

Abstract
All living organisms exist in a precarious state of homeostasis that requires constant maintenance. A wide variety of stresses, including hypoxia, heat, and infection by pathogens perpetually threaten to imbalance this state. Organisms use a battery of defenses to mitigate damage and restore normal function. Previously, we described a Caenorhabditis elegans-Pseudomonas aeruginosa assay (Liquid Killing) in which toxicity to the host is dependent upon the secreted bacterial siderophore pyoverdine. Although pyoverdine is also indispensable for virulence in mammals, its cytological effects are unclear. We used genetics, transcriptomics, and a variety of pathogen and chemical exposure assays to study the interactions between P. aeruginosa and C. elegans. Although P. aeruginosa can kill C. elegans through at least 5 different mechanisms, the defense responses activated by Liquid Killing are specific and selective and have little in common with innate defense mechanisms against intestinal colonization. Intriguingly, the defense response utilizes the phylogenetically-conserved ESRE (Ethanol and Stress Response Element) network, which we and others have previously shown to mitigate damage from a variety of abiotic stresses. This is the first report of this networks involvement in innate immunity, and indicates that host innate immune responses overlap with responses to abiotic stresses. The upregulation of the ESRE network in C. elegans is mediated in part by a family of bZIP proteins (including ZIP-2, ZIP-4, CEBP-1, and CEBP-2) that have overlapping and unique functions. Our data convincingly show that, following exposure to P. aeruginosa, the ESRE defense network is activated by mitochondrial damage, and that mitochondrial damage also leads to ESRE activation in mammals. This establishes a role for ESRE in a phylogenetically-conserved mitochondrial surveillance system important for stress response and innate immunity.
Author summary
It is increasingly clear that living organisms surveil their cellular structures and physiology to gain early warning about the presence and activity of pathogens or abiotic threats (like hypoxia, hyper- or hypothermia, oxidative damage, etc.). Upon recognizing dysfunction, host defense networks are engaged to prevent and mitigate the damage. Here we report the involvement of a phylogenetically-conserved defense network, called ESRE, in mitochondrial surveillance in C. elegans and mammals. Our report expands the function of this network to include innate immunity in addition to its role in abiotic stress response. We also observed that a variety of mitochondrial poisons, and the opportunistic human pathogen Pseudomonas aeruginosa, activate the ESRE network, suggesting that mitochondrial surveillance is a key facet of its regulation. Since mitochondrial health is an important factor in many diseases, having a better understanding of how cells perceive and respond to damage to this organelle is critical for understanding numerous aspects of human health.
Introduction
All living organisms encounter unfavorable stimuli, including both abiotic stressors and pathogens that interfere with their fitness and survival. To recover their homeostatic balance, they activate conserved defense networks. These include classical immune (innate and adaptive) pathways to respond to pathogens and other defenses to mitigate damage from heat shock, hypoxia, DNA damage, and unfolded protein responses in the endoplasmic reticulum (ER) and mitochondria. Each pathway triggers the production of a myriad of effectors to minimize further damage, mitigate and repair existing damage, and restore organismal health.
Intensive study has begun to shed light on many host defense pathways, helping to identify both the signals detected and the master transcriptional regulators that coordinate the defense response. For example, most organisms use pattern recognition receptors like the well-known Toll-Like Receptor family to detect a variety of structural determinants (such as lipopolysaccharide, flagellin, dsRNA, etc., commonly known as 'Pathogen-Associated Molecular Patterns', or PAMPS) shared by large numbers of pathogens [1]. Pattern recognition receptors are also triggered by normal biomolecules out of their proper context (e.g., extracellular ATP is recognized as a danger signal, as it typically indicates the lysis of nearby cells [2–4]). A wide variety of ligands, including ATP, uric acid, DNA, dsRNA, and misfolded proteins have been identified to act in this fashion [5–7], and are generally referred to as 'Damage-Associated Molecular Patterns', or DAMPs.
DAMPs are also released when organelles are damaged. For example, mitochondria release N-formylated peptides and mitochondrial DNA into the cytosol, which are recognized by the formyl peptide receptor and TLR9, respectively [8–11]. In this fashion, DAMPs serve an important role in organellar surveillance. These programs can monitor organelle function and integrity, as shown by the unfolded protein responses of the ER and mitochondria or mitochondrial dynamics that maintain homeostasis. It is thought that surveillance systems of this type may have developed as an early warning system to identify surreptitious pathogen activity [12], but these surveillance programs also provide invaluable feedback when normal metabolism has gone awry.
C. elegans is widely used as a model for investigating host defenses and surveillance programs [12–19]. We developed a new, liquid-based assay using C. elegans to facilitate the identification of novel treatments for the opportunistic human pathogen Pseudomonas aeruginosa [20–23]. Characterization of the assay demonstrated that pathogenesis strongly depended upon production of the bacterial siderophore pyoverdine [24,25], which is indispensable for virulence in several mammalian infection models [26–28]. We showed that pyoverdine damages mitochondria, targeting them for mitophagic turnover, and activating a hypoxia-like defense response; similar phenomena were observed when worms were exposed to the chemical chelator 1,10-phenanthroline [24,29].
In this study, we aimed to elucidate the defense mechanisms used by C. elegans to respond to pyoverdine exposure. We determined that the transcriptional response to pyoverdine is very different from classical, intestinal colonization-related immune responses in C. elegans. Moreover, we show that the conserved Ethanol and Stress Response Element (ESRE) master stress response network is vital for defense against pyoverdine, and that a family of bZIP transcription factors mediate this activity. Finally, we show that disrupting mitochondrial function in C. elegans and in mammals activates the ESRE network, demonstrating that it plays a key role in surveillance immunity, specifically by assessing mitochondrial health.
Results
C. elegans response to P. aeruginosa is contingent upon context of exposure
P. aeruginosa uses a remarkable variety of virulence factors to colonize and infect many hosts [30–32]. Even within a single host, a myriad of virulence factors is often utilized, including quorum sensing, biofilms, and a variety of toxins [33–35]. At least five different P. aeruginosa-C. elegans pathosystems have been described [36], including our recently developed liquid-based model (referred to as 'Liquid Killing') that requires the siderophore pyoverdine [23,24]. Pyoverdine damages host mitochondria (triggering mitophagy in the process) and induces a hypoxic response [24,29]. It is worth noting that the involvement of pyoverdine in Liquid Killing makes it unique amongst P. aeruginosa invertebrate pathogenesis models. The differences between this pathosystem and the others make it an excellent model to continue the process of elucidating whether the host detects the presence of a pathogen using PAMPS or DAMPS.
To address this question, we performed microarray-based transcriptional profiling of C. elegans glp-4(bn2), exposed to P. aeruginosa PA14 in liquid (Liquid Killing) and on agar plates (Slow Killing) as well as corresponding E. coli controls (see S1 Table and Methods for details). Microarray analysis correlated well with qRT-PCR results (S1 Fig). As the Liquid Killing assay uses conditionally sterile glp-4(bn2) worms, we verified that glp-4(bn2) and wild-type cohorts showed similar transcriptional profiles (S2 Fig). Of the 159 genes upregulated under Liquid Killing conditions, only 8 were also upregulated under Slow Killing conditions (Fig 1A, p = 6.1E-05, see also S2 Table for a list of the genes). Although the overlap is statistically significant, it is likely that the biological significance is limited. For example, Slow Killing (like most other agar-based infections in C. elegans) strongly relies on activation of the PMK-1/p38 MAPK pathway for defense [37–39], while activation of this same pathway is detrimental in Liquid Killing [29]. Correspondingly, gene ontology (GO) enrichment for upregulated genes during Slow Killing predominantly involved genes with CUB-like regions and defense response categories (similar to what was published by Troemel and colleagues [39], while Liquid Killing was dominated by genes involved in endoplasmic reticulum stress and small heat shock proteins (Fig 1B).
Fig 1. Host defense against P. aeruginosa-mediated killing in liquid is mediated via different factors than infection on plates.

(A) Venn diagram of genes upregulated in Slow Killing (i.e., agar-based infection) and Liquid Killing (i.e., siderophore mediated intoxication). The number of genes in each category is presented and p-values (based on the hypergeometric distribution) are shown. (B) Lists of Gene Ontology (GO) groups overrepresented in the transcriptional responses against P. aeruginosa in Slow Killing (top) or Liquid Killing (bottom).
We compared genes upregulated under Liquid Killing to similar lists from other infection conditions (including P. aeruginosa on solid media, Y. pestis, and S. aureus). Generally, there was little overlap between Liquid Killing and other pathogens. Similarly, PMK-1-dependent genes, which are frequently responsive to pathogenic bacteria (including being enriched in our Slow Killing microarray), showed little overlap with genes upregulated during Liquid Killing (Table 1). In contrast, a significant number of both ZIP-2-dependent and hypoxic responsive genes were upregulated under Liquid Killing conditions (p = 10−25 and p = 10−24, respectively).
Table 1. Host response to P. aeruginosa is dependent on pathogen virulence.
| Condition 1 | # Genes | Condition 2 | # Genes | Overlap | p-value |
|---|---|---|---|---|---|
| Slow Killing | 159 | PMK-1 Dependent (PMID: 17096597) |
101 | 11 | 10−10 |
| Liquid Killing | 167 | 2 | 0.21 | ||
| Slow Killing | 159 | ZIP-2 Dependent (PMID: 20133860) |
25 | 4 | 10−5 |
| Liquid Killing | 167 | 15 | 10−25 | ||
| Slow Killing | 159 | Hypoxia Responsive (PMID: 15781453) |
110 | 2 | 0.22 |
| Liquid Killing | 167 | 22 | 10−24 | ||
| Slow Killing | 159 |
S. aureus Infection (PMID: 20617181) |
189 | 42 | 10−49 |
| Liquid Killing | 167 | 4 | 0.077 | ||
| Slow Killing | 159 |
Y. pestis Infection (PMID: 20133945) |
114 | 16 | 10−16 |
| Liquid Killing | 167 | 0 | 1 |
Genes upregulated by Liquid Killing belong to the ESRE network
We were interested in identifying a transcriptome response specific to exposure to P. aeruginosa under Liquid Killing conditions, particularly in its distinction from more generic infection responses. We performed a meta-analysis of transcriptome profiles using our data for Liquid Killing and Slow Killing and published data from C. elegans exposed to other pathogens (S3A Fig). 167 genes upregulated in Liquid Killing were chosen for analysis, and normalized expression data from each transcriptional profile were used to generate clusters of genes with similar expression patterns. One cluster, containing approximately 50 genes (S3B Fig, Methods) showed upregulation only in Liquid Killing. We hypothesized that the coexpression of these genes may indicate the presence of one or more shared transcriptional regulators controlling their expression. Therefore, we searched for enrichment of nucleotide motifs that could represent potential binding sites in their promoters. Computational analysis of the upstream regions identified a strongly conserved, 11-nucleotide sequence (Fig 2A). This motif, called the ESRE [40] has been shown to work with the PBAF chromatin remodeling complex to control the response to pleiotropic abiotic stresses [40–43]. Statistical analysis demonstrated overrepresentation of this motif in the previously described cluster of genes strongly upregulated specifically in Liquid Killing (present in >40% of genes, p = 1.1 E-10) and in the 167 genes upregulated by Liquid Killing; but not in genes upregulated by P. aeruginosa on agar or infection with other pathogens (S3 Table). This suggested that involvement of the ESRE network was likely specific to exposure to P. aeruginosa in liquid.
Fig 2. Activation of the ESRE defense network requires the presence of the siderophore pyoverdine.
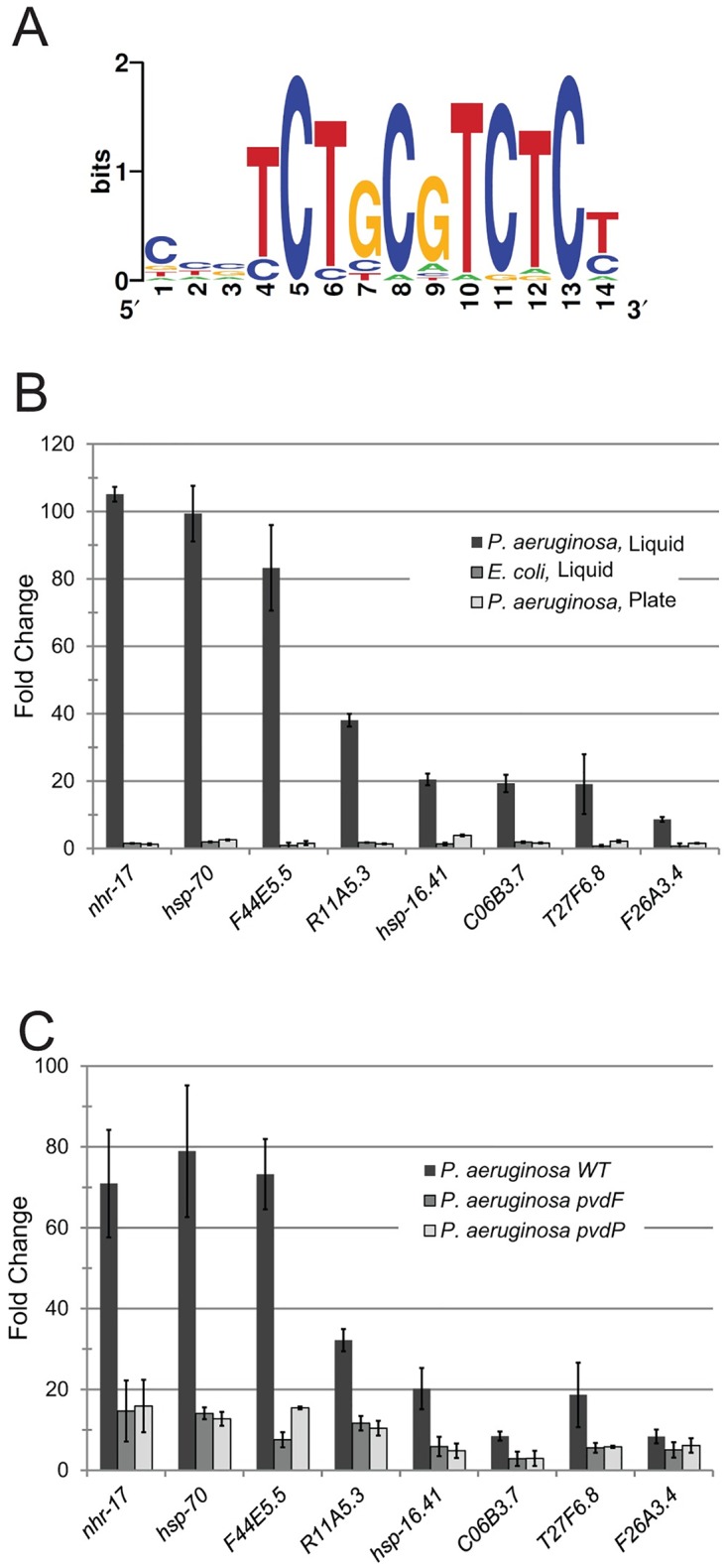
(A) Logogram of an 11-nucleotide motif identified in a cluster of genes specifically upregulated in Liquid Killing, as compared to other infectious conditions. (B) Expression of ESRE-containing genes in worms exposed to: P. aeruginosa in liquid, their normal (E. coli) bacterial food in liquid, or infected with P. aeruginosa on plates. Fold changes were determined by qPCR and were normalized to a cohort fed with E. coli on solid media. (C) Expression of ESRE genes after exposure to either wild-type P. aeruginosa or P. aeruginosa mutants with defects in pyoverdine biosynthesis. Fold changes were determined by qPCR and were normalized to a cohort exposed to E. coli under Liquid Killing conditions. Error bars represent SEM.
We used two methods to validate the importance of the ESRE motif in the P. aeruginosa Liquid Killing assay. First, qRT-PCR was used to evaluate expression of ESRE genes in worms exposed to either P. aeruginosa on agar, E. coli in liquid, or P. aeruginosa in liquid. Significant upregulation of ESRE genes was found only in worms exposed to P. aeruginosa in liquid (Fig 2B). Second, we identified 42 ESRE genes specifically upregulated in response to Liquid Killing for which correct RNAi constructs were available [44,45] and that did not compromise growth on E. coli. These 42 genes were tested for involvement in resistance to Liquid Killing. Ten genes showed increased sensitivity in at least three out of four replicates, reinforcing the importance of the ESRE motif (S4 Fig).
P. aeruginosa-mediated activation of the ESRE network requires pyoverdine
Liquid Killing is largely contingent upon the production of pyoverdine, as virulence is attenuated in P. aeruginosa mutants with compromised pyoverdine biosynthesis [24]. This lead to the obvious hypothesis that pyoverdine is necessary for the activation of ESRE genes. To test this, we assayed ESRE gene expression in worms exposed to P. aeruginosa pvdF and pvdP mutants, which are incapable of producing pyoverdine. Upregulation of ESRE genes was substantially compromised (Fig 2C), supporting the idea that the ESRE pathway responds to pyoverdine. This raised an intriguing question: was the host recognizing pyoverdine (indicating that the siderophore is a PAMP) or is the host detecting iron removal (i.e., the damage caused by the toxin, implying a DAMP-mediated response).
Exposure to phenanthroline also induces ESRE gene expression
We have previously demonstrated that another chemical chelator, 1,10-phenanthroline (hereafter referred to as "phenanthroline") recapitulates many key aspects of pyoverdine intoxication, such as activation of hypoxic response, imbalance of C. elegans’ mitochondrial homeostasis with subsequent fragmentation of mitochondrial network, and activation of mitophagy [24,29]. When we tested whether exposure to 1 mM phenanthroline would similarly induce ESRE gene expression, we saw strong gene upregulation (Fig 3A). As this chelator is structurally distinct from pyoverdine, it is likely that at least part of the ESRE response occurs via a DAMP/surveillance mechanism.
Fig 3. The ESRE gene network is activated by acute iron removal.

(A) Expression of ESRE-containing genes in worms exposed to 1 mM 1,10-phenanthroline. Fold changes were normalized to vehicle (DMSO) control. (B-C) Venn diagrams of genes upregulated by exposure to 1 mM 1,10-phenanthroline (Chelator) and either P. aeruginosa exposure in liquid (B) or P. aeruginosa infection on agar plates (C). In each case, the number of genes in each category is shown, as is the p-value (based on hypergeometric distribution). (D) Fluorescence microscopy of reporters with (left) or without (right) the ESRE element that were treated with 1 mM 1,10-phenanthroline (Phe) or vehicle (DMSO). Representative images are shown; three biological replicates (with ~50 worms / replicate) were analyzed. A schematic of the promoter region for the construct is shown for convenience. (E) Quantification of GFP fluorescence for a Phsp-16.1::GFP reporter with or without the ESRE element that was treated with vehicle (DMSO) or 1 mM 1,10-phenanthroline (Phe). Error bars represent SEM.
To widen these findings to include the entire genome, we profiled transcriptomes of worms exposed to phenanthroline (S4 Table). We saw a dramatic overlap: ~2/3 genes upregulated by P. aeruginosa in Liquid Killing were also upregulated after phenanthroline treatment (p = 10−122, Fig 3B). Interestingly, the 108 genes in this group were associated with GO categories reminiscent of stress response (S5 Table). In contrast, the 59 genes that were specific to Liquid Killing displayed enrichment in a category related to metabolism (Transferases), while the genes specifically upregulated after phenanthroline exposure exhibited a strong detoxification signature (S6 and S7 Tables). This difference could be due to the relatively higher concentration of phenanthroline (compared to pyoverdine) used, resulting in a larger number of effected genes. Alternatively, it is also possible that phenanthroline, for some reason, more effectively activates detoxification genes.
As was observed for Liquid Killing, P. aeruginosa infection on agar upregulated a disparate set of genes from phenanthroline exposure (p = 0.011, Fig 3C). We also examined how many genes upregulated by phenanthroline treatment were dependent upon PMK-1, ZIP-2, or were responsive to hypoxia, S. aureus, and Y. pestis. In each case, the comparison to phenanthroline was quite similar to what we observed for Liquid Killing (S8 Table). ESRE genes were also overrepresented in genes upregulated after phenanthroline treatment (14.3%, p = 10−7). These results support our previous conclusions that host response to P. aeruginosa in Liquid Killing is activated after recognition of pyoverdine-mediated damage.
Although ESRE genes were overrepresented in the response to Liquid Killing and phenanthroline, the possibilities remained that the motif was a computational artifact or that the motif was irrelevant for gene expression in this context. To rule these out, we used GFP reporter strains based on the hsp-16.1 promoter [43]. The hsp-16.1 promoter includes two ESRE motifs, and drives transient expression of a small heat shock protein upon exposure to various stresses [41]. Exposure of the Phsp-16.1::GFP reporter to phenanthroline significantly increased GFP expression (Fig 3D). Efficient expression required intact ESRE sites, as GFP expression was abolished in an otherwise identical promoter where ESRE sites were removed (Fig 3D and 3E). P. aeruginosa also activated the Phsp-16.1::GFP reporter (S5 Fig). Combined, these results emphasize that the ESRE element is necessary for gene activation after acute iron removal.
Compromising mitochondrial activity induces the ESRE network in C. elegans and mammals
Despite their structurally disparate triggers, the transcriptional responses to Liquid Killing and to a small molecule iron chelator were very similar, suggesting that C. elegans may be directly monitoring intracellular iron stores. Alternatively, the removal of iron from host organelles, such as mitochondria, may disrupt their structure and/or function. We surmise that one or both of these events are recognized as a DAMP, triggering the ESRE host defense network. Therefore, we tested whether non-chelating toxins that induce mitochondrial damage activate ESRE genes’ transcription. glp-4(bn2) worms were exposed to either rotenone or antimycin A; both of which compromise oxidative phosphorylation, either by disrupting electron transfer from Complex I to ubiquinol or by preventing ubiquinol oxidation, respectively. Both compounds caused significant upregulation of ESRE genes (Fig 4A), indicating that compromising mitochondrial activity is sufficient to cause ESRE network induction.
Fig 4. Mitochondrial disruption specifically induces the ESRE network.

(A) Expression of ESRE-containing genes in worms exposed to 50 μM rotenone or 50 μM antimycin A. Fold changes are normalized to vehicle (DMSO) control. (B) Expression of ESRE-containing and non-ESRE containing genes (red label) after treatment with 1 mM 1,10-phenanthroline (Phe) or with 80 μg/mL hygromycin B (Hygro). irg-1 is a hygromycin-responsive gene used as a control. Fold changes are normalized to vehicle (DMSO) control. Error bars represent SEM.
The core cellular process surveillance paradigm suggests that a variety of cellular events are monitored for disruption or dysfunction, possibly as an early pathogen warning system [6,46–48]. To assess the specificity of ESRE gene upregulation, we treated worms with hygromycin, a well-known inhibitor of translation elongation. Although hygromycin caused significant upregulation of irg-1, an established target [49,50], ESRE genes in general showed little to no response (Fig 4B). In addition, neither tunicamycin (which induces ER stress [51]), bortezomib (a proteasome inhibitor [52]), nor ToxA (a different toxin produced by P. aeruginosa that hampers translation, but has no known effect on mitochondria [49]) caused upregulation of ESRE genes (S6 Fig). This strongly indicates that the ESRE network is specifically activated by mitochondrial damage, and is not merely a response to general disruption of cellular processes.
The ESRE network is evolutionarily conserved [42,53], and chelators have been demonstrated to damage mammalian mitochondria [29,54,55]. Therefore, we were interested in determining whether synthetic chelators also activate the ESRE network in mammals, indicating a conservation of its function in mitochondrial surveillance. We used a previously described process to identify human orthologs of C. elegans ESRE genes [42] (Fig 5A). In brief, we identified putative human orthologs by using C. elegans ESRE genes that were upregulated by pyoverdine as queries for reciprocal BLAST. We verified the presence of at least one ESRE consensus motif in the promoter regions (see S9 Table for a list of selected gene pairs). For putative orthologs retaining ESRE consensus sites, we used qRT-PCR to measure gene expression in HEK293T cells treated with the iron chelating agents ciclopirox olamine or phenanthroline (Fig 5B). The compounds significantly induced expression of all 11 ESRE genes tested.
Fig 5. Mitochondrial surveillance and ESRE defense network activation are evolutionarily conserved.

(A) Scheme used to identify human ESRE genes orthologous to C. elegans ESRE genes. (B) Expression of human ESRE genes in HEK293T cells after treatment with 0.5 mM 1,10-phenanthroline or with 0.1 mM ciclopirox olamine for 12 h. Fold changes were normalized to vehicle (DMSO) control. Error bars represent SEM.
A family of bZIP transcription factors are involved in responding to mitochondrial damage
The overlap between genes dependent upon ZIP-2 for their expression and upregulation by either phenanthroline or Liquid Killing, along with the remarkable sensitivity of ZIP-2 mutants to Liquid Killing [29], suggested that ZIP-2 may be involved in host defense against mitochondrial damage. Therefore, we hypothesized that ZIP-2 may be necessary for proper expression of ESRE genes. We tested this prediction by using qRT-PCR to assay ESRE gene activation in zip-2(tm4248) mutants after exposure to phenanthroline. Upregulation of all genes tested was attenuated (Fig 6A). However, neither an intragenic deletion (tm4248) nor zip-2 RNAi in a glp-4(bn2); zip-2(tm4248) background were sufficient to completely abolish expression of ESRE genes (Figs 6A and 7B). It should be noted that zip-2(RNAi) has been reported to be at least as effective as either of the deletion alleles (tm4248 or tm4067) in disrupting ZIP-2 function [56]. However, it remains at least formally possible that even the combination of the deletion and the RNAi knockdown do not result in a null phenotype.
Fig 6. ESRE network transcription requires bZIP transcription factors.

(A) Expression of ESRE-containing genes in glp-4(bn2) or glp-4(bn2); zip-2(tm4248) mutants treated with 1 mM 1,10-phenanthroline. Fold changes were normalized to untreated genotypic cohorts. (B) glp-4(bn2) worms were grown on a panel of RNAi strains that knock down expression of zip-2 or one of three closely related bZIP transcription factors (zip-4, cebp-1, and cebp-2). Proportion of worms killed by P. aeruginosa is shown for each condition. (C, D) Survival of a panel of bZIP mutants exposed to Liquid Killing (C) or 1 mM 1,10-phenanthroline (D). (E) Expression of ESRE-containing genes in a panel of bZIP mutants exposed to Liquid Killing. Expression for each gene was first normalized to untreated genotypic cohort and then normalized to fold changes in glp-4(bn2) controls. Yellow indicates WT expression level; darker shades reflect downregulation relative to wild type. Numbers in the “Fold” column show the arithmetic mean for the entire set of raw fold change for gene expression in worms exposed to Liquid Killing compared to untreated genotypic cohorts (first round of normalization). Statistical significance was determined via Student’s t-test, error bars represent SEM, asterisks represent p-value < 0.01.
Fig 7. ZIP-2 and CEBP-1 have independent and overlapping innate immune functions.

(A) Survival of glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants grown on zip-2(RNAi) after exposure to P. aeruginosa in liquid. Statistical significance was determined via Student’s t-test, error bars represent SEM, hash represents p-value < 0.05, N.S. represents p>0.05. (B) Expression of ESRE genes in the same panel of worms as in (A) after exposure to Liquid Killing conditions for 12h. Fold changes were normalized to untreated genotypic cohorts.
Another possibility is that ZIP-2 exhibits at least partial genetic redundancy. Therefore, we searched the genome of C. elegans for other bZIP proteins that were closely related to ZIP-2 and found three additional C/EBP subfamily members: zip-4, cebp-1, and cebp-2. Interestingly, CEBP-2 was recently reported to function with ZIP-2 to mediate host defense against Slow Killing [57]. RNAi targeting any of these four bZIP family members significantly compromised survival in Liquid Killing (Fig 6B). To confirm the RNAi phenotype, and eliminate the possibility of off-target RNAi effects, we generated glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) double mutants. cebp-2 and glp-4 are both located on LGI, complicating construction of the cebp-2(tm5421) glp-4(bn2) double mutant. Each of the three double mutants showed increased sensitivity to Liquid Killing and phenanthroline (Fig 6C and 6D). Loss of ZIP-2, ZIP-4, or CEBP-1 individually was sufficient to compromise ESRE gene induction (Fig 6E), confirming their involvement in this process.
To further test the interrelationship of zip-2, zip-4, and cebp-1, we used RNAi to knock down zip-2 in glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) strains. We assayed survival in Liquid Killing (Fig 7A) and ESRE gene induction (Fig 7B) in these backgrounds. Adding zip-2(RNAi) into double mutant backgrounds only showed a negative additive effect on survival of glp-4(bn2); cebp-1(tm2807) double mutants. At the same time, there was no evidence of any additive or synergistic interaction between zip-2 and cebp-1 in regulating the expression of ESRE genes. This suggests that at least some CEBP-1 function(s) relevant for survival in Liquid Killing are not shared with ZIP-2 and are independent of ESRE regulation. In the same vein, all of ZIP-4’s functions in Liquid Killing are within the same pathway(s) (or even the same complex) as those of ZIP-2.
Finally, we assayed glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants for the expression of a panel of genes that were upregulated under Slow Killing conditions (i.e., infections with P. aeruginosa on agar). As previously noted, ZIP-2 is critical for defense under these conditions [56]. While zip-4 mutation failed to affect the expression of any of the genes tested, cebp-1 mutation disrupted expression of one-third of the genes (the same number showed attenuation after zip-2 mutation, S7A Fig). We further tested whether this gene family plays a role in defense by performing classical Slow Killing assays with mutant animals (S7B Fig). Mutation of either zip-2 or cebp-1 caused substantial decreases in survival, demonstrating a previously unappreciated role for CEBP-1 in promoting innate immune function.
Regulation of ESRE genes by bZIP transcription factors is not a consequence of glp-4(bn2) mutation
In order to confirm that the phenotypes observed are not a consequence of the glp-4(bn2) allele, we assayed expression of the same panel of 8 ESRE genes in wild-type, zip-4(tm1359), zip-2(tm4248), and cebp-1(tm2807) worms after exposure to P. aeruginosa (Table 2, and see S10 Table for detailed data analysis). Consistent with our prior observations, ESRE genes were upregulated under Liquid Killing conditions. Upregulation was attenuated in each of the bZIP mutants tested.
Table 2. ESRE genes’ expression in wild-type background.
| Gene | Name | N2/LK | zip-2/LK | zip-4/LK | cebp-1/LK |
|---|---|---|---|---|---|
| C02B4.2 | nhr-17 | 40.6 | 10.2 | N.S. | N.S. |
| C12C8.1 | hsp-70 | 118.7 | 19.3 | N.S. | N.S. |
| F44E5.5 | F44E5.5 | 733.4 | 207.8 | 127.1 | 35.8 |
| R11A5.3 | R11A5.3 | 85.9 | N.S. | N.S. | N.S. |
| Y46H3A.2 | hsp-16.41 | 378.2 | 115.9 | 71.1 | N.S. |
| C06B3.7 | C06B3.7 | Below Detection Threshold in All Conditions | |||
| T27F6.8 | T27F6.8 | Below Detection Threshold in All Conditions | |||
| F26A3.4 | F26A3.4 | 4.5 | 2.8 | N.S. | N.S. |
LK–Liquid Killing;
N.S.–did not pass significance criteria
Next, we assayed survival of single, double, and triple bZIP mutants during Liquid Killing. Each genotype tested showed significant susceptibility to pathogen when compared to wild-type worms (S8A Fig), but only one double mutant (cebp-2(tm5421); zip-2 (tm4248)) was significantly more sensitive to the pathogen than the individual mutants comprising it (S8A Fig, marked with grey arrow). However, this double mutant strain appeared sickly (smaller size and with reduced fecundity) (S8B Fig). All other mutants were indistinguishable from wild-type worms when fed E. coli. These observations make it impossible to unambiguously claim that the increase in death under Liquid Killing conditions is specifically due to increases in sensitivity to P. aeruginosa.
Discussion
The opportunistic, multi-host pathogen P. aeruginosa exhibits a variety of virulence mechanisms, and has been shown to cause at least five different types of killing in C. elegans [36]. Amongst these is Liquid Killing, a lethal, pyoverdine-mediated intoxication characterized by mitochondrial damage and a hypoxic response [24,29]. Since pyoverdine is also an essential component of mammalian infections [26–28,58], we assayed the response to C. elegans Liquid Killing to gain understanding about how hosts defend themselves against this type of insult.
Our data clearly demonstrate that the responses of C. elegans to pyoverdine intoxication or phenanthroline exposure are very different from genes activated by classical intestinal colonization (Slow Killing). Although there was a statistically significant overlap in transcriptional responses between Slow and Liquid Killing, the biological significance of this overlap is likely very low. For example, only eight genes were differentially regulated under both conditions and the Gene Ontology profiles are quite disparate. Perhaps most meaningfully, the PMK-1/p38 MAPK pathway, which is most responsible for resistance to intestinal colonization compromises the host's ability to survive pyoverdine intoxication [29].
In contrast, the host response to Liquid Killing was more similar to conventional stress responses, such as those induced by hypoxia or by heat shock. Much of the response to Liquid Killing, can be mimicked by phenanthroline, which has the same molecular function, but is structurally unrelated (Fig 3B). Combined, these data indicate that the host's defensive mechanisms are focused on mitigating the damage caused by iron removal that is catalyzed by these different compounds. This demonstrates a particularly nuanced response to P. aeruginosa exposure, wherein the host defense identifies and attempts to ameliorate the most important virulence determinants.
Analysis of the promoter regions of genes upregulated specifically in response to Liquid Killing revealed a previously-identified, 11-nucleotide motif referred to as ESRE [40]. This motif has an interesting history, as it has seemingly been independently discovered at least seven times, and is responsive to hypoxia, ethanol, heat, and oxidative stress [40,42,53,59–62]. Intriguingly, all nine ESRE-activating conditions identified in this and other papers (pyoverdine, phenanthroline, rotenone, antimycin A, ethanol, hypoxia, heat shock, oxidative stress, and spg-7 (RNAi)) disrupt mitochondrial function. In contrast, stressors with other modalities like hygromycin B, tunicamycin, bortezomib, or exotoxin A did not result in ESRE genes’ activation. The identity of the DAMP(s) (e.g., substrates or products of oxidative phosphorylation, defective protein import or overproduction of reactive oxygen species) remains to be elucidated.
A recent report observed a strong similarity between the ESRE motif and the consensus binding site for the EOR-1 transcription factor, and speculated that EOR-1 may play a previously unknown role in longevity after mitochondrial disruption [61]. There are several compelling reasons to doubt that EOR-1 is required for upregulation of ESRE genes, however. First, although 8 of the 11 nucleotides in ESRE match the EOR-1 consensus, the 3 nucleotides in the center showed much higher conservation in ESRE than are observed in the EOR-1 consensus, suggesting the presence of an additional specificity determinant for ESRE binding. Second, small HSPs, such as the HSP-16 family, are enriched in ESRE sites and are amongst the most strongly upregulated genes after Liquid Killing. EOR-1 and EOR-2 have clearly been shown to repress, rather than induce, the expression of this gene family [63]. Third, expression of ESRE-containing genes was indistinguishable between strains with or without mutations in eor-1 or eor-2 (S9 Fig). Combined, these observations make it unlikely that EOR-1 is the factor driving expression of ESRE genes after mitochondrial insult.
In contrast, the bZIP family transcription factors ZIP-2, ZIP-4, and CEBP-1 do seem to regulate the expression of ESRE genes, as mutation of any one of them significantly diminishes the upregulation of ESRE genes after exposure to iron chelating compounds (Fig 6E). These three transcription factors also confer resistance to both Liquid Killing and phenanthroline (Fig 6C and 6D). It is worth noting that bZIP family members have distinct individual roles. For example, ZIP-2 and CEBP-1 are both required for survival in Liquid Killing and play additional, unique roles independent of ESRE regulation. This is evidenced by the enhanced sensitivity to P. aeruginosa in Liquid Killing observed for a glp-4(bn2); zip-2(RNAi); cebp-1(tm2807) triple mutant. ZIP-2 and CEBP-1, but not ZIP-4, are also required for survival during infection with P. aeruginosa on agar plates. This is, to our knowledge, the first report suggesting that CEBP-1 promotes host defense against P. aeruginosa infection in Slow Killing. Studies using single, double, and triple mutants in a wild-type background eliminated the possibility that the phenomena we observed were related to the absence of the germline (caused by the glp-4(bn2) lesion). Further study of the ESRE network, its mechanisms, and this bZIP protein family are currently underway in our lab.
Finally, our data support the existence of a phylogenetically conserved mitochondrial surveillance network that promotes organismal survival in the context of innate immune and stress response functions. In contrast to ER surveillance, where a wealth of knowledge has been discovered (including pathways monitoring protein folding or glycosylation [64], etc.), mitochondrial surveillance is relatively new, especially in mammals. Due to its diverse functions, its extracellular (i.e., non-self) origins, and its roles in many cell death pathways, mitochondrial surveillance is almost certainly under intricate and detailed control. This research has been spearheaded in C. elegans, where important discoveries about surveillance of the mitochondrial unfolded protein response [46,65–68] have been made. In many cases, activation of these pathways results in transcription of pathogen defense pathways [48,66], a process called cellular-Surveillance-Activated Detoxification and Defenses (cSADD) [48].
cSADD has been hypothesized to help identify otherwise covert microbial activities compromising host health. We favor an alternative hypothesis. The cSADD pathways and the ESRE network may have originally arisen to monitor intracellular processes and only later were integrated into the innate immune system. Although stress response and innate immunity are generally conceived of as separate phenomena (abiotic vs biotic), this dichotomy is a conceptual device, and may not accurately reflect events occurring at the cellular level. In practice, both stress response and innate immunity recognize the same perturbations: molecules that are out of their normal context (e.g., misfolded proteins, disruptions of mitochondrial integrity and function, dsRNA of an inappropriate length, etc.). This is evidenced by the extensive overlap of the cellular processes involved in both defense mechanisms. Broadening our understanding to include both processes under the same conceptual framework is likely to lead to new hypotheses and discoveries.
Methods
C. elegans strains
All C. elegans strains were maintained on nematode growth medium (NGM) seeded with Escherichia coli strain OP50 as a food source and were maintained at 15°C [69], unless otherwise noted. C. elegans strains used in this study included N2 Bristol (wild-type), SS104 [glp-4(bn2)] [70], CZ8920 [cebp-1(tm2807)], ERT061 [zip-2(tm4248)], ERT214 [cebp-2(tm5421)], ERT216 [cebp-2(tm5421); zip-2(tm4248)], NVK56 [zip-4(tm1359)], NVK77 [zip-4(tm1359); zip-2(tm4248)], NVK81 [zip-2(tm4248); cebp-1(tm2807)], NVK83 [zip-4(tm1359); cebp-1(tm2807)], NVK87 [zip-4(tm1359); zip-2(tm4248); cebp-1(tm2807)], NVK98 [glp-4(bn2); houIs002 |pJY323(Phsp-16.1::GFP); pRF4|], NVK214 [glp-4(bn2); zip-2(tm4248)], NVK215 [zip-4(tm1359) glp-4(bn2)], NVK216 [glp-4(bn2); cebp-1(tm2807)], WY753 (fdEx142 |pJY323 [Phsp-16.1::GFP]; pRF4 [rol-6(gf)]|) [43], WY756 (fdEx139 |pJY312 [Phsp-16.1(dd)::GFP]; pRF4|) [43].
Media conditions include NGM [69], standard nematode growth medium; SK, modified NGM used for plate-based infection with P. aeruginosa [71]; LK, modified liquid NGM used for Liquid Killing [71]. Prior to experiments, worms were synchronized by hypochlorite isolation of eggs from gravid adults, followed by hatching of eggs in S basal. L1 larvae were transferred to NGM plates seeded with OP50 or NGM plates supplemented with 25 μg/ml carbenicillin and 1mM IPTG that were seeded with appropriate RNAi strains. After transfer, worms were grown overnight at 15°C, and then shifted to 25°C for 44–48 hours prior to use. For strains that did not have a sterile background, worms were reared on cdc-25.1(RNAi) prior to use in assays to guarantee sterility. Young adult worms were used for all assays. RNAi treatment was performed by seeding 4500 synchronized L1 larvae onto 10 cm NGM plates supplemented with carbenicillin and 1 mM IPTG. Worms were used when they reached the young adult stage.
Bacterial strains
Pseudomonas aeruginosa strain PA14 is a previously described clinical isolate [31]. PA14 pyoverdine mutants (pvdF and pvdP) were obtained from a transposon insertion library [72] and were verified by DNA sequencing. RNAi experiments in this study were performed by using RNAi-competent OP50(xu363) [73], except for RNAi of ESRE-containing effectors, which were performed using HT115(DE3)-based strains. RNAi clones were obtained by purifying appropriate plasmids from the Ahringer RNAi library [44,45] and were sequenced prior to use.
C. elegans pathogenesis and chemical exposure assays
Slow and Liquid Killing assays were performed essentially as previously described, except that synchronized young adult worms were used for Slow Killing [71]. To determine phenanthroline sensitivity, the Liquid Killing assay was modified as follows: first, P. aeruginosa PA14 was substituted with E. coli OP50 at a final density of OD600 = 0.03. Second, 1,10-phenanthroline was added at a final concentration of 1 mM.
Prior to exposure to small molecules, synchronized young adult worms were washed from NGM plates seeded with OP50, and resuspended in S basal supplemented with 50 μM antimycin A (Sigma), 50 μM rotenone (Sigma), 80 μg/mL hygromycin B, 60 μM tunicamycin (TOCRIS Bioscience), or 12.5 μM bortezomib (LC Laboratories) in the presence of OP50. Worms were exposed for 8 hours prior to RNA harvest (see below).
The ToxA sensitivity assay was performed as previously described, except that worms were reared on the toxin-expressing bacteria for 8 hours. E. coli strain expressing empty pET100 vector and pET100 expressing ToxA were grown, induced, and seeded on NGM plates containing carbenicillin and IPTG as described [49]. Synchronized young adult worms were washed from NGM plates seeded with OP50 and were rinsed three times to remove residual bacteria. Approximately 5000 worms were added to each lawn and were incubated for 8 hours at 25°C and then RNA was harvested (see below).
At least three biological replicates (defined as three independent trials) were performed for each experiment. p-values were determined via Student’s t-test.
Microarray and motif discovery
Microarray analysis and motif discovery were performed essentially as previously described [74]. Briefly, RNA was isolated from worms after 8 h exposure to: E. coli strain OP50 on NGM media plates, E. coli OP50 on SK media plates, E. coli OP50 in LK media, infected with P. aeruginosa strain PA14 on SK media plates, exposed to P. aeruginosa PA14 in liquid, treated with vehicle (1% v/v DMSO) in LK media, or treated with 1 mM 1,10-phenanthroline in LK media. RNA was harvested via phenol-chloroform extraction and cleaned up with an RNEasy kit (Qiagen). Triplicate samples were hybridized to Affymetrix C. elegans genome arrays. Gene expression levels were determined via GCRMA in Bioconductor. Genes were considered differentially expressed on the basis of a modified Wilcoxon rank test, as previously described (i.e., fold change > 2, modified Wilcoxon rank > 1.5) [74].
For conditions reported elsewhere (Table 1, S3 Fig), differentially regulated genes were defined by the original publication. p-values for Venn diagrams were calculated using hypergeometric probability. p-values for overrepresentation of Gene Ontology categories were derived by DAVID v. 6.7 (david.abcc.ncifcrf.gov/), * denotes multiple hypothesis correction using Benjamini-Hochberg method. Microarray data has been deposited in the GEO database with the access number GSE55422. Access link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wtgzsmcojjwdjmr&acc=GSE55422
Quantitative Reverse Transcriptase PCR (qPCR)
RNA purification and qPCR were performed as previously described [24]. For human ESRE genes, targets were identified using a reciprocal BLAST search as previously described [42]. Prior to RNA extraction, human cells were treated with 1,10-phenanthroline, ciclopirox olamine, or DMSO. Primer sequences are available upon request. For each experiment at least three biological replicates (defined as three independent trials) were used. p-values were derived from Student’s t-test.
Imaging
Fluorescence and bright-field micrographs of WY753 and WY756 reporter strains [43] were captured from slide-mounted, young adults that had previously been exposed to 1% DMSO (v/v) or 1 mM 1,10-phenanthroline in 1% DMSO (v/v) for 12 hours in S basal in the presence of E. coli OP50. Experiments were performed at 20°C to minimize any fluorescence resulting from heat shock. Images were acquired and fluorescence was quantified using a Zeiss AXIO Imager ZI microscope with a Zeiss AxioCam HRm camera and Axiovision 4.6 (Zeiss) software. Fluorescence was quantified from at least 50 worms per condition per biological replicate. At least three biological replicates (defined as three independent trials) were performed. p-values were determined by Student’s t-test.
For visualization of NVK98 young adults exposed to P. aeruginosa or E. coli in 96-well plates, a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek Instruments) was used. All imaging was performed with identical settings.
Supporting information
Expression levels of 11 upregulated genes as determined by microarray (x-axis) and qPCR (y-axis). The line of best-fit and its correlation coefficient are shown. Fold changes were normalized to untreated genotypic cohorts.
(PDF)
(A,C) Expression levels of 12 genes upregulated in Liquid Killing in either wild-type (N2) background or in the presence of the sterility-inducing glp-4(bn2) allele after exposure to P. aeruginosa in liquid (A) or on agar plates (C). (B,D) Expression levels of 7 genes upregulated in Slow Killing in either wild-type (N2) background or in the presence of the sterility-inducing glp-4(bn2) allele after exposure to P. aeruginosa in liquid (B) or on agar plates (D). Fold changes were normalized to untreated genotypic cohorts. Error bars represent SEM.
(PDF)
(A) Expression patterns of genes were used for hierarchical clustering across eleven different microarray conditions. Host and microbial strains, media conditions (see Methods for details), and references are shown. (B) A cluster showing enrichment of genes upregulated specifically in Liquid Killing is shown. Conditions (x-axis) correspond to the eleven data sets listed in (A). The heavy red line indicates mean expression level of genes within the cluster.
(PDF)
A panel of ESRE-containing genes were disrupted via RNAi and worms were subsequently exposed to P. aeruginosa (Liquid Killing). Asterisks represent RNAi knockdowns with significantly higher death rate (p < 0.05) in 4/4 replicates. Hashes represent RNAi knockdowns with significantly higher death rate (p < 0.05) in 3/4 replicates. Average survival is plotted for all biological replicates. Error bars represent standard deviation.
(PDF)
NVK98 young adult worms expressing phsp-16::GFP were exposed to either E. coli OP50 or P. aeruginosa PA14 under Liquid Killing conditions for 24 h (A) or 48 h (B) in 96-well plates. Images were taken under identical settings. Scale bars represent 1000 μm.
(PDF)
Expression of ESRE-containing and non-ESRE containing genes (red label) after treatment with a proteasomal inhibitor (bortezomib), an activator of the ER unfolded protein response (tunicamycin), or a translational inhibitor (E. coli expressing Exotoxin A from P. aeruginosa). Fold changes are normalized to the solvent control.
(PDF)
(A) Expression of a panel of genes upregulated during P. aeruginosa infection on agar (i.e., Slow Killing) in glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants. Expression values were normalized to untreated genotypic cohorts. Error bars represent SEM, asterisks show p-value < 0.01, hashes show p-value < 0.05. (B) Slow kill assays of glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants. A representative replicate (one of three) is shown.
(PDF)
(A) Survival of a panel of bZIP mutants exposed to P. aeruginosa (Liquid Killing). (B) Representative images of bZIP mutants on a non-pathogenic food source (E. coli OP50). Statistical significance was determined via Student’s t-test, error bars represent SEM, asterisks represent p-value < 0.01. The grey arrow indicates the cebp-2; zip-2 double mutant, which shows significantly more death than either single mutant.
(PDF)
Expression of a panel of ESRE genes in glp-4(bn2), glp-4(bn2); eor-1(cs28), and glp-4(bn2); eor-2(cs42). Expression values were normalized to untreated genotypic cohorts. Error bars represent SEM.
(PDF)
(XLSX)
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Acknowledgments
We are grateful to Fred Ausubel, Emily Troemel, and Eyleen O’Rourke for providing bacterial strains used in this study. C. elegans strains used were provided by David Fay or obtained from CGC. We thank Daniel Kirienko, David Fay, Fred Ausubel, and Gary Ruvkun for helpful discussion.
Data Availability
All relevant data are within the paper and its Supporting Information files, except for the microarray data which are available from the Gene Expression Omnibus database, accession number GSE55422.
Funding Statement
This work was supported by grants K22 AI110552 from the National Institutes of Health (https://www.niaid.nih.gov/) and RR150044 from Cancer Prevention and Research Institute of Texas (http://www.cprit.state.tx.us/) to NVK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Medzhitov R (2009) Approaching the asymptote: 20 years later. Immunity 30: 766–775. doi: 10.1016/j.immuni.2009.06.004 [DOI] [PubMed] [Google Scholar]
- 2.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC (2006) Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112: 358–404. doi: 10.1016/j.pharmthera.2005.04.013 [DOI] [PubMed] [Google Scholar]
- 3.Jacob F, Perez Novo C, Bachert C, Van Crombruggen K (2013) Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal 9: 285–306. doi: 10.1007/s11302-013-9357-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laliberte RE, Eggler J, Gabel CA (1999) ATP treatment of human monocytes promotes caspase-1 maturation and externalization. J Biol Chem 274: 36944–36951. [DOI] [PubMed] [Google Scholar]
- 5.Kono H, Rock KL (2008) How dying cells alert the immune system to danger. Nat Rev Immunol 8: 279–289. doi: 10.1038/nri2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12: 991–1045. doi: 10.1146/annurev.iy.12.040194.005015 [DOI] [PubMed] [Google Scholar]
- 7.Vance RE, Isberg RR, Portnoy DA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6: 10–21. doi: 10.1016/j.chom.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, et al. (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255. doi: 10.1038/nature10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, et al. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107. doi: 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM (2015) N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 46: 975–984. doi: 10.1016/j.injury.2015.03.028 [DOI] [PubMed] [Google Scholar]
- 11.Raoof M, Zhang Q, Itagaki K, Hauser CJ (2010) Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma 68: 1328–1332; discussion 1332–1324. doi: 10.1097/TA.0b013e3181dcd28d [DOI] [PubMed] [Google Scholar]
- 12.Shore DE, Ruvkun G (2013) A cytoprotective perspective on longevity regulation. Trends Cell Biol 23: 409–420. doi: 10.1016/j.tcb.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irazoqui JE, Urbach JM, Ausubel FM (2010) Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol 10: 47–58. doi: 10.1038/nri2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powell JR, Ausubel FM (2008) Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol 415: 403–427. doi: 10.1007/978-1-59745-570-1_24 [DOI] [PubMed] [Google Scholar]
- 15.Cohen LB, Troemel ER (2015) Microbial pathogenesis and host defense in the nematode C. elegans. Curr Opin Microbiol 23: 94–101. doi: 10.1016/j.mib.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrusiak MG, Jin Y (2016) Context Specificity of Stress-activated Mitogen-activated Protein (MAP) Kinase Signaling: The Story as Told by Caenorhabditis elegans. J Biol Chem 291: 7796–7804. doi: 10.1074/jbc.R115.711101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, Isik M (2015) SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic Biol Med 88: 290–301. doi: 10.1016/j.freeradbiomed.2015.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denzel MS, Antebi A (2015) Hexosamine pathway and (ER) protein quality control. Curr Opin Cell Biol 33: 14–18. doi: 10.1016/j.ceb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- 19.Nussbaum-Krammer CI, Morimoto RI (2014) Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis Model Mech 7: 31–39. doi: 10.1242/dmm.013011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Govan JR, Deretic V (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60: 539–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieberman D (2003) Pseudomonal infections in patients with COPD: epidemiology and management. Am J Respir Med 2: 459–468. [DOI] [PubMed] [Google Scholar]
- 22.Lyczak JB, Cannon CL, Pier GB (2000) Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2: 1051–1060. [DOI] [PubMed] [Google Scholar]
- 23.Conery AL, Larkins-Ford J, Ausubel FM, Kirienko NV (2014) High-throughput screening for novel anti-infectives using a C. elegans pathogenesis model. Curr Protoc Chem Biol 6: 25–37. doi: 10.1002/9780470559277.ch130160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirienko NV, Kirienko DR, Larkins-Ford J, Wahlby C, Ruvkun G, et al. (2013) Pseudomonas aeruginosa Disrupts Caenorhabditis elegans Iron Homeostasis, Causing a Hypoxic Response and Death. Cell Host Microbe 13: 406–416. doi: 10.1016/j.chom.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirienko DR, Revtovich AV, Kirienko NV (2016) A High-Content, Phenotypic Screen Identifies Fluorouridine as an Inhibitor of Pyoverdine Biosynthesis and Pseudomonas aeruginosa Virulence. mSphere 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA (1996) Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64: 518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, et al. (2016) Role of Iron Uptake Systems in Pseudomonas aeruginosa Virulence and Airway Infection. Infect Immun 84: 2324–2335. doi: 10.1128/IAI.00098-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takase H, Nitanai H, Hoshino K, Otani T (2000) Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun 68: 1834–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirienko NV, Ausubel FM, Ruvkun G (2015) Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112: 1821–1826. doi: 10.1073/pnas.1424954112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chand NS, Lee JS, Clatworthy AE, Golas AJ, Smith RS, et al. (2011) The sensor kinase KinB regulates virulence in acute Pseudomonas aeruginosa infection. J Bacteriol 193: 2989–2999. doi: 10.1128/JB.01546-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, et al. (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268: 1899–1902. [DOI] [PubMed] [Google Scholar]
- 32.Tan MW, Mahajan-Miklos S, Ausubel FM (1999) Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 96: 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM (1995) Microbial biofilms. Annu Rev Microbiol 49: 711–745. doi: 10.1146/annurev.mi.49.100195.003431 [DOI] [PubMed] [Google Scholar]
- 34.Smith RS, Iglewski BH (2003) P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol 6: 56–60. [DOI] [PubMed] [Google Scholar]
- 35.Tang HB, DiMango E, Bryan R, Gambello M, Iglewski BH, et al. (1996) Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun 64: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Utari PD, Quax WJ (2013) Caenorhabditis elegans reveals novel Pseudomonas aeruginosa virulence mechanism. Trends Microbiol 21: 315–316. doi: 10.1016/j.tim.2013.04.006 [DOI] [PubMed] [Google Scholar]
- 37.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, et al. (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297: 623–626. doi: 10.1126/science.1073759 [DOI] [PubMed] [Google Scholar]
- 38.Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, et al. (2004) Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A 101: 10990–10994. doi: 10.1073/pnas.0403546101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, et al. (2006) p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2: e183 doi: 10.1371/journal.pgen.0020183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon JY, Hong M, Choi MS, Kang S, Duke K, et al. (2004) Ethanol-response genes and their regulation analyzed by a microarray and comparative genomic approach in the nematode Caenorhabditis elegans. Genomics 83: 600–614. doi: 10.1016/j.ygeno.2003.10.008 [DOI] [PubMed] [Google Scholar]
- 41.Hong M, Kwon JY, Shim J, Lee J (2004) Differential hypoxia response of hsp-16 genes in the nematode. J Mol Biol 344: 369–381. doi: 10.1016/j.jmb.2004.09.077 [DOI] [PubMed] [Google Scholar]
- 42.Kirienko NV, Fay DS (2010) SLR-2 and JMJC-1 regulate an evolutionarily conserved stress-response network. EMBO J 29: 727–739. doi: 10.1038/emboj.2009.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuzmanov A, Karina EI, Kirienko NV, Fay DS (2014) The conserved PBAF nucleosome-remodeling complex mediates the response to stress in Caenorhabditis elegans. Mol Cell Biol 34: 1121–1135. doi: 10.1128/MCB.01502-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. doi: 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- 45.Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14: 2162–2168. doi: 10.1101/gr.2505604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haynes CM, Ron D (2010) The mitochondrial UPR—protecting organelle protein homeostasis. J Cell Sci 123: 3849–3855. doi: 10.1242/jcs.075119 [DOI] [PubMed] [Google Scholar]
- 47.Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, et al. (1997) Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 17: 3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melo JA, Ruvkun G (2012) Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149: 452–466. doi: 10.1016/j.cell.2012.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McEwan DL, Kirienko NV, Ausubel FM (2012) Host translational inhibition by Pseudomonas aeruginosa Exotoxin A Triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11: 364–374. doi: 10.1016/j.chom.2012.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER (2012) C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11: 375–386. doi: 10.1016/j.chom.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richardson CE, Kinkel S, Kim DH (2011) Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet 7: e1002391 doi: 10.1371/journal.pgen.1002391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehrbach NJ, Ruvkun G (2016) Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pignataro L, Varodayan FP, Tannenholz LE, Protiva P, Harrison NL (2013) Brief alcohol exposure alters transcription in astrocytes via the heat shock pathway. Brain Behav 3: 114–133. doi: 10.1002/brb3.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allen GF, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 14: 1127–1135. doi: 10.1038/embor.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park SJ, Shin JH, Kim ES, Jo YK, Kim JH, et al. (2012) Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett 586: 4303–4310. doi: 10.1016/j.febslet.2012.10.035 [DOI] [PubMed] [Google Scholar]
- 56.Estes KA, Dunbar TL, Powell JR, Ausubel FM, Troemel ER (2010) bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc Natl Acad Sci U S A 107: 2153–2158. doi: 10.1073/pnas.0914643107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy KC, Dunbar TL, Nargund AM, Haynes CM, Troemel ER (2016) The C. elegans CCAAT-Enhancer-Binding Protein Gamma Is Required for Surveillance Immunity. Cell Rep 14: 1581–1589. doi: 10.1016/j.celrep.2016.01.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, et al. (2013) Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc Natl Acad Sci U S A 110: 7458–7463. doi: 10.1073/pnas.1222706110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaudet J, Muttumu S, Horner M, Mango SE (2004) Whole-genome analysis of temporal gene expression during foregut development. PLoS Biol 2: e352 doi: 10.1371/journal.pbio.0020352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.GuhaThakurta D, Palomar L, Stormo GD, Tedesco P, Johnson TE, et al. (2002) Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res 12: 701–712. doi: 10.1101/gr.228902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munkacsy E, Khan MH, Lane RK, Borror MB, Park JH, et al. (2016) DLK-1, SEK-3 and PMK-3 Are Required for the Life Extension Induced by Mitochondrial Bioenergetic Disruption in C. elegans. PLoS Genet 12: e1006133 doi: 10.1371/journal.pgen.1006133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruvinsky I, Ohler U, Burge CB, Ruvkun G (2007) Detection of broadly expressed neuronal genes in C. elegans. Dev Biol 302: 617–626. doi: 10.1016/j.ydbio.2006.09.014 [DOI] [PubMed] [Google Scholar]
- 63.Rongo C (2011) Epidermal growth factor and aging: a signaling molecule reveals a new eye opening function. Aging (Albany NY) 3: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18: 716–731. doi: 10.1016/j.semcdb.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37: 529–540. doi: 10.1016/j.molcel.2010.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Samuel BS, Breen PC, Ruvkun G (2014) Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508: 406–410. doi: 10.1038/nature13204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58: 123–133. doi: 10.1016/j.molcel.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337: 587–590. doi: 10.1126/science.1223560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stiernagle T (2006) Maintenance of C. elegans. WormBook: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beanan MJ, Strome S (1992) Characterization of a germ-line proliferation mutation in C. elegans. Development 116: 755–766. [DOI] [PubMed] [Google Scholar]
- 71.Kirienko NV, Cezairliyan BO, Ausubel FM, Powell JR (2014) Pseudomonas aeruginosa PA14 pathogenesis in Caenorhabditis elegans. Methods Mol Biol 1149: 653–669. doi: 10.1007/978-1-4939-0473-0_50 [DOI] [PubMed] [Google Scholar]
- 72.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, et al. (2006) An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103: 2833–2838. doi: 10.1073/pnas.0511100103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao R, Chun L, Ronan EA, Friedman DI, Liu J, et al. (2015) RNAi Interrogation of Dietary Modulation of Development, Metabolism, Behavior, and Aging in C. elegans. Cell Rep 11: 1123–1133. doi: 10.1016/j.celrep.2015.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kirienko NV, Fay DS (2007) Transcriptome profiling of the C. elegans Rb ortholog reveals diverse developmental roles. Dev Biol 305: 674–684. doi: 10.1016/j.ydbio.2007.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression levels of 11 upregulated genes as determined by microarray (x-axis) and qPCR (y-axis). The line of best-fit and its correlation coefficient are shown. Fold changes were normalized to untreated genotypic cohorts.
(PDF)
(A,C) Expression levels of 12 genes upregulated in Liquid Killing in either wild-type (N2) background or in the presence of the sterility-inducing glp-4(bn2) allele after exposure to P. aeruginosa in liquid (A) or on agar plates (C). (B,D) Expression levels of 7 genes upregulated in Slow Killing in either wild-type (N2) background or in the presence of the sterility-inducing glp-4(bn2) allele after exposure to P. aeruginosa in liquid (B) or on agar plates (D). Fold changes were normalized to untreated genotypic cohorts. Error bars represent SEM.
(PDF)
(A) Expression patterns of genes were used for hierarchical clustering across eleven different microarray conditions. Host and microbial strains, media conditions (see Methods for details), and references are shown. (B) A cluster showing enrichment of genes upregulated specifically in Liquid Killing is shown. Conditions (x-axis) correspond to the eleven data sets listed in (A). The heavy red line indicates mean expression level of genes within the cluster.
(PDF)
A panel of ESRE-containing genes were disrupted via RNAi and worms were subsequently exposed to P. aeruginosa (Liquid Killing). Asterisks represent RNAi knockdowns with significantly higher death rate (p < 0.05) in 4/4 replicates. Hashes represent RNAi knockdowns with significantly higher death rate (p < 0.05) in 3/4 replicates. Average survival is plotted for all biological replicates. Error bars represent standard deviation.
(PDF)
NVK98 young adult worms expressing phsp-16::GFP were exposed to either E. coli OP50 or P. aeruginosa PA14 under Liquid Killing conditions for 24 h (A) or 48 h (B) in 96-well plates. Images were taken under identical settings. Scale bars represent 1000 μm.
(PDF)
Expression of ESRE-containing and non-ESRE containing genes (red label) after treatment with a proteasomal inhibitor (bortezomib), an activator of the ER unfolded protein response (tunicamycin), or a translational inhibitor (E. coli expressing Exotoxin A from P. aeruginosa). Fold changes are normalized to the solvent control.
(PDF)
(A) Expression of a panel of genes upregulated during P. aeruginosa infection on agar (i.e., Slow Killing) in glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants. Expression values were normalized to untreated genotypic cohorts. Error bars represent SEM, asterisks show p-value < 0.01, hashes show p-value < 0.05. (B) Slow kill assays of glp-4(bn2), glp-4(bn2); zip-2(tm4248), zip-4(tm1359) glp-4(bn2), and glp-4(bn2); cebp-1(tm2807) mutants. A representative replicate (one of three) is shown.
(PDF)
(A) Survival of a panel of bZIP mutants exposed to P. aeruginosa (Liquid Killing). (B) Representative images of bZIP mutants on a non-pathogenic food source (E. coli OP50). Statistical significance was determined via Student’s t-test, error bars represent SEM, asterisks represent p-value < 0.01. The grey arrow indicates the cebp-2; zip-2 double mutant, which shows significantly more death than either single mutant.
(PDF)
Expression of a panel of ESRE genes in glp-4(bn2), glp-4(bn2); eor-1(cs28), and glp-4(bn2); eor-2(cs42). Expression values were normalized to untreated genotypic cohorts. Error bars represent SEM.
(PDF)
(XLSX)
(DOCX)
(DOCX)
(XLSX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files, except for the microarray data which are available from the Gene Expression Omnibus database, accession number GSE55422.