

Abstract
Exercise elicits coordinated multi-organ responses including skeletal muscle, vasculature, heart and lung. In the short term, the output of the heart increases to meet the demand of strenuous exercise. Long term exercise instigates remodeling of the heart including growth and adaptive molecular and cellular re-programming. Signaling pathways such as the insulin-like growth factor 1/PI3K/Akt pathway mediate many of these responses. Exercise-induced, or physiologic, cardiac growth contrasts with growth elicited by pathological stimuli such as hypertension. Comparing the molecular and cellular underpinnings of physiologic and pathologic cardiac growth has unveiled phenotype-specific signaling pathways and transcriptional regulatory programs. Studies suggest that exercise pathways likely antagonize pathological pathways, and exercise training is often recommended for patients with chronic stable heart failure or following myocardial infarction. Herein, we summarize the current understanding of the structural and functional cardiac responses to exercise as well as signaling pathways and downstream effector molecules responsible for these adaptations.
Introduction
The mammalian heart is a pump, supplying blood and a constant supply of oxygen and nutrients to itself and peripheral organs (Olver et al., 2015). This workload can account for 20% of total oxygen consumption and is supported by a complex cellular metabolic and contractile system of the heart to sustain maximal pump efficiency. This is especially true during intense bouts of exercise where oxygen consumption by the heart may increase up to 10-fold (Olver et al., 2015). In a remarkable display of metabolic flexibility, cardiac myocytes can match this increased demand illustrating an extraordinary energy potential and reserve capacity (Poole et al., 2012). Often referred to as the “athlete's heart,” the heart remodels in response to chronic exercise to match the increased workload by increasing in size in the absence of cardiac myocyte proliferation. This physiologic growth is accompanied by increases in energy production capacity, primarily mitochondrial. This hypertrophic growth of the heart is characterized by normal contractile function at rest. This is in contrast to pathologic growth that results from prolonged hypertension or ischemic heart disease, where cardiac contractile function as well as energy metabolic production declines (Doenst et al., 2013; Ingwall, 2008, 2009; Maillet et al., 2013; Scheuer and Buttrick, 1987). Historically, investigators have quantified the extent of cardiac growth by measuring how much the heart wall thickens (Fulton and Rajiah, 2017). However, wall thickening can occur with either an increase, decrease or no change in ventricular volume. To account for the proportionality between wall thickness and ventricular volume, cardiac growth is also characterized as either concentric or eccentric, reflecting the precise morphological and geometrical features of the enlarged heart (Figure 1) (Blomqvist and Saltin, 1983; Scheuer and Tipton, 1977). Both physiologic and pathologic growth show changes in heart wall thickness and volume, yet pathologic growth often progresses to severe cardiac remodeling and heart failure, whereas physiologic growth does not.
Figure 1. Concentric compared to eccentric cardiac growth due to physiologic or pathologic stress.
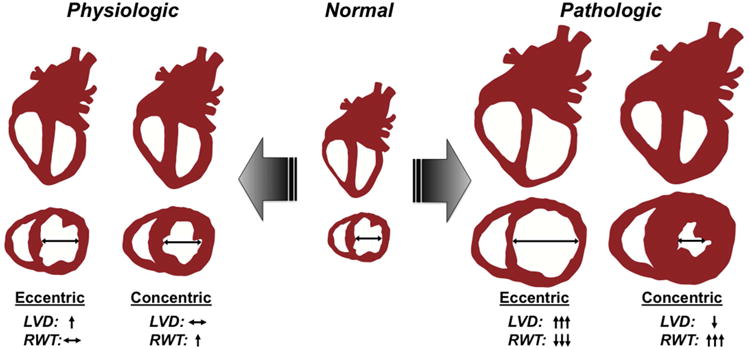
The normal heart will adapt to an increase in hemodynamic demand whether it is due to physiologic (e.g. exercise) or to pathologic (e.g. hypertension) stimuli. This hypertrophic growth, follows 2 typical patterns of growth determined by geometric relationship between ventricular internal diameter (LVD) and ventricular wall thickness, or relative wall thickness (RWT): (1) a concomitant increase in ventricular wall thickness and LVD (eccentric), usually driven by volume overload; (2) a disproportionate increase in wall thickness compared to LVD (concentric), driven by pressure overload. Typically, eccentric or concentric growth due exercise (Physiologic) is limited to a 12-15% increase in overall heart weight and does not progress to heart failure. In contrast, cardiac growth due to disease such as hypertension, myocardial infarction, or hypertrophic cardiomyopathy (Pathologic) usually exhibits a more robust hypertrophic response (concentric or eccentric) and often progress to a heart failure state. Pathologic eccentric growth may represent early transition to a dilated state; pathologic concentric growth results in profound thickening of the ventricular wall with a reduction in LVD.
In addition to direct effects on the heart, it is well-established that regular exercise results in improved quality of life and increased lifespan (Arem et al., 2015; Moore et al., 2012). Furthermore, exercise and particularly aerobic exercise, is a central component of cardiac rehabilitation and reduces cardiovascular morbidity and mortality in patients with heart disease (Gasiorowski and Dutkiewicz, 2013; Gayda et al., 2016; Guyatt and Devereaux, 2004; Ribeiro et al., 2016; Sharma et al., 2001). While the benefits of regular physical activity are clearly multifactorial reflecting the integrated responses of many organ systems, the specific cellular and molecular mechanisms of how exercise attenuates and often reverses much of the metabolic and contractile derangements in the diseased heart remain ill-defined (Hirai et al., 2015; Poole et al., 2012).
At the cellular and molecular level, it is clear that activation of upstream signaling pathways and resultant transcriptional responses are distinct between physiologic and pathologic cardiac growth (O'Neill and Abel, 2005). In this review, we focus on the morphologic, cellular, metabolic, and molecular adaptations of the heart to chronic exercise. For the first part of this discussion, we take a “systems” approach to define adaptions of the heart to exercise with consideration of an intact cardiovascular (CV) system (cardiac, neural, and vascular systems). We will demonstrate that the heart has an innate ability to identify and distinguish between the types of hemodynamic stress/exercise (dynamic vs static) but also whether and how the heart senses physiologic versus pathologic signals. We describe the morphological characteristics of cardiac growth and the implications for cardiac disease progression. Finally, we will incorporate a description of how the response to exercise compares among human subjects and laboratory animals and males and females.
In the second part of this review, we outline the molecular and cellular events that result in physiologic cardiac adaptation. Many of the molecular mechanisms that mediate the heart's response to exercise have been identified. For instance, the insulin-like growth factor 1 (IGF-1) and downstream signaling pathway represents one of the critical mediators of physiologic growth. In addition, considerable evidence from mouse genetic models demonstrates that activation of physiologic growth directly antagonizes pathologic growth. Finally, we examine how delineation of the mechanisms and molecular events involved in the response of the heart to exercise may unveil new therapeutic approaches for heart failure.
Cardiovascular system response to exercise
The CV system displays remarkable adaptability during acute and long-term bouts of physical exertion. Central to this adaptability is the coordinated integration of systemic, cellular and molecular signaling pathways within the CV system to meet the body's needs. For example, during acute bouts of exercise, the heart can increase its pumping capacity by 4-8 times as measured by the amount of blood pumped per unit time (cardiac output). This increase in cardiac function is driven mainly by a 3-4 fold increase in heart rate (60 bpm to 200 bpm) with a small contribution (2-fold) from the strength of contraction or contractility (Evans, 1985; Hellsten and Nyberg, 2015; Xiang and Hester, 2011). Changes in cellular signaling as a result of sympathetic but not parasympathetic activation underlie this increase in heart rate and contractility to acutely impact cardiac output. Because the CV system is a closed network, the immense increase in blood flow raises blood pressure, which serves to maintain adequate flow and nutrient delivery to both exercising and non-exercising tissues. Despite an early increase in vascular resistance, exercising skeletal muscles can increase its blood flow through the release of local factors causing blood vessels to dilate, shunting oxygen-rich blood to metabolically active tissues (primarily working skeletal muscle). In the short term, these local cues initiate cellular signaling pathways in the vascular system that can redistribute blood flow to working muscle up to 10 fold, illustrating the power of these local regulatory control mechanisms. Accordingly, sustained exercise ultimately leads to a global reduction in vascular resistance (Fagard, 2003; Lauschke and Maisch, 2009).
The response of the heart to acute exercise demonstrates the extraordinary reserve capacity of the CV system. Because the end-goal of these hemodynamic adaptations is to meet the prevailing needs of increased oxygen and metabolic demand, sustained or long-term exercise training (endurance or strength) induces physiologic and morphological adaptive changes in the CV system and heart. Typically, athletic conditioning can be categorized as either endurance/dynamic training or power/strength training (Fagard, 2003; Fagard, 1996; Pluim et al., 2000). This categorization is largely based on the type of load imposed on the heart; dynamic sports such as running require a sustained elevation in cardiac function (volume overload) whereas static sports such as power lifting produce brief instances of elevated pressure loads (pressure overload). On the other hand, activities such as cycling and rowing continuously invoke both dynamic and static conditioning throughout the exercise period. In general, many training programs will often incorporate elements of both dynamic and static activities, and, as a result, instigate a mixed-type load on the CV system.
Depending on the type of exercise, the CV system will accommodate the physiologic need and adapt based on the dominant type of hemodynamic load. Therefore, categorizing exercise “type” can also be based on the resultant physiologic and morphological changes in the heart and CV system. In general, long term changes that occur with endurance exercise training are enhanced aerobic capacity in skeletal muscle and reduced resting cardiac output, which is paralleled by slower heart rate and increased strength of contraction. Strength training, on the other hand, may not induce a change in basal cardiac function even though blood pressure and heart rate can increase dramatically during training maneuvers (MacDougall et al., 1985; Pluim et al., 2000). In addition, because strength training results in skeletal muscle growth, the overall perfusion to the skeletal muscles is higher due to the augmentation of skeletal cross-sectional area. The suggestion is that the heart and CV system have the innate ability to distinguish between exercise types presumably due to differences in hemodynamic load.
Both types of exercise result in cardiac growth. The cardiac myocyte is terminally differentiated and increases in size following exercise. Therefore, it is generally accepted that the growth of the heart due to exercise is from the collective increase in cardiac myocyte cell size and not from an increase in cardiac cell number. Along with an increase in cardiac mass, the heart undergoes changes in cardiac morphology that are dependent on the type of exercise training (Blomqvist and Saltin, 1983; Dickhuth et al., 1994; Fagard, 2003; Scheuer and Tipton, 1977; Teske et al., 2010). Physiologic cardiac growth, or more appropriately, adaptive remodeling (to signify the non-pathologic nature of this type of cardiac growth) can also be categorized as either concentric or eccentric (Figure 1) (Fagard, 1996; Lauschke and Maisch, 2009; Pluim et al., 2000). Designation of cardiac growth in this way depends on the geometric relationship between ventricular chamber size and ventricular wall thickness, or relative wall thickness (RWT). Typically, eccentric remodeling describes an increase in cardiac mass where RWT is maintained implicating a coordinated increase in chamber size and wall thickness. Alternatively, concentric remodeling is characterized by an increase in cardiac mass accompanied by a disproportionate increase in wall thickness relative to chamber size. Although both types of cardiac growth normalize (eccentric) or reduce (concentric) ventricular wall stress, concentric cardiac growth is correlated with worse clinical outcomes when coupled to cardiac disease (see below).
As mentioned above, athletic conditioning may be dynamic (running) or static (strength training), which in turn dictates the type of hemodynamic load imposed unto the heart. Although heart rate is the predominant determinant of cardiac function, dynamic conditioning increases venous return to the heart leading to increase contractility via the so-called Starling effect, enhancing the amount of blood ejected by the heart. This type of chronic “volume overload” induces eccentric remodeling. On the other hand, strength training imposes a sudden elevation in pressure inducing a brief, but dramatic increase in blood pressure, which in turn increases the impedance against which the heart must pump. For example, peak pressures of 480/350 and 320/250 mmHg have been recorded in subjects performing strength training (MacDougall et al., 1985). This profound increase in blood pressure, though intermittent and brief, provokes concentric remodeling of the heart (Blomqvist and Saltin, 1983; Devereux and Reichek, 1977; Fagard, 2003; Lauschke and Maisch, 2009). Again, these designations cannot be universally applied because athletic training is rarely purely dynamic or static. For example, exercise training programs which evoke elements of both dynamic and static stimuli lead to a mixed-type of hypertrophic remodeling (Fagard, 2003).
Dynamic (intermittent) changes in cardiac structure and geometry in response to exercise training is distinct from that due to chronic hemodynamic stress such as hypertension. Regardless of the type of hypertrophic response, exercise-induced remodeling is generally considered adaptive in that progression to heart failure does not occur. For example, physiologic (exercise) cardiac growth is fundamentally distinct from pathologic (hypertension) cardiac growth in that the former results in preserved or enhanced cardiac function and adaptive geometry. Hypertrophic hearts resulting from cardiac disease steadily decline in function over time progressing from concentric growth (elevated RWT) to a dilated state (decreased RWT) (Fagard, 1996; Frey et al., 2004). Moreover, physiologic growth may be limited in magnitude compared to pathologic growth. Perhaps the most critical feature of physiologic growth is its reversibility; growth due to exercise is fully reversible without apparent adverse consequences (Maillet et al., 2013; Shiojima et al., 2005). Although anti-hypertensive therapy may reverse cardiac growth, it is unclear whether this regression is without lasting effect, given that underlying cellular responses such as fibrosis may not resolve (Fagard et al., 2009). It should also be noted that other types of physiologic growth include cardiac growth due to pregnancy or normal postnatal growth and development.
Animal models of physiologic growth
While it is clear that physiologic cardiac growth is fundamentally distinct from pathologic cardiac growth, a major line of research has focused on the question of whether cardiac remodeling following exercise training can evolve into a pathologic form (Lauschke and Maisch, 2009; Maron, 2009; Pelliccia et al., 2012). This issue is especially relevant in the case of hypertrophic cardiomyopathy (HCM), an inherited disorder with a 0.2-0.5% prevalence in the general population. In many instances, the hypertrophic phenotype due to exercise overlaps with HCM, or differences may be subtle that cannot be distinguished by measures of cardiac geometry. Specifically, in most instances, HCM does not evolve into a dilated dysfunctional heart. However, HCM patients are at significant risk for sudden death. Indeed, published reports indicate that 35-50% of sudden cardiac deaths in young athletes is attributable to HCM. Recognizing the critical need to demarcate clear differences between training-induced and diseased-induced cardiac growth, animal models of exercise have been employed to study cardiac remodeling and adaption to exercise. As discussed below, the results of these studies have identified distinct molecular and metabolic signatures that distinguish physiologic from pathologic forms of cardiac growth.
Animal models, especially rodents, hold many practical advantages including short gestation periods, large litters, compressed lifespans, identical genetic backgrounds if studying a single wild type strain, and inexpensive housing. However, other animal species have been used such as horses, rabbits, dogs and swine (DiCarlo and Bishop, 1988; Evans and Rose, 1988; Laughlin et al., 1991; Stepien et al., 1998; White et al., 1987). Currently, however, the genetically modified mouse model is the cornerstone for interrogating cellular and molecular pathways underlying cardiac biology. Apart from important strain differences, rodents undergo physiologic adaptations to exercise with the type and extent of adaptation dependent on the exercise type. Three types of rodent exercise training protocols dominate the literature: (1) treadmill running, (2) voluntary wheel running and (3) swim training. The majority of investigations have used a motorized treadmill, subjecting rodents to treadmill protocols that vary in intensity, incline and duration (Kasper, 2013; Perrino et al., 2011; Wang et al., 2010). These early, foundational studies in treadmill-induced adaptation employed endurance running protocols, where treadmill speed, duration or incline was progressively increased or remained fixed. The detail of such protocols has been comprehensively outlined in other reviews (Perrino et al., 2011; Wang et al., 2010). In most instances, rats and mice demonstrate a predictable physiologic adaptation to an endurance exercise regimen that recapitulates human physiology, regardless of the training protocol. Similar to human counterparts, rats and mice also undergo exercise-induced cardiac growth ranging from a 5-24% increase in cardiac mass (Diffee and Nagle, 2003; Fenning et al., 2003; Kemi et al., 2002; Moore et al., 1993). Existing data suggest that a training period of at least 3-4 weeks is necessary to elicit a significant increase in cardiac mass, again mimicking the requirement for minimum conditioning period in humans to induce physiologic cardiac growth.
An alternative treadmill training protocol uses intervals of high-intensity with low-intensity treadmill at varying inclines (Haram et al., 2009). Studies using this type of high-intensity interval training report a more robust cardiac hypertrophic response than continuous protocols with increases in cardiac mass up to 30% (Haram et al., 2009; Kemi et al., 2005; Kemi et al., 2002). This approach may also prove relevant to humans given that in the clinical literature, there is an emerging interest in high-intensity interval training over that of standard exercise protocols, which incorporates a longer training period (20-60 minutes) at lower exercise intensities for patients with cardiovascular disease (Gayda et al., 2016; Pattyn et al., 2014; Ribeiro et al., 2016). Again, a more detailed description of specific interval training protocols is documented elsewhere (Cardozo et al., 2015; Perrino et al., 2011; Wang et al., 2010). Although this is an ongoing area of research, patients undergoing high-intensity interval training show greater or equivalent benefits as compared with established cardiac rehabilitation programs for most of the parameters, with overall lower exercise times (Ribeiro et al., 2016). Many factors contribute to these improved benefits such as the starting fitness level of the patient. A major confounder, however, is that patients may simply prefer this type of interval training and exhibit improved adherence to the exercise prescription confounding interpretation of comparative studies. Accordingly, pre-clinical studies may provide valuable insight into the distinct cellular and molecular responses to these different protocols.
Swim training is another well-established aerobic exercise modality to study the physiologic response of the heart to exercise (Scheinowitz et al., 2003; Tatsuguchi et al., 2004) and for the assessment of the potential protective impact on cardiac disease (McMullen et al., 2007; McMullen et al., 2003). For these studies, mice or rats are placed in a swim tank for a given duration. The fundamental difference between swim and treadmill exercise is how exercise intensity is modified; swim training protocols vary intensity by progressively adding weight “carried” by the animal (Almeida et al., 2009). When comparing cardiac growth from swim or treadmill exercise training, swim training induces an impressive 30-50% increase in cardiac mass (Almeida et al., 2009; McMullen et al., 2007; McMullen et al., 2003), which is, on average, more than that from treadmill exercise.
Considering the forced nature of swim and treadmill exercise protocols, there is concern that the type of cardiac growth may not be purely physiologic given the stress involved. Although mice are typically acclimated to the exercise environment, it can be argued that swim or treadmill conditioning deviate from naturally-occurring rodent activities and initiate, at least in part, a stress-induced cardiac growth. Interestingly, the mouse heart is able to recognize an intermittent, pathological overload meant to mimic the intermittent nature of an exercise load as distinctly pathological (Perrino et al., 2006). For this reason, we and others have employed a voluntary exercise paradigm, where rodents have free, unobstructed access to a cage wheel (Allen et al., 2001; Eldomiaty et al., 2016; Konhilas et al., 2015; Konhilas et al., 2004; McMullan et al., 2016). There is ample evidence that voluntary wheel running is a complex behavior resulting from natural rodent tendencies and laboratory environment (Konhilas et al., 2015; Meijer and Robbers, 2014; Richter et al., 2014). Activity from cage wheel exercise generally improves depressive-like behaviors (Eldomiaty et al., 2016), although this is challenged by studies suggesting an anxiolytic or addictive-like component to wheel running (Duman et al., 2008; Fuss et al., 2010; Richter et al., 2014). Apart from the impact on neural or behavioral function, cage wheel running provides sufficient aerobic training to induce physiologic cardiac growth in both rats and mice (Allen et al., 2001; Konhilas et al., 2004; Natali et al., 2001). Similar to humans and forced training, a minimum of 3-4 weeks of training is required to instigate a 5-20% increase in cardiac mass depending on strain, sex, diet, age or any combination of these variables (Allen et al., 2001; Konhilas et al., 2015; Konhilas et al., 2004; Lerman et al., 2002; McMullan et al., 2016). Interestingly, long-term or loaded cage wheel exercise does not increase the amount of cardiac growth (Konhilas et al., 2006; Konhilas et al., 2005; Natali et al., 2001).
Regardless of exercise type, the pre-clinical exercise training models have been invaluable for determining putative cellular and molecular mechanism underlying cardiac adaptation as will be described below. Still, there are important limitations when attempting to translate rodent models of exercise to humans. Apart from the obvious size differences, the complex hemodynamic response with regards to heart rate, vascular and sympathetic/parasympathetic systems to increased workload is unique to each species. Consequently, the type of cardiac remodeling cannot be identical in rodents when compared to humans. In addition, just as human genotype influences exercise performance, the exercise response in rodents has a significant genetic component, varying across inbred mouse strains. Therefore, choice of model or strain may confound or bias results towards the particular model. Despite the limitations, including some specific for transgenic mice(Davis et al., 2012), this work has clearly facilitated our understanding of the cardiac exercise response.
Sex dimorphisms in cardiac adaptation to exercise
A major limitation of many published human exercise studies is that males and females are typically grouped, or only one sex is studied, when comparing trained and untrained subjects. Whether examining humans or rodents, the anatomical arrangement of the heart and peripheral cardiovascular system is identical between males and females. However, the size of the female heart is smaller than age- and race-matched men (Vasan et al., 1997). Moreover, the exercise response is likely influenced by sex-specific hormonal and metabolic factors. Looking at a human study population comparing men and women (average age of 57 years) free of clinically overt cardiovascular disease, all volumetric parameters measured by magnetic resonance imaging (MRI) were higher in males, even when adjusted for subject height, weight or body surface area (Cain et al., 2009; Salton et al., 2002). In these same studies, no differences in LV functional parameters were evident although these data are not always consistent (Cain et al., 2009; Huxley, 2007; Salton et al., 2002). In a large-scale prospective study of female athletes, investigators reported that LV thickness and cavity dimensions rarely exceed the upper, clinical limit (Pelliccia et al., 1996). Subsequently, many reports have emerged showing that females do indeed undergo exercise-induced growth, especially when adjusted for body mass (George et al., 1999; George et al., 1995; Maron, 2009). A recent echocardiographic analysis of over 1,000 elite athletes stratified male and female athletes by type of exercise (Finocchiaro et al., 2016). In this comprehensive assessment of cardiac adaptation, the study demonstrated increased chamber dimensions in female athletes compared to male counterparts, only when adjusted for body surface area. The suggestion is that a greater percentage of female athletes undergoing dynamic training exhibit eccentric cardiac growth compared to male athletes. Further, female athletes rarely demonstrate concentric cardiac growth (Finocchiaro et al., 2016).
Animal models of exercise training also exhibit sex-dependent patterns in cardiac adaptation/growth. In both rats and mice, the proportionate increase in cardiac mass following treadmill exercise is greater in females than males (Kemi et al., 2002; Wisloff et al., 2001b). Compared to males, swim-trained female rats exhibit a greater increase in absolute heart mass (Mole, 1978). Our group found that female mice exposed to a voluntary cage wheel exhibit approximately 3-fold greater percent increase in heart mass than their male counterparts. Because females spent more time running on the cage wheel, we normalized the extent of growth to distance run; females still demonstrate a great hypertrophic response even when the extent of growth is adjusted for activity (Konhilas et al., 2004). A similar sex difference was observed with treadmill exercise in mice, where females demonstrated more robust cardiac growth than male counterparts (Foryst-Ludwig et al., 2011). Our subsequent work provided evidence that the sex dimorphism in cardiac adaptation to exercise is dependent on many factors. For example, simply switching from a traditional soy-based diet to a calorically similar casein-based diet eliminated the sex difference such that males responded to cage wheel running similarly to females eating either diet (Konhilas et al., 2015). Taken together, the results of numerous studies indicate that exercise training in rodents (and humans) is a complex behavior and that cardiac adaptation is impacted by many factors including sex and diet.
Cellular adaptions to exercise
As noted above, the presence of hypertrophic remodeling in athletes presents a unique problem considering the significant rate of sudden cardiac death, particularly in young athletes (Maron, 2009). The diagnosis is made even more problematic because, whether induced by exercise or by an inherited mutation, cardiac growth may be modest or not evident (Tardiff, 2011). Adding to the complexity, cardiac growth/remodeling from disease is a progressive process making prognostic predictions difficult without prior knowledge of pre-disease cardiac architecture. Therefore, delineation of the cellular manifestations of exercise-induced cardiac remodeling will not only help delineate mechanisms involved in the adaptive responses, but could provide predictive tools relevant to the more precise diagnosis of a clinical hypertrophic phenotype. However, to date, only a few cellular features distinguishing physiologic from pathologic cardiac growth have been defined, the most robust of which is interstitial fibrosis.
During early postnatal development, cardiac myocytes lose their ability to proliferate and become terminally differentiated. As a consequence, enlargement of the heart during development and in response to exercise or pathologic conditions is caused by an increase in cardiac myocyte size. It has been suggested that changes in myocyte width, length or both depends on the specific type of cardiac growth (concentric vs eccentric), and whether cardiac growth is induced by physiologic (exercise) or pathologic (cardiac disease) stimuli (Maillet et al., 2013; Wisloff et al., 2001a). However, a direct correlation is not always consistent (Gerdes et al., 1992; Gerdes et al., 1996; Wisloff et al., 2002). Independent of the specific changes in myocyte morphology, cardiac growth fundamentally normalizes wall stress regardless of the physiologic or pathologic origin of the hemodynamic load. As a result, cardiac function is dependent on the 3-dimensional geometry and solely dictated by myocyte function. Incidentally, male and female hearts begin with an equivalent number of cardiac myocytes and, therefore, the difference in cardiac size, although proportional to body mass, is due to larger myocyte size in males compared to females (de Simone, 1995; Luczak and Leinwand, 2009; Olivetti et al., 1995). As discussed previously, cardiac growth can be designated as either concentric or eccentric based on RWT, a measure of ventricular geometry. The pathological nature of this assignment depends on the extent and origin of cardiac growth. However, pathologic growth is accompanied by several histological features that are distinct from physiologic growth. Pathologic growth may be accompanied by an accumulation of interstitial and perivascular fibrosis. In addition, pathologic cardiac growth may manifest non-uniform myocyte alignment, termed myocyte disarray (Maron et al., 1986; Varnava et al., 2001). Myocyte disarray is particularly prominent in HCM and other inherited forms of cardiomyopathy. In contrast, fibrosis and myocyte disarray has not been observed following chronic exercise in rats or mice of either sex (Burgess et al., 1996; Konhilas et al., 2006; Perez et al., 2013; Thomas et al., 2000) and, presumably, human athletes. Although collagen accumulation is absent in exercising rodents, previous studies have shown that treadmill exercise training increases collagen turnover and alters collagen cross-linking and sub-type in the aging rat (Takala et al., 1991; Thomas et al., 1992; Thomas et al., 2000). It has also been shown that specific patterns in collagen cross-linking can markedly influence ventricular diastolic (relaxation) properties (Todaka et al., 1997).
Molecular and metabolic responses in exercise-induced adaptive cardiac growth: Gene regulatory mechanisms
Numerous studies have documented cardiac transcriptomic changes following exercise training (Budiono et al., 2012; Chung et al., 2012; Iemitsu et al., 2005; Solskov et al., 2012; Strom et al., 2005). The results of these studies indicate that re-progamming of cardiac structure and function during cardiac growth, whether due to exercise or pathologic stimuli, involves a cascade of gene regulatory mechanisms. Comparison of the transcriptome of pathologic forms of cardiac growth with that of physiologic growth has proven informative. By and large, the transcriptional signatures of physiologic cardiac growth are distinct from pathologic hypertrophic response (Strom et al., 2005). Pathologic growth is characterized by activation of the so-called “fetal gene” program including induction of natriuretic peptides and fetal sarcomeric isoform genes (Figure 2). This pattern is largely not observed in exercise transcriptomes (Strom et al., 2005). Early studies in mice demonstrated that swimming exercise and aortic banding (a pathologic hypertrophic stimulus) displayed distinct gene expression pattern changes (McMullen et al., 2003). In contrast to robust changes in atrial or brain natriuretic peptide (ANP, BNP) seen with banding, no change was observed with swimming. Similarly, the fetal β-myosin heavy chain was markedly upregulated by aortic constriction but not by swimming (McMullen et al., 2003). Metabolic gene expression patterns are also distinct in pathologic versus physiologic cardiac growth (Figure 2). Expression of genes involved in fuel metabolism and bioenergetics undergo a well-characterized re-programming during pathologic cardiac hypertrophic growth, and in the failing heart. This metabolic gene regulatory re-programming is also reminiscent of a “fetal” shift in that the capacity for burning the main fuel, fatty acids, is reduced and the expression of nuclear and mitochondrial genes involved in many mitochondrial energy transduction and respiratory pathways are downregulated (Aubert et al., 2013). Conversely, genes involved in some glucose utilization pathways are upregulated. Significant genetic and experimental evidence suggests that this energy metabolic re-programming may contribute to the development of heart failure. The alterations in fuel metabolic genes are of particular interest. Whereas the expression of genes involved in mitochondrial fatty acid oxidation (FAO), the chief source of ATP in the normal adult heart, are downregulated in disease (Aubert et al., 2013), this program remains unaltered in response to exercise (Beisvag et al., 2009). In addition, expression of cardiac genes encoding several fatty acid transporters, fatty acid binding proteins, and related lipid metabolic pathways are increased in response to exercise training but not in pathologic cardiac growth or heart failure (Riehle et al., 2014; Strom et al., 2005). These gene regulatory re-programming events are also reflected in fuel oxidation rates. Specifically, palmitate oxidation rates, as a measure of FAO, are increased in isolated working heart preparations from exercise-trained rats (Burelle et al., 2004). Conversely, FAO rates are reduced in pathologic cardiac growth and heart failure (Vega et al., 2015). A mitochondrial biogenic response is also observed in the heart following exercise training in contrast to the reduction of oxidative capacity in the diseased heart. Components of the electron transport chain, mtDNA, and citrate synthase activity are all increased in mouse hearts with swimming (Riehle et al., 2014). Consistent with the changes in expression of fatty acid utilization genes, mitochondrial respiration in isolated cardiac strips from trained mice is also increased when palmitoylcarnitine is used as a substrate (O'Neill et al., 2007).
Figure 2. Molecular and metabolic signatures distinguish pathologic and physiologic cardiac remodeling.
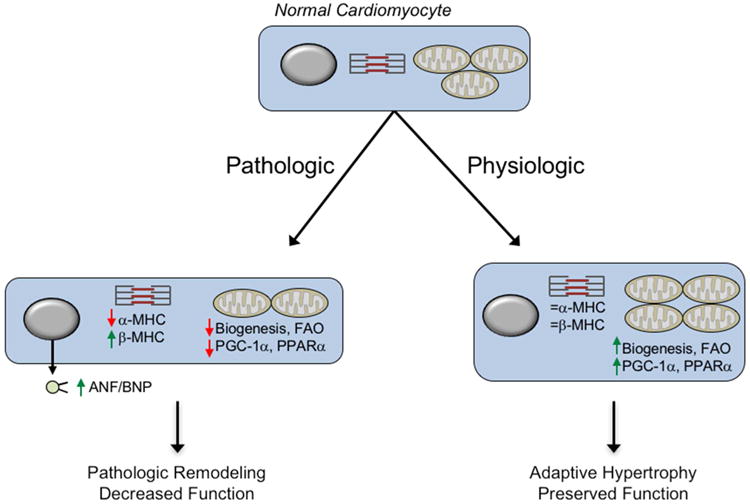
Different external stimuli trigger distinct growth programs in the cardiomyocyte. In response to hypertension or pressure overload (Pathologic), the cardiomyocyte activates a growth program characterized by the induction of a fetal gene program including increased natriuretic peptide expression and changes in sarcomere isoform gene expression. This program eventually leads to a more global pathologic remodeling including left ventricular dilation and diminished cardiac function en route to the syndrome of heart failure. In contrast, exercise (Physiologic) elicits a growth program without induction of the fetal-gene program and an increase in energy metabolic capacity that matches the increase energy demands imposed by chronic exercise. This latter program maintains normal cardiac function. MHC, myosin heavy chain; ANF, atrial natriuretic factor; BNP, brain natriuretic peptide; FAO, fatty acid oxidation; PPARα, peroxisome proliferator activated receptor α; PGC-1, PPARγ coactivator-1α.
Quantitative assessment of metabolites representative of major energy metabolic pathways (targeted metabolomics) provides additional correlates to the cardiac metabolic gene expression changes that occur in response to exercise versus pathologic stimuli. Just as transcriptomics can provide a signature representative of alterations in gene expression, metabolomics provides a signature reflective of the changes in energy metabolism by measuring metabolite intermediate pools. Several studies have reported cardiac metabolomic measurements in experimental and clinical heart failure samples (Bedi et al., 2016; Lai et al., 2014; Sansbury et al., 2014). These studies have consistently shown changes in acylcarnitine levels reflective of alterations in mitochondrial FAO rates. Interestingly, direct comparison of pre-clinical models of heart failure and exercise reveals striking differences. Levels of acylcarnitines were increased in the early stages of a pressure-overload cardiac growth/myocardial infarct model in mice while voluntary wheel running mice displayed lower acylcarnitine abundance (Lai et al., 2014). A similar pattern was also observed with TCA cycle intermediates. Collectively, combined with gene expression changes discussed above, these results have unveiled robust differences in metabolic re-programming in pathologic versus exercise-trained physiologic cardiac growth. The maintenance of high capacity for FAO and, thus, ATP production, is consistent with an adaptive response to the increased workload. In contrast, pathologic cardiac growth results in a marked shift towards the lower capacity fetal program, likely a maladaptive energetic response.
What factors mediate the cardiac gene regulatory responses to exercise? Some lessons have been learned from the delineation of the transcriptional regulatory circuitry involved in mitochondrial biogenesis in heart and the effects of exercise on skeletal muscle gene expression. The peroxisome proliferator activated receptor γ coactivator-1a (PGC-1a) is a transcriptional coactivator originally identified as a cold-inducible factor involved in mitochondrial biogenesis in the brown adipocyte (Puigserver et al., 1998). In skeletal muscle, expression of PGC-1a is dramatically induced by an acute bout of exercise (Pilegaard et al., 2003). PGC-1a has been shown to mediate the mitochondrial adaptations in muscle in response to exercise. Targeted disruption of PGC-1a and the related PGC-1b in skeletal muscle markedly alters mitochondrial function and diminishes exercise performance (Wende et al., 2007; Zechner et al., 2010). PGC-1a is also activated by exercise in the heart (O'Neill et al., 2007; Riehle et al., 2014). Some evidence suggests that PGC-1a mediates the mitochondrial biogenic response and changes in fuel and energy metabolism in heart following exercise. Specifically, mouse models with altered exercise-induced activation of PGC-1a expression exhibit a defect in the induction of mtDNA content and palmitate oxidation in heart (Riehle et al., 2014). However, additional studies are needed in heart-specific PGC-1 deficient mice to confirm the role of PGC-1 as being necessary for the mitochondrial and energy metabolic adaptations observed with exercise.
The pleiotropic actions of cardiac PGC-1α are mediated through direct interaction and activation of a downstream cascade of transcription factors including nuclear receptors (Figure 3) (Vega et al., 2015). For instance, PGC-1α binds and co-activates the peroxisome proliferator activated receptor a (PPARα) to regulate fatty acid import, storage and oxidation in the heart (Djouadi et al., 1999; Finck et al., 2002; Gilde et al., 2003). Importantly, PPARα and PGC-1a signaling is inhibited in pathologic growth and heart failure (Aubert et al., 2013), consistent with the downstream fuel and energy metabolic changes described above. PGC-1a also promotes a broader mitochondrial biogenic response through its interaction with the estrogen-related receptor (ERR) and nuclear receptor factor 1 (NRF-1) (Scarpulla et al., 2012). In the cardiac myocyte, ERR directly activates expression of genes involved in virtually all aspects of mitochondrial energy metabolism including the TCA cycle, electron transport and oxidative phosphorylation, in addition to FAO enzymes (Dufour et al., 2007). ERRα, the major isoform in the heart, is also required for maximal ATP synthesis and adaptation to pressure overload (Huss et al., 2007). Notably however, despite being a logical candidate, the precise role for ERR in cardiac adaptation to exercise has not been determined.
Figure 3.
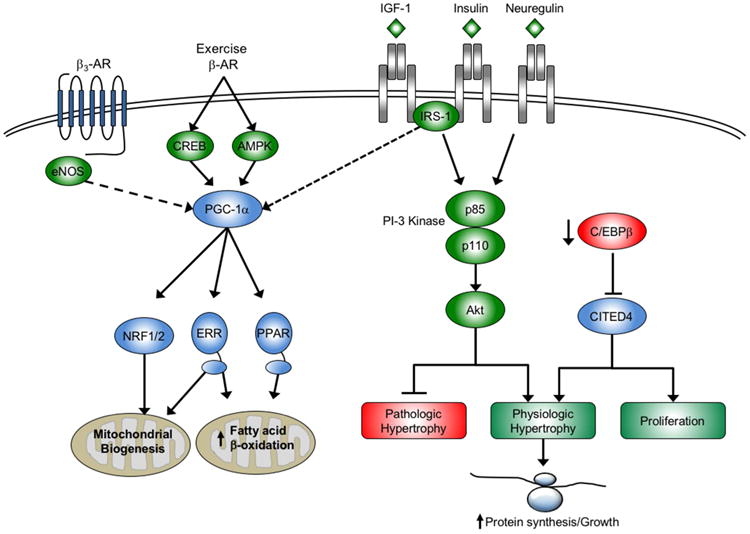
Cellular signaling pathways and transcriptional regulatory circuits mediating physiologic cardiac hypertrophic growth. Multiple growth factor pathways feed into the PI3K(p110α)-Akt signaling pathway including insulin growth factor 1 (IGF-1) and insulin to promote the physiologic hypertrophic response to exercise. These pathways directly antagonize pathologic growth growth. Exercise also enhances capacity for fuel oxidation and ATP production through peroxisome proliferator activated γ coactivator-1α (PGC-1α) regulated pathways that increase mitochondrial biogenesis and expression of genes involved in fatty acid β-oxidation. There is also evidence that cross-talk from growth factor signaling or eNOS to PGC-1α coordinates growth and metabolic pathways through unknown mechanisms (dotted line). Growth factors and signaling molecules that promote physiologic growth are shown in green; shown in red are those factors that antagonize physiologic growth. Transcription factors and coregulators are shown in blue. IRS-1, insulin receptor substrate 1; CREB, cAMP response element binding protein; eNOS, endothelial nitric oxide synthase; NRF, nuclear respiratory factor; ERR, estrogen-related receptor; PPAR, peroxisome proliferator activated receptor; C/EBPβ, CCAAT-enhancer binding protein β; CITED4, CBP/p300–interacting transactivator with ED-rich carboxy-terminal domain-4.
Unbiased profiling studies have identified several other transcriptional regulators, in addition to PGC-1a, to be involved in the response of the heart to exercise. One of these is CCAAT-enhancer binding protein β (C/EBPβ). C/EBPβ was found to be downregulated in the heart following a chronic swimming protocol (Bostrom et al., 2010). Interestingly, knockdown of C/EBPβ resulted in cardiac myocyte growth and activation of genes similar to what is observed with exercise, including PGC-1a (Bostrom et al., 2010). These effects are due, at least in part, to increased levels of the CBP/p300–interacting transactivator with ED-rich carboxy-terminal domain-4 (CITED4). CITED4 promoted a cellular proliferation response to exercise in the heart (Bostrom et al., 2010). Overexpression of CITED4 in the heart resulted in growth with preserved function in the absence of activation of the fetal gene program (Bezzerides et al., 2016). CITED4 also protected the heart against ischemia/reperfusion injury (Bezzerides et al., 2016). At this point, the precise targets of CITED4 in mediating this response are not known. Acute exercise also induces expression of the nuclear factor-erythroid 2 p45-related factor 2 (NFE2L2 or Nrf2) (Muthusamy et al., 2012). Nrf2 activates antioxidant gene expression and provides protection against oxidative stress. Moderate exercise in older mice also activates the Nrf2-antioxidant pathway(Gounder et al., 2012).
Non-coding RNAs have also been implicated in the molecular regulatory circuitry that contributes to the response of the heart to exercise. A large number of miRNAs have now been shown to be critical regulators of cardiac health and disease (van Rooij and Olson, 2012). Not surprisingly, numerous miRNAs are regulated in the heart with exercise (Fernandes et al., 2015; Liu et al., 2015). To date, these miRNAs have been shown to regulate genes involved in cardiac structure and function on multiple levels. For instance, a miRNA expressed in cardiac fibroblasts, miR-29, was shown to repress expression of genes involved in the fibrotic response and is activated by exercise in the heart (Soci et al., 2011; van Rooij et al., 2008). Profiling of exercise models also identified miR-222 as a critical mediator of physiologic growth (Liu et al., 2015). miR-222 promoted a physiologic growth phenotype in cardiac myocytes in vitro with increased α-MHC/β-MHC ratio and inhibited fetal gene markers such as ANF and BNP. And consistent with a recurrent theme, miR-222 was necessary for physiologic growth growth in vivo and was cardioprotective following ischemic injury (Liu et al., 2015).
Cellular signaling pathways involved in the cardiac exercise response
A key question in understanding the molecular adaptions to exercise in the heart is the identification of the cellular signaling pathways responsible for transmitting the physiologic stimuli to downstream transcriptional and other regulatory mechanisms. The use of genetic mouse models has identified key cellular receptor and downstream signaling pathway mediators of the cardiac response to exercise and physiologic growth (summarized in Table 1). Arguably, the key central signaling pathway in this regard is the phosphoinositide 3-kinase (PI3K)/Akt pathway (Table 1 and Figure 3). A significant finding was the identification of PI3K(p110a) as a critical mediator of physiologic but not pathologic hypertrophic growth (McMullen et al., 2003). Notably, signaling through PI3K(p110a) was shown to be necessary for hypertrophic growth by exercise training but not pressure overload resultant from aortic banding in mice (McMullen et al., 2003). Activation of PI3K(p110a), either by exercise or genetically, was also cardioprotective and directly antagonized pathologic cardiac growth (McMullen et al., 2007; Weeks et al., 2012). Akt serves as an effector kinase downstream of PI3K signaling and is critical for the physiologic growth program. Akt1-deficient mice displayed impaired cardiac growth in response to swimming exercise (DeBosch et al., 2006). Interestingly, Akt1-/- mice also exhibited an exaggerated hypertrophic response to aortic banding further demonstrating the antagonism between physiologic and pathologic growth programs (DeBosch et al., 2006). The PI3K/Akt pathway is primarily responsible for transmitting signals from growth factors, namely insulin and insulin-like growth factor 1 (IGF-1). Interestingly, cardiac IGF-1 expression and levels are associated with physiologic growth in athletes (Serneri et al., 1999). The IGF-1 receptor (IGF-1R) has also been shown to be both necessary and sufficient to induce physiologic growth in mice (Kim et al., 2008; McMullen et al., 2004).
Table 1. Mouse models of physiologic and pathologic growth.
Multiple models of transgenic overexpression or genetic deletion have been subjected to pathologic or physiologic hypertrophic cardiac growth. In many instances, the models were subjected to both pathologic and physiologic stimuli. The response of each model is summarized in the table indicating the type and extent of cardiac hypertrophy.
| Model | Physiologic | Pathologic | Reference |
|---|---|---|---|
| C/EBPβ+/- | Promotes physiologic growth and markers of proliferation | Inhibits pathologic remodeling following pressure overload | Boström et al., 2010 |
| CITED4 cardiac Tg | Promotes physiologic growth | Protects against l/R injury | Bezzerides et al., 2016 |
| miR-222 knockdown | Necessary for physiologic growth | n.d. | Liu et al., 2015 |
| miR-222 cardiac Tg | No change in cardiac size | Inhibits pathologic remodeling following l/R injury | Liu et al., 2015 |
| caPI3K(p110α) | Promotes physiologic growth | Inhibits pathologic remodeling following pressure overload | McMullen et al., 2007, 2004 |
| dnPI3K(p110α) | Prevents physiologic growth to swimming | Exaggerated pathologic remodeling following pressure overload | McMullen et al., 2003 |
| Aktl-/- | Prevents physiologic growth to swimming | Exaggerated pathologic remodeling following pressure overload | DeBosch et al., 2006 |
| IGF1R cardiac Tg | Promotes physiologic growth | n.d. | McMullen et al., 2004 |
| IGF1R cardiac KO | Prevents physiologic growth to swimming | n.d. | Kim et al., 2008 |
| IR cardiac KO | Normal growth, inhibits metabolic adaptation | n.d. | Boudina et al., 2009 |
| IRS-1/IRS-2 cardiac KO | Prevents physiologic growth to swimming | n.d. | Riehle et al., 2014 |
| ERRB2 cardiac KO | Necessary for CM proliferation during development | n.d. | D'Uva et al., 2015 |
| ERRB2 cardiac Tg | Promotes CM proliferation | n.d. | D'Uva et al., 2015 |
| eNOS-/- | No impact on physiologic growth | Necessary for protective effects of wheel running post-MI | de Waard et al., 2010 |
Additional evidence supports the idea that signaling through IGF-1R, as well as the insulin receptor (IR), transduces key components of the cardiac metabolic adaptation to exercise. Interestingly, cardiac-specific IR knockout mice displayed impaired mitochondrial respiration and ATP synthesis rates (Boudina et al., 2009). Surprisingly, these changes occur in the absence of altered PGC-1a levels although PPARα expression is reduced (Boudina et al., 2009). However, deletion of the insulin receptor substrate (IRS), required for both IGF-1R and IR signaling, prevented PGC-1a activation and increased mitochondrial capacity following exercise (Riehle et al., 2014). These results provide evidence that coordination of growth programs and metabolic re-programming may occur by IGF-1- and insulin-triggered signaling pathways.
Other cellular signaling events are capable of activating the PI3K/Akt pathway. For instance, activation of the ErbB2/ErbB4 tyrosine kinase receptors by the growth factor neuregulin-1 can stimulate PI3K signaling (D'Uva et al., 2015; Fukazawa et al., 2003). Notably, the neuregulin-1/ErbB2/ErbB4 pathway regulates cardiomyocyte proliferation and differentiation during development, and may affect a cardiac regenerative response in the adult heart (Bersell et al., 2009; D'Uva et al., 2015). Neuregulin-1 expression is upregulated in the heart with exercise training (Cai et al., 2016; Waring et al., 2014). Interestingly, exercise has been shown, at least in some contexts, to induce markers of cardiomyocyte proliferation (Bostrom et al., 2010; Cai et al., 2016). However, the precise role for neuregulin-1 signaling in the adaptation to exercise, including cardiomyocyte proliferation, is unknown.
Nitric oxide (NO) has also been shown to be a mediator of the beneficial effects of exercise in the heart. NO generated by endothelial nitric oxide synthase (eNOS) activates soluble guanylate cyclase (sGC) leading to increases in cGMP and activation of protein kinase G (PKG). The sGC/PKG pathway has been shown to be cardioprotective (Rainer and Kass, 2016). There is substantial evidence for a link between these pathways and the cardiac benefits of exercise. Exercise was shown to protect against ischemia/reperfusion injury through activation of endothelial nitric oxide synthase (eNOS) and subsequent increase in cardiac and circulating levels of NO (Calvert et al., 2011). The benefits of exercise following myocardial infarction are lost in eNOS knockout animals (de Waard et al., 2010). Interestingly, these effects may also be dependent on b3-adrenergic receptors. eNOS activation by exercise also prevents cardiac remodeling in response to chronic adrenergic activation (Yang et al., 2014). Finally, there is evidence that eNOS contributes to the cardiac metabolic remodeling with exercise. Specifically, increases in mitochondrial biogenesis as well as PGC-1a levels were absent in eNOS deficient mice following swim training (Vettor et al., 2014).
Translational and Therapeutic Implications
There is evidence, largely from pre-clinical models, to suggest that exercise and the physiologic cardiac hypertrophic program antagonizes pathologic growth and improves cardiac function in the context of pathologic conditions that typically lead to heart failure. Indeed, most of the murine genetic models that have been identified to control physiologic growth also impact pathologic growth, demonstrating the inherent dichotomy of these growth programs (Table 1). Apart from these genetic manipulations of signaling pathways and other factors, exercise training has a direct effect on cardiac pathologic remodeling. Low intensity exercise training has been shown to delay the onset of heart failure in the spontaneously hypertensive heart failure rat without effects on hypertension (Emter et al., 2005). Training also improved cardiac function in surgical and genetic models of heart failure or hypertrophic cardiomyopathy in mice (Konhilas et al., 2006; McMullen et al., 2007; Vanzelli et al., 2013). In humans, the largest study to examine the effects of exercise in heart failure patients was the HF-ACTION trial that examined the safety and efficacy of exercise in over 2300 patients with heart failure with reduced ejection fraction (HFrEF) (O'Connor et al., 2009). The study examined the effects of a combination of walking, treadmill and stationary bike exercise at prescribed percentages of maximal heart rate five times per week. The results of HF-ACTION demonstrated non-significant reductions in cardiovascular mortality or heart failure hospitalization in those patients prescribed exercise as compared to the usual care group (O'Connor et al., 2009). However, these findings were significant when adjusted for baseline characteristics. Importantly, there were significant improvements in physical fitness and 6-minute walk test that indicate improvements in quality of life (Flynn et al., 2009). It is important to note that the HF-ACTION trial only enrolled patients with reduced ejection fraction. It is now well recognized that a significant proportion of HF patients have heart failure with preserved ejection fraction (HFpEF). While a large randomized trial assessing the impact of exercise in HFpEF patients has not been performed, a number of smaller studies have been reported. A retrospective analysis of these latter studies has shown improvements in cardiorespiratory fitness and quality of life that were not associated with changes in either diastolic or systolic function in HFpEF patients (Pandey et al., 2015). This suggests that exercise-mediated improvements in these patients may not be due to primary effects on cardiac function but rather peripheral effects on skeletal muscle, endothelial function or arterial stiffness (affecting peripheral vascular resistance). Given the low number of patients enrolled in the HFpEF trials to date, larger controlled and randomized studies are needed to confirm these findings. In addition, assessment of the type of exercise most beneficial in both HFpEF and HFrEF cohorts is warranted.
As noted above, virtually every signaling pathway or factor that promotes physiologic growth also provides cardioprotection or antagonizes pathologic growth in pre-clinical studies (Table 1) (Bezzerides et al., 2016; Bostrom et al., 2010; Liu et al., 2015; Weeks et al., 2012). This rationale, together with the HF-ACTION trial results, cardiac rehabilitation may now be prescribed for HFrEF patients. However, significant barriers exist for exercise therapy in these patients. Historically, adherence to a strict exercise regimen has been difficult. Even in HF-ACTION, overall adherence and the amount of time spent exercising fell over the trial period (O'Connor et al., 2009). These patients are typically older and have additional co-morbidities that prevent participation or adherence to an exercise regimen. Therefore, targeted therapies that promote exercise-signaling pathways in the heart hold promise to complement exercise regimens. However, finding safe and efficacious targets to promote physiologic hypertrophic pathways is a challenge. The IGF-1/PI3K pathway is a well-validated target in pre-clinical models. In addition, growth hormone (GH) replacement therapy represents one strategy to increase circulating IGF-1 levels. Indeed, early pre-clinical studies demonstrated a benefit of GH in experimental heart failure (Isgaard et al., 1997; Yang et al., 1995). Unfortunately, however, clinical trials with GH have been mixed and have not provided strong rationale for this approach (reviewed in (Arcopinto et al., 2013)). Targeting energy metabolism and mitochondrial dysfunction would seem to be another attractive option, possibly in combination with agents that enhance upstream signaling events. Targeting factors within the downstream PGC-1 cascade may prove effective. For example, PPARδ shares many targets with PPARα in the heart but also activates glucose oxidation driving a more balanced fuel utilization profile (Burkart et al., 2007). PPARδ also mediates the response to exercise in cooperation with AMPK signaling in skeletal muscle (Narkar et al., 2008). Therefore, selective PPARδ ligands seem an attractive target for promoting exercise-like effects in the setting of heart failure. This study also demonstrated that AMPK synergized with PPARδ in skeletal muscle suggesting that direct AMPK activators could function as exercise mimetics. AMPK activation has a variety of metabolic effects that could be of benefit in heart failure (Kim and Dyck, 2015). PGC-1a is also a direct target of AMPK providing a direct link to mitochondrial biogenesis and energy production (Jäger, PMID: 17609368). However, concerns over AMPK activation and excessive glycogen storage in cardiac myocytes have dampened enthusiasm for this approach (Arad et al., 2007). ERR may also prove to be an effective target, although to date potent activators have not been identified. In addition, the mitochondrial targeted antioxidant molecule SS-31 interacts with the mitochondrial-specific phospholipid cardiolipin (Birk et al., 2013). SS-31, also known as elamipretide, promotes mitochondrial respiration and improves cardiac function in a canine microembolization heart failure model (Birk et al., 2014; Sabbah et al., 2016). This molecule is now being tested in the clinic.
Summary and future perspectives
A greater appreciation and understanding of the specific and quantifiable benefits of exercise on cardiac structure and function has emerged over the last decade or so. Physical activity and reversal of sedentary behavior has a clear benefit on mortality risk, attributable in large part to a reduction of death from cardiovascular disease. Much of the work in this area has focused on the adaptations of the heart to exercise training. Endurance training produces growth of cardiac myocyte size with preserved contractile function. This occurs in the absence of interstitial or perivascular fibrosis or myocyte disarray commonly associated with pathologic forms of growth. In this regard, we now recognize that specific growth factors and signaling pathways orchestrate a “physiologic” or adaptive hypertrophic program that is characterized by increased cardiac myocyte size in the absence of activation of the fetal gene program. In addition, exercise training results in metabolic adaptive cardiac remodeling enhanced capacity for fuel utilization and a mitochondrial biogenic response coordinated, at least in part, by PGC-1a. The same signaling pathways responsible for cardiac myocyte growth also regulate mitochondrial adaptations demonstrating a coordinated evolved response to ensure adequate energy production to meet the increased demands of exercise. Importantly, these adaptations also antagonize pathologic signaling in the setting of heart failure or provide cardioprotection from ischemic insult. Despite mounting evidence that activation of pathways associated with physiologic growth would provide therapeutic benefit, issues related to druggability, non-cardiac side effects and efficacy remain major challenges. However, we believe that the potential benefit of the elusive “exercise mimetic” remains high enough for continued research in this area.
Acknowledgments
The authors would like to acknowledge the following funding sources. To J.K., National Institutes of Health Independent Scientist Award (K02 HL105799); American Heart Association (16GRNT31390006); Sarver Heart Center at the University of Arizona and the Steven M. Gootter Foundation. To L.A.L., NIH HL117138. To D.P.K., NIH grants R01 DK045416 (D.P.K.), R01 HL058493 (D.P.K.), and R01 HL128349 (D.P.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC, Leinwand LA. Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J Appl Physiol. 2001;90 doi: 10.1152/jappl.2001.90.5.1900. [DOI] [PubMed] [Google Scholar]
- Almeida PW, Gomes-Filho A, Ferreira AJ, Rodrigues CE, Dias-Peixoto MF, Russo RC, Teixeira MM, Cassali GD, Ferreira E, Santos IC, et al. Swim training suppresses tumor growth in mice. J Appl Physiol (1985) 2009;107:261–265. doi: 10.1152/japplphysiol.00249.2009. [DOI] [PubMed] [Google Scholar]
- Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circulation research. 2007;100:474–488. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- Arcopinto M, Bobbio E, Bossone E, Perrone-Filardi P, Napoli R, Sacca L, Cittadini A. The GH/IGF-1 axis in chronic heart failure. Endocr Metab Immune Disord Drug Targets. 2013;13:76–91. doi: 10.2174/1871530311313010010. [DOI] [PubMed] [Google Scholar]
- Arem H, Moore SC, Patel A, Hartge P, Berrington de Gonzalez A, Visvanathan K, Campbell PT, Freedman M, Weiderpass E, Adami HO, et al. Leisure time physical activity and mortality: a detailed pooled analysis of the dose-response relationship. JAMA Intern Med. 2015;175:959–967. doi: 10.1001/jamainternmed.2015.0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta. 2013;1833:840–847. doi: 10.1016/j.bbamcr.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi KC, Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation. 2016;133:706–716. doi: 10.1161/CIRCULATIONAHA.115.017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisvag V, Kemi OJ, Arbo I, Loennechen JP, Wisloff U, Langaas M, Sandvik AK, Ellingsen O. Pathological and physiological hypertrophies are regulated by distinct gene programs. Eur J Cardiovasc Prev Rehabil. 2009;16:690–697. doi: 10.1097/HJR.0b013e32833158a2. [DOI] [PubMed] [Google Scholar]
- Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- Bezzerides VJ, Platt C, Lerchenmuller C, Paruchuri K, Oh L, Xiao C, Cao Y, Mann N, Spiegelman BM, Rosenzweig A. CITED4 induces physiologic hypertrophy and promotes functional recovery after ischemic injury. JCI Insight. 2016;1 doi: 10.1172/jci.insight.85904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomqvist CG, Saltin B. Cardiovascular adaptations to physical training. Annu Rev Physiol. 1983;45:169–189. doi: 10.1146/annurev.ph.45.030183.001125. [DOI] [PubMed] [Google Scholar]
- Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budiono BP, See Hoe LE, Peart JN, Sabapathy S, Ashton KJ, Haseler LJ, Headrick JP. Voluntary running in mice beneficially modulates myocardial ischemic tolerance, signaling kinases, and gene expression patterns. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1091–1100. doi: 10.1152/ajpregu.00406.2011. [DOI] [PubMed] [Google Scholar]
- Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2004;287:H1055–1063. doi: 10.1152/ajpheart.00925.2003. [DOI] [PubMed] [Google Scholar]
- Burgess ML, Buggy J, Price RL, Abel FL, Terracio L, Samarel AM, Borg TK. Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. Am J Physiol. 1996;270:H151–159. doi: 10.1152/ajpheart.1996.270.1.H151. [DOI] [PubMed] [Google Scholar]
- Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai MX, Shi XC, Chen T, Tan ZN, Lin QQ, Du SJ, Tian ZJ. Exercise training activates neuregulin 1/ErbB signaling and promotes cardiac repair in a rat myocardial infarction model. Life Sci. 2016;149:1–9. doi: 10.1016/j.lfs.2016.02.055. [DOI] [PubMed] [Google Scholar]
- Cain PA, Ahl R, Hedstrom E, Ugander M, Allansdotter-Johnsson A, Friberg P, Arheden H. Age and gender specific normal values of left ventricular mass, volume and function for gradient echo magnetic resonance imaging: a cross sectional study. BMC Med Imaging. 2009;9:2. doi: 10.1186/1471-2342-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL, Sindler AL, Gundewar S, Seals DR, Barouch LA, et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circulation research. 2011;108:1448–1458. doi: 10.1161/CIRCRESAHA.111.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo GG, Oliveira RB, Farinatti PT. Effects of high intensity interval versus moderate continuous training on markers of ventilatory and cardiac efficiency in coronary heart disease patients. Scientific World Journal. 2015;2015:192479. doi: 10.1155/2015/192479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Heimiller J, Leinwand LA. Distinct cardiac transcriptional profiles defining pregnancy and exercise. PLoS One. 2012;7:e42297. doi: 10.1371/journal.pone.0042297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- Davis J, Maillet M, Miano JM, Molkentin JD. Lost in transgenesis: a user's guide for genetically manipulating the mouse in cardiac research. Circulation research. 2012;111:761–777. doi: 10.1161/CIRCRESAHA.111.262717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Simone Gea. Gender differences in left ventricular growth. Hypertension. 1995;26:979–983. doi: 10.1161/01.hyp.26.6.979. [DOI] [PubMed] [Google Scholar]
- de Waard MC, van Haperen R, Soullie T, Tempel D, de Crom R, Duncker DJ. Beneficial effects of exercise training after myocardial infarction require full eNOS expression. J Mol Cell Cardiol. 2010;48:1041–1049. doi: 10.1016/j.yjmcc.2010.02.005. [DOI] [PubMed] [Google Scholar]
- DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- Devereux RB, Reichek N. Echocardiographic determination of left ventricular mass in man. Anatomic validation of the method. Circulation. 1977;55:613–618. doi: 10.1161/01.cir.55.4.613. [DOI] [PubMed] [Google Scholar]
- DiCarlo SE, Bishop VS. Exercise training attenuates baroreflex regulation of nerve activity in rabbits. Am J Physiol. 1988;255:H974–979. doi: 10.1152/ajpheart.1988.255.4.H974. [DOI] [PubMed] [Google Scholar]
- Dickhuth HH, Rocker K, Hipp A, Heitkamp HC, Keul J. Echocardiographic findings in endurance athletes with hypertrophic non-obstructive cardiomyopathy (HNCM) compared to non-athletes with HNCM and to physiological hypertrophy (athlete's heart) Int J Sports Med. 1994;15:273–277. doi: 10.1055/s-2007-1021059. [DOI] [PubMed] [Google Scholar]
- Diffee GM, Nagle DF. Regional differences in effects of exercise training on contractile and biochemical properties of rat cardiac myocytes. J Appl Physiol (1985) 2003;95:35–42. doi: 10.1152/japplphysiol.00951.2002. [DOI] [PubMed] [Google Scholar]
- Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP. The role of the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 1999;60:339–343. doi: 10.1016/s0952-3278(99)80009-x. [DOI] [PubMed] [Google Scholar]
- Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345–356. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Russell DS, Duman RS. Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Res. 2008;1199:148–158. doi: 10.1016/j.brainres.2007.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldomiaty MA, Almasry SM, Desouky MK, Algaidi SA. Voluntary Running Improves Depressive Behaviours and the Structure of the Hippocampus in Rats: A Possible Impact of Myokines. Brain Res. 2016 doi: 10.1016/j.brainres.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Emter CA, McCune SA, Sparagna GC, Radin MJ, Moore RL. Low-intensity exercise training delays onset of decompensated heart failure in spontaneously hypertensive heart failure rats. Am J Physiol Heart Circ Physiol. 2005;289:H2030–2038. doi: 10.1152/ajpheart.00526.2005. [DOI] [PubMed] [Google Scholar]
- Evans DL. Cardiovascular adaptations to exercise and training. Vet Clin North Am Equine Pract. 1985;1:513–531. doi: 10.1016/s0749-0739(17)30748-4. [DOI] [PubMed] [Google Scholar]
- Evans DL, Rose RJ. Cardiovascular and respiratory responses to submaximal exercise training in the thoroughbred horse. Pflugers Arch. 1988;411:316–321. doi: 10.1007/BF00585121. [DOI] [PubMed] [Google Scholar]
- Fagard R. Athlete's heart. Heart. 2003;89:1455–1461. doi: 10.1136/heart.89.12.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagard RH. Athlete's heart: a meta-analysis of the echocardiographic experience. Int J Sports Med. 1996;17(3):S140–144. doi: 10.1055/s-2007-972915. [DOI] [PubMed] [Google Scholar]
- Fagard RH, Celis H, Thijs L, Wouters S. Regression of left ventricular mass by antihypertensive treatment: a meta-analysis of randomized comparative studies. Hypertension. 2009;54:1084–1091. doi: 10.1161/HYPERTENSIONAHA.109.136655. [DOI] [PubMed] [Google Scholar]
- Fenning A, Harrison G, Dwyer D, Rose'Meyer R, Brown L. Cardiac adaptation to endurance exercise in rats. Mol Cell Biochem. 2003;251:51–59. doi: 10.1007/978-1-4419-9238-3_8. [DOI] [PubMed] [Google Scholar]
- Fernandes T, Barauna VG, Negrao CE, Phillips MI, Oliveira EM. Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am J Physiol Heart Circ Physiol. 2015;309:H543–552. doi: 10.1152/ajpheart.00899.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finocchiaro G, Dhutia H, D'Silva A, Malhotra A, Steriotis A, Millar L, Prakash K, Narain R, Papadakis M, Sharma R, et al. Effect of Sex and Sporting Discipline on LV Adaptation to Exercise. JACC Cardiovasc Imaging. 2016 doi: 10.1016/j.jcmg.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Flynn KE, Pina IL, Whellan DJ, Lin L, Blumenthal JA, Ellis SJ, Fine LJ, Howlett JG, Keteyian SJ, Kitzman DW, et al. Effects of exercise training on health status in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA. 2009;301:1451–1459. doi: 10.1001/jama.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foryst-Ludwig A, Kreissl MC, Sprang C, Thalke B, Bohm C, Benz V, Gurgen D, Dragun D, Schubert C, Mai K, et al. Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am J Physiol Heart Circ Physiol. 2011;301:H115–122. doi: 10.1152/ajpheart.01222.2010. [DOI] [PubMed] [Google Scholar]
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- Fukazawa R, Miller TA, Kuramochi Y, Frantz S, Kim YD, Marchionni MA, Kelly RA, Sawyer DB. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardiol. 2003;35:1473–1479. doi: 10.1016/j.yjmcc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Fulton N, Rajiah P. Utility of magnetic resonance imaging in the evaluation of left ventricular thickening. Insights Imaging. 2017;8:279–293. doi: 10.1007/s13244-017-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss J, Ben Abdallah NM, Vogt MA, Touma C, Pacifici PG, Palme R, Witzemann V, Hellweg R, Gass P. Voluntary exercise induces anxiety-like behavior in adult C57BL/6J mice correlating with hippocampal neurogenesis. Hippocampus. 2010;20:364–376. doi: 10.1002/hipo.20634. [DOI] [PubMed] [Google Scholar]
- Gasiorowski A, Dutkiewicz J. Comprehensive rehabilitation in chronic heart failure. Ann Agric Environ Med. 2013;20:606–612. [PubMed] [Google Scholar]
- Gayda M, Ribeiro PA, Juneau M, Nigam A. Comparison of Different Forms of Exercise Training in Patients With Cardiac Disease: Where Does High-Intensity Interval Training Fit? Can J Cardiol. 2016;32:485–494. doi: 10.1016/j.cjca.2016.01.017. [DOI] [PubMed] [Google Scholar]
- George KP, Gates PE, Birch KM, Campbell IG. Left ventricular morphology and function in endurance-trained female athletes. J Sports Sci. 1999;17:633–642. doi: 10.1080/026404199365669. [DOI] [PubMed] [Google Scholar]
- George KP, Wolfe LA, Burggraf GW, Norman R. Electrocardiographic and echocardiographic characteristics of female athletes. Med Sci Sports Exerc. 1995;27:1362–1370. [PubMed] [Google Scholar]
- Gerdes AM, Kellerman SE, Moore JA, Muffly KE, Clark LC, Reaves PY, Malec KB, McKeown PP, Schocken DD. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation. 1992;86:426–430. doi: 10.1161/01.cir.86.2.426. [DOI] [PubMed] [Google Scholar]
- Gerdes AM, Onodera T, Wang X, McCune SA. Myocyte remodeling during the progression to failure in rats with hypertension. Hypertension. 1996;28:609–614. doi: 10.1161/01.hyp.28.4.609. [DOI] [PubMed] [Google Scholar]
- Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, Staels B, van Bilsen M. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circulation research. 2003;92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- Gounder SS, Kannan S, Devadoss D, Miller CJ, Whitehead KJ, Odelberg SJ, Firpo MA, Paine R, 3rd, Hoidal JR, Abel ED, et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One. 2012;7:e45697. doi: 10.1371/journal.pone.0045697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyatt GH, Devereaux PJ. A review of heart failure treatment. Mt Sinai J Med. 2004;71:47–54. [PubMed] [Google Scholar]
- Haram PM, Kemi OJ, Lee SJ, Bendheim MO, Al-Share QY, Waldum HL, Gilligan LJ, Koch LG, Britton SL, Najjar SM, et al. Aerobic interval training vs. continuous moderate exercise in the metabolic syndrome of rats artificially selected for low aerobic capacity. Cardiovasc Res. 2009;81:723–732. doi: 10.1093/cvr/cvn332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M. Cardiovascular Adaptations to Exercise Training. Compr Physiol. 2015;6:1–32. doi: 10.1002/cphy.c140080. [DOI] [PubMed] [Google Scholar]
- Hirai DM, Musch TI, Poole DC. Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. Am J Physiol Heart Circ Physiol. 2015;309:H1419–1439. doi: 10.1152/ajpheart.00469.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Huxley VH. Sex and the cardiovascular system: the intriguing tale of how women and men regulate cardiovascular function differently. Adv Physiol Educ. 2007;31:17–22. doi: 10.1152/advan.00099.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iemitsu M, Maeda S, Miyauchi T, Matsuda M, Tanaka H. Gene expression profiling of exercise-induced cardiac hypertrophy in rats. Acta Physiol Scand. 2005;185:259–270. doi: 10.1111/j.1365-201X.2005.01494.x. [DOI] [PubMed] [Google Scholar]
- Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–419. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgaard J, Kujacic V, Jennische E, Holmang A, Sun XY, Hedner T, Hjalmarson A, Bengtsson BA. Growth hormone improves cardiac function in rats with experimental myocardial infarction. Eur J Clin Invest. 1997;27:517–525. doi: 10.1046/j.1365-2362.1997.1430692.x. [DOI] [PubMed] [Google Scholar]
- Kasper CE. Animal models of exercise and obesity. Annu Rev Nurs Res. 2013;31:1–17. doi: 10.1891/0739-6686.31.1. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Haram PM, Loennechen JP, Osnes JB, Skomedal T, Wisloff U, Ellingsen O. Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function. Cardiovasc Res. 2005;67:161–172. doi: 10.1016/j.cardiores.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Loennechen JP, Wisloff U, Ellingsen O. Intensity-controlled treadmill running in mice: cardiac and skeletal muscle hypertrophy. J Appl Physiol. 2002;93:1301–1309. doi: 10.1152/japplphysiol.00231.2002. [DOI] [PubMed] [Google Scholar]
- Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TT, Dyck JR. Is AMPK the savior of the failing heart? Trends Endocrinol Metab. 2015;26:40–48. doi: 10.1016/j.tem.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Konhilas JP, Chen H, Luczak E, McKee LA, Regan J, Watson PA, Stauffer BL, Khalpey ZI, McKinsey TA, Horn T, et al. Diet and sex modify exercise and cardiac adaptation in the mouse. Am J Physiol Heart Circ Physiol. 2015;308:H135–145. doi: 10.1152/ajpheart.00532.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Maass AH, Luckey SW, Stauffer BL, Olson EN, Leinwand LA. Sex modifies exercise and cardiac adaptation in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2768–2776. doi: 10.1152/ajpheart.00292.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Watson PA, Maass A, Boucek DM, Horn T, Stauffer BL, Luckey SW, Rosenberg P, Leinwand LA. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circulation research. 2006;98:540–548. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- Konhilas JP, Widegren U, Allen DL, Paul AC, Cleary A, Leinwand LA. Loaded wheel running and muscle adaptation in the mouse. Am J Physiol Heart Circ Physiol. 2005;289:H455–465. doi: 10.1152/ajpheart.00085.2005. [DOI] [PubMed] [Google Scholar]
- Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin MH, Hale CC, Novela L, Gute D, Hamilton N, Ianuzzo CD. Biochemical characterization of exercise-trained porcine myocardium. J Appl Physiol (1985) 1991;71:229–235. doi: 10.1152/jappl.1991.71.1.229. [DOI] [PubMed] [Google Scholar]
- Lauschke J, Maisch B. Athlete's heart or hypertrophic cardiomyopathy? Clin Res Cardiol. 2009;98:80–88. doi: 10.1007/s00392-008-0721-2. [DOI] [PubMed] [Google Scholar]
- Lerman I, Harrison BC, Freeman K, Hewett TE, Allen DL, Robbins J, Leinwand LA. Genetic variability in forced and voluntary endurance exercise performance in seven inbred mouse strains.PG -2245–55. J Appl Physiol. 2002;92 doi: 10.1152/japplphysiol.01045.2001. [DOI] [PubMed] [Google Scholar]
- Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F, Xiao C, Bezzerides V, Bostrom P, Che L, et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584–595. doi: 10.1016/j.cmet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczak ED, Leinwand LA. Sex-based cardiac physiology. Annu Rev Physiol. 2009;71:1–18. doi: 10.1146/annurev.physiol.010908.163156. [DOI] [PubMed] [Google Scholar]
- MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol (1985) 1985;58:785–790. doi: 10.1152/jappl.1985.58.3.785. [DOI] [PubMed] [Google Scholar]
- Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ. Distinguishing hypertrophic cardiomyopathy from athlete's heart physiological remodelling: clinical significance, diagnostic strategies and implications for preparticipation screening. Br J Sports Med. 2009;43:649–656. doi: 10.1136/bjsm.2008.054726. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Wolfson JK, Epstein SE, Roberts WC. Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1986;8:545–557. doi: 10.1016/s0735-1097(86)80181-4. [DOI] [PubMed] [Google Scholar]
- McMullan RC, Kelly SA, Hua K, Buckley BK, Faber JE, Pardo-Manuel de Villena F, Pomp D. Long-term exercise in mice has sex-dependent benefits on body composition and metabolism during aging. Physiol Rep. 2016;4 doi: 10.14814/phy2.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase(p110{alpha}) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003 doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer JH, Robbers Y. Wheel running in the wild. Proceedings Biological sciences/The Royal Society. 2014;281 doi: 10.1098/rspb.2014.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole PA. Increased contractile potential of papillary muscles from exercise-trained rat hearts. Am J Physiol. 1978;234:H421–425. doi: 10.1152/ajpheart.1978.234.4.H421. [DOI] [PubMed] [Google Scholar]
- Moore RL, Musch TI, Yelamarty RV, Scaduto RC, Jr, Semanchick AM, Elensky M, Cheung JY. Chronic exercise alters contractility and morphology of isolated rat cardiac myocytes. Am J Physiol. 1993;264:C1180–1189. doi: 10.1152/ajpcell.1993.264.5.C1180. [DOI] [PubMed] [Google Scholar]
- Moore SC, Patel AV, Matthews CE, Berrington de Gonzalez A, Park Y, Katki HA, Linet MS, Weiderpass E, Visvanathan K, Helzlsouer KJ, et al. Leisure time physical activity of moderate to vigorous intensity and mortality: a large pooled cohort analysis. PLoS Med. 2012;9:e1001335. doi: 10.1371/journal.pmed.1001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy VR, Kannan S, Sadhaasivam K, Gounder SS, Davidson CJ, Boeheme C, Hoidal JR, Wang L, Rajasekaran NS. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med. 2012;52:366–376. doi: 10.1016/j.freeradbiomed.2011.10.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natali AJ, Turner DL, Harrison SM, White E. Regional effects of voluntary exercise on cell size and contraction-frequency responses in rat cardiac myocytes. J Exp Biol. 2001;204:1191–1199. doi: 10.1242/jeb.204.6.1191. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, et al. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA. 2009;301:1439–1450. doi: 10.1001/jama.2009.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest. 2005;115:2059–2064. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti G, Giordano G, Corradi D, Melissari M, Lagrasta C, Gambert SR, Anversa P. Gender differences and aging: effects on the human heart. J Am Coll Cardiol. 1995;26:1068–1079. doi: 10.1016/0735-1097(95)00282-8. [DOI] [PubMed] [Google Scholar]
- Olver TD, Ferguson BS, Laughlin MH. Molecular Mechanisms for Exercise Training-Induced Changes in Vascular Structure and Function: Skeletal Muscle, Cardiac Muscle, and the Brain. Prog Mol Biol Transl Sci. 2015;135:227–257. doi: 10.1016/bs.pmbts.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Pandey A, Parashar A, Kumbhani DJ, Agarwal S, Garg J, Kitzman D, Levine BD, Drazner M, Berry JD. Exercise training in patients with heart failure and preserved ejection fraction: meta-analysis of randomized control trials. Circ Heart Fail. 2015;8:33–40. doi: 10.1161/CIRCHEARTFAILURE.114.001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn N, Coeckelberghs E, Buys R, Cornelissen VA, Vanhees L. Aerobic interval training vs. moderate continuous training in coronary artery disease patients: a systematic review and meta-analysis. Sports Med. 2014;44:687–700. doi: 10.1007/s40279-014-0158-x. [DOI] [PubMed] [Google Scholar]
- Pelliccia A, Maron BJ, Culasso F, Spataro A, Caselli G. Athlete's heart in women. Echocardiographic characterization of highly trained elite female athletes. JAMA. 1996;276:211–215. doi: 10.1001/jama.276.3.211. [DOI] [PubMed] [Google Scholar]
- Pelliccia A, Maron MS, Maron BJ. Assessment of left ventricular hypertrophy in a trained athlete: differential diagnosis of physiologic athlete's heart from pathologic hypertrophy. Prog Cardiovasc Dis. 2012;54:387–396. doi: 10.1016/j.pcad.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Perez JN, Chen H, Regan JA, Emert A, Constantopoulos E, Lynn M, Konhilas JP. Effects of chemically induced ovarian failure on voluntary wheel-running exercise and cardiac adaptation in mice. Comparative medicine. 2013;63:233–243. [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Gargiulo G, Pironti G, Franzone A, Scudiero L, De Laurentis M, Magliulo F, Ilardi F, Carotenuto G, Schiattarella GG, et al. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am J Physiol Heart Circ Physiol. 2011;300:H1983–1989. doi: 10.1152/ajpheart.00784.2010. [DOI] [PubMed] [Google Scholar]
- Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete's heart. A meta-analysis of cardiac structure and function. Circulation. 2000;101:336–344. doi: 10.1161/01.cir.101.3.336. [DOI] [PubMed] [Google Scholar]
- Poole DC, Hirai DM, Copp SW, Musch TI. Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance. Am J Physiol Heart Circ Physiol. 2012;302:H1050–1063. doi: 10.1152/ajpheart.00943.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rainer PP, Kass DA. Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc Res. 2016;111:154–162. doi: 10.1093/cvr/cvw107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro PA, Boidin M, Juneau M, Nigam A, Gayda M. High-intensity interval training in patients with coronary heart disease: Prescription models and perspectives. Ann Phys Rehabil Med. 2016 doi: 10.1016/j.rehab.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Richter SH, Gass P, Fuss J. Resting Is Rusting: A Critical View on Rodent Wheel-Running Behavior. Neuroscientist. 2014;20:313–325. doi: 10.1177/1073858413516798. [DOI] [PubMed] [Google Scholar]
- Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. 2014;34:3450–3460. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN, Gupta RC, Kohli S, Wang M, Hachem S, Zhang K. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ Heart Fail. 2016;9:e002206. doi: 10.1161/CIRCHEARTFAILURE.115.002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton CJ, Chuang ML, O'Donnell CJ, Kupka MJ, Larson MG, Kissinger KV, Edelman RR, Levy D, Manning WJ. Gender differences and normal left ventricular anatomy in an adult population free of hypertension. A cardiovascular magnetic resonance study of the Framingham Heart Study Offspring cohort. J Am Coll Cardiol. 2002;39:1055–1060. doi: 10.1016/s0735-1097(02)01712-6. [DOI] [PubMed] [Google Scholar]
- Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634–642. doi: 10.1161/CIRCHEARTFAILURE.114.001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinowitz M, Kessler-Icekson G, Freimann S, Zimmermann R, Schaper W, Golomb E, Savion N, Eldar M. Short- and long-term swimming exercise training increases myocardial insulin-like growth factor-I gene expression. Growth Horm IGF Res. 2003;13:19–25. doi: 10.1016/s1096-6374(02)00137-5. [DOI] [PubMed] [Google Scholar]
- Scheuer J, Buttrick P. The cardiac hypertrophic responses to pathologic and physiologic loads. Circulation. 1987;75:I63–68. [PubMed] [Google Scholar]
- Scheuer J, Tipton CM. Cardiovascular adaptations to physical training. Annu Rev Physiol. 1977;39:221–251. doi: 10.1146/annurev.ph.39.030177.001253. [DOI] [PubMed] [Google Scholar]
- Serneri GG, Modesti PA, Boddi M, Cecioni I, Paniccia R, Coppo M, Galanti G, Simonetti I, Vanni S, Papa L, et al. Cardiac growth factors in human hypertrophy. Relations with myocardial contractility and wall stress. Circulation research. 1999;85:57–67. doi: 10.1161/01.res.85.1.57. [DOI] [PubMed] [Google Scholar]
- Sharma S, Firoozi S, McKenna WJ. Value of exercise testing in assessing clinical state and prognosis in hypertrophic cardiomyopathy. Cardiol Rev. 2001;9:70–76. doi: 10.1097/00045415-200103000-00005. [DOI] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soci UP, Fernandes T, Hashimoto NY, Mota GF, Amadeu MA, Rosa KT, Irigoyen MC, Phillips MI, Oliveira EM. MicroRNAs 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiol Genomics. 2011;43:665–673. doi: 10.1152/physiolgenomics.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solskov L, Magnusson NE, Kristiansen SB, Jessen N, Nielsen TT, Schmitz O, Botker HE, Lund S. Microarray expression analysis in delayed cardioprotection: the effect of exercise, AICAR, or metformin and the possible role of AMP-activated protein kinase (AMPK) Mol Cell Biochem. 2012;360:353–362. doi: 10.1007/s11010-011-1075-z. [DOI] [PubMed] [Google Scholar]
- Stepien RL, Hinchcliff KW, Constable PD, Olson J. Effect of endurance training on cardiac morphology in Alaskan sled dogs. J Appl Physiol (1985) 1998;85:1368–1375. doi: 10.1152/jappl.1998.85.4.1368. [DOI] [PubMed] [Google Scholar]
- Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J. 2005;272:2684–2695. doi: 10.1111/j.1742-4658.2005.04684.x. [DOI] [PubMed] [Google Scholar]
- Takala TE, Ramo P, Kiviluoma K, Vihko V, Kainulainen H, Kettunen R. Effects of training and anabolic steroids on collagen synthesis in dog heart. Eur J Appl Physiol Occup Physiol. 1991;62:1–6. doi: 10.1007/BF00635624. [DOI] [PubMed] [Google Scholar]
- Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circulation research. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuguchi M, Hiratsuka E, Machida S, Nishikawa T, Imamura S, Shimizu S, Nishimura M, Komuro I, Furutani Y, Furutani M, et al. Swimming exercise in infancy has beneficial effect on the hearts in cardiomyopathic Syrian hamsters. J Muscle Res Cell Motil. 2004;25:69–76. doi: 10.1023/b:jure.0000021353.82449.9e. [DOI] [PubMed] [Google Scholar]
- Teske AJ, Prakken NH, De Boeck BW, Velthuis BK, Doevendans PA, Cramer MJ. Echocardiographic deformation imaging reveals preserved regional systolic function in endurance athletes with left ventricular hypertrophy. Br J Sports Med. 2010;44:872–878. doi: 10.1136/bjsm.2008.054346. [DOI] [PubMed] [Google Scholar]
- Thomas DP, McCormick RJ, Zimmerman SD, Vadlamudi RK, Gosselin LE. Aging- and training-induced alterations in collagen characteristics of rat left ventricle and papillary muscle. Am J Physiol. 1992;263:H778–783. doi: 10.1152/ajpheart.1992.263.3.H778. [DOI] [PubMed] [Google Scholar]
- Thomas DP, Zimmerman SD, Hansen TR, Martin DT, McCormick RJ. Collagen gene expression in rat left ventricle: interactive effect of age and exercise training. J Appl Physiol. 2000;89:1462–1468. doi: 10.1152/jappl.2000.89.4.1462. [DOI] [PubMed] [Google Scholar]
- Todaka K, Wang J, Yi GH, Knecht M, Stennett R, Packer M, Burkhoff D. Impact of exercise training on ventricular properties in a canine model of congestive heart failure. Am J Physiol. 1997;272:H1382–1390. doi: 10.1152/ajpheart.1997.272.3.H1382. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzelli AS, Medeiros A, Rolim N, Bartholomeu JB, Cunha TF, Bechara LR, Gomes ER, Mattos KC, Sirvente R, Salemi VM, et al. Integrative effect of carvedilol and aerobic exercise training therapies on improving cardiac contractility and remodeling in heart failure mice. PLoS One. 2013;8:e62452. doi: 10.1371/journal.pone.0062452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275–279. doi: 10.1016/s0002-9149(01)01640-x. [DOI] [PubMed] [Google Scholar]
- Vasan RS, Larson MG, Levy D, Evans JC, Benjamin EJ. Distribution and categorization of echocardiographic measurements in relation to reference limits: the Framingham Heart Study: formulation of a height- and sex-specific classification and its prospective validation. Circulation. 1997;96:1863–1873. doi: 10.1161/01.cir.96.6.1863. [DOI] [PubMed] [Google Scholar]
- Vega RB, Horton JL, Kelly DP. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circulation research. 2015;116:1820–1834. doi: 10.1161/CIRCRESAHA.116.305420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettor R, Valerio A, Ragni M, Trevellin E, Granzotto M, Olivieri M, Tedesco L, Ruocco C, Fossati A, Fabris R, et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: role in adaptation of glucose metabolism. Am J Physiol Endocrinol Metab. 2014;306:E519–528. doi: 10.1152/ajpendo.00617.2013. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wisloff U, Kemi OJ. Animal models in the study of exercise-induced cardiac hypertrophy. Physiol Res. 2010;59:633–644. doi: 10.33549/physiolres.931928. [DOI] [PubMed] [Google Scholar]
- Waring CD, Vicinanza C, Papalamprou A, Smith AJ, Purushothaman S, Goldspink DF, Nadal-Ginard B, Torella D, Ellison GM. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. Eur Heart J. 2014;35:2722–2731. doi: 10.1093/eurheartj/ehs338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KL, Gao X, Du XJ, Boey EJ, Matsumoto A, Bernardo BC, Kiriazis H, Cemerlang N, Tan JW, Tham YK, et al. Phosphoinositide 3-kinase p110alpha is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ Heart Fail. 2012;5:523–534. doi: 10.1161/CIRCHEARTFAILURE.112.966622. [DOI] [PubMed] [Google Scholar]
- Wende AR, Schaeffer PJ, Parker GJ, Zechner C, Han DH, Chen MM, Hancock CR, Lehman JJ, Huss JM, McClain DA, et al. A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J Biol Chem. 2007;282:36642–36651. doi: 10.1074/jbc.M707006200. [DOI] [PubMed] [Google Scholar]
- White FC, McKirnan MD, Breisch EA, Guth BD, Liu YM, Bloor CM. Adaptation of the left ventricle to exercise-induced hypertrophy. J Appl Physiol (1985) 1987;62:1097–1110. doi: 10.1152/jappl.1987.62.3.1097. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Helgerud J, Kemi OJ, Ellingsen O. Intensity-controlled treadmill running in rats: VO(2 max) and cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2001a;280:H1301–1310. doi: 10.1152/ajpheart.2001.280.3.H1301. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res. 2002;54:162–174. doi: 10.1016/s0008-6363(01)00565-x. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, Ellingsen O. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. 2001b;50:495–508. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- Xiang L, Hester R. Cardiovascular Responses to Exercise. San Rafael: Biota Publishing; 2011. [Google Scholar]
- Yang L, Jia Z, Yang L, Zhu M, Zhang J, Liu J, Wu P, Tian W, Li J, Qi Z, et al. Exercise protects against chronic beta-adrenergic remodeling of the heart by activation of endothelial nitric oxide synthase. PLoS One. 2014;9:e96892. doi: 10.1371/journal.pone.0096892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Bunting S, Gillett N, Clark R, Jin H. Growth hormone improves cardiac performance in experimental heart failure. Circulation. 1995;92:262–267. doi: 10.1161/01.cir.92.2.262. [DOI] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC, et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010;12:633–642. doi: 10.1016/j.cmet.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]