

Publisher's Note: There is an Inside Blood Commentary on this article in this issue.
Key Points
Diffuse myocardial fibrosis is a common and novel mechanism of heart disease in SCA that can be detected noninvasively.
Diffuse myocardial fibrosis is strongly associated with diastolic dysfunction in individuals with SCA.
Abstract
Sickle cell anemia (SCA)–related cardiomyopathy is characterized by diastolic dysfunction and hyperdynamic features. Diastolic dysfunction portends early mortality in SCA. Diastolic dysfunction is associated with microscopic myocardial fibrosis in SCA mice, but the cause of diastolic dysfunction in humans with SCA is unknown. We used cardiac magnetic resonance measurements of extracellular volume fraction (ECV) to discover and quantify diffuse myocardial fibrosis in 25 individuals with SCA (mean age, 23 ± 13 years) and determine the association between diffuse myocardial fibrosis and diastolic dysfunction. ECV was calculated from pre– and post–gadolinium T1 measurements of blood and myocardium, and diastolic function was assessed by echocardiography. ECV was markedly increased in all participants compared with controls (0.44 ± 0.08 vs 0.26 ± 0.02, P < .0001), indicating the presence of diffuse myocardial fibrosis. Seventeen patients (71%) had diastolic abnormalities, and 7 patients (29%) met the definition of diastolic dysfunction. Participants with diastolic dysfunction had higher ECV (0.49 ± 0.07 vs 0.37 ± 0.04, P = .01) and N-terminal pro–brain natriuretic peptide (NT-proBNP; 191 ± 261 vs 33 ± 33 pg/mL, P = .04) but lower hemoglobin (8.4 ± 0.3 vs 10.9 ± 1.4 g/dL, P = .004) compared with participants with normal diastolic function. Participants with the highest ECV values (≥0.40) were more likely to have diastolic dysfunction (P = .003) and increased left atrial volume (57 ± 11 vs 46 ± 12 mL/m2, P = .04) compared with those with ECV <0.4. ECV correlated with hemoglobin (r = −0.46, P = .03) and NT-proBNP (r = 0.62, P = .001). In conclusion, diffuse myocardial fibrosis, determined by ECV, is a common and previously underappreciated feature of SCA that is associated with diastolic dysfunction, anemia, and high NT-proBNP. Diffuse myocardial fibrosis is a novel mechanism that appears to underlie diastolic dysfunction in SCA.
Introduction
Cardiac disease is a leading cause of adult mortality and morbidity in sickle cell anemia (SCA).1 Diastolic dysfunction, pulmonary hypertension (PH), and increased levels of N-terminal pro–brain natriuretic peptide (NT-proBNP), a marker of myocardial wall stress and cardiac dysfunction, are all associated with early mortality in SCA.2-4 We recently reported that cardiac dysfunction in SCA can be explained by a cardiomyopathy with features of both restrictive and hyperdynamic physiology,5,6 characterized by diastolic dysfunction, left atrial (LA) enlargement, and normal systolic function (restrictive physiology) superimposed on left ventricular (LV) dilation and hypertrophy (hyperdynamic physiology). Like other restrictive cardiomyopathies, the cardiomyopathy of SCA may lead to PH and elevated tricuspid regurgitant jet velocity (TRV) and predisposes to dysrhythmias and sudden cardiac death, all of which are recognized but unexplained complications of SCA.
The etiology of diastolic dysfunction underlying the restrictive physiology of SCA-related cardiomyopathy is unknown. Myocardial fibrosis is known to cause diastolic dysfunction and restrictive physiology in non-SCA populations.7 We found that diastolic dysfunction in SCA mice was associated with microscopic myocardial fibrosis, abnormal electrophysiology, and transcriptome changes.6 Whether this same pathology occurs in humans with SCA is unknown. Postmortem studies have shown a varying extent of myocardial fibrosis in SCA.8,9 In addition, cardiac magnetic resonance (CMR) studies using late gadolinium enhancement (LGE) have identified focal myocardial fibrosis in a few SCA patients.8,10-12 LGE imaging detects dense, focal fibrosis based on differences in gadolinium volume of distribution between diseased and normal myocardium.13 Therefore, LGE is insensitive to diffuse myocardial fibrosis that is seen in nonischemic cardiomyopathies and some SCA autopsy studies.8,13 Recently, CMR measurement of myocardial T1 relaxation times before and after gadolinium administration has been used to quantify the myocardial extracellular volume fraction (ECV).13 In the absence of amyloid deposition or edema, increased ECV results from increased myocardial collagen fraction. CMR-measured ECV correlates strongly with histologically quantified myocardial collagen.14,15
To test our hypothesis that diffuse myocardial fibrosis underlies the diastolic dysfunction of SCA-related cardiomyopathy, we sought to discover the presence and extent of diffuse myocardial fibrosis in SCA patients using CMR T1 mapping and define the relationship between ECV and diastolic dysfunction.
Methods
Participants and study design
Participants with SCA were enrolled in a prospective, longitudinal CMR study to characterize SCA-related cardiomyopathy (www.clinicaltrials.gov #NCT02410811). Twenty-six participants with SCA were enrolled in 3 predefined strata, defined by age and TRV between 2014 and 2015 (Figure 1). Only participants who could undergo CMR and echocardiography without sedation were included. Exclusion criteria were current chronic transfusion therapy, ventricular septal defect, estimated glomerular filtration rate <60 mL/min per 1.73 m2, and any contraindication to magnetic resonance imaging. Study participants were evaluated by an investigator to ensure adequate hydration (using a standardized questionnaire and physical examination) and renal function (serum creatinine and estimated glomerular filtration rate) prior to CMR. Participants were monitored for the development of adverse events in the 30 days following the study visit for CMR. The investigators reviewed and classified all adverse events, including pain or other vasoocclusive complications, according to the Common Terminology Criteria for Adverse Events v4.0.
Figure 1.

Study design. Scheme of study enrollment, grouping, and final evaluable participants.
The study was approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center. Informed consent was obtained from adults or parents of minor participants, and an assent was obtained from minors ≥11 years of age. T1-mapping results (T1 and ECV) were compared with normal values from 16 healthy individuals (mean age, 15.2 years) obtained using the same scanner. Laboratory data included complete blood count, reticulocyte count, bilirubin, lactate dehydrogenase, fetal hemoglobin, NT-proBNP, serum creatinine, and cystatin C.
CMR protocol and image analysis
The CMR protocol is detailed in supplemental Methods (available on the Blood Web site). Briefly, CMR was performed on a 1.5T scanner. Vectorcardiographic gated sequences were performed in the ventricular long- and short-axis planes for chamber sizes, ejection fraction, and ventricular mass. LGE imaging was performed with a standard phase-sensitive inversion recovery sequence protocol 10 minutes after injection with gadolinium-diethylenetriamine penta-acetic acid.
ECV was measured from T1 maps acquired with a modified Look-Locker inversion recovery sequence in the short- and long-axis planes precontrast and 10 minutes postcontrast. The accuracy of the T1-relaxation measurements of the scanner was determined at the beginning of the study using a standard, commercially available relaxation phantom at room temperature. To ensure accurate T1 measurements and exclusion of blood T1 pixels that may affect the accuracy of the measurements, the regions of interest (ROIs) for analysis were drawn very conservatively, keeping 60% to 75% of the myocardium within the ROI and excluding blood pool, papillary muscles, chordae, and trabeculations (supplemental Figure 1A).13 In addition, we created a parameter error map (R2 map) for each image acquisition that was visually evaluated to determine the need for reacquisition during the scan session. During analysis, the parameter errors were assessed for all pixels within ROIs, and areas of the parametric map with R2 < 0.6 were excluded from the contour (supplemental Figure 1B).
All planimetric and T1 analyses were done with CMR42 (Circle Imaging, Alberta, Canada). ECV was calculated using the formula:
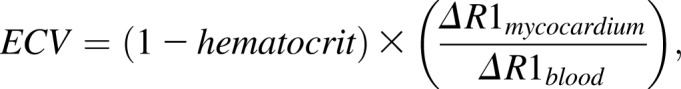 |
where R is relaxation time.
All contours for atrial and ventricular volumes and masses and T1 values were drawn by a single experienced observer blinded to the clinical status of the patients. Twenty percent of the contours were repeated by a second experienced observer. The agreement between measurements estimated by the intraclass correlation coefficient for each parameter was excellent (>0.75) and ranged between 0.78 and 0.98. Intraclass correlation coefficient values for each parameter are provided in supplemental Methods.
Echocardiography
Transthoracic echocardiography was performed with a Philips iE-33 system (Philips Electronics, Andover, MA). Measurements were analyzed using a Cardiology Analysis System (Digisonics, Houston, TX). Pulsed-wave Doppler was used to measure mitral and tricuspid inflow velocity peak at early (E) and late filling (A). Tissue Doppler imaging was used to determine mitral and tricuspid valve annular velocities in early (e′) and late diastole (a′) at both the septal and lateral annulus. Continuous-wave Doppler sampling of the peak TRV was used. TRV was repeated during the administration of agitated saline, and the higher value was used for analysis.
Statistical analysis
CMR and echocardiographic values were compared with normal values for age and sex.16-18 Comparisons across study groups were performed using one-way analysis of variance followed by Tukey multiple comparisons for normally distributed variables; otherwise, Kruskal-Wallis analysis of variance followed by Dunn test was used. A Student t test or Mann-Whitney U test was used to compare 2 groups of continuous variables as indicated or Fisher’s exact test for categorical variables. Associations between normally distributed variables were calculated using the Pearson correlation coefficient. All P values were 2 tailed, and differences were considered significant when P < .05. Statistical analyses were performed using Prism 6.0h (GraphPad Software, La Jolla, CA).
Results
Characteristics of participants
Twenty-six participants with SCA (25 HbSS and 1 HbSβ0-thalassemia) were enrolled. One patient did not complete CMR and was replaced by another participant in the same stratum, leaving 25 evaluable participants (Figure 1). Median age was 19 years; 56% were female. Participants’ characteristics are shown in Table 1. One participant experienced a grade 2 reaction to contrast administration (nausea and vomiting). Three participants with a history of recurrent vaso-occlusive episodes had a painful event 3 to 24 days after CMR that resolved without sequelae. None had a grade 3 or 4 adverse event related to the study procedures.
Table 1.
Baseline clinical and laboratory characteristics of study participants
| Characteristic | Value |
|---|---|
| Age, mean ± SD (range), y | 23 ± 13 (6-61) |
| Female, n (%) | 14 (56) |
| Receiving hydroxyurea, n (%) | 19 (76) |
| Heart rate, bpm | 78 ± 12 |
| Systolic blood pressure, mmHg | 125 ± 16 |
| Diastolic blood pressure, mmHg | 63 ± 9 |
| White blood cell count, 103/mm3 | 9.5 ± 3.4 |
| Hemoglobin, g/dL | 9.7 ± 1.5 |
| Hematocrit, % | 27.7 ± 4.2 |
| Reticulocyte count, % | 8.3 ± 5.2 |
| Platelet count, 103/mm3 | 341 ± 94 |
| Bilirubin, mg/dL | 2.4 ± 1.5 |
| AST, U/L | 48 ± 28 |
| LDH, U/L | 556 ± 267 |
| Plasma free hemoglobin, mg/dL | 75.3 ± 102 |
| Nucleated RBC, cell/100 WBC | 6.2 ± 17 |
| Fetal hemoglobin, % | 17.8 ± 12.5 |
| Mean corpuscular volume, fL | 94.3 ± 19 |
| Absolute neutrophil count, K/μL | 4.9 ± 2.5 |
| Creatinine, mg/dL | 0.58 ± 0.2 |
| Cystatin C, mg/L | 0.67 ± 0.18 |
| GFR, mL/min per 1.73 m2 | 139.7 ± 38 |
| Aldosterone, ng/dL | 8.8 ± 6.1 |
| Plasma renin activity, ng/mL per hour | 1.86 ± 1.4 |
| NT-proBNP, pg/mL | 127.1 ± 182.8 |
Values are shown as mean ± standard deviation unless otherwise specified.
AST, aspartate aminotransferase; GFR, glomerular filtration rate; LDH, lactate dehydrogenase; RBC, red blood cell; SD, standard deviation; WBC, white blood cell.
CMR chamber characteristics
Most participants had LA enlargement (68%) and ventricular enlargement (LV, 72%; right ventricle [RV], 56%). Most (76%) had increased cardiac index due to chronic anemia. Indeed, LV cardiac index negatively correlated with hemoglobin (r = −0.64, P < .001) (supplemental Figure 2A). Five (20%) had eccentric LV hypertrophy consistent with anemia-related volume overload. Only one had LV and RV systolic dysfunction (LV ejection fraction [LVEF], 47%; RV ejection fraction, 49%) (supplemental Table 1).
T1 mapping: ECV is markedly increased in SCA
ECV was markedly increased in all SCA participants (0.44 ± 0.08 in SCA vs 0.26 ± 0.02 in normal controls; P < .001), indicating the presence of diffuse myocardial fibrosis (Figure 2A; supplemental Table 1). One also had focal fibrosis detected by LGE. In addition, native T1 values, which also correlate with collagen volume fraction,19 were significantly increased in SCA compared with controls (1008 ± 67 vs 942 ± 25 ms, P < .001) (Figure 2B). No participant had a native T1 value below the lower limit of normal, indicating absence of significant cardiac iron overload in this cohort.20 ECV values in SCA, in this study, were higher than most previously published ECV values in other fibrotic heart diseases (Figure 2C),21-27 demonstrating the significance and severity of diffuse myocardial fibrosis in SCA.
Figure 2.

Increased myocardial extracellular volume fraction and native T1 in sickle cell anemia. Scatterplots showing increased (A) ECV and (B) native T1 in SCA compared with normal controls. Dotted lines represent the reported lower and upper normal limits. (C) Bar graph demonstrating the mean ECV values in previously published studies of fibrotic heart diseases (shown in green),21-27 in comparison with normal controls (shown in blue) and SCA patients in this study (shown in red). Dashed line represents the upper limit of normal. CMP, cardiomyopathy; MI, myocardial infarction; TM, thalassemia major (+iron, indicates presence of myocardial iron overload).
Association between ECV and diastolic dysfunction
To determine the association between myocardial fibrosis and diastolic dysfunction that underlies the SCA-related cardiomyopathy, we categorized participants with normal systolic function (n = 24) into 3 groups: normal diastolic function (group 1), diastolic dysfunction (group 3), or inconclusive classification (group 2) (supplemental Methods).17 Seven participants (29%) had diastolic dysfunction, 7 (29%) had normal diastolic function, and 10 (42%) had inconclusive classification (Table 2). ECV was significantly higher in group 3 compared with group 1 (0.49 ± 0.07 vs 0.37 ± 0.04, P = .01). In addition, LA volume index (LAVi) (P = .01) and LV end-diastolic volume index (LVEDVi) (P = .01) were significantly higher in group 3; however, LV mass index (LVMi), LVEF, and cardiac index were not different (Table 2; Figure 3D). Participants in group 3 had lower hemoglobin concentration (8.4 ± 0.3 vs 10.9 ± 1.4 g/dL, P = .004) and higher reticulocyte count (12.5% ± 7.4% vs 6% ± 2.8%, P = .04) compared with group 1 (Table 2; Figure 3A-C). When participants were grouped by ECV, those with higher ECV (≥0.40) had higher native T1 values (P = .005), lower hemoglobin (P = .02), and higher LV stroke volume index (P = .04), and were more likely to have LV diastolic dysfunction (P = .03) (Table 3). LAVi was the only significant chamber enlargement in participants with higher ECV (65 ± 13 vs 54 ± 15 mL/m2, P = .04), consistent with diastolic dysfunction (with LA enlargement resulting from restrictive physiology).
Table 2.
Clinical, echocardiographic, and CMR characteristics of study participants based on diastolic function
| Characteristic | Group 1 (N = 7) | Group 2 (N = 10) | Group 3 (N = 7) | P |
|---|---|---|---|---|
| Age, y | 21 ± 11 | 17 ± 6 | 30 ± 18 | .39 |
| Female, n (%) | 2 (29) | 7 (70) | 5 (71) | .29 |
| Receiving hydroxyurea, n (%) | 4 (57) | 9 (90) | 5 (71) | .99 |
| TRV, m/s | 2.3 ± 0.3 | 2.7 ± 0.6 | 2.5 ± 0.4 | .69 |
| E/A ratio | 1.8 ± 0.4 | 2.1 ± 0.8 | 1.6 ± 0.6 | .84 |
| Lateral e′, cm/s | 16.7 ± 4 | 16.6 ± 2.7 | 10.3 ± 2.8 | .003 |
| Lateral E/e′ ratio | 6.5 ± 1.8 | 6.7 ± 1.6 | 9.9 ± 1.6 | .01 |
| Septal e′, cm/s | 11.7 ± 2.5 | 11.6 ± 1.3 | 9.7 ± 2.7 | .11 |
| Septal E/e′ ratio | 8.7 ± 0.9 | 9.4 ± 1.5 | 10.6 ± 1.3 | .04 |
| Tricuspid E/A ratio | 1.7 ± 0.2 | 1.7 ± 0.3 | 1.4 ± 0.6 | .21 |
| Hemoglobin, g/dL | 10.9 ± 1.4 | 9.8 ± 1.3 | 8.4 ± 0.3 | .004 |
| Reticulocyte count, % | 6.0 ± 2.8 | 6.8 ± 3.1 | 12.5 ± 7.4 | .04 |
| Bilirubin, mg/dL | 2.6 ± 1.3 | 1.8 ± 0.7 | 3.3 ± 2.2 | .65 |
| AST, U/L | 57 ± 30 | 44 ± 33 | 48 ± 20 | .84 |
| LDH, U/L | 651 ± 292 | 521 ± 314 | 548 ± 169 | .77 |
| Plasma free hemoglobin, mg/dL | 135 ± 173 | 64 ± 49 | 38 ± 45 | .34 |
| Fetal hemoglobin, % | 23 ± 18 | 18 ± 11 | 12 ± 8 | .27 |
| Mean corpuscular volume, fL | 93 ± 23 | 96 ± 18 | 89 ± 16 | .90 |
| Creatinine, mg/dL | 0.6 ± 0.1 | 0.5 ± 0.2 | 0.7 ± 0.2 | .86 |
| GFR, mL/min per 1.73 m2 | 154 ± 35 | 143 ± 29 | 120 ± 51 | .24 |
| NT-proBNP, pg/mL | 33 ± 33 | 146 ± 180 | 191 ± 261 | .04 |
| Native T1, ms | 963 ± 75 | 1021 ± 41 | 1034 ± 79 | .10 |
| ECV | 0.37 ± 0.04 | 0.45 ± 0.07 | 0.49 ± 0.07 | .01 |
| LAVi, mL/m2 | 50 ± 11 | 66 ± 15 | 64 ± 12 | .01 |
| LVEDVi, mL/m2 | 92 ± 20 | 111 ± 20 | 125 ± 18 | .01 |
| LVESVi, mL/m2 | 35 ± 9 | 42 ± 11 | 49 ± 10 | .06 |
| LVSVi, mL/m2 | 57 ± 12 | 67 ± 15 | 76 ± 10 | .03 |
| LVEF, % | 62 ± 3.6 | 62 ± 5 | 61 ± 4 | .86 |
| LVMi, g/m2 | 60 ± 17 | 63 ± 11.5 | 72 ± 23 | .37 |
| RVEDVi, mL/m2 | 97 ± 22.5 | 116 ± 29 | 121 ± 18.5 | .18 |
| RVESVi, mL/m2 | 39 ± 11.8 | 51 ± 17 | 47 ± 11 | .57 |
| RVSVi, mL/m2 | 58 ± 12 | 66 ± 13 | 74 ± 12 | .06 |
| RVEF, % | 60 ± 4.8 | 57 ± 4.6 | 61 ± 5.8 | .87 |
| LV cardiac index, L/min per m2 | 4.7 ± 1.2 | 4.9 ± 1.4 | 5.5 ± 1.3 | .50 |
| RV cardiac index, L/min per m2 | 4.8 ± 1.2 | 4.3 ± 2 | 5.3 ± 1.2 | .78 |
Values are presented as mean ± standard deviation unless otherwise indicated. P values were calculated using Tukey or Dunn’s multiple comparisons tests as indicated and Fisher’s exact test for categorical variables comparing group 1 and group 3. Statistically significant measurements are shown in bold.
A, late diastolic velocity; E, early diastolic velocity; e′, early annular diastolic velocity; LVEDVi, left ventricular end-diastolic volume index; LVESVi, left ventricular end-systolic volume index; LVSVi, left ventricular stroke volume index; RVEDVi, right ventricular end-diastolic volume index; RVEF, right ventricular ejection fraction; RVESVi, right ventricular end-systolic volume index; RVSVi, right ventricular stroke volume index. Other abbreviations are defined in Table 1.
Figure 3.

Associations among ECV, diastolic dysfunction, and markers of restrictive physiology. Box-and-whisker plots showing differences in (A) ECV, (B) hemoglobin, (C) LAVi, (D) LVEF, and (E) NT-proBNP between participants with diastolic dysfunction and normal diastolic function (normal). (F) Difference in TRV between participants with diastolic dysfunction and normal diastolic function. Participants >21 years are shown in green. P value for the difference in the overall group is shown in black, and P value for the difference among participants >21 years is shown in green. Each box in panels A-F extends from the 25th to 75th percentile, and the line in the box is plotted at the median while the whiskers extend from the smallest to the largest value. (G-I) Linear regression of ECV and (G) lateral E/e′ ratio, (H) tricuspid E/A ratio, and (I) log-NT-proBNP.
Table 3.
Clinical, echocardiographic, and CMR characteristics of study participants based on ECV
| Characteristic | ECV <0.40 (N = 9) | ECV ≥0.40 (N = 16) | P |
|---|---|---|---|
| Age, y | 21.4 ± 8 | 23.8 ± 16 | .68 |
| Female, n (%) | 4 (44) | 10 (63) | .43 |
| Receiving hydroxyurea, n (%) | 5 (56) | 14 (88) | .14 |
| Normal diastolic function, n (%) | 6 (67) | 1 (6) | .003 |
| TRV, m/s | 2.4 ± 0.4 | 2.5 ± 0.6 | .83 |
| Tricuspid E/A ratio | 1.9 ± 0.3 | 1.4 ± 0.4 | .006 |
| Tricuspid E/e′ ratio | 8.4 ± 2.4 | 8.3 ± 2.2 | .93 |
| Hemoglobin, g/dL | 10.6 ± 1.4 | 9.3 ± 1.4 | .02 |
| Reticulocyte count, % | 6 ± 2.5 | 9.6 ± 5.9 | .08 |
| NT-proBNP, pg/mL | 38 ± 40 | 177 ± 216 | .005 |
| Native T1, ms | 960 ± 56 | 1034 ± 58 | .005 |
| ECV | 0.36 ± 0.02 | 0.49 ± 0.06 | <.0001 |
| LAVi, mL/m2 | 54 ± 15 | 65 ± 13 | .04 |
| LVEDVi, mL/m2 | 99 ± 22 | 118 ± 22 | .057 |
| LVESV index, mL/m2 | 38 ± 10 | 47 ± 14 | .11 |
| LVSVi index, mL/m2 | 59 ± 15 | 71 ± 12 | .04 |
| LVEF, % | 62 ± 4 | 61 ± 6 | .54 |
| LVMi, g/m2 | 59 ± 15 | 70 ± 19 | .16 |
| RVEDVi, mL/m2 | 103 ± 24 | 118 ± 25 | .17 |
| RVESV, mL/m2 | 42 ± 11 | 50 ± 16 | .19 |
| RVSV index, mL/m2 | 62 ± 14 | 68 ± 13 | .25 |
| RVEF, % | 60 ± 4 | 58 ± 6 | .50 |
| LV cardiac index, L/min per m2 | 4.7 ± 0.9 | 5.2 ± 1.4 | .31 |
| RV cardiac index, L/min per m2 | 4.7 ± 1 | 4.7 ± 1.8 | .92 |
Values are presented as mean ± standard deviation unless otherwise indicated. P values were calculated using the Student t test, Mann-Whitney U test, or Fisher’s exact test as appropriate. Statistically significant measurements are shown in bold. The abbreviations used are defined in Table 2.
Together, these data demonstrate that myocardial fibrosis, LV diastolic dysfunction, and severe anemia are concordant in SCA.
ECV was also associated with RV diastolic dysfunction (Figure 3G-H). Low tricuspid E/A ratio, a marker of RV diastolic function,28 was associated with high ECV (r = −0.53, P = .009) (Figure 3H; Table 3). Although RV systolic function was normal in all participants with normal LV systolic function, most participants (17/19) with available RV tissue Doppler measurements, had increased tricuspid E/e′ ratio (>6) (supplemental Table 2), consistent with elevated right atrial pressure. RV diastolic dysfunction was common in this cohort, which may portend cardiac impairment.28
Association between ECV and markers of restrictive physiology
NT-proBNP is associated with PH and diastolic dysfunction in SCA.3,29 Participants in group 3 had significantly higher NT-proBNP levels than those in group 1 (191 ± 261 vs 33 ± 33 pg/mL, P = .04; Table 2; Figure 3E). High ECV and native T1 values were associated with high NT-proBNP levels, which remained significant for log-transformed levels (r = 0.61, P = .001 and r = 0.52, P = .009, respectively) (Figure 3I; supplemental Figure 3A). Consistent with previous studies,3 NT-proBNP was associated with LAVi (r = 0.42, P = .04), but not LVEF, LVEDVi, or LVMi (supplemental Figure 3B-E). Collectively, NT-proBNP was associated with diastolic dysfunction, LA enlargement, and increased myocardial fibrosis. Consequently, NT-proBNP is likely also a marker of restrictive physiology in SCA.
Increased TRV is associated with early mortality in adults with SCA,4 but not in children.30 TRV was associated with LAVi (r = 0.50, P = .01), RV end-diastolic volume index (r = 0.45, P = .03), and NT-proBNP (r = 0.52, P = .009), but not with LVEF or LVMi (supplemental Figure 4A-E). While TRV was not statistically different between group 3 and group 1 when participants of all ages were included, adults (≥21 years) with diastolic dysfunction had significantly higher TRV compared with those with normal diastolic function (2.8 ± 0.2 vs 2.2 ± 0.3 m/s, P = .01) (Figure 3F). In addition, the strength of association between ECV and TRV improved (from r = −0.10 to r = 0.37) when children were excluded, albeit not statistically significantly (supplemental Figure 4F). Moreover, native T1 values were significantly higher among adults with TRV ≥2.8 m/s (1059 ± 33 vs 975 ± 60 ms, P = .04) (supplemental Table 3). These data suggest that fibrosis-mediated diastolic dysfunction may contribute to elevated TRV in older individuals by causing pulmonary venous hypertension.
More severe anemia was associated with myocardial fibrosis (higher ECV and native T1; P = .03 and .003, respectively) (supplemental Figure 2B-C). Although anemia was associated with both diastolic dysfunction and myocardial fibrosis in our study, hemoglobin concentration had a stronger association with markers of volume overload, LVEDVi, cardiac index, and LV stroke volume index than LAVi or TRV (supplemental Figure 2D-H).
Clinical correlates of ECV
ECV was significantly associated with the historical number of acute transfusions (r = 0.42 and P = .04), but not with the number of painful events or acute chest syndrome. We did not find an association between ECV and systolic blood pressure (P = .24), diastolic blood pressure (P = .17), baseline heart rate (P = .84), baseline oxygen saturation (P = .89), or pulse pressure (P = .27). There was no sex difference in ECV measurements (P = .17). There were no deaths during the study.
Discussion
Why relatively mild PH and diastolic dysfunction are associated with increased mortality in SCA is not known. Given our findings in a murine model of the SCA cardiomyopathy,6 we sought to determine the presence and extent of diffuse myocardial fibrosis in humans with SCA and its association with diastolic dysfunction. Indeed, we found that diffuse myocardial fibrosis is a common feature in SCA that appears to predate the development of diastolic dysfunction: ECV was markedly increased in all participants, including young children, and even in the absence of focal fibrosis (LGE). In addition, higher fibrosis was associated with diastolic dysfunction and the restrictive physiology features of the SCA-related cardiomyopathy. Our results highlight an important, novel, and previously underrecognized mechanism of heart pathology in SCA and demonstrate the feasibility of noninvasive myocardial fibrosis assessments in children and adults with SCA.
The association between myocardial fibrosis and diastolic dysfunction has been established in other heart diseases.7 Here, we also identified a strong link between myocardial fibrosis and diastolic dysfunction that underlies the cardiomyopathy of SCA.5 Besides the known anemia-related hyperdynamic features, in this study, most participants had some evidence of diastolic dysfunction, and 29% met diagnostic criteria for diastolic dysfunction17; indeed, those with diastolic dysfunction had the highest ECV values.
Different patterns of myocardial fibrosis, including diffuse myocardial fibrosis and focal fibrosis involving the conduction system, have been reported in some autopsy studies in SCA.8,9 LGE studies have found focal/macroscopic fibrosis in few individuals with SCA.8,10-12 However, diffuse myocardial fibrosis has not been studied systematically before.
Measurement of ECV is a validated CMR technique to quantify myocardial fibrosis that correlates strongly with histologic myocardial collagen.14 ECV is a robust measure of cardiac remodeling associated with diastolic dysfunction in other cardiac diseases31,32 and has more prognostic significance than LGE in cardiomyopathies.33 LGE, however, was reported not to be associated with diastolic measures in one SCA study.8 In addition, high ECV is associated with adverse cardiac events, because fibrosis increases myocardial predisposition to dysrhythmias, heart failure, and sudden death.34,35 Diffuse myocardial fibrosis in SCA may similarly decrease myocardial tolerance to microvascular ischemia, causing conduction abnormalities, which could be the cause of the unexplained high frequency of dysrhythmias and sudden death in SCA patients.36 ECV values can also be increased if inflammation or tissue edema is present. SCA-related tissue injury causing inflammation and edema are possible early factors that contribute to increased ECV in some individuals that eventually culminate in tissue fibrosis. Because T1 changes do not account for vascular changes,13 we cannot rule out a possible role for vascular remodeling in increasing ECV values in our study. Vascular remodeling and intimal thickening are known complications of SCA that appear later in life and correlate with age.37,38 Therefore, it is unlikely that vascular remodeling is the sole explanation of this degree of ECV elevation in our study, especially in young patients with high ECV. Notably, ECV values in this cohort, which lacks other known risk factors for myocardial fibrosis (hypertension, diabetes, autoimmune diseases, or coronary artery disease), approach values seen in cardiac amyloidosis and are among the highest reported ECV values in any disease associated with myocardial fibrosis, especially when LGE is absent. This suggests that diffuse myocardial fibrosis is both severe and common in SCA.
The cause of myocardial fibrosis in SCA is unknown. In thalassemia, myocardial fibrosis may be associated with cardiac siderosis.24 However, cardiac siderosis is rare in SCA, even when systemic iron overload is present.8,12,39 In our study, we excluded individuals receiving chronic transfusions, and no participant had low native T1 values, which reasonably excludes significant cardiac siderosis.20 Instead, most had high native T1 values consistent with myocardial fibrosis.19 The role of anemia in the development of myocardial fibrosis in SCA is unknown. We observed a correlation between ECV and both severity of anemia and increased stroke volume. However, the association between anemia and indices of hyperdynamic physiology was stronger than its association with ECV, TRV, or diastolic indices, suggesting that mechanisms besides anemia, the cause of hyperdynamic physiology, may contribute to myocardial fibrosis and restrictive physiology in SCA. The severity of anemia is associated with many adverse outcomes in SCA, so these correlations may not be causal. In a murine model, microscopic fibrosis and diastolic dysfunction of the SCA-related cardiomyopathy were caused by sickle hematopoiesis and not chronic anemia or myocardial iron.6 In addition, thalassemia patients without cardiac siderosis had normal ECV values, supporting the notion that myocardial fibrosis is unlikely to result from anemia alone.24 Whether anemia worsens myocardial fibrosis is also undetermined. Increased oxygen demand and extraction in hypertrophied ventricles due to chronic anemia may increase vaso-occlusion and microvascular ischemia leading to worsening fibrosis. The interaction between anemia and many cardiac complications in SCA is intricate and incompletely understood, and isolating the effects of anemia from other concurrent pathologic processes of SCA in the heart, such as vaso-occlusion and inflammation, is challenging.40 Many processes, including ischemia, inflammation, and microvascular disease, may predispose to myocardial fibrosis.41 Ischemia-reperfusion injury, increased reactive oxygen species, and dysregulated transforming growth factor β have all been described in SCA42-44 and could also be implicated in the development of myocardial fibrosis.41
We observed a strong association between ECV and NT-proBNP levels, an adverse prognostic marker in adults with SCA.3 NT-proBNP correlates with RV dysfunction in patients with PH and has been validated as a marker of pulmonary vascular disease and PH in SCA.45 Also, NT-proBNP is increased in diastolic dysfunction and correlates with disease severity and prognosis in cardiac diseases.46 NT-proBNP has also been associated with markers of diastolic dysfunction in SCA.29 We also found an association between diastolic dysfunction and NT-proBNP, and both were additionally associated with myocardial fibrosis, suggesting that NT-proBNP may also be a marker of the restrictive physiology of SCA-related cardiomyopathy. Moreover, high ECV was associated with RV diastolic dysfunction, which was common in our study and may result directly from LV diastolic dysfunction or RV fibrosis or be secondary to PH. RV remodeling was recently recognized as a prognostic factor in SCA.11 RV diastolic dysfunction should be further studied as a potential early marker of cardiac dysfunction that may predict poor outcome.28,47 Increased ECV is associated with 2 markers of early mortality in SCA, diastolic dysfunction and elevated NT-proBNP, suggesting that diffuse myocardial fibrosis may predispose to cardiac death in SCA.
Our study has several limitations. First, this is a relatively small observational study with 25 participants. Nevertheless, we found 2 large effects in this study: ECV was significantly increased in all participants, and the degree of elevation was higher than most other cardiomyopathies. However, the conclusions regarding the presence or absence of an association between ECV and clinical, laboratory, and echocardiographic outcomes in patients with SCA are limited by this small sample size. Consequently, the relationship between ECV and diastolic dysfunction in our study should be interpreted as hypothesis generating. This novel finding needs to be studied in independent and larger cohorts to confirm these associations. We did not encounter any deaths in this cross-sectional sample with a young median age. Longer studies will confirm any association between myocardial fibrosis and the major clinical outcomes of SCA. A longitudinal study to evaluate the progress of diffuse myocardial fibrosis in SCA is currently ongoing at our institution. Second, classification of diastolic dysfunction is challenging in children. In this study, we used contemporary consensus criteria for diastolic dysfunction in adults,17 because pediatric guidelines are lacking. Eight of 10 participants with inconclusive classification were <21 years of age, reflecting the difficulty of classifying diastolic dysfunction in young individuals. When available, we compared results to normal values for age, size, and sex. Third, we used an averaged ECV value, folding the regional heterogeneity into a single metric. Potentially, additional information could be gleaned from analysis of the regional variability of ECV. Fourth, the inadvertent inclusion of blood pool in calculating myocardial T1 values decreases the accuracy of ECV measurements, especially in anemic patients who have thin ventricular walls. Therefore, we carefully identified myocardial regions of interest maintaining large margins of separation (60% to 75% of myocardial ROI included) and excluding blood contamination. In our study, T1 values were stable over an ROI width ranging between 2 mm and 6 mm, indicating that an accurate T1 measurement is reliable in patients with SCA and thin ventricular walls (supplemental Figure 5).
The use of gadolinium-based contrast agents was theorized to potentiate vaso-occlusion and hemolysis of sickle red cells in vitro. However, these concerns have not been supported in vivo in several observational studies.48,49 In our study, gadolinium-enhanced CMR was well tolerated in adult and pediatric individuals with SCA. Similar to non-SCA populations, SCA patients should be carefully selected for gadolinium administration to avoid the rare, but serious, nephrogenic systemic fibrosis that can occur in individuals with severe renal failure.50
There are insufficient data to determine the role of disease-modifying therapies (hydroxyurea and transfusions) in ameliorating the cardiac complications of SCA. In our study, most participants were prescribed hydroxyurea, and chronic transfusion therapy was an exclusion criterion. Any protective effect of hydroxyurea on diffuse myocardial fibrosis cannot be determined from this study; however, patients with diastolic dysfunction had numerically lower fetal hemoglobin levels. Specific therapies for the cardiac complications of SCA are lacking altogether, but if the association between high myocardial fibrosis and cardiac dysfunction is confirmed with additional study, a study of antifibrotic agents may be warranted.
In summary, we identified diffuse myocardial fibrosis as a novel mechanism underlying the diastolic dysfunction of the SCA-related cardiomyopathy with restrictive physiology. Diffuse myocardial fibrosis appears to be common and severe in both children and adults with SCA. Individuals with the most severe fibrosis have concurrent diastolic dysfunction. SCA-related myocardial injury, profibrotic pathways, and possibly volume overload may predispose to myocardial fibrosis, which can be assessed non-invasively using CMR T1 mapping. Therapeutic targeting of myocardial fibrosis should be investigated to ameliorate cardiac disease, a leading cause of mortality in adults with SCA.
Acknowledgments
The authors thank Courtney Little, Eileen Beckman, and Amy Shova for assistance with recruitment of participants and collection of clinical data.
This study is supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (Excellence in Hemoglobinopathy Research Award program U01HL117709) (J.A.T., P.M., and C.T.Q.). O.N. was the recipient of a Translational Research Scholar Award (U01HL117709).
Footnotes
Presented in abstract form at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2016.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.T.Q., M.D.T., R.F., P.M., and J.A.T. designed research; C.T.Q., O.N., and P.D. recruited participants and collected clinical data; M.D.T., R.F., F.M., and T.A. acquired CMR and echocardiography data; O.N. analyzed data and wrote the first draft of the manuscript; and all authors critically revised, edited, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Omar Niss, 3333 Burnet Ave, MLC 7015, Cincinnati, OH 45229; e-mail: omar.niss@cchmc.org.
References
- 1.Fitzhugh CD, Lauder N, Jonassaint JC, et al. . Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol. 2010;85(1):36-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sachdev V, Machado RF, Shizukuda Y, et al. . Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49(4):472-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Machado RF, Anthi A, Steinberg MH, et al. ; MSH Investigators. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296(3):310-318. [DOI] [PubMed] [Google Scholar]
- 4.Gladwin MT, Sachdev V, Jison ML, et al. . Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886-895. [DOI] [PubMed] [Google Scholar]
- 5.Niss O, Quinn CT, Lane A, et al. . Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc Imaging. 2016;9(3):243-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakeer N, James J, Roy S, et al. . Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci USA. 2016;113(35):E5182-E5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94(12):1533-1542. [DOI] [PubMed] [Google Scholar]
- 8.Desai AA, Patel AR, Ahmad H, et al. . Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging. 2014;7(3):430-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James TN, Riddick L, Massing GK. Sickle cells and sudden death: morphologic abnormalities of the cardiac conduction system. J Lab Clin Med. 1994;124(4):507-520. [PubMed] [Google Scholar]
- 10.Junqueira FP, Fernandes JL, Cunha GM, et al. . Right and left ventricular function and myocardial scarring in adult patients with sickle cell disease: a comprehensive magnetic resonance assessment of hepatic and myocardial iron overload. J Cardiovasc Magn Reson. 2013;15:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen KL, Tian X, Alam S, et al. . Elevated transpulmonary gradient and cardiac magnetic resonance-derived right ventricular remodeling predict poor outcomes in sickle cell disease. Haematologica. 2016;101(2):e40-e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westwood MA, Shah F, Anderson LJ, et al. . Myocardial tissue characterization and the role of chronic anemia in sickle cell cardiomyopathy. J Magn Reson Imaging. 2007;26(3):564-568. [DOI] [PubMed] [Google Scholar]
- 13.Moon JC, Messroghli DR, Kellman P, et al. ; Society for Cardiovascular Magnetic Resonance Imaging; Cardiovascular Magnetic Resonance Working Group of the European Society of Cardiology. Myocardial T1 mapping and extracellular volume quantification: a Society for Cardiovascular Magnetic Resonance (SCMR) and CMR Working Group of the European Society of Cardiology consensus statement. J Cardiovasc Magn Reson. 2013;15:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flett AS, Hayward MP, Ashworth MT, et al. . Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation. 2010;122(2):138-144. [DOI] [PubMed] [Google Scholar]
- 15.Sibley CT, Noureldin RA, Gai N, et al. . T1 Mapping in cardiomyopathy at cardiac MR: comparison with endomyocardial biopsy. Radiology. 2012;265(3):724-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawel-Boehm N, Maceira A, Valsangiacomo-Buechel ER, et al. . Normal values for cardiovascular magnetic resonance in adults and children. J Cardiovasc Magn Reson. 2015;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagueh SF, Smiseth OA, Appleton CP, et al. . Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2016;29(4):277-314. [DOI] [PubMed] [Google Scholar]
- 18.Eidem BW, McMahon CJ, Cohen RR, et al. . Impact of cardiac growth on Doppler tissue imaging velocities: a study in healthy children. J Am Soc Echocardiogr. 2004;17(3):212-221. [DOI] [PubMed] [Google Scholar]
- 19.Bull S, White SK, Piechnik SK, et al. . Human non-contrast T1 values and correlation with histology in diffuse fibrosis. Heart. 2013;99(13):932-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sado DM, Maestrini V, Piechnik SK, et al. . Noncontrast myocardial T1 mapping using cardiovascular magnetic resonance for iron overload. J Magn Reson Imaging. 2015;41(6):1505-1511. [DOI] [PubMed] [Google Scholar]
- 21.Barison A, Del Torto A, Chiappino S, et al. . Prognostic significance of myocardial extracellular volume fraction in nonischaemic dilated cardiomyopathy. J Cardiovasc Med (Hagerstown). 2015;16(10):681-687. [DOI] [PubMed] [Google Scholar]
- 22.Barison A, Gargani L, De Marchi D, et al. . Early myocardial and skeletal muscle interstitial remodelling in systemic sclerosis: insights from extracellular volume quantification using cardiovascular magnetic resonance. Eur Heart J Cardiovasc Imaging. 2015;16(1):74-80. [DOI] [PubMed] [Google Scholar]
- 23.Hammer-Hansen S, Bandettini WP, Hsu LY, et al. . Mechanisms for overestimating acute myocardial infarct size with gadolinium-enhanced cardiovascular magnetic resonance imaging in humans: a quantitative and kinetic study. Eur Heart J Cardiovasc Imaging. 2016;17(1):76-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanneman K, Nguyen ET, Thavendiranathan P, et al. . Quantification of myocardial extracellular volume fraction with cardiac MR imaging in thalassemia major. Radiology. 2016;279(3):720-730. [DOI] [PubMed] [Google Scholar]
- 25.Ho CY, Abbasi SA, Neilan TG, et al. . T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6(3):415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kellman P, Wilson JR, Xue H, et al. . Extracellular volume fraction mapping in the myocardium, part 2: initial clinical experience. J Cardiovasc Magn Reson. 2012;14:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ntusi NA, Piechnik SK, Francis JM, et al. . Diffuse myocardial fibrosis and inflammation in rheumatoid arthritis: insights from CMR T1 mapping. JACC Cardiovasc Imaging. 2015;8(5):526-536. [DOI] [PubMed] [Google Scholar]
- 28.Rudski LG, Lai WW, Afilalo J, et al. . Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23(7):685-713; quiz 786-688. [DOI] [PubMed] [Google Scholar]
- 29.Takatsuki S, Ivy DD, Nuss R. Correlation of N-terminal fragment of B-type natriuretic peptide levels with clinical, laboratory, and echocardiographic abnormalities in children with sickle cell disease. J Pediatr. 2012;160(3):428-433, e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee MT, Small T, Khan MA, Rosenzweig EB, Barst RJ, Brittenham GM. Doppler-defined pulmonary hypertension and the risk of death in children with sickle cell disease followed for a mean of three years. Br J Haematol. 2009;146(4):437-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rommel KP, von Roeder M, Latuscynski K, et al. . Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2016;67(15):1815-1825. [DOI] [PubMed] [Google Scholar]
- 32.Dusenbery SM, Jerosch-Herold M, Rickers C, et al. . Myocardial extracellular remodeling is associated with ventricular diastolic dysfunction in children and young adults with congenital aortic stenosis. J Am Coll Cardiol. 2014;63(17):1778-1785. [DOI] [PubMed] [Google Scholar]
- 33.Florian A, Ludwig A, Rösch S, Yildiz H, Sechtem U, Yilmaz A. Myocardial fibrosis imaging based on T1-mapping and extracellular volume fraction (ECV) measurement in muscular dystrophy patients: diagnostic value compared with conventional late gadolinium enhancement (LGE) imaging. Eur Heart J Cardiovasc Imaging. 2014;15(9):1004-1012. [DOI] [PubMed] [Google Scholar]
- 34.Schelbert EB, Piehler KM, Zareba KM, et al. . Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. 2015;4(12):e002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neilan TG, Mongeon FP, Shah RV, et al. . Myocardial extracellular volume expansion and the risk of recurrent atrial fibrillation after pulmonary vein isolation. JACC Cardiovasc Imaging. 2014;7(1):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manci EA, Culberson DE, Yang YM, et al. ; Investigators of the Cooperative Study of Sickle Cell Disease. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123(2):359-365. [DOI] [PubMed] [Google Scholar]
- 37.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11(2):129-151. [PubMed] [Google Scholar]
- 38.Hadeed K, Hascoet S, Castex MP, Munzer C, Acar P, Dulac Y. Endothelial function and vascular properties in children with sickle cell disease. Echocardiography. 2015;32(8):1285-1290. [DOI] [PubMed] [Google Scholar]
- 39.Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD. Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood. 2004;103(5):1934-1936. [DOI] [PubMed] [Google Scholar]
- 40.Wood JC. The heart in sickle cell disease, a model for heart failure with preserved ejection fraction. Proc Natl Acad Sci USA. 2016;113(35):9670-9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28(2):181-198. [DOI] [PubMed] [Google Scholar]
- 43.Heimlich JB, Speed JS, O’Connor PM, et al. . Endothelin-1 contributes to the progression of renal injury in sickle cell disease via reactive oxygen species. Br J Pharmacol. 2016;173(2):386-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fertrin KY, Costa FF. Genomic polymorphisms in sickle cell disease: implications for clinical diversity and treatment. Expert Rev Hematol. 2010;3(4):443-458. [DOI] [PubMed] [Google Scholar]
- 45.Casserly B, Klinger JR. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Des Devel Ther. 2009;3:269-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirk V, Bay M, Parner J, et al. . N-terminal proBNP and mortality in hospitalised patients with heart failure and preserved vs. reduced systolic function: data from the prospective Copenhagen Hospital Heart Failure Study (CHHF). Eur J Heart Fail. 2004;6(3):335-341. [DOI] [PubMed] [Google Scholar]
- 47.Sallach JA, Tang WH, Borowski AG, et al. . Right atrial volume index in chronic systolic heart failure and prognosis. JACC Cardiovasc Imaging. 2009;2(5):527-534. [DOI] [PubMed] [Google Scholar]
- 48.Meloni A, Favilli B, Positano V, et al. . Safety of cardiovascular magnetic resonance gadolinium chelates contrast agents in patients with hemoglobinopathies. Haematologica. 2009;94(11):1625-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dillman JR, Ellis JH, Cohan RH, et al. . Safety of gadolinium-based contrast material in sickle cell disease. J Magn Reson Imaging. 2011;34(4):917-920. [DOI] [PubMed] [Google Scholar]
- 50.Perazella MA. Nephrogenic systemic fibrosis, kidney disease, and gadolinium: is there a link? Clin J Am Soc Nephrol. 2007;2(2):200-202. [DOI] [PubMed] [Google Scholar]