

Abstract
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder that leads to slowness of movement, tremor, rigidity and in the later stages of PD, cognitive impairment. Pathologically PD is characterized by the accumulation of α-synuclein in Lewy bodies and neurites. There is degeneration of neurons throughout the nervous system with the degeneration of dopamine neurons in the substantia nigra pars compacta leading to the major symptoms of PD.
RATIONALE
In the brains of PD patients, pathologic α-synuclein seems to spread from cell-to-cell via self-amplification, propagation, and transmission in a stereotypical and topographical pattern among neighboring cells and/or anatomically connected brain regions. The spread or transmission of pathologic α-synuclein is emerging as potentially important driver of PD pathogenesis. The underlying mechanisms and molecular entities responsible for the transmission of pathologic α-synuclein from cell-to-to cell are not known, but the entry of pathologic α-synuclein into neurons is thought to occur, in part through an active clathrin-dependent endocytic process.
RESULTS
Using recombinant α-synuclein pre-formed fibrils (PFF) as a model system to study the transmission of misfolded α-synuclein from neuron to neuron, we screened a library encoding transmembrane proteins for α-synuclein-biotin PFF binding candidates via detection by streptavidin-AP (alkaline phosphatase) staining. Three positive clones were identified that bind α-synuclein PFF and include lymphocyte-activation gene 3 (LAG3), neurexin 1β and amyloid beta precursor-like protein 1 (APLP1). Of these three transmembrane proteins, LAG3 demonstrated the highest ratio of selectivity for α-synuclein PFF over the α-synuclein monomer. α-Synuclein PFF binds to LAG3 in a saturable manner (Kd = 77 nM), while the α-synuclein monomer does not bind to LAG3. Co-immunoprecipitation also suggests that pathological α-synuclein PFF specifically binds to LAG3. Tau PFF, β-amyloid oligomer and β-amyloid PFF do not bind LAG3 indicating that LAG3 is specific for α-synuclein PFF. The internalization of α-synuclein PFF involves LAG3 since deletion of LAG3 reduces the endocytosis of α-synuclein PFF. LAG3 colocalizes with the endosomal GTPases, Rab5 and Rab7 and co-endocytoses with pathologic α-synuclein. Neuron-to-neuron transmission of pathologic α-synuclein and the accompanying pathology and neurotoxicity is substantially attenuated by deletion of LAG3 or by LAG3 antibodies. The lack of LAG3 also substantially delayed α-synuclein PFF induced loss of dopamine neurons, as well as biochemical and behavioral deficits in vivo.
CONCLUSION
We discovered that pathologic α-synuclein transmission and toxicity is initiated by binding to LAG3 and that neuron-to-neuron transmission of pathological α-synuclein involves the endocytosis of exogenous α-synuclein PFF by the engagement of LAG3 on neurons. Depletion of LAG3 or antibodies to LAG3 substantially reduce the pathology set in motion by the transmission of pathologic α-synuclein. The identification of LAG3 as an α-synuclein PFF binding protein provides a new target for developing therapeutics designed to slow the progression of PD and related α-synucleinopathies.
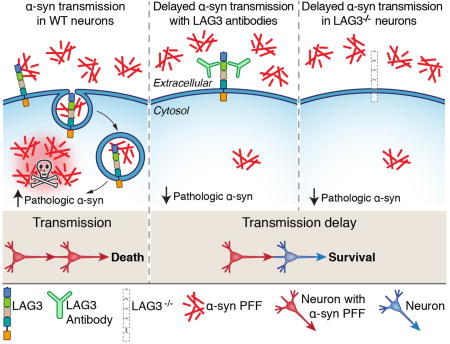
LAG3 deletion or antibodies delays α-synuclein PFF transmission. Compared to wild-type neurons, binding and endocytosis of α-synuclein PFF is dramatically reduced with LAG3 antibodies or when LAG3 is deleted, resulting in delayed pathologic α-synuclein transmission and toxicity. I-Hsun Wu created figure.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder that is characterized clinically by motor dysfunction and pathologically by the aggregation and accumulation of α-synuclein (α-syn) (1–3). Emerging evidence suggests that α-syn spreads from neuron to neuron via self-amplification, propagation, and transmission in the pathogenesis of PD (4–6). In the brains of PD patients, α-syn aggregates seem to spread in a stereotypical and topographical pattern (7, 8). Postmortem examination of fetal grafts in patients with PD found α-syn positive Lewy bodies, suggestive of spread of α-syn from host to graft (9, 10). Other proteins such as β-amyloid and tau in Alzheimer’s disease are also thought to propagate and spread and contribute to the onset and progression of these disorders (11, 12). Pathological α-syn has been shown to spread among neighboring cells and/or anatomically connected brain regions. Recently recombinant α-syn pre-formed fibrils (PFF) provide a model system enabling the study of the transmission of misfolded α-syn from neuron to neuron both in vitro (13) and in vivo (14). How pathological α-syn exits cells and transmits to neighboring neurons is not known, but entry into neurons is thought to occur, in part through an active clathrin-dependent endocytic process (15–17).
Identification of α-syn PFF binding proteins
Mouse α-syn was synthesized and conjugated to biotin (α-syn-biotin), and then aggregated over seven days followed by sonication to form α-syn PFF of relatively uniform size (18). Size exclusion chromatography was used to separate α-syn PFF from α-syn monomers (fig. S1A). Recombinant α-syn-biotin monomers and α-syn PFF were validated by immunoblot analysis (fig. S1B). α-Syn-biotin monomers and PFF were examined by atomic force microscopy (AFM) and transmission electron microscopy. α-Syn-biotin monomers exhibited no regular structure whereas α-syn-biotin PFF exhibited short fibrillar structures (fig. S1, C and D). We then sought to investigate the interaction between extracellular α-syn and neurons. α-Syn-biotin PFF binds to cortical neurons as detected by streptavidin-AP (alkaline phosphatase) staining (19–21), whereas α-syn-biotin monomers weakly bind nonspecifically to neurons (fig. S2, A to C). α-Syn-biotin PFF binding to neurons is saturable with an apparent disassociation constant (Kd) of 374 nM (fig. S2, A to C), suggesting the existence of receptor(s) or binding site(s) for α-syn PFF. We screened a library of transmembrane proteins for potential α-syn-biotin PFF binding candidates via detection by streptavidin-AP staining (fig. S3A) (19–21). A key requirement for expression cloning is the existence of a cell line with low α-syn-biotin PFF background binding. SH-SY5Y cells exhibited less than 8% of the binding levels of α-syn-biotin PFF as compared to cortical neurons, whereas COS7 and HeLa cells exhibited relatively high binding, and HEK293FT cells exhibited mild binding (fig. S3, B and C). Complimentary DNAs encoding 352 transmembrane proteins (TMGW10001, GFC-transfection array panel, Origene) were expressed in SH-SY5Y cells and screened for α-syn-biotin PFF binding candidates. This screening approach is a well-established method to identify ligand receptor interactions (19, 20), but it will not identify receptors composed of heteromeric subunits. Three positive clones were identified that bind α-syn-biotin PFF and include lymphocyte-activation gene 3 (LAG3), neurexin 1β and amyloid beta precursor-like protein 1 (APLP1) (Fig. 1A). Our screen provides Z-factor co-efficients of (0.82, 0.84, and 0.84) through three independent experiments suggesting that the screen was within the optimal signal window to preclude false positives or negatives.
Fig. 1. α-Syn PFF binds to LAG3.

(A) Individual clones from a library consisting of 352 individual cDNAs encoding transmembrane proteins (GFC-transfection array panel, Origene) were transfected into SH-SY5Y cells, and the relative binding signals of human α-syn PFF to individual transmembrane proteins are shown. Positive candidates are LAG3 (NM_002286), NRNX1 (NM_138735) and APLP1 (NM_005166). (B) Mouse α-syn-biotin monomer and α-syn-biotin PFF binding affinity to SH-SY5Y cells expressing the indicated proteins. LAG3* Kd assessment was performed without Triton X-100. All other experiments were performed with 0.1% Triton X-100. Transmembrane proteins similar to the candidates were also tested. Quantification of bound α-syn-biotin PFF to the candidates was performed with ImageJ. Kd values are means ± SEM and are based on monomer equivalent concentrations. Selectivity was calculated by dividing Kd (monomer) by Kd (PFF). Binding of α-syn-biotin monomer was detected at a concentration of 3000 nM, but binding was not saturable. (C) α-Syn-biotin monomer or α-syn-biotin PFF binding to LAG3-overexpressing SH-SY5Y cells as a function of total α-syn concentration in 0% Triton X-100 (TX-100) or 0.1% TX-100 conditions (monomer equivalent for PFF preparations, top panel). Scatchard analysis (bottom panel). Kd= 71 nM (0% TX-100) and 77 nM (0.1% TX-100), data are the means ± SEM, n = 3. (D) Binding of α-syn-biotin PFF to cultured cortical neurons (21 days in vitro (DIV)) is reduced by LAG3 knockout (LAG3−/−), as assessed by alkaline phosphatase assay. α-Syn-biotin PFF WT-Kd = 374 nM, LAG3−/−-Kd = 449 nM, estimated Kd for neuronal LAG3 [dashed line: ΔLAG3 = wild-type (WT) minus LAG3−/−] is 103 nM. Data are the means ± SEM, n = 3. * P < 0.05, Student’s t-test. Power (1-β err prob) = 1. (E) Specificity of LAG3 binding with α-syn-biotin PFF (Fig. S4). Tau-biotin PFF (Fig. S8), β-amyloid-biotin oligomer and β-amyloid-biotin PFF (Fig. S9) are negative controls.
α-Syn PFF preferentially binds LAG3
The selectivity of LAG3, neurexin 1β and APLP1 and related transmembrane proteins for α-syn-biotin PFF versus α-syn-biotin monomers was determined via the ratio of Kd values (Fig. 1B). LAG3 exhibited the highest selectivity with a ratio of 38 followed by neurexin 1β with a ratio of 11 and APLP1 with a ratio of 7. The binding of α-syn-biotin PFF to LAG3 was specific since α-syn-biotin PFF does not bind to the CD4 receptor, which has 20% homology to LAG3 (Fig. 1B and fig. S4). In addition to α-syn-biotin PFF binding to neurexin 1β, it also binds to neurexin 3β and mildly binds to neurexin 1α and neurexin 2β (Fig. 1B). α-Syn-biotin PFF does not bind the amyloid precursor protein (APP) or the amyloid precursor like protein 2 (APLP2) suggesting that the binding to APLP1 was specific (Fig. 1B). Since LAG3 exhibited the highest selectivity for α-syn-biotin PFF, it was advanced for further study. No LAG3 immunoreactive band was observed in HEK293FT and SH-SY5Y cells consistent with the absence of a streptavidin signal in these cell lines (fig. S5A). Immunoblot analysis with a LAG3 antibody 410C9 revealed that LAG3 was expressed in wild-type (WT), but not LAG3 knockout (LAG3−/−) cortical cultures, confirming the specificity of the LAG3 antibody. LAG3 was primarily expressed in neurons since LAG3 was not detectable in astrocytes or microglia (fig. S5B). α-Syn-biotin PFF binds to LAG3 in a saturable manner with a Kd of 77 nM (Fig. 1, B and C and fig. S4). α-Syn-biotin monomers did not demonstrate specific binding to LAG3 expressing SH-SY5Y cells up to 3000 nM (Fig. 1, B and C and fig. S4). In the absence of cell permeabilization by excluding TX-100, α-syn-biotin PFF binding still binds with a Kd of 71 nM (Fig. 1, B and C and fig. S4). WT mouse cortical neurons demonstrated α-syn-biotin PFF binding whereas LAG3−/− mouse cortical neurons had reduced α-syn-biotin PFF binding (Fig. 1D and fig. S6). Specific binding of α-syn-biotin PFF to LAG3 in cortical neurons was determined by subtracting the binding in WT cultures from the binding in LAG3−/− cultures and revealed a Kd of 103 nM (Fig. 1D and fig. S6). These results taken together suggest that α-syn-biotin PFF binds to extracellular LAG3 on neurons.
In vitro co-immunoprecipitation (Co-IP) studies showed that α-syn-biotin PFF but not α-syn-biotin monomers pulls down LAG3 (fig. S7A) and conversely LAG3 pulls down α-syn-biotin PFF but not α-syn-biotin monomers (fig. S7B). Moreover, in vivo Co-IP studies showed that misfolded α-syn from aged transgenic mice pulls down LAG3, but not monomeric α-syn from young transgenic mice overexpressing human A53T α-syn mutant protein (22) or α-syn monomers from WT aged/young mice (fig. S7C), which suggests that LAG3 binds specifically to pathological species of α-syn. We further confirmed that α-syn-biotin PFF binds to human recombinant LAG3 directly with a Kd of 2.7 nM by using an enzyme-linked immunosorbent assay (ELISA) assay (fig. S7D).
LAG3 specifically binds to α-syn PFFs
To test the specificity of α-syn-biotin PFF binding to LAG3, tau-biotin PFF and β-amyloid-biotin oligomer and β-amyloid PFF were tested in non-transfected and LAG3 transfected SH-SY5Y cells via the streptavidin-AP staining assay (19, 20) (Fig. 1E and fig. S8 and S9). Tau-biotin PFF binds to non-transfected SH-SY5Y cells in a saturable manner while overexpression of LAG3 failed to increase the binding of tau-biotin PFF (Fig. 1E and fig. S8, A and B). β-amyloid-biotin oligomer and β-amyloid PFF binds to both non-transfected and LAG3 overexpressing SH-SY5Y cells at high concentrations in a non-specific manner (Fig. 1E and fig. S9, A and B). Taken together these results indicate the LAG3 specifically binds to α-syn PFF.
Like other immunoglobulin (Ig) superfamily molecules, LAG3 contains an ectodomain composed of four Ig-like domains (D1–D4) (23). To determine the α-syn-biotin PFF binding domain, we sequentially deleted each Ig-like domain of LAG3 and performed the cell surface-binding assay with overexpression of LAG3 deletion mutants. These experiments revealed that α-syn-biotin PFF preferentially binds to the D1 domain, whereas deletion of the D2, D3 or the intracellular domain (ICD) substantially weakens binding, but not the D4 domain (fig. S10). Since α-syn-biotin PFF binding to LAG3 is eliminated by deletion of the D1 domain, additional deletions of D1 subdomains (Δdel1-5-D1) were examined. Δdel2(aa 52–80)-D1 and Δdel3(aa 81–109)-D1 significantly reduced α-syn-biotin PFF binding to LAG3, while Δdel1(aa23–51)-D1, Δdel4(aa110–138)-D1 and Δdel5(aa139–167)-D1 moderately reduced binding of LAG3 to α-syn-biotin PFF (fig. S10), suggesting that LAG3 residues 52–109 in the D1 domain are responsible for the LAG3 interaction with α-syn-biotin PFF.
The endocytosis of α-syn PFF involves LAG3
To determine whether LAG3 is involved in the endocytosis of α-syn PFF, pHrodo red was conjugated to α-syn PFF. pHrodo red is a pH dependent dye that increases in fluorescence as pH decreases from the neutral cytosolic pH to the acidic pH of the endosome (fig. S11A) (24). Conjugation of α-syn PFF with pHrodo red does not appreciably change the properties of the α-syn PFF as assessed by immunoblot and AFM (fig. S11, A and B). α-Syn-pHrodo PFF undergoes endocytosis in WT cortical neuron cultures while LAG3−/− neurons exhibited minimal α-syn-pHrodo PFF endocytosis (Fig. 2, A and B and fig. S11C). Overexpression of LAG3 in WT cultures enhanced the endocytosis of α-syn-pHrodo PFF and overexpression of LAG3 in the LAG3−/−cortical cultures restored endocytosis of α-syn-pHrodo PFF (Fig. 2, A and B and fig. S11C). Adeno associated virus serotype 2 (AAV2) expressing EGFP via a synapsin promoter was used to identify neurons and confirmed that α-syn-pHrodo PFF was endocytosed in WT neurons, but much less in LAG3−/− neurons (fig. S11D). Examination of overexpression of the deletion mutants in LAG3−/− cortical cultures showed that the D1 domain deletion mutant failed to restore the endocytosis of α-syn-pHrodo PFF (Fig. S11, E and F). However, introduction of LAG3 deletion mutants (D2, D3 or D4) restored the internalization of α-syn-pHrodo PFF (Fig. S11, E and F).
Fig. 2. Endocytosis of α-syn PFF is dependent on LAG3.

(A) Live image analysis of the endocytosis of α-syn-pHrodo PFF. α-Syn PFF was conjugated with a pH dependent dye (pHrodo red), in which fluorescence increases as pH decreases from neutral to acidic environments. White triangles indicate non-transfected wild-type (WT) or LAG3−/− neurons and white arrows indicate LAG3 transfected neurons. Scale bar, 10 µm. (B) Quantification of panel A, cell number (5–46) from n = 3. (C) Internalized α-syn-biotin PFF co-localizes with Rab5. Co-localization of internalized α-syn-biotin PFF and Rab5 was assessed by confocal microscopy, scale bar, 10 µm. (D) Quantification of panel C, cell number (13–32) from n = 4. One-way ANOVA with Tukey’s correction. Data in B and D are as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Power (1-β err prob) = 1.
The Rab5 GTPase is an early endosomal marker and helps mediate endocytosis (25). As such, we sought to confirm the endocytosis of α-syn-biotin PFF into endosomes by measuring the intensity of co-localized α-syn-biotin PFF with Rab5. We found that α-syn-biotin PFF was co-localized with Rab5 in WT cortical neurons (Fig. 2, C and D). In contrast there was less α-syn-biotin PFF in LAG3−/− cortical neurons (Fig. 2, C and D). Overexpression of LAG3 in WT and LAG3−/− cortical neurons enhanced the intensity of α-syn-biotin PFF co-localizing with Rab5 (Fig. 2, C and D). Rab5 appears to be up-regulated following LAG3 over-expression. LAG3 co-endocytoses with α-syn PFF as LAG3, Rab5 and α-syn-biotin PFF were co-localized (fig. S12A). α-Syn-biotin PFF also colocalized with Rab5 in dendrites and showed a similar pattern in WT, WT + LAG3, LAG3−/−, LAG3−/− + LAG3 neurons (fig. S12, B and C). The endosome markers Rab7 and LAMP1 were also co-localized with α-syn-biotin PFF (fig. S12D).
Examination of overexpression of the deletion mutants in LAG3−/− neuronal cultures showed that the D1 domain deletion mutants failed to increase the colocalization of α-syn-biotin PFF with Rab5 (fig. S13). However, introduction of LAG3 deletion mutants (D2, D3, D4 and the ICD) enhanced the intensity α-syn-biotin PFF co-localizing with Rab5 (fig. S13). Deletions of D1 subdomains (Δdel(1-5)-D1) were also examined for α-syn-biotin PFF colocalization with Rab5. Consistent with our binding assays, Δdel2-D1 and Δdel3-D1 exhibited the greatest effect on reducing the intensity α-syn-biotin PFF co-localizing with Rab5 (fig. S13).
Endosome enriched fractions were isolated via differential centrifugation (26, 27) from WT and LAG3−/− cultures following treatment with α-syn-biotin PFF (fig. S14A). Both monomeric and higher molecular weight forms of α-syn-biotin PFF were found in the endosome enriched fraction of WT neuron cultures, while there was significantly less of both forms in LAG3−/− cultures (fig. S14, B and C). Lenti-viral mediated overexpression of LAG3 in WT cultures enhanced the levels of α-syn-biotin PFF in the endosome enriched fractions and restored the levels of α-syn-biotin PFF in LAG3−/− culture endosome enriched fractions (fig. S14, B and C). LAG3 exists in the endosome enriched fraction (fig. S14D), which is consistent with the data that LAG3, Rab5 and α-syn-biotin PFF are co-localized (fig. S12A). To exclude the possibility that deletion of LAG3 causes a generalized defect in endocytosis, the internalization of latex beads were studied and there is no difference in the internalization of latex beads between WT and LAG3−/− cultures (fig. S14, E and F). LAG3 also specifically recognized α-syn PFF, since tau-biotin PFF as characterized by transmission electron microscopy (TEM) (fig. S15A) were internalized in LAG3−/− neurons and failed to show greater internalization in neurons overexpressing LAG3 (fig. S15B) consistent with the observation of the lack of tau-biotin PFF binding to LAG3 (Fig. 1E and fig. S8, A and B). Taken together these results indicate that LAG3 can mediate the endocytosis of α-syn-biotin PFF into neurons.
The absence of LAG3 prevents α-syn PFF pathology
We then asked whether knocking out LAG3 prevents the pathology induced by α-syn PFF. Phosphorylation of α-syn at serine 129 (P-α-syn) is associated with pathology in α-synucleinopathies. Its levels increase following treatment of α-syn PFF to neuronal cultures (13, 14). Accordingly, we added α-syn PFF to WT and LAG3−/− cortical cultures at seven days in vitro (DIV). Ten days after α-syn PFF treatment, the levels of P-α-syn were markedly increased in WT cultures, while the levels of P-α-syn in LAG3−/− cultures were barely detectable (Fig. 3, A and B). Overexpression of LAG3 enhanced the level of P-α-syn in WT cultures and restored P-α-syn levels in LAG3−/− cultures (Fig. 3, A and B). Furthermore, the accumulation of P-α-syn co-localized with LAG3 (fig. S16A), which is consistent with binding (fig.S16B) and the co-endocytosis of LAG3 and α-syn PFF (fig. S14D). Overexpressing the D1 domain deletion mutant in LAG3−/− neuron cultures failed to restore P-α-syn levels (fig. S17). Overexpression of the D2, D3 and D4 domain deletion mutants in LAG3−/− cortical cultures recaptured the α-syn pathology as monitored by P-α-syn (fig. S17).
Fig. 3. α-Syn PFF induced pathology is reduced by deletion of LAG3 in vitro.

(A) WT and LAG3−/− primary cortical neurons at 7 DIV were treated with α-syn PFF or PBS. LAG3 was overexpressed via Lenti-virus (LV) transduction in WT or LAG3−/− neurons at 4 DIV. 3 days after transduction, 7 DIV cultures were treated with α-syn PFF or PBS. All the cultures were fixed 10-day post-treatment in 4% PFA. Neurons were stained with rabbit mAb MJF-R13 (8-8) for P-α-syn. Scale bar, 40 µm. (B) Quantification of panel A, n = 5 independent experiments, each performed in duplicate. Values are given as the means ± SEM. Statistical significance was determined using one-way ANOVA followed with Tukey’s correction, ***P < 0.001. Power (1-β err prob) = 1. (C) Immunoblots in WT and LAG3−/− neuron lysates of misfolded α-syn, P-α-syn, synapsin II, SNAP25 and LAG3. β-actin served as a loading control. WT and LAG3−/− neuron lysates were sequentially extracted in 1% TX-100 (TX-soluble) followed by 2% SDS (TX-insoluble) 14 days after α-syn PFF treatment. α-Syn PFF recruited endogenous α-Syn into TX-insoluble and hyperphosphorylated aggregates, which was ameliorated by deletion of LAG3. α-Syn PFF caused a reduction in levels of SNAP25 and synapsin II compared to PBS 14 days post-treatment. Deletion of LAG3 prevented PFF-induced synaptic protein loss. (D–G) Quantification of panel C. Values are given as means ± SEM, n = 3 independent experiments. Statistical significance was determine using one-way ANOVA followed by Tukey’s correction, *P < 0.05, **P < 0.01, ***P < 0.001.
Two weeks after α-syn PFF treatment of cortical neurons, immunoblots of α-syn and P-α-syn from lysates sequentially extracted in 1% Triton X-100 (TX-soluble) followed by 2% SDS (TX-insoluble) were examined. α-Syn PFF led to an accumulation of α-syn and P-α-syn in the TX-insoluble fraction in WT cultures, while there was significantly less accumulation in LAG3−/− cultures (Fig. 3, C and D). α-Syn PFF also caused a reduction in SNAP25 and synapsin II levels compared to PBS 14 days post-treatment as previously described (13). Deletion of LAG3 prevented the α-syn PFF-induced synaptic protein loss (Fig. 3, C to G). Overexpression of LAG3 in WT cultures caused an increase accumulation of α-syn and P-α-syn in the TX-insoluble fraction in WT cultures and a further reduction in SNAP25 and synapsin II levels, whereas it prevented the sparing in LAG3−/− cultures (Fig. 3, C to G).
Cell-to-cell transmission of α-syn PFF is reduced in LAG3−/− neurons
To examine the transmission of α-syn PFF and to establish the role of LAG3 in the interneuron transmission of α-syn, we used a microfluidic neuronal culture device with three chambers connected in tandem by a series of microgrooves separating the chambers (TCND1000, Xona Microfluidics, LLC). The medium volume in chamber 1 (C1) is 50-µL lower than the one in chamber 2 (C2), and 100-µL lower than the one in chamber 3 (C3) to prevent diffusion of α-syn PFF to adjacent chambers. Cortical neurons were cultured in each chamber. To ensure that α-syn PFF cannot diffuse between chambers, primary WT cortical neurons in C1 were treated with α-syn-biotin PFF. 14 days post-treatment, the neurons were fixed in 4% paraformaldehyde (PFA) and stained with streptavidin-568 fluorescence dye. Only neurons in C1 exhibited immunofluorescence, indicating that α-syn-biotin PFF cannot transmit from chamber to chamber through diffusion (fig. S18A).
α-Syn transmission from C1 to C3 requires intermediating neurons in C2, since α-syn PFF administration to C1 failed to induce P-α-syn accumulation in C3 when C2 was left empty (Fig. 4, A to C). Using this system, the transmission of α-syn PFF was monitored via P-α-syn levels in WT and LAG3−/− cultures (Fig. 4, D to F and fig. S18B). The microfluidic neuron culture device was then set up to contain WT cultures in C1 and C3, whereas C2 either contained WT or LAG3−/− cultures. In another set of chambers, LAG3 was overexpressed in the C2 chamber containing either WT or LAG3−/− cultures. Administration of α-syn PFF to C1 led to increased P-α-syn levels (Fig. 4, E and F and fig. S18B). To assess the propagation of α-syn PFF along dendrites and axons as well as transmission of misfolded α-syn, the levels of P-α-syn were monitored in C2 and C3. When C2 contains WT cultures, P-α-syn was observed in both C2 and C3 and LAG3 overexpression in C2 neurons enhanced the levels of P-α-syn in both chambers (Fig. 4, E and F and fig. S18B). In contrast, when C2 contains LAG3−/− cultures, P-α-syn levels were significantly reduced in C2 and were absent in C3 (Fig. 4, E and F, fig. S18B). LAG3 overexpression in C2 neurons restored the propagation of α-syn PFF as assessed by similar levels of P-α-syn compared to WT cultures (Fig. 4, E and F and fig. S18B). These results taken together indicate that LAG3 is required for the propagation and transmission of pathologic α-syn.
Fig. 4. α-Syn PFF transmission is reduced by deletion of LAG3 in vitro.

(A) Schematic representation of the 3 chambers in which neurons were cultured in chamber 1 (C1) chamber 2 (C2) and chamber 3 (C3) (top) or C1 and C3 (bottom). (B) α-Syn PFF was added to C1 of the microfluidic device. On day 14, α-syn-biotin PFF was detected via P-α-syn in C2 and C3 when neurons were present in all three chambers. Transmission to C3 is not detectable when neurons are not present in C2. (C) Quantification of immunofluorescent images in B. Data are the means ± SEM, n = 3. one-way ANOVA followed by Sidak’s correction. ***P < 0.001 versus C1. Power (1-β err prob) = 1. Scale bars, 100 µm. (D) Schematic of microfluidic neuron device with three chambers to separate neurons seeded in three chambers. (E)Transmission of pathologic P-α-syn from chamber 1 (C1) to chamber 2 (C2) to chamber 3 (C3) 14 days post-addition of α-syn PFF in C1. The different combinations of neurons tested in C2, listed as C1-(C2)-C3, are: WT-(WT)-WT, WT-(WT+LAG3)-WT, WT-(LAG3−/−)-WT, WT-(LAG3−/−+LAG3)-WT. Scale bar, 10 µm. (F) Quantification of panel E. Values are given as means ± SEM, n = 3. Statistical significance was determine using one-way ANOVA followed by Tukey’s correction, *P < 0.05, **P < 0.01, ***P < 0.001. Power (1-β err prob) = 1.
α-Syn PFF toxicity is reduced in LAG3−/− neurons
Treatment of WT cortical cultures with α-syn PFF cause neuronal cell death as previously described (13) (fig. S19). α-Syn PFF treatment led to substantial cell death compared to PBS treated cultures as assessed by propidium iodide staining (fig. S19, A and B). LAG3−/− cultures exhibited significantly less cell death and overexpression of LAG3 restored the toxicity to α-syn PFF (fig. S19, A and B). Neuronal nuclei (NeuN) antibody staining was also performed to assess neuronal number. α-Syn PFF treatment caused a significant loss of NeuN immunoreactivity and overexpression of LAG3 enhanced neuronal loss (fig. S19, C and D). NeuN immunoreactivity was preserved in LAG3−/− cultures after α-syn PFF treatment, whereas overexpression of LAG3 in LAG3−/− cultures led to a loss of NeuN immunoreactivity (fig. S19 C and D). NeuN immunostaining of deletion mutants (ΔD1-D4, ΔICD) overexpression in LAG3−/− neurons indicated that deletion of the D1 domain failed to exhibit cell death, but deletion of D2, D3, D4 or the ICD domains still led to cell death (fig. S19, E and F). Since deletion of the LAG3 ICD domain may reduce α-syn PFF toxicity, signaling through the ICD of LAG3 may contribute to α-syn PFF toxicity. Furthermore, since calcium might be involved in α-synuclein-induced neurotoxicity (13, 28–32), calcium influx was monitored in response to α-syn PFF. Perfusion of α-syn PFF (500 nM) onto WT cortical neurons led to a gradual increase in intracellular calcium levels (fig. S20). Lack of LAG3 caused a significant decrease of α-syn PFF-induced calcium influx, which may account for the reduced toxicity in LAG3−/− cultures (fig. S20).
LAG3 antibodies reduce α-syn PFF toxicity and cell-to-cell transmission
Two different anti-LAG3 antibodies C9B7W (50 µg/mL) (33) and 410C9 (50 µg/mL) (34) blocked the binding of α-syn-biotin PFF (500 nM) in SH-SY5Y cells expressing LAG3 (Fig. 5A). The anti-LAG3 antibodies also reduced the enrichment of α-syn-biotin PFF (1 µM) in the endosomal-enriched fraction of primary cortical neurons 12 DIV (Fig. 5B) consistent with reduced endocytosis in LAG3−/− neuron culture. Both antibodies reduced the subsequent α-syn PFF pathology detected by phosphorylated α-syn Ser129 (P-α-syn) (Fig. 5C). Rat or mouse IgG had no effect in these assays. Cell-to-cell transmission of α-syn PFF as detected by P-α-syn was significantly reduced by 410C9 while mouse IgG had no effect after 14-days of α-syn PFF treatment (Fig. 5D and fig. S21).
Figure 5. Anti-LAG3 antibodies block α-syn PFF binding, endocytosis, pathology and transmission.

(A) Anti-LAG3 antibodies C9B7W and 410C9 (both 50 µg/mL) block the binding of α-syn-biotin PFF (500 nM) on SH-SY5Y cells expressing LAG3, Scale bar, 50 µm. Right panel, quantification of images in Panel A. Data are the means ± SEM, n =3, Student’s t-test. ***P < 0.001. (B) Anti-LAG3 antibodies C9B7W and 410C9 (both 50 µg/mL) reduced the endocytosis of α-syn-biotin PFF (1 µM) in 12 DIV primary cortical neurons. Rab7 was used to confirm the isolation of endosomes. (C) Phosphorylated α-syn (P-α-syn) as detected by rabbit mAb MJF-R13 (8-8) was reduced by anti-LAG3 antibodies in primary cortical neurons. Scale bar, 50 µm. Right panel, quantification of images in Panel C. Data are the means ± SEM, n = 3, one-way ANOVA followed by Tukey’s correction. **P < 0.01. (D) Anti-LAG3 410C9 antibody delays α-syn PFF transmission in neurons. Left panel is a schematic representation of the 3 microchambers in which neurons were cultured in C1, C2 and C3. α-syn PFF was added to C1 of the microfluidic device on day 7. Mouse IgG or anti-LAG3 410C9 antibody (both 50 µg/mL) was added to C2 on day 7. α-syn PFF transmission was detected via P-α-syn immunostaining in C2 and C3 on day 21. Scale bar, 10 µm. Right panel, quantification of images in bottom panel. Data are the means ± SEM, N =3, one-way ANOVA followed by Tukey’s correction. ***P < 0.001, n.s., non-significant.
To determine whether LAG3 mediates the pathology induced by human α-syn PFF, we assessed P-α-syn immunoreactivity in cortical cultures from mouse prion promoter transgenic mice overexpressing human A53T mutant α-syn (22) in the presence or absence of the LAG3 antibody, 410C9, and in human A53T mutant α-syn cultures lacking LAG3. Human α-syn PFF treatment of A53T α-syn neuron cultures increased P-α-syn, while the absence of LAG3 or the anti-LAG3 antibody significantly reduced P-α-syn immunoreactivity (fig. S22, A and B). Brain homogenates from 10 month old symptomatic human A53T transgenic mice were used to assess endogenous α-syn aggregates and we found that they also increased the level of P-α-syn, while deletion of LAG3 or treatment with the anti-LAG3 410C9 antibody reduced the level of P-α-syn induced by A53T transgenic brain homogenates (fig. S22, C and D). Human α-syn PFF induced toxicity in human A53T α-syn transgenic neuronal cultures while deletion of LAG3 or anti-LAG3 410C9 antibody significantly reduced the toxicity (fig. S22, E and F). These results taken together suggest that LAG3 can mediate the pathology of human α-syn aggregates.
The absence of LAG3 reduces α-syn PFF transmission and toxicity in vivo
To determine whether LAG3 is necessary for α-syn PFF transmission and toxicity in vivo, α-syn PFF was stereotactically injected into the dorsal striatum of WT and LAG3−/− mice (35). Representative maps (red dots, fig. S23A) and representative images and quantification (fig. S23, B and C) of the distribution of LB/LN-like pathology of P-α-syn accumulation and the stereotaxic injection site indicated by gray circles in the α-syn PFF-injected hemisphere are shown for mice sacrificed at 30 and 180 days post-injection (dpi) (fig. S23). P-α-syn immunoreactivity was monitored in substantia nigra pars compacta (SNpc) tyrosine hydroxylase (TH)-positive neurons 30 and 180 days after α-syn PFF injection. We observed substantial P-α-syn staining in WT SNpc TH positive neurons at 30 and 180 days (Fig. 6A). In LAG3−/−, SNpc TH positive neurons, P-α-syn staining is reduced by greater than 50% at both time points. Accompanying the α-syn pathology, stereologic counting of SNpc TH and Nissl positive neurons revealed significant loss of dopamine (DA) neurons in WT mice at 180 days post-injection (Fig. 6B). There was a dramatic preservation of DA neurons in α-syn PFF injected LAG3−/− mice (Fig. 6B). HPLC analysis demonstrated a significant reduction in dopamine and its metabolites DOPAC, HVA and 3MT in WT mice and a sparing of the reduction in LAG3−/− mice (Fig. 6C and fig. S24, A to C). Immunoblot analysis demonstrated a significant reduction in TH and DAT in WT mice and a sparing of the reduction in LAG3−/− mice (fig. S24D). At 180 days post-α-syn PFF injection, WT mice exhibited robust clasping behavior when suspended by their tail similar to prior reports (14, 36), whereas LAG3−/− mice demonstrated a response similar to PBS injected mice (fig. S24E). WT mice injected with α-syn PFF showed significant impairment in the pole test, which is thought to be a sensitive behavioral indicator of dopaminergic function (37), with increased time to turn and time to reach the base, whereas LAG3−/− mice injected with α-syn PFF showed no significant impairments (Fig. 6D). Grip strength analysis indicated that WT mice have reduced forelimb and forelimb plus hindlimb strength after α-syn PFF injection similar to prior reports (14, 36), while the LAG3−/− mice showed no significant loss in grip strength (Fig. 6E). Therefore, LAG3 is crucial for α-syn PFF induced neurodegeneration and the development of PD related motor defects.
Fig. 6. α-Syn PFF induced pathology is reduced by deletion of LAG3 in vivo.

(A) Representative P-α-syn immunostaining and quantification in the substantia nigra par compacta (SNpc) of WT and LAG3−/− mice sacrificed at 30 and 180 days after intrastriatal α-syn PFF injection. Data are the means ± SEM, n = 5–9 mice per group, one-way ANOVA with Sidak’s correction. (B) Stereology counts from TH immunostaining and Nissl staining of SNpc DA neurons of WT and LAG3−/− mice at 180 days after intrastriatal α-syn PFF, α-syn monomer or PBS injection. Data are the mean number of cells per region ± SEM, n = 5–9 mice per group, one-way ANOVA with Dunnett’s correction. (C) DA concentrations in the striatum of α-syn PFF-injected mice and PBS-treated controls measured at 180 days by HPLC. Data are the means ± SEM, n = 5–8 mice per group, one-way ANOVA with Tukey’s correction. (D, E) 180 days after α-syn PFF injection, the pole test and grip strength was performed in WT or LAG3−/− mice injected with PBS or α-syn PFF. Behavioral abnormalities in the pole test and grip strength induced by α-syn PFF injection were ameliorated in LAG3−/− mice. Data are the means ± SEM, n = 7–9 mice per group for behavioral studies. Statistical significance was determined using one-way ANOVA with Tukey’s correction, * P < 0.05, ** P < 0.001, *** P < 0.001, n.s., nonsignificant. Power (1-β err prob) = 1.
Conclusion
The major finding of this paper is that α-syn PFF transmission and toxicity is initiated by binding LAG3. We isolated LAG3 via an unbiased screen for α-syn PFF binding sites. Although our data indicate that LAG3 is not the sole α-syn PFF binding site, it plays a major role in α-syn PFF endocytosis and transmission. Moreover, mice lacking LAG3 exhibit delayed α-syn PFF induced pathology and reduced toxicity.
Consistent with our observations that the absence of LAG3 does not completely reduce α-syn PFF binding and pathology we observed that α-syn PFF also binds to APLP1 and neurexins. A recent study also identified neurexin 1α and 2α as α-syn fibril binding partners (32). The Toll-like receptor 2 (TLR2) on microglia was shown to be involved in the activation of microglia due to exposure to oligomeric α-syn from conditioned neuronal media (38). Heparan sulfate proteoglycans (HSPGs) can mediate α-syn aggregate uptake and seeding via micropinocytosis (39) and the Α3-subnit of the Na+/K+-ATPase (α3-NKA) binds to α-syn fibrils and oligomers (32). Whether these alternative α-syn binding partners contribute to α-syn transmission and pathogenesis and how they might interact with LAG3 requires further study.
LAG3 is enriched not only in the thymus and spleen, but the brain as well (33). Immunoblot analysis indicates that LAG3 is expressed predominantly in neurons, consistent with the observation that LAG3 mediates the transmission of misfolded α-syn from neuron to neuron. According to the Allen Brain Atlas, LAG3 is localized to neurons throughout the central nervous system including DA neurons. It is becoming increasing clear that proteins that were thought to function primarily in the immune system also have important roles in the nervous system including the functional requirement for class I major histocompatibility complex (MHC) in central nervous system development and plasticity (40, 41). Class II MHC is also expressed in the nervous system (42). The function of LAG3 in the CNS is not known and whether misfolded α-syn activates downstream signaling following engagement of LAG3 requires further study.
In summary, we propose a novel mechanism for cell-to-cell transmission of misfolded α-syn that involves the endocytosis of exogenous α-syn PFF by the engagement of LAG3 on neurons. The interaction between LAG3 and α-syn PFF provides a new target for the development of therapeutics designed to slow the progression of PD and related α-synucleinopathies. This could potentially be quickly translated in PD as many pharmaceutical companies are developing agents that block LAG3 (43). The prion-like spread of α-syn PFF and other amyloidgenic proteins is a multi-step process involving the uptake, propagation and release of pathological amyloid proteins. USP19 was recently shown to promote α-syn secretion suggesting a pathway by which pathologic α-syn exits cells in α-synucleinopathies (44). Combined with LAG3 playing a major role in the internalization of pathologic α-syn, we speculate that more mediators will be discovered that are involved in the cell-to-cell transmission of pathologic α-syn.
Materials and methods
α-Syn purification and α-Syn PFF preparation
Recombinant α-syn proteins were purified as previously described (18). α-Syn PFF were prepared by agitating α-syn in a transparent glass vial with a magnetic stirrer (350 rpm at 37° C). After 7 days of incubation the α-syn aggregates were sonicated for 30 seconds at 10% amplitude (Branson Digital Sonifier, Danbury, CT, USA) and the α-syn monomer and α-syn PFF were separated by FPLC using a Superose 6 10/300GL column (GE Healthcare, Life Sciences, Wauwatosa, WI, USA) and fractions containing the α-syn monomer and α-syn PFF were kept at −80° C. To characterize α-syn PFF mediators, recombinant α-syn monomer was purified and labeled with sulfo-NHS-LC-Biotin (Thermo Scientific, Grand Island, NY, USA. EZ-link Sulfo-NHS-LC-Biotin, 21435). The molar ratio of biotin to α-syn was 2~3. After conjugation, α-syn-biotin monomer and α-syn-biotin PFF was prepared as mentioned above.
Preparation and characterization of tau fibrils
Recombinant Tau protein was obtained from rPeptide (T-1001-2, Bogart, GA, USA). Tau was labeled with sulfo-NHS-LC-Biotin as described above for α-syn. In vitro fibrillization of full-length Tau (2N4R) was prepared by mixing 50 µM low molecular weight heparin and 2 mM DTT with 300 µM recombinant Tau in 100 mM sodium acetate buffer (pH 7.0) under constant orbital agitation (1,000 rpm) at 37 °C for 5–7 days (45). Successful fibrillization was confirmed using the thioflavin T fluorescence assay and transmission electron microscopy. The fibrils were mechanically broken down into small fragments by sonication (30 sec, 10% amplitude).
Preparation of synthetic β-amyloid1–42 oligomers
β-amyloid1–42 peptide was purchased from Anaspec (AS-64129-1). 2.2 mM β-amyloid1–42 was dissolved in DMSO, and diluted in PBS to obtain a 250 µM stock solution. β-amyloid1–42 was labeled with sulfo-NHS-LC-Biotin as described above for α-syn. The solution was incubated at 4° C for at least 24 hours to cross-link the peptides (46). The solution was aliquoted after cross-linking and stored at −80° C until use. Before usage, the solution was centrifuged at 12,000 g for 10 minutes to remove the fibril forms of β-amyloid1–42. The supernatant, which contains the dissolved oligomeric β-amyloid1–42 was used for experiments.
Preparation of synthetic β-amyloid1–42 fibrils and PFF
β-amyloid1–42 peptide was purchased from Anaspec (AS-64129-1). β-amyloid1–42 monomer was freshly resuspended in DMSO at concentration of 2.2 mM. The stock solution was further dissolved in PBS to a final concentration of 250 µM and labeled with sulfo-NHS-LC-Biotin, then incubated in 37° C for 24 h. After forming β-amyloid fibrils, the solution was sonicated for 2 minutes to fragment and form the pre-formed fibrils (PFF) before treatment to neurons.
Atomic force microscopy (AFM) measurements
An atomic force microscope (AFM) (Asylum MFP-3D-BIO™, Santa Barbara, CA, USA) was used to perform AFM experiments. Under ambient condition using Si cantilevers with nominal resonance frequency of 330 kHz and nominal spring constant 20–80 N/m (Veeco, Horsham, PA, USA) AFM was performed in the tapping mode.
Transmission electron microscopy (TEM) measurements
Samples were adsorbed to glow discharged 400 mesh carbon coated copper grids (EMS) for 2 minutes. The grids were quickly transferred through three drops of Tris-HCl (50 mM pH 7.4) rinse, then floated upon two consecutive drops of 0.75% uranyl formate 30 seconds each. Stain was either aspirated or blotted off with #1 Whatman filter paper triangles. Grids were allowed to dry before imaging on a Phillips CM 120 TEM operating at 80 kV. Images were captured and digitized with an ER-80 CCD (8 megapixel) by advanced microscopy techniques.
Cell line selection for expression cloning
COS-7, HeLa, HEK293FT and SH-SY5Y cells were screened for α-syn-biotin PFF binding using a similar method that was used to identify binding partners of β-amyloid-biotin oligomer (21). Cells were incubated with α-syn-biotin PFF (0 µM, 0.5 µM and 1 µM total α-syn-biotin monomer concentrations) in DMEM media with 10% FBS at room temperature for 2 hours. We removed unbound α-syn-biotin PFF by thoroughly washing (4–6 times, 20 minutes each) with DMEM media with 10% FBS followed by fixation with 4% paraformaldehyde in PBS. The cells were then washed three times with PBS, blocked for 30 minutes with 10% horse serum and 0.1% Triton X-100 in PBS. Using alkaline-phosphatase-conjugated streptavidin (final dilution 1:2000) in PBS supplemented with 5% horse serum and 0.05% Triton X-100, the cells were incubated for 16 hours and then bound alkaline phosphatase was visualized by 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium reaction (19, 20). α-Syn-biotin PFF binding to mouse cortical neurons was used as a positive control. Quantification of bound α-syn-biotin PFF to cells was performed with ImageJ.
Screening strategy, expression cloning and SH-SY5Y cell surface binding assays
A focused set of experiments to identify α-syn PFF mediator(s) was performed. We transfected a library consisting of 352 individual cDNA clones encoding transmembrane proteins (TMGW10001, GFC-transfection array panel, Origene, Rockville, MD, USA) into SH-SY5Y cells (21). Two days after transfection, the cells were incubated for 2 hours with α-syn-biotin PFF (1 µM total α-syn-biotin monomer concentration) in DMEM media with 10% FBS at room temperature. We removed unbound α-syn-biotin PFF by thoroughly washing (4–6 times, 20 minutes each) with DMEM media with 10% FBS and then fixed with 4% paraformaldehyde in PBS. The cells were then washed three times with PBS, blocked for 30 minutes with 10% horse serum and 0.1% Triton X-100 in PBS. Using alkaline-phosphatase-conjugated streptavidin (final dilution 1:2000) in PBS supplemented with 5% horse serum and 0.05% Triton X-100, the cells were incubated for 16 hours and then bound alkaline phosphatase was visualized by 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium reaction (19–21). Bound α-syn-biotin PFF to LAG3-transfected SH-SY5Y cells was quantified with ImageJ. The Z-factor was calculated by the value of the positive control (neuron) and the negative control (SH-SY5Y cells expressed GFP) in each plate.
ImageJ analysis
Threshold was selected under Image/Adjust in order to achieve a desired range of intensity values for each experiment. Once determined, this threshold was applied to all the images in each experiment. The threshold setting was also used to exclude the background. After exclusion of the background, the selected area in the signal intensity range of the threshold was measured using the measurement option under the Analyze/Measure menu. The area values with different concentrations of α-syn-biotin (monomer or PFF) were input into the Prism program to obtain Kd.
Primary neuronal cultures, α-syn PFF transduction and neuron binding assays
CD1 mice were obtained from the Jackson Laboratories (Bar Harbor, ME). Primary cortical neurons were prepared from E15.5 pups and cultured in Neurobasal media supplemented with B-27, 0.5 mM L-glutamine, penicillin and streptomycin (all from Invitrogen, Grand Island, NY, USA) on tissue culture plates coated with poly-L-lysine. The neurons were maintained by changing medium every 3–4 days. α-Syn PFF (final concentration 5 µg/mL) was added at 7 days in vitro (DIV) and α-syn PFF was incubated for 10–21 days followed by biochemical experiments or toxicity assays. Each experiment was performed in duplicate and repeated 3–6 times. Neurons were harvested for indirect immunofluorescence and sequential extraction. To determine the amount of bound α-syn-biotin PFF in WT and LAG3−/− neuronal cultures, α-syn-biotin PFF with different concentrations were used. Quantification of bound α-syn-biotin PFF to WT and LAG3−/− neurons were performed with ImageJ.
Primary microglial and astrocyte culture
Primary microglial and astrocyte cultures were performed as described previously (47). Whole brains from mouse pups at postnatal day age 1 (P1) were obtained. After removal of the meninges the brains were washed in DMEM-F12 supplemented with 10% heat-inactivated FBS, 50 U/mL penicillin, 50 µg/mL streptomycin, 2 mM L-glutamine, 100 µM non-essential amino acids, and 2 mM sodium pyruvate (DMEM-F12 complete medium) three times. The brains were transferred to 0.25% Trypsin-EDTA followed by 10 minutes of gentle agitation. DMEM-F12 complete medium was used to stop the trypsinization. The brains were washed three times in this medium again. A single cell suspension was obtained by trituration. Cell debris and aggregates were removed by passing the single cell suspension through a 100 µm nylon mesh. The final single cell suspension thus achieved was cultured in T-75 flasks for 13 days, with a complete medium change on day 6. The mixed glial cell population was separated into astrocyte rich and microglia rich fractions using the EasySep Mouse CD11b Positive Selection Kit (StemCell). The magnetically separated fraction containing microglia and the pour-off fraction containing astrocytes were cultured separately, and harvested 48 hours after plating by scraping.
Mouse strains
C57BL6 and CD1 mice were obtained from the Jackson Laboratories (Bar Harbor, ME). LAG3−/− mice (48) were kindly provided Dr. Charles Drake (Johns Hopkins University) and were maintained on a C57BL6 background. The mice do not develop any autoimmune or inflammatory phenotype. However, they do have a defect in T cell homeostasis and at 3–4 months they have enlarged spleens and lymph nodes (43). All housing, breeding, and procedures were performed according to the NIH Guide for the Care and Use of Experimental Animals and approved by the Johns Hopkins University Animal Care and Use Committee.
Human A53T α-synuclein transgenic mice, neuron culture and brain homogenates
LAG3−/− mice were crossbred to G2-3 human A53T α-synuclein transgenic mice to obtain A53T α-synuclein and LAG3−/−/A53T α-synuclein neuron cultures. Substantial neurodegeneration is observed in the human A53T α-synuclein transgenic mice including serine 129 phosphorylation and the formation of α-synuclein fibrils (22). Brain areas that exhibit neuropathology including the brainstem and cerebellum of the human A53T α-synuclein transgenic mice were dissected and stored at −80°C. The brain tissue was sonicated in sterile PBS (100 mg per 1 ml of buffer) and centrifuged for 5 minutes (3000 g at 4° C). The resultant supernatant was taken and stored at −80° C until further use.
Enzyme-linked immunosorbent assay (ELISA) analysis
The binding affinity between α-syn-biotin PFF and LAG3 was analyzed using a sandwich ELISA kit (Sigma, St. Louis, MO, USA) according to the manufacturer instructions. The lyophilized human LAG3 protein was added into a human LAG3 antibody-coated ELISA plate and left overnight at 4° C with gentle shaking. Followed by 5 washes, 20 minutes each. Different concentrations of α-syn-biotin PFF (0.1 nM to 100 nM) were added to each well and were incubated for 2 hours at 22° C with gentle shaking. HRP-streptavidin solution was incubated for 45 minutes at 22° C with gentle shaking followed by 5 washes, 20 minutes each. Finally, the ELISA colorimetric TMB (3,3',5,5'-tetramethylbenzidine) reagent was added and incubated for 10 minutes at 22° C in the dark with gentle shaking.
Plasmids
Human and mouse LAG3 cDNA clones were kindly obtained from Dr. Charles Drake at the Johns Hopkins University, School of Medicine. pcDNA3.1-APLP1, APP and APLP2 cDNA clone were obtained from Dr. Yasushi Shimoda at Nagaoka University of Technology and Dr. Gopal Thinakaran at The University of Chicago. pCAG-Neurexin cDNA clones were obtained from Dr. Thomas C. Südhof at Stanford University and Dr. Peter Scheiffele at Basel University. The pMX-CD4 plasmid #14614 was obtained from Addgene (Cambridge, MA, USA).
Deletion mutants
LAG3 deletion mutants with a HA tag were constructed by PCR using herculase polymerase (Agilent Technologies, Wilmington, DE, USA) and primers were designed to flank the sequences to be deleted. The DNA was separated on a 1% agarose gel and the appropriate band was excised and isolated using a gel extraction kit (Qiagen, Valencia, CA, USA). 100 ng of DNA was phosphorylated at the 5’ end using T4 polynucleotide kinase (Invitrogen, Grand Island, NY, USA) for 30 mins at 37° C and ligated overnight at room temperature using T4 DNA ligase (Invitrogen, Grand Island, NY, USA). Reactions were purified with a PCR purification kit (Qiagen, Valencia, CA, USA) and transformed into competent Stbl3 cells (Invitrogen, Grand Island, NY, USA). Integrity of the constructs was verified by sequencing.
Live images
α-Syn PFF was labeled with pHrodo red (Invitrogen, Grand Island, NY, USA). pHrodo red is weakly fluorescent at neutral pH, but fluorescence increases as the pH drops. α-syn-pHrodo PFF was directly added to LAG3 WT and LAG3−/− neuron groups. For the WT + LAG3 and LAG3−/− + LAG3 groups, neurons were co-transfected with LAG3 and GFP expression vector 2 days prior to the addition of α-syn-pHrodo PFF. Live images were recorded every 0.5~1 minute for 20~30 minutes using Microscope Axio Observer Z1 (Zeiss, Dublin, CA, USA). 1~4 minutes later after α-syn-pHrodo treatment, the cells were suitable for recording. Treatment with calcein AM (C1430, ThermoFisher Scientific, USA) was used to identify the outline of neurons for quantification. Transduction with AAV2-eSYN-EGFP-WPRE in some experiments (VB4870, Vector Biosystems Inc., USA) was also used to identify neurons. This AAV is a EGFP construct driven by the neuron E/SYN promoter, a hybrid promoter consisting of a 0.4 Kb CMV enhancer (E) and the 0.45 Kb human Synapsin I promoter fragment (SYN). Outlining the neuron via the Zeiss Zen Software and then subtracting the background determined the signal intensity of the α-syn-pHrodo PFF. The baseline was established as the fluorescence intensity of the neuron at 2–3 minutes after α-syn-pHrodo PFF treatment. The percentage of internalized α-syn-pHrodo PFF signal at each time point was obtained by calculating the ratio to the baseline in each experiment. The live images in Fig. 2A were normalized to reduce background.
Co-localization of Rab5 and α-syn-biotin PFF
The images were obtained using the same exposure time and treated in the same way for analysis. The signal of α-syn-biotin PFF co-localized with Rab5 was measured and quantified by the Zeiss Zen software, by outlining the co-localization and measuring the signal intensity and area of the co-localized signals. The overall signal was determined by multiplying the signal intensity by the area to determine the overall value.
Neuronal internalization of Latex beads
Latex beads (Sigma, USA) were applied to 12–14 DIV wild type and LAG3−/− neuron culture (1 µL/mL) for 4 hours at 37° C. The numbers of internalized latex beads were quantified by confocal microscopy.
Calcium imaging
Intracellular calcium levels were monitored with the fluorescent calcium indicator, Fluo-4 acetoxymethyl (AM) ester (Thermo Fisher Scientific, Waltham, MA, USA). Primary cortical neurons derived from wild-type and LAG3−/− embryo mouse brains were plated on polyornithine-coated glass coverslips for 18 days, and then were loaded with Fluo-4 AM for 30 minutes at 1 µM final concentration. After one wash with Hank’s balanced salt solution (HBSS) (with 2 mM Calcium chloride), the cells were placed in a 37° C heated adaptor on a confocal microscope (Carl Zeiss, Germany). Live imaging was performed with an excitation wavelength of 485 nm and an emission wavelength of 525 nm. Regions of interest (ROI) were selected in a field having usually more than 10 neurons. PFF (0.5 µM) was added with perfusion when the baseline fluorescent signals had been steady for 5 minutes, and recordings continued for an additional 70 min. Images were acquired at 30 second intervals, and were analyzed with Zen (Carl Zeiss, Germany) and ImageJ software.
LAG3 antibody blocking experiments
Anti-LAG3 antibodies (C9B7W and 410C9) were administered to cell cultures (50 µg/mL) 30 minutes before α-syn PFF treatment (33, 34). Rat IgG and mouse IgG were applied as negative controls at the same concentration. SH-SY5Y cells overexpressing LAG3 were used for the binding assay 2 days after transfection of LAG3. Mouse primary cortical neurons (12 DIV) were used for the endocytosis endosome enrichment assay. Neurons (7 DIV) were treated with α-syn PFF for pathology and transmission assays. The antibodies were added to chamber 2 of the microfluidic chambers in the transmission assay.
Endosome enrichment
α-Syn-biotin PFF was administered to neuron (12 DIV) cultures and incubated for 1.5 h. To remove the bound α-syn-biotin PFF, trypsin was added for 30 seconds and followed by three brief washes with culture medium. Endosomes were enriched as previously described (26, 27). The neurons were harvested with PBS and prepared with lysis buffer (250 mM sucrose, 50 mM Tris-Cl (pH 7.4), 5 mM MgCl2, 1 mM EDTA, 1mM EGTA) with a protease inhibitor cocktail (Roche, New York, NY, USA). The suspended cell lysates were pipetted 6 times and passed through a syringe 20 times (1 ml TB Syringe, BD, Franklin Lakes, NJ, USA). The endosomes were harvested in the third pellet followed by three steps of centrifugation 1st(1000 g, 10min), 2nd (16,000 g, 20 min) 3rd (100,000 g, 1h) for immunoblot analysis.
Biochemical analysis
Dissected brain regions of interest or culture samples were prepared with sequential lysis buffers. For the soluble fraction, samples were homogenized in the following TX-soluble buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton-100) containing protease and phosphatase inhibitors (Roche, New York, NY, USA) and samples were centrifuged and the soluble supernatant was collected. The insoluble pellet was resuspended in TX-insoluble buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton X-100, 2% SDS) containing protease and phosphatase inhibitors (Roche, New York, NY, USA). Samples were sonicated and centrifuged at 20,000 g for 20 min. Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL, USA) and samples (10 µg total proteins) were separated on SDS-polyacrylamide gels (13.5%) and transferred onto nitrocellulose membranes. Blots were blocked in 5% non-fat milk or 7.5% BSA in TBS-T (Tris-buffered saline-Tween 20) and probed using various primary antibodies. Target antigens were incubated with appropriate secondary antibodies and were detected using ECL substrate and imaged by ImageQuant LAS 4000mini scanner (GE Healthcare Life Sciences, Wauwatosa, WI, USA) or via film.
Lentiviral vector construction, production and transduction
Lentiviral vectors were generated as previously described (49). LAG3 or deletion mutants of LAG3 with HA tag were subcloned into a lentiviral cFugw vector by Age I restriction sites. The human ubiquitin C (hUBC) promoter was used to drive expression. The recombinant cFugw vector was transiently transfected into HEK293FT cells together with three packaging vectors: pLP1, pLP2, and pVSV-G (1.3:1.5:1:1.5) to generate the lentiviruses. 48 hours and 72 hours after transfection, the viral supernatants were collected and concentrated by ultracentrifugation for 3 hours at 50,000 g. Viral particles were resuspended into serum free medium and stored at −80 °C. At DIV 4 to 5, neurons were infected by lentivirus carrying LAG3, deletion mutants of LAG3, or empty vector as a control [1 × 109 transduction units (TU)/ml] for 72 hours.
In vitro co-immunoprecipitation (co-IP)
HEK293FT cells were transfected with cFUGW-LAG3 or cFUGW-GFP by lipofectamine 2000. 2 days later the cultures were treated with α-syn-biotin monomer or α-syn-biotin PFF (final concentration 1 µM) for 2–3 hours. The cells were washed 2 times with PBS and harvested with lysis buffer containing 50 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton X-100, and protease inhibitors (Roche, New York, NY, USA). Samples were frozen and thawed three times, followed by centrifugation at 20627 × g for 20 minutes. Protein concentrations of the supernatants were determined using the BCA assay (Pierce, Rockford, IL, USA). Aliquots of the samples containing 500 µg of protein were incubated with Dynabeads MyOneTM Streptavidin T1 (Invitrogen, Grand Island, NY, USA) overnight at 4° C for α-syn-biotin co-IP assay. In LAG3 co-IP, aliquots of the samples were pre-cleared with 10 µL of Dynabeads® Protein G (Life Technologies, Grand Island, NY, USA) for 1 hour. Simultaneously, 50 µL of Dynabeads® were incubated for one hour with 4 µL of either mouse 410C9 antibody or mouse IgG (Santa Cruz, Dallas, TX, USA). Pre-cleared samples were incubated with Dynabead®-antibody/IgG overnight at 4° C. The IP complexes were washed 5 times with IP buffer and then denatured by adding 2 × Laemlli Buffer plus β-mercaptoethanol, followed by boiling for 5 minutes.
In vivo co-immunoprecipitation
Transgenic mice overexpressing human A53T α-synuclein (22), and WT littermate controls were euthanized at 4 months and 10 months of age. The brainstem was removed and lysates were prepared with brain lysis buffer containing 50 mM Tris [pH 8.0], 150 mM NaCl, 1% NP-40, and protease inhibitors (Roche, New York, NY, USA). Samples were frozen and thawed three times, followed by centrifugation at 20627 × g for 20 minutes. Protein concentrations of the supernatants were determined using the BCA assay (Pierce, Rockford, IL, USA). Aliquots of the samples containing 500 µg of protein were pre-cleared with 10 µL of Dynabeads® Protein G (Life Technologies, Grand Island, NY, USA) for one hour. Simultaneously, 50 µL of Dynabeads® were incubated for 1 hour with 4 µL of either rabbit α-synuclein antibody (Cell Signaling, Beverly, MA, USA) or rabbit IgG (Santa Cruz, Dallas, TX, USA). Pre-cleared samples were incubated with Dynabead®-antibody/IgG overnight at 4° C. The immunocomplexes were washed 5 times with IP buffer and then denatured by adding 2 × Laemlli Buffer plus β-mercaptoethanol, followed by boiling for 5 minutes.
Microfluidic chambers
Triple compartment microfluidic devices (TCND1000) were obtained from Xona Microfluidic, LLC (Temecula, CA, USA). Glass coverslips were prepared and coated as described, before being affixed to the microfluidic device (13). Approximately 100,000 neurons were plated per chamber. At 4 DIV, WT and LAG3−/− neurons were transduced with lentivirus LAG3 to create WT + LAG3 and LAG3−/− + LAG3 neurons. At 7 DIV, 0.5 µg α-syn PFF was added into chamber 1. To control for direction of flow, a 75 µL difference in media volume was maintained between chamber 1 and chamber 2 and chamber 2 and chamber 3 according to the manufacturers’ instructions. Neurons were fixed on day 14 after α-syn PFF treatment using 4% paraformaldehyde in PBS. The chambers were then processed for immunofluorescence staining.
α-Syn fibril and Stereotaxic procedure
Purification of recombinant of α-syn proteins and in vitro fibril generation was performed as published (14). Assembly reactions of α-syn were performed by continuous agitation of α-syn for 7 days in an amber glass vial with a magnetic stirrer (350 rpm at 37° C). α-Syn PFF was harvested and evaluated for the quality of the fibrils. To avoid repeated freeze and thaw, the PFFs were aliquoted and stored in −80° C. On the day of intrastriatal injections, preparations were diluted in sterile PBS and briefly sonicated in a temperature controlled sonicator water bath. Three month old mice were anesthetized with pentobarbital and PBS, recombinant α-syn PFF (5 µg/2 µL) or recombinant α-syn monomer (5 µg/2 µL) was stereotactically delivered into one striatum. The following reference coordinates for the dorsal neostriatum were used: +0.2 mm Medial-lateral (ML); +2.0 mm antero-posterior (AP) and +2.8 mm dorso-ventral (DV) from bregma. Injections were performed using a 2 µL syringe (Hamilton, Reno, NV, USA) at a rate of 0.1 µL per minutes with the needle left in place for ≥ 5 minutes before slow withdrawal of the needle. After surgery, animals were monitored and post-surgical care was provided. Animal behavior was performed at 30 or 180 days and mice were euthanized for biochemical, neurochemical and histological studies. For biochemical studies, tissues were immediately frozen after removal and stored at −80° C. For histological studies, mice were perfused transcardially with PBS and 4% PFA and brains were removed, followed by overnight fixation in 4% PFA and transfer to 30% sucrose for cryoprotection.
HPLC for DA, DOPAC, HVA and 3MT
High-performance liquid chromatography with electrochemical detection (HPLC-ECD) was used to measure biogenic amine concentrations. Briefly, mice were sacrificed by decapitation. After rapid removal of the striatum, it was weighed and sonicated in 0.2 ml ice cold 0.01 mM perchloric acid containing 0.01% EDTA. 60 ng 3,4-dihydroxybenzylamine (DHBA) was included as an internal standard. This was followed by centrifugation (15,000 × g, 30 min, 4°C) and passing the supernatant through a 0.2 µm filter. Twenty µl of the supernatant was analyzed in the HPLC column (3 mm × 150 mm, C-18 reverse phase column, Acclaim™ Polar Advantage II, Thermo Scientific, USA) by a dual channel coulochem III electrochemical detector (Model 5300, ESA, Inc. Chelmsford, MA, USA). The BCA protein assay kit (Pierce, Rockford, IL, USA) was used to measure protein concentration of the tissue homogenates. Data were normalized to protein concentrations and expressed in ng/mg protein.
Immunohistochemistry, immunofluorescence and mapping of α-syn pathology
Immunohistochemistry (IHC) and immunofluorescence (IF) was performed on 50 µm thick serial brain sections. Primary antibodies and working dilutions are detailed in Table S1. For histological studies, coronal sections were incubated in primary antibodies for P-α-syn or Tyrosine Hydroxylase followed by incubation with biotin-conjugated anti-mouse or anti-Rabbit antibody respectively (Vector Labs, Burlingame, CA, USA), ABC reagents (Vector Labs, Burlingame, CA, USA), and SigmaFast DAB Peroxidase Substrate (Sigma-Aldrich, St. Louis, MO, USA). Sections were counterstained with Nissl (0.09% thionin). Immunoreactivity in double-labeled sections was labeled using appropriate fluorescent secondary antibodies conjugated to Alexa-fluor 488, 594 or 647 (Invitrogen, Carlsbad, CA, USA). Images (IHC) were captured on AxioCam Mrc camera connected to Observer. Z1 microscope (Zeiss, Oberkochen, Germany). Images (IF) were obtained by confocal scanning microscopy (LSM710, Zeiss, Dublin, CA, USA). Photoshop CS6 (Adobe Systems) was used to assemble montages. Fine mapping of P-α-syn pathology was performed by tracing all visible immunoreactive inclusions/cells and neurites at 10×magnification from sections at multiple rostrocaudal levels corresponding to coronal sections at approximately +1.92, +0.74, −0.82, and −2.30, and −3.52 mm relative to Bregma.
Animals
All procedures involving animals were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Johns Hopkins University. Animals were housed in a 12 h dark and light cycle with free access to water and food. All mice were acclimatized in the procedure room before starting any animal experiments. We have taken great measure to reduce the number of animal used in these studies and also taken effort to reduce animal suffering from pain and discomfort.
Behavioral analysis
To evaluate α-syn PFF-induced behavioral deficits, control and α-syn PFF injected mice were tested for pole test, grip strength, and clasping behavior 1-week prior to sacrifice. The experimenter was blinded to treatment group for all behavioral studies. All tests were performed between 10:00–16:00 in the lights-on cycle.
Pole test
A 9 mm diameter 2.5 foot metal rod wrapped with bandage gauze was used as the pole. Mice were placed on the top of the pole (3 inch from the top of the pole) facing head-up. The time taken to turn and total time taken to reach the base of the pole was recorded. Before the actual test the mice were trained for two consecutive days and each training session consisted of three test trials. The maximum cutoff of time to stop the test was 120 seconds. Results were expressed in turn and total time (in seconds) (37).
Grip strength
Neuromuscular strength was measured by maximum holding force generated by the mice (Biosed, USA). Mice were allowed to grasp a metal grid with either by their fore and/or hind limbs or both. The tail was gently pulled and the maximum holding force recorded by the force transducer when the mice released their grasp on the grid. The peak holding strength was digitally recorded and displayed as force in grams (50).
Hindlimb clasping
Hindlimb clasping is a marker of disease progression in neurodegeneration. The procedure was by performed by grasping the mouse-tail and hindlimb clasping was monitored for 10 seconds. Mice with neuropathologic deficits exhibit retracted hindlimbs and grasping of the abdominal area. In addition the mice were observed for a hunchbacked posture (51).
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH NS38377 and the JPB Foundation. P.G. was supported by the Summer Student Fellowship PDF-SFW-1572 from the Parkinson’s Disease Foundation. Y.X. is supported by NIH/NIA grant K01-AG046366 and The William N. & Bernice E. Bumpus Foundation Innovation Awards. H.K. is supported by NIH/NINDS grant NS082205. C.J.W. and D.A.A.V. are supported by NIH P01-AI108545. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program M-2014. Dr. Charles Drake from Johns Hopkins University generously provided LAG3−/− mice. I-Hsun Wu created figure S3A and figure S10A. 410C9 hybridoma cell line is available from Dr. Dario Vignali under a material agreement with the University of Pittsburgh. Dario Vignali and Creg Workman are inventors on issued patents (US [8,551,481]; Europe [1897548]; Australia [2004217526], Hong Kong [1114339]) held by St Jude Children’s Research Hospital and Johns Hopkins University that cover LAG3. Additional applications pending. X. Mao, V.L. Dawson, H. Ko, C. Workman, D. Vignali and T.M. Dawson are inventors on a patent application submitted by Johns Hopkins University that covers the use of inhibitors of LAG3 to treat Parkinson’s disease. Supplement contains additional data.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nature reviews. Neuroscience. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nature reviews. Neurology. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 3.Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 4.Dehay B, Vila M, Bezard E, Brundin P, Kordower JH. Alpha-synuclein propagation: New insights from animal models. Mov Disord. 2016;31:161–168. doi: 10.1002/mds.26370. [DOI] [PubMed] [Google Scholar]
- 5.Lee SJ, Desplats P, Lee HJ, Spencer B, Masliah E. Cell-to-cell transmission of alpha-synuclein aggregates. Methods Mol Biol. 2012;849:347–359. doi: 10.1007/978-1-61779-551-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyson T, Steiner JA, Brundin P. Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. Journal of neurochemistry. 2015 doi: 10.1111/jnc.13449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 8.Del Tredici K, Braak H. Review: Sporadic Parkinson's disease: development and distribution of alpha-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42:33–50. doi: 10.1111/nan.12298. [DOI] [PubMed] [Google Scholar]
- 9.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nature Medicine. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 10.Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nature Medicine. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 11.Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015;38:87–103. doi: 10.1146/annurev-neuro-071714-033828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volpicelli-Daley LA, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben Gedalya T, et al. Alpha-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic. 2009;10:218–234. doi: 10.1111/j.1600-0854.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng F, et al. alpha-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. Journal of Neurochemistry. 2011;119:815–825. doi: 10.1111/j.1471-4159.2011.07460.x. [DOI] [PubMed] [Google Scholar]
- 17.Oh SH, et al. Mesenchymal Stem Cells Inhibit Transmission of alpha-Synuclein by Modulating Clathrin-Mediated Endocytosis in a Parkinsonian Model. Cell Reports. 2016;14:835–849. doi: 10.1016/j.celrep.2015.12.075. [DOI] [PubMed] [Google Scholar]
- 18.Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nature Protocols. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng HJ, Flanagan JG. Identification and cloning of ELF-1, a developmentally expressed ligand for the Mek4 and Sek receptor tyrosine kinases. Cell. 1994;79:157–168. doi: 10.1016/0092-8674(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 20.Cheng HJ, Flanagan JG. Cloning and characterization of RTK ligands using receptor-alkaline phosphatase fusion proteins. Methods Mol Biol. 2001;124:313–334. doi: 10.1385/1-59259-059-4:313. [DOI] [PubMed] [Google Scholar]
- 21.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MK, et al. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proceedings of the National Academy of Sciences, U.S.A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huard B, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proceedings of the National Academy of Sciences, U.S.A. 1997;94:5744–5749. doi: 10.1073/pnas.94.11.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J, Burgess K. Fluorescent indicators for intracellular pH. Chemical Reviews. 2010;110:2709–2728. doi: 10.1021/cr900249z. [DOI] [PubMed] [Google Scholar]
- 25.Huotari J, Helenius A. Endosome maturation. The EMBO Journal. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mu FT, et al. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine "fingers" and contains a calmodulin-binding IQ motif. The Journal of Biological Chemistry. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 27.Taha MS, et al. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PloS One. 2014;9:e91465. doi: 10.1371/journal.pone.0091465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angelova PR, et al. Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. Journal of Cell Science. 2016;129:1792–1801. doi: 10.1242/jcs.180737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hettiarachchi NT, et al. alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. Journal of Neurochemistry. 2009;111:1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x. [DOI] [PubMed] [Google Scholar]
- 30.Melachroinou K, et al. Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiology of Aging. 2013;34:2853–2865. doi: 10.1016/j.neurobiolaging.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Nath S, Goodwin J, Engelborghs Y, Pountney DL. Raised calcium promotes alpha-synuclein aggregate formation. Molecular and Cellular Neurosciences. 2011;46:516–526. doi: 10.1016/j.mcn.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Shrivastava AN, et al. alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. The EMBO Journal. 2015;34:2408–2423. doi: 10.15252/embj.201591397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DA. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3) European Journal of Immunology. 2002;32:2255–2263. doi: 10.1002/1521-4141(200208)32:8<2255::AID-IMMU2255>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 34.Woo SR, et al. Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. European Journal of Immunology. 2010;40:1768–1777. doi: 10.1002/eji.200939874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazaki T, Dierich A, Benoist C, Mathis D. Independent modes of natural killing distinguished in mice lacking Lag3. Science. 1996;272:405–408. doi: 10.1126/science.272.5260.405. [DOI] [PubMed] [Google Scholar]
- 36.Luk KC, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karuppagounder SS, et al. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson's disease. Scientific Reports. 2014;4:4874. doi: 10.1038/srep04874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim C, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nature Communications. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmes BB, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proceedings of the National Academy of Sciences, U.S.A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huh GS, et al. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- 43.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nature Cell Biology. 2016;18:765–776. doi: 10.1038/ncb3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo JL, Lee VMY. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. Journal of Biological Chemistry. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kam TI, et al. FcgammaRIIb mediates amyloid-beta neurotoxicity and memory impairment in Alzheimer's disease. The Journal of Clinical Investigation. 2013;123:2791–2802. doi: 10.1172/JCI66827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panicker N, et al. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson's Disease. The Journal of Neuroscience. 2015;35:10058–10077. doi: 10.1523/JNEUROSCI.0302-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. European Journal of Immunology. 2003;33:970–979. doi: 10.1002/eji.200323382. [DOI] [PubMed] [Google Scholar]
- 49.Andrabi SA, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nature Medicine. 2011;17:692–699. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park HY, et al. Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson's disease. The Journal of Neuroscience. 2014;34:11744–11753. doi: 10.1523/JNEUROSCI.0864-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor TN, Greene JG, Miller GW. Behavioral phenotyping of mouse models of Parkinson's disease. Behav Brain Res. 2010;211:1–10. doi: 10.1016/j.bbr.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.