

 Formation of dianionic 2,2′-bipyridyl-bridged dinuclear yttrium complexes proceeded upon treatment of (ArNCH2CH2NAr)Y(CH2SiMe3)(THF)2 with 2-arylpyridine, in which mononuclear (2-pyridylphenyl)yttrium complexes were detected as key intermediates.
Formation of dianionic 2,2′-bipyridyl-bridged dinuclear yttrium complexes proceeded upon treatment of (ArNCH2CH2NAr)Y(CH2SiMe3)(THF)2 with 2-arylpyridine, in which mononuclear (2-pyridylphenyl)yttrium complexes were detected as key intermediates.
Abstract
An alkylyttrium complex supported by an N,N′-bis(2,6-diisopropylphenyl)ethylenediamido ligand, (ArNCH2CH2NAr)Y(CH2SiMe3)(THF)2 (1, Ar = 2,6- i Pr2C6H3), activated an ortho-phenyl C–H bond of 2-phenylpyridine (2a) to form a (2-pyridylphenyl)yttrium complex (3a) containing a five-membered metallacycle. Subsequently, a unique C(sp2)–C(sp2) coupling of 2-phenylpyridine proceeded through a bimetallic yttrium intermediate, derived from an intramolecular shift of the yttrium center to an ortho-position of the pyridine ring in 3a, to yield a bimetallic yttrium complex (4a) bridged by two-electron reduced 6,6′-diphenyl-2,2′-bipyridyl. Aryl substituents at the ortho-position of the pyridine ring were key in order to destabilize the μ,κ2-(C,N)-pyridyldiyttrium intermediate prior to the C(sp2)–C(sp2) bond formation.
Introduction
Transition metal-catalyzed homo-coupling reactions of two arenes are an important category of C–C bond forming reactions to construct π-conjugated biaryl skeletons. 1,2 For example, in the Ullmann coupling reaction, the activation of aryl C–X bonds of heteroarenes with a low-valent metal species, such as Cu powder or Ni(cod)2, produces the corresponding biaryl compounds. Although these reductive homo-coupling reactions are frequently used, the formation of salt waste is inevitable, and thus there is a demand for a more atom-economical method to synthesize biaryl skeletons. The most direct protocol for C(sp2)–C(sp2) bond formation is through C–H bond activation of aromatic compounds. To date, various transition metal complexes have been applied to such dehydrogenative biaryl coupling reactions.3 The most well-established mechanism for biaryl C(sp2)–C(sp2) bond formation mediated by a mononuclear species is reductive elimination of mononuclear di(aryl)metal species. However, some monoarylated metal species undergo biaryl C(sp2)–C(sp2) coupling reactions. In this reaction, two mechanisms are proposed to be involved, i.e. disproportionation to produce a di(aryl)metal and low-valent metal species,4 and an associative C–C bond formation mediated by two metal centers. 5,6 In the associative mechanism, a bridged dimer species A is first formed through π-coordination of the aryl moiety to another metal center, followed by the formation of species B, which contains a 3-centered-2-electron bridging aryl moiety (Fig. 1). Subsequent C–C bond formation from species B produces the corresponding biaryl compound. A closely related reaction is the Glaser diyne coupling reaction of terminal alkynes using a Cu catalyst, the mechanism of which involves a stepwise process through π-coordination of the C C bond to a different metal center and a 3-centered-2-electron C(sp)-bridging dinuclear intermediate before the C–C bond-forming step.7 Synthesis of the C C π-coordination-bridged multimetallic species and mechanistic studies of the bimetallic aggregation-assisted C(sp)–C(sp) bond formation are feasible due to the strong coordinating ability of the alkyne moiety to the metal center; however, corresponding studies of arylmetal species and the mechanism of associative biaryl C(sp2)–C(sp2) bond formation have not been established due to the weaker π-aromatic coordination to the metal center compared with C C π-coordination.
Fig. 1. Aggregated monoarylated and monoalkynylated metal species. A: π-coordination-bridged bimetallic species. B: 3-centered-2-electron Ph-bridged bimetallic species. C: dianionic biphenyl-bridged bimetallic lanthanide complex. D: dianionic diyne-bridged bimetallic lanthanide complex.

Rare-earth metal complexes containing a mono(aryl)- or mono(alkynyl)metal moiety, generated by C–H bond activation of arenes and terminal alkynes using an alkylmetal species, also mediate C(sp2)–C(sp2) and C(sp)–C(sp) bond formation through the aggregation of two metal species. Because of the stability of the +3 oxidation state of the rare-earth metal center, C–C bond formation products, biaryls and diynes, have been trapped in their dianionic form to give bimetallic complexes such as C and D as reported by Fryzuk et al. (Fig. 1), 5,8,9 even though access to low-valent rare-earth metal complexes has been reported by Evans et al. 10 In our studies on the C–H bond activation of heteroaromatic compounds by rare-earth metal and early transition metal complexes,11 we found that σ-bond metathesis and subsequent 2,2′-bipyridyl formation from 2-arylpyridines proceeded upon treatment of an alkyl complex of (ethylenediamido)yttrium (1) with 2-arylpyridine (2). During this transformation, the C–H bond adjacent to the nitrogen atom of the pyridine ring was selectively functionalized. Mononuclear (2-pyridylphenyl)yttrium complex 3 was detected and isolated as an intermediate in the formation of dianionic 2,2′-bipyridyl-bridged dinuclear yttrium complex 4 (Scheme 1). This is the first example of 2,2′-bipyridyl formation through bimetallic aggregation, even though dimerization of pyridine via C–H bond activation and insertion reactions has previously been reported by Teuben and Diaconescu, respectively.12 Catalytic 2,2′-bipyridyl formation via C–H bond activation has only been achieved using heterogeneous Pd/C and Ru cluster catalysts.13 In addition, steric and electronic tuning of the pyridine derivatives led to the isolation of dinuclear bis(μ,κ2-(C,N)-pyridyl)diyttrium, mononuclear κ2-(C,N)-pyridylyttrium, and 5-membered metallacycle complexes as possible intermediates in the C–C bond formation.
Scheme 1. Pyridylyttrium-mediated 2,2′-bipyridyl formation. [Y] = (ArNCH2CH2NAr)Y.

Results and discussion
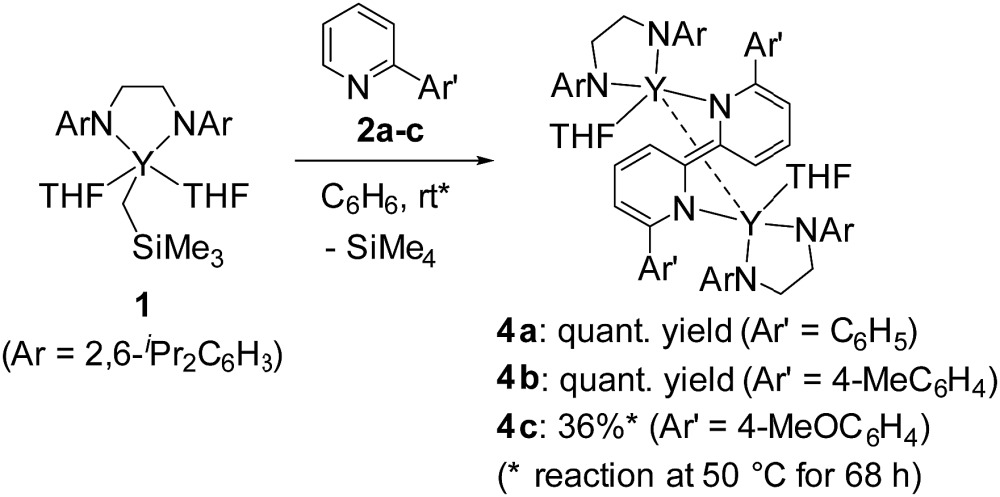
We first treated an alkylyttrium complex (ArNCH2CH2NAr)Y(CH2SiMe3)(THF)2 (1, Ar = 2,6- i Pr2C6H3)14 with 1 equiv. of 2-phenylpyridine (2a) in benzene at room temperature. The color of the solution changed immediately from pale yellow to orange and then to dark green, and green-colored crystals of 4a were precipitated (eqn (1)). The green crystals were sparingly soluble in aromatic and aliphatic solvents. The molecular structure of 4a was determined by X-ray diffraction studies, and its ORTEP drawing is shown in Fig. 2. During the reaction, 6,6′-diphenyl-2,2′-bipyridyl was formed as a biaryl coupling product of 2-phenylpyridine. Two (ethylenediamido)yttrium moieties are bridged by the two-electron reduced 6,6′-diphenyl-2,2′-bipyridyl ligand. The nitrogen atoms in the 2,2′-bipyridyl moiety are located on opposite sides, and the 2,2′-bipyridyl ligand coordinates to the two yttrium atoms in a μ–η4:η4-coodination mode. The bond length of Y1–N1 (2.344 Å) is shorter than the typical yttrium–nitrogen dative bond (ca. 2.5 Å),15 but longer than the Y1–N2 and Y1–N3 bonds (ca. 2.19 Å). The Y1–C2* bond (2.661 Å) is much longer than the yttrium–carbon covalent bond (ca. 2.45 Å). 5b,15c The C1–C1* bond (1.396 Å) of the central 2,2′-bipyridyl moiety is similar in length to the analogous bond in two-electron reduced 2,2′-bipyridyl bound to two alkali metal centers with alternate planes (1.400 Å, rubidium).16
Fig. 2. Molecular structure of complex 4a with 30% thermal ellipsoids. All hydrogen atoms and isopropyl groups are omitted for clarity. Selected bond lengths (Å) and angles (°): Y1–N1, 2.344(5); Y1–C1, 2.746(5); Y1–C1*, 2.857(5); Y1–C2*, 2.661(5); N1–C1, 1.424(6); C1–C2, 1.476(7); C1–C1*, 1.396(10); Y1–N2, 2.194(4); Y1–N3, 2.192(5); N1–Y1–C2*, 70.68(16); N2–Y1–N3, 80.78(17). Dihedral angle between N1–Y1–C2* and N1–C1–C1*–C2* planes, 109.6.

 |
1 |
In addition to 2-phenylpyridine, 2-arylpyridines with methyl and methoxy groups at the para-position of the phenyl group were applicable to the 2,2′-bipyridyl formation. Complex 4b was isolated in quantitative yield after treatment of 1 with 1 equiv. of 2-(4-methylphenyl)pyridine (2b) at room temperature for 48 h. When 2-(4-methoxyphenyl)pyridine (2c) was used as the substrate, heating the reaction mixture at 50 °C for 68 h led to the formation of green crystals of 4c in 36% yield. The low isolated yield of 4c was attributed to decomposition during the C–C bond forming process. Because complexes 4b and 4c had poor solubility in aliphatic and aromatic solvents and low stability in coordinating solvents, characterization of 4b and 4c was based only on X-ray diffraction studies and combustion analyses.17
We next conducted a deuterium labelling experiment. The addition of C6D5 derivative 2a-d 5 to complex 1 in C6H6 resulted in the formation of the same green crystals together with a mixture of SiMe4 and SiMe4-d 1, the former indicating direct C–H bond activation at the ortho-position of the pyridine ring by the alkylyttrium moiety, and the latter indicating C–D bond activation of the ortho-C6D5 position by Y–CH2SiMe3 followed by an intramolecular shift of the yttrium atom to the ortho-position of the pyridyl before the C–C bond forming process (vide infra). These processes are consistent with subsequent oxidative quenching of the crystalline compound by CCl4 to give a mixture of d 8-, d 9-, and d 10-6,6′-diphenyl-2,2′-bipyridyl, as evidenced by the intensity (69%-H) of the singlet signal corresponding to the ortho-position of the phenyl ring at δ H 8.18 (Scheme 2).
Scheme 2. Deuterium labelling experiment.

In addition, when 1 was reacted with 2a at room temperature in THF for 9 days to allow complete C–H bond activation, 5-membered metallacyclic complex 3a was isolated in 98% yield. Complex 3a was stable and no further coupling reaction was detected in THF, but dissolution of 3a in C6D6 afforded the bimetallic compound 4a quantitatively. This clearly indicated that the 5-membered metallacyclic complex 3a is a metastable species in benzene that could lead to a subsequent intramolecular shift of the yttrium center to the ortho-position of the pyridine ring, followed by C–C bond formation to afford 4a (Scheme 3).
Scheme 3. Stepwise metallacycle and C–C bond formation.

When 2-arylpyridines 2c and 2d were treated with 1 in C6D6 at room temperature, C–H bond activation of the aryl ring proceeded to form five-membered metallacyclic complexes 3c and 3d (Scheme 4). On heating the C6D6 solution of 3c at 50 °C, green microcrystals were precipitated from the reaction mixture, as determined by eqn (1). In contrast, 3d was stable in C6D6 at 50 °C. We thus presumed that the C–C bond formation was affected by electron-donating/-withdrawing substituents meta to the metallated carbon; THF coordination to yttrium for 3a and 3b was weaker than that for 3c and 3d in benzene, leading to easy dissociation of THF from yttrium for 3a and 3b and C–C bond formation to form 4a and 4b at room temperature.
Scheme 4. Effect of substituents on the aryl ring of 2-arylpyridine on the C–C bond formation step.

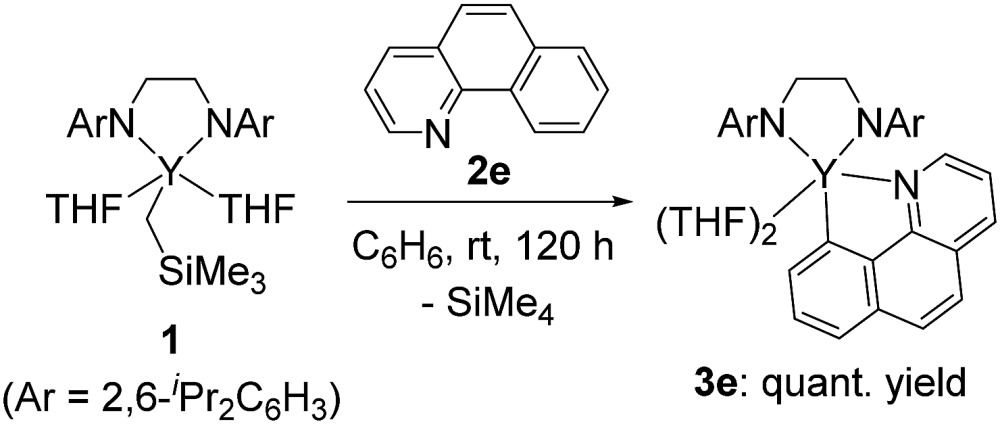
In addition to the isolation of stable five-membered metallacyclic complex 3d, the reaction of 1 with benzo[h]quinolone at room temperature gave 3e in quantitative yield (eqn (2)). Complex 3e was isolated as microcrystals suitable for X-ray diffraction studies. Although the quality of the crystallographic data from the X-ray diffraction studies was insufficient, we determined the overall structure of 3e, in which a C–H bond of benzo[h]quinolone was activated to form a five-membered metallacycle as shown in the ESI (Fig. S1†). Complex 3e was not converted to the C–C bond formation product analogous to 4a–c, probably due to the low flexibility of the benzo[h]quinolone scaffold.
 |
2 |
In sharp contrast to the reaction of 1 with 2-arylpyridines 2a–2c, treatment of 1 with 1 equiv. of pyridine (5a) or 4- or 3-substituted pyridines (5b–d) afforded bis(μ,κ2-(C,N)-pyridyl)diyttrium complexes 6a–d as poorly soluble yellow microcrystals (Scheme 5). The ORTEP drawing of 6a is shown in Fig. 3. The μ,κ2-(C,N)-pyridyl ligand is positioned at the bridging part of the bimetallic structure. The bond lengths of Y–N1 (2.328(4) Å) and Y*–C1 (2.560(5) Å) are longer than those found for mononuclear κ(C,N)-pyridylyttrium complexes. 17,18 Two yttrium atoms, two bridging carbons, and the two nitrogen atoms of the bridging pyridines are located in the same plane. The 1H NMR spectrum of 6a displays four resonances corresponding to the bridging pyridine ring at δ H 9.18 (3-py), 8.81 (6-py), 7.46 (4-py), and 6.80 (5-py). A significant downfield shift of the resonance at the 3-py position might be due to the proximity of the C–H bond to the metal fragment. When 3,5-dimethylpyridine (5e) was used as the substrate, mononuclear yttrium complex 7e was isolated in 97% yield. In the 13C NMR spectrum, a doublet signal was observed for the carbon atom attached to the yttrium center at δ C 219.6 (1 J Y–C = 35.2 Hz), which is in the typical range for mononuclear arylyttrium complexes.18 Even after heating solutions of complexes 6a–d and 7e, which contained a 2-pyridylyttrium moiety in the molecular structure, C–C coupling products were not detected in the reaction mixture; decomposition of the complexes was observed, and no single species was isolated from the reaction mixture. 2-Trimethylsilylpyridine (5f) was also reacted with yttrium complex 1 to form (dimethylpyridylsilyl)methylyttrium complex 8f via C(sp3)–H bond activation. In this case, an intramolecular shift of the yttrium center to form 2-pyridylyttrium species or 6,6′-bis(trimethylsilyl)-2,2′-bipyridyl formation was not observed.
Scheme 5. Reactions of alkylyttrium complex 1 with pyridine derivatives.

Fig. 3. Molecular structure of complex 6a with 30% thermal ellipsoids. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Y1–N1, 2.328(4); Y1–C1, 2.681(5); Y1–C1*, 2.560(5); Y1–N2, 2.216(4); Y1–N3, 2.219(4); Y1–O1, 2.422(3).

Based on our findings for the alkylyttrium-mediated C–H bond activation and C–C coupling reaction, we propose a mechanism for 2,2′-bipyridyl formation as shown in Scheme 6. First, alkylyttrium complex 1 cleaves a C–H bond at the ortho-position on the phenyl ring of 2a to produce five-membered metallacycle complex 3a. Complex 3a is isomerized to three-membered metallacycle intermediate E. Initial formation of the five-membered metallacycle prior to formation of the three-membered metallacycle, as the major pathway, was confirmed by the deuterium labelling experiment as shown in Scheme 2, where mono-deuterated SiMe4-d 1 was generated and one H atom was incorporated into the phenyl ring. Direct formation of the intermediate E from complex 1 was plausible as a minor pathway, and this was confirmed by the detection of SiMe4 in the deuterium labelling experiment. Isomerization between three- and five-membered metallacycles was similarly reported by Diaconescu et al. for rare-earth metal complexes. Although the isomerization trend is opposite to the report by Diaconescu et al., they mentioned that the pyridyl carbanion is more stable (2.8 kcal mol–1) than the phenyl carbanion for the phenylpyridyl anion. We presume that the relative stabilities of the three- and five-membered metallacycles are significantly affected by the attached metal fragment.19 The effect of substituents of the 2-arylpyridines on the C–C bond formation as shown in Scheme 4 indicates that the dissociation of the coordinating THF from the yttrium is key for further isomerization. Although the three-membered metallacycle intermediate E dimerized as a doubly μ,κ2-(C,N)-bridged dinuclear structure, similar to diyttrium complexes 6a–d, introduction of aryl groups at the ortho-position of the pyridine ring might destabilize the μ,κ-(C,N)-bridging mode of the pyridine moiety to afford 4a through 3-centered-2-electron aryl-bridged intermediate F.
Scheme 6. Plausible mechanism for the reductive dimerization of 2-phenylpyridine.

Conclusions
We demonstrated that yttrium-mediated 2,2′-bipyridyl formation proceeded through a bimetallic pyridylyttrium intermediate. Introduction of aryl substituents at the ortho-position of the pyridine ring destabilized the μ,κ2-(C,N)-bridged intermediate to accelerate associative bimetallic C(sp2)–C(sp2) bond formation. Further application of such bimetallic-mediated coupling reactions with not only rare-earth metal complexes but also early transition metal complexes is ongoing in our laboratory.
Acknowledgments
Y. S. acknowledges the financial support from the JSPS Postdoctoral Fellowship. H. N. expresses his special thanks for the financial support provided by the JSPS Research Fellowships for Young Scientists. H. T. acknowledges financial support from a Grant-in-Aid for Young Scientists (A) of the Ministry of Education, Culture, Sports, Science, and Technology, Japan. This work was supported by the Core Research for Evolutional Science and Technology (CREST) Program of the Japan Science and Technology Agency.
Footnotes
References
- (a) Nelson T. D., Crouch R. D. Org. React. 2004;63:265. [Google Scholar]; (b) Yamamoto Y., Copper-mediated aryl-aryl bond formation leading to biaryls. A century after the Ullmann breakthrough, in Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014, p. 335. [Google Scholar]
- (a) Bringmann G., Walter R., Weirich R. Angew. Chem., Int. Ed. Engl. 1990;29:977. [Google Scholar]; (b) Hassan J., Sevignon M., Gozzi C., Schulz E., Lemaire M. Chem. Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- For recent reviews of dehydrogenative coupling reactions, see: ; (a) Yeung C. S., Dong V. M. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; (b) Sun C.-L., Li B.-J., Shi Z.-J. Chem. Rev. 2011;111:1293. doi: 10.1021/cr100198w. [DOI] [PubMed] [Google Scholar]; (c) Liu C., Zhang H., Shi W., Lei A. Chem. Rev. 2011;111:1780. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]; (d) Cho S. H., Kim J. Y., Kwak J., Chang S. Chem. Soc. Rev. 2011;40:5068. doi: 10.1039/c1cs15082k. [DOI] [PubMed] [Google Scholar]; (e) Ashenhurst J. A. Chem. Soc. Rev. 2010;39:540. doi: 10.1039/b907809f. [DOI] [PubMed] [Google Scholar]; (f) Ackermann L., Vicente R., Kapdi A. R., Angew. Chem., Int. Ed., 2009, 48 , 9792 , . For recent reports of dehydrogenative 2,2′-bipyridine formation, see . [DOI] [PubMed] [Google Scholar]; (g) Kawashima T., Takao T., Suzuki H. J. Am. Chem. Soc. 2007;129:11006. doi: 10.1021/ja074224u. [DOI] [PubMed] [Google Scholar]; (h) Takao T., Kawashima T., Kanda H., Okamura R., Suzuki H. Organometallics. 2012;31:4817. [Google Scholar]
- (a) Nakamura A., Otsuka S. Tetrahedron Lett. 1974;15:463. [Google Scholar]; (b) Tsou T. T., Kochi J. K. J. Am. Chem. Soc. 1979;101:7547. [Google Scholar]
- For reports of biaryl formation from phenyllanthanide complexes, see: ; (a) Fryzuk M. D., Love J. B., Rettig S. J. J. Am. Chem. Soc. 1997;119:9071. [Google Scholar]; (b) Fryzuk M. D., Jafarpour L., Kerton F. M., Love J. B., Patrick B. O., Rettig S. J. Organometallics. 2001;20:1387. [Google Scholar]
- For reports of biaryl formation from arylmetal complexes of late transition metals, see: ; (a) Konze W. V., Scott B. L., Kubas G. J. J. Am. Chem. Soc. 2002;124:12550. doi: 10.1021/ja020798h. [DOI] [PubMed] [Google Scholar]; (b) Ramakrishna T. V., Sharp P. R. Organometallics. 2004;23:3079. [Google Scholar]; (c) Wallasch M. W., Weismann D., Riehn C., Ambrus S., Wolmershauser G., Lagutschenkov A., Niedner-Schatteburg G., Sitzmann H. Organometallics. 2010;29:806. [Google Scholar]
- (a) Mizuno N., Kamata K., Nakagawa Y., Oishi T., Yamaguchi K. Catal. Today. 2010;157:359. [Google Scholar]; (b) Zhang G., Yi H., Zhang G., Deng Y., Bai R., Zhang H., Miller J. T., Kropf A. J., Bunel E. E., Lei A. J. Am. Chem. Soc. 2014;136:924. doi: 10.1021/ja410756b. [DOI] [PubMed] [Google Scholar]; (c) Bai R., Zhang G., Yi H., Huang Z., Qi X., Liu C., Miller J. T., Kropf J., Bunnel E. E., Lan Y., Lei A. J. Am. Chem. Soc. 2014;136:16760. doi: 10.1021/ja5097489. [DOI] [PubMed] [Google Scholar]
- For related dianionic diyne-ligated rare-earth metal complexes, see: ; (a) Evans W. J., Keyer R. A., Ziller J. W. Organometallics. 1990;9:2628. [Google Scholar]; (b) Heeres H. J., Nijhoff J., Teuben J. H., Rogers R. D. Organometallics. 1993;12:2609. [Google Scholar]; (c) Dube T., Guan J., Gambarotta S., Yap G. P. A. Chem.–Eur. J. 2001;7:374. doi: 10.1002/1521-3765(20010119)7:2<374::aid-chem374>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- For related dianionic biaryl-ligated rare-earth metal complexes, see: ; (a) Huang W., Dulong F., Wu T., Khan S. I., Miller J. T., Cantat T., Diaconescu P. L. Nat. Commun. 2013;4:1448. doi: 10.1038/ncomms2473. [DOI] [PubMed] [Google Scholar]; (b) Huang W., Diaconescu P. L. Eur. J. Inorg. Chem. 2013:4090. [Google Scholar]; (c) Huang W., Abukhalil P. M., Khan S. I., Diaconescu P. L. Chem. Commun. 2014;50:5221. doi: 10.1039/c3cc47505k. [DOI] [PubMed] [Google Scholar]
- (a) MacDonald M. R., Ziller J. W., Evans W. J. J. Am. Chem. Soc. 2011;133:15914. doi: 10.1021/ja207151y. [DOI] [PubMed] [Google Scholar]; (b) MacDonald M. R., Bates J. E., Fieser M. E., Ziller J. W., Furche F., Evans W. J. J. Am. Chem. Soc. 2012;134:8420. doi: 10.1021/ja303357w. [DOI] [PubMed] [Google Scholar]; (c) MacDonald M. R., Bates J. E., Ziller J. W., Furche F., Evans W. J. J. Am. Chem. Soc. 2013;135:9857. doi: 10.1021/ja403753j. [DOI] [PubMed] [Google Scholar]; (d) Kotyk C. M., MacDonald M. R., Ziller J. W., Evans W. J. Organometallics. 2015;34:2287. [Google Scholar]; (e) Meyer G. Angew. Chem., Int. Ed. 2014;53:3550. doi: 10.1002/anie.201311325. [DOI] [PubMed] [Google Scholar]
- (a) Tsurugi H., Yamamoto K., Mashima K. J. Am. Chem. Soc. 2011;133:732. doi: 10.1021/ja1100118. [DOI] [PubMed] [Google Scholar]; (b) Kaneko H., Nagae H., Tsurugi H., Mashima K. J. Am. Chem. Soc. 2011;133:19626. doi: 10.1021/ja208293h. [DOI] [PubMed] [Google Scholar]
- For reports of related 2,2′-bipyridyl formation through an insertion-dimerization pathway, see: ; (a) Deelman B.-J., Stevels W. M., Teuben J. H., Lakin M. T., Spek A. L. Organometallics. 1994;13:3881. [Google Scholar]; (b) Soo H. S., Diaconescu P. L., Cummins C. Organometallics. 2004;23:498. [Google Scholar]; (c) Carver C. T., Diaconescu P. L. J. Am. Chem. Soc. 2008;130:7558. doi: 10.1021/ja802728k. [DOI] [PubMed] [Google Scholar]; (d) Carver C. T., Benitez D., Miller K. L., Williams B. N., Tkatchouk E., Goddard III W. A., Diaconescu P. L. J. Am. Chem. Soc. 2009;131:10269. doi: 10.1021/ja902794w. [DOI] [PubMed] [Google Scholar]; (e) Carver C. T., Williams B. N., Ogilby K. R., Diaconescu P. L. Organometallics. 2010;29:835. [Google Scholar]; (f) Jie S., Diaconescu P. L. Organometallics. 2010;29:1222. [Google Scholar]; (g) Williams B. N., Huang W., Miller K. L., Diaconescu P. L. Inorg. Chem. 2010;49:11493. doi: 10.1021/ic101493k. [DOI] [PubMed] [Google Scholar]
- (a) Kelly N. R., Goetz S., Batten S. R., Kruger P. E. CrystEngComm. 2008;10:68. [Google Scholar]; (b) Shaw A. P., Ghosh M. K., Törnroos K. W., Wragg D. S., Tilset M., Sawng O., Heyn R. H., Jakobsen S. Organometallics. 2012;31:7093. [Google Scholar]; (c) Kawashima T., Takao T., Suzuki H. J. Am. Chem. Soc. 2007;129:11006. doi: 10.1021/ja074224u. [DOI] [PubMed] [Google Scholar]
- Eppinger J., Nikolaides K. R., Zhang-Presse M., Riederer F. A., Rabe G. W., Rheingold A. L. Organometallics. 2008;27:736. [Google Scholar]
- (a) Roitershtein D., Domingos A., Pereira L. C. J., Ascenso J. R., Marques N. Inorg. Chem. 2003;42:7666. doi: 10.1021/ic034838+. [DOI] [PubMed] [Google Scholar]; (b) Karpov A. V., Shavyrin A. S., Cherkasov A. V., Fukin G. K., Trifonov A. A. Organometallics. 2012;31:5349. [Google Scholar]; (c) Zhang Y., Zhang J., Hong J., Zhang F., Weng L., Zhou X. Organometallics. 2014;33:7052. [Google Scholar]; (d) Nagae H., Shibata Y., Tsurugi H., Mashima K. J. Am. Chem. Soc. 2015;137:640. doi: 10.1021/ja511964k. [DOI] [PubMed] [Google Scholar]
- (a) Bock H., Lehn J.-M., Pauls J., Holl S., Krenzel V. Angew. Chem., Int. Ed. 1999;38:952. doi: 10.1002/(SICI)1521-3773(19990401)38:7<952::AID-ANIE952>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; (b) Gore-Randall E., Irwin M., Denning M. S., Goicoechea J. M. Inorg. Chem. 2009;48:8304. doi: 10.1021/ic9009459. [DOI] [PubMed] [Google Scholar]
- Molecular structures of 4b and 4c can be found in the ESI.
- (a) Deelman B.-J., Stevels W. M., Teuben J. H., Lakin M. T., Spek A. L. Organometallics. 1994;13:3881. [Google Scholar]; (b) Duchateau R., Brussee E. A. C., Meetsma A., Teuben J. H. Organometallics. 1997;16:5506. [Google Scholar]; (c) Elvidge B. R., Arndt S., Zeimentz P. M., Spaniol T. P., Okuda J. Inorg. Chem. 2005;44:6777. doi: 10.1021/ic0511165. [DOI] [PubMed] [Google Scholar]; (d) Arndt S., Elvidge B. R., Zeimentz P. M., Spaniol T. P., Okuda J. Organometallics. 2006;25:793. [Google Scholar]
- Williams B. N., Benitez D., Miller K. L., Tkatchouk E., Goddard III W. A., Diaconescu P. L. J. Am. Chem. Soc. 2011;133:4680. doi: 10.1021/ja110505r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.