

Abstract
Purpose
Metaplastic breast carcinoma (MBC) is a rare and aggressive histologic type of breast cancer predominantly of triple-negative phenotype, and characterized by the presence of malignant cells showing squamous and/or mesenchymal differentiation. We sought to define the repertoire of somatic genetic alterations and the mutational signatures of MBCs.
Experimental Design
Whole-exome sequencing was performed in 35 MBCs, with 16, ten and nine classified as harboring chondroid, spindle and squamous metaplasia as the predominant metaplastic component. The genomic landscape of MBCs was compared to that of triple-negative invasive ductal carcinomas of no special type (IDC-NSTs) from The Cancer Genome Atlas. Wnt and PI3K/AKT/mTOR pathway activity was assessed using a quantitative PCR assay.
Results
MBCs harbored complex genomes with frequent TP53 (69%) mutations. In contrast to triple-negative IDC-NSTs, MBCs more frequently harbored mutations in PIK3CA (29%), PIK3R1 (11%), ARID1A (11%), FAT1 (11%) and PTEN (11%). PIK3CA mutations were not found in MBCs with chondroid metaplasia. Compared to triple-negative IDC-NSTs, MBCs significantly more frequently harbored mutations in PI3K/AKT/mTOR pathway-related (57% vs 22%) and canonical Wnt pathway-related (51% vs 28%) genes. MBCs with somatic mutations in PI3K/AKT/mTOR or Wnt pathway-related genes displayed increased activity of the respective pathway.
Conclusion
MBCs are genetically complex and heterogeneous, and are driven by a repertoire of somatic mutations distinct from that of triple-negative IDC-NSTs. Our study highlights the genetic basis and the importance of PI3K/AKT/mTOR and Wnt pathway dysregulation in MBCs, and provides a rationale for the metaplastic phenotype and the reported responses to PI3K/AKT/mTOR inhibitors in these tumors.
Keywords: Massively parallel sequencing, metaplastic breast cancer, copy number alterations, somatic mutations, Wnt pathway
INTRODUCTION
Metaplastic breast carcinoma (MBC) is a rare histologic type of breast cancer, representing approximately 0.2%–5% of invasive breast cancers (1, 2). Histologically, MBCs are characterized by the presence of malignant cells showing squamous and/or mesenchymal differentiation such as spindle, chondroid, osseous or rhabdoid cells (1, 2). MBCs are predominantly of triple-negative phenotype (i.e. lack expression of estrogen receptor (ER), progesterone receptor (PR) and HER2 overexpression/gene amplification) but seem to exhibit a prognosis worse than that of triple-negative invasive ductal carcinomas of no special type (IDC-NSTs) (3, 4). Furthermore, unlike other forms of triple-negative breast cancers, MBCs have been reported not to respond well to conventional chemotherapy regimens (3–5).
The genetic basis for the distinctive histologic and transcriptomic features of MBCs is currently unknown. Copy number profiling studies have revealed that MBCs harbor complex patterns of copy number alterations (CNAs), which are almost indistinguishable from those of histologic grade- and ER-matched IDC-NSTs, with frequent losses of 1p, 3p, 5q, 8p, 14 and 17 and gains of 1q, 3q and 8q (6). The genetic analyses of MBCs performed to date have revealed recurrent TP53 mutations, CDKN2A deletions and epidermal growth factor receptor (EGFR) amplifications (5, 7, 8), all of which are again not uncommonly found in triple-negative IDC-NSTs (9). By contrast, studies have suggested that the PI3K/AKT/mTOR pathway could be more frequently affected by genetic alterations in MBCs than in triple-negative IDC-NSTs (10, 11). Intriguingly, whilst a previous study reported on the presence of CTNNB1 activating mutations in 26% of MBCs (12), we and others found that MBCs frequently display β-catenin nuclear expression/Wnt pathway activation (13, 14), but did not detect CTNNB1 somatic mutations (10, 13, 14). At variance with colorectal cancers, in which Wnt pathway activation is frequently caused by somatic mutations in APC, CTNNB1, TCF7L2 and FAM123B among others (15), mutations affecting these genes are exceedingly rare in breast cancers (9). Interestingly, a recent study showed that inactivating FAT1 mutations induce Wnt pathway activation in multiple cancers, providing a genetic basis for the aberrant signaling in cancer types other than colorectal (16). Whether the epithelial-mesenchymal transition (EMT) phenotype observed in a subset of MBCs is associated with Wnt pathway activation and if this activation is underpinned by somatic genetic alterations remain to be explored.
MBCs are, however, a heterogeneous group of lesions. In fact, we have recently shown that MBCs with chondroid, spindle and squamous metaplasia displayed distinct transcriptomic profiles (17). For instance, spindle cell MBCs are invariably classified as of the claudin-low molecular subtype, whereas MBCs with chondroid metaplasia are frequently of basal-like subtype (17). These observations are consistent with the notion that MBCs with distinct histologic features may be underpinned by distinct somatic genetic alterations. In agreement with this hypothesis, distinct gene CNAs have been reported in histologically distinct components of MBCs (7).
Given the distinctive histologic features and clinical behavior of MBCs, here we sought to define the genetic landscape of 35 MBCs based on whole-exome sequencing analysis. As a hypothesis-generating, exploratory aim, we compared the somatic genetic alterations that underpin MBCs of distinct histologic subtypes (chondroid, spindle cell and squamous). These analyses confirmed the genomic complexity of MBCs and the high frequency of TP53 mutations, demonstrated that mutations affecting genes related to the PI3K/AKT/mTOR and Wnt pathways are recurrent in MBCs, and provided evidence that the Wnt and PI3K/AKT/mTOR pathways are more frequently activated in MBCs than in triple-negative IDC-NSTs.
MATERIAL AND METHODS
Cases and histopathologic review
Thirty-five cases diagnosed as MBCs were retrieved from the tissue banks and/or pathology files of the authors’ institutions. Diagnostic slides were reviewed by at least two pathologists with an interest in breast pathology (FCG, CAE, YHW, AV-S and/or JSR-F), subsequently centrally reviewed by two pathologists (FCG and JSR-F) and diagnosed according to the latest World Health Organization classification (1) into MBCs with squamous metaplasia, MBCs with mesenchymal elements, and spindle cell MBCs (Supplementary Methods). Representative sections of each MBC used for DNA extraction were reviewed, and the tumor cell content and composition of the metaplastic elements were estimated (i.e., chondroid metaplasia, spindle cell metaplasia and squamous metaplasia). In each sample, the metaplastic component most abundantly present was defined as previously described (17) (Table 1 and Supplementary Table S1), and this classification was used for subsequent analyses. Tumors were graded according to the Nottingham grading system (18). Samples were anonymized prior to analysis. This study was approved by the local institutional review boards of the authors’ institutions. Copy number profiling and/or microarray gene expression profiling results for nine samples (with prefix META) were reported elsewhere (17).
Table 1.
Clinico-pathologic features of the 35 metaplastic breast carcinomas included in this study.
| Clinico-pathologic features | MBC (n=35) | MBC with chondroid metaplasia* (n=16) |
MBC with spindle cell metaplasia* (n=10) |
MBC with squamous metaplasia* (n=9) |
|---|---|---|---|---|
| Histologic grade** | ||||
| 1 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 2 | 5 (14%) | 3 (19%) | 1 (10%) | 1 (11%) |
| 3 | 30 (86%) | 13 (81%) | 9 (90%) | 8 (89%) |
| ER status | ||||
| Negative | 34 (97%) | 16 (100%) | 10 (100%) | 8 (89%) |
| Positive | 1 (3%) | 0 (0%) | 0 (0%) | 1 (11%) |
| PR status | ||||
| Negative | 34 (97%) | 16 (100%) | 10 (100%) | 8 (89%) |
| Positive | 1 (3%) | 0 (0%) | 0 (0%) | 1 (11%) |
| HER2 status | ||||
| Negative | 34 (97%) | 16 (100%) | 10 (100%) | 8 (89%) |
| Positive | 1 (3%) | 0 (0%) | 0 (0%) | 1 (11%) |
| Triple-negative phenotype*** | 33 (94%) | 16 (100%) | 10 (100%) | 7 (78%) |
Immunohistochemistry and fluorescence in situ hybridization (FISH)
Immunohistochemistry for ER, PR and HER2 and FISH for HER2 amplification were performed according to the American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) guidelines (19–21) (Supplementary Methods).
Microdissection and DNA extraction
Eight-μm-thick representative sections of the snap-frozen (samples with prefixes META or MTC) or formalin-fixed paraffin-embedded (samples with prefix MP, Supplementary Table S1) blocks of MBCs were microdissected with a needle under a stereomicroscope (Olympus SZ61), to ensure >70% of tumor cell content as previously described (17) (Supplementary Methods). Matched germline DNA was microdissected from adjacent normal breast tissue for each case; to avoid the possibility of morphologically appearing non-neoplastic cells harboring somatic mutations, we prioritized the microdissection of stromal cells and avoided normal breast ducts and lobules.
Whole-exome massively parallel sequencing
DNA from MBC and matched germline of the 35 cases was subjected to whole-exome capture using the SureSelect Human All Exon v4 (Agilent) platform and to massively parallel sequencing on an Illumina GAIIx or HiSeq 2000 at the Institute of Cancer Research, UK (GAIIx) or Memorial Sloan Kettering Cancer Center (MSKCC) Integrated Genomics Operation (IGO, HiSeq 2000) following validated protocols (22). Paired-end 75/76/101-bp reads were generated (Supplementary Table S1). Sequencing data have been deposited in the NCBI Sequence Read Archive under the accession SRP073692.
Whole-exome sequencing analysis was performed as described (22) with modifications. CNAs were identified using FACETS (23) and the cancer cell fraction (CCF) of each mutation using ABSOLUTE (v1.0.6) (24). Mutations were classified as likely pathogenic, of indeterminate pathogenicity or likely passenger based on mutation function predictors (25–28), cancer gene lists (29–31), hotspot residues (32) and loss of heterozygosity status (Supplementary Methods).
Sanger sequencing
Sanger sequencing was performed as previously described (22). All mutations subjected to Sanger sequencing were confirmed (Supplementary Methods and Supplementary Table S2).
Mutational significance, mutational signature and analysis of genomic scars
Significantly mutated genes were defined using MutSigCV (33) and the Youn and Simon algorithm (34). Mutational signature (35, 36) was defined using deconstructSigs (37) for samples with at least 30 somatic mutations. Microsatellite instability detection was performed using MSIsensosr (38). Large-scale state transitions (LSTs) were employed to define homologous recombination (HR) deficiency as described in (39) (Supplementary Methods).
Comparative analysis with triple-negative IDC-NSTs and statistical analysis
Clinical and molecular data for triple-negative IDC-NSTs from The Cancer Genome Atlas (TCGA, n=69) (9) were obtained from the TCGA Data Portal (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/). Comparisons of continuous and categorical features were performed using Mann-Whitney U and Fisher’s exact tests as appropriate. p<0.05 were considered statistically significant. All tests were two-sided. Statistical analyses were performed with R v3.1.2 and SPSS v24 (IBM, Supplementary Methods).
Unsupervised hierarchical clustering
Hierarchical clustering of somatic mutations from MBCs and triple-negative IDC-NSTs from TCGA were performed using an asymmetric binary distance metric and complete-linkage (Supplementary Methods).
Wnt and PI3K-AKT signaling pathway RT2 profiler PCR arrays
The levels of Wnt and PI3K/AKT/mTOR signaling pathway activation were assessed using the Human WNT Signaling Pathway Plus and Human PI3K-AKT Signaling RT2 Profiler™ PCR Array kits (catalog numbers: PAHS-043Y and PAHS-058Z; Qiagen; Supplementary Methods).
RESULTS
Pathologic features of MBCs
A central histopathologic review of the 35 MBCs included in this study revealed that, according to the predominant cellular type, 16 were MBCs with chondroid metaplasia, of which 13 were matrix-producing, nine were MBCs with squamous metaplasia and ten were spindle cell MBCs (Table 1 and Supplementary Table S1). Low-grade variants of MBCs were not included in this study. Assessment of histologic grade according to the Nottingham grading system (18) revealed that 14% (5/35) and 86% (30/35) were of grades 2 and 3, respectively. Immunohistochemical analysis revealed that 94% (33/35) were of triple-negative phenotype. Of the two cases that did not display a triple-negative phenotype, MTC18 was ER-positive (1%), PR-positive (20%) and HER2-negative (Supplementary Fig. S1a–c) and MTC23 was ER-negative, PR-negative and HER2-positive (confirmed by FISH and whole-exome sequencing, Supplementary Fig. S1d–g). The frequency of ER, PR and HER2 positivity in this cohort of MBCs is not different from that of previously reported studies (40, 41).
Landscape of somatic mutations in MBCs
To define the repertoire of somatic genetic alterations in MBCs, we subjected the 35 MBCs to whole-exome sequencing to median depths of 246× (range 48×–843×) and 95× (range 51×–160×) for the tumors and matched germline samples, respectively (Supplementary Table S1). Our analysis revealed a median of 27 (range 4–95) and 76 (range 11–249) somatic synonymous and non-synonymous somatic mutations per case, respectively. The mutation rate in our cohort of MBCs (median 103, range 15–344) was not statistically different from that of triple-negative IDC-NSTs from TCGA (median 76, range 14–233, p>0.05, Mann-Whitney U-test, Fig. 1).
Figure 1. Repertoire of non-synonymous somatic mutations of metaplastic breast cancers (MBCs) and triple-negative invasive carcinomas of no special type (IDC-NSTs).
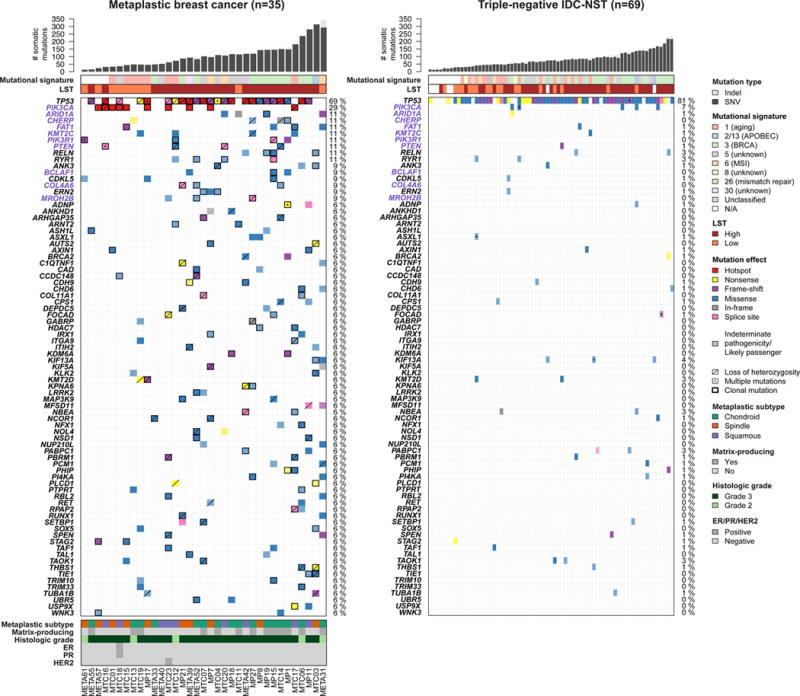
Clinico-pathologic and immunohistochemical features, and non-synonymous somatic mutations identified in 35 MBCs subjected to whole-exome sequencing (left), and in 69 triple-negative IDC-NSTs from TCGA breast cancer study (right) (9). The effects of the mutations are color-coded according to the legend, with hotspots (32) colored in red. Likely passenger mutations and mutations of indeterminate pathogenicity are marked using a hatched pattern. The presence of multiple non-synonymous mutations in the same gene is represented by an asterisk. For MBCs, the presence of loss of heterozygosity of the wild-type allele of a mutated gene is represented by a diagonal bar, and mutations found to be clonal by ABSOLUTE (24) are indicated by a black box. Genes recurrently mutated in MBCs and displaying at least one likely pathogenic mutation are presented. Gene names highlighted in purple were significantly more frequently altered in MBCs as compared to triple-negative IDC-NSTs. Percentages to the right of the mutation heatmaps indicate the percentage of cases affected by non-synonymous somatic mutations in a given gene. Bar charts (top) indicate the number of non-synonymous and synonymous somatic single nucleotide variants (SNVs) and the number of somatic insertions and deletions (indels) for each sample. The dominant mutational signatures (35, 36) were assigned using deconstructSigs (37). Large-scale transitions (LST)-high and LST-low status was determined in accordance with Popova et al. (39). TCGA, The Cancer Genome Atlas.
Analysis of the recurrently mutated genes identified 285 genes with non-synonymous mutations and 33 genes with likely pathogenic mutations in at least 2/35 MBCs (Fig. 1, Supplementary Fig. S2a and Supplementary Tables S2 and S3). The genes that frequently harbored non-synonymous somatic mutations were TP53 (69%, all likely pathogenic) and PIK3CA (29%, all likely pathogenic; Fig. 1). Although TTN (31%) and FAT3 (20%) were recurrently mutated in MBCs (Supplementary Fig. S2a, Supplementary Tables S2 and S3), somatic genetic alterations in these genes are known to constitute passenger alterations; consistent with this notion, none of the mutations affecting these genes were predicted to be ‘likely pathogenic’. TP53 was significantly mutated based on MutSigCV (33) and TP53, PIK3CA, PTEN (11%, all likely pathogenic) and PIK3R1 (11%, all likely pathogenic) were significantly mutated based on Youn and Simon (34) (Supplementary Table S4). In all cases that harbored TP53 mutation, both alleles were affected, either by a somatic mutation coupled with loss of heterozygosity (LOH, n=22) or two distinct somatic TP53 mutations (n=2), and in all TP53-mutant cases, at least one TP53 mutation was found to be clonal as defined by ABSOLUTE (24) (i.e. present in virtually 100% of the neoplastic cells, Fig. 1 and Supplementary Table S2). All PIK3CA mutations identified were clonal activating missense mutations and/or affected hotspots (C420R, E542K, H1047R and H1047L). Several other cancer genes were also recurrently mutated, including ARID1A, KMT2C, FAT1 (all 11%), BCLAF1 (9%) and AXIN1, BRCA2, KMT2D, NCOR1, RUNX1 and SPEN (all 6%, Fig. 1). Similar to the TP53 mutations, all PTEN mutations detected were clonal and were either associated with LOH of the wild-type allele (n=3) or a second PTEN somatic mutation (n=1). Of the 27 TP53, PIK3CA and PTEN mutations tested, all were confirmed using Sanger sequencing (Supplementary Table S2 and Supplementary Fig. S1h).
Compared to triple-negative IDC-NSTs in TCGA, 31 genes were found to be mutated more frequently in MBCs, including PCLO (14% vs 0%, p=0.0035, Fisher’s exact test), PIK3CA (29% vs 7%, p=0.0064, Fisher’s exact test), CHERP and PIK3R1 (both 11% vs 0%, p=0.0114, Fisher’s exact test), BCLAF1, FANCM and USP5 (all 9% vs 0%, p=0.0360, Fisher’s exact tests), and ARID1A, KMT2C, FAT1, LRP1B and PTEN (all 11% vs 1%, p=0.0428, Fisher’s exact tests, Fig. 1, Supplementary Fig. S2a and Supplementary Table S3). On the other hand, TP53, the gene most frequently mutated in both MBCs and triple-negative IDC-NSTs, was mutated at comparable frequencies (69% vs 81%, p=0.2174, Fisher’s exact test, Fig. 1 and Supplementary Table S3). Since some of the genes found to be differentially mutated harbored primarily likely passenger mutations and mutations of indeterminate pathogenicity, we further restricted the comparison to likely pathogenic mutations. This analysis revealed seven genes in which MBCs more frequently harbored likely pathogenic mutations than triple-negative IDC-NSTs, with the differences in PIK3CA (29% vs 7%, p=0.0064, Fisher’s exact test), PIK3R1 (11% vs 0%, p=0.0114, Fisher’s exact tests), BCLAF1 (9% vs 0%, p=0.0360, Fisher’s exact test) and ARID1A, KMT2C, FAT1 and PTEN (all 11% vs 1%, p=0.0429, Fisher’s exact tests) remaining statistically significant (Fig. 1 and Supplementary Table S3).
As previously reported (6), the overall landscape of CNAs of MBCs was similar to that of triple-negative IDC-NSTs, with frequent copy number gains of 1q, 3q and 8q, copy number losses of 1p, 3p, 5q, 8p and 17p, as well as high-level copy number gains/amplifications of 8q (Supplementary Fig. S3a–d).
Given that two of the MBCs did not display a triple-negative phenotype, we repeated the analyses using the 33 triple-negative MBCs and came to similar conclusions in relation to the differences in the mutational repertoires and CNAs (Supplementary Table S3). In fact, when clustered with triple-negative MBCs and triple-negative IDC-NSTs from the TCGA study based on the repertoire of somatic mutations and CNAs, non-triple-negative MBCs were indistinguishable from either triple-negative MBCs or triple-negative IDC-NSTs on the genomic level (Supplementary Fig. S3e–g).
To gain additional insights into the mutational processes that underpin the somatic mutational landscape (35, 36) of MBCs and to define whether a subset of MBCs would display genomic scars indicative of homologous recombination (HR) DNA repair deficiency, we employed the algorithm deconstructSigs (37) and the assessment of LSTs (39), respectively. Of the 31 MBCs with at least 30 somatic mutations, eight (26%) and 11 (35%) displayed mutational signatures associated with aging (Signature 1) and with BRCA1/2 mutations (Signature 3). These results are consistent with our previous observation that 31% of MBCs displayed the BRCA-like genomic signature (17), and that MBCs were not different from the 16/60 (26%) and 25/60 (42%) triple-negative IDC-NSTs displaying Signatures 1 and 3, respectively (p>0.05, Fisher’s exact tests, Fig. 1). Indeed, the MBCs that showed a dominant Signature 3 were all LSThigh and were associated with the presence of large (>3bp) indels and/or microhomology at breakpoint junctions (35), suggesting these cases were likely HR-deficient (39). Of the cases displaying Signature 3, only MP1 harbored a pathogenic somatic BRCA2 mutation (heterozygous Q222fs) coupled with LOH of the wild-type allele (Supplementary Table S2). The two cases with the overall highest mutation rate, however, displayed mutational signatures consistent with microsatellite instability (MSI, Signature 6, META31) and APOBEC cytidine deaminase activity (Signatures 2 and 13, MTC03, Fig. 1 and Supplementary Fig. S4a). In META31, an analysis of microsatellite instability detection using MSIsensor (38) revealed a score of 9.5, well above the previously published threshold of 3.5, indicating MSI. Consistent with these observations, 14.8% of the somatic mutations found in META31 were insertions and deletions (indels) and META31 was found to harbor a frameshift K383fs mutation in MSH3, although it was not associated with LOH of the wild-type allele (Supplementary Table S2). In MTC03, low-level copy number gains were identified in APOBEC1 and APOBEC4, but no somatic mutations or amplifications/homozygous deletions were identified in AID (encoded by AICDA) or in the genes in the APOBEC family. Of the mutational signatures less frequently observed in breast cancer, we identified one case each harboring Signatures 5 and 30 (Fig. 1).
Taken together, our results demonstrated that, akin to triple-negative IDC-NSTs (9), MBCs constitute a genomically heterogeneous group of lesions that often display aging or BRCA1/2 signatures and frequently harbor TP53 mutations coupled with LOH of the wild-type allele. Importantly, however, MBCs were found to harbor PIK3CA activating mutations and mutations in several other known cancer genes, including PIK3R1, ARID1A, FAT1 and PTEN, more frequently than triple-negative IDC-NSTs.
Mutational landscape of MBCs associated with distinct histologic features
Given the distinct transcriptomic profiles associated with different histologic components of MBCs (17), as an exploratory, hypothesis-generating analysis, we sought to define whether MBCs of different histologic subtypes would differ in their repertoires of somatic genetic alterations. Based on the predominant metaplastic component present in each sample, 16, ten and nine were classified as chondroid, spindle cell and squamous, respectively (Supplementary Table S1). This analysis revealed that MBCs with chondroid metaplasia had numerically, though not statistically significant, higher number of somatic mutations (median 117, range 16–344) than MBCs with spindle cell metaplasia (median 70, range 15–315, p=0.0730, Mann-Whitney U-test), whereas the number of somatic mutations in MBCs with squamous metaplasia (median 73, range 31–287) did not differ from that of MBCs with chondroid or spindle cell metaplasia (p>0.05, Mann-Whitney U-tests).
TP53 was found to be significantly mutated in all three histologic subtypes of MBCs using the method by Youn and Simon (34) and in MBCs with chondroid or squamous metaplasia by MutSigCV (33) (chondroid 75%, spindle 50% and squamous 78%, Fig. 2, Supplementary Table S4). Additionally, PIK3CA was significantly mutated in spindle cell and squamous MBCs (60% and 44%, respectively), PTEN and USP5 in squamous MBCs (both 33%), as well as CHERP and CD84 in chondroid MBCs (25% and 19%, respectively, Fig. 2, Supplementary Fig. S2b, Supplementary Tables S4 and S5). Interestingly, PIK3CA mutations were not found in MBCs with chondroid metaplasia (0% vs 53% in non-chondroid MBCs, p<0.00001, Fisher’s exact test) and were significantly more frequent in MBCs with spindle cell metaplasia than MBCs of other histologic subtypes (60% vs 16%, p=0.0161, Fisher’s exact test, Fig. 2, Supplementary Fig. S2b and Supplementary Table S5). Mutations affecting distinct PI3K/AKT/mTOR pathway-related genes (e.g., PTEN and PIK3R1) were, however, identified in chondroid MBCs. On the other hand, mutations in CHERP, a gene involved in calcium homeostasis, were found exclusively in MBCs with chondroid metaplasia (25% vs 0% in non-chondroid MBCs, p=0.0348, Fisher’s exact test, Fig. 2 and Supplementary Table S5) and all of which were associated with the matrix-producing phenotype. Of the four mutations identified in CHERP, one was a likely pathogenic truncating Q320* mutation associated with LOH, with the other three of indeterminate significance (S166N and W472C missense mutations and an in-frame deletion Q797_S800del not associated with LOH, Fig. 2 and Supplementary Table S2). Mutations in USP5, all of which were clonal but were of indeterminate pathogenicity, were found only in MBCs with squamous metaplasia (33% vs 0% in non-squamous MBCs, p=0.0128, Fisher’s exact test, Supplementary Fig. S2b and Supplementary Table S5).
Figure 2. Repertoire of somatic non-synonymous mutations of metaplastic breast cancers (MBCs) of different histologic subtypes.
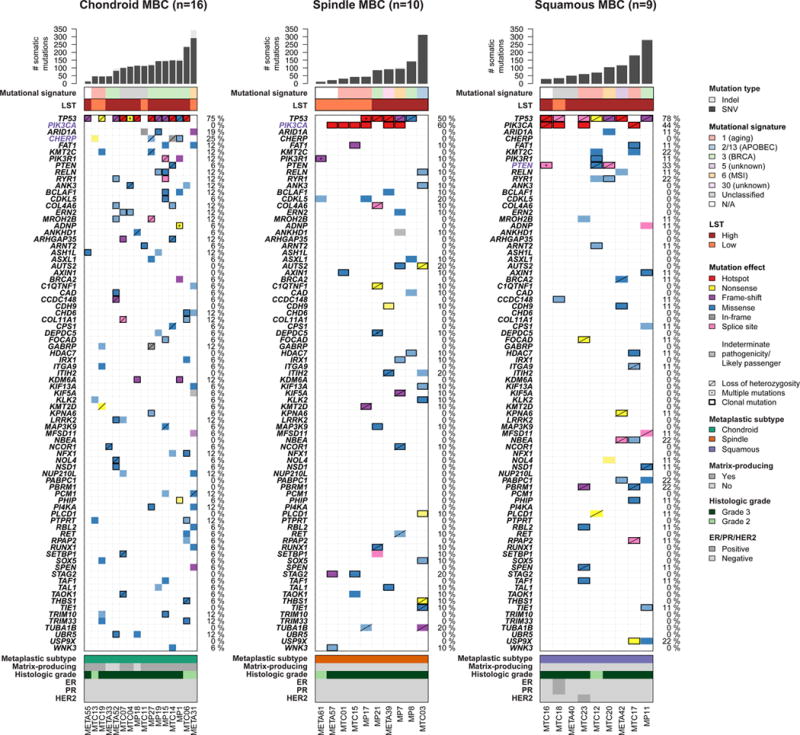
Non-synonymous somatic mutations identified in 16, ten and nine chondroid, spindle and squamous MBCs, respectively, by whole-exome sequencing are color-coded by their effect according to the legend, with hotspots (32) colored in red. Likely passenger mutations and mutations of indeterminate pathogenicity are marked using a hatched pattern. The presence of multiple non-synonymous mutations in the same gene is represented by an asterisk. The presence of loss of heterozygosity of the wild-type allele of a mutated gene is represented by a diagonal bar, and mutations found to be clonal by ABSOLUTE (24) are indicated by a black box. Genes recurrently mutated in MBCs and displaying at least one likely pathogenic mutation are presented. Genes highlighted in purple were significantly differentially altered in the given subtype of MBC. Percentages to the right of the mutation heatmaps indicate the percentage of cases affected by non-synonymous somatic mutations in a given gene. Bar charts (top) indicate the number of non-synonymous and synonymous somatic single nucleotide variants (SNVs), and the number of somatic insertions and deletions (indels) for each sample. The dominant mutational signatures (35, 36) were assigned using deconstructSigs (37). Large-scale transitions (LST)-high and -low status was determined in accordance with Popova et al (39).
A comparison of the mutational signatures found in the different subtypes revealed that all three MBC subtypes were heterogeneous in terms of the mutational signatures, with three, five and four distinct signatures being found in chondroid, spindle cell and squamous MBCs, respectively (Fig. 2). A numerically higher frequency of signature 3, associated with BRCA1/2 mutations, was observed in chondroid MBCs compared to other subtypes of MBCs (p=0.07, Fisher’s exact test). In terms of LSTs, 12 (75%), five (50%) and eight (89%) of chondroid, spindle cell and squamous MBCs, respectively, were LSThigh and there was no difference between its prevalence between the different histologic components assessed (p>0.05, Fisher’s exact test, Fig. 2). Our results demonstrate that the different morphologic subtypes of MBCs are similar in terms of LSTs, mutational signatures and TP53 mutation rates, but differ in the mutational frequency in other genes, including PIK3CA and PTEN.
Somatic mutations affecting genes in the Wnt and PI3K/AKT/mTOR signaling pathways are enriched in MBCs and result in pathway activation
To determine whether the somatic mutations identified in MBCs may represent functional recurrence, we mapped the mutated genes to KEGG pathways and Gene Ontology (Supplementary Table S6). Specifically, given that previous studies have identified PI3K/AKT/mTOR and Wnt pathway dysregulation in MBCs (10, 11, 13, 14), and that we observed more frequent FAT1 mutations in MBCs than in triple-negative IDC-NSTs (11% vs 1%, P=0.0428, Fisher’s exact test, Fig. 1 and Supplementary Table S3), we hypothesized that somatic mutations in these pathways would be more frequent in MBCs than in triple-negative IDC-NSTs. We observed that 57% (20/35) of MBCs harbored at least one somatic non-synonymous mutation in the KEGG PI3K/AKT/mTOR pathway, including PIK3CA (n=10), PIK3R1 and PTEN (each n=4), ERBB4, IGF1R, KIT, RICTOR, RPS6KB1 and RPTOR (each n=1), with 17/20 cases harboring at least one likely pathogenic mutation in the PI3K/AKT/mTOR pathway (Fig. 3a). The KEGG PI3K/AKT/mTOR pathway was significantly more frequently affected by non-synonymous and likely pathogenic mutations in MBCs than in triple-negative IDC-NSTs (57% vs 22% and 49% vs 20%, P=0.0004 and P=0.0058, respectively, Fisher’s exact tests, Supplementary Table S6). Similar results were obtained when these analyses were repeated using the PI3K signaling gene set from Gene Ontology (Supplementary Table S6). In addition, 51% (18/35) of MBCs harbored at least one non-synonymous mutation in FAT1 (33) or genes associated with the Gene Ontology canonical Wnt signaling pathway, including AXIN1, BCL9L, KDM6A, LRRK2, RYR2 and WNT5A (each n=2), as well as APC, FZD2, FZD7, LRP5, PORCN, SMO (each n=1), with 13/18 cases harboring at least one likely pathogenic mutation (Fig. 3a and Supplementary Table S6). Compared to triple-negative IDC-NSTs, MBCs significantly more frequently harbored somatic or likely pathogenic mutations in the Gene Ontology canonical Wnt pathway (including FAT1, 51% vs 28% and 37% vs 9%, P=0.0190 and P=0.0008, respectively, Fisher’s exact tests, Fig. 3a and Supplementary Table S6), with similar results obtained when the analyses were repeated using the KEGG canonical Wnt signaling gene set (including FAT1, Supplementary Table S6). As an additional exploratory and hypothesis-generating analysis, we examined whether MBCs of different histologic subtypes would differ in the repertoire of mutations affecting the PI3K/AKT/mTOR and Wnt signaling pathways. No difference was identified between the subtypes in regards to the KEGG PI3K/AKT/mTOR and KEGG/Gene Ontology Wnt pathways (P>0.05, Fisher’s exact test, Supplementary Table S6). Non-chondroid MBCs were enriched for mutations affecting the Gene Ontology PI3K/AKT/mTOR pathway (P=0.0065, Fisher’s exact test, Supplementary Table S6).
Figure 3. Repertoire of non-synonymous somatic mutations affecting genes associated with the PI3K/AKT/mTOR and Wnt pathways in metaplastic breast cancers (MBCs) and triple-negative invasive carcinomas of no special type (IDC-NSTs).

(a) Non-synonymous somatic mutations identified in 35 MBCs subjected to whole-exome sequencing and 69 triple-negative IDC-NSTs from TCGA (9) are color-coded by their effect according to the legend, with hotspots (32) colored in red. Likely passenger mutations and mutations of indeterminate pathogenicity are marked using a hatched pattern. Genes associated with the KEGG PI3K/AKT/mTOR and Gene Ontology Wnt pathways (including FAT1, see Supplementary Table S6) and mutated in at least one MBC or triple-negative IDC-NST are included and ordered in decreasing order of mutational frequency in MBCs. The presence of multiple non-synonymous mutations in the same gene is represented by an asterisk. For MBCs, the presence of loss of heterozygosity of the wild-type allele of a mutated gene is represented by a diagonal bar, and mutations found to be clonal by ABSOLUTE (24) are indicated by a black box. Percentages to the right indicate the percentage of cases affected by non-synonymous somatic mutations in a given gene. Gene names highlighted in purple were significantly more frequently altered in MBCs. (b) Overall survival of patients with MBCs or triple-negative IDC-NSTs from TCGA that harbored and did not harbor somatic non-synonymous or likely pathogenic mutations in the KEGG PI3K/AKT/mTOR and Gene Ontology Wnt pathways (including FAT1, see Supplementary Table S6) using the Kaplan–Meier method. TCGA, The Cancer Genome Atlas.
An exploratory, hypothesis-generating comparison of the clinicopathologic features of MBCs and triple-negative IDC-NSTs that harbored mutations in genes of the PI3K/AKT/mTOR or the Wnt signaling pathways did not reveal any difference other than MBCs with likely pathogenic mutations in the Wnt pathway having better overall survival than the equivalent group of triple-negative IDC-NSTs (p=0.037, log-rank test, Fig. 3b and Supplementary Table S6). Caution should be exercised in the interpretation of these findings, given the limited number of mutation-positive triple-negative IDC-NSTs (n=6).
A further exploratory, hypothesis-generating comparison between MBCs with and without Wnt pathway-related mutations revealed that MBCs with likely pathogenic mutations had greater extent of stromal tumor infiltrating lymphocytes (p=0.0175, Mann-Whitney U-test) and MBCs with non-synonymous mutations in the same pathway had lower rates of distant metastasis (p=0.0076, Fisher’s exact test, Supplementary Table S6) and better overall survival (p=0.003, log-rank test, Fig. 3b). Mutation status in the Wnt pathway, however, was not an independent prognostic indicator in a multivariate analysis incorporating lymphocytic infiltration (data not shown). For the PI3K/AKT/mTOR pathway, mutation-positive MBCs had greater extent of chondroid metaplastic elements and were older than mutation-negative MBCs (p=0.0380 and p=0.0048, Mann-Whitney U-tests, Supplementary Table S6). No other differences in the clinicopathologic features between MBCs with and without mutations in either pathway were observed (Supplementary Table S6).
Given that the genomic analysis of MBCs revealed an enrichment of mutations affecting genes in the Wnt and PI3K/AKT/mTOR pathways, to define whether these mutations would result in activation of the respective signaling pathways, we subjected 30 and 24 MBCs for which sufficient RNA was available to the Wnt and PI3K-AKT signaling pathway RT2 profiler PCR arrays, respectively (Qiagen, Supplementary Table S1). This analysis revealed that MBCs with somatic non-synonymous mutations or likely pathogenic mutations in genes associated with the canonical Wnt pathway had significantly higher pathway activation scores than wild-type MBCs (p=0.0136 and p=0.0133, respectively, Mann-Whitney U-tests, Fig. 4a–b). Similarly, the RT2 profiler PCR assay revealed higher levels of PI3K/AKT and mTOR signaling pathway activation in MBCs with somatic non-synonymous mutations or likely pathogenic mutations in genes in the PI3K/AKT/mTOR pathway (p=0.0173 and p=0.0323, respectively, for the PI3K/AKT pathway and p=0.0034 and p=0.0067, respectively, for the mTOR pathway, Mann-Whitney U-tests, Fig. 4c–f). Taken together, these results provide evidence that mutations affecting genes related to the canonical Wnt and PI3K/AKT/mTOR signaling pathways result in the aberrant activation of the pathways in MBCs.
Figure 4. Activation of Wnt/β-catenin and PI3K/AKT/mTOR pathways in metaplastic breast cancers (MBCs).
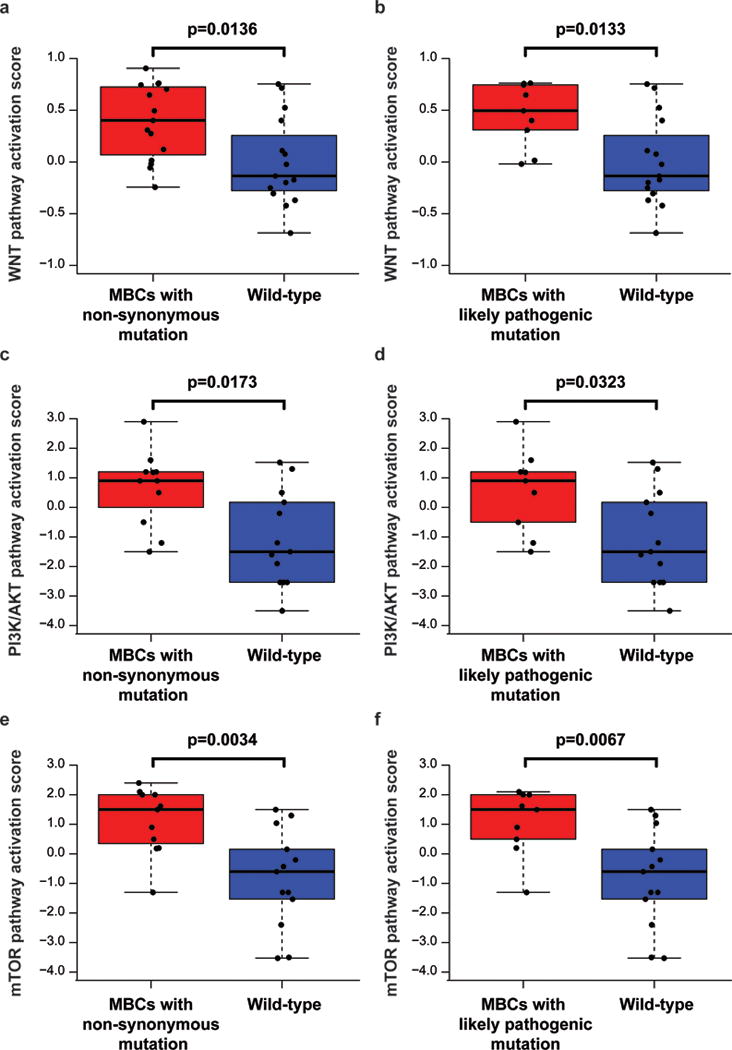
Wnt signal transduction pathway activity scores (a) between MBCs with somatic mutations in genes related to the Wnt signaling pathway (Gene Ontology GO:0060070, including FAT1) and wild-type MBCs and (b) between MBCs with likely pathogenic mutations in genes related to the Wnt signaling pathway (Gene Ontology GO:0060070, including FAT1) and wild-type MBCs (Supplementary Table S6). PI3K/AKT pathway activation scores (c) between MBCs with somatic mutations in genes related to the PI3K/AKT/mTOR (KEGG pathways hsa04150, hsa05151 and hsa04012) and wild-type MBCs and (d) between MBCs with likely pathogenic mutations in genes related to the PI3K/AKT/mTOR (KEGG pathways hsa04150, hsa05151 and hsa04012) and wild-type MBCs, and mTOR pathway activation scores (e) between MBCs with somatic mutations in genes related to the PI3K/AKT/mTOR (KEGG pathways hsa04150, hsa05151 and hsa04012) and wild-type MBCs and (f) between MBCs with likely pathogenic mutations in genes related to the PI3K/AKT/mTOR (KEGG pathways hsa04150, hsa05151 and hsa04012) and wild-type MBCs (Supplementary Table S6). Significance was assessed using Mann-Whitney U-test.
DISCUSSION
Here we demonstrate that, akin to triple-negative IDC-NSTs, MBCs comprise a group of complex, genetically heterogeneous tumors. In terms of mutation burden, mutational signatures, LSTs and TP53 mutational frequency, MBCs are heterogeneous and similar to triple-negative IDC-NSTs. Despite the heterogeneity and similarities with triple-negative IDC-NSTs, PIK3CA, PIK3R1 and PTEN were found to be significantly mutated in MBCs, and MBCs significantly more frequently harbored likely pathogenic mutations in these genes than triple-negative IDC-NSTs, in agreement with previous studies reporting on recurrent alterations affecting PI3K pathway-related genes in MBCs (10, 11). In fact, 57% of MBCs harbored at least one non-synonymous somatic mutation affecting PI3K/AKT/mTOR pathway-related genes, compared to 22% of triple-negative IDC-NSTs. Given that most of these clonal mutations detected in MBCs are known to dysregulate the PI3K/AKT/mTOR pathway and pathway activation was observed in mutation-positive MBCs based on the RT2 profiler analysis, our findings are consistent with the notion that this pathway is frequently dysregulated in MBCs (10, 11). These results also provide a genetic basis for the efficacy of the mTOR inhibitor temsirolimus in a recent study in which 6/19 (32%) metastatic MBC patients treated with temsirolimus and anthracycline-based regimen achieved complete/partial response (11).
MBCs also more frequently harbored mutations affecting the Wnt pathway than triple-negative IDC-NSTs. Consistent with studies by our group and others (10, 14), Wnt pathway activation in MBCs was not driven by CTNNB1 somatic mutations; rather, 51% of MBCs harbored somatic mutations in FAT1 and/or canonical Wnt pathway genes other than CTNNB1. Previous analyses demonstrated that truncating mutations in FAT1 and missense mutations in the cytoplasmic domain promote Wnt signaling and tumorigenesis (16). In our cohort, we identified four (11%) MBCs with FAT1 mutations, including a clonal heterozygous frameshift mutation in the cadherin repeats (V2168fs) and a clonal missense mutation in the cytoplasmic domain (E4283Q), in a pattern reminiscent of the previously reported mutations (16). Moreover, the mutations in diverse Wnt-related genes (e.g., APC, FZD2 and WNT5A) suggest that Wnt pathway activation constitutes a convergent phenotype in MBCs. This notion was further corroborated by the RT2 profiler analysis, which revealed increased signaling pathway activation in MBCs harboring Wnt pathway-related mutations than those without. It should be noted, however, that a subset of MBCs devoid of Wnt pathway genetic alterations detected by the approaches employed also displayed pathway activation, suggesting that alternative genetic or epigenetic activation mechanisms, such as methylation of CDH1, APC or of genes in the secreted frizzled-related protein (SFRP) and the Dickkopf (DKK) families (42, 43), may be operational in these MBCs. Our findings, however, provide a genetic basis for Wnt activation in a subset of MBCs and provide the rationale for the potential use of Wnt pathway small molecule inhibitors (44) in a substantial proportion of MBCs. Further studies to define optimal biomarkers and/or a functional Wnt pathway activation assays to identify the MBCs that would benefit from these targeted agents are warranted.
In this study, 74% (26/35) of MBCs and 71% (44/62) of triple-negative IDC-NSTs were LSThigh on the basis of a whole-exome based research version of LST assessment, and 17% (6/35) and 23% (14/62) of MBCs and triple-negative IDC-NSTs, respectively, harbored genetic alterations resulting in the bi-allelic inactivation of HR-related genes (Supplementary Fig. S4b). Importantly, all MBCs harboring bi-allelic inactivation of HR-related genes displayed high LST scores. It is plausible that other genetic and/or epigenetic alterations not detected by the approach employed (e.g. loss-of-function somatic rearrangements, genetic alterations affecting non-coding regulatory elements and epigenetic mechanisms) may contribute to the LSThigh phenotype of MBCs. Based on the relatively frequent alterations affecting HR-related genes and the similarly high frequency of LSThigh in MBCs and triple-negative IDC-NSTs, clinical studies testing the efficacy of platinum salts and PARP inhibitors, and predictive tests for these agents should also include patients with MBC together with those with triple-negative IDC-NST (45–47). Importantly, however, the clinical utility of LST and genomic surrogates that include LST to detect HR deficiency and predict sensitivity to platinum salts or PARP inhibitors remains to be fully established (45–48), especially for breast cancers lacking BRCA1 or BRCA2 mutations.
The analysis of the repertoire of somatic genetic alterations in the distinct histologic subtypes of MBC profiled, namely chondroid, spindle cell or squamous, revealed that whilst 44% of MBCs with chondroid metaplasia harbored somatic mutations in the PI3K/AKT/mTOR pathway, none affected PIK3CA. This is in stark contrast to MBCs with spindle and squamous metaplasia, where PIK3CA mutations were detected in 60% and 44% of cases, respectively. Furthermore, we found that 25% of MBCs with chondroid metaplasia harbored CHERP mutations. CHERP encodes the calcium homeostasis endoplasmic reticulum protein. Perturbed calcium homeostasis has important implications in cell migration and proliferation (reviewed in (49)). Of note, however, three of the four CHERP mutations identified were subclonal and only one was predicted to be likely pathogenic. Additionally, three of nine MBCs with squamous metaplasia were found to harbor clonal mutations of indeterminate pathogenicity in USP5, the gene encoding ubiquitin carboxyl-terminal hydrolase 5, a ubiquitin-specific protease required for double-strand break repair (50), and two of these cases were classified as LSThigh. Notably, CHERP and USP5 mutations were not found in triple-negative IDC-NSTs from TCGA. Our exploratory analysis has generated the hypothesis that specific histologic subtypes of MBCs may be underpinned by distinct repertoire of somatic mutations, and warrants further investigation of our findings in the context of a large, multi-institutional collaborative effort.
Our study has several limitations. First, owing to the relative rarity of MBCs, our sample size is limited. It should be noted, however, that our study constitutes the largest series of MBCs subjected to whole-exome sequencing to date. Second, although we have not identified pathognomonic somatic mutations that define each subtype of MBCs, we cannot rule out that whole-genome sequencing and/or RNA-sequencing may result in the identification of either non-coding genetic alterations or fusion genes that define each subtype. Further studies are warranted to define the genetic and/or epigenetic basis of the different subtypes of the disease.
Despite these limitations, our study revealed that MBCs more frequently harbor somatic mutations affecting the PI3K/AKT/mTOR and canonical Wnt signaling pathways than triple-negative IDC-NSTs. Our results support the recent pre-clinical observations (44) and clinical trial results (11) that Wnt and PI3K pathway inhibition may be beneficial for a subset of patients with these aggressive and chemotherapy-resistant triple-negative breast cancers.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Metaplastic breast carcinoma (MBC) is a rare histologic type of breast cancer that typically lacks the expression of estrogen receptor and progesterone receptor and HER2 overexpression/gene amplification (i.e. triple-negative). MBCs do not respond well to conventional chemotherapy and have been reported to be associated with a worse prognosis than triple-negative invasive ductal carcinomas of no special type (IDC-NSTs). Although similar in the mutational frequency in TP53, our study reveals that MBCs are genetically distinct from triple-negative IDC-NSTs, with more frequent mutations in PIK3CA, PIK3R1, PTEN and Wnt pathway genes. In fact, 57% and 51% of MBCs harbored somatic mutations affecting PI3K/AKT/mTOR pathway and Wnt pathway related genes, respectively. Our study provides a molecular basis for the recent pre-clinical and clinical observations that Wnt and PI3K/AKT/mTOR pathway inhibition may be beneficial for a subset of patients with these aggressive and chemotherapy-resistant triple-negative breast cancers.
Acknowledgments
The authors would like to acknowledge Dr Deepu Alex and Dr Snjezana Dogan for their assistance with the HER2 FISH analysis.
Financial support: JSR-F is funded in part by the Breast Cancer Research Foundation. SP is currently funded by Swiss National Foundation (Ambizione grant number PZ00P3_168165). Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: F.C. Geyer, L. Norton, A. Vincent-Salomon, B. Weigelt and J.S. Reis-Filho.
Development of methodology: C.K.Y. Ng, S. Piscuoglio, N. Riaz, B. Weigelt and J.S. Reis-Filho.
Acquisition, analysis and interpretation of data: C.K.Y. Ng, S. Piscuoglio, F.C. Geyer, K.A. Burke, F.A. Pareja, C.A. Eberle, R.S. Lim, R. Natrajan, N. Riaz, O. Mariani, A. Vincent-Salomon, Y.H. Wen, B. Weigelt and J.S. Reis-Filho.
Writing, review, and/or revision of the manuscript: C.K.Y. Ng, S. Piscuoglio, F.C. Geyer, K.A. Burke, F.A. Pareja, C.A. Eberle, R.S. Lim, R. Natrajan, N. Riaz, O. Mariani, L. Norton, A. Vincent-Salomon, Y.H. Wen, B. Weigelt and J.S. Reis-Filho.
Study supervision: L. Norton, A. Vincent-Salomon, B. Weigelt and J.S. Reis-Filho.
References
- 1.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ. WHO Classification of Tumours of the Breast. Lyon: IARC Press; 2012. [Google Scholar]
- 2.Weigelt B, Eberle C, Cowell CF, Ng CKY, Reis JS. Metaplastic breast carcinoma: more than a special type. Nature Reviews Cancer. 2014;14:147–8. doi: 10.1038/nrc3637. [DOI] [PubMed] [Google Scholar]
- 3.Jung SY, Kim HY, Nam BH, Min SY, Lee SJ, Park C, et al. Worse prognosis of metaplastic breast cancer patients than other patients with triple-negative breast cancer. Breast Cancer Res Treat. 2010;120:627–37. doi: 10.1007/s10549-010-0780-8. [DOI] [PubMed] [Google Scholar]
- 4.Nelson RA, Guye ML, Luu T, Lai LL. Survival outcomes of metaplastic breast cancer patients: results from a US population-based analysis. Ann Surg Oncol. 2015;22:24–31. doi: 10.1245/s10434-014-3890-4. [DOI] [PubMed] [Google Scholar]
- 5.Hennessy BT, Giordano S, Broglio K, Duan Z, Trent J, Buchholz TA, et al. Biphasic metaplastic sarcomatoid carcinoma of the breast. Ann Oncol. 2006;17:605–13. doi: 10.1093/annonc/mdl006. [DOI] [PubMed] [Google Scholar]
- 6.Horlings HM, Weigelt B, Anderson EM, Lambros MB, Mackay A, Natrajan R, et al. Genomic profiling of histological special types of breast cancer. Breast Cancer Res Treat. 2013;142:257–69. doi: 10.1007/s10549-013-2740-6. [DOI] [PubMed] [Google Scholar]
- 7.Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–23. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reis-Filho JS, Pinheiro C, Lambros MB, Milanezi F, Carvalho S, Savage K, et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J Pathol. 2006;209:445–53. doi: 10.1002/path.2004. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moulder S, Helgason T, Janku F, Wheler J, Moroney J, Booser D, et al. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann Oncol. 2015;26:1346–52. doi: 10.1093/annonc/mdv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes MJ, Thomas D, Emmons A, Giordano TJ, Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res. 2008;14:4038–44. doi: 10.1158/1078-0432.CCR-07-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, et al. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. 2011;24:209–31. doi: 10.1038/modpathol.2010.205. [DOI] [PubMed] [Google Scholar]
- 14.Lacroix-Triki M, Geyer FC, Lambros MB, Savage K, Ellis IO, Lee AH, et al. beta-catenin/Wnt signalling pathway in fibromatosis, metaplastic carcinomas and phyllodes tumours of the breast. Mod Pathol. 2010;23:1438–48. doi: 10.1038/modpathol.2010.141. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45:253–61. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weigelt B, Ng CK, Shen R, Popova T, Schizas M, Natrajan R, et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected] Mod Pathol. 2015;28:340–51. doi: 10.1038/modpathol.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–10. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 19.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–95. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 21.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138:241–56. doi: 10.5858/arpa.2013-0953-SA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CK, Perez-Ordonez B, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–9. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 26.Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–7. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–81. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–62. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 40.Lee H, Jung SY, Ro JY, Kwon Y, Sohn JH, Park IH, et al. Metaplastic breast cancer: clinicopathological features and its prognosis. J Clin Pathol. 2012;65:441–6. doi: 10.1136/jclinpath-2011-200586. [DOI] [PubMed] [Google Scholar]
- 41.Reis-Filho JS, Milanezi F, Steele D, Savage K, Simpson PT, Nesland JM, et al. Metaplastic breast carcinomas are basal-like tumours. Histopathology. 2006;49:10–21. doi: 10.1111/j.1365-2559.2006.02467.x. [DOI] [PubMed] [Google Scholar]
- 42.Prasad CP, Mirza S, Sharma G, Prashad R, DattaGupta S, Rath G, et al. Epigenetic alterations of CDH1 and APC genes: relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008;83:318–25. doi: 10.1016/j.lfs.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98:1147–56. doi: 10.1038/sj.bjc.6604259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jang GB, Hong IS, Kim RJ, Lee SY, Park SJ, Lee ES, et al. Wnt/beta-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015;75:1691–702. doi: 10.1158/0008-5472.CAN-14-2041. [DOI] [PubMed] [Google Scholar]
- 45.Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33:1902–9. doi: 10.1200/JCO.2014.57.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodler ET, Kurland BF, Griffin M, Gralow JR, Porter P, Yeh RF, et al. Phase I Study of Veliparib (ABT-888) Combined with Cisplatin and Vinorelbine in Advanced Triple-Negative Breast Cancer and/or BRCA Mutation-Associated Breast Cancer. Clin Cancer Res. 2016;22:2855–64. doi: 10.1158/1078-0432.CCR-15-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22:3764–73. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.http://www.ascopost.com/issues/february-25-2015/tnt-trial-supports-platinums-in-brca-mutated-breast-cancer/
- 49.Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol. 2014;171:945–60. doi: 10.1111/bph.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakajima S, Lan L, Wei L, Hsieh CL, Rapic-Otrin V, Yasui A, et al. Ubiquitin-specific protease 5 is required for the efficient repair of DNA double-strand breaks. PLoS One. 2014;9:e84899. doi: 10.1371/journal.pone.0084899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.