that blocking motor experience or motor cortex activity causes defects in CS axon remodeling in the spinal cord, leading to permanent impairments in skilled movements (Friel et al., 2007; Friel and Martin, 2005; Martin et al., 2000). Additionally, feline motor maps for interjoint muscle synergies develop during postnatal stages, presumably due to the contemporaneous reorganization of CS circuitry (Chakrabarty and Martin, 2000). Although activity-dependent CS axon remodeling strongly suggests that CS connectivity is also reorganized during postnatal development, how this occurs during maturation to control precise flexor and extensor muscle activity remains unclear.
Here we explore the connectivity between CS neurons in the motor cortex and functionally related muscle pairs in early postnatal and adult mice. Anatomical analyses at circuit levels using retrograde trans-synaptic viruses (PRVs) reveal that in early postnatal mice, relatively distinct populations of CS neurons establish circuits with individual muscles of a related antagonistic pair, whereas in adult mice, cortical circuits for these muscles diverge from common CS neurons. Accordingly, muscle activity in response to cortical stimulation shows co-activation of antagonistic muscle pairs in early postnatal mice, but not in adults. This indicates that CS circuits controlling antagonistic muscle pairs undergo dramatic synaptic reorganization during early postnatal development. Furthermore, PRV-based circuit analyses reveal that this circuit rearrangement of antagonistic muscle pairs is dependent upon signaling by the activity-dependent, non-apoptotic Bax/Bak-caspase pathway. Aberrant co-activation of flexor and extensor muscles, similar to that observed in early postnatal mice, is seen in adult Bax/Bak double mutant mice. Finally, these Bax/Bak double mutant mice show deficits in skilled grasping behaviors without affecting early reaching or retrieval behaviors in a pellet-reaching task. Our findings therefore reveal that acquisition of appropriate flexor and extensor muscle activities for skilled movements requires synaptic reorganization of antagonistic CS circuits driven by the activity-dependent, Bax/Bak-caspase pathway during development.
Results
Visualization of CS circuits by PRV-tracing assays
To visualize the CS neurons that form disynaptic connections with selective MN pools through INs (CS-IN-MN), we used retrograde, trans-synaptic PRVs expressing fluorescent proteins or LacZ (PRV-152/PRV-eGFP, PRV-614/PRV-RFP, or PRV-BaBlu/PRV-LacZ) (Banfield et al., 2003; Kerman et al., 2003; Smith et al., 2000). When PRV-eGFP was injected intramuscularly into the rectus femoris muscle (Rf) at postnatal (P) day 14, it was retrogradely transported to infect MNs that innervate the muscle (1st order neurons at 1 day post injection (dpi), then INs (2nd order neurons at 2 dpi) that synapse on the 1st order MNs, and finally Ctip2+ CS neurons (3rd order neurons at 3.5 dpi) in layer V of the motor cortex (Figures 1A–1E). At 1–3 dpi (P15-P17), we did not find any neurons labeled in the cortex (Figures S1B–S1D), red nucleus, or vestibular nucleus, and only observed a few labeled neurons within the medullary reticular formation at 3 dpi (P17) (54 ± 12, n=3 animals; data not shown). At 3.5 dpi (P18), PRV+ neurons were detected in the red nucleus, vestibular nucleus, and medullary reticular formation (Figures S1E and S1J–S1L). These data suggest that the majority of labeled neurons in the cortex at 3.5 dpi are CS neurons that form disynaptic connections with MNs in the spinal cord. We found that PRV-labeled CS neurons were restricted to the motor area in the cortex (Figure S2). Similar time-course studies were performed for various forelimb and hindlimb muscles in adult mice to visualize precisely disynaptic CS circuits by PRVs (Figures S1F–S1I, S1M–S1O, and S1S–S1V). The number of MNs, INs, and CS neurons labeled by PRVs were highly correlated across different injected mice (Figures S1P–S1Q), indicating that PRV-tracing assays are reliable and reproducible.
Figure 1. Anatomical reorganization of IN-MN connectivity controlling functionally related muscle pairs during development.
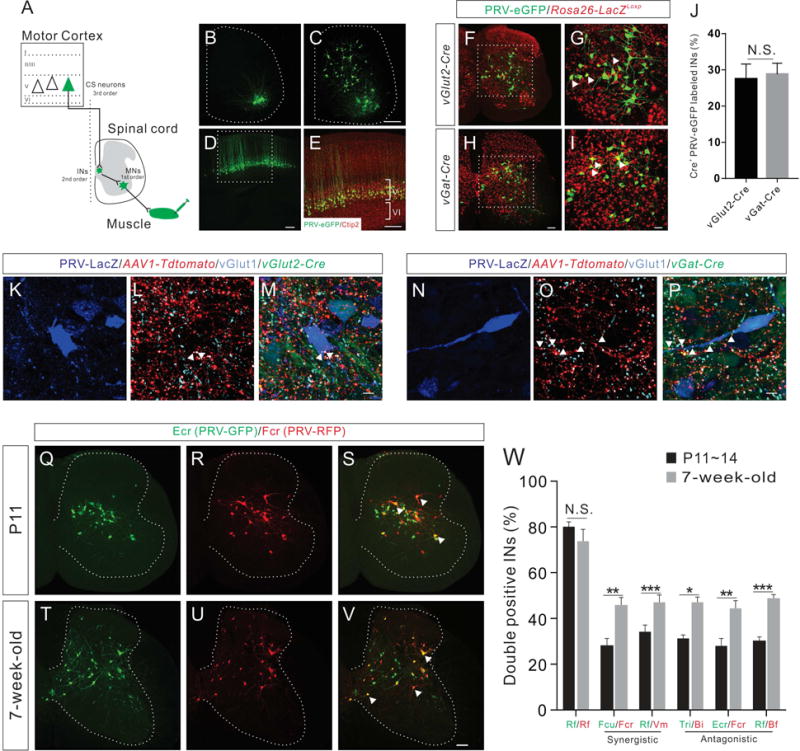
(A) Schematic diagram showing the PRV tracing strategy to label CS circuit pathways. Intramuscular injection of retrograde PRV labels muscles, then first order MNs, second order INs, and finally third order CS neurons.
(B–E) Representative images showing PRV-labeled first order MNs (B, 1 day post injection, dpi, n=6), second order INs (C, 2 dpi, n=5), and third order CS neurons (D, 3.5 dpi, n=8) following PRV-eGFP injection into the Rf muscle at P14. (E) Higher magnification view of the boxed area in (D) showing that PRV-labeled cortical neurons are positive for the layer V marker Ctip2 (red).
(F–I) Representative images showing PRV-labeled INs following PRV-eGFP injection into the Rf muscle of vGlut2-Cre-Rosa26-LacZLoxp (F) and vGat-Cre-Rosa26-LacZLoxp (H) mice at P14. (G and I) Higher magnification views of the boxed area in (F and H) showing that a subset of PRV-labeled INs is also LacZ+ (red, representative INs are indicated by white arrowheads).
(J) The percentage of PRV-eGFP+/LacZ+ INs in vGlut2-Cre-Rosa26-LacZLoxp mice (n=9) is similar (P=0.7952) to that of vGat-Cre-Rosa26-LacZLoxp mice (n=9).
(K–P) Representative cervical spinal cord images showing that vGlut2-Cre-expressing excitatory (K–M) and vGat-Cre-expressing inhibitory (N–P) PRV-LacZ+ INs receiving putative CS inputs (tdTomato+/vGlut1+, indicated by white arrowheads) from 5-week-old mice.
(Q–V) Representative images of cervical spinal cord showing INs labeled by dual-color PRV tracing from Ecr/Fcr antagonistic muscle pair at P11 and 7-week-old stages (representative double-labeled INs are indicated by white arrowheads).
(W) Percentage of double-labeled INs in P11~14 (black bar) and 7-week-old adult mice (grey bar) by dual-color PRV tracing for different synergistic and antagonistic muscle pairs showing the significant increase of double positive INs in adult animals for all muscle pairs except Rf/Rf control: Rf/Rf, P14 (n=6), adult (n=6), P=0.3312; Fcu/Fcr, P11 (n=8), adult (n=8), P=0.0061; Rf/Vm, P14 (n=8), adult (n=8), P=0.0004; Tri/Bi, P11 (n=8), adult (n=4), P=0.0434; Ecr/Fcr, P11 (n=8), adult (n=8), P=0.0085; and Rf/Bf, P14 (n=8), adult (n=8), P<0.0001. Where “n” represents the number of mice used in each experiment. Scale bars, 10 μm (M and P), 50 μm (I), 100 μm (H and V), 200 μm (C–E). See also Figures S1–S3.
To test whether PRVs cross inhibitory synapses in addition to excitatory synapses, we genetically marked inhibitory INs and excitatory INs by crossing vGat-Cre or vGlut2-Cre mice to LacZ reporter mice, respectively (Vong et al., 2011). By injecting PRV-eGFP into the Rf muscle at P14 to label INs, we found that the number of PRV-eGFP labeled inhibitory INs was similar to the number of excitatory INs (Figures 1F–1J), suggesting that PRVs cross inhibitory synapses as efficiently as excitatory synapses. Next, to determine whether PRV-labeled inhibitory and excitatory INs receive direct CS inputs, we injected adeno-associated virus 1 (AAV1)-CAG-tdTomato into the motor cortex of 5-week-old adult vGlut2-Cre and vGat-Cre mice that received injections of PRV-LacZ into their forelimb muscles (biceps brachii (Bi) and triceps brachii (Tri)). We found that both excitatory (Figures 1K–1M) and inhibitory (Figures 1N–1P) INs received tdTomato+ putative direct CS inputs that also express presynaptic excitatory marker vGlut1. Together, these data indicate that PRVs are unique and effective tools for visualizing CS circuits in mice.
Anatomical reorganization of IN-MN connectivity during development
To determine how CS neurons establish circuits with functionally related (synergistic or antagonistic) muscle pairs, we examined CS circuit connectivity in early postnatal and adult animals using a dual-color PRV tracing assay (Figures S3A–S3K). We first examined the developmental changes in connectivity between INs and MNs innervating synergistic or antagonistic muscle pairs. When we injected PRV-eGFP and PRV-RFP into an antagonistic muscle pair (extensor carpi radialis (Ecr)/flexor carpi radialis (Fcr) in the forelimb) of P9 mice, approximately 30% of the PRV-infected INs were labeled with both markers (eGFP+/RFP+) at 2 dpi (P11) (Figures 1Q–1S and 1W). These double-labeled INs represent shared INs projecting to both Ecr and Fcr MNs. Interestingly, in 7-week-old adult mice, Ecr/Fcr muscle injections of PRVs resulted in ~ 44% double-labeled INs (Figures 1T–1V and 1W). Similarly, we observed age-related increases in double-labeled INs in all functionally related synergistic and antagonistic muscle pairs that were tested using our dual-color PRV tracing assay (Figure 1W).
Anatomical reorganization of CS-IN-MN connectivity for antagonistic muscle pairs during development
To examine further the developmental changes in connectivity between CS neurons and IN/MN networks in the spinal cord, we injected PRV-eGFP and PRV-RFP into a synergistic muscle pair (flexor carpi ulnaris (Fcu)/Fcr in the forelimb or Rf/vastus medialis (Vm) in the hindlimb) of early postnatal (P9~14) and 7-week-old adult mice. PRV-eGFP and PRV-RFP injections into the same muscle (Rf) were performed as a control (Figures S3L–S3Q and S3S). For the synergistic muscle pair, approximately 80% of the PRV-infected CS neurons were labeled with both markers in early postnatal and adult mice (Figures 2A and 2C). These double-labeled CS neurons likely control the synergistic muscles simultaneously. We then injected PRV-eGFP and PRV-RFP into an antagonistic forelimb muscle pair in P9 mice, the elbow extensor of Tri and the elbow flexor of Bi, respectively. In contrast to the synergistic muscles, only about a half of the PRV-infected CS neurons were both eGFP+ and RFP+ at 3 dpi (P12) (Figures 2B and 2C), indicating that the singly-labeled CS neurons form circuits with only one muscle of the antagonistic pair in early postnatal mice (Figure 2D, right panel, muscle specific CS neurons). Intriguingly, in 7-week-old adult mice, Tri/Bi muscle injections of PRVs produced about 80% double-labeled CS neurons (Figures 2B and 2C). We observed similar age-related increases in double-labeled CS neurons for other pairs of antagonistic muscles (Ecr/Fcr, and Rf/biceps femoris (Bf) in the hindlimb) (Figures 2C and S3R). Taken together, these data suggest that CS synaptic reorganization within antagonistic circuits occurs during development.
Figure 2. Anatomical reorganization of CS connectivity for antagonistic muscle pairs during development.
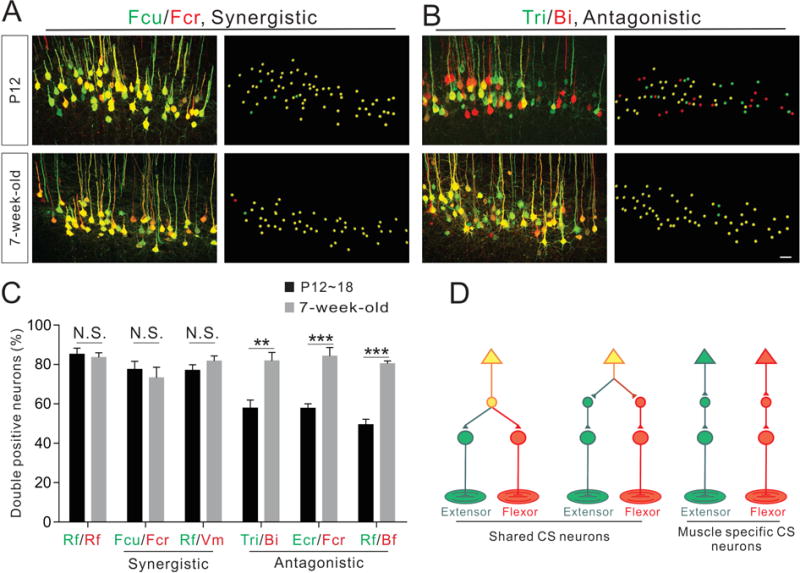
(A–B) Representative images (left: confocal and right: imaris reconstruction) of CS neurons labeled by dual-color PRV tracing from a pair of synergistic muscles (Fcu/Fcr, A) and a pair of antagonistic muscles (Tri/Bi, B) at P12 (top panels) and 7-week-old (bottom panels) wild-type mice.
(C) Percentage of double-labeled CS neurons in early postnatal (black bar) and adult mouse (grey bar) muscle pairs (synergistic and antagonistic) following dual-color PRV tracing. Significant increases in double-positive CS neurons were observed in adult animals for all antagonistic muscle pairs, but not synergistic muscle pairs or the Rf/Rf control injections: Rf/Rf, P18 (n=3), adult (n=6), P=0.7017; Fcu/Fcr, P12 (n=4), adult (n=4), P=0.5495; Rf/Vm, P18 (n=6), adult (n=4), P=0.2748; Tri/Bi, P12 (n=4), adult (n=5), P=0.0053; Ecr/Fcr, P12 (n=6), adult (n=6), P=0.0003; and Rf/Bf, P18 (n=6), adult (n=6), P<0.0001.
(D) Model summarizing the connectivity of disynaptic CS circuits. Where “n” represents the number of mice used in each experiment. Scale bar, 50 μm (B). See also Figure S2.
Functional reorganization of CS connectivity to antagonistic muscle pairs during development
To test the functionality of the CS circuits connecting the motor cortex to various muscle pairs before and after circuit rearrangement, we performed intracortical microstimulation (ICMS) in the motor cortex and recorded electromyograms (EMGs) from pairs of synergistic (Fcu/Fcr) and antagonistic (Bi/Tri) forelimb muscles in early postnatal (P16~19) and adult 3-month-old mice (Figures 3A and 3I). EMG recordings from the synergistic muscle pair following threshold ICMS evoked co-activation in early postnatal wild-type mice (69 sites from 4 mice) (Figure 3E). We calculated the integrated EMG values for each muscle and derived a co-activation index for the synergistic muscle pair Fcu/Fcr (co-activation index = the integrated EMG value for the flexor Fcu muscle divided by the integrated EMG value for the flexor Fcr muscle) for individual sites from each animal. A plot of this index revealed widespread co-activation of this muscle pair in the forelimb motor cortex in early postnatal mice (Figure 3B). We then recorded EMGs from adult wild-type mice (67 sites from 3 mice) and observed similar co-activation patterns for the same synergistic muscles (Fcu/Fcr) (Figure 3G), resulting in a co-activation map that closely resembled that of early postnatal mice (Figures 3B and 3C). Furthermore, cumulative frequency curves of the synergistic muscle co-activation index (plot of all sites from both groups) from early postnatal and adult mice were very similar (Figure 3D). Therefore, these electrophysiological assays showed co-activation of this synergistic muscle pair both in early postnatal and adult mice (Figures 3F and 3H).
Figure 3. Functional reorganization of CS connectivity for antagonistic muscle pairs during development.
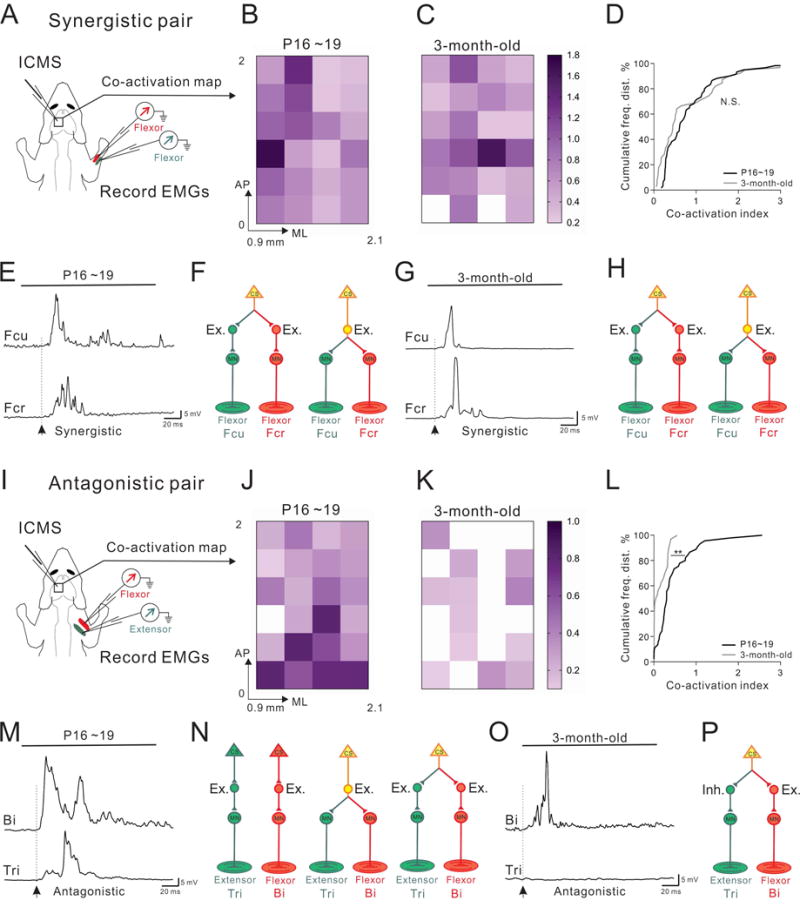
(A) Experimental strategy for recording ICMS-evoked muscle EMGs from the synergistic Fcr/Fcu muscle pair.
(B–C) Color-coded plots of the co-activation indices of Fcu and Fcr muscles in P16~19 (B, n=69 sites from 4 mice) and 3-month-old adult mice (C, n=67 sites from 3 mice). AP: Anterior-Posterior, ML: Medial-lateral, relative to the Bregma.
(D) Cumulative histograms showing similar (P=0.074) distributions of co-activation indices in early postnatal (black) and adult (gray) mice.
(E and G) Representative EMG traces from the synergistic Fcu/Fcr muscle pair in early postnatal (E) and adult (G) mice, showing co-activation of Fcu/Fcr in both cohorts (arrows indicate the onsets of EMG responses).
(F, H) Predominant CS circuit connectivity patterns proposed for the synergistic muscle pair Fcu/Fcr in early postnatal (F) and adult mice (H). These diagrams show how excitatory muscle-specific or shared INs could be recruited by CS neurons to elicit co-activation of synergistic muscles.
(I) Experimental strategy for recording ICMS-evoked muscle EMGs from the antagonistic Tri/Bi muscle pair.
(J–K) Color-coded plots of the co-activation indices of Bi and Tri muscles from P16~19 (J, n=47 sites from 4 mice) and adult mice (K, n=53 sites from 3 mice).
(L) Cumulative histograms of co-activation in antagonistic muscle pairs showing a shift from significant (P=0.001) co-activation in early postnatal mice (black) to little or no co-activation in adult mice (gray).
(M and O) Representative EMG traces from the antagonistic Bi/Tri muscle pair in postnatal (M) and adult (O) mice, showing co-activation of Bi/Tri only in early postnatal mice (arrows indicate the onsets of EMG responses).
(N and P) Predominant CS circuit connectivity patterns proposed for the antagonistic muscle pair Bi/Tri in early postnatal (N) and adult mice (P). These diagrams show how excitatory muscle-specific or shared INs could be recruited by CS neurons in early postnatal stages to elicit co-activation of antagonistic muscles. In adults, excitatory and inhibitory INs could be recruited by CS neurons to excite the flexor and inhibit the extensor muscles, respectively (P).
We then recorded EMGs from antagonistic muscle pairs in early postnatal mice (Figure 3I; 47 sites from 4 mice) and found co-activation of the antagonistic Bi/Tri muscle pair (Figure 3M). A plot of the co-activation index for Bi/Tri revealed a high degree of co-activation throughout the forelimb motor cortex (Figure 3J), which was similar to the co-activation index obtained for the Fcu/Fcr synergistic muscle pair (Figures 3B and 3C). In contrast, EMG recordings from the Bi/Tri muscle pair in adult mice (53 sites from 3 mice) demonstrated a preponderance of singular activation of only the flexor Bi muscle, with little or no activation of the antagonistic extensor Tri muscle at threshold stimulation levels (Figures 3K and 3O). Moreover, cumulative frequency distribution analysis resulted in distinct co-activation curves for early postnatal and adult mice, showing a shift from extensive muscle co-activation in early postnatal mice to little or no co-activation in adult animals (Figure 3L). These data suggest that CS circuit reorganization occurs in developing antagonistic muscle circuits (Figures 3N and 3P).
Bax/Bak are required for neuronal activity-dependent caspase activation in descending CS axons
Recent studies have shown non-apoptotic roles for the activity-dependent Bax-caspase signaling cascade in promoting formation and reorganization of neural circuits as well as synaptic and structural plasticity in neural circuits (Jiao and Li, 2011; Li et al., 2010; Nikolaev et al., 2009; Ohsawa et al., 2010; Simon et al., 2012). This prompted us to examine the Bax-caspase pathway as a possible mediator of CS circuit remodeling. By examining the expression of the inactive (full length) and active (cleaved) form of caspase-3, we found that inactive caspase-3 was detected in somas of Ctip2+ CS neurons in the motor cortex of P14 mice, but the cleaved, active form of caspase-3 was not found (Figures 4A–4E). We also did not observe any terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)+ apoptotic nuclei in Ctip2+ CS neurons in early postnatal mice (data not shown), demonstrating that this pathway is not involved in neuronal death or apoptosis at postnatal stages. We then determined whether caspase-3 is activated in CS axons in the spinal cord. To visualize CS axons, we crossed Emx1-Cre mice with an eGFP reporter line (CAG-CAT-eGFP; ccGFP) (Bareyre et al., 2005; Gorski et al., 2002; Nakamura et al., 2006). In Emx1-Cre; ccGFP mice, descending CS axons as well as their terminals were selectively labeled by eGFP in the spinal cord (Figures 4F and 4I). Active caspase-3 was strongly expressed in eGFP+ CS axons in both the dorsal funiculus and gray matter of the spinal cord at various postnatal stages (Figures 4F–4K and S4A–S4F).
Figure 4. Activity-dependent activation of the Bax/Bak-Caspase pathway in descending CS axons.
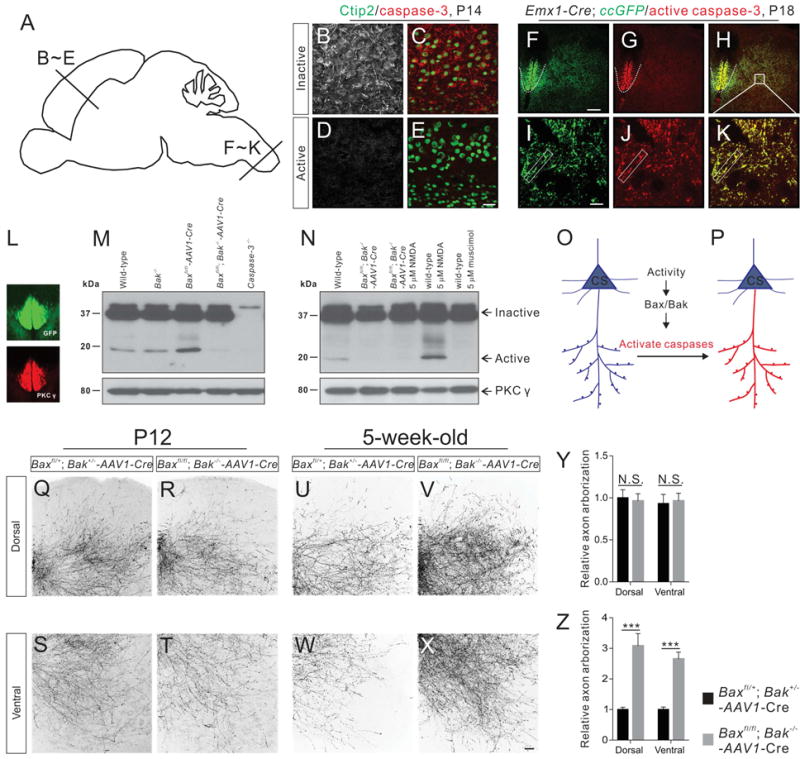
(A) Sagittal view of the mouse brain showing section locations of images shown in panels B–E.
(B–E)Immunofluorescent labeling of P14 motor cortex sections using antibodies targeting the inactive and active forms of caspase-3 (red) or Ctip2 (green). The inactive form of caspase-3 (B and C) but not active caspase-3 (D and E) was expressed in the soma of CS neurons. Sections are taken from the motor cortex as shown in A.
(F–K) Immunofluorescent labeling of P18 cervical spinal cord sections of Emx1-Cre; ccGFP mice using antibodies against GFP (green) and the active form of caspase-3 (red). Selective activation of caspase-3 was observed in GFP+ CS axons in the dorsal funiculus (indicated by white dashed lines in F–H) and CS axon collaterals in the gray matter (indicated by rectangular white boxes in I–K).
(L) Immunostaining for PKCγ in a P14 cervical spinal cord showing the selective expression of PKCγ in GFP+ CS axons.
(M and N) Western blot analyses of protein lysates from the cervical dorsal funiculus of P14 mice using antibodies against full-length (inactive) caspase-3 and PKCγ. (M) Protein lysates from P14 wild-type, Bak−/−, Baxfl/fl-AAV1-Cre, Baxfl/fl; Bak−/−-AAV1-Cre, and caspase-3−/− mice. (N) Protein lysates from wild-type mice, Baxfl/fl; Bak−/−-AAV1-Cre, Baxfl/fl; Bak−/−-AAV1-Cre mice treated with NMDA (5 μM, 2 hours), wild-type mice treated with NMDA (5 μM, 2 hours), and wild-type mice treated with muscimol (5 μM, 2 hours).
(O and P) Diagram of caspase-3 activation in CS neurons. The inactive form of caspase-3 (blue) is present in CS neurons but its activation (red) is selectively restricted to descending axons and their collaterals.
(Q–X) Representative images of cervical spinal cords showing GFP+ CS axon collaterals from P12 and 5-week-old adult Baxfl/+; Bak+/− (control, Q, S, U, and W) and Baxfl/fl; Bak−/− (R, T, V, and X) mice.
(Y and Z) Comparison of levels of axon arborization in Bax/Bak double-mutant and control mice. CS axon collaterals in Bax/Bak double-mutant mice (n=8) are similar (dorsal, P=0.7880; ventral, P=0.8214) to that of control mice (n=8) at P12 (Y). Levels of axon arborization in adult Bax/Bak double-mutant mice (n=8) are significantly (dorsal, P<0.0001; ventral, P=0.0001) higher than that of control mice (n=8). Where “n” represents the number of mice used in each experiment. Scale bars, 10 μm (I), 30 μm (E), 50 μm (X), and 100 μm (F). See also Figures S4 and S5.
We then determined whether Bax and its related protein Bak (Lindsten et al., 2000; Zong et al., 2001) are required for caspase-3 activation both in vitro and in vivo. To generate Bax and Bak single- and double-deficient neurons, we cultured cortical neurons from P0 Baxfl/+; Bak+/− (for controls), Baxfl/fl (for Bax single-mutants), and Baxfl/fl; Bak−/− (for Bak single- or Bax/Bak double-mutants) mice. These neurons were then infected with either AAV1-hSyn-GFP/AAV1-GFP (for controls and Bak single-mutants) or AAV1-hSyn-CreGFP/AAV1-CreGFP to delete Bax (for Bax single-mutants and Bax/Bak double-mutants) (Figures S5A–S5D). We observed axon-localized activation of caspase-3 in control, Bak and Bax single-deficient cortical neurons, but not in Bax/Bak double-deficient neurons (Figures S5A–S5D), demonstrating that both Bax and Bak are required for the activation of caspase-3 within cortical axons in vitro. We then examined whether caspase-3 was activated in CS axons in vivo in control, caspase-3−/−, Bak−/−, Baxfl/fl-AAV1-Cre, and Baxfl/fl; Bak−/−-AAV1-Cre mice. Postnatal deletion of floxed Bax alleles in mice were successfully generated by bilaterally injecting AAV1-hSyn-Cre (or AAV1-Cre) into the motor cortex of P2 Baxfl/fl or Baxfl/fl; Bak−/− mice to preserve the apoptotic functions of Bax during embryonic and fetal development (Chen et al., 2013; White et al., 1998). We stained cervical spinal cord sections from these mice at P14 using antibodies against active caspase-3 and found that active caspase-3 was absent only from caspase-3−/− and Bax/Bak double mutant (Baxfl/fl; Bak−/−-AAV1-Cre) mice (Figures S5E–S5J). We obtained similar results by Western blot analysis using protein lysates prepared from the dorsal funiculus of P14 cervical cords (Figures 4L and 4M). These data demonstrate that Bax and Bak compensate for each other in CS axons, and that removal of both proteins is required to eliminate caspase-3 activation both in vitro and in vivo.
To assess whether neuronal activity is involved in the localized activation of caspase-3 within CS axons, we elevated or suppressed neuronal activity by bilaterally injecting NMDA or the GABAA receptor agonist, muscimol, into the motor cortex, respectively. Protein lysates for Western blot analyses were harvested from the dorsal funiculus of the cervical spinal cord two hours after injections. Elevating neuronal activity in the motor cortex through NMDA injections resulted in increased activation of caspase-3 in wild-type CS axons, while suppression of neuronal activity with muscimol reduced active caspase-3 to undetectable levels (Figure 4N). In Bax/Bak double mutant mice, active caspase-3 was not observed in CS axons, even with cortical injections of NMDA (Figure 4N). Together, these data demonstrate that neuronal activity can activate caspase-3 through the Bax and Bak proteins in the CS axons of early postnatal mice (Figures 4O and 4P).
Remodeling of CS axon collaterals in the spinal cord is abolished in Bax/Bak double mutant mice
Previous studies have shown that some CS axon branches are pruned in the spinal cords of early postnatal rats and cats (Curfs et al., 1994; Li et al., 2001). To assess whether activation of the Bax/Bak-caspase pathway is involved in remodeling CS axon collateral branches in the spinal cord during CS circuit maturation in mice, we deleted floxed Bax alleles in CS neurons by injecting AAV1-CAG-Flex-eGFP and AAV1-Cre into one hemisphere of the motor cortex of Baxfl/+; Bak+/− (n=16 animals) or Baxfl/fl; Bak−/− (n=16 animals) mice at P4 to create Baxfl/+; Bak+/−-AAV1-Cre (control) or Baxfl/fl; Bak−/−-AAV1-Cre (Bax/Bak double mutant) mice. CS axonal arbors were analyzed at P12 and 5-week-old mice (Figures S6A–S6C). We found that similar numbers of CS neurons were labeled in the injected hemispheres of control (Baxfl/+; Bak+/−-AAV1-Cre) and Bax/Bak double mutant mice (Figures S6D–S6I and S6P). Similar CS axon collateral branches were observed in the spinal gray matter in the cervical spinal cord from both cohorts at P12 (Figures 4Q–4T, and 4Y). In contrast, we found significantly more CS axon collateral branches in the spinal gray matter of adult Bax/Bak double mutant mice compared to control mice (Figures 4U–4X and 4Z). Corpus callosum axons and their projections to the contralateral cortex in Bax/Bak double mutant mice were not noticeably different from those of control mice (Figures S6J–S6O, S6Q and S6R), consistent with the lack of active caspase-3 in axons within the cortex (Figure 4D). Taken together, these data suggest that the activity-dependent, Bax/Bak-caspase pathway regulates the remodeling of CS axon collaterals in the spinal cord during development.
Reorganization of CS-IN connections is disrupted in Bax/Bak double mutant mice
To compare the connectivity of CS circuits in control and Bax/Bak double mutant mice, we first visualized the CS-IN connections by injecting both AAV1-Cre and AAV1-CAG-flex-eGFP viruses into the motor cortex of Baxfl/+; Bak+/− and Baxfl/fl; Bak−/− mice at P4 to delete floxed Bax alleles in a subset of CS neurons, and simultaneously label their descending CS axons (Figure 5A). We then injected PRV-LacZ and PRV-RFP into antagonistic muscle pairs (Tri/Bi and Ecr/Fcr) at P12 and 5-week-old mice (Figure 5A). This approach combining anterograde axon tracing with retrograde PRV tracing enabled us to identify the INs that receive putative direct CS inputs and, in turn, connect to extensor, flexor or both MNs (Figures 5A–5F). We found that the majority of muscle-specific or shared INs in control (Baxfl/+; Bak+/−) mice received putative direct CS connections in the spinal cord of P14 mice (Figures 5G and 5I), whereas in 5-week-old adult control mice, there were significantly fewer muscle-specific or shared INs receiving putative direct CS inputs (Figures 5K and 5M). In addition, the numbers of putative CS synapses on soma and proximal dendrites of INs substantially decreased as CS circuits matured in control mice (Figures 5J and 5N). These data identified reorganization of CS-IN connectivity during development. We also found that the number of INs that received putative direct CS inputs in Bax/Bak double mutant mice was similar to that of control (Baxfl/+; Bak+/−) mice at P14 (Figures 5G–5I). In contrast, the developmental reduction in numbers of INs that received putative CS inputs was not observed in adult Bax/Bak double mutant mice (Figures 5H, 5I, 5L, and 5M). We also observed that individual INs in adult Bax/Bak double mutant mice had significantly more synapses than control mice (Figure 5N). These results strongly indicate that the Bax/Bak-caspase pathway is essential for the developmental reorganization of CS-IN connectivity.
Figure 5. Developmental refinement of CS-IN connectivity is impaired in Baxfl/fl; Bak−/−-AAV1-Cre mice.
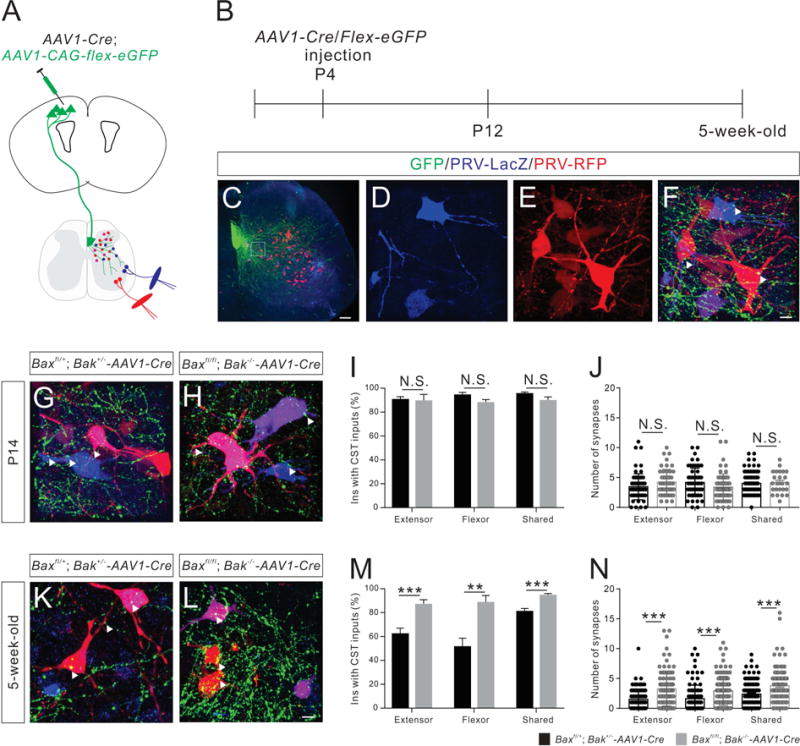
(A and B) Experimental design. (A) A mixed virus solution (comprised of AAV1-Cre and a Cre-dependent eGFP reporter AAV) was injected into the mouse motor cortex at P4 to delete the floxed Bax allele and label CS axons. (B) Dual-color PRV tracing were performed at P12 and 5-week-old stages.
(C–F) Representative images of the cervical spinal cord from virus-injected mice at 2 dpi/P14 (C). (D–F) Higher magnification views of the boxed area in (C), showing PRV-labeled INs that receive putative CS inputs (representative synapses are indicated by white arrowheads).
(G and H) Images of the cervical spinal cord from control (G, Baxfl/+; Bak+/−-AAV1-Cre, n=8) and Bax/Bak double-mutant (H, n=7) mice at P14, showing PRV-labeled INs that are receiving putative CS inputs.
(I) Percentages of INs that receive putative CS inputs in P14 Bax/Bak double-mutant mice are similar (extensor, P=0.7371; flexor, P=0.0664; shared, P=0.1086) to those of control mice.
(J) P14 Bax/Bak double-mutant mice have similar numbers of putative CS synapses on PRV-labeled INs (extensor, P=0.0719; flexor, P=0.0652; shared, P=0.7161) as control mice.
(K and L) Cervical spinal cord images from 5-week-old adult control (K, n=10) and Bax/Bak double-mutant (L, n=9) mice showing PRV-labeled INs that have putative CS inputs.
(M) The percentage of INs that have putative CS inputs are significantly higher in 5-week-old adult Bax/Bak double-mutant mice (extensor, P=0.0005; flexor, P=0.0058; shared, P=0.0002) than same-age control mice.
(N) Five-week-old adult Bax/Bak double-mutant mice have significantly higher numbers of putative CS synapses on PRV-labeled INs (extensor, P<0.0001; flexor, P<0.0001; shared, P<0.0001) than same-age control mice. Where “n” represents the number of mice used in each experiment. Scale bars, 10 μm (F and L) and 100 μm (C).
Developmental CS circuit reorganization is impaired in Bax/Bak double mutant mice
Using two different assays, we examined CS-IN-MN connectivity in Bax/Bak double mutant (Baxfl/fl; Bak−/−-AAV1-Cre) and control (Baxfl/+; Bak+/−-AAV1-Cre) mice. First, to compare the overall segregation and convergence of IN-MN connections in the spinal cords, we injected PRV-eGFP and PRV-RFP into Ecr/Fcr muscles of P12 and 7-week-old adult mice. We found that the number of double-labeled INs in Bax/Bak double mutant mice was similar to that observed in control mice for both early postnatal and adult mice (Figures 6E–6G). In addition, we observed an increase in double-labeled INs in adult Bax/Bak double mutant mice compared to early postnatal mice that paralleled the increases observed in wild-type and control mice (Figures 1Q–1W and 6E–6G). Thus, deletion of Bax and Bak in mice does not appear to affect IN-MN connectivity during development.
Figure 6. Anatomical reorganization of CS circuits is abolished in Bax/Bak double-mutant mice.
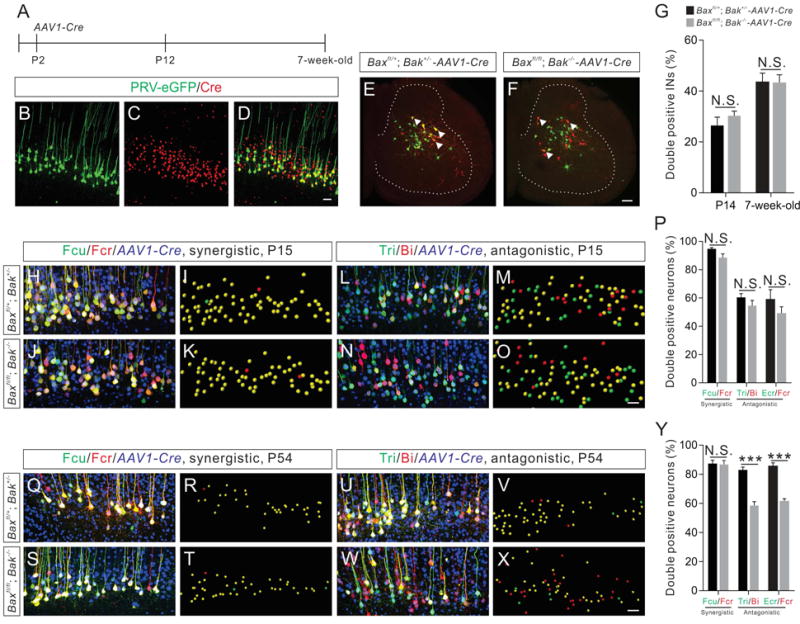
(A) Experimental design of the temporally-specific deletion of the floxed Bax alleles in the motor cortex. AAV1-Cre was injected into the motor cortex at P2 to conditionally delete floxed Bax alleles. PRV injections were performed on P12 and 7-week-old adult mice.
(B–D) Representative images of a brain section from the motor cortex showing that most of the PRV-eGFP+ CS neurons were also Cre+ (yellow).
(E and F) Cervical spinal cord images from P14 control (Baxfl/+; Bak+/−-AAV1-Cre) (E) and Bax/Bak double-mutant (F) mice showing INs labeled by dual-color PRV tracing from the antagonistic Ecr/Fcr muscle pair (three double positive INs are indicated by white arrowheads).
(G) Percentages of double positive INs labeled by PRVs in Bax/Bak double-mutant mice (P14, n=12; adult, n=12) are similar (P14, P=0.3328; adult, P=0.9486) to those in control (P14, n=9; adult, n=11) mice.
(H–O and Q–X) Representative images (left: confocal and right: imaris reconstruction) from dual-color PRV tracing assays for a synergistic (H–K and Q–T, Fcr/Fcu) and an antagonistic (L–O and U–X, Tri/Bi) muscle pair from control and Bax/Bak double-mutant mice at P15 (H–O) and adult (Q–X) stages.
(P) Percentages of double positive CS neurons labeled by PRVs in P15 Bax/Bak double-mutant mice (Fcu/Fcr, n=9; Tri/Bi, n=17; Ecr/Fcr, n=13) are similar (Fcu/Fcr, P=0.0555; Tri/Bi, P=0.2841; Ecr/Fcr, P=0.2654) to those in P15 control mice (Fcu/Fcr, n=13; Tri/Bi, n=15; Ecr/Fcr, n=10).
(Y) Percentages of double positive CS neurons labeled by PRVs from the synergistic Fcu/Fcr muscle pair in adult Bax/Bak double-mutant mice (n=12) is similar (P=0.8946) to those in control mice (n=9), while percentages of double positive CS neurons labeled by PRVs from antagonistic muscle pairs in adult Bax/Bak double-mutant mice (Tri/Bi, n=8; Ecr/Fcr, n=12) are significantly (Tri/Bi, P<0.0001; Ecr/Fcr, P<0.0001) lower than those of control mice (Tri/Bi, n=9; Ecr/Fcr, n=12). Where “n” represents the number of mice used in each experiment. Scale bars, 50 μm (D, O, and X), 100 μm (F).
Next, we examined the PRV-labeled INs to determine whether they were excitatory or inhibitory in nature. To identify the excitatory INs, we crossed Baxfl/fl; Bak−/− mice to a vGlut2-ChR2-YFP transgenic line that specifically marks vGlut2+ excitatory INs in the spinal cord (Hagglund et al., 2010). We performed bilateral injections of AAV1-Cre to the motor cortex of P2 Baxfl/+; Bak+/− and Baxfl/fl; Bak−/− mice that carried the vGlut2-ChR2-YFP transgene. PRV-LacZ and PRV-RFP were then injected into Bi/Tri muscles of these mice either at P12 or 5-week-old adults to identify Bi- or Tri-specific INs, and those shared by both Bi and Tri muscles. We found that deletion of Bax and Bak in mice caused no detectable effects on the composition of PRV-labeled vGlut2+ INs (Figures S7A–S7C). To characterize PRV-labeled INs in P14 or 5-week-old adult control and Bax/Bak double mutant mice further, transverse sections of cervical spinal cords were stained with antibodies to glutamate and GABA to identify putative excitatory and inhibitory INs, respectively (Hagglund et al., 2010). We found no differences in IN profiling between control and Bax/Bak double mutant mice (Figures S7D–S7I). Together, these results suggest that there are no obvious defects in IN-MN connectivity in Bax/Bak double mutant mice.
We next injected PRV-eGFP and PRV-RFP into various muscle pairs of control and Bax/Bak double mutant mice at P12 and 7-week-old adult stages, and analyzed CS neurons. At P15, similar numbers of double-labeled CS neurons were observed in both cohorts for all muscle pairs, regardless of whether the muscles were synergistic or antagonistic (Figures 6H–6P). However, when we injected PRV-eGFP and PRV-RFP into antagonistic muscle pairs (Tri/Bi and Ecr/Fcr) of adult animals, the ratios of eGFP+/RFP+ to all labeled CS neurons were markedly lower in Bax/Bak double mutant mice compared to controls (wild-type and Baxfl/+; Bak+/− mice that had received AAV1-Cre injections at P2) (Figures 6U–6Y). These ratios resembled those observed in wild-type immature mice (Figures 2B, 2C and 6L–6P). In contrast, eGFP+ and RFP+ CS neurons from synergistic Fcu/Fcr muscle pairs showed a similarly high degree of overlap in both control and Bax/Bak double mutant mice (Figures 6Q–6T and 6Y). Collectively, these PRV-tracing assays suggest that CS-IN but not IN-MN connectivity is impaired in the antagonistic CS circuits of adult Bax/Bak double mutant mice.
Aberrant co-activation of antagonistic muscle pairs in adult Bax/Bak double mutant mice
To assess whether the anatomical defects in CS circuits in Bax/Bak double mutant (Baxfl/fl; Bak−/− -AAV1-Cre) mice affect functional connectivity, we performed ICMS in the motor cortex and recorded EMGs from pairs of forelimb antagonistic muscles (Bi/Tri and Ecr/Fcr) in 3-month-old adult wild-type (received AAV1-Cre injections at P2), Baxfl/fl; Bak−/− (Bak single mutant) and Bax/Bak double mutant mice (Figure 7A). In adult control (wild-type and Bak single mutant) mice, threshold ICMS evoked activation of flexor (Bi and Fcr) agonist muscles only, with little or no co-activation of antagonistic muscles (Figures 7B and 7H). Co-activation was observed only for synergistic muscle pairs in adult wild-type mice as shown earlier (Figures 3C and 3G). In contrast, there was aberrant co-activation of antagonistic muscle pairs evoked by ICMS at virtually all sites in adult Bax/Bak double mutant mice (Figures 7E and 7I). Cumulative frequency distribution analyses of the co-activation indices resulted in two distinct curves for control and Bax/Bak double mutants, showing predominant co-activation for Bax/Bak double mutants (Figure 7D). Moreover, we observed high current thresholds (207 ± 9.374 μA) in adult Bax/Bak double mutant mice compared to adult control mice (25.46 ± 1.852 μA), which were very similar to the high thresholds observed in early postnatal control mice (204.5 ± 10.74 μA; Figure 7J). This further suggests that proper maturation of CS circuits is impaired in Bax/Bak double mutant mice. In contrast, we did not find any obvious defects in the resting membrane potentials, rheobase, membrane resistance, firing frequencies, or action potential thresholds in layer V neurons in the Bax/Bak double mutant mice (Figure S8), indicating that intrinsic CS neuron excitability in these mutants is not altered. Thus, our electrophysiological data reveal that Bax/Bak-dependent reorganization of CS circuits is essential for appropriate patterns of antagonistic muscle activation.
Figure 7. Aberrant co-activation of antagonistic muscle pairs in adult Bax/Bak double mutant mice.
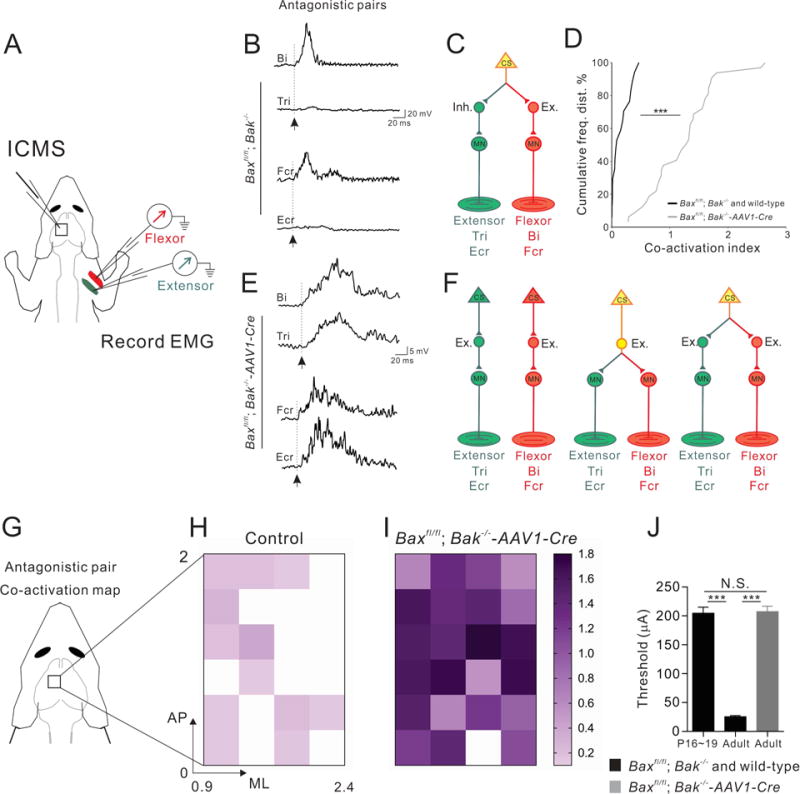
(A) Experimental setup of ICMS-evoked muscle EMGs.
(B and E) Representative traces of EMG recordings from the antagonistic muscle pairs Tri/Bi (top) and Fcr/Ecr (bottom) from adult control (Baxfl/fl; Bak−/−) (B) and Bax/Bak double-mutant mice (E), showing co-activation of antagonistic muscle pairs in Bax/Bak double-mutant mice but not in control mice (arrows indicate the onsets of EMG responses).
(C and F) Predominant CS circuit connectivity patterns proposed for adult control (C; 2 wild-type mice and 4 Baxfl/fl; Bak−/− mice) and Bax/Bak double-mutant mice (F). There may be predominantly one type of connectivity in adult control mice (C) whereas multiple connectivity scenarios may be present in Bax/Bak double mutants (F), much like what we see in juvenile wild-type mice (see Fig 3N)
(D) Cumulative histograms showing prevalent (P<0.0001) co-activation in adult Bax/Bak double-mutant mice (gray, right) and very little co-activation in control (black, left) mice.
(G–I) Color-coded plots of co-activation indices of antagonistic muscle pairs in control (n=17 sites from 2 wild-type mice, and n=31 sites from 4 Baxfl/fl; Bak−/− mice) and Bax/Bak double-mutant mice (n=32 sites from 5 mice).
(J) Quantification showing thresholds (μA) for evoking muscle responses in (P16~19) control (n=37 sites from 4 mice) is significantly (P<0.0001) higher than that of adult control (n=41 sites from 3 mice) but is similar (P=0.8505) to adult Bax/Bak double-mutant (n=32 sites from 5 mice). See also Figure S8, which shows normal cortical neuronal excitability in adult Bax/Bak double-mutant mice.
Adult Bax/Bak double mutant mice exhibit skilled behavioral deficits
To determine whether the anatomical and physiological defects in CS circuit maturation in Bax/Bak double mutant (Baxfl/fl; Bak−/−-AAV1-Cre) mice affected motor behaviors, we first examined their locomotor abilities. The treadmill locomotor test did not reveal any obvious deficits in adult Bax/Bak double mutant mice (data not shown), consistent with previous studies showing that CS circuits are not required for basic locomotor behaviors (Drew et al., 2004). In addition, we did not find any changes in either forelimb or hindlimb grip strength in Bax/Bak double mutant mice (Figure 8A).
Figure 8. Adult Bax/Bak double mutant mice exhibit skilled behavioral deficits.
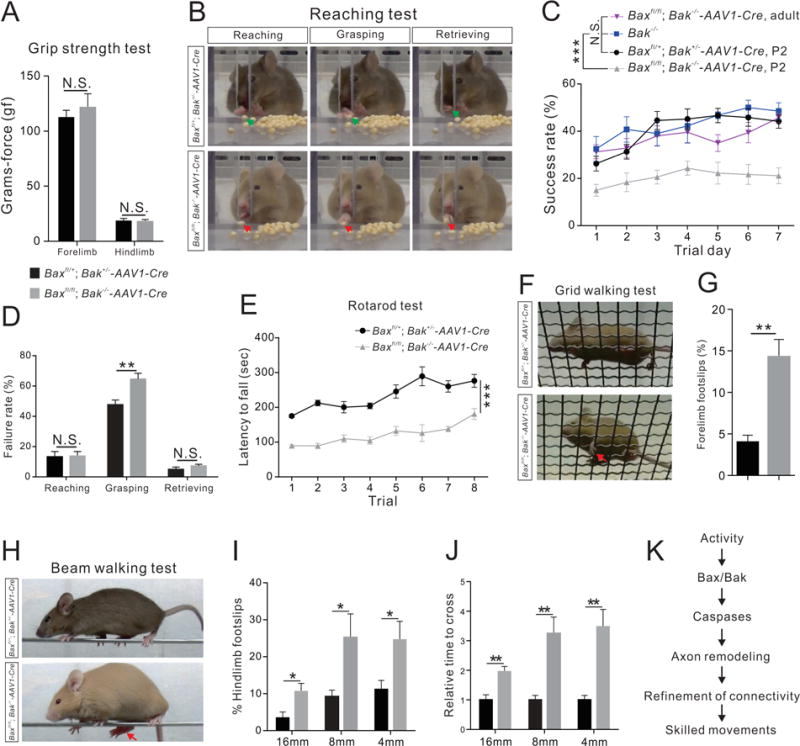
(A–J) Performance on behavior tests by adult Bax/Bak double-mutant (n=8) and control (Baxfl/+; Bak+/−-AAV1-Cre) mice (n=8).
(A) Grip strength test showing the strength of forelimbs (P=0.1634) and hindlimbs (P=0.6901) were similar between Bax/Bak double-mutant and control mice.
(B) Video snapshots depicting the reaching, grasping and retrieving phases of the reaching test. A firm grip of the seed involving the thumb and middle finger by a control mouse during the grasping phase leads to a successful retrieval, while a Bax/Bak double-mutant mouse uses a combination of index, middle, and ring fingers and do not exhibit a proper grip.
(C) Success rates of the reaching test from Baxfl/+; Bak+/−-AAV1-Cre, P2 (n=8), Bak−/− (n=6), Baxfl/fl; Bak−/−-AAV1-Cre, P2 (AAV1-Cre injections at P2, n=8), and Baxfl/fl; Bak−/−-AAV1-Cre, adult (AAV1-Cre injections at P42, n=8) mice showing that the success rates of Baxfl/fl; Bak−/−-AAV1-Cre, P2 mice were significantly (P<0.0001) lower than those of other groups.
(D) Failure percentages for different phases of the reaching behavior on experimental day 7 showing selective impairment of Bax/Bak double-mutant mice during the grasping phase (reaching, P=0.9136; grasping, P=0.0057; and retrieving, P=0.2287).
(E) Bax/Bak double-mutant mice fell off the rotarod sooner (P<0.0001) than control mice in the rotarod test.
(F) Video snapshots of the grid walking test. A forelimb footslip is indicated by the red arrow.
(G) Bax/Bak double-mutant mice exhibited more forelimb footslips during the grid walking test than control mice (P=0.0005).
(H–J) Performance of control and Bax/Bak double-mutant mice on the beam walking test.
(H) Video snapshots of control and Bax/Bak double-mutant mice crossing an 8 mm beam.
(I) Quantification of hindlimb footslips during the beam walking test showing that Bax/Bak double-mutant mice had significantly more footslips than control mice at every beam width tested (16 mm, P=0.0230, 8 mm, P=0.0340, 4 mm, P=0.0327).
(J) Bax/Bak double-mutant mice required greater time to cross the beams than control mice (16 mm, P=0.0021, 8 mm, P=0.0023, 4 mm, P=0.0014).
(K) Proposed mechanism of CS circuit remodeling during development. Where “n” represents the number of mice used in each experiment.
Bax/Bak double mutant mice were then subjected to skilled behavioral assays. First, we carried out a single-seed reaching task (Figure 8B) (Xu et al., 2009). Both control (Bax fl/+; Bak+/−-AAV1-Cre) and Bax/Bak double mutant mice had similar learning curves over the 7-day trial period (n=8 for each cohort; Figure 8C), however, success rates for the double mutants during each training day were significantly lower than control (Bax fl/+; Bak+/−-AAV1-Cre) and Bak−/− mice throughout the testing period (Figure 8C, and Movies S1–S2). Strikingly, we found significant impairments only during the grasping phase of seed retrieval in Bax/Bak double mutant mice compared to controls (Figure 8D). Early reaching or retrieving behaviors by the double mutant mice were similar to those observed in control mice (Figure 8D). To further determine if these impairments were caused by defects in the Bax/Bak-mediated CS circuit reorganization during early postnatal development, we deleted Bax in the motor cortex after CS circuits had fully matured by injecting AAV1-Cre into the motor cortex of P42 Baxfl/fl; Bak−/− mice (Baxfl/fl; Bak−/−-AAV1-Cre, adult). The success rates of these mutant mice in the single-seed reaching task were very similar to those of control and Bak single mutants (Figure 8C), suggesting that the defects observed in Baxfl/fl; Bak−/−-AAV1-Cre mice in which Bax was deleted at P2 were due to defects in CS circuit reorganization occurring during early postnatal development.
We then used an accelerating rotarod to examine skill acquisition and motor coordination (Daily et al., 2011). The performance of control mice on the rotarod test improved progressively over the course of 8 trials (Figure 8E). A similar improvement in performance was observed in Bax/Bak double mutant mice, however, the falling latencies for the double mutants were significantly shorter than that of controls (Figure 8E).
Next, we performed a grid-walking test to further evaluate forelimb behaviors. The grid test assesses the ability to place forepaws accurately on an elevated grid during spontaneous exploration by analyzing the frequency with which the mice failed to grasp the rungs accurately (Starkey et al., 2005; Z’Graggen et al., 1998). Bax/Bak double mutant mice exhibited more forelimb footslips than controls during grid exploration (Figures 8F–8G).
Finally, fine motor coordination and hindlimb balance were assessed by the beam-walking test (Figure 8H) in which mice traversed narrow, elevated beams (4, 8, and 16 mm widths) to a safe platform. Performance on this task was quantified by measuring the number of hindlimb footslips during crossing and the time taken to cross the beam (Luong et al., 2011). Bax/Bak double mutant mice exhibited more hindlimb footslips and longer crossing times than control mice (Figures 8I–8J). Taken together, these data demonstrate that activity-dependent, Bax/Bak-mediated CS circuit reorganization during development is essential for the execution of skilled movements (Figure 8K).
Discussion
During adolescence, young mammals gradually acquire precise control over their flexor and extensor muscles to allow them to perform skilled motor behaviors much like their adult counterparts. CS circuits drive the muscle activation patterns controlling skilled movements, and it has been postulated that CS circuits are reorganized during development. However, how CS circuits are reorganized and by what underlying mechanisms was unclear. In this study, by visualizing the neural circuits between CS neurons and flexor/extensor muscles using two color variants of PRVs, we characterized the reorganization of CS circuitry during development. Our findings show that the CS circuits controlling antagonistic muscle pairs are reorganized during immature stages by a process regulated by the activity-dependent, Bax/Bak-caspase pathway.
Reorganization of CS circuits for antagonistic muscle pairs
Our PRV-tracing assays show that a higher portion of CS neurons connected to antagonistic muscle pairs is segregated in early postnatal mice compared to that observed in adult mice. Electrophysiological studies further show that cortical stimulation induces co-activation of antagonistic muscle pairs only in early postnatal, but not, adult mice. These findings demonstrate that CS synaptic reorganization occurs for controlling antagonistic muscle pairs during development. How does the CS circuit reorganization occur? Segregated CS circuits may be selectively eliminated during development to acquire mature shared CS circuits. Another possibility is that inhibitory INs could be recruited by segregated CS circuits during synaptic reorganization to mediate feed-forward inhibition of antagonistic muscles. Having these feed-forward inhibitory CS circuits, co-activation of antagonistic muscle pairs may not be observed by cortical stimulation and EMG recording in adult mice. However, an antagonistic muscle is not always inhibited, but it is indeed co-contracted to stabilize joints at certain time-points or phases during skilled movements (Hansen et al., 2002; Hyland and Jordan, 1997; Nielsen and Kagamihara, 1992, 1993; Smith, 1981; Suzuki et al., 2001). Suppression of reciprocal inhibition of spinal inhibitory INs has been suggested to be involved in this switch from the exclusive activation of an antagonistic muscle pair to co-activation (Ethier et al., 2007; Pierrot-Deseilligny and Burke, 2012). Detailed analysis of spinal IN subtypes connected to CS circuits during development will give us some hints for these questions. Despite these uncertainties, our study lays the groundwork for the identification and functional analysis of postnatal circuit reorganization in antagonistic CS circuits and its integral role in the development of a functioning CS system in mammals.
The activity-dependent, non-apoptotic Bax/Bak-caspase pathway drives maturation of CS circuits
Previous studies showed that CS axon branches are eliminated in a neuronal activity-dependent manner within the spinal cord in cats (Friel and Martin, 2005; Martin, 2005; Salimi et al., 2008). However, how this axon remodeling relates to synaptic reorganization, and what underlying mechanisms drive the remodeling, have not been determined. We show that deletion of the Bax/Bak proteins selectively in the mouse motor cortex results in significantly more CS axon collaterals in the spinal cord in adult, but not early postnatal, mice compared to control mice, which suggests that the Bax/Bak pathway regulates axon elimination to remove exuberant or inappropriate axon collaterals and their associated synapses in an activity-dependent manner. This corroborates the findings of other studies in which Bax-caspase-3 activation is required for proper elimination of inappropriate axon collaterals in the mouse retinocollicular system (Nikolaev et al., 2009; Simon et al., 2012). Alternatively, the Bax/Bak-caspase pathway may inhibit the formation of new axon branches in wild-type mice. Moreover, interestingly, the caspase pathway has also been shown to play a role in adding new axon arbor branches and growth in the zebrafish retinal ganglion cell system (Campbell and Okamoto, 2013). This raises the possibility that the Bax/Bak-caspase pathway could also be involved in the addition of new CS axon branches in the spinal cord during development, although synapse elimination is likely the dominant function of the Bax/Bak pathway, considering that more CS axon branches were observed in the spinal cords of adult Bax/Bak mutant mice compared to control mice.
Consistent with the PRV tracing data showing that there are more segregated CS circuits in Bax/Bak mutant mice compared to controls (Figure 6), cortical stimulation with EMG recordings show aberrant co-activation of antagonistic muscle pairs in adult Bax/Bak double mutant mice (Figure 7). This strongly suggests that Bax/Bak-caspase-mediated synaptic reorganization is required to generate precise patterns of flexor and extensor muscle activation during skilled movements. This study, in agreement with previous studies (Asante and Martin, 2013; Jiang et al., 2013), shows that threshold cortical stimulation mainly evokes flexor but not extensor muscle activation in adult rats and mice. However, we do not yet know how these CS circuits develop to differentially control flexor and extensor muscles. This will be another interesting subject for future investigations.
Finally, we found that the threshold currents required to evoke EMG activity after cortical stimulation in adult Bax/Bak double mutant mice were significantly higher than those required in adult control mice. Similar high thresholds were also observed in early postnatal wild-type mice, further suggesting that CS circuits in Bax/Bak double mutant mice have failed to mature. High thresholds have also been implicated in the induction of EMG activity in early postnatal cats with lower thresholds observed in adult cats (Chakrabarty and Martin, 2000) and humans (Nezu et al., 1997).
Bax/Bak double mutant mice exhibit compromised skilled motor behaviors
Reciprocal or feed-forward inhibition suppresses the activity of antagonist muscles to ensure that movement is not impeded (Cheney, 1997). When antagonistic muscles in the hand are aberrantly co-activated, voluntary movement of fingers is impaired (Matsuo et al., 1990). In similar fashion, our findings suggest that the impairment of fine motor skills in Bax/Bak double mutant mice is also due to aberrant co-activation of antagonistic muscle pairs and the resulting failure to generate appropriate muscle activation patterns. Intriguingly, genetic deletion of Bax/Bak in mice caused impairment of the grasping phase in a single-seed reaching task without influencing the early reaching and retrieving phases. In contrast, complete CS axon deletion by pyramidal lesion causes much more severe motor deficits, which includes impairments in early reaching (Starkey et al., 2005). It has been shown that both propriospinal neurons (PNs) and segmental INs (sINs) are innervated by CS neurons at cervical levels (Alstermark and Isa, 2012; Lemon, 2008). The lesion experiments show that PNs play a role in the early reaching phase, whereas sINs are involved in the grasping phase in cats (Alstemark et al., 1981a, b; Alstermark and Sasaki, 1985). Another recent study in mice also showed that ablation of Chx10+ PNs caused deficits in skilled reaching behaviors only (Azim et al., 2014). Therefore, considering the grasping phase deficits in Bax/Bak double mutant mice, synaptic reorganization defects may occur mainly between CS neurons and sINs.
Our findings provide new insights into how the acquisition of sophisticated skilled movements is developmentally regulated by a neuronal activity-dependent molecular process. As much as 6% of children around the world suffer from developmental motor disabilities affecting skilled motor control and some of these patients show atypical co-activation of antagonistic muscle pairs as adults (Eliasson et al., 1991; Gottlieb et al., 1982; Matsuo et al., 1990; Nashner et al., 1983; Spaulding et al., 1990; Vaivre-Douret, 2014; Zwicker et al., 2012). CS circuit impairment has been suggested to be responsible for this co-activation (Cheney, 1997). Future studies will determine whether disruptions in the activity-dependent Bax/Bak-caspase pathway is implicated in some of these patients with skilled motor disabilities and whether this pathway also regulates synaptic reorganization of other circuits in different regions of the mammalian central nervous system.
STAR★Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Yutaka Yoshida (yutaka.yoshida@cchmc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures were approved by the Institutional Animal Care and Use Committee at the Cincinnati Children`s Hospital Medical Center, and the Institutional Animal Care and Use Committee of the City College of New York. All procedures were conducted in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. All mice were housed in a specific pathogen-free barrier facility in ventilated polysulfone cages, up to 4 mice per cage. The cages, food and bedding were autoclaved. The bedding used was ¼ inch corn cob. The rooms were maintained at 72 degrees Fahrenheit plus or minus 2 degrees and humidity between 30-70%. Male and female mice were both used in this study depending on availability. Mice were group housed and kept on a 12 hr light/dark cycle (lights on at 6:00 am) with ad libitum access to food and water. We collect data from multiple mice (n≥3, the numbers of each experiment are documented in individual figure legends) for each experiment. The following mouse strains were used in this study: Emx1- Cre (Gorski et al., 2002), CAG-CAT-eGFP (ccGFP) (Nakamura et al., 2006), caspase-3−/− (Kuida et al., 1996), and Baxfl/fl; Bak−/− (Takeuchi et al., 2005), vGlut2-Cre, vGat-Cre (Vong et al., 2011), and vGlut2-ChR2-YFP (Hagglund et al., 2010). Wild-type mice were purchased from Charles River Laboratories.
METHOD DETAILS
We did not use strategy for randomization and/or stratification, blinding, and sample-size estimation and statistical method of computation. Data collect from all experimental subjects are included in this study.
Virus production and injections
Three recombinant viruses (expressing eGFP, RFP or LacZ) of the attenuated PRV strain, PRV-bartha, were used in this study. PRV-eGFP (PRV152) (Smith et al., 2000), PRV-RFP (PRV614) (Banfield et al., 2003) and PRV-LacZ (PRV-BaBlu) (d’Avella et al., 2003) were harvested from cultured pig kidney cells (PK15) at a titer of 108 plaque forming units/ml (pfu/ml) and diluted to a titer of 5 × 107 pfu/ml for injections. Viral stocks were aliquoted in 50~100 μl volumes and stored at −80°C. Individual muscles of P9~P14 mice were injected with 0.5~2 μl of PRV solution. Brains and spinal cords from postnatal mice were collected at 1~3.5 dpi (Figures S1S and S1T). Adult mice received 5~15 μl injections of PRV solution into individual muscles. Brains and spinal cords from adult mice were collected at 2~5 dpi (Figures S1U and S1V). AAV solutions were diluted to a final titer of 1 × 1012 genome copy/ml (GC/ml) for injections. AAV1-Cre/AAV1-hSyn-Cre and AAV1-Flex-eGFP/AAV1-CAG-Flex-eGFP were purchased from the Penn Vector Core, University of Pennsylvania.
Bilateral injections of AAV1-Cre into the motor cortex of Baxfl/fl; Bak−/− mice were performed at P2 to generate Baxfl/fl; Bak−/−-AAV1-Cre mice (Bax/Bak double mutants related to Figures 4N, 6, 7, 8, and S7). Two injections were administered to each hemisphere resulting in four injections per mouse using the following coordinates: 1 mm rostral to Bregma, 0.7 mm lateral to the midline, at a depth of 0.4 mm; 0 mm to Bregma, 0.7 mm lateral to the midline, at a depth of 0.4 mm. For P2 mice, 0.3 μl of AAV1-Cre was injected at each site. Baxfl/+; Bak+/− or wild-type P2 mice with AAV1-Cre injections were used as controls. Similar injections were also performed at P42 to generate (Baxfl/fl; Bak−/−-AAV1-Cre) adult mice. The coordinates for adult injections were 1 mm rostral to Bregma, 1.5 mm lateral to the midline, at a depth of 0.5 mm; 0 mm to Bregma, 1.5 mm lateral to the midline, at a depth of 0.4 mm. For P42 mice, 0.5 μl of AAV1-Cre was injected at each site.
0.3 μl of virus mixture (2.5 × 1011 GC/ml of each virus) combining AAV1-Cre and AAV1-Flex-eGFP was injected into the motor cortex of Baxfl/fl; Bak−/− and Baxfl/+; Bak+/− mice at P4 using the following coordinates: 1 mm to Bregma, 0.7 mm lateral to the midline, at a depth of 0.4 mm. Brains and spinal cords were then fixed at P12 or P38 (related to Figures 4Q–4Z, 5, S6).
Immunohistochemistry, confocal imaging, and axon collateral quantification
Perfusion fixation (ice-cold 1X phosphate buffered saline (PBS) followed by 4% paraformaldehyde, PFA) was used during brain and spinal cord harvests. Upon dissection, brains and spinal cords were fixed overnight at 4°C. Brains and spinal cord were cryoprotected by immersion in 30% sucrose/1X PBS for two nights and sectioned to 50 μm and 80 μm slices respectively using a cryostat. Free-floating immunohistochemistry was performed by incubating brain and spinal cord sections with primary antibodies overnight at room temperature, then with fluorophore-conjugated secondary antibodies for 4 hours at room temperature. Sections were mounted with VECTASHIELD Mounting Media (Vector Labs) and coverslipped for imaging. Confocal images were taken with a Nikon A1R GaAsP or Carl Zeiss LSM 510 Laser Scanning Confocal Microscope. Reconstructions of PRV-labeled CS neurons were performed using Imaris software (Bitplane). Axon density measurements following AAV-labeling were conducted using Nikon Elements AR software as follows: 1) images of CS axon collaterals in dorsal and medial cervical spinal cord sections were acquired using a 20X objective; 2) confocal images were exported using Nikon Elements software; 3) the General Analysis function included in the Nikon Elements software was used to quantify the area covered by CS axons (the default threshold was manually adjusted to match the area defined by the General Analysis with the area covered by GFP); 4) three or more representative sections were analyzed for each animal; 5) the relative axon arborization surface of Baxfl/fl; Bak−/−-AAV1-Cre mice corresponded to the area of each image divided by the average area derived from Baxfl/+; Bak+/− -AAV1-Cre mice.
Primary antibodies used in this study were: chicken anti-GFP (1:2000, Abcam); sheep anti-GFP (1:2000, AbD Serotec); rabbit anti-DsRed (1:1000, Clontech); goat anti-mCherry (1:1000, Biorbyt); goat anti-ChAT (1:100, Millipore); rat anti-Ctip2 (1:1000, Abcam); chicken anti- Beta III Tubulin (1:1000, Millipore); rabbit anti-full caspase-3 (1:1000, Cell Signaling); rabbit anti-cleaved caspase-3 (1:200, Cell Signaling); rabbit anti-PKCγ (1:500, Santa Cruz); rabbit anti-Cre (1:4000, Covance); chicken anti-beta Galactosidase (1:1000, Abcam); rabbit anti-Glutamate (1:1000, ImmunoSolution); and rabbit anti-GABA (1:1000, ImmunoSolution); chicken anti-Beta III Tubulin (1:500, Millipore). Fluorophore-conjugated secondary antibodies were purchased from Jackson ImmunoResearch and Invitrogen.
Cortical neuron culture
At P0, motor cortices were dissected from 1) Baxfl/+; Bak+/−, 2) Baxfl/fl; Bak−/−, 3) and Baxfl/fl mice in hibernation buffer as previous described (Qian et al., 1998). 3 × 104 neurons were plated into each chamber of a 2-well chamber slide (Fisher Scientific, Catalog Number: 154461) and cultured for 7 days using cortical neuron culture media (neurobasal medium, B-27 supplement (200 μl/10 ml), 200 mM L-Glutamine, 1 M Glucose, 50 units/ml penicillin, 50 ug/ml streptomycin). All culture reagents were purchased from Invitrogen. Culture media was changed daily. 5 μl of AAV1-CreGFP/AAV1-hSyn-Cre-eGFP (Penn Vector Core, University of Pennsylvania) or AAV1-GFP/AAV1-hSyn-eGFP (Penn Vector Core, University of Pennsylvania) at titers of 1 × 1012 GC/ml were added into each chamber on day 1.
In vivo pharmacological manipulations
Bilateral injections of 5 μM NMDA (Sigma) and 5 μM muscimol (Sigma) were performed on P14 mice using the following coordinates: 1 mm to Bregma, ± 1.2 mm lateral to the midline, at a depth of 0.5 mm; 0 mm to Bregma, ± 1.2 mm lateral to the midline, at a depth of 0.5 mm. Two injections were performed in each hemisphere resulting in four injections per mouse. 0.4 μl of solution was injected at each site. 1X PBS was injected as a control. Cervical CS axons were dissected two hours after injections.
Isolation of axonal proteins and Western blot analysis
The dorsal funiculus and associated CS axons were dissected from the C1 to C6 levels of the cervical spinal cords of P14 mice. Tissues were homogenized and lysed in lysis buffer (50 mM Tris [pH 8.0], 1 mM MgCl2, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, and protease inhibitors (Roche)) at 4Υ for 30 min. The lysates were centrifuged at 12,000 rpm at 4℃ for 10 min, then supernatants were collected. Western blot analyses were performed using standard methods with rabbit anti-full caspase-3 (1:1000, Cell Signaling) and rabbit anti-PKCγ (1:200, Santa Cruz).
In vitro brain slice preparations and patch clamp recordings
Injections of the AAV virus mixture (AAV1-Cre and AAV1-Flex-eGFP, 1 × 1012 GC/ml of each virus) into the left motor cortex of Baxfl/fl; Bak−/− mice were performed at P2 to generate Baxfl/fl; Bak−/−-AAV1-Cre mice (Bax/Bak double mutant mice). Two injection sites with the following coordinates were used: 1 mm rostral to Bregma, 0.7 mm lateral to the midline, at a depth of 0.4 mm; 0 mm to Bregma, 0.7 mm lateral to the midline, at a depth of 0.4 mm. 0.3 μl of AAV virus was injected at each site. Two day-old wild-type mice (P2) with AAV1-Cre and AAV1-Flex-eGFP injections were used as controls. 1 month-old mice were deeply anesthetized with sodium pentobarbital (60 mg/kg) and perfused with ice-cold dissection solution (250 mM sucrose, 2.5 mM KCl, 25 mM NaHCO3, 1.0 mM NaH2PO4, 6 mM MgCl2, 0.5 mM CaCl2, and 25 mM glucose) continuously bubbled with 95% O2/5% CO2. Brains were isolated and transverse slices (300 μm) containing the motor cortex were cut using a vibrating microtome (7000smz-2; Campden Instruments, Lafayette, IN). Slices were incubated for 15–20 min in a recovery solution at room temperature (92 mM NMDG, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 5 mM sodium ascorbate, 2 mM thiourea, 3 mM sodium pyruvate, 10 mM MgSO4, and 0.5 mM CaCl2) and then allowed to recover further in an oxygenated artificial CSF (aCSF) solution (125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.0 mM NaH2PO4, 1.0 mM MgCl2, 2.0 mM CaCl2, and 25 mM glucose) for ≥ 1 hour at room temperature.
After recovery, brain slices were transferred to a submersion-type recording chamber (RC-22; Warner Instruments, Hamden, CT) and mounted onto the stage of an upright microscope (BX51WI, Olympus, Center Valley, PA) equipped with optical filters to allow for the identification of eGFP-labeled CS neurons. Slices were then perfused at room temperature with oxygenated aCSF at a rate of 1–3 ml/min.
Patch electrodes were constructed from thin-walled single-filamented borosilicate glass (1.5 mm outer diameter; World Precision Instruments) using a microelectrode puller (P-97; Sutter Instruments, Novato, CA) and filled with an intracellular solution (130 mM K-gluconate, 10 mM KCl, 10 mM HEPES, 10 mM sodium phosphocreatine, 4 mM MgATP, and 0.3 mM Na2-GTP, pH 7.2, 295–300 mOsm). Pipette resistances ranged from 4 to 6 MΩ and seal resistances were >1 GΩ.
Patch clamp recordings were obtained from eGFP-labeled cortical neurons in layer V of the motor cortex using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Membrane voltages were adjusted for liquid junction potentials (approximately −14 mV) calculated using JPCalc software (P. Barry, University of New South Wales, Sydney, Australia; modified for Molecular Devices). Signals were filtered at 4–6 kHz through a −3 dB, four-pole low-pass Bessel filter, and digitally sampled at 20 kHz using a commercially available data acquisition system (Digidata 1440A with pClamp 10.3 software; Molecular Devices).
ICMS and EMG recordings
Electrical responses were recorded from various muscle pairs (Bi/Tri, Ecr/Fcr, and Fcu/Fcr) using percutaneous Ni-chrome wire electrodes (deinsulated 1 mm from the tip) in response to motor cortex threshold stimulation as previously reported (Serradj et al., 2014). EMG recording wires were inserted using a 27-gauge hypodermic needle guide. We recorded differentially, with two wire electrodes within each muscle and a separate ground, and verified the adequacy of the EMG recordings and muscle placements by noting increased EMG activity with passive joint rotation. EMG recordings were made with a differential AC amplifier with low- and high-pass filtration (Model 1700; A–M Systems).
EMG signals were acquired using an analog-to-digital converter (CED) and processed using Signal software (version 5.01; CED). Averages of rectified EMGs were generated from individual sites from each animal. To quantify results, we developed a co-activation index that corresponded to the integrated EMG value of the extensor muscle divided by the integrated EMG value for the flexor muscle. Strongly flexor-driven responses have a co-activation value that is close to zero and co-active/extensor responses, close to one or larger. For the synergistic muscles Fcu and Fcr, a co-activation index for each site corresponded to the integrated EMG value of the Fcu muscle divided by the integrated EMG value for the Fcr muscle. For each cortical stimulation site, the time window used for integration of EMG responses was the same for both muscles in the pair. The time window varied for each animal depending on the latency and form of the response. Co-activation indices from all sites in a given condition were used to plot the cumulative frequency distribution (Figures 3D, 3L and 7D). To illustrate the co-activation status of different cortical areas, a 2 mm × 2.1 mm (Figures 3B, 3C, 3J, and 3K) or 2 mm × 2.4 mm (Figures 7H and 7I) portion of the motor cortex was divided equally into 24 squares. Individual sites were assigned to different squares based on their anterior to posterior and medial to lateral coordinates (relative to Bregma). An average co-activation index was calculated from all sites in each square. Co-activation maps were generated from plots of the average co-activation indices from all squares in each map (Figures 3B, 3C, 3J, 3K, 7H, and 7I).
Behavioral experiments
Grip strength analysis
Forelimb and hindlimb grip strengths of mice were measured using a grip strength meter from Columbus Instruments. Average force was calculated for each mouse based on three trials and expressed as grams-force (gf, 1 gf = 9.8 × 10−3 Newtons).
Single-pellet reaching test
We assessed the performance of 7-week-old mice in a single-pellet reaching test as previously described with minor modifications (Xu et al., 2009). Briefly, mice were food-restricted to maintain 90% of their free feeding weight before training. We determined the preferred limb for each mouse during the shaping phase (2~4 days), which was followed by a 7-day training phase. We recorded 30 reaches from each mouse per day during training. One episode of reaching behavior was divided into three steps: early reaching, grasping, and retrieving. A failure in any one of the steps meant an overall attempt failure: if the mouse missed the pellet, it was a reaching failure; if the mouse failed to grasp the pellet with its digits, it was a grasping failure; if the mouse retrieved the pellet but dropped it before putting it into its mouth, it was a retrieval failure. Only when the mouse successfully retrieved the seed and put it into its mouth was the attempt considered a success.
Accelerating rotarod
The accelerating rotarod was used to assess motor coordination and motor learning (Med Associates Inc.). Mice were placed on a 3 cm diameter rod with an initial rotation of 4 rpm that accelerated to 40 rpm over 5 min. Mice were tested for tumble latency (time before falling off the rod) in 8 trials over 2 consecutive days as previously described (Daily et al., 2011).
Grid walking test
The grid walking test was carried out as previously described (Starkey et al., 2005). Briefly, mice were placed on a square wire grid (15 mm × 15 mm grid squares) and allowed to freely explore for 3 minutes. Sessions were videotaped and scored for the percentage of footslips (when paws missed a rung or slipped off a rung) in the first 50 steps taken with forepaws and hindpaws.
Beam walking test
The beam walking test was carried out as previously described (Luong et al., 2011) with a few modifications. Beams with flat surfaces of 16 mm, 8 mm, and 4 mm widths were used in this test. Performance on each beam was quantified by measuring the time required for the mouse to traverse the beam and the number of footslips that occurred in the process. Sessions were videotaped and scored for the percentage of footslips in the first 50 hindlimb steps.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
We collect data from multiple mice (n≥3) for each experiment. The numbers of animal for each experiment are documented in individual figure legends. Results are expressed as the mean ± s.e.m. We do not use any methods to determine whether the data met assumptions of the statistical approach. Two-way repeated-measures ANOVA analysis was used for Figures 8C, 8E (followed by post hoc comparisons) and Figure S8C. Cumulative frequency distributions of co-activation indices (Figures 3D, 3L, and 7F) were analyzed by the Kolmogorov-Smirnov test (KS-test). Student t-tests were used for other statistical analyses. Error bars indicate the mean ± s.e.m. Significance levels are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Acknowledgments
We would like to thank L. Enquist and the Center for Neuroanatomy with Neurotropic Viruses (CNNV; NIH grant P40RR018604) at Princeton University and J. Kalambogias for providing PRVs and technical assistance for the EMG study, respectively. We would also like to thank M. Nakafuku, D. Ladle, K. Campbell, and S. Crone for critical comments on the manuscript. J.H.M. and Y.Y. are supported by grants from R01 NS093002 and NS079569, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and two movies and can be found with this article online at
Author Contributions
Z.G and Y.Y. conceived of the project and contributed to overall experimental design and interpretation. Z.G., Y.Y., and J.H.M. contributed to the design and interpretation of EMG experiments. Z.G. performed most of the experiments. Z.G., N.S., and J.H.M. collected and analyzed EMG data. J.L. and M.B performed electrophysiology using cortical slices. M.L. contributed to behavioral analyses. M.U. assisted with brain surgeries and behavioral assays. Y. Y. supervised all aspects of the work. Z.G. and Y.Y. drafted the paper with contributions from J.H.M, N.S., M.U., J.L., and M.B.
References
- Alstermark B, Isa T. Circuits for skilled reaching and grasping. Annual review of neuroscience. 2012;35:559–578. doi: 10.1146/annurev-neuro-062111-150527. [DOI] [PubMed] [Google Scholar]
- Arber S. Motor circuits in action: specification, connectivity, and function. Neuron. 2012;74:975–989. doi: 10.1016/j.neuron.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Asante CO, Martin JH. Differential joint-specific corticospinal tract projections within the cervical enlargement. PloS one. 2013;8:e74454. doi: 10.1371/journal.pone.0074454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim E, Jiang J, Alstermark B, Jessell TM. Skilled reaching relies on a V2a propriospinal internal copy circuit. Nature. 2014;508:357–363. doi: 10.1038/nature13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. Journal of virology. 2003;77:10106–10112. doi: 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Misgeld T, Sanes JR. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat Med. 2005;11:1355–1360. doi: 10.1038/nm1331. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, Pfaff SL. Motor axon pathfinding. Cold Spring Harb Perspect Biol. 2010;2:a001735. doi: 10.1101/cshperspect.a001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Okamoto H. Local caspase activation interacts with Slit-Robo signaling to restrict axonal arborization. J Cell Biol. 2013;203:657–672. doi: 10.1083/jcb.201303072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty S, Martin JH. Postnatal development of the motor representation in primary motor cortex. Journal of neurophysiology. 2000;84:2582–2594. doi: 10.1152/jn.2000.84.5.2582. [DOI] [PubMed] [Google Scholar]
- Chen SK, Chew KS, McNeill DS, Keeley PW, Ecker JL, Mao BQ, Pahlberg J, Kim B, Lee SC, Fox MA, et al. Apoptosis regulates ipRGC spacing necessary for rods and cones to drive circadian photoentrainment. Neuron. 2013;77:503–515. doi: 10.1016/j.neuron.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney PD. Pathophysiology of the corticospinal system and basal ganglia in cerebral palsy. Mental Retardation and Developmental Disabilities Research Reviews. 1997;3:153–167. [Google Scholar]
- d’Avella A, Saltiel P, Bizzi E. Combinations of muscle synergies in the construction of a natural motor behavior. Nature neuroscience. 2003;6:300–308. doi: 10.1038/nn1010. [DOI] [PubMed] [Google Scholar]
- Daily JL, Nash K, Jinwal U, Golde T, Rogers J, Peters MM, Burdine RD, Dickey C, Banko JL, Weeber EJ. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PloS one. 2011;6:e27221. doi: 10.1371/journal.pone.0027221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici N, Ivanenko YP, Cappellini G, d’Avella A, Mondi V, Cicchese M, Fabiano A, Silei T, Di Paolo A, Giannini C, et al. Locomotor primitives in newborn babies and their development. Science. 2011;334:997–999. doi: 10.1126/science.1210617. [DOI] [PubMed] [Google Scholar]
- Drew T, Prentice S, Schepens B. Cortical and brainstem control of locomotion. Progress in brain research. 2004;143:251–261. doi: 10.1016/S0079-6123(03)43025-2. [DOI] [PubMed] [Google Scholar]
- Eliasson AC, Gordon AM, Forssberg H. Basic co-ordination of manipulative forces of children with cerebral palsy. Dev Med Child Neurol. 1991;33:661–670. doi: 10.1111/j.1469-8749.1991.tb14943.x. [DOI] [PubMed] [Google Scholar]
- Ethier C, Brizzi L, Giguere D, Capaday C. Corticospinal control of antagonistic muscles in the cat. Eur J Neurosci. 2007;26:1632–1641. doi: 10.1111/j.1460-9568.2007.05778.x. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annual review of neuroscience. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel KM, Drew T, Martin JH. Differential activity-dependent development of corticospinal control of movement and final limb position during visually guided locomotion. Journal of neurophysiology. 2007;97:3396–3406. doi: 10.1152/jn.00750.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel KM, Martin JH. Role of sensory-motor cortex activity in postnatal development of corticospinal axon terminals in the cat. The Journal of comparative neurology. 2005;485:43–56. doi: 10.1002/cne.20483. [DOI] [PubMed] [Google Scholar]
- Gerber RJ, Wilks T, Erdie-Lalena C. Developmental milestones: motor development. Pediatrics in review/American Academy of Pediatrics. 2010;31:267–276. doi: 10.1542/pir.31-7-267. quiz 277. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb GL, Myklebust BM, Penn RD, Agarwal GC. Reciprocal excitation of muscle antagonists by the primary afferent pathway. Experimental brain research. 1982;46:454–456. doi: 10.1007/BF00238640. [DOI] [PubMed] [Google Scholar]
- Goulding M. Circuits controlling vertebrate locomotion: moving in a new direction. Nature reviews Neuroscience. 2009;10:507–518. doi: 10.1038/nrn2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagglund M, Borgius L, Dougherty KJ, Kiehn O. Activation of groups of excitatory neurons in the mammalian spinal cord or hindbrain evokes locomotion. Nature neuroscience. 2010;13:246–252. doi: 10.1038/nn.2482. [DOI] [PubMed] [Google Scholar]
- Hansen S, Hansen NL, Christensen LO, Petersen NT, Nielsen JB. Coupling of antagonistic ankle muscles during co-contraction in humans. Experimental brain research. 2002;146:282–292. doi: 10.1007/s00221-002-1152-3. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Jordan VM. Muscle activity during forelimb reaching movements in rats. Behavioural brain research. 1997;85:175–186. doi: 10.1016/s0166-4328(97)87582-1. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nature reviews Genetics. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Williams PT, Martin JH. Rapid and persistent impairments of the forelimb motor representations following cervical deafferentation in rats. Eur J Neurosci. 2013;38:3702–3711. doi: 10.1111/ejn.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaska JF. From intention to action: motor cortex and the control of reaching movements. Advances in experimental medicine and biology. 2009;629:139–178. doi: 10.1007/978-0-387-77064-2_8. [DOI] [PubMed] [Google Scholar]
- Kerman IA, Enquist LW, Watson SJ, Yates BJ. Brainstem substrates of sympatho-motor circuitry identified using trans-synaptic tracing with pseudorabies virus recombinants. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:4657–4666. doi: 10.1523/JNEUROSCI.23-11-04657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn O. Development and functional organization of spinal locomotor circuits. Curr Opin Neurobiol. 2011;21:100–109. doi: 10.1016/j.conb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Lemon RN. Descending pathways in motor control. Annual review of neuroscience. 2008;31:195–218. doi: 10.1146/annurev.neuro.31.060407.125547. [DOI] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Molecular cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. Journal of visualized experiments: JoVE. 2011 doi: 10.3791/2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH. The corticospinal system: from development to motor control. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2005;11:161–173. doi: 10.1177/1073858404270843. [DOI] [PubMed] [Google Scholar]
- Martin JH, Donarummo L, Hacking A. Impairments in prehension produced by early postnatal sensory motor cortex activity blockade. Journal of neurophysiology. 2000;83:895–906. doi: 10.1152/jn.2000.83.2.895. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Lai T, Tayama N. Combined flexor and extensor release for activation of voluntary movement of the fingers in patients with cerebral palsy. Clin Orthop Relat Res. 1990:185–193. [PubMed] [Google Scholar]
- Miura M. Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myklebust BM, Gottlieb GL. Development of the stretch reflex in the newborn: reciprocal excitation and reflex irradiation. Child Dev. 1993;64:1036–1045. [PubMed] [Google Scholar]
- Myklebust BM, Gottlieb GL, Agarwal GC. Stretch reflexes of the normal infant. Dev Med Child Neurol. 1986;28:440–449. doi: 10.1111/j.1469-8749.1986.tb14281.x. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circulation research. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Nashner LM, Shumway-Cook A, Marin O. Stance posture control in select groups of children with cerebral palsy: deficits in sensory organization and muscular coordination. Experimental brain research. 1983;49:393–409. doi: 10.1007/BF00238781. [DOI] [PubMed] [Google Scholar]
- Nezu A, Kimura S, Uehara S, Kobayashi T, Tanaka M, Saito K. Magnetic stimulation of motor cortex in children: maturity of corticospinal pathway and problem of clinical application. Brain Dev. 1997;19:176–180. doi: 10.1016/s0387-7604(96)00552-9. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Kagamihara Y. The regulation of disynaptic reciprocal Ia inhibition during co-contraction of antagonistic muscles in man. The Journal of physiology. 1992;456:373–391. doi: 10.1113/jphysiol.1992.sp019341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Kagamihara Y. The regulation of presynaptic inhibition during co-contraction of antagonistic muscles in man. The Journal of physiology. 1993;464:575–593. doi: 10.1113/jphysiol.1993.sp019652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ohsawa S, Hamada S, Kuida K, Yoshida H, Igaki T, Miura M. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13366–13371. doi: 10.1073/pnas.0910488107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang MY, Yang JF. Interlimb co-ordination in human infant stepping. The Journal of physiology. 2001;533:617–625. doi: 10.1111/j.1469-7793.2001.0617a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrot-Deseilligny E, Burke DJ. The circuitry of the human spinal cord: spinal and corticospinal mechanisms of movement. Cambridge, England; New York: Cambridge University Press; 2012. [Google Scholar]
- Porter R, Lemon R. Corticospinal function and voluntary movement. Oxford, Eng. New York: Clarendon Press; Oxford University Press; 1993. [Google Scholar]
- Qian X, Goderie SK, Shen Q, Stern JH, Temple S. Intrinsic programs of patterned cell lineages in isolated vertebrate CNS ventricular zone cells. Development. 1998;125:3143–3152. doi: 10.1242/dev.125.16.3143. [DOI] [PubMed] [Google Scholar]
- Salimi I, Friel KM, Martin JH. Pyramidal tract stimulation restores normal corticospinal tract connections and visuomotor skill after early postnatal motor cortex activity blockade. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:7426–7434. doi: 10.1523/JNEUROSCI.1078-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serradj N, Paixao S, Sobocki T, Feinberg M, Klein R, Kullander K, Martin JH. EphA4-mediated ipsilateral corticospinal tract misprojections are necessary for bilateral voluntary movements but not bilateral stereotypic locomotion. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:5211–5221. doi: 10.1523/JNEUROSCI.4848-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Weimer RM, McLaughlin T, Kallop D, Stanger K, Yang J, O’Leary DD, Hannoush RN, Tessier-Lavigne M. A caspase cascade regulating developmental axon degeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:17540–17553. doi: 10.1523/JNEUROSCI.3012-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM. The coactivation of antagonist muscles. Canadian journal of physiology and pharmacology. 1981;59:733–747. doi: 10.1139/y81-110. [DOI] [PubMed] [Google Scholar]
- Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. Pseudorabies virus expressing enhanced green fluorescent protein: A tool for in vitro electrophysiological analysis of transsynaptically labeled neurons in identified central nervous system circuits. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9264–9269. doi: 10.1073/pnas.97.16.9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding SJ, White SC, McPherson JJ, Schild R, Transon C, Barsamian P. Electromyographic analysis of reach in individuals with cerebral palsy. Electromyogr Clin Neurophysiol. 1990;30:109–115. [PubMed] [Google Scholar]
- Starkey ML, Barritt AW, Yip PK, Davies M, Hamers FP, McMahon SB, Bradbury EJ. Assessing behavioural function following a pyramidotomy lesion of the corticospinal tract in adult mice. Experimental neurology. 2005;195:524–539. doi: 10.1016/j.expneurol.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Shiller DM, Gribble PL, Ostry DJ. Relationship between cocontraction, movement kinematics and phasic muscle activity in single-joint arm movement. Experimental brain research. 2001;140:171–181. doi: 10.1007/s002210100797. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teulier C, Sansom JK, Muraszko K, Ulrich BD. Longitudinal changes in muscle activity during infants’ treadmill stepping. Journal of neurophysiology. 2012;108:853–862. doi: 10.1152/jn.01037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaivre-Douret L. Developmental coordination disorders: state of art. Neurophysiologie clinique = Clinical neurophysiology. 2014;44:13–23. doi: 10.1016/j.neucli.2013.10.133. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nature neuroscience. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Z’Graggen WJ, Metz GA, Kartje GL, Thallmair M, Schwab ME. Functional recovery and enhanced corticofugal plasticity after unilateral pyramidal tract lesion and blockade of myelin-associated neurite growth inhibitors in adult rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:4744–4757. doi: 10.1523/JNEUROSCI.18-12-04744.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes & development. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker JG, Missiuna C, Harris SR, Boyd LA. Developmental coordination disorder: a review and update. European journal of paediatric neurology: EJPN: official journal of the European Paediatric Neurology Society. 2012;16:573–581. doi: 10.1016/j.ejpn.2012.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.