

Abstract
RNA-binding proteins (RBPs) play essential roles in RNA biology, responding to cellular and environmental stimuli to regulate gene expression. Important advances have helped to determine the (near) complete repertoires of cellular RBPs. However, identification of RBPs associated with specific transcripts remains a challenge. Here, we describe “specific ribonucleoprotein (RNP) capture,” a versatile method for the determination of the proteins bound to specific transcripts in vitro and in cellular systems. Specific RNP capture uses UV irradiation to covalently stabilize protein–RNA interactions taking place at “zero distance.” Proteins bound to the target RNA are captured by hybridization with antisense locked nucleic acid (LNA)/DNA oligonucleotides covalently coupled to a magnetic resin. After stringent washing, interacting proteins are identified by quantitative mass spectrometry. Applied to in vitro extracts, specific RNP capture identifies the RBPs bound to a reporter mRNA containing the Sex-lethal (Sxl) binding motifs, revealing that the Sxl homolog sister of Sex lethal (Ssx) displays similar binding preferences. This method also revealed the repertoire of RBPs binding to 18S or 28S rRNAs in HeLa cells, including previously unknown rRNA-binding proteins.
Keywords: RNA-binding proteins, RNA interactome, ribosomal RNA, RNA–protein interactions, specific RNP capture
INTRODUCTION
RNA-binding proteins (RBPs) associating with RNAs form ribonucleoproteins (RNPs) that influence RNA's fate (Glisovic et al. 2008). Over the past few years, major technical advances in mass spectrometry and next-generation sequencing have driven the development of system-wide approaches for global analysis of RNPs. Native RNA–protein interactions in cultured cells can be covalently immobilized by short exposure to ultraviolet light at 254 nm (UV254). Nucleotide bases absorb UV light at this wavelength, generating free radicals that react with amino acids at very short range, forming covalent bonds. After immunoprecipitation, RNAs bound to the targeted RBP can be identified by next-generation sequencing (crosslinking, immunoprecipitation and sequencing, CLIP-seq) (Licatalosi et al. 2008; Milek et al. 2012). In a complementary fashion, the repertoire of proteins that bind to polyadenylated RNAs in living cells can be determined by RNA interactome capture (Baltz et al. 2012; Castello et al. 2012, 2013). This protocol uses UV irradiation, stringent oligo(dT) isolation of polyadenylated RNAs, and mass spectrometry of the crosslinked proteins. Applied to HeLa and HEK293 cells, RNA interactome capture identified more than a thousand RBPs, hundreds of which were previously unknown to bind RNA. This method was successfully used to determine the poly(A)-RNA-binding proteome of different human cell types, including human (Baltz et al. 2012; Castello et al. 2012; Beckmann et al. 2015) and murine (Kwon et al. 2013; Liao et al. 2016; Liepelt et al. 2016) cells, as well as from organisms such as S. cerevisiae (Mitchell et al. 2013; Beckmann et al. 2015; Matia-González et al. 2015), C. elegans (Matia-González et al. 2015), D. melanogaster (Sysoev et al. 2016; Wessels et al. 2016), A. thaliana (Marondedze et al. 2016; Reichel et al. 2016), and the parasites plasmodium and trypanosoma (Bunnik et al. 2016; Lueong et al. 2016). However, the identification of the RBPs bound to specific RNAs remains challenging. Approaches using MS2 loops or aptamers (Bachler et al. 1999; Srisawat and Engelke 2001; Hartmuth et al. 2002; Youngman and Green 2005; Hogg and Collins 2007; Leppek and Stoecklin 2014) require modification of the targeted RNA sequence using genetic engineering or in vitro approaches, excluding native endogenous RNAs. These approaches are also incompatible with the use of denaturing isolation conditions. The CRISPR system was recently also used to establish a specific RNA isolation protocol with RNA-guided, biotin-tagged Cas9 under physiological conditions (O'Connell et al. 2014). As this method also relies on the structural integrity and function of the Cas9 protein, it is incompatible with the highly stringent, denaturing conditions required to isolate direct RNA–protein interactions at high purity.
Antisense oligonucleotides have been extensively used to purify specific RNAs and their protein partners (Lamond et al. 1989; West et al. 2014; Chu et al. 2015; McHugh et al. 2015), as exemplified by the recent determination of the Xist RNA-bound proteome using biotinylated DNA oligonucleotides covering the entire transcript length (referred to as “tiling approach”) (Chu et al. 2015; McHugh et al. 2015). While such a strategy enables targeting of RNA lacking full integrity, the “tiling approach” cannot be used to analyze distinct transcript isoforms or particular regions of interest from within a given RNA. Some of these tiling approaches used extensive formaldehyde crosslinking, copurifying indirect RNA interactors via protein–protein interactions (West et al. 2014; Chu et al. 2015).
We describe here a method for the identification of RBPs directly bound to an endogenous, native RNA of interest under physiological conditions. This method, referred to as specific RNP capture, makes use of UV crosslinking and specific hybridization with specifically designed LNA/DNA mixmer oligonucleotides covalently coupled to a magnetic resin and mass spectrometry. This method offers notable advantages over existing methods: (i) it can be applied to endogenous transcripts; (ii) UV crosslinking allows the implementation of stringent biochemical conditions for removal of nonspecific interactions; and (iii) LNA probes increase the melting temperature and thus hybridization specificity, allowing the selective isolation of the RNA of interest by just one short probe.
RESULTS
Experimental strategy and implementation of “specific RNP capture”
To establish a method for the identification of RBPs bound to a specific transcript, we adapted the RNA interactome capture protocol (Castello et al. 2012, 2013). Specific RNP capture also uses “zero distance” UV (254 nm) protein–RNA crosslinking, but replaces oligo(dT) mediated by sequence-specific capture using antisense oligonucleotide probes (Greenberg 1979; Pashev et al. 1991). We chose locked nucleic acid (LNA)-containing 20-mer probes with full complementarity to the target RNA sequence (Fig. 1). LNA nucleosides carry a ribose ring that is “locked” by a methylene bridge connecting the 2′-O atom and the 4′-C atom. By “locking” the molecule, the LNA is constrained into a conformation that facilitates Watson–Crick base-pairing, increasing the duplex melting temperature by 2°C–10°C per LNA nucleotide (Wahlestedt et al. 2000). Our hybrid LNA/DNA probes were designed to permit selective, highly stringent hybridization conditions in spite of their limited (∼20 nucleotide [nt]) lengths. Moreover, LNAs render the probe resistant to nucleases including RNase H, when avoiding gaps of six or more consecutive deoxynucleotides (Kurreck et al. 2002). Probes are also designed to include a 3′ extension with a flexible C6 linker and a primary amine for the covalent coupling to magnetic beads. Covalent coupling between the capture probe and the beads withstands high salt concentrations and chaotropic detergents, supporting the efficient removal of noncovalently bound contaminants. Covalent coupling also facilitates the recycling of the capture probes, saving costs.
FIGURE 1.

Schematic overview of specific RNP capture. Specific RNP capture uses antisense LNA/DNA mixmer oligonucleotides that are covalently coupled to a magnetic resin. By applying UV crosslinking on living cells or extracts at 254 nm, direct RNA–protein interactions are covalently fixed. After cellular lysis or within the extracts, hybridization with the covalently coupled oligonucleotides is performed under stringent denaturing conditions at elevated temperatures. The RNA-bound proteins are identified by quantitative mass spectrometry in comparison to negative controls.
We designed LNA/DNA probes targeting the Renilla luciferase (Rluc) open reading frame (ORF) (LNA1) and human 18S and 28S rRNA (18S LNA and 28S LNA), following established LNA design guidelines (Exiqon) and adjusting the melting temperature of the oligonucleotide–RNA hybrids to ∼80°C (scheme in Fig. 2A). As a control, we used an LNA/DNA mixmer with a randomized sequence (LNAscr). The sequence of the probe on the RNA was selected to avoid complementarity with sequences present in other transcripts. When a limited number of nucleotides exhibited complementarity with a nontarget RNA, we avoided insertion of LNA at these positions. In addition, probes were designed to hybridize to regions lacking detectable stable secondary structures, as determined with mfold (Zuker 2003) or from published structural information for the rRNAs (Anger et al. 2013).
FIGURE 2.

Specific RNP capture in vitro using recombinant Sxl. (A) Schematic summary of the experimental design of specific RNP capture in cell-free systems supplemented with recombinant Sxl protein. HeLa in vitro translation extracts (Cilbiotech) are programmed with recombinant GST-tagged Sxl protein and Rluc reporter RNAs harboring either the U-rich Sxl-binding sites (wt) or a mutated version of these. After UV crosslinking, specific RNP capture was performed with LNA1 targeting the Rluc ORF, with the LNAscr as a negative control. (B) RT-qPCR analysis of the RNA isolated with the different LNAs. RNA levels in eluates are displayed relative to the input sample. (C) After RNase treatment, proteins copurified with the target RNA were separated by SDS-PAGE and detected by silver staining. Recombinant Sxl-GST was loaded as a control. (D) Western blot analysis of the eluates using an antibody against Sxl.
Specific RNP capture in vitro
We first tested the specific RNP capture protocol in vitro, using the well-studied interaction between Drosophila Sex lethal (Sxl) and the polyuridine (U) stretches located within the 5′ (A and B site) and 3′ UTR (E and F sites) of the male specific lethal-2 (msl-2) mRNA (Gebauer et al. 2003; Beckmann et al. 2005). The interaction of Sxl with poly(U) motifs has been well-characterized biochemically and structurally (Inoue et al. 1990; Valcárcel et al. 1993; Handa et al. 1999; Hennig et al. 2014). In addition to its interaction with msl-2 mRNA (Bashaw and Baker 1995; Kelley et al. 1995, 1997; Bashaw and Baker 1997), Sxl also binds to poly(U) motifs present in its own mRNA (Cline 1984; Bell et al. 1991) and in the transformer mRNA (tra) (Boggs et al., 1987). It is predicted to interact with hundreds of Drosophila RNAs containing analogous motifs (Medenbach et al. 2011). We cloned the Sxl-binding sites of msl-2 mRNA upstream of and downstream from an Rluc open reading frame, resembling the architecture of native msl-2 mRNA (scheme in Fig. 2A). This construct harbors a U16 stretch in the 5′ UTR (B site) and two U7 stretches in the 3′ UTR (E and F sites). A mutant with every second U replaced by a cytidine (C) in all these binding sites (B, E, and F) was used as negative control for Sxl binding (Gebauer et al. 2003).
First, we performed specific RNP capture in HeLa cytoplasmic extracts supplemented with recombinant Sxl and programmed with wild-type (wt) or mutant (mut) Rluc RNAs. RNA analysis by RT-qPCR showed that LNA1 highly enriches the targeted Rluc RNAs (dark gray bars; Fig. 2B), over other RNAs (β-actin and 18S rRNA, petrol blue and cyan bars; Fig. 2B). After selection, the background level of these nontargeted RNAs is comparable to that with the LNAscr control mixmer. Thus, the target RNAs are specifically enriched by the designed antisense LNA/DNA probes. Silver staining and Western blotting analyses show that LNA1 only captures recombinant Sxl from HeLa extracts when these are programmed with wt Rluc RNA (Fig. 2C,D). In contrast to LNA1, the scrambled LNA (LNAscr) fails to purify Sxl. Importantly, LNA1 does not purify Sxl when its binding sites are mutated (“mut”). The capture of Sxl hence depends on (i) specific targeting of the mRNA with a complementary probe and (ii) the presence of a functional Sxl-binding site within the captured mRNA. Pull-down of endogenous polyadenylated mRNAs from HeLa extracts with oligo(dT) beads captures Sxl irrespective of whether wt and mut Rluc RNA is added to the extract. This result implicates the presence of Sxl-binding sequences (U-rich tracts) within endogenous HeLa cell mRNAs. Taken together, the results show that specific RNP capture successfully re-identifies the binding of Sxl to the msl-2 mRNA with high specificity.
Identification of novel RBPs by specific in vitro RNP capture combined with quantitative mass spectrometry
To determine whether specific RNP capture can be used to identify proteins binding to an RNA of interest in vitro, we programmed Drosophila embryo extracts with the Rluc mRNA bearing the Sxl-binding sites or its mutant version, and, after RNA capture, bound proteins were analyzed by MS. As before, we observed consistent and specific enrichment of the target RNAs (Fig. 3A) and of their bound proteins using the specific LNA1 capture probe compared to the LNAscr control (Fig. 3B,C; Supplemental Table S1). Only minor differences in the pool of copurified proteins could be observed between the wt and the mut Rluc reporter RNAs using LNA1 (Fig. 3D; Supplemental Table S1), which reflect the near identity of these two mRNAs. However, the sister of Sex lethal (Ssx), heterogeneous nuclear ribonucleoprotein at 98DE (Hrb98DE, Hrp38) and heterogeneous nuclear ribonucleoprotein at 87F (Hrb87F, Hrp36) were reproducibly enriched with the wt over the mut Rluc RNA (Fig. 3D). Unexpectedly, Ssx rather than Sxl, which was previously identified as a regulatory protein that binds to the msl-2 mRNA regulatory sequences (Fig. 2C,D; Gebauer et al. 2003), was identified as the specific binder to the wt mRNA. Ssx and Sxl are paralogs that arose from a gene duplication event (Traut et al. 2006; Cline et al. 2010). Ssx is highly homologous to Sxl, and was previously identified as an RBP without assigned binding targets (Sysoev et al. 2016; Wessels et al. 2016). The similarity between the two RBPs is especially high across the two RNA recognition motifs (RRMs) as well as in the glycine-rich region in the N terminus (52% identity; 72% similarity via LALIGN Readseq version 2.1.30). Taken together, the results identify Ssx as an RBP with similar preference for the U-rich motifs of msl-2 mRNA as its paralog Sxl.
FIGURE 3.

Specific RNP capture in Drosophila melanogaster extracts. (A) RT-qPCR analysis of RNA isolated with the different LNAs. RNA levels in eluates are displayed relative to the input sample. The error bars represent standard deviation from four biological replicates. (B,C) Volcano plots comparing the fold change (x-axis) and the P-value (y-axis) of each identified protein in eluates from LNA1 capture in extracts programmed Rluc wt mRNA (B), or its mutated counterpart (C), versus a capture performed with the LNAscr control. (D) As in B and C, but comparing extracts programmed with Rluc wt mRNA compared to its mutated version. Data shown in B,C correspond to four biological replicates. Orange and red dots represent proteins with 5% FDR and 1% FDR, respectively.
To understand the unexpected lack of Sxl from the specific RNP purification, we analyzed the whole proteome of the Drosophila embryo extracts used for these experiments (Supplemental Table S2). While Ssx was identified with a medium intensity score, Sxl was not detected. Thus, the concentration of Sxl in the 0–12 h, mixed male/female embryo extract is very low, and Sxl appears to have been outcompeted by its ubiquitously expressed paralog Ssx. Sxl has previously been identified by mass spectrometry as a component of the D. melanogaster RNA interactome, but found to change during maternal-to-zygotic transition (Sysoev et al. 2016). Thus, the lack of Sxl from the specifically captured RNP appears to have biological rather than technical reasons.
In addition to Ssx, Hrb87F and Hrb98DE were also significantly enriched in the wt Rluc mRNA sample. These are splicing factors that share a high degree of homology with each other (Haynes et al. 1991; Blanchette et al. 2009). Sxl can interact with Hrb87F, most likely via its glycine-rich region, and it increases the affinity of Hrb87F for U-rich sequences (Wang et al. 1997). As the glycines in the glycine-rich region of Ssx and Sxl are conserved, Ssx may also be able to recruit Hrb87F as its paralog Sxl.
Determination of the rRNA interactomes of HeLa cells by specific RNP capture
We next adapted the protocol to living cells. First, we aimed to estimate the minimal quantity of captured RNA required to obtain high-quality proteomic information. To avoid bias by the specific nature of an individual RNP, we used the analogous RNA interactome capture in HeLa cells, which uses UV crosslinking and oligo(dT) selection to purify the proteins bound to poly(A) RNA (Baltz et al. 2012; Castello et al. 2012). We performed serial dilutions of the eluates and analyzed these by proteomics (Fig. 4A). More than 300 peptides were identified when using 2 µg of eluted RNA. In contrast, only a dozen of the peptides were detected with 0.5 µg of eluted RNA or with a nonirradiated control. Therefore, more than 0.5 µg of eluted RNA may be required to separate signal from noise, and the use of 2 µg of eluted RNA notably improved the MS output to a satisfactory level.
FIGURE 4.

Specific RNP captures of 18S and 28S rRNAs from HeLa cells. (A) HeLa cells were UV-irradiated and poly(A) RNA was subsequently purified with oligo(dT). Two micrograms or 0.5 µg of eluted RNA was analyzed by mass spectrometry. A nonirradiated sample (NocCl) was used as control. The number of identified peptides is shown. (B) RNA eluates using probes against 28S rRNA or 18S rRNA were analyzed by bioanalyzer. Panels show the electropherograms of input (upper panels), 28S capture (middle upper panels), 18S capture (middle bottom panels), and capture with LNAscr of one representative experiment. The two peaks observed in the input samples correspond to 18S (2000 nt) and 28S (4000 nt) rRNAs. The y-axes display the fluorescence intensity derived from the dye labeling the RNA.
We selected the ribosomal 18S and 28S RNAs because of their high copy number per cell and their biological relevance for ribosome biogenesis and translation. Little is known about nonribosomal proteins binding to the ribosome, especially those that interact with the so-called expansion segments that distinguish mammalian rRNAs from those of lower eukaryotes and bacteria (Gerbi 1996; Melnikov et al. 2012; Anger et al. 2013). We designed specific LNA/DNA probes targeting regions of the 18S and 28S rRNAs that are predicted to be mostly single-stranded. Bioanalyzer traces of the captured RNAs revealed a profound enrichment of the targeted rRNA (18S or 28S rRNA) while its rRNA counterpart (i.e., 28S rRNA for the 18S rRNA capture and vice versa) was strongly depleted (Fig. 4). The LNAscr control served to define a nonspecific background.
A number of proteins were reproducibly and strongly enriched in 28S and 18S rRNA captures over the LNAscr and non-UV-irradiated negative controls (Tables 1, 2; Supplemental Table S3). Most of the proteins listed in Tables 1 and 2 as specific interactors of 18S and/or 28S rRNA are undetectable in the eluates from the scrambled control probe (Fig. 5A–C; Supplemental Table S3). As expected, the components of the large ribosomal subunit Rpl29, Rpl31, Rpl5, and Rpl4 were identified only in the 28S rRNA interactome; whereas the small ribosomal subunit members RpsA, Rps15, and Rps24 were identified only in the 18S rRNA interactome. The 18S and 28S rRNA interactomes thus displayed the expected differences from each other. Most of the proteins present in either of the two rRNA interactomes were known components of the cytoplasmic ribosome, associated with ribosomal function, or involved in ribosome biogenesis (Tables 1, 2). However, the proteins identified in the 18S and 28S rRNA interactomes constitute only a subset of all ribosomal proteins. The lack of some ribosomal proteins from the data sets is likely due to the inefficient crosslinking of proteins bound to double-stranded (ds)RNA, which involves much of the ribosomal RNA (Ricci et al. 2014). The ribosomal proteins captured here are likely to interact mostly with single-stranded stretches of the rRNAs.
TABLE 1.
RBPs enriched by 28S rRNP capture
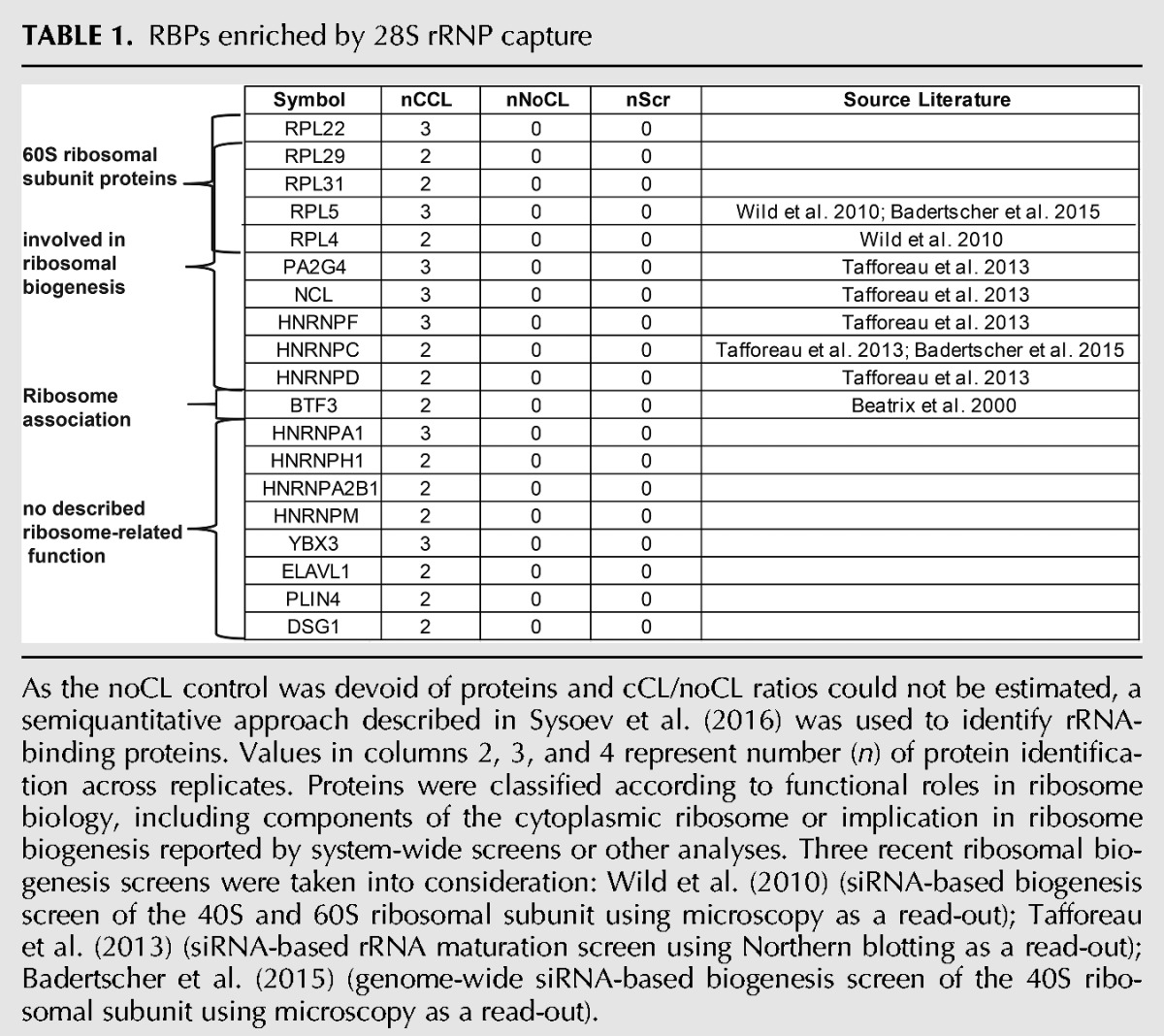
As the noCL control was devoid of proteins and cCL/noCL ratios could not be estimated, a semiquantitative approach described in Sysoev et al. (2016) was used to identify rRNA-binding proteins. Values in columns 2, 3, and 4 represent number (n) of protein identification across replicates. Proteins were classified according to functional roles in ribosome biology, including components of the cytoplasmic ribosome or implication in ribosome biogenesis reported by system-wide screens or other analyses. Three recent ribosomal biogenesis screens were taken into consideration: Wild et al. (2010) (siRNA-based biogenesis screen of the 40S and 60S ribosomal subunit using microscopy as a read-out); Tafforeau et al. (2013) (siRNA-based rRNA maturation screen using Northern blotting as a read-out); Badertscher et al. (2015) (genome-wide siRNA-based biogenesis screen of the 40S ribosomal subunit using microscopy as a read-out).
TABLE 2.
RBPs enriched by 18S rRNP capture
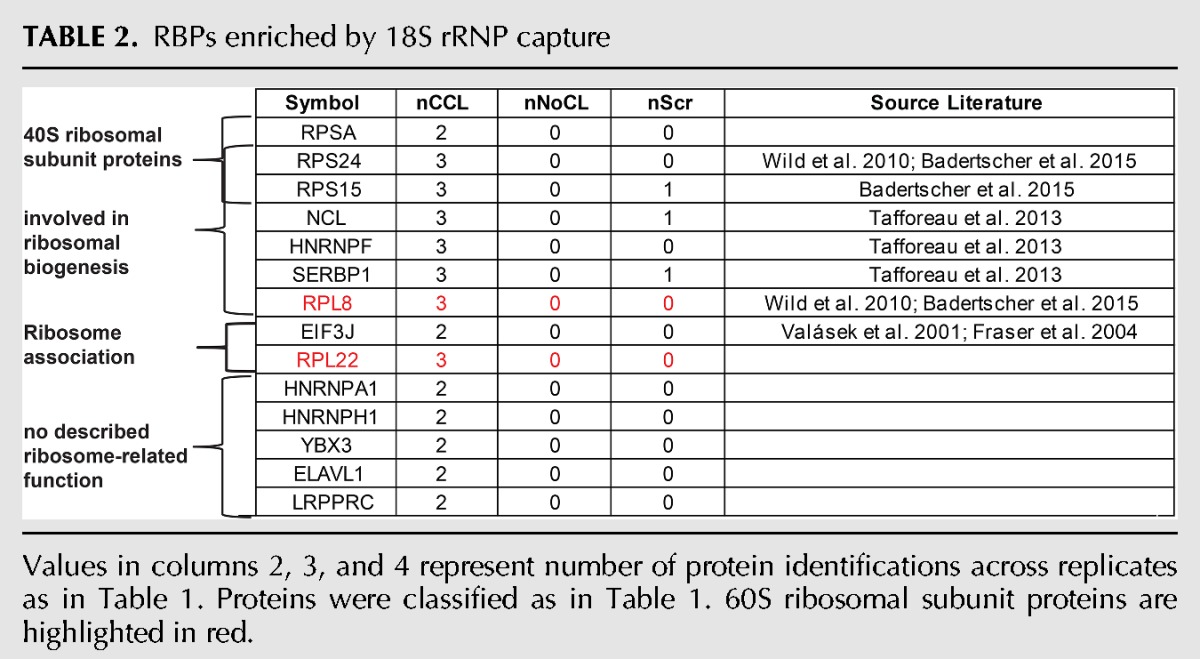
FIGURE 5.

Mass spectrometric analysis of the 18S and 28S captures. (A) Bar plot showing the MS intensities (Supplemental Table S3) of the small ribosomal subunit proteins in eluates from the 18S, 28S, and scrambled capture probes. 2E15 is used as the lower value in the y-axis because it corresponds to the approximate noise level in these MS experiments. (B) As in A, but for the large ribosomal subunit proteins. (C) As in A, but for other abundant RBPs.
Two large ribosomal proteins, Rpl8 and Rpl22, were enriched in both 18S and 28S rRNA interactomes. Rpl8 was identified in previous screens as a potential ribosome biogenesis factor (Wild et al. 2010; Badertscher et al. 2015) and could potentially interact with both the 18S and the 28S rRNA by residing close to the intersubunit space (Anger et al. 2013). However, the signal intensity for Rpl8 in the 18S rRNA interactome is similar to that in the 28S scrambled control data set (Fig. 5B), and it is substantially lower in the 18S than in the 28S rRNA interactome data set. Thus, it is also possible that Rpl8 is a contaminant of the 18S rRNA interactome. In contrast, Rpl22 protein intensity is higher in the 28S than in the 18S rRNA interactome; however, in both cases the protein intensity was significantly higher than the measured intensities in the scrambled control probes. The identification of Rpl22 in the 18S rRNA interactome may reflect an interaction with the 45S pre-rRNA prior to the processing into the mature 28S, 5S, and 18S rRNAs.
In addition to the expected ribosomal proteins, several nonribosomal proteins were enriched in the 28S and 18S rRNA interactomes. eiF3J, SERBP1, and LRPPRC were specifically enriched in the 18S rRNA interactome data sets, whereas PA2G4, HNRNPC, HNRNPD, BTF3, HNRNPA2B1, HNRNPM, PLIN4, and DSG1 were identified as specific components of the 28S rRNA interactome. SERBP1 (also known as plasminogen activator inhibitor 1 RNA-binding protein) was previously identified within the structure of the 80S ribosome, mostly in association with the 40S subunit (Anger et al. 2013). This is consistent with the observed interaction of SERBP1 with the 18S rRNA. The protein eIF3J is a component of the eukaryotic translation initiation factor 3 (eIF3) complex that associates with the 40S ribosomal subunit (Fraser et al. 2004). In Saccharomyces cerevisiae, eIF3J has also been shown to be required for the processing of 20S pre-rRNA into 18S rRNA and to bind the 18S rRNA (Valásek et al. 2001). These data are consistent with the interaction of eIF3J with 18S rRNA observed here.
The 28S rRNP-binding protein PA2G4 (proliferation-associated 2G4; other name = EBP1) is a double-stranded RNA-binding protein (Squatrito et al. 2006) that is present in preribosomal ribonucleoprotein complexes and has been implicated in ribosome maturation and in the regulation of rRNA processing (Squatrito et al. 2004). Its ortholog in yeast, Arx1, was identified to be a nuclear export receptor for the 60S ribosomal subunit (Hung et al. 2008). Structural data in yeast show that Arx1 interacts with the 28S rRNA expansion segment ES27L (Greber et al. 2012). It binds the ribosome close to the peptide exit tunnel, where it inhibits the association of translation factors before the maturation of the ribosome is complete. BTF3 (also known as nascent polypeptide-associated complex subunit β) is part of the nascent polypeptide-associated complex (Rospert et al. 2002). It was previously described to be associated with the ribosome and to prevent inappropriate ribosome binding to ER membranes (Beatrix et al. 2000). As the nascent peptide exit tunnel resides within the large ribosomal subunit, finding BTF3 as a component of the 28S rRNA interactome but not in the 18S rRNA interactome meets the expectations.
Identification of previously unrecognized ribosomal RBPs
Both rRNA interactomes harbor proteins that were previously unknown to interact with rRNAs or to be involved in rRNA or ribosome biology. While some of these RBPs are abundant, their absence or low abundance in the eluates from the scrambled control probes suggests specific interactions with rRNA in vivo (Fig. 5C). YBX3, ELAVL1, HNRNPA1, and HNRNPH are present in both data sets, while LRPPRC is specifically enriched in the 18S rRNA interactome, and Perilipin-4 (PLIN4) and Desmoglein (DSG1) in the 28S rRNA interactome. Proteins present in both rRNA interactomes with nuclear localization may play a role in rRNA biogenesis.
ELAVL1 (also known as HUR) is a RBP known to bind to AU-rich elements (AREs) (Herdy et al. 2015). An interesting aspect of ELAVL1 is that it preferentially binds mRNAs that lack N6-methyladenosine (m6A) sites, stabilizing targeted mRNAs (Wang et al. 2014). As human 18S and 28S rRNAs are m6A modified (Motorin and Helm 2011), ELAVL1 could potentially bind to rRNA depending on the presence or absence of this modification during biogenesis. LRPPRC (leucine-rich pentatricopeptide repeat containing) is a RBP that associates with both nuclear and mitochondrial mRNAs (Mili and Piñol-Roma 2003). Some evidence implicates LRPPRC in nuclear mRNA export, while mitochondrial LRPPRC controls RNA processing and translation (Sasarman et al. 2010; Chujo et al. 2012). Thus, LRPPRC could be involved in rRNP export from the nucleus, akin to what has been described for mRNA export.
The occurrence of Perilipin-4 (PLIN4) and Desmoglein-1 (DSG1) in the 28S rRNA interactome is surprising, because neither of these has been associated with RNA binding before nor has played a biological role in the ribosome's life or function. DSG1 is associated with cell junctions (Ohsugi et al. 1997), whereas PLIN4 is involved in the biogenesis of lipid droplets (Heid et al. 2014). Thus, their unexpected association with 28S rRNA may point to possible noncanonical functions to be explored in the future.
The HNRNP family meets rRNA
Interestingly, several heterogenous ribonucleoproteins (HNRNPs) were identified in both rRNA interactome data sets, suggesting a role of these proteins in the biogenesis of ribosomal RNA. The identification of some of the enriched HNRNPs as regulators of the ribosomal biogenesis in system-wide screens provides strong evidence of their involvement in rRNA life. HNRNPC/F knockdown was previously shown to affect the early processing of 18S and 28S rRNAs (Tafforeau et al. 2013). HNRNPC may thus interact with the pre-rRNA before cleavage of the internal transcribed spacer 1 (ITS1) that separates 18S and 28S rRNA. A recent study revealed that RNA binding of HNRNPC is enhanced upon addition of m6A modifications (Liu et al. 2015), and it is possible that HNRNPC interacts with rRNA in an m6A-dependent manner. Furthermore, HNRNPD was identified in the same study as being involved in large ribosomal subunit maturation (Henras et al. 2015). This finding agrees with the strong enrichment of HNRNPD in the 28S rRNA interactome and its absence in the 18S rRNA interactome.
DISCUSSION
Based on the data obtained in Drosophila embryo extracts and human HeLa cells, specific RNP capture complements currently existing protocols and offers notable advantages. The recently described CHART-MS (capture hybridization analysis of RNA targets) (West et al. 2014), CHIRP-MS (chromatin isolation by RNA purification) (Chu et al. 2015), and RAP-MS (RNA antisense purification) (McHugh et al. 2015) are all based on so-called “tiling approaches,” which use sets of overlapping biotinylated DNA oligonucleotides that cover the length of the whole transcript. The advantage of the tiling approaches is that RNA integrity is less critical than for specific RNP capture, which uses just one oligonucleotide to capture the RNA of interest. However, the use of multiple oligonucleotides can aggravate background problems. In fact, application of the three tiling methods to Xist RNA has yielded notably inconsistent results. As the tiling probes are made of DNA, they require less stringent and selective biochemical conditions than LNA/DNA mixmers. Finally, tiling approaches cannot be used for targeting distinct transcript isoforms or particular regions of an RNA of interest.
A protocol suitable for the isolation of endogenous mRNA, called TRIP (tandem RNA isolation procedure), has also been described (Matia-González et al. 2017). TRIP uses short (21–24 nt) 3′-biotinylated 2′-O-methylated antisense RNA oligonucleotides. The 2′-O-methyl groups increase stability against general base hydrolysis and nucleases. In contrast to the tiling methods, this approach has not yet been combined with mass spectrometric analyses. A current drawback of this method is the requirement of a poly(A) enrichment step and the reported limited enrichment of the target RNAs.
The above-mentioned methods all share the use of streptavidin–biotin interactions to link the oligonucleotides to a support resin in a noncovalent manner. Streptavidin-based pull-down can lead to the copurification of naturally biotinylated proteins present in many cell extracts, since biotin is a naturally occurring molecule.
Notably, specific RNP capture has some limitations of its own. UV crosslinking is inefficient by nature (1%–5%) (Darnell 2010), especially for proteins that bind double-stranded RNA (Ricci et al. 2014) or interact with RNA transiently. Using HeLa cell-derived RNA interactomes, we estimated that ∼2 µg of isolated RNA are required to obtain high-quality MS data from UV crosslinked RNPs (Fig. 4A). This quantity translates into a requirement for high RNA expression levels and/or biological material for purification. Together with a limited RNA recovery of typically ∼20%, specific RNP capture is not applicable to transcripts of low abundance. To increase RNA recovery, multiple LNA/DNA mixmer probes could be combined. However, it may be important to conduct independent analyses of the individual probes to ascertain similar specificity and stringency as with the single probe approach. To increase the yield of the protein components of the RNPs, this protocol could be combined with more highly efficient crosslinkers such as formaldehyde (Solomon and Varshavsky 1985; Niranjanakumari et al. 2002).
Finally, specific RNP capture could be extended by combination with RNase H digestion (e.g., Uyeno et al. 2004) to enrich for specific regions of a target RNA, such as ribosomal expansion segments. Continued improvements in sample preparation and mass spectrometry technologies will also extend the range of target RNAs for specific RNP capture.
MATERIALS AND METHODS
Plasmids and in vitro transcription
The plasmids pBluescript-BLEF and p-Bluescript-BmL(EF)m were described before (Gebauer et al. 2003). The Renilla luciferase version of these, i.e., p-Bluescript-BREF and p-Bluescript-BmR(EF)m, were generated by exchanging the firefly luciferase (Fluc) ORF by the Renilla version derived from the pRL-null plasmid (Promega), using XmaI and HpaI restriction sites. The plasmids bearing the Fluc ORF or chloramphenicol acetyl transferase (CAT) under the transcriptional control of T3 and T7 polymerases were previously described (Iizuka et al. 1994; Gebauer et al. 1999). Rluc, Fluc, and CAT reporter mRNAs were transcribed using the linearized (HindIII) plasmids in the presence of m7GpppG cap (KEDAR S.C.) using T7 or T3 in vitro transcription kits (MEGAscript; Thermo Fisher Scientific) following the manufacturer's recommendations. RNA was purified using the Quick-RNA MiniPrep kit (Zymo), RNA integrity was confirmed by gel electrophoresis, and translational activity was analyzed by in vitro translation assays followed by a dual luciferase activity assay.
In vitro translational extracts
Translation-competent Drosophila melanogaster embryo extracts were prepared as previously described (Gebauer et al. 1999), omitting treatment with micrococcal nuclease. Translation reactions (Gebauer et al. 1999; Rakotondrafara and Hentze 2011) consist of 40% Drosophila embryo or HeLa cytoplasmic extracts, 16 mM HEPES/KOH (pH 7.4), 50 µM Spermidine, 2% recombinant RNase A Inhibitor (prepared in house), 100 µM GTP, 800 µM ATP, 80 mM KOAc, 2 mM Mg(OAc)2, 60 μM amino acids mix, 20 mM creatine phosphate, 400 ng creatine kinase. Reactions were incubated for 90 min at 25°C for Drosophila embryo extracts and for 30 min at 37°C for HeLa cytoplasmic extracts. Luciferase activities were measured with the Dual luciferase assay system (Promega) in a microplate luminometer (Berthold).
Cell culture
Adherent Kyoto HeLa cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 4.5 mg/L D-glucose (Sigma-Aldrich), 10% heat inactivated fetal bovine serum (Gold, GE Healthcare), 2 mM L-glutamine (Sigma-Aldrich), and 10 mL/L penicillin/streptomycin mix (Sigma-Aldrich; P4333) at 37°C in 5% CO2. Cells were seeded in 500 cm2 dishes (Corning) and grown overnight to reach 70%–80% confluence.
LNA/DNA mixmer oligonucleotides
Antisense 21–23 nt LNA/DNA mixmer oligonucleotides were designed to avoid regions of strong secondary structure within the target RNA. Secondary structures were either predicted using prediction tools (mfold) (Zuker 2003), or the experimentally determined RNA secondary structure of rRNAs was used (Anger et al. 2013). To minimize off-target effects, the specificity of the antisense sequence was checked by performing BLAST searches against the genome and the transcriptome of interest. We used the general design tools provided by Exiqon (https://www.exiqon.com/oligo-tools) to determine the melting temperature (Tm), self-complementarity and dimerization potential. At the 3′ end of the mixmers, a flexible C6 linker carrying a primary amino group was introduced to couple the probes covalently to beads. All probes (Exiqon) were HPLC purified.
LNA scramble: A+CAC+TTAAC+CGTA+TAT+TCC+TA/3AmMO
LNA1: ACC+TATA+AGA+AC+CAT+TACC+AGA/3AmMO/
LNA 18S rRNA: T+TAA+TCA+TGG+CCTC+AGTT+CCGA/3AmMO/
LNA 28S rRNA: CC+AA+TCC+TTAT+CCCG+AAGTT+AC/3AmMO/
Oligonucleotide coupling to carboxylated magnetic beads
Probes were coupled in DNA/RNA low binding tubes (Eppendorf) to the carboxy-activated surface of magnetic beads (PerkinElmer MPVAC11) via the 3′ aminomodifier. First, 50 mg/mL of bead slurry were washed three times in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer, pH 6. The coupling activator N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl; Sigma-Aldrich) was freshly prepared as a 20 mg/mL solution in MES buffer. For each 100 µL of bead slurry, 10 nmol of probe in 100 µL of H2O and 500 µL of EDC-HCl solution was added. Coupling was performed in a thermomixer for 5 h at 50°C and at 800 rpm. After collecting the beads with a magnet, coupling performance was controlled by NanoDrop measurements of the supernatant. Upon coupling, beads were washed three times with PBS, and residual carboxyl residues were inactivated by incubating the beads with 200 mM ethanolamine pH 8.5 for 1 h at 37°C. Beads were finally washed three times with 1 M NaCl and stored in PBS supplemented with 0.1%Tween.
Specific RNP capture with in vitro transcribed reporter RNAs
Specific RNP capture was performed in 7.5 mL of translation mix containing 3 mL of cleared HeLa cytoplasmic extracts (Cilbiotech), 65 µg Rluc reporter mRNA, and a fourfold molar excess of recombinant, GST-tagged RNA-binding region of Sxl (a generous gift from the Medenbach laboratory). Translation reactions were incubated at 25°C for 1 h. Next, reactions were evenly spread out onto 500 cm2 cell culture dishes and irradiated with 300 mJ/cm2 of UV light at 254 nm. Two milliliters of 5× lysis buffer (100 mM Tris pH 7.5; 2.5M LiCl; 5 mM EDTA; 25 mM DTT; LiDS at a final concentration of 0.5% was added separately to the sample due to precipitation) supplemented with protease inhibitor (complete EDTAfree, Roche) were added to the crosslinked extracts. Of note, 250 µL LNA/DNA mixmer oligonucleotide-coupled magnetic beads were washed three times with TE buffer (10 mM Tris pH 7.5; 1 mM EDTA) and then blocked with 100 µg yeast tRNA. Subsequently, probes were washed in lysis buffer (20 mM Tris pH 7.5; 0.5 M LiCl; 0.5% LiDS, 1 mM EDTA, 5 mM DTT) followed by a short incubation for 3′ at 95°C. Probes were then added to the lysate and incubated for 2 h in a water bath at 41°C, mixing the samples every 15 min by inverting the tubes four times. Control oligo(dT) was incubated for 2 h at 25°C. The beads were then washed with buffers containing decreasing concentrations of LiCl and LiDS, as described previously (Castello et al. 2013; Lysis Buffer: 20 mM Tris-HCl [pH 7.5], 500 mM LiCl, 0.5% LiDS [wt/vol, stock 10%], 1 mM EDTA and 5 mM DTT; Buffer 1: 20 mM Tris-HCl [pH 7.5], 500 mM LiCl, 0.1% LiDS [wt/vol], 1 mM EDTA, and 5 mM DTT; Buffer 2: 20 mM Tris-HCl [pH 7.5], 500 mM LiCl, 1 mM EDTA, and 5 mM DTT; Buffer 3: 20 mM Tris-HCl [pH 7.5], 200 mM LiCl, 1 mM EDTA, and 5 mM DTT). RNAs and crosslinked proteins were eluted with TE buffer (10 mM Tris pH 7.5; 1 mM EDTA) by incubation in a thermomixer for 3 min at 90°C and 800 rpm. For RNA analyses, samples were digested with proteinase K (Roche) as previously described (Castello et al. 2013) followed by the isolation of RNA with Tri Reagent (Sigma-Aldrich). For protein analyses, samples were treated with RNase T1 and RNase A (Sigma-Aldrich) as previously described (Castello et al. 2013), and released proteins were analyzed by silver staining and Western blotting.
For capture of Rluc mRNA from Drosophila embryo extracts, 10 µg (32 pmol) of in vitro transcribed reporter mRNA was transferred to 2 mL low binding tubes (Eppendorf) and denatured at 95°C for 3 min. Next, the reporter RNAs were added to 2 mL of Drosophila melanogaster embryo extracts and incubated for 30 min at 25°C. Next, the extracts were evenly dispersed on a 15 cm circular cell culture dish (Corning) placed on ice, and irradiated with 300 mJ/cm2 of UV light at 254 nm. After crosslinking, the sample was supplemented with 2 mL of 2× lysis buffer (composition, include the RNase and protease inhibitors in there). Probes were prepared as described above. Samples were incubated with 200 µL of beads for 2 h at 41°C, the pull-down was performed as described above. Eluted RNA was analyzed by RT-qPCR, the crosslinked proteins by mass spectrometry.
Specific RNP capture in HeLa cells
RNA capture from cells was performed as follows. For each of three replicates, three samples were prepared: one for the specific 18S or 28S rRNA pull-downs subjected to crosslinking, one with the specific anti-18S or 28S LNA probes with noncrosslinked lysate, and one with a scrambled LNA control on crosslinked lysate. Per sample, four 70% confluent 500 cm2 dishes of HeLa cells at 70% confluence were washed twice with 30 mL of PBS. PBS was removed and the cell monolayer was irradiated on ice with 150 mJ/cm2 of UV light at 254 nm. Cells were subsequently harvested and lysed into 4 mL of 1× lysis buffer (20 mM Tris-HCl [pH 7.5], 500 mM LiCl, 0.5% LiDS [wt/vol, stock 10%], 1 mM EDTA and 5 mM DTT) supplemented with protease inhibitors (complete EDTAfree, Roche) and RNAsIn. Lysates were homogenized through a narrow gauge needle (0.4 mm diameter). After prewarming the lysate to 60°C, 15% v/v ethylenecarbonate (Sigma-Aldrich) was added as a hybridization enhancer (Matthiesen and Hansen 2012). Three-hundred microliters LNA/DNA mixmer oligonucleotide coupled to magnetic beads, prepared as described above, were added to the lysate and incubated for 2 h at 41°C, inverting the tubes every 15 min, four times. After hybridization, beads were washed three times in 1.9 mL of lysis buffer, subsequently twice in 1.9 mL of buffers with decreasing concentrations of LiDS and LiCl, as previously described (see above and Castello et al. 2013). Captured RNA was eluted with 150 µL RNase free H2O for 3 min at 90°C and 800 rpm shaking. Six rounds of pull-down were performed for each sample and the eluates were pooled. For RNA analysis, RNA from input and eluates were isolated with the RNA Microprep Kit (Zymo Research) after proteinase K (Roche) treatment, as previously described (Castello et al. 2013), and analyzed with the RNA Pico Bioanalyzer kit (Agilent) and by RT-qPCR. For mass spectrometry, samples were treated for 1 h at 37°C with Benzonase (25U; Novagen) followed by alkaline hydrolysis for 1 h at pH 12 to remove residual RNA. After adjusting the pH to 7.5 with HCl, protein samples were subjected to mass spectrometric analyses.
Real-time PCR
Isolated and proteinase K-digested RNAs as well as total RNA from whole-cell lysate were purified using the Quick-RNA MicroPrep kit (Zymo), and reverse transcribed into cDNA (SuperScript II, Life Technologies) using random primers (Life Technologies), following the manufacturer's recommendations. CAT RNA was spiked in as a control for cDNA preparation. Reverse transcriptase quantitative PCR (RT-qPCR) was performed with SYBR green (Applied Biosystems) and specific primers for the different RNAs.
RT-qPCR primers used (all from 5′ to 3′; f:forward; r:reverse):
CAT: f:GCAAGATGTGGCGTGTTACG; r:AAAACGGGGGCGAAGAAGTT
Primers for the quantification of human 18S rRNA and GAPDH (Castello et al. 2012) and Drosophila melanogaster 18S rRNA and GAPDH (Sysoev et al. 2016) and Rluc and Fluc (Sanchez et al. 2007) were previously described.
Western blotting and silver staining
Detection of proteins by Western blotting was performed using antibodies against Sxl (polyclonal rabbit 1:2000; produced in house) and α-tubulin (mouse 1:10000; Sigma T5168).
Sample preparation for mass spectrometry
Samples captured from Drosophila embryo extracts were supplemented with 1× protease inhibitor cocktail (Roche) and treated with 25 U of Benzonase (Novagen) for 1 h at 37°C. Samples were prepared for MS following the single-pot solid-phase-enhanced Sample Preparation (SP3) (Hughes et al. 2014). Peptides were dimethyl labeled following a standardized protocol (Boersema et al. 2009).
The eluates of the rRNA captures in 50 mM HEPES buffer pH7.5 were subjected to standard trichloroacetic acid (TCA) precipitation and then processed as described above.
LC-MS/MS
Peptides were separated using the nanoAcquity ultra performance liquid chromatography (UPLC) system (Waters) as previously described (Sysoev et al. 2016) with the following modifications: The samples were loaded with a constant flow of solvent A (0.1% formic acid) at 5 μL/min onto the trapping column. Trapping time was 6 min. Peptides were eluted via the analytical column with a constant flow of 0.3 μL/min. During the elution step, the percentage of solvent B (acetonitrile, 0.1% formic acid) increased in a linear fashion from 3% to 7% within 10 min, then increased to 25% within 100 min (60 min for human rRNA interactomes), and finally to 40% for a further 10 min (5 min for human rRNA interactome).
Proteomic data analysis
All proteomic data were analyzed using MaxQuant (v 1.4.1.2). Carbamidomethylation of cysteine was specified as a fixed modification, acetylation of the protein N terminus and oxidation of methionine as variable. More variable modifications of the N terminus and lysine were specified for masses of +28.0313 (light dimethyl), +32.0564 (medium dimethyl), +36.0757 (heavy dimethyl), and oxidation of methionine in all searches. The data were searched against the Uniprot_Drosophila_20140519 database. For all searches, trypsin was specified with two missed cleavages allowed.
For the experiments in Drosophila embryo extracts, peptides from four replicates were mapped back to the Uniprot protein database (Version 2014_06) and only uniquely mapped peptides were used to identify proteins. Protein log2-intensity ratios were computed by averaging the log2-intensity ratios of each peptide mapping to it, dividing by the median deviation to compensate for scaling effects. For comparisons between wild type to mutant reporter RNA-bound proteins, the median ratio was subtracted to center the ratios to “zero.” Protein ratios were tested against zero by a moderated t-test (Lönnstedt and Speed 2002) implemented in the R/Bioconductor package limma (Smyth 2004). The method of Benjamini–Hochberg (Benjamini and Hochberg 1995) was used to control for false discovery rate.
HeLa ribosomal RNA-binding proteins (rRBP) were identified as above. However, to compare the cCL fraction to the nonirradiated control, we followed a semiquantitative approach as described in Sysoev et al. (2016) due to the high incidence of zero values in the control and the sample.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
Bernd Fischer unexpectedly passed away during the time in which this manuscript was revised. We dedicate this work to his memory. We want to thank Jan Medenbach for discussions and reagents. We acknowledge Simone Pellegrino from the Yusupov laboratory for insightful discussions. We also thank the EMBL Genecore facility for their support. M.W.H. acknowledges support by the European Research Council Advanced Grant ERC-2011-ADG_20110310. A.C. is funded by the Medical Research Council Career Development Award #MR/L019434/1.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060798.117.
Freely available online through the RNA Open Access option.
REFERENCES
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. 2013. Structures of the human and Drosophila 80S ribosome. Nature 497: 80–85. [DOI] [PubMed] [Google Scholar]
- Bachler M, Schroeder R, von Ahsen U. 1999. StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA 5: 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badertscher L, Wild T, Montellese C, Alexander LT, Bammert L, Sarazova M, Stebler M, Csucs G, Mayer TU, Zamboni N, et al. 2015. Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep 13: 2879–2891. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Bashaw GJ, Baker BS. 1995. The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex-lethal. Development 121: 3245–3258. [DOI] [PubMed] [Google Scholar]
- Bashaw GJ, Baker BS. 1997. The regulation of the Drosophila msl-2 gene reveals a function for Sex-lethal in translational control. Cell 89: 789–798. [DOI] [PubMed] [Google Scholar]
- Beatrix B, Sakai H, Wiedmann M. 2000. The α and β subunit of the nascent polypeptide-associated complex have distinct functions. J Biol Chem 275: 37838–37845. [DOI] [PubMed] [Google Scholar]
- Beckmann K, Grskovic M, Gebauer F, Hentze MW. 2005. A dual inhibitory mechanism restricts msl-2 mRNA translation for dosage compensation in Drosophila. Cell 122: 529–540. [DOI] [PubMed] [Google Scholar]
- Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume A-M, Schwarzl T, Curk T, Foehr S, Huber W, et al. 2015. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun 6: 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LR, Horabin JI, Schedl P, Cline TW. 1991. Positive autoregulation of sex-lethal by alternative splicing maintains the female determined state in Drosophila. Cell 65: 229–239. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 57: 289–300. [Google Scholar]
- Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, Eisen MB, Rio DC. 2009. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell 33: 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJR. 2009. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4: 484–494. [DOI] [PubMed] [Google Scholar]
- Boggs RT, Gregor P, Idriss S, Belote JM, McKeown M. 1987. Regulation of sexual differentiation in D. melanogaster via alternative splicing of RNA from the transformer gene. Cell 50: 739–747. [DOI] [PubMed] [Google Scholar]
- Bunnik EM, Batugedara G, Saraf A, Prudhomme J, Florens L, Le Roch KG. 2016. The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biol 17: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. [DOI] [PubMed] [Google Scholar]
- Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, Krijgsveld J, Hentze MW. 2013. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc 8: 491–500. [DOI] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. 2015. Systematic discovery of Xist RNA binding proteins. Cell 161: 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chujo T, Ohira T, Sakaguchi Y, Goshima N, Nomura N, Nagao A, Suzuki T. 2012. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res 40: 8033–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline TW. 1984. Autoregulatory functioning of a Drosophila gene product that establishes and maintains the sexually determined state. Genetics 107: 231–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline TW, Dorsett M, Sun S, Harrison MM, Dines J, Sefton L, Megna L. 2010. Evolution of the Drosophila feminizing switch gene Sex-lethal. Genetics 186: 1321–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB. 2010. HITS-CLIP: panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip Rev RNA 1: 266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Lee JY, Mayeur GL, Bushell M, Doudna JA, Hershey JWB. 2004. The j-subunit of human translation initiation factor eIF3 is required for the stable binding of eIF3 and its subcomplexes to 40 S ribosomal subunits in vitro. J Biol Chem 279: 8946–8956. [DOI] [PubMed] [Google Scholar]
- Gebauer F, Corona DF, Preiss T, Becker PB, Hentze MW. 1999. Translational control of dosage compensation in Drosophila by Sex-lethal: cooperative silencing via the 5′ and 3′ UTRs of msl-2 mRNA is independent of the poly(A) tail. EMBO J 18: 6146–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Grskovic M, Hentze MW. 2003. Drosophila sex-lethal inhibits the stable association of the 40S ribosomal subunit with msl-2 mRNA. Mol Cell 11: 1397–1404. [DOI] [PubMed] [Google Scholar]
- Gerbi SA. 1996. Expansion segments: regions of variable size that interrupt the universal core secondary structure of ribosomal RNA. In Ribosomal RNA: structure, evolution, processing and function in protein synthesis (ed. Zimmermann RA, Dahlberg AE), pp. 71–87. Telford-CRC Press, Boca Raton, FL. [Google Scholar]
- Glisovic T, Bachorik JL, Yong J, Dreyfuss G. 2008. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 582: 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Montellese C, Ban N. 2012. Cryo-EM structures of Arx1 and maturation factors Rei1 and Jjj1 bound to the 60S ribosomal subunit. Nat Struct Mol Biol 19: 1228–1233. [DOI] [PubMed] [Google Scholar]
- Greenberg JR. 1979. Ultraviolet light-induced crosslinking of mRNA to proteins. Nucleic Acids Res 6: 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. 1999. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature 398: 579–585. [DOI] [PubMed] [Google Scholar]
- Hartmuth K, Urlaub H, Vornlocher HP, Will CL, Gentzel M, Wilm M, Lührmann R. 2002. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc Natl Acad Sci 99: 16719–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SR, Johnson D, Raychaudhuri G, Beyer AL. 1991. The Drosophila Hrb87F gene encodes a new member of the A and B hnRNP protein group. Nucleic Acids Res 19: 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid H, Rickelt S, Zimbelmann R, Winter S, Schumacher H, Dörflinger Y, Kuhn C, Franke WW. 2014. On the formation of lipid droplets in human adipocytes: the organization of the perilipin-vimentin cortex. PLoS One 9: e90386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig J, Militti C, Popowicz GM, Wang I, Sonntag M, Geerlof A, Gabel F, Gebauer F, Sattler M. 2014. Structural basis for the assembly of the Sxl–Unr translation regulatory complex. Nature 515: 287–290. [DOI] [PubMed] [Google Scholar]
- Henras AK, Plisson-Chastang C, O'Donohue MF, Chakraborty A, Gleizes PE. 2015. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA 6: 225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdy B, Karonitsch T, Vladimer GI, Tan CSH, Stukalov A, Trefzer C, Bigenzahn JW, Theil T, Holinka J, Kiener HP, et al. 2015. The RNA-binding protein HuR/ELAVL1 regulates IFN-β mRNA abundance and the type I IFN response. Eur J Immunol 45: 1500–1511. [DOI] [PubMed] [Google Scholar]
- Hogg JR, Collins K. 2007. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13: 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. 2014. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10: 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung NJ, Lo KY, Patel SS, Helmke K, Johnson AW. 2008. Arx1 is a nuclear export receptor for the 60S ribosomal subunit in yeast. Mol Biol Cell 19: 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka N, Najita L, Franzusoff A, Sarnow P. 1994. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol Cell Biol 14: 7322–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Hoshijima K, Sakamoto H, Shimura Y. 1990. Binding of the Drosophila Sex-lethal gene product to the alternative splice site of transformer primary transcript. Nature 344: 461–463. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI. 1995. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell 81: 867–877. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Wang J, Bell L, Kuroda MI. 1997. Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature 387: 195–199. [DOI] [PubMed] [Google Scholar]
- Kurreck J, Wyszko E, Gillen C, Erdmann VA. 2002. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res 30: 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN. 2013. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20: 1122–1130. [DOI] [PubMed] [Google Scholar]
- Lamond AI, Sproat B, Ryder U, Hamm J. 1989. Probing the structure and function of U2 snRNP with antisense oligonucleotides made of 2′-OMe RNA. Cell 58: 383–390. [DOI] [PubMed] [Google Scholar]
- Leppek K, Stoecklin G. 2014. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res 42: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Castello A, Fischer B, Leicht S, Föehr S, Frese CK, Ragan C, Kurscheid S, Pagler E, Yang H, et al. 2016. The cardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Rep 16: 1456–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepelt A, Vries ISN, Simons N, Eichelbaum K, Föhr S, Archer SK, Castello A, Usadel B, Krijgsveld J, Preiss T, et al. 2016. Identification of RNA-binding proteins in macrophages by interactome capture. Mol Cell Proteomics 15: 2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. 2015. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönnstedt I, Speed T. 2002. Replicated microarray data. Stat Sin 12: 31–46. [Google Scholar]
- Lueong S, Merce C, Fischer B, Hoheisel JD, Erben ED. 2016. Gene expression regulatory networks in Trypanosoma brucei: insights into the role of the mRNA-binding proteome. Mol Microbiol 100: 457–471. [DOI] [PubMed] [Google Scholar]
- Marondedze C, Thomas L, Serrano NL, Lilley KS, Gehring C. 2016. The RNA-binding protein repertoire of Arabidopsis thaliana. Sci Rep 6: 29766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matia-González AM, Laing EE, Gerber AP. 2015. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat Struct Mol Biol 22: 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matia-González AM, Iadevaia V, Gerber AP. 2017. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 118–119: 93–100. [DOI] [PubMed] [Google Scholar]
- Matthiesen SH, Hansen CM. 2012. Fast and non-toxic in situ hybridization without blocking of repetitive sequences. PLoS One 7: e40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. 2015. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medenbach J, Seiler M, Hentze MW. 2011. Translational control via protein-regulated upstream open reading frames. Cell 145: 902–913. [DOI] [PubMed] [Google Scholar]
- Melnikov S, Ben-Shem A, Garreau de Loubresse N, Jenner L, Yusupova G, Yusupov M. 2012. One core, two shells: bacterial and eukaryotic ribosomes. Nat Struct Mol Biol 19: 560–567. [DOI] [PubMed] [Google Scholar]
- Milek M, Wyler E, Landthaler M. 2012. Transcriptome-wide analysis of protein–RNA interactions using high-throughput sequencing. Semin Cell Dev Biol 23: 206–212. [DOI] [PubMed] [Google Scholar]
- Mili S, Piñol-Roma S. 2003. LRP130, a pentatricopeptide motif protein with a noncanonical RNA-binding domain, is bound in vivo to mitochondrial and nuclear RNAs. Mol Cell Biol 23: 4972–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Jain S, She M, Parker R. 2013. Global analysis of yeast mRNPs. Nat Struct Mol Biol 20: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Helm M. 2011. RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2: 611–631. [DOI] [PubMed] [Google Scholar]
- Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26: 182–190. [DOI] [PubMed] [Google Scholar]
- O'Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. 2014. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516: 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsugi M, Larue L, Schwarz H, Kemler R. 1997. Cell-junctional and cytoskeletal organization in mouse blastocysts lacking E-cadherin. Dev Biol 185: 261–271. [DOI] [PubMed] [Google Scholar]
- Pashev IG, Dimitrov SI, Angelov D. 1991. Crosslinking proteins to nucleic acids by ultraviolet laser irradiation. Trends Biochem Sci 16: 323–326. [DOI] [PubMed] [Google Scholar]
- Rakotondrafara AM, Hentze MW. 2011. An efficient factor-depleted mammalian in vitro translation system. Nat Protoc 6: 563–571. [DOI] [PubMed] [Google Scholar]
- Reichel M, Liao Y, Rettel M, Ragan C, Evers M, Alleaume A-M, Horos R, Hentze MW, Preiss T, Millar AA. 2016. In planta determination of the mRNA-binding proteome of Arabidopsis etiolated seedlings. Plant Cell 28: 2435–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci EP, Kucukural A, Cenik C, Mercier BC, Singh G, Heyer EE, Ashar-Patel A, Peng L, Moore MJ. 2014. Staufen1 senses overall transcript secondary structure to regulate translation. Nat Struct Mol Biol 21: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rospert S, Dubaquié Y, Gautschi M. 2002. Nascent-polypeptide-associated complex. Cell Mol Life Sci 59: 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M, Galy B, Muckenthaler MU, Hentze MW. 2007. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat Struct Mol Biol 14: 420–426. [DOI] [PubMed] [Google Scholar]
- Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA, Consortium L. 2010. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol Biol Cell 21: 1315–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Varshavsky A. 1985. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc Natl Acad Sci 82: 6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squatrito M, Mancino M, Donzelli M, Areces LB, Draetta GF. 2004. EBP1 is a nucleolar growth-regulating protein that is part of pre-ribosomal ribonucleoprotein complexes. Oncogene 23: 4454–4465. [DOI] [PubMed] [Google Scholar]
- Squatrito M, Mancino M, Sala L, Draetta GF. 2006. Ebp1 is a dsRNA-binding protein associated with ribosomes that modulates eIF2α phosphorylation. Biochem Biophys Res Commun 344: 859–868. [DOI] [PubMed] [Google Scholar]
- Srisawat C, Engelke DR. 2001. Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA 7: 632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sysoev VO, Fischer B, Frese CK, Gupta I, Krijgsveld J, Hentze MW, Castello A, Ephrussi A. 2016. Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nat Commun 7: 12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries J-L, Mullineux S-T, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DLJ. 2013. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of pre-rRNA processing factors. Mol Cell 51: 539–551. [DOI] [PubMed] [Google Scholar]
- Traut W, Niimi T, Ikeo K, Sahara K. 2006. Phylogeny of the sex-determining gene Sex-lethal in insects. Genome 49: 254–262. [DOI] [PubMed] [Google Scholar]
- Uyeno Y, Sekiguchi Y, Sunaga A, Yoshida H, Kamagata Y. 2004. Sequence-specific cleavage of small-subunit (SSU) rRNA with oligonucleotides and RNase H: a rapid and simple approach to SSU rRNA-based quantitative detection of microorganisms. Appl Environ Microbiol 70: 3650–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L, Hasek J, Nielsen KH, Hinnebusch AG. 2001. Dual function of eIF3j/Hcr1p in processing 20 S pre-rRNA and translation initiation. J Biol Chem 276: 43351–43360. [DOI] [PubMed] [Google Scholar]
- Valcárcel J, Singh R, Zamore PD, Green MR. 1993. The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature 362: 171–175. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, Broberger C, Porreca F, Lai J, Ren K, et al. 2000. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci 97: 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dong Z, Bell LR. 1997. Sex-lethal interactions with protein and RNA. Roles of glycine-rich and RNA binding domains. J Biol Chem 272: 22227–22235. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. 2014. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels H-H, Imami K, Baltz AG, Kolinski M, Beldovskaya A, Selbach M, Small S, Ohler U, Landthaler M. 2016. The mRNA-bound proteome of the early fly embryo. Genome Res 26: 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, Kingston RE. 2014. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55: 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U. 2010. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8: e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, Green R. 2005. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods 36: 305–312. [DOI] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.