

Abstract
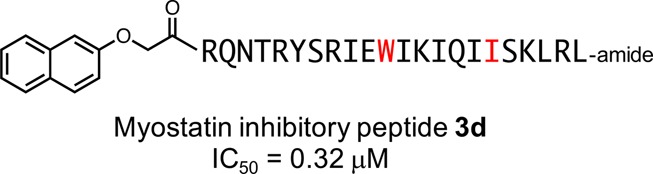
Myostatin, a negative regulator of skeletal muscle growth, is a promising target for treating muscle atrophic disorders. Recently, we discovered a minimal myostatin inhibitor 1 (WRQNTRYSRIEAIKIQILSKLRL-amide) derived from positions 21–43 of the mouse myostatin prodomain. We previously identified key residues (N-terminal Trp21, rodent-specific Tyr27, and all aliphatic amino acids) required for effective inhibition through structure–activity relationship (SAR) studies based on 1 and characterized a 3-fold more potent inhibitor 2 bearing a 2-naphthyloxyacetyl group at position 21. Herein, we performed 1-based SAR studies focused on all aliphatic residues and Ala32, discovering that the incorporations of Trp and Ile at positions 32 and 38, respectively, enhanced the inhibitory activity. Combining these findings with 2, a novel peptide 3d displayed an IC50 value of 0.32 μM, which is 11 times more potent than 1. The peptide 3d would have the potential to be a promising drug lead to develop better peptidomimetics.
Keywords: Inhibitor, peptide, muscle atrophic disorder, myostatin, structure−activity relationship
Myostatin is a member of the transforming growth factor β (TGF-β) superfamily and is specifically expressed in skeletal muscle to negatively regulate muscle growth.1 Previous reports showed that a genetic defect in myostatin can significantly increase muscular mass, whereas its overexpression induces cachexia.1,2 Hence, myostatin is a worthy target for treating muscle atrophic disorders, such as muscular dystrophy, cancer cachexia, and sarcopenia. A variety of myostatin inhibitory strategies have been investigated to induce muscular growth, including neutralizing antibodies,3 prodomain proteins,4,5 soluble decoys of active type II receptors (ActRII),6 interacting proteins (growth and differentiation factor-associated serum protein),7 follistatin,8 and follistatin-related protein.5,9 Neutralizing antibodies increased muscle mass and strength in Duchenne muscular dystrophy (DMD) model mdx mice,3 whereas treatment of a decoy ActRII prevented muscle wasting and cancer-induced cardiac atrophy in tumor-bearing mice.6
A prodomain protein derived from a myostatin precursor has been shown to have inhibitory properties through the formation of an inactive complex with mature myostatin in the extracellular matrix and in serum.2,5,10 In 2010, Walton et al. reported that aliphatic (Ile and Leu) residues in the N-terminal α-helical region of the prodomain plays a key role in TGF-β1 inactivation through the formation of a complex.11 The crystal structure of pro-TGF-β1 revealed that the N-terminal α-helical region of the prodomain was positioned in the type I receptor-binding pocket of the mature TGF-β1.12 This interactive domain is highly conserved throughout the TGF-β superfamily. A study of the myostatin prodomain, by Jiang et al.,4 found that a glutathione S-transferase fusion protein bearing a 74-mer fragment (positions: 19–92 in the prodomain) displayed inhibitory properties. Recently, our group identified a mouse myostatin prodomain-derived minimum peptide 1 (23 residues, including a conserved α-helical region spanning positions 21–43, Figure 1, derivative 7 in ref. (13)) based on an earlier discovery of a core inhibitory fragment (p29) consisting of 29 residues (position: 19–47).14 These prodomain-derived peptides, including p29, significantly increased muscle mass in mouse model of muscular dystrophy. Next, we carried out a structure–activity relationship (SAR) study of 1 to develop a potent peptidic inhibitor. Ala scanning clarified the importance of the Trp (position 21), the rodent-specific Tyr (position 27), and all aliphatic residues (positions 30, 33, 35, 37, 38, 41, and 43) in 1 using a cell-based luciferase reporter assay.15 Our SAR study focused on the N-terminal Trp of 1 identified the derivative 2 (Figure 1, derivative 36 in ref. (16)), which was three times as potent as 1, and registered an IC50 value of 1.19 μM on the reporter assay.16 Additionally, we demonstrated that the aromatic hydrophobic side chain structures of Tyr at position 27 were important for achieving effective myostatin inhibitory activity.15 Hence, the human-derived peptide possessing Ser at position 27 showed a weaker inhibitory activity.13 However, a SAR study directed specifically toward the aliphatic residues (Ile30, Ile33, Ile35, Ile37, Leu38, Leu41, and Leu43) among the key residues of 1 has not yet been carried out, and the function of the central Ala32 residue remains unclear. Herein, we performed structural modifications directed toward all hydrophobic residues and Ala32 of 1 to obtain more potent myostatin inhibitors.
Figure 1.

Structures and myostatin inhibitory activities (IC50 values) of the peptides 1 and 2.13,15 The numbers above each amino acid indicate the position in the prodomain sequence of mouse myostatin. X = 2-naphthyloxyacetyl.
Because the previous Ala-scanning results demonstrated that both Ile and Leu (Ile30, Ile33, Ile35, Ile37, Leu38, Leu41, and Leu43) were important residues,15 the number of structures that required testing during optimization were limited. We chose to examine the effects of modifying the amino acid branching structure by inverting Ile with Leu (and Leu with Ile) in the peptide 1 sequence. This was initially investigated by synthesizing a series of double-substituted peptides (experimental detail of peptide synthesis is described in the Supporting Information), as shown in Figure 2A (the first five sequences are I(30,33)L, I(33,35)L, I(35,37)L, L(38,41)I, and L(41,43)I). Proximal Ile and Leu residue pairs were swapped (Ile30 and Ile33 were swapped, Ile33 and Ile35, and so on). Figure 2B shows the results obtained from these double-substituted peptides using the luciferase reporter assay, compared to peptide 1 and mouse myostatin prodomain (prodomain) as a reference (experimental detail of cell-based assay is described in the Supporting Information). Interestingly, all peptides displayed weaker myostatin inhibitory activities, with the exception of the L(38,41)I derivative, which was about twice as effective at a 3 μM concentration.
Figure 2.

(A) Structures of the aliphatic residue-substituted peptides. The numbers above each amino acid indicate the position in the prodomain sequence of mouse myostatin. (B,C) Luciferase reporter assay experiments determined the activities of the double-substituted peptides (B) or the Leu-substituted peptides (C) toward myostatin inhibition relative to peptide 1. Peptide concentration: 3 μM. Results are presented as mean values ± SD (n = 3).
These results led to the synthesis of peptides modified specifically at positions 38, 41, and 43, at which the Leu residues were individually substituted with Ile, to identify which of these positions were most important (Figure 2A, last three sequences: L38I, L41I, and L43I). The in vitro luciferase reporter assay revealed that a single change from Leu to Ile at position 38 (L38I) was sufficient to provide the previously observed increase in myostatin inhibition at a 3 μM peptide concentration (Figure 2C). Additionally, substitutions of Leu38 to Phe or Trp (Figure S1A) did not allow improvement of the inhibitory potency over L38I, although the myostatin inhibitory activities of these peptides proved to be better than that of peptide 1 in the reporter assay (Figure S1B). These results suggested that Ile at position 38 was suitable for effective myostatin inhibition. We additionally investigated the individual substitutions of Ile at positions 30, 33, 35, and 37 with Val or Phe by synthesizing a series of peptides, as shown in Figure S2A. The inhibitory activities of I30F and I37F were similar to that of 1; however, all other peptides showed weaker inhibitory activities compared to 1 (Figure S2B).
With the results of the SAR study of the C-terminal region in hand, we next examined the incorporation of a variety of amino acids at position 32, as substitutes for Ala. These substitutions were not investigated in our more recent Ala-scanning study.15 Ala32 was replaced with a variety of amino acids possessing various side chains and steric hindrance or polarity (Figure 3A). All of the modified peptides were subjected to the luciferase reporter assay. Interestingly, all of the tested peptides provided better inhibitory activities than the original peptide 1 (Figure 3B), implying that there is a relatively large chemical space available at this position. The introduction of valine, tryptophan, or glutamic acid at position 32 gave the best results, with comparable myostatin inhibitory potencies. Trp was eventually selected as the residue of choice at this position to occupy chemical space, to provide a better hydrophobic surface compared to Val,17 and to conserve the total charge of the peptide.
Figure 3.

(A) Structures of the Ala-substituted peptides. The numbers above each amino acid indicate its position in the prodomain sequence of mouse myostatin. (B) Luciferase reporter assay to determine the activities of the Ala-substituted peptides toward myostatin inhibition, relative to peptide 1. Peptide concentration: 1 μM. Results are presented as mean values ± SD (n = 3).
Next, combinations of the modifications were systematically examined. Previous studies revealed that the 2-naphthyloxyacetyl group at the N-terminal position was a suitable replacement for Trp21,15 and single-point modifications described herein established that Trp32 and Ile38 were appropriate substitutions. Consequently, peptides comprising two and three modifications were synthesized, and the sequences are shown in Figure 4A. The reporter assay results demonstrated that the peptide with all three modifications (3d) was the best peptide candidate, with a relative luciferase activity at a 1 μM concentration on par with that of the mouse prodomain at a 10 nM concentration (Figure 4B).
Figure 4.

(A) Structures of the double- or triple-substituted peptides. The numbers above each amino acid indicate its position in the prodomain sequence of mouse myostatin. (B) The luciferase reporter assay determined the activities of the double- and triple-substituted peptides toward myostatin inhibition relative to peptide 1. Peptide concentration: 1 μM. Results are presented as mean values ± SD (n = 3).
To confirm that peptide 3d was a more potent myostatin inhibitor, a dose-dependent study was performed comparing peptides 1 and 3d at concentrations between 0.04 and 20 μM (peptide 1, 0.63–20 μM; peptide 3d, 0.04–5 μM) (Figure 5). The difference of activity between the two peptides is significant. At the highest tested concentration (5 μM) for peptide 3d, peptide 1 inhibited only 75% of the luciferase activity, whereas 3d provided 100% inhibition. At 2.5 μM, this difference increased to only 25% inhibition for 1 compared to 3d at 100%. The greatest gap was observed at 1.25 μM (almost no inhibition for 1, 90% for 3d). A lower concentration (0.63 μM) of 1 did not provide any inhibition in the luciferase reporter assay. By contrast, 3d was a much more satisfactory inhibitor: 75% at 0.63 μM, 50% at 0.32 μM, and 20% at 0.16 μM. Compound 3d failed to detectably inhibit myostatin at dose levels of 0.08 μM and below. These data revealed that the IC50 values for peptides 1 and 3d were calculated to be 3.56 ± 0.25 and 0.32 ± 0.05 μM, respectively (95% confidence intervals for IC50 are shown in Table S1, Supporting Information). The inhibitory potency of peptide 2 (IC50 value: 1.19 μM) showed a 3.7-fold increase by Trp32- and Ile38-substitutions.16
Figure 5.
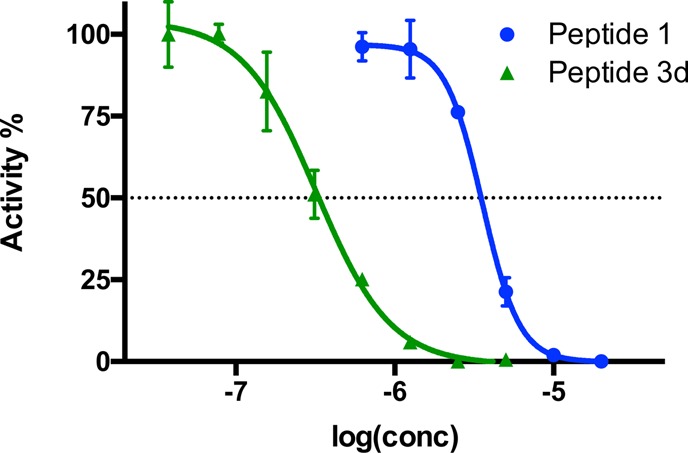
Normalized dose-dependent myostatin inhibitory activities of peptides 1 and 3d at respective doses of 0.63, 1.25, 2.5, and 5 μM. The results obtained from doses of 0.04, 0.08, 0.16, and 0.32 μM for peptide 3d, and 10 and 20 μM for peptide 1 are included for comparison (the lowest concentrations correspond to the most negative values on the log scale). Concentrations are given as the logarithmic values along the X-axis, and the percentage of luciferase activity is indicated on the Y-axis. A four-parametric nonlinear regression was used as a model for curve fitting (see the Experimental Section for more details, Supporting Information). Each experiment was carried out in triplicate. Results are presented as mean values ± SD (n = 3).
In addition, we measured circular dichroic spectra of peptides 1 and 3d in 20 mM sodium phosphate buffer (pH 7.4) containing 10% 2,2,2-trifluoroethanol (Figure S3). As reported previously,13,151 showed a propensity to form α-helix structure with a positive band at 192 nm and negative bands at 208 and 222 nm. We reported that the α-helical structure formed around positions 32–36 and two hydrophobic faces are important for effective myostatin inhibition of 1.15 In the present study, 3d displayed a propensity to form a β-sheet structure due to the appearance of a negative band near 216 nm and decrease of positive band at 192 nm compared with 1 (Figure S3). This result implies that 3d may provide more adequate hydrophobic interaction with the β-sheet structure-forming property to obtain a more potent inhibitory activity. Further investigation to address an accurate 3D-binding mode is ongoing.
Finally, we examined whether peptide 3d could increase the total weight of a skeletal muscle. As reported previously,13 the peptide solution (30 nmol) or saline were intramuscularly administered into the tibialis anterior (TA) muscle of mdx mice (5-week-old males) at days 0 and 14. At day 42, the muscles were collected and weighed. In mdx mice, peptide 3d increased the weight of the TA muscles by 10–19% compared to saline-treated muscles (Figure 6A,B). Despite in vitro improvement, the in vivo results were similar to those obtained from peptide 1.13 We additionally investigated the possibility that our peptide 3d increased the muscular mass of the wild-type ICR mice (5-week-old males). The same protocol was used to evaluate the effects of 3d in the TA and gastrocnemius (GAS). In ICR mice, peptide 3d increased the weight of the TA and GAS muscles by 10–34% and 11–35% compared to saline-treated muscles, respectively (Figure 6C,D). In a prior report of p29 (20 nmol, single intramuscular dose), the TA muscle weight of wild-type mice was increased by 10.6%, smaller than the 20.6% obtained in caveolin 3-deficient limb-girdle muscular dystrophy 1C model mice.14 In the present study, we confirmed similar in vivo efficacies of the peptide derivative 3d both in the wild-type and pathological model mice. Moreover, histological analyses based on hematoxylin and eosin staining were carried out in mdx mice (Figure 6E,F), suggesting that 3d induced an increase of muscle fiber sizes compared with saline. An optimized dosing schedule of 3d is currently under investigation.
Figure 6.

(A) Photographs of the tibialis anterior (TA) of mdx mice (n = 2) at day 42 after treatment with 3d (left TA) or saline (right TA) at days 0 and 14. The peptide solution (0.75 mM peptide 3d in saline, 40 μL) was intramuscularly injected into the TA muscle. (B) Weight of the treated muscles in mdx mice (n = 2). Respective increases in ratios compared to control values (= 100) are provided above the red bars. (C) Photographs of the TA and gastrocnemius (GAS) of ICR mice (n = 2) at day 42 after treatment with 3d (left TA and GAS) or saline (right TA and GAS) at days 0 and 14. The peptide solution (0.75 mM peptide 3d in saline, 40 μL) was intramuscularly injected into the TA and GAS muscles. (D) Weight of the treated muscles in ICR mice (n = 2). Respective increases in ratios compared to control values (= 100) are provided above the red bars. (E) Hematoxylin and eosin staining of GAS muscles of mdx mice (n = 3) at day 42 after treatment with 3d or saline at days 0 and 14. Scale bars = 50 μm. (F) Cross-sectional areas and distributions of myofiber sizes in the 3d or saline treated muscles.
In conclusion, we synthesized a variety of myostatin inhibitory peptides, focusing particularly on a set of key positions in the sequence of mouse myostatin prodomain-derived minimum peptide 1. This was determined through amino acid scanning and allowed to produce myostatin inhibitory peptides with two or three relevant modifications (positions 21, 32, and 38). These peptides were all tested in an in vitro luciferase reporter assay to evaluate their inhibitory potencies. The results revealed that the best myostatin inhibitory peptide 3d was the one with three modifications. A dose-dependent study of 3d against the original peptide 1 revealed IC50 values of 3.56 ± 0.25 and 0.32 ± 0.05 μM, respectively, indicating that peptide 3d was 11 times more potent than its parent peptide, with a submicromolar IC50. Peptide 3d induced muscular growth in both DMD-model mdx as well as wild-type ICR mice. Undoubtedly, this peptide is a promising lead for myostatin inhibition drug development efforts targeted to combat muscular wasting.
Acknowledgments
This research was supported by the Japan Society for the Promotion of Sciences (JSPS) KAKENHI, including Grants-in-Aid for Scientific Research (B) 15H04658 (to K.T. and Y.H.) and JSPS Fellows 16F16413 (to Y.H.), MEXT-supported Program for the Strategic Research Foundation at Private Universities, and Intramural Research Grant (29-4) for Neurological and Psychiatric Disorders on NCNP (to Y.H.). The authors thank Mr. Shota Takayama for peptide synthesis and cell-based assay.
Glossary
ABBREVIATIONS
- ActRII
activin type II receptor
- DMD
Duchenne muscular dystrophy
- GAS
gastrocnemius
- SAR
structure–activity relationship
- TA
tibialis anterior
- TGF-β
transforming growth factor-β
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00168.
Materials, experimental procedure, analytical data for all peptide derivatives, analytical HPLC chromatograms, Table S1, and Figures S1–S3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- McPherron A. C.; Lawler A. M.; Lee S.-J. Regulation of skeletal muscle in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Zimmers T. A.; Davies M. V.; Koniaris L. G.; Haynes P.; Esquela A. F.; Tomkinson K. N.; McPherron A. C.; Wolfman N. M.; Lee S.-J. Induction of cachexia in mice by systemically administrated myostatin. Science 2002, 296, 1486–1488. 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S.; Krag T. O.; Barton E. R.; Morris L. D.; Whittemore L. A.; Ahima R. S.; Khurana T. S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Jiang M.-S.; Liang L.; Wang S.; Ratovitski T.; Holmstrom J.; Barker C.; Stotish R. Characterization and identification of the inhibitory domain of GDF-8 propeptide. Biochem. Biophys. Res. Commun. 2004, 315, 525–531. 10.1016/j.bbrc.2004.01.085. [DOI] [PubMed] [Google Scholar]
- Hill J. J.; Davies M. V.; Pearson A. A.; Wang J. W.; Hewick R. M.; Wolfman N. M.; Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002, 277, 40735–40741. 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Wang J. L.; Lu J.; Song Y.; Kwak K. S.; Jiao Q.; Rosenfeld R.; Chen Q.; Boone T.; Simonet W. S.; Lacey D. L.; Goldberg A. L.; Han H. Q. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Lee S.-J.; Lee Y.-S. Regulation of GDF-11 and myostatin by GASP-1 and GASP-2. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3713–3722. 10.1073/pnas.1309907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-S.; Lee S.-J.; Zimmers T. A.; Soleimani A.; Matzuk M. M.; Tsuchida K.; Cohn R. D.; Barton E. R. Regulation of muscle mass by follistatin and activins. Mol. Endocrinol. 2010, 24, 1998–2008. 10.1210/me.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani M.; Takehara Y.; Sugino H.; Matsumoto M.; Hashimoto O.; Hasegawa Y.; Murakami T.; Uezumi A.; Takeda S.; Noji S.; Sunada Y.; Tsuchida K. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008, 22, 477–487. 10.1096/fj.07-8673com. [DOI] [PubMed] [Google Scholar]
- Anderson S. B.; Goldberg A. L.; Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J. Biol. Chem. 2008, 283, 7027–7035. 10.1074/jbc.M706678200. [DOI] [PubMed] [Google Scholar]
- Walton K. L.; Makanji Y.; Chen J.; Wilce M. C.; Chan K. L.; Robertson D. M.; Harrison C. A. Two distinct region of latency-associated peptide coordinate stability of the latent transforming growth factor-β1 complex. J. Biol. Chem. 2010, 285, 17029–17037. 10.1074/jbc.M110.110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M.; Zhu J.; Wang R.; Chen X.; Mi L.; Walz T.; Springer T. A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K.; Noguchi Y.; Aoki S.; Takayama S.; Yoshida M.; Asari T.; Yakushiji F.; Nishimatsu S.; Ohsawa Y.; Itoh F.; Negishi Y.; Sunada Y.; Hayashi Y. Identification of the minimum peptide from mouse myostatin prodomain for human myostatin inhibition. J. Med. Chem. 2015, 58, 1544–1549. 10.1021/jm501170d. [DOI] [PubMed] [Google Scholar]
- Ohsawa Y.; Takayama K.; Nishimatsu S.; Okada T.; Fujino M.; Fukai Y.; Murakami T.; Hagiwara H.; Itoh F.; Tsuchida K.; Hayashi Y.; Sunada Y. The inhibitory core of the myostatin prodomain: its interaction with both type I and II membrane receptors, and potential to treat muscle atrophy. PLoS One 2015, 10, e0133713. 10.1371/journal.pone.0133713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asari T.; Takayama K.; Nakamura A.; Shimada T.; Taguchi A.; Hayashi Y. Structural basis for the effective myostatin inhibition of the mouse myostatin prodomain–derived minimum peptide. ACS Med. Chem. Lett. 2017, 8, 113–117. 10.1021/acsmedchemlett.6b00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K.; Nakamura A.; Rentier C.; Mino Y.; Asari T.; Saga Y.; Taguchi A.; Yakushiji F.; Hayashi Y. Effect of N-terminal acylation on the activity of myostatin inhibitory peptides. ChemMedChem 2016, 11, 845–849. 10.1002/cmdc.201500533. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.