

Abstract
The Epidermal Growth Factor Receptor (EGFR) is a cell surface receptor with primary implications in cell growth in both normal and malignant tissue. Paradoxically, cell lines that hyperexpress the EGFR have been documented to undergo receptor-mediated apoptosis. The underlying mechanism by which EGF-induced apoptosis occurs however remains inexplicit. In an attempt to identify this mechanism, we assessed downstream effectors of EGFR in MDA-MB-468 cells during conditions of EGF-induced apoptosis. The effector assessment revealed STAT3 as a potential mediator of EGF-induced apoptosis. Alternative strategies for activating STAT3, independent of EGFR stimulation, resulted in the induction of the apoptotic pathways. A reduction in STAT3 expression via RNAi resulted in a significant attenuation of EGF-induced PARP cleavage. Our findings support STAT3 as a positive mediator of EGF-induced apoptosis in MDA-MB-468 cells.
Keywords: Epidermal Growth Factor Receptor, STAT3, Apoptosis, MDA-MB-468 Cells, A431 Cells, Oncostatin M, Stattic, S3I-201
Introduction
The Epidermal Growth Factor Receptor (EGFR) is a transmembrane receptor tyrosine kinase that plays critical roles in cell growth, tissue development, and overall cellular homeostasis [1, 2]. Despite these critical physiological roles, there are a myriad of human malignancies [3–5] that are characterized by hyper-activated EGFR signaling. This is due to either receptor overexpression or activating mutations of the receptor [4, 6]. The FDA has approved pharmacological agents that antagonize ligand binding and inhibit EGFR kinase activity for the treatment of patients with cancers characterized by hyper-activated EGFR signaling [7–9]; however, these drugs have off-target effects (i.e. colitis, corneal erosions and dermatitis) and cancers often become desensitized to these agents over time [10–12]. Thus, there remains a need to develop drugs that more aggressively and specifically target those cancers with enhanced EGFR signaling, but allow normal, healthy cells to perform their biological roles.
One potential strategy for doing this is to hijack intrinsic signaling pathways and exploit them to compromise the cell’s health. Cell lines with EGFR overexpression either naturally (A431 and MDA-MB-468 cells [13, 14]) or via bioengineering (Rat-1 fibroblasts) [15], undergo EGF-induced apoptosis [4, 16, 17]. Understanding the molecular mechanism by which this occurs will identify new potential pharmacological targets that can induce death in cells that overexpress the EGFR (i.e. cancer cells).
Through an effector screen, Signal Transducer and Activator of Transcription 3 (STAT3) was identified as a plausible mediator of EGFR-induced apoptosis. STAT3 is one of seven members of the STAT family of transcription factors, namely, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT7. STAT activation is critical for a number of biological processes, including cell proliferation, survival, differentiation, development and inflammation [18]. STAT3 is a DNA-binding transcription factor known to mediate normal cellular processes upon activation by growth factors, ligands and cytoplasmic cytokines, such as cytokine receptor-associated Janus kinases (JAKs) [19–22]. These upstream protein components phosphorylate STAT3 at its unique, critical tyrosine residues, primarily Tyr705 [19, 20]. The EGFR has also been reported to directly tyrosine phosphorylate STAT3 [23, 24]. STAT phosphorylation occurs at the cytoplasm, which induces STAT homo and heterodimer formation between two monomers via their Src homology 2 (SH2) domain interactions. From the cytoplasm, activated STAT dimers translocate and accumulate in the nucleus, where they initiate and mediate gene transcription by binding to DNA response elements [19]. This results in either upregulation or downregulation of the biological effectors and subsequent cellular processes that are critical for cellular homeostasis.
STAT3 has been implicated in the post-transcriptional modification of cellular processes, positively and negatively affecting cell growth and proliferation. Aberrant STAT3 signaling and constitutive activity has been reported in a number of cancers [18, 25]. Elevated basal STAT3 activity has been reported in 30–60% of primary mammary malignancies, with reports of it being required for tumor cell progression and metastasis [23]. However, prior to being implicated in cancer, STAT3-mediated programmed cellular death was found to be associated with and required for mammary gland involution [26, 27]. Additionally, STAT3 has been previously identified as a key mediator of apoptosis in murine pro-B cells [28], myeloid leukemia [29], and prostate cancer [30]. These conflicting roles for STAT3 in the context of normal and malignant cellular biology highlight the need for a better understanding of this protein in EGFR signaling mechanisms.
The overarching goal of this study was to identify intermediates in EGFR-mediated apoptosis. STAT3 was identified as being specifically activated under apoptosis-inducing conditions. Utilizing siRNA targeting STAT3 resulted in a significant attenuation of EGF-induced apoptosis. Additionally, EGFR-independent activation of STAT3 through cytokine stimulation promoted apoptosis, as measured by PARP and Caspase-3 cleavage. From these findings, we conclude that STAT3 is required for EGFR-mediated cell death via PARP cleavage and subsequent activation.
Materials and Methods
Cell Lines–
MDA-MB-468 and A431 cell lines were acquired from the American Type Culture Collection (ATCC, Manassas, VA). PC3 cells were generously gifted from Dr. Geoffrey Clark (University of Louisville, Louisville, KY, USA). All cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 1% penicillin, 1% streptomycin, and L glutamine (2 mM). The cells were maintained at incubation conditions of 37 °C in 5% CO2.
Cell Viability Analyses–
Alamar Blue (ThermoFisher Scientific, Waltham, MA) cell viability assays were performed according to the manufacturer’s instructions and as previously described [31]. Cells were plated at 10,000 cells per well in in a 96-well dish. After a 24-hour recovery period, the cells were treated with the indicated reagents for the indicated times. Alamar Blue reagent was added at 10% of the sample volume, an incubated for 2 hours. Fluorescence was measured in a BioTek (Winooski, VT) fluorescence microplate reader at 530 nm/590 nm (excitation/emission).
Cell Lysate Preparation and Immunoblot Analyses–
In all experiments, MDA-MB-468 cells were serum starved overnight in serum free DMEM. All treatments were made in serum free DMEM. Because A431 cells are not viable for prolonged periods of time in DMEM alone, these cells were serum starved for 2 hours in DMEM supplemented with 1.25% FBS. All reagents used within this cell line were made in DMEM supplemented with 1.25% FBS. Cell lysates were generated as previously documented [31, 32]. Protein lysates were resolved by SDS PAGE, transferred to nitrocellulose, and immunoblotted with the indicated antibodies according the manufacturer’s directions. Antibody sources are as follows: EGFR and GAPDH (Santa Cruz Biotechnology, Dallas, TX); pY1045, pMAPK (Thr202/Ty204), pSRC(Tyr416), pAKT(Thr308), pBAD(Ser112), pSTAT3(Tyr705), STAT3, PARP, and cleaved Caspase-3(Asp175) (Cell Signaling Technology, Danvers, M.A.); HRP-conjugated secondary antibodies (ThermoFisher Scientific, Waltham, MA). Enhanced chemimmunofluoresence (ECL) reagent and a Fotodyne imaging system (Hartland, WI) were used to visualize the bands. Immunoblot data were quantified using ImageJ software.
Pharmacological Agents–
STAT3 antagonists, Stattic and S3I-201, were both acquired from Sigma-Aldrich (St. Louis, MO). They were prepared in Dimethyl Sulfoxide (DMSO) and were employed at concentrations specified in each figure. The Oncostatin M (OSM) cytokine was acquired from PeproTech (Rocky Hill, NJ), and EGF ligand was acquired from ProSpec Protein Specialist (East Brunswick, NJ). Staurosporine (STS) was used as a positive control for apoptosis, and was acquired from Sigma-Aldrich.
siRNA knockdown of STAT3–
Two siRNA oligonucleotides targeting STAT3 (5′-GGAGAAGCAUCGUGAGUGA-3′) (5′-CCAUUUGGUGUUUCAUAA-3′) were acquired from Dharmacon (Lafayette, CO). Scramble control siRNA (siCON) was acquired from IDTDNA (Coralville, IA). MDA-MB-468 cells were transfected with final concentrations of 200 nM STAT3 siRNA or 200 nM siCON with INTERFERin (Polyplus Transfection, Strasbourg, France). Transfection protocol and reagents were employed as previously reported [31]. MDA-MB-468 cells were exposed to siRNA for 72 hours, and to treatment various reagents to induce cell growth or cell death for a total of 24 hours. Cell viability or apoptosis was then assessed as previously described.
Statistical Analyses–
An unpaired, student t-test was performed for the determination of significance. A p value of less than 0.05 is designated significant, and is indicated by a single asterisk (*). A p value of less than 0.01 is designated significant, and is indicated by two asterisks (**). A p value of less than 0.001 is designated very significant, and is indicated by three asterisks (***). A p value of less than 0.0001 is designated extremely significant, and is indicated by four asterisks (****).
Results
It is known that cell lines that hyperexpress the EGFR, such as MDA-MB-468 and A431, undergo EGFR-mediated apoptosis [4, 16, 17]; however, the molecular mechanism(s) by which this occurs have not yet been fully elucidated. Previously, it has been documented that in MDA-MB-468 cells, low concentrations of EGF (0.16 nM) promote cell growth, whereas high concentrations (16 nM) induce apoptosis [4]. With this in mind, we screened MDA-MB-468 cells for the ability to activate five effector proteins that have been previously associated with cell growth and/or apoptosis in cancer: MAPK [33, 34], SRC [35, 36], AKT [33], BAD [37], and STAT3 [29, 38] (Figs. 1 and 2). MDA-MB-468 cells were treated with either low (0.16 nM) or high (16 nM) concentrations of EGF for 0–120 minutes, and assessed the phosphorylation status (activity) of these effectors by immunoblot (Fig. 1A and Fig 2A). MAPK, SRC, AKT, and BAD displayed no significant differences in phosphorylation patterns with the high and low concentrations of ligand (Figs. 1B–1E). However, STAT3 phosphorylation (Tyrosine 705), was undetectable at all time points following stimulation with low concentrations of ligand, but was robustly increased over time following treatment with high levels of ligand (Figs. 2A and B).
Figure 1. Phosphorylation of MAPK, SRC, AKT, and BAD in response to high and low EGF concentrations.
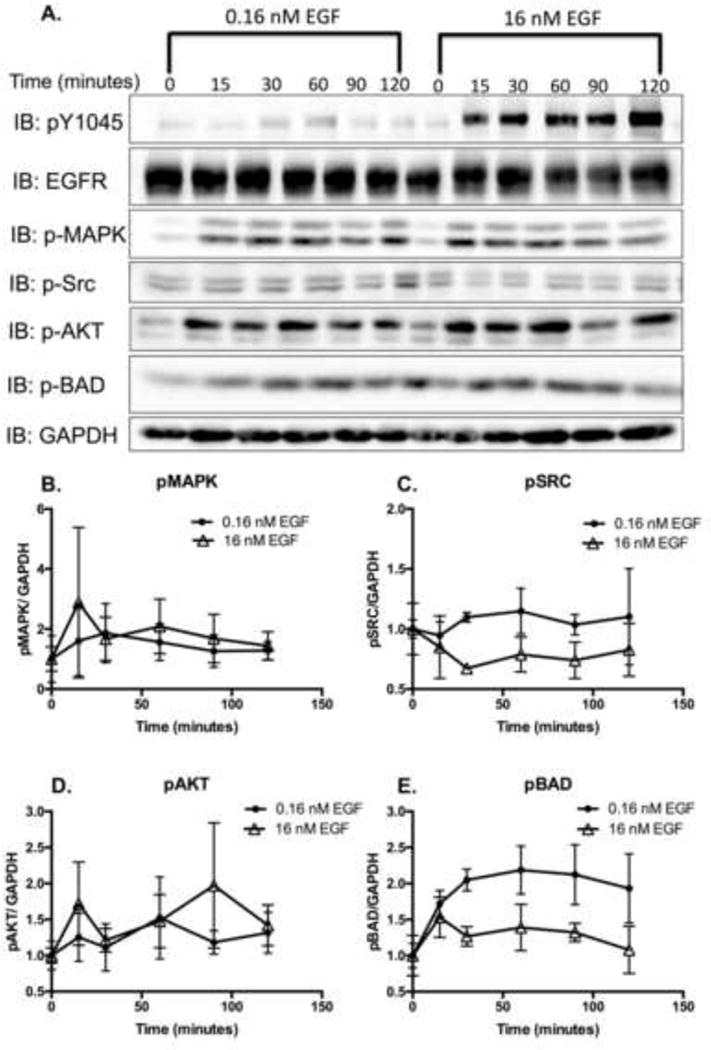
A. Serum-starved MDA-MB-468 cells were stimulated with low (0.16 nM) or high (16 nM) EGF ligand for 0–120 minutes. Cell lysates (20 μg) were resolved by 12% SDS-PAGE and were assessed for the indicated proteins via immunoblot analysis. Band intensities from the immunoblot data of pMAPK (B.), pSRC (C.), pAKT (D.) and pBAD (E.) were quantified, normalized to GAPDH levels, and plotted as the relative level compared to cells with no treatment. Data are expressed as the average ± Standard Error of the Mean (SEM; n=3).
Figure 2. STAT3 is tyrosine phosphorylated in response to high, but not low, EGF concentrations.
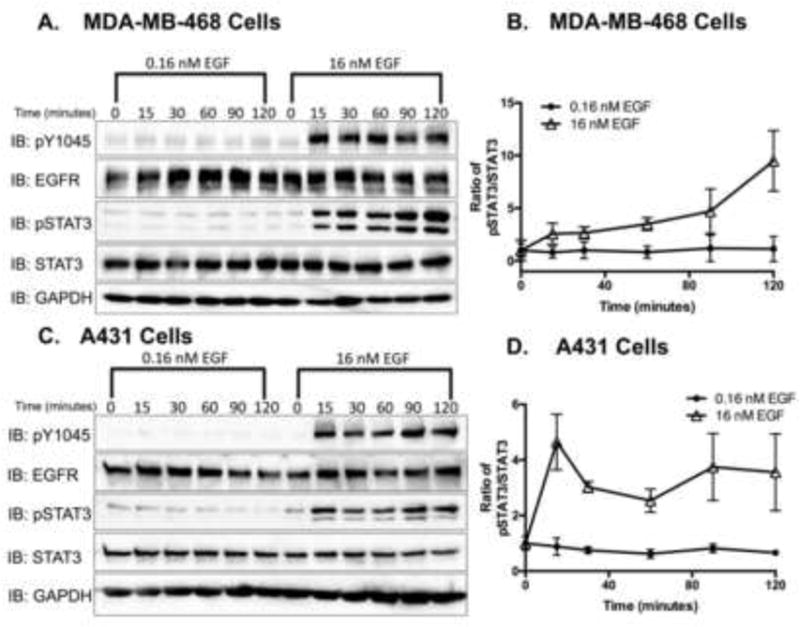
Serum-starved A. MDA-MB-468 and B. A431 cells were stimulated with 0.16 nM and 16 nM EGF ligand for 0–120 minutes. Cell lysates (20 μg and 15 μg, respectively) were resolved by 12% SDS-PAGE and were assessed for phospho-EGFR (pY0145), EGFR, pSTAT3 (Tyr705), STAT3 and GAPDH, via immunoblot analysis. C. MDA-MB-468 and D. A431 immunoblot data were quantified, and pSTAT3 was normalized to total STAT3. Data are expressed as the average ± SEM (n=3) relative to untreated samples.
To determine if STAT3 phosphorylation is cell type specific, A431 cells, a metastatic epidermoid cell line, were employed. MDA-MB-468 and A431 cell lines both induce apoptosis upon EGF stimulation [16] and express comparable levels of EGFR [39, 40]. Like MDA-MB-468 cells, A431 cells treated with low concentrations of EGF did not yield significant increases in pSTAT3, but cells treated with high concentrations did (Figs. 2C and 2D).
Next, we monitored STAT3 phosphorylation in MDA-MB-468 and A431 cells following treatment with increasing concentrations of EGF ligand. Data from the dose-response experiments supported the findings of our time course experiments, displaying a dose-dependent increase in STAT3 phosphorylation in both cell lines (Fig. 3). These experiments, in two different cell lines, confirmed that higher concentrations of EGF were required for STAT3 phosphorylation. Importantly, the concentrations of EGF required for STAT3 phosphorylation were co-incident with the concentrations required to induce apoptosis.
Figure 3. EGF phosphorylates STAT3 in a dose-dependent manner in MDA-MB-468 and A431 cell lines.
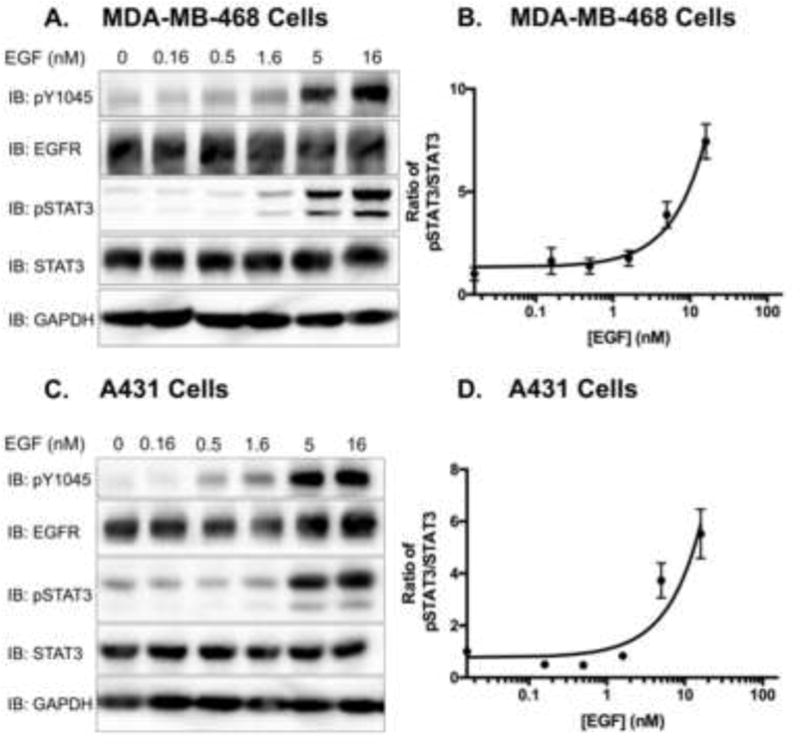
Serum-starved A. MDA-MB-468 cells and B. A431 cells were treated with varying concentrations of EGF (0–16 nM) for 30 minutes. Cell lysates (40 μg) were resolved by 10% SDS PAGE and immunobloted for the indicated proteins. C. and D. Immunoblot data were quantified, and pSTAT3 was normalized to total STAT3. Data are expressed as the average ± SEM (n=3) relative to untreated samples.
To determine if the association between STAT3 activity and apoptosis was unique for the EGFR, we examined another receptor family that stimulates STAT3 to see if it, too, promoted apoptosis in MDA-MB-468 cells. Oncostatin M (OSM) belongs to the Interleukin-6 (IL-6) family of cytokines [41]. Like the EGFR, it has established roles in promoting cancer and tumor formation in malignant prostate [42], lung, head and neck, and cervical [43] tissue. In addition, OSM has been shown in some cells to induce apoptosis [44–46]. Thus, stimulation of the OSM receptor was a logical candidate for testing our hypothesis that STAT3 is a mediator apoptosis, independent of EGFR activity.
MDA-MB-468 cells were stimulated with 100 ng/mL OSM over a course of 96 hours and were assessed for STAT3 phosphorylation as well as Caspase-3 and PARP cleavage (Fig. 4). Caspase-3 is a well-established executioner of apoptosis [47]. Upon its cleavage and subsequent activation, it cleaves and activates other downstream effectors that promote the induction of apoptosis (i.e. poly ADP-ribose polymerase or PARP). In addition to stimulating pSTAT3, OSM activated the apoptotic pathways, as evident by increases in cleaved PARP and cleaved Caspase-3 (Fig. 4A). Quantification of multiple assays indicates that OSM-induced PARP cleavage (Fig. 4B) and Caspase-3 cleavage (Fig. 4C) are comparable to that induced by EGF, strengthening the correlation between STAT3 and apoptosis.
Figure 4. EGFR-independent activation of STAT3 promotes apoptosis in MDA-MB-468 cells.
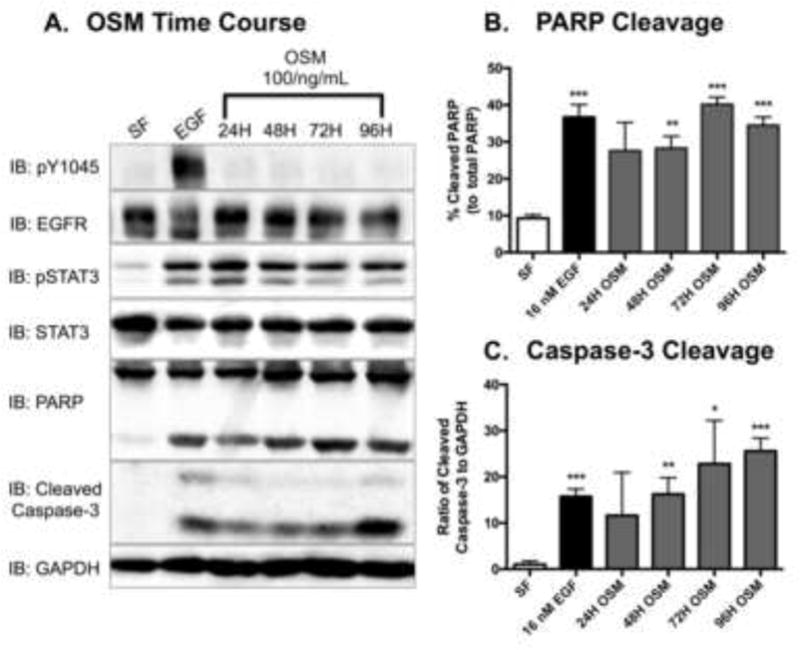
A. Serum-starved MDA-MB-468 cells were stimulated with 100 ng/mL of Oncostatin M (OSM) for the indicated times. Control cells were exposed to serum free DMEM (SF) and or 16 nM EGF for 24 hours. After harvesting, the cell lysates (40 μg) were resolved by SDS PAGE, and were immunoblotted for the indicated proteins. Densitometry quantification of western blot data of cleaved PARP (B.) and cleaved Caspase-3 (C.). The cleaved PARP band intensities were normalized to and plotted as a percentage of total PARP bands. Cleaved Caspase-3 bands were quantified to total protein for each respective sample (GAPDH). Data are expressed as the average ± SEM (n=3).
Having established that STAT3 phosphorylation correlates with high concentrations of EGF ligand, we hypothesized that EGFR-mediated apoptosis occurred in a STAT3-dependent manner. On the contrary, STAT3 could have been have been upregulated with high ligand stimulation as a compensatory mechanism, in an attempt to rescue the cells from an EGFR-mediated apoptotic fate. To distinguish between these two possibilities, we antagonized STAT3 with pharmacological inhibitors, Stattic and S3I-201, and then stimulated MDA-MB-468 and A431 cells with EGF (Figs. 5 and 6). These cells were assessed for cell viability and for their ability to induce apoptotic pathways. If STAT3 were in fact promoting apoptosis, we anticipated an attenuation of PARP and Caspase-3 cleavage, coincident with enhanced viability in STAT3 antagonized cells. Conversely, if STAT3 were being activated as a compensatory mechanism, inhibition of STAT3 would not affect induction of the apoptotic pathways and would likely further decrease cell viability.
Figure 5. STAT3 inhibitors attenuate cell viability and EGFR activity in MDA-MB-468 cells.
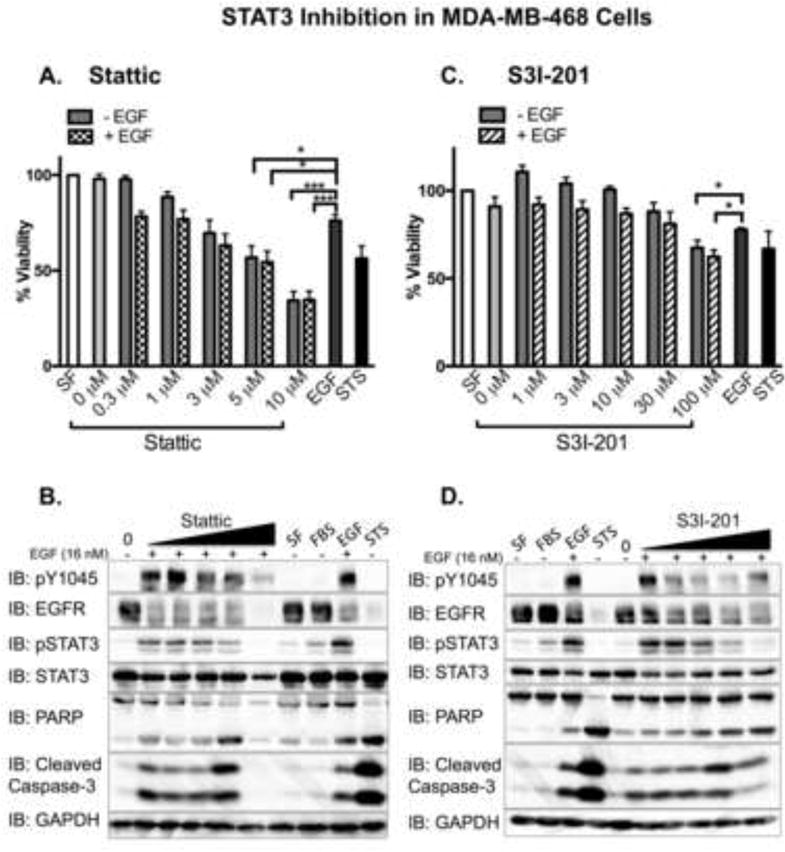
MDA-MB-468 cells, plated and treated in parallel, were assessed for cell viability using an Alamar Blue assay. Data from A. Stattic or C. S31-201 treated cells are expressed as the average ± SEM (n=3) relative to cells treated without serum (SF). Serum-starved cells were pre-treated for 1 hour with 0 (0.025% DMSO), 0.3, 1, 3, 5, or 10 μM Stattic (IC50 = 5.1 μM; panel B.), or with 0 (0.5% DMSO), 1, 3, 10, 30, and 100 μM S3I-201(IC50 = 86 μM; panel D.), followed by the addition of 16 nM EGF (+) for 24 hours. Control cells were incubated with serum free media alone (SF), media containing 10% fetal bovine serum (FBS), or 16 nM EGF (EGF) for 24 hours. As a positive control cells were treated with 1 μM staurosporine (STS) for 4 hours. Cell lysates were prepared and 40 μg were resolved by SDS-PAGE before being assessed for the indicated proteins. For STS treated samples, 15 μg of cell lysates were loaded to keep the signals in the dynamic range. Shown is a representative experimental replicate of three.
Figure 6. STAT3 inhibitors attenuate EGFR activity in A431 cells.
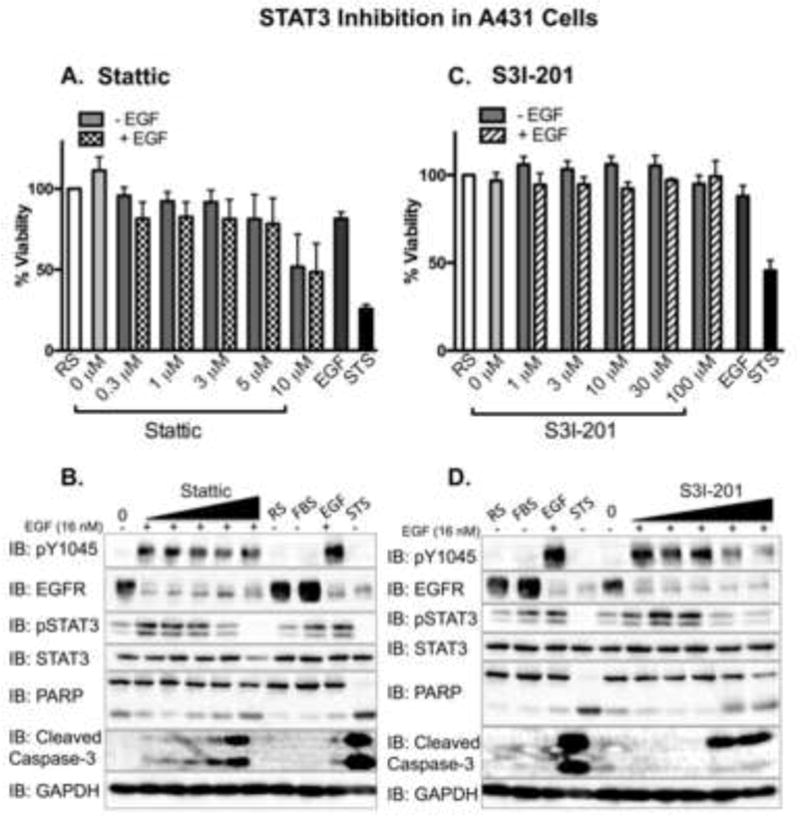
A431 cells, plated and treated in parallel, were assessed for cell viability using an Alamar Blue assay. Data from A. Stattic or B. S31-201 treated cells are expressed as the average ± SEM (n=3) relative to cells incubated in 1.25% FBS in DMEM (reduced serum – RS). Serum-starved A431 cells were pre-treated for 1 hour with 0 (0.025% DMSO), 0.3, 1, 3, 5, or 10 μM Stattic (IC50 = 5.1 μM; panel A.), or with 0 (0.5% DMSO), 1, 3, 10, 30, and 100 μM S3I-201 (IC50 = 86 μM; panel C.), followed by the addition of 16 nM EGF (+) for 24 hours. Control cells were incubated with reduced serum media alone (RS), media containing 10% fetal bovine serum (FBS), or 16 nM EGF (EGF) for 24 hours. As a positive control cells were treated with 1 μM staurosporine (STS) for 4 hours. Cell lysates were prepared, 40 μg were resolved by SDS-PAGE, and immunoblotted for the indicated proteins. Fifteen μg of the STS sample were used to keep the immunoblot analysis in the dynamic range. Shown is a representative experimental replicate of three.
Upon exposure to both inhibitors, cell viability analyses revealed a dose-dependent decrease in viability in both MDA-MB-468 and A431 cells, independent of the presence of EGF (Figs. 5A, 5C, 6A, 6C). Although Stattic was more potent than S31-201, and MDA-MB-468 cells were more sensitive to inhibitor treatments than A431 cells, the trends held in both cell lines. Complementary with the viability data, we observed a dose-dependent increase in cleaved PARP and cleaved Capsase-3 phosphorylation. This finding was observed in both cell lines when cells were treated with 16 nM EGF in combination with the STAT3 inhibitors. However, coincident with a dose-dependent decrease in phosphorylated STAT3, both cell lines displayed a corresponding decrease in EGFR phosphorylation (Figs. 5B, 5D, 6B, and 6D).
We wanted to determine whether these decreases in cell viability were due to STAT3 inhibition or from a toxicity mediated by the compounds themselves. To assess whether the compounds were inherently cytotoxic, we employed the PC3 cell line, a prostatic carcinoma cell line lacking STAT3 expression [30]. Our biochemistry analysis confirmed that PC3 cells do not express STAT3 and have very low levels of EGFR expression (Fig. 7A). PC3 cells exhibited a dose-dependent decrease in viability upon stimulation with Stattic and S3I-201 (Fig. 7B). These data indicate the STAT3 inhibitors had off-target activity, and an alternative method was needed to attenuate STAT3 activity and determine its ultimate role in EGFR-mediated apoptosis.
Figure 7. STAT3 inhibitors reduce cell viability in STAT3-null, PC3 cells.
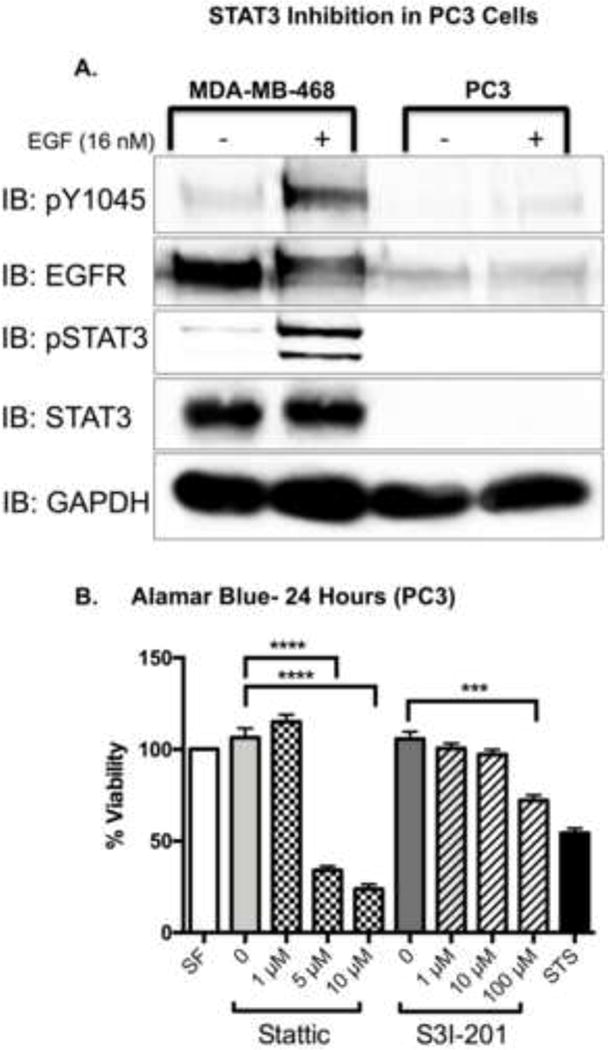
A. Serum-starved MDA-MB-468 and PC3 cells were treated with either DMEM alone (−), or DMEM supplemented with 16 nM EGF (+) for 1 hour. Cell lysates (20 μg) were resolved by 10% SDS PAGE, and immunoblotted for the indicated proteins. Shown is a representative experiment repeated three times. B. PC3 cells were exposed to treatments of serum free media (SF), DMEM with 0.025% DMSO (0), 1, 5, 10 μM Stattic; 0.5% DMSO (0), 1, 10, 100 μM S31-201; or 168 nM Staurosporine (STS) for a total of 24 hours. Cell viability was assessed with an Alamar Blue assay. Data are normalized to cell viability in serum free media (SF) and expressed as the average ± SEM (n=3).
Next, we employed STAT3 siRNA as a more selective mode of reducing STAT3 expression in the MDA-MB-468 cell line (Fig. 8). As compared to control siRNA (siCON), STAT3 siRNA reduced STAT3 expression by 78% (Fig. 8A). STAT3 knock down cells were slightly more viable than control cells following treatment with the high concentrations of EGF required to induce apoptosis (Fig. 8B). Biochemically, STAT3 knock down cells had reduced EGF-induced cleaved PARP and Caspase-3 (Fig 8C–E).
Figure 8. Knockdown of STAT3 attenuates EGFR-mediated apoptosis in MDA-MB-468 cells.
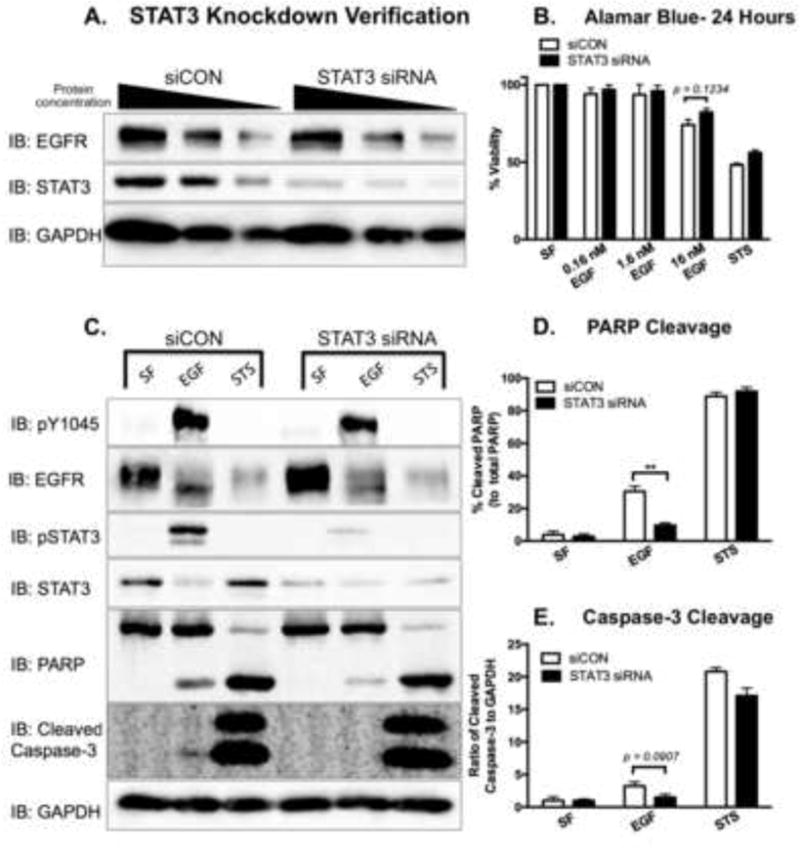
A. MDA-MB-468 cells were transfected with either 200 nM control siRNA (siCON) or 200 nM STAT3 siRNA for 72 hours. Cell lysates were prepared, and decreasing protein concentrations (40, 20, and 10 μg) were resolved by 10% SDS PAGE and immunoblotted for EGFR, STAT3, and GAPDH. B. Fourty-eight hours post-transfection, siCON and STAT3 siRNA-transfected cells were treated for 24 hours with 0 (SF), 0.16, 1.6, 16 nM EGF or 168 nM Staurosporine (STS) as a positive control. Cell viability was assessed by an Alamar Blue assay. Data are expressed as the average ± SEM (n=3). C. Cleaved PARP and cleaved Caspase-3 were assessed in siCON and STAT3 siRNA transfected cells. Seventy-two hours post-transfection, cells were coincidently exposed to SF DMEM and 16 nM EGF for 24 hours or 1 μM STS for 4 hours prior to harvesting. Cell lysates (40 μg) were resolved on either a 10% SDS PAGE (pY1045, PARP, and GAPDH) or a 15% SDS PAGE (pSTAT3 and cleaved Caspase-3), and were assessed for the indicated proteins via immunoblot analysis. Densitometry quantification of western blot data of cleaved PARP (D.) and cleaved Caspase-3 (E.). The cleaved PARP band intensities were normalized to and plotted as a percentage of total PARP. The intensity of cleaved Caspase-3 was normalized to the levels of GAPDH. Data are expressed as the average ± SEM (n=3).
Discussion
EGFR-mediated apoptosis is a paradox for growth factor receptors. However, dissecting how a receptor that normally mediates cell growth and proliferation induces cell death when overexpressed provides an opportunity to understand receptor signaling and develop strategies for attenuating the growth of cancer cells with elevated EGFR levels. In this study, we assessed effector candidates with documented affiliation with EGFR activation [33, 36, 48] and implication in human malignancies by either promoting proliferation [33, 34, 38, 49] and/or attenuating it [29, 34, 35, 37]. We assessed MAPK, SRC, AKT, BAD, and STAT3 for phosphorylation following treatment with low (0.16 nM) versus high (16 nM) concentrations of EGF (Figs. 1 and 2). Among these, only STAT3 was activated when ligand concentrations were sufficient to induce the apoptotic pathways. This was not only true for the EGFR, but cytokine-mediated activation of STAT3 via OSM resulted in the induction of apoptosis as well (Fig. 4).
Pharmacological inhibitors of STAT3 were employed to ascertain the role of STAT3 on EGFR-mediated apoptosis. As expected, STAT3 inhibitors in MDA-MB-468 and A431 cells resulted in a dose-dependent decrease in STAT3 phosphorylation. Unexpectedly, these inhibitors also antagonized EGFR phosphorylation and decreased cell viability (Figs. 5 and 6). This cytotoxicity cannot be attributed only to STAT3 inhibition; STAT3-null, PC3 cells were also less viable in response to these inhibitors (Fig. 7). RNAi proved to be a more useful tool for studying the role of STAT3. Knock down of STAT3 with siRNA significantly attenuated EGF-induced PARP cleavage and also reduced Caspase-3 cleavage and cell death. These data provide evidence that support STAT3 as a direct mediator of EGF-induced apoptosis. These findings are consistent with reports that OSM receptor-mediated apoptosis in mammary tissue is lost in STAT3 knock out mice [50].
Since it was first reported by Armstrong et al in 1994 [51], the molecular mechanism by which EGFR-mediated apoptosis occurs has remained controversial. A number of signaling molecules have been reported to be involved in this process including Etk [52], STAT1 [53], Caspase-1 [54], and Protein Kinase G [31]. Dissecting the signaling events that emanate from a hyperexpressed receptor and culminate in cell death are challenging for a number of reasons. First, high levels of receptors are going to favor the formation of receptor:effector complexes that may not occur with physiological levels of receptor expression. Second, cells with elevated receptor expression have altered kinetics of receptor down regulation through the endocytic pathway [55, 56]. In cells with physiological levels of receptor expression, the EGFR will traffic to the lysosome for degradation; in MDA-MB-468 cells, the activated EGFR internalizes to the early endosome and can be retained there for 24 hours [57]. This increases the duration of total receptor activity, as well as any receptor:effector interactions in the early endosome. Finally, the kinetics of EGFR-mediated apoptosis provide a large window (0–16 hours) to monitor the many effectors activated by the receptor.
Despite these limitations to studying apoptosis, we were able to identify a role for STAT3. There are multiple lines of evidence that support this role. First, the concentrations of EGF needed to activate STAT3 in MDA-MB-468 and A431 cells (Fig. 3) are consistent with the concentrations of EGF required to induce apoptosis. Next, the kinetics of EGFR-mediated apoptosis (over the course of 16–48 hours [31, 58]) are consistent with a transcriptionally regulated event. Last, STAT3 is a well-documented regulator of cell growth, albeit more frequently having a positive effect. STAT3 has been associated with proliferative activity in numerous malignancies, including glioblastoma [59], non-small cell lung [60], and colorectal cancer [61]. In addition, STAT3-induced tumorigenesis is linked to its constitutive activity in certain cancer cells [62–64]. Constitutive STAT3 activity was not observed in our MDA-MB-468 or A431 cells (Figs. 2 and 3). There are also data, consistent with ours, which contradict these findings and implicate STAT3 as a mediator of cell death [65–67].
The contradiction between our experimental results regarding STAT3 activity under pro-apoptotic conditions and the published data regarding the pro-growth events of STAT3 led us to propose two opposing hypothesis. The first hypothesis was that STAT3 is a mediator of apoptosis and that inhibition of the protein would enhance cell viability. The second hypothesis was that the induction of STAT3 activity was a compensatory reaction to the induction of apoptosis via another pathway. Our subsequent experiments indicate that STAT3 mediates EGF-induced apoptosis and is not a compensatory, cell growth mechanism.
Previous reports have shown that the EGFR kinase directly activates STAT3 [68], and that is likely the case in the MDA-MB-468 and A431 cells, particularly based on the kinetics of STAT3 phosphorylation. Given these interactions require high doses of EGF and high receptor levels, this is likely a pathological event rather than a physiological.
How STAT3 progresses to apoptosis is more ambiguous. It has been previously reported that STAT3 activity directly promotes apoptosis in some cancers [29, 30, 69, 70], and suppresses cancer cell colony formation and invasion [71]. One possibility is that in MDA-MB-468 cells, like LNCaP cells, STAT3 transcription is causing growth arrest and a block in cell cycle progression [30]. This, coupled with strong proliferative EGFR signals, may result in a signaling conflict for the cell that results in an apoptotic response. A second option is that the high levels of STAT3 phosphorylation may increase the magnitude and/or duration gene transcription that disrupts cell homeostasis and causes cell death. Alternatively, high levels of activated STAT3 may lead to aberrant STAT heterodimer formation. Previous studies have reported a role for STAT1 in the induction of apoptosis [34, 53, 72]. More specifically, STAT1 has been implicated in EGFR-mediated apoptosis in A431 cells [53, 73] and MDA-MB-468 cells [52]. Increased STAT3 activity may drive STAT1:STAT3 heterodimer formation which in turn allows STAT1 to drive apoptosis.
These changes in signaling may reflect mis-regulation of a normal physiological process. STAT3-mediated apoptosis occurs during mammary gland involution [26, 27], a complex process that returns mammary tissue to its pre-lactation, morphological state [74]. The initiation of this process is dependent on the presence of milk in the alveoli after weaning, which elicits the synthesis of cytokines and growth factors that activate the STAT3 pathway to then subsequently induce apoptosis [75–77]. The high concentrations of EGF ligand in milk [78] are indirect evidence that point to the EGFR’s role within this process. EGF-induced STAT3 activity and the subsequent induction of apoptosis in MDA-MB-468 cells may therefore employ the same molecular mechanisms required for mammary gland involution. Kreuzaler et al. have reported that STAT3-mediated apoptosis in mammary gland involution entails enhancing cathepsin B and L activity, while also downregulating their endogenous inhibitor, serine protease inhibitor 2A (spi2A), in a process that occurs independently of caspase 3, 6 and 7 activity [50]. Similarly, our data indicate that caspase 3 is not a major factor in EGF-induced, STAT3-mediated apoptosis in MDA-MB-468 cells. Together, the cathepsin proteases, spi2A, and other effectors implicated in mammary gland involution may be worth assessing in the context of EGF-induced, STAT3-mediated apoptosis.
Another plausible explanation for this phenomenon is the pathway in which cell death is induced. Caspase-1 activation promotes a particular form of cell death that results from inflammatory responses, in a process known as pyroptosis [79]. Pyroptosis has been documented to induce DNA fragmentation [80] and has also been shown to cleave and activate PARP [81]. Given that Caspase-1 inhibition is capable of blocking EGFR-mediated cell death in MDA-MB-468 cells [54], pyroptosis may be an additional mode of cell death employed by the EGFR. In addition to eliciting STAT3 activity, the Interleukin family of cytokines has been well documented to promote inflammation and further progress inflammatory diseases [82–84]. Interferon-beta, a member of the interferon family of cytokines, promotes apoptosis in pro-B cells in a STAT3-dependent manner [28]. Thus, EGF-induced STAT3 activation and the subsequent induction of pyroptosis would not be an unexpected finding.
These signaling differences must also be considered in the context of the subcellular location in which they occur. There has been a well-established role for spatial regulation of EGFR signaling by the endocytic pathway [85]. In MDA-MB-468 cells, retention of the activated EGFR on the plasma membrane promotes cell proliferation whereas when the EGF:EGFR complex internalizes the cell undergoes apoptosis [58]. Further, the induction of apoptotic pathways is linked to the endosomal accumulation of the EGF:EGFR complex [57]. Since high concentrations of ligand are required for apoptosis, one must consider the contribution of different routes of EGFR internalization. Sigismund et al. have shown that low EGF ligand stimulation (1.5 ng/mL) primarily results in a clathrin-mediated manner of EGFR internalization, whereas high ligand stimulation (20 ng/mL) promotes a clathrin-independent, lipid raft-mediated mode of endocytosis [86]. These differing routes may also impact the composition of EGFR-containing endosomes and which down stream signaling molecules are present.
Although all models are possible, in summarizing the literature, we favor the notion that apoptosis results from the convergence of multiple signaling events. Hyper-activated signaling, due to an increased number of active receptors coupled with slowed down regulation, results in increased metabolic and proliferative cell biology. The cell cannot accommodate these demands, resulting in a disrupted cellular homeostasis that cannot be restored. Ultimately, the cell succumbs due to the stress of the saturated endocytic pathway. While this does not identify a specific, pharmacological target, it suggests the potential benefit of a multi-faceted approach that employs several low level, non-toxic insults.
Highlights.
In MDA-MB-468 cells, STAT3 phosphorylation associates with apoptotic activity.
STAT3 knockdown significantly attenuates EGFR-mediated apoptosis.
STAT3 antagonists, Stattic and S3I-201, exhibit off-target cytotoxicity, in vitro.
Acknowledgments
This work was supported in part by NIH grant GM092874 (B.P.C.) and Southern Regional Education Board – State Doctoral Scholars Program (N.M.J.). We thank members of the Ceresa lab for their critical insights on this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider MR, et al. Beyond wavy hairs: the epidermal growth factor receptor and its ligands in skin biology and pathology. Am J Pathol. 2008;173(1):14–24. doi: 10.2353/ajpath.2008.070942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong DK, et al. Epidermal growth factor-mediated apoptosis of MDA-MB-468 human breast cancer cells. Cancer Res. 1994;54(20):5280–3. [PubMed] [Google Scholar]
- 5.Zhang X, et al. Mutations of epidermal growth factor receptor in colon cancer indicate susceptibility or resistance to gefitinib. Oncol Rep. 2008;19(6):1541–4. [PubMed] [Google Scholar]
- 6.Brabyn CJ, Kleine LP. EGF causes hyperproliferation and apoptosis in T51B cells: involvement of high and low affinity EGFR binding sites. Cell Signal. 1995;7(2):139–50. doi: 10.1016/0898-6568(94)00073-k. [DOI] [PubMed] [Google Scholar]
- 7.Lou YF, et al. Combination of gefitinib and DNA methylation inhibitor decitabine exerts synergistic anti-cancer activity in colon cancer cells. PLoS One. 2014;9(5):e97719. doi: 10.1371/journal.pone.0097719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sordella R, et al. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 9.Wong SF. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther. 2005;27(6):684–94. doi: 10.1016/j.clinthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Janjigian YY, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–45. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maemondo M, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 12.Oxnard GR, et al. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17(17):5530–7. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanton P, et al. Epidermal growth factor receptor expression by human squamous cell carcinomas of the head and neck, cell lines and xenografts. Br J Cancer. 1994;70(3):427–33. doi: 10.1038/bjc.1994.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson NE, et al. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol Endocrinol. 1987;1(3):216–23. doi: 10.1210/mend-1-3-216. [DOI] [PubMed] [Google Scholar]
- 15.Hognason T, et al. Epidermal growth factor receptor induced apoptosis: potentiation by inhibition of Ras signaling. FEBS Lett. 2001;491(1–2):9–15. doi: 10.1016/s0014-5793(01)02166-4. [DOI] [PubMed] [Google Scholar]
- 16.Gill GN, Lazar CS. Increased phosphotyrosine content and inhibition of proliferation in EGF-treated A431 cells. Nature. 1981;293(5830):305–7. doi: 10.1038/293305a0. [DOI] [PubMed] [Google Scholar]
- 17.Kottke TJ, et al. Comparison of paclitaxel-, 5-fluoro-2′-deoxyuridine-, and epidermal growth factor (EGF)-induced apoptosis. Evidence for EGF-induced anoikis. J Biol Chem. 1999;274(22):15927–36. doi: 10.1074/jbc.274.22.15927. [DOI] [PubMed] [Google Scholar]
- 18.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18(1):45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell JE., Jr STATs and gene regulation. Science. 1997;277(5332):1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 20.Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets. 2004;8(5):409–22. doi: 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77(1):63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 22.Wegenka UM, et al. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol Cell Biol. 1993;13(1):276–88. doi: 10.1128/mcb.13.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia R, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 24.Park OK, Schaefer TS, Nathans D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci U S A. 1996;93(24):13704–8. doi: 10.1073/pnas.93.24.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Jove R. The STATs of cancer–new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 26.Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. 2008;135(6):995–1003. doi: 10.1242/dev.005439. [DOI] [PubMed] [Google Scholar]
- 27.Watson CJ, Neoh K. The Stat family of transcription factors have diverse roles in mammary gland development. Semin Cell Dev Biol. 2008;19(4):401–6. doi: 10.1016/j.semcdb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Gamero AM, et al. Activation of Tyk2 and Stat3 is required for the apoptotic actions of interferon-beta in primary pro-B cells. J Biol Chem. 2006;281(24):16238–44. doi: 10.1074/jbc.M509516200. [DOI] [PubMed] [Google Scholar]
- 29.Minami M, et al. STAT3 activation is a critical step in gp130-mediated terminal differentiation and growth arrest of a myeloid cell line. Proc Natl Acad Sci U S A. 1996;93(9):3963–6. doi: 10.1073/pnas.93.9.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced growth inhibition in the human prostate cancer cell line LNCaP. Prostate. 2000;42(2):88–98. doi: 10.1002/(sici)1097-0045(20000201)42:2<88::aid-pros2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 31.Jackson NM, Ceresa BP. Protein Kinase G facilitates EGFR-mediated cell death in MDA-MB-468 cells. Exp Cell Res. 2016;346(2):224–32. doi: 10.1016/j.yexcr.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rush JS, et al. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem. 2012;287(1):712–22. doi: 10.1074/jbc.M111.294470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Messersmith W, et al. Assessment of Epidermal Growth Factor Receptor (EGFR) signaling in paired colorectal cancer and normal colon tissue samples using computer-aided immunohistochemical analysis. Cancer Biol Ther. 2005;4(12):1381–6. doi: 10.4161/cbt.4.12.2287. [DOI] [PubMed] [Google Scholar]
- 34.Kozyulina PY, et al. p38 MAP kinase enhances EGF-induced apoptosis in A431 carcinoma cells by promoting tyrosine phosphorylation of STAT1. Biochem Biophys Res Commun. 2013;430(1):331–5. doi: 10.1016/j.bbrc.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 35.Gingras MC, et al. Cytoplasmic death signal triggered by SRC-mediated phosphorylation of the adenovirus E4orf4 protein. Mol Cell Biol. 2002;22(1):41–56. doi: 10.1128/MCB.22.1.41-56.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.She QB, et al. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8(4):287–97. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang E, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80(2):285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–64. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filmus J, et al. MDA-468, a human breast cancer cell line with a high number of epidermal growth factor (EGF) receptors, has an amplified EGF receptor gene and is growth inhibited by EGF. Biochem Biophys Res Commun. 1985;128(2):898–905. doi: 10.1016/0006-291x(85)90131-7. [DOI] [PubMed] [Google Scholar]
- 40.Kumar G, et al. Involvement of Janus kinases, p52shc, Raf-1, and MEK-1 in the IL-6-induced mitogen-activated protein kinase cascade of a growth-responsive B cell line. J Immunol. 1994;153(10):4436–47. [PubMed] [Google Scholar]
- 41.Heinrich PC, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374(Pt 1):1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman RC, et al. Resonance assignments for Oncostatin M, a 24-kDa alpha-helical protein. J Biomol NMR. 1996;7(4):273–82. doi: 10.1007/BF00200429. [DOI] [PubMed] [Google Scholar]
- 43.Kucia-Tran JA, et al. Overexpression of the oncostatin-M receptor in cervical squamous cell carcinoma is associated with epithelial-mesenchymal transition and poor overall survival. Br J Cancer. 2016;115(2):212–22. doi: 10.1038/bjc.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auernhammer CJ, et al. The oncostatin M receptor/gp130 ligand murine oncostatin M induces apoptosis in adrenocortical Y-1 tumor cells. J Endocrinol. 2004;180(3):479–86. doi: 10.1677/joe.0.1800479. [DOI] [PubMed] [Google Scholar]
- 45.Chipoy C, et al. Sensitization of osteosarcoma cells to apoptosis by oncostatin M depends on STAT5 and p53. Oncogene. 2007;26(46):6653–64. doi: 10.1038/sj.onc.1210492. [DOI] [PubMed] [Google Scholar]
- 46.Negoro S, et al. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation. 2001;103(4):555–61. doi: 10.1161/01.cir.103.4.555. [DOI] [PubMed] [Google Scholar]
- 47.Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269(49):30761–4. [PubMed] [Google Scholar]
- 48.Luttrell DK, et al. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc Natl Acad Sci U S A. 1994;91(1):83–7. doi: 10.1073/pnas.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lutz MP, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243(2):503–8. doi: 10.1006/bbrc.1997.8043. [DOI] [PubMed] [Google Scholar]
- 50.Kreuzaler PA, et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011;13(3):303–9. doi: 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- 51.Armstrong DK, et al. Epidermal growth factor-mediated apoptosis of MDA-MB-468 human breast cancer cells. Cancer Res. 1994;54(20):5280–5283. [PubMed] [Google Scholar]
- 52.Chen KY, et al. The role of tyrosine kinase Etk/Bmx in EGF-induced apoptosis of MDA-MB-468 breast cancer cells. Oncogene. 2004;23(10):1854–62. doi: 10.1038/sj.onc.1207308. [DOI] [PubMed] [Google Scholar]
- 53.Grudinkin PS, et al. EGF-induced apoptosis in A431 cells is dependent on STAT1, but not on STAT3. Eur J Cell Biol. 2007;86(10):591–603. doi: 10.1016/j.ejcb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Chin YE, et al. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol Cell Biol. 1997;17(9):5328–37. doi: 10.1128/mcb.17.9.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.French AR, et al. Postendocytic Trafficking of Epidermal Growth Factor-Receptor Complexes is Mediated through Saturable and Specific Endosomal Interactions. J Biol Chem. 1994;269(22):15749–15755. [PubMed] [Google Scholar]
- 56.Wiley HS. Anomalous Binding of Epidermal Growth Factor to A431 Cells Is Due to the Effect of High Receptor Densities and a Saturable Endocytic System. J Cell Biol. 1988;1–7:801–810. doi: 10.1083/jcb.107.2.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rush JS, et al. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. The Journal of biological chemistry. 2012;287(1):712–22. doi: 10.1074/jbc.M111.294470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hyatt DC, Ceresa BP. Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp Cell Res. 2008;314(18):3415–25. doi: 10.1016/j.yexcr.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sherry MM, et al. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27(10):2383–92. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alvarez JV, et al. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66(6):3162–8. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 61.Berthenet K, et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene. 2016 doi: 10.1038/onc.2016.403. [DOI] [PubMed] [Google Scholar]
- 62.Azare J, et al. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol Cell Biol. 2007;27(12):4444–53. doi: 10.1128/MCB.02404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burke WM, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene. 2001;20(55):7925–34. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 64.Xie TX, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23(20):3550–60. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- 65.Li L, Shaw PE. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J Biol Chem. 2002;277(20):17397–405. doi: 10.1074/jbc.M109962200. [DOI] [PubMed] [Google Scholar]
- 66.Tan Q, et al. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015;106(8):1023–32. doi: 10.1111/cas.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.West NR, Watson PH. S100A7 (psoriasin) is induced by the proinflammatory cytokines oncostatin-M and interleukin-6 in human breast cancer. Oncogene. 2010;29(14):2083–92. doi: 10.1038/onc.2009.488. [DOI] [PubMed] [Google Scholar]
- 68.Ruff-Jamison S, Chen K, Cohen S. Induction by EGF and interferon-gamma of tyrosine phosphorylated DNA binding proteins in mouse liver nuclei. Science. 1993;261(5129):1733–6. doi: 10.1126/science.8378774. [DOI] [PubMed] [Google Scholar]
- 69.Skov S, et al. Activation of Stat-3 is involved in the induction of apoptosis after ligation of major histocompatibility complex class I molecules on human Jurkat T cells. Blood. 1998;91(10):3566–73. [PubMed] [Google Scholar]
- 70.Zhang J, et al. STAT3 exerts two-way regulation in the biological effects of IL-6 in M1 leukemia cells. Leuk Res. 2001;25(6):463–72. doi: 10.1016/s0145-2126(00)00157-0. [DOI] [PubMed] [Google Scholar]
- 71.Pencik J, et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun. 2015;6:7736. doi: 10.1038/ncomms8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sironi JJ, Ouchi T. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem. 2004;279(6):4066–74. doi: 10.1074/jbc.M307774200. [DOI] [PubMed] [Google Scholar]
- 73.Chin YE, et al. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science. 1996;272(5262):719–22. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 74.Stein T, Salomonis N, Gusterson BA. Mammary gland involution as a multi-step process. J Mammary Gland Biol Neoplasia. 2007;12(1):25–35. doi: 10.1007/s10911-007-9035-7. [DOI] [PubMed] [Google Scholar]
- 75.Abell K, et al. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7(4):392–8. doi: 10.1038/ncb1242. [DOI] [PubMed] [Google Scholar]
- 76.Kritikou EA, et al. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130(15):3459–68. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 77.Rieanrakwong D, et al. Prolactin Suppression of Gonadotropin-Releasing Hormone Initiation of Mammary Gland Involution in Female Rats. Endocrinology. 2016;157(7):2750–8. doi: 10.1210/en.2016-1180. [DOI] [PubMed] [Google Scholar]
- 78.Beardmore JM, Richards RC. Concentrations of epidermal growth factor in mouse milk throughout lactation. J Endocrinol. 1983;96(2):287–92. doi: 10.1677/joe.0.0960287. [DOI] [PubMed] [Google Scholar]
- 79.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–59. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 80.Mariathasan S, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 81.Malireddi RK, et al. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol. 2010;185(6):3127–30. doi: 10.4049/jimmunol.1001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alonzi T, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187(4):461–8. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsutsui H, et al. Cytokine-induced inflammatory liver injuries. Curr Mol Med. 2003;3(6):545–59. doi: 10.2174/1566524033479618. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto M, et al. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164(9):4878–82. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 85.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nature reviews Molecular cell biology. 2009;10(9):609–22. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sigismund S, et al. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci U S A. 2005;102(8):2760–5. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]