

Abstract
Small interfering RNA (siRNA) is a promising molecule for gene therapy, but its therapeutic administration remains problematic. Among the recently proposed vectors, cell-penetrating peptides show great promise in in vivo trials for siRNA delivery. Human protein DMBT1 (deleted in malignant brain tumor 1) is a pattern recognition molecule that interacts with polyanions and recognizes and aggregates bacteria. Taking advantage of these properties, we investigated whether specific synthetic DMBT1-derived peptides could be used to formulate nanoparticles for siRNA administration. Using an electrophoretic mobility shift assay and UV spectra, we identified two DMBT1 peptides that could encapsulate the siRNA with a self- and co-assembly mechanism. The complexes were stable for at least 2 hr in the presence of either fetal bovine serum (FBS) or RNase A, with peptide-dependent time span protection. ζ-potential, circular dichroism, dynamic light scattering, and transmission electron microscopy revealed negatively charged nanoparticles with an average diameter of 10–800 nm, depending on the reaction conditions, and a spherical or rice-shaped morphology, depending on the peptide and β-helix conformation. We successfully transfected human MCF7 cells with fluorescein isothiocyanate (FITC)-DMBT1-peptide-Cy3-siRNA complexes. Finally, DMBT1 peptides encapsulating an siRNA targeting a fluorescent reporter gene showed efficient gene silencing in MCF7-recombinant cells. These results lay the foundation for a new research line to exploit DMBT1-peptide nanocomplexes for therapeutic siRNA delivery.
Keywords: DMBT1, cell-penetrating peptides, cellular uptake, transfection, siRNA delivery, RNAi, silencing, gene knockdown, nanoparticles, nanotherapy
Graphical Abstract
Introduction
Therapeutic use of synthetic small interfering RNAs (siRNAs) might allow targeted knockdown of selected genes and thus could be key to successful gene therapy for the treatment of several diseases such as cancer, viral infections, and genetic disorders.1 Despite RNAi being a very efficient mechanism, siRNAs are poorly deliverable in vivo as naked molecules, primarily because of low stability in the bloodstream due to degradation by RNases as well as low cell and tissue permeability.2, 3, 4, 5, 6 Nevertheless, there is increasing interest in the development of efficient siRNA carriers.7, 8, 9, 10, 11
Among the recently developed siRNA delivery systems such as viral vectors, cationic lipids, nanoparticles, and peptides, the so-called cell-penetrating peptides have shown great promise.12, 13 Cell-penetrating peptides contain fewer than 40 amino acids and have the ability to cross the cell membrane by either endocytosis or direct translocation, thus potentially facilitating the translocation of cargo (e.g., nucleic acids and drugs) that can be encapsulated in either a covalent or non-covalent manner.14, 15 Cell-penetrating peptides can be classified as either protein derived, consisting of truncated versions of proteins, or as de novo designed when the sequence is predicted in silico and subsequently validated to allow cellular uptake.16 Although the latter may have better cell penetration because it is usually arginine rich and strongly cationic, designed exogenic sequences may potentially trigger cell toxicity or immunogenicity and thus raise concerns regarding their biocompatibility.17, 18, 19, 20 For this reason, low-arginine and less cationic protein-derived cell-penetrating peptides, especially when derived from human proteins, may be more attractive tools for siRNA delivery.
Protein DMBT1 (deleted in malignant brain tumor 1) is a pattern recognition molecule that plays a key role within the innate immune system, recognizing and aggregating bacteria.21, 22 This protein was initially identified by its absence in malignant brain tumor cells.23 The primary sites of expression of DMBT1 are epithelia and associated glands. The protein is secreted into the lumen of mucosal tissues, such as the respiratory and gastrointestinal tract, and to the surface of non-mucosal tissues, such as the skin.24, 25, 26, 27 Subsequently, this protein has also been identified in the ECM (extracellular matrix) of some tissues.28 The DMBT1 sequence contains a repetition of SRCR (scavenger receptor cysteine-rich) domains alternated with SID (SRCR-interspersed domain) regions and ends with two CUB (complement C1r/C1s-Uegf-Bmp1) domains and a final ZP (zona pellucida) domain.22, 23, 29, 30 To investigate the DMBT1-bacteria interaction mechanism, a consensus sequence of the SRCR domains was split into several peptides, and the SRCRP2 peptide was shown to interact with polyanionic molecules, such as lipoteichoic acid (LTA) of Gram-positive bacteria and lipopolysaccharide (LPS) of Gram-negative bacteria, a property responsible for the protein’s pattern recognition ability.21, 31 Subsequently, an 11-amino-acid subsequence named SRCRP2-11 was discovered to be the smallest functional sequence of DMBT1 and, interestingly, it was found to interact with several polyanionic molecules, including nucleic acids.32, 33
In the present study, we designed a potentially non-immunogenic DMBT1-derived non-covalent siRNA delivery system that is promising for in vivo therapies.
Results
DMBT1 Synthetic Peptides Interact with siRNA by Electrostatic Complexation
We used the electrophoretic mobility shift assay to screen synthetic peptides from the consensus sequence of the SRCR domain of DMBT1 to determine their interaction with tdTomato1, a 21-mer siRNA targeting the fluorescent protein tdTomato. Constant amounts of siRNA were incubated with increasing quantities of the DMBT1-derived peptides. The peptide SRCRP2-11 (sequence: GRVEVLYRGSW) bound to tdTomato1 siRNA efficiently (Figure 1A). A tdTomato1 siRNA interaction was also observed for the peptide SRCRP2-11-R (sequence: GRVRVLYRGSW), which arose from the substitution of the glutamic acid in position 4 of the sequence of SRCRP2-11 with an arginine. Analogous to similar peptide-nucleic acid delivery systems,34 interaction of the peptides with the siRNA was dependent on the molar ratio. In the conditions tested, SRCRP2-11 started to clearly shift the siRNA band at a peptide/siRNA molar ratio of 60:1, while a complete shift was observed at a molar ratio of 100:1. In contrast, SRCRP2-11-R at increasing peptide/siRNA molar ratios showed a more gradual shift of the siRNA band at higher molar ratios (150:1–600:1, Figure 1B).
Figure 1.

Interaction Analysis of DMBT1 Peptides with tdTomato1 siRNA
(A and B) Electrophoretic mobility shift assay of SRCRP2-11 (A) and SRCRP2-11-R (B) incubating tdTomato1 siRNA (5 μM) with increasing amounts of peptide. (C and D) UV spectra analysis shows hypochromicity of the siRNA peak at 260 nm. (E and F) Hypochromicity can be highlighted by plotting the relative change in absorbance, ΔODr, against the peptide/siRNA molar ratio.
UV absorbance spectra were used to investigate in detail the interactions between the peptide and siRNA at a low molar ratio. Similar to the electrophoretic mobility shift assay, increasing amounts of peptide were added to a constant amount of siRNA in solution (0.05 nmol). In this assay, the hypochromic effect of the 260-nm siRNA peak indicated peptide-siRNA interaction.35 Hypochromicity was observed at a molar ratio of 6:1 for both peptides (Figures 1C and 1D) and the hypochromic effect on siRNA at 260 nm was quantified by the relative change in absorbance,35 , defined as follows:
where OD0 is the initial absorbance of the free siRNA and OD is the absorbance of the siRNA-peptide complexes. Plotting ΔODr allows detection of the interaction of SRCRP2-11 or SRCRP2-11-R with the siRNA at a molar ratio of 6:1 for both peptides (Figures 1D and 1E). Complete hypochromicity was observed at peptide/siRNA molar ratios of 28:1–35:1.
To examine the nature of the interaction, we incubated siRNA-peptide complexes at 37°C for 2 hr with an excess of dextran sulfate sodium, which mimics the negatively charged backbone of the siRNA. No electrophoretic shift occurred under these conditions (Figure S1), indicating that siRNA encapsulation is dependent on the electrostatic interaction between the negatively charged siRNA backbone and the positively charged arginines of the peptides.
SRCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA Complexes Are Stable in the Presence of PBS, RNase, or Fetal Bovine Serum
To evaluate the stability of the SRCRP2-11-tdTomato1 siRNA and SCRP2-11-R-tdTomato1 siRNA complexes, peptide-siRNA complexes were incubated in PBS at 37°C for 30, 60, 90, and 120 min. Molar ratios of 200:1 and 800:1 were used for SRCRP2-11 and SRCRP2-11-R, respectively. Molar ratios were chosen based on complete shifts observed in the electrophoretic mobility shift assay. Our results showed that the complexes were stable for at least 120 min, as determined by the lack of free siRNA and the signal strength of the siRNA-shifted bands (Figure 2).
Figure 2.
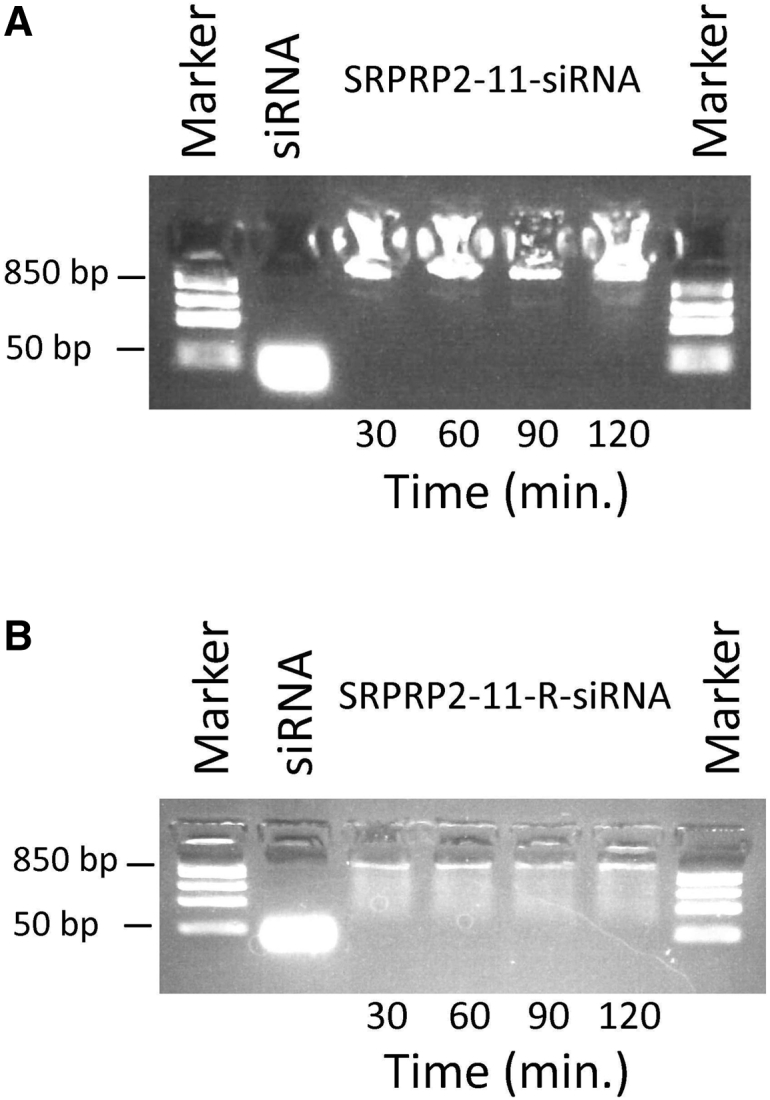
Stability of DMBT1-Peptide-tdTomato1 siRNA Complexes over Time
(A and B) SRCRP2-11-siRNA (A) and SRCRP2-11-R-siRNA (B) complexes at molar ratios of 200:1 and 800:1, respectively, were incubated in PBS for 30, 60, 90, and 120 min. Stability was evaluated as a lack of release of siRNA.
To mimic in vivo conditions, we also tested the stability of the complexes in the presence of either RNase A or fetal bovine serum (FBS). A previous study showed that the blood concentration of RNase A may be increased in patients with cancer36; therefore, SCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA complexes were incubated for 2 hr at 37°C with increasing amounts of RNase A in a range between that of normal blood RNase A and the maximum level observed in these patients. Increasing peptide/siRNA molar ratios were also tested in this assay, and complex stability was related to the strength of siRNA-shifted band signals. As shown in Figures 3A and 3B, RNase A-treated DMBT1-derived peptide-siRNA complexes were more stable than free siRNA in the presence of RNase A. This stability was shown to increase concomitant with higher amounts of peptide, indicating that the peptide protected the siRNA against RNase A degradation.
Figure 3.

Evaluation of DMBT1-Peptide-tdTomato1 siRNA Complex Stability in the Presence of RNase A or FBS
(A and B) SRCRP2-11-siRNA (A) and SRCRP2-11-R-siRNA (B) complexes were formed at peptide-siRNA molar ratios between 200 and 800, corresponding to increasing amounts of peptide. The complexes were incubated 2 hr at 37°C in the presence of RNase A in concentrations corresponding to serum levels ranging from normal subjects to patients with cancer (100–800). Stability was evaluated as the lack of free siRNA and the strength of the shifted band signal. (C and D) The experiment was repeated analogously in the presence of 10%–50% FBS.
The assay was additionally performed in the presence of increasing amounts of FBS (Figures 3C and 3D). Analogous to observations in the presence of RNase A, the SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes were more stable than free siRNA in the presence of 10%, 25%, and 50% FBS after 2-hr incubation at 37°C. This effect was greater at higher peptide/siRNA molar ratios corresponding to higher peptide amounts. The unspecific band at about 400 bp corresponded to FBS, as shown in Figure S2.
Characterization of SRCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA Complexes
To further characterize the peptide-siRNA complexes, we used dynamic light scattering to measure particle size and charge. The size of the SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes was not only molar ratio dependent but also increased as the peptide/siRNA ratio increased (Figures 4A and 4B). Moreover, the complex size obtained with both DMBT1-derived peptides was dependent on the concentration of the siRNA. In fact, a particle size shift was observed when complexes were obtained using a reaction mixture containing increasing siRNA concentrations (0.5, 5, and 10 μM). This sensitive dependence on both factors allowed control of the final average diameters between 10 and 800 nm (Figures 4A and 4B). However, analogous to other self-assembled peptide-siRNA nanocomplexes, the polydispersity index was larger than 0.4 for both peptide-siRNA complexes in all conditions tested, showing the broad size range of the particles. While the dimensions of the two peptide-siRNA complexes changed similarly with the increasing peptide/siRNA molar ratio and siRNA concentration, the charge trend of the two peptides at pH 7.0 differed (Figures 4C and 4D). The SRCRP2-11-siRNA complexes showed a negative charge (minimum at −30 mV) at a low molar ratio, which became positive (maximum at +30 mV) when the molar ratio was increased by adding more peptide to the mixture (Figure 4C). SRCRP2-11-R-siRNA complexes, however, had a slightly negative charge at a 1:5 molar ratio with a minimum at −14 mV, which slightly increased at −7 mV when the molar ratio was increased and remained constant until the highest amount of peptide tested (Figure 4D).
Figure 4.

Dynamic Light Scattering, ζ-Potential, and Circular Dichroism Characterization of DMBT1 Peptide-tdTomato1 siRNA Complexes and the Proposed 3D Model
(A and B) Dynamic light scattering for SRCRP2-11-siRNA (A) and SRCRP2-11-R-siRNA (B) complexes showed dependence of the average diameter on the molar ratio and on the siRNA concentration. (C and D) ζ-potential was also sensitive to these two parameters. (E and F) Circular dichroism showed that conformation of both peptides changed from a random coil to a β-helix when the siRNA was gradually added. (G and H) Based on the conformational analysis, we built a 3D model for SRCRP2-11-siRNA (G) and SRCRP2-11-R-siRNA (H), which assumed a central siRNA surrounded by peptide molecules disposed parallel to each other. d.nm., diameter in nanometers.
Circular dichroism was used to evaluate the secondary structure of the complexes (Figures 4E and 4F), and the system was analyzed dynamically under progressive addition of siRNA. As expected, the free peptide solution (SRCRP2-11 or SRCRP2-11-R) showed circular dichroism spectra typical of a random coil conformation. The addition of increasing amounts of siRNA to either SRCRP2-11 or SRCRP2-11-R induced a conformational change of the peptide toward a β-helix conformation, with a solenoidal structure.37 Based on the size and conformational studies, we elaborated a 3D model of the complexes (Figures 4G and 4H).
Transmission electron microscopy (TEM) was performed on SRCRP2-11-R-siRNA and SRCRP2-11-siRNA nanocomplexes, which were obtained from reaction conditions allowing particles with an average diameter of 100 nm, as determined by dynamic light scattering. This analysis revealed a different morphology of the two nanocomplexes and confirmed their polydispersity. While SCRCP2-11-siRNA complexes (Figures 5A and 5B) were round and 20–50 nm in size under the tested conditions (a few aggregates had a maximum size of 350 nm), SRCRP2-11-R-siRNA were more rice shaped with a length of 10–20 nm, which seemed to reassemble in amorphous aggregates of different shapes and sizes between 100 and 500 nm (Figures 5C and 5D).
Figure 5.

Morphology of DMBT1-Peptide-tdTomato1 siRNA Complexes
(A–D) Transmission electron microscopy showed a spherical shape of SRCRP2-11-siRNA (A and B) and a rice shape for SRCRP2-11-R-siRNA (C and D). While the SRCRP2-11-siRNA monomer diameter was about 20–50 nm, the SRCRP2-11-R-siRNA monomer was about 10–20 nm in length and tended to aggregate in bigger clusters of 100–500 nm.
SRCRP2-11 and SRCRP2-11-R Nanocomplexes with siRNA Are Internalized by Breast Cancer MCF7 Cells and Induce Target Gene Knockdown
Next, we evaluated the transfection efficiency of fluorescein isothiocyanate (FITC)-DMBT1-derived peptides and their complexes with Cy3-siRNA in MCF7 cells. FITC-SRCRP2-11 and FITC-SRCRP2-11R and their nanocomplexes with Cy3-siRNA were administered to MCF7 cells at a constant concentration of Cy3-siRNA but at an increasing concentration of peptides (0.1–1.8 mM for FITC-SRCRP2-11; 1.5–12 mM for FITC-SRCRP2-11R), corresponding to increasing peptide-siRNA molar ratios, ranging from 10:1 to 180:1 for FITC-SRCRP2-11-Cy3-siRNA complexes and from 150:1 to 1,200:1 for FITC-SRCRP2-11R-Cy3-siRNA complexes. After 24-hr culture, cells were washed with PBS to remove the complexes from the surface, and FITC and Cy3 fluorescence was measured to evaluate the cellular content of peptide and siRNA. For both peptide variants, dose-dependent cellular uptake of the peptides was observed. Interestingly, for both peptide variants, the fluorescence of Cy3 was augmented in parallel with the FITC-peptide amount, although the concentration of Cy3-siRNA was kept constant. This effect was maximal at peptide/siRNA molar ratios of 60:1 or 150:1 for FITC-SRCRP2-11 or the FITC-SRCRP2-11-R variant, respectively, suggesting that these were the optimal molar ratios forcing the cellular uptake of Cy3-siRNA (Figure 6). Beyond the optimal molar ratios, a gradual decrease in the Cy3-siRNA signal was observed in parallel with the expected size increase of the siRNA-peptide complexes. On the contrary, peptide internalization seemed to increase in parallel with the amount of peptide without apparent saturation, since internalization of the excess peptide is not affected by complex size.
Figure 6.
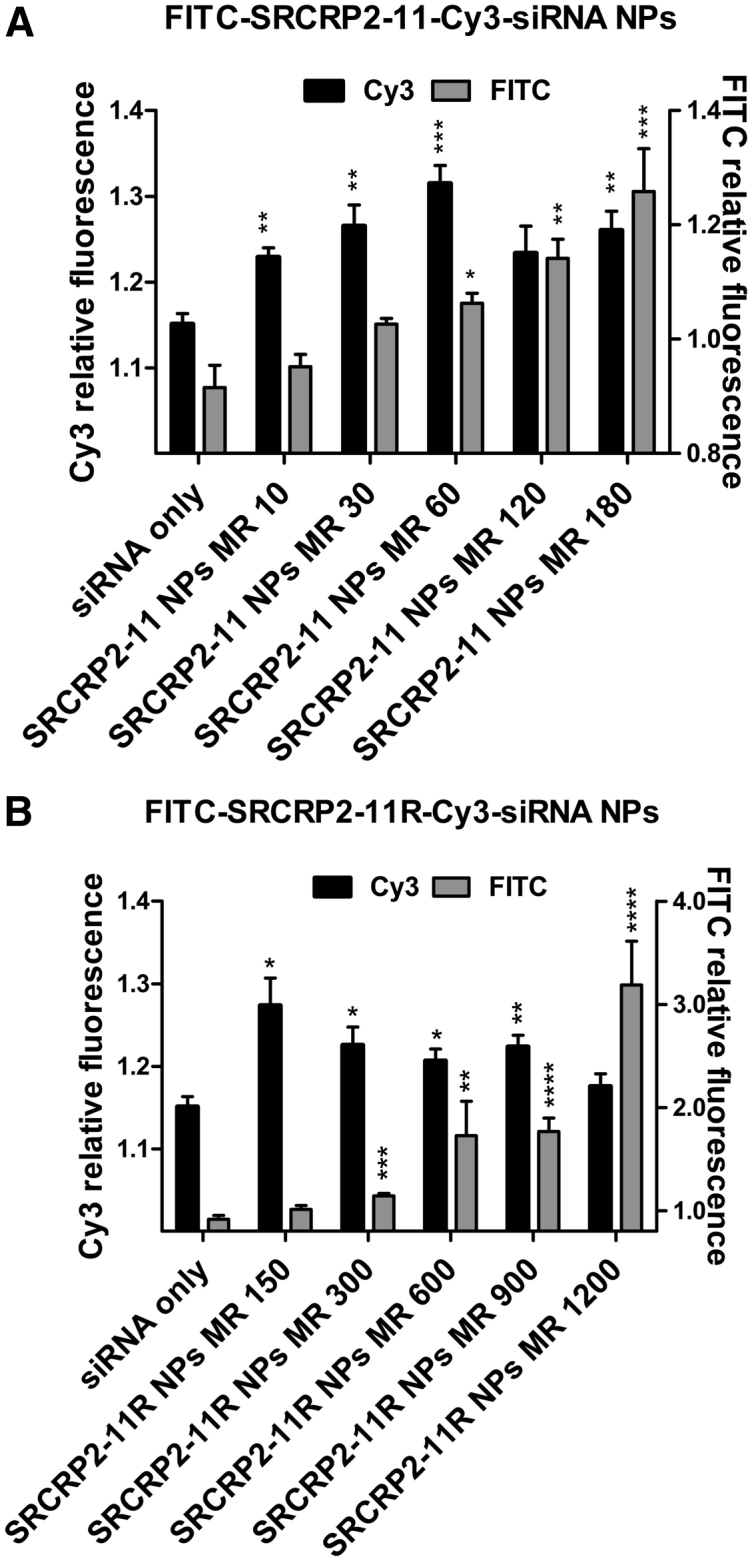
Transfection Efficiency of FITC-SRCRP2-11-Cy3-siRNA Complexes in MCF7 Cells
FITC fluorescence (gray) and Cy3 fluorescence (black) was measured after 24-hr culturing of the cells in the presence of either FITC-SRCRP2-11-Cy3-siRNA (A) or FITC-SRCRP2-11R-Cy3-siRNA (B) complexes. The amount of FITC-peptides was increased and the amount of Cy3-siRNA was kept constant. Values are the average of three experiments; bars are SEM. Asterisks indicate the significance level, based on the Student’s t test (*p = 0.0021–0.04332, **p = 0.0002–0.0021, ***p = 0.0001–0.0002, ****p < 0.0001).
Subsequently, the FITC-SRCRP2-11-Cy3-siRNA and FITC-SRCRP2-11R-Cy3-siRNA complexes were administered to MCF7 cells at 60:1 and 150:1 molar ratios (corresponding to 0.6 and 1.6 mM), respectively, and the cells were incubated 24 hr before they were imaged by confocal microscopy. Imaging confirmed the internalization of the peptides (Figures 7A and 7C) and peptide-siRNA complexes (Figures 7B and 7D) and the increase in siRNA uptake when administered after peptide complexation compared to the naked-siRNA control (Figure 7E). Higher magnification of these images, as shown in Figure S3, revealed that the morphology of cells that took up FITC-SRCRP2-11-Cy3-siRNA and FITC-SRCRP2-11-R-Cy3-siRNA is similar to that of healthy untreated cells, a finding supported by the well-defined shape of the nuclei visualized by DAPI stain. Successfully transfected cells showed a diffused green signal and green vesicles (Figure S4, image at enhanced magnification for FITC-SRCRP2-11 transfected cells).
Figure 7.

Internalization of FITC-DMBT1-Peptide and FITC-DMBT1-Peptide-siRNA Complexes by MCF7 Breast Cancer Cells
(A–E) Confocal microscopy images of MCF7 cells treated with FITC-SRCRP2-11 peptide (A), FITC-SRCRP2-11-Cy3-siRNA complex (B), FITC-SRCRP2-11-R peptide (C), FITC-SRCRP2-11-R-Cy3-siRNA complex (D), and naked Cy3-siRNA (E). Colors indicate the following: red, CellMask Deep Red plasma membrane staining of plasma membranes; blue, DAPI staining of nuclei; green, FITC-peptide; yellow, Cy3-siRNA. Scale bars represent 50 μm.
The possibility of using unlabeled SRCRP2-11-siRNA and SRCRP2-11-R-siRNA nanocomplexes to induce target gene knockdown was investigated by transfecting recombinant MCF7 cells stably expressing the fluorescent protein tdTomato. SRCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA complexes were administered to the cells, and tdTomato fluorescence after 96 hr was compared to that of the untreated cells. For this purpose, we evaluated peptide/siRNA complexes at three different molar ratios as follows: 6:1, 60:1, and 600:1 for SRCRP2-11 and 15:1, 150:1, and 1,500:1 for SRCRP-11-R. Complexes were diluted with PBS to peptide concentrations of 6–600 μM for SRCRP2-11-tdTomato1 and 15–1,500 μM for SRCRP2-11-R-tdTomato1, corresponding to an siRNA concentration of 1 μM. Finally, after the complex solutions were added to a 96-well plate, FBS-free medium containing MCF7 tdTomato recombinant cells was added to reach a final peptide concentration of 0.6–60 μM and 1.5–150 μM, respectively, and a final siRNA concentration of 100 nM. As a negative control, complexes at the same molar ratios were generated with a non-targeting siRNA. Furthermore, complexes of tdTomato1 with the cell-penetrating peptides TAT or CADY, or with Lipofectamine, were used as positive controls. tdTomato knockdown was evaluated as the decrease in cell fluorescence after 96 hr. The knockdown efficiency of SRCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA was optimal at peptide/siRNA molar ratios of 60:1 and 150:1 for SRCRP2-11 and SRCRP2-11-R, respectively. In these conditions, knockdown was 48% and 55% for SRCRP2-11 and SRCRP2-11-R, respectively, at 96 hr, which was higher than that for TAT-tdTomato1 (18%) and CADY-tdTomato1 (21%) and also approached the knockdown measured for Lipofectamine-tdTomato1 (66%). Complexes of SRCRP2-11 and SRCRP2-11-R with a non-targeting siRNA did not significantly change the fluorescence (Figure 8).
Figure 8.

Knockdown Efficiency of MCF7-tdTomato Cells Transfected with DMBT1-Peptide-tdTomato1 siRNA Complexes
(A) Relative fluorescence of MCF7-tdTomato cells in the presence of a range of SRCRP2-11-tdTomato1 or SRCRP2-11-non-targeting (NT) siRNAs. (B) Relative fluorescence of MCF7-tdTomato cells in the presence of a range of SRCRP2-11-R-tdTomato1 or SRCRP2-11-R-non-targeting siRNAs. TAT-tdTomato1, CADY-tdTomato1, and Lipofectamine-tdTomato1 were used as positive controls. tdTomato fluorescence was normalized to cell viability. Knockdown was observed at molar ratios of 60:1 and 600:1 for SRCRP2-11-siRNA and at all molar ratios for SRCRP2-11-R-siRNA. Furthermore, knockdown was observed for TAT-tdTomato1 and CADY-tdTomato1 complexes and siRNA combined with Lipofectamine. Values are the average of two biological replicates and three technical replicates, with bars indicating SEMs. Asterisks indicate the level of significance, based on the Student t test (*p = 0.0021–0.04332, **p = 0.0002–0.0021, ***p = 0.0001–0.0002, ****p < 0.0001).
Discussion
siRNA interference and its potential therapeutic application is currently a subject of great interest,2, 8, 38 despite challenges of its use in vivo.39 Among the possible novel delivery systems, self-assembly of siRNA with arginine-rich cell-penetrating peptides is one of the most promising.40, 41 Compared to designed arginine-rich cell-penetrating peptides that are often immunogenic, low-arginine-containing protein-derived peptides have higher biocompatibility potential,17, 18, 19, 20 which is a fundamental requirement for their use in vivo. In this study, we used protein-derived peptides from the pattern recognition protein DMBT1, known to recognize and aggregate bacteria.31, 42 The smallest sequence of this protein that retains these pattern recognition properties is the peptide SRCRP2-11, which is also shown to interact with DNA.32, 33 We successfully used this peptide and its mutant SRCRP2-11-R for non-covalent encapsulation of siRNA. The UV spectra of the siRNA at increasing amounts of peptides resulted in a hypochromic effect of the 260-nm peak. This suggests that both SRCRP2-11 and SRCRP2-11-R peptides begin to interact with the siRNA at a peptide/siRNA molar ratio of 1:7, with complete interaction at molar ratios in the range of 28:1–35:1. The electrophoretic mobility shift assay only showed a shift of the siRNA band at molar ratios of 60:1 for SRCRP2-11 and 150:1 for SRCRP2-11-R under the same experimental conditions as for the UV spectra analysis. The difference in the quantitative results of these two experiments is due to the fact that the molecular interaction between the siRNA and the peptide at a low molar ratio is not detectable by the electrophoretic mobility shift assay, since the low gel resolution does not allow detection of slight changes in molecular weight (MW). At a low molar ratio, the UV spectrum showed hypochromicity of the 260-nm siRNA peak, indicating that the interaction begins at a much lower molar ratio than that revealed by gel shift analysis. Conversely, at high molar ratios where the electrophoretic mobility shift assay revealed a shift of the band, the formation of polydisperse complexes made it impossible to reliably measure UV absorbance. We believe that the siRNA band shift in the electrophoretic mobility shift assay arises from a second-level interaction of the siRNA-peptide complexation in which small preformed complexes interact with each other. Interestingly, the band shift trend differed for the two peptide-siRNA complexes (SRCRP2-11 and SRCRP2-11-R), with the former exhibiting a snap shift of the siRNA band and the latter showing a more gradual band shift. This different behavior is similar to that observed for viral capsid siRNA encapsulation.43, 44 Snap band shift is typical of “high cooperative encapsulation” in which the peptide molecules interact with both the siRNA (co-assembly) and other peptide molecules (self-assembly). Gradual band shift is observed for “low cooperative encapsulation,” where the interaction is mainly between the peptide and the siRNA molecules (co-assembly). Our competition assay showed that polyanion dextran sulfate sodium competes with the siRNA and disassembles the complexes, which strongly suggests that the electrostatic interactions between the backbone of the siRNA and the positively charged arginine are responsible for the complexation.
The SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes were shown to be stable for at least 2 hr at 37°C in the electrophoretic mobility shift assay, which would be sufficient to allow cell internalization. Moreover, peptides SRCRP2-11 and SRCRP2-11-R protected the siRNA from FBS (50%) or RNase A degradation at RNase A concentrations ranging between the serum concentration of healthy human subjects and that of patients with cancer.36 Moreover, the protective effect was stronger when peptide concentrations were increased.
Structural characterization of the peptide-siRNA complexes was done with dynamic light scattering and TEM for size, morphology, and ζ-potential and with circular dichroism for the conformational studies. Although high polydispersity was observed after complexation, both peptides SRCRP2-11 and SRCRP2-11-R allowed the generation of nanocomplexes with the siRNA of the desired average diameter in a range of 10–800 nm. This was achieved by adjusting parameters such as the molar ratio and the reagent concentration. The particle sizes found by dynamic light scattering were confirmed by TEM, which also showed a different morphology for the two peptide-siRNA complexes. SRCRP2-11-siRNA formed 20- to 50-nm nanospheres with a morphology resembling virus-like particles of enterovirus stained in a similar protocol and imaged by TEM,45 a similarity that fits with our virus-like self- and co-assembly hypothesis. SRCRP2-11-R-siRNA complexes exhibited rice shapes and smaller spheres, starting from 10 nm in diameter, with sizes and shapes that are reported to be favorable for direct cellular translocation.46, 47, 48
The circular dichroism spectra suggest that the SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes may have a β-helix conformation characterized by solenoid protein domains, similar to antifreeze proteins.49, 50, 51 Solenoid domains are common protein patterns formed by parallel stacking of repeated units forming a solenoid.52 The circular dichroism spectra suggest that the siRNA forms a central structure that is surrounded by multiple peptide molecules. Based on this assumption, we constructed 3D models of the nanocomplexes where the inner core of the solenoid is probably the siRNA, surrounded by multiple peptides. The 3D models we designed did not show the possibility that some siRNA might surround the external layer of the particles, which would be consistent with the negative ζ-potential measured in PBS for the nanocomplexes.
FITC-conjugated peptides alone and complexed with Cy3-siRNA were successfully delivered to MCF7 cells, and internalization of the complexes was dependent on the FITC-peptide dose. Moreover, despite the constant Cy3-siRNA concentration, the Cy3 signal was increased in parallel with the FITC-conjugated peptide concentration, indicating that Cy3-siRNA transfection efficiency was improved in the presence of the peptides. The siRNA internalization was maximal at peptide-siRNA molar ratios of 60:1 and 150:1 for the SRCRP2-11 and SRCRP2-11R variant, respectively, and decreased after these molar ratios (Figures 7A and 7B). Indeed, at very high molar ratios, the siRNA-peptide complexes are expected to increase in size and aggregate proportionally to the amount of peptide (Figures 4A and 4B). Large and aggregating complexes will prevent encapsulated siRNA from being internalized, which explains the gradual decrease in the Cy3-siRNA signal after optimal molar ratios are reached. In contrast, the peptide internalization consistently increased in parallel with the amount of peptide. At high molar ratio, the peptide was in large excess; thus, the amount of non-complexed free peptide was much higher than peptide complexed with siRNA. As a consequence, the FITC-peptide internalization did not appear to be affected by the increased size of the complexes. Considering the combination of compartmentalization and diffusion of the green signal observed in transfected cells, we hypothesize that either the internalization occurs via endocytosis and direct membrane translocation together or that the nanocomplexes are endocytosed and then able to diffuse through the endosomes.
Both SRCRP2-11-tdTomato1 and SRCRP2-11-R-tdTomato1 were able to promote knockdown of the gene tdTomato1, albeit in the absence of FBS. In the case of SRCRP2-11-R-tdTomato1, the discrepancy between the transfection and the knockdown efficiency can be explained by an improved capability of peptide SRCRP2-11-R in promoting endosomal escape.
Although the optimal peptide/siRNA molar ratios allowing a biological effect (either internalization or knockdown) were apparently high (60:1 or 150:1 for either SRCRP2-11 or SRCRP2-11-R, respectively), it is important to consider that DMBT1-derived peptides are much smaller than the siRNA and much less charged. Consequently, the molar ratios of 60:1 for SRCRP2-11-siRNA and 150:1 for SRCRP2-11-R-siRNA correspond to charge ratios of 1.5:1 and 10:1, respectively. We assume that the larger charge ratio needed for the peptide SRCRP2-11R is related to its smaller steric hindrance, justified by the absence of negative amino acids compared to the peptide SRCRP2-11, which has a glutamic acid in the sequence, allowing self-interaction of the peptides. On the other hand, the advantage of peptide SRCRP2-11R over SRCRP2-11 is the possibility of more compact and smaller nanocomplexes, as confirmed by TEM and a slightly higher knockdown efficiency.
In conclusion, DMBT1-derived peptides SRCRP2-11 and SRCRP2-11-R form stable siRNA complexes exhibiting different stoichiometry and assembly kinetics. They allow the formation of nanocomplexes in a diameter range of 10–800 nm, which can be controlled by modulating reaction conditions (e.g., peptide/siRNA molar ratios and siRNA amounts). Both peptides protect the siRNA against RNase A activity and form complexes that are stable in the presence of FBS. SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes have suitable morphology and size to allow successful cellular internalization by MCF7 cells. Furthermore, the nanocomplexes can be successfully used to allow a target gene knockdown after 96 hr in the absence of FBS. As DMBT1-derived peptides are derived from an endogenous protein and are not strongly cationic, we expect high biocompatibility with low toxicity in vivo and low immunogenicity.17, 18, 19, 20 This study opens promising investigations on novel siRNA delivery systems designed from DMBT1. Future work will focus on increasing the stability of the DMBT1-derived peptide-siRNA complexes in the presence of FBS in the cell culture to evaluate the biological effect of the designed nanocomplexes in vivo, and engineering DMBT1-derived peptide sequences to increase knockdown efficiency at lower incubation times and in the presence of FBS. Should DMBT1-derived peptide-siRNA nanocomplexes succeed in targeting in vivo, they would have the advantage in clinical trials of being “human derived” as opposed to most cell-penetrating peptides, which are either of viral origin or designed de novo.
Materials and Methods
Peptides and siRNA
Peptides SRCRP2-11 (sequence: GRVEVLYRGSW) and SRCRP2-11-R (sequence: GRVRVLYRGSW) and their FITC conjugates were chemically synthesized with >95% purity by GenScript USA. Peptides were resuspended in UltraPure DNase/RNase-Free Distilled Water (Gibco/Thermo Fisher Scientific). The siRNA used for the characterization and for the knockdown experiments was a tdTomato targeting siRNA (tdTomato1 sequence: UUGGUGUCCACGUAGUAGUAG) kindly provided by Prof. Jesper Wengel (Biomolecular Nanoscale Engineering Center, Department of Physics, Chemistry, and Pharmacy, University of Southern Denmark). Silencer Cy3-labeled Negative Control No. 1 siRNA (Ambion/Thermo Fisher Scientific) was used for the internalization experiments. siRNAs were resuspended in siRNA buffer (GE Dharmacon), while dilutions of peptides and siRNA were made in PBS filtered using a Whatman Anotop 25 Plus syringe filter with a 0.02-μm pore size (Whatman).
Electrophoretic Mobility Shift Assay
siRNA (5 μM) was incubated for 30 min at 37°C with increasing amounts of SRCRP2-11 or SRCRP2-11-R, corresponding to peptide/siRNA molar ratios of 20:1–300:1 or 20:1–1,000:1, respectively, in PBS. Preformed complexes were exposed to electrophoresis on agarose gel [3% (w/v)] stained with ethidium bromide and analyzed by UV light. Tris-ethylenediaminetetraacetic acid (TE) buffer was used as a running buffer and to prepare the agarose gels to ensure a pH of 7.
UV Spectrophotometry
siRNA (5 μM) was incubated for 30 min at 37°C with increasing amounts of either SRCRP2-11 or SRCRP2-11-R at peptide/siRNA molar ratios of 0:1–35:1, and UV spectra were measured using a NanoDrop One/Onec UV-Vis Spectrophotometer (Thermo Fisher Scientific) in the custom function with an absorbance range of 190–290 nm.
Dextran Sulfate Sodium Competition Assay
SRCRP2-11-siRNA or SRCRP2-11-R-siRNA prepared at peptide/siRNA molar ratios of 20:1–300:1 or 20:1–1,000:1, respectively, were incubated with 10 mg/mL dextran sulfate sodium (Sigma-Aldrich) for 2 hr at 37°C. The samples were run on a 3% (w/v) agarose gel with ethidium bromide and analyzed by UV light.
Complex Stability over Time and in the Presence of RNase or FBS
To analyze stability over time, SRCRP2-11-siRNA and SRCRP2-11-R-siRNA were prepared at peptide/siRNA molar ratios of 200:1 and 800:1, respectively. The complexes were then incubated in PBS for 30, 60, 90, and 120 min at 37°C and analyzed in comparison with naked siRNA by electrophoresis on a 3% (w/v) agarose gel. For the RNase protection assay, naked siRNA, SRCRP2-11-siRNA, or SRCRP2-11-R-siRNA complexes (peptide/siRNA molar ratios of 200:1–800:1) were incubated for 2 hr at 37°C in PBS containing increasing RNase A (QIAGEN) activity (0–800 U/mL). The samples were analyzed on a 3% (w/v) agarose gel. Similarly, the stability in FBS (Sigma-Aldrich) was examined by incubating the complexes for 2 hr at 37°C in PBS containing 0%, 25%, and 50% FBS. After this treatment, the complexes were analyzed in 3% (w/v) agarose gels.
Hydrodynamic Diameter and ζ-Potential Measurements
siRNA (0.5, 5, or 10 μM) was added with increasing amounts of the peptide SRCRP2-11 or SRCRP2-11-R corresponding to peptide/siRNA molar ratios of 0:20. For each sample and molar ratio, the particle size or ζ-potential was measured at the equilibrium by dynamic light scattering with a Zetasizer Nano ZS (Malvern Instruments), and the average diameter was plotted against the molar ratio. All measurements were performed at 25°C at a measurement angle of 173°. siRNA and peptide stock solutions were filtered separately through 0.02-μm non-protein binding syringe filters (Whatman) before they were used for complex formation. The size and ζ-potential are presented as the mean from three measurements of at least 10 runs per measurement.
Circular Dichroism
Circular dichroism measurements were performed with a J810 Spectropolarimeter (Jasco). Spectra were acquired from samples in a 55-μL, 3-mm path-length quartz cuvette at 25°C. Spectra were scanned from 240 to 200 nm at 200 nm/min with a 2-s response time and a 1-nm pitch. The conformational change of the peptide was detected by measuring the circular dichroism of a peptide solution at constant concentration (0.25 μM for SRCRP2-11 or 0.1 μM for SRCRP2-11-R) at increasing concentrations of siRNA.
TEM
To remove the free peptide (MW 1.3 kDa), SRCRP2-11-siRNA and SRCRP2-11-R-siRNA complexes at peptide/siRNA molar ratios of 200:1 and 800:1, respectively, were formed as described earlier. The complexes were then washed in a dialysis chamber (Spectra/Por Float-A-Lyzer G2 CE, Spectrum Labs) with a MW cutoff (MWCO) of 8–10 kDa against PBS for 24 hr, and the buffer was replaced with fresh PBS after 12 hr. Samples for TEM imaging were prepared at room temperature by pipetting 20 μL of the colloidal dispersion in water on a sheet of Parafilm and placing a 200-mesh Formvar-coated copper grid (Ted Pella) on top of the sample drop for 30 min. The grid was then washed three times in PBS, and drops were spotted on Parafilm and subsequently placed on a drop of 2.5% glutaraldehyde for 10 min. Fixation was followed by five washes with ultrapure water and incubation for 15 min with 2% uranyl acetate as a contrast agent. Finally, the grid was rinsed on a drop of 0.13% methyl cellulose and the excess liquid was gently removed using filter paper, followed by air drying for 30 min. Samples were imaged using a FEI Tecnai G2 20 Twin transmission electron microscope operated at an accelerating voltage of 200 kV.
Modeling
Three-dimensional models of proteins were obtained by manual docking using Swiss PDB Viewer software in combination with Chimera software.53, 54
Internalization Studies
MCF7 cells were obtained from American Type Culture Collection (ATCC) and cultured in DMEM containing 10% FBS, 10 μg/mL insulin, and 100 U/μL penicillin/streptomycin (all from Sigma-Aldrich) at 37°C in humidified 5% CO2.
To quantitatively evaluate transfection efficiency, cells were trypsinized with TrypLE Express trypsin (Invitrogen) when 80% confluence was reached, transferred to 96-well plates at 20,000 cells/well, and cultured overnight. Cells were then transfected with FITC-SRCRP2-11 (1–18 μM), FITC-SRCRP2-11-R (15–120 μM), or their complexes with Cy3-siRNA. The peptide/siRNA molar ratios used ranged between 10 and 180 for FITC-SRCRP2-11-Cy3-siRNA or 150 and 1,200 for FITC-SRCRP2-11R-Cy3-siRNA (corresponding to 1–18 μM and 15–120 μM final peptide concentrations, respectively, and a 100 nM final siRNA concentration). Cells were cultured for 24 hr and then washed three times with PBS, and FITC and Cy3 fluorescence was measured with a VICTOR3 multilabel plate reader (PerkinElmer).
For the imaging experiments, cells were trypsinized with TrypLE Express trypsin (Invitrogen) when 80% confluence was reached, transferred to a Nunc Lab-Tek 12-Chambered Coverglass (Thermo Fisher Scientific) at 30,000 cells/well, and cultured overnight. Cells were then transfected with FITC-SRCRP2-11 or FITC-SRCRP2-11-R at 6 or 15 μM, respectively, or their complexes with Cy3-siRNA at peptide/siRNA molar ratios of 60:1 and 150:1, respectively (corresponding to a 6 or 15 μM peptide concentration and a 0.1 μM siRNA concentration). After 24-hr culture, cells were washed three times with PBS and membranes were stained with CellMask Deep Red plasma membrane stain (Thermo Fisher Scientific) according to the manufacturer’s protocol. Subsequently, cells were fixed with 4% paraformaldehyde (10-min incubation at room temperature), and the chambers were disassembled. Slides were mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Samples were imaged using an FV1000MPE Olympus confocal multiphoton laser scanning microscope, equipped with a 20× numerical aperture (NA) 0.96 objective.
Knockdown Studies
The original MCF7 cell line was obtained from ATCC. The cell line was genetically modified in the research group to establish the MCF7 N107 acceptor cell line, which served in the construction of the tdTomato-MCF7 stable recombinant cell line.55 Cells were cultured as previously described with the addition of hygromycin (300 μg/mL). When 80% confluence was reached, cells were trypsinized and seeded in 96-well plates pretreated with 10 μL peptide-siRNA complexes or controls at 10,000 cells/well in 100 μL FBS-free DMEM containing 10% FBS, supplemented with 10 μg/mL insulin, 100 U/μL penicillin/streptomycin, and 0.5% ITS Liquid Media Supplement (all from Sigma-Aldrich) and were cultured for 96 hr at 37°C in humidified 5% CO2.
SRCRP2-11-tdTomato1 siRNA and SRCRP2-11-R-tdTomato1 siRNA were formed at peptide/siRNA molar ratios of 6:1–600:1 and 15:1–150:1, respectively. The complexes were then diluted with PBS to peptide concentrations of 6–600 μM and 15–1,500 μM, respectively, corresponding to an siRNA concentration of 1 μM for both complexes. The complexes of SRCRP2-11 and SRCRP2-11-R with the non-targeting siRNA (ON-TARGETplus Non-targeting Pool; Dharmacon) were formed using the same process.
The complexes of TAT (sequence: GRKKRRQRRRPQ) or CADY (sequence: Ac-GLWRALWRLLRSLWRLLWRA-cysteamide; both from GenScript USA) with tdTomato1 siRNA were formed as described elsewhere, at peptide/siRNA molar ratios of 10:1 and 80:1, respectively.56, 57 Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) was used to encapsulate the tdTomato1 siRNA following the manufacturer’s instructions.
After the complexes were formed, the volumes were regulated with PBS to 10 μL and added to the plates before the cells were seeded (reverse transfection protocol). tdTomato-MCF7 cells treated in the same way were used as blanks. The final peptide concentration in cell culture was 0.6–60 μM for SRCRP2-11-siRNA and 1.5–150 μM for SRCRP2-11-R-siRNA. The final tdTomato1 concentration in the cell culture was 100 nM for the siRNA-containing samples. Red fluorescence was measured at 560 nm after 96 hr using a VICTOR3 multilabel plate reader. Thereafter, the CellTiter-Blue Cell Viability Assay (Promega) was performed following the manufacturer’s instructions. Fluorescence was measured at 560 nm after 2 hr using a VICTOR3 multilabel plate reader. For all assays, the measured fluorescence was normalized by the cell viability.
Author Contributions
M.T. contributed to the design of the work and performed the majority of the experiments as well as data interpretation, critical revision, and manuscript preparation. C.C. contributed to data interpretation of the in vitro experiments on peptide-siRNA interaction and stability assays. P.L.H. contributed to data interpretation and manuscript writing and editing. E.P. contributed to data interpretation in the characterization of the peptide-siRNA complexes. L.M.H. contributed to the TEM images and manuscript editing. K.A.D. was responsible for funding acquisition and intellectual support for data interpretation and experimental design in the characterization of the peptide-siRNA complexes. H.J.D. contributed to data interpretation and experimental design and made substantial contributions to manuscript writing and editing. J.M. conceived and designed the initial outline of the work and contributed to data interpretation and experimental design.
Conflicts of Interest
J.M. holds a patent related to this topic. All other authors declare no conflict of interest.
Acknowledgments
We gratefully acknowledge funding support from the European Union PathChooser Marie-Curie Initial Training Network (ITN) (PITN-GA-2013-608373), the Lundbeckfonden Center of Excellence NanoCAN, the University of Southern Denmark (SDU) DAWN-2020 project of the SDU Presidents SDU2020 program, the SFI (Science Foundation Ireland), and the A.P. Moeller Foundation. Moreover, we acknowledge the Danish Molecular Biomedical Imaging Center (DaMBIC; University of Southern Denmark) for the use of the bioimaging facilities, Ms. Tiina O’Neill (Conway Institute of Biomolecular and Biomedical Research, University College Dublin [UCD]) for technical support in TEM sample preparation, and Prof. Jesper Wengel (Biomolecular Nanoscale Engineering Center, University of Southern Denmark) for providing the tdTomato1 siRNA. We also thank M. Kat Occhipinti for editorial review.
Footnotes
Supplemental Information includes four figures and one table can be found with this article online at http://dx.doi.org/10.1016/j.omtn.2017.06.020.
Contributor Information
Martina Tuttolomondo, Email: mtuttolomondo@health.sdu.dk.
Henrik J. Ditzel, Email: hditzel@health.sdu.dk.
Supplemental Information
References
- 1.Das S., Marwal A., Choudhary D.K., Gupta V.K., Gaur R.K. Mechanism of RNA interference (RNAi): current concept. Proceeding Food Eng. Biotechnol. 2011;9:244–245. [Google Scholar]
- 2.Aagaard L., Rossi J.J. RNAi therapeutics: principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007;59:75–86. doi: 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 4.Whitehead K.A., Langer R., Anderson D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner J.J., Jones S.W., Moschos S.A., Lindsay M.A., Gait M.J. MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity. Mol. Biosyst. 2007;3:43–50. doi: 10.1039/b611612d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupenthal J., Baehr C., Kiermayer S., Zeuzem S., Piiper A. Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem. Pharmacol. 2006;71:702–710. doi: 10.1016/j.bcp.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 7.Lee J.M., Yoon T.J., Cho Y.S. Recent developments in nanoparticle-based siRNA delivery for cancer therapy. BioMed Res. Int. 2013;2013:782041. doi: 10.1155/2013/782041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts T.C., Ezzat K., El Andaloussi S., Weinberg M.S. Synthetic SiRNA delivery: progress and prospects. Methods Mol. Biol. 2016;1364:291–310. doi: 10.1007/978-1-4939-3112-5_23. [DOI] [PubMed] [Google Scholar]
- 9.Oh Y.K., Park T.G. siRNA delivery systems for cancer treatment. Adv. Drug Deliv. Rev. 2009;61:850–862. doi: 10.1016/j.addr.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 10.Wang J., Lu Z., Wientjes M.G., Au J.L.-S. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shajari N., Mansoori B., Davudian S., Mohammadi A., Baradaran B. Overcoming the challenges of siRNA delivery: nanoparticle strategies. Curr. Drug Deliv. 2017;14:36–46. doi: 10.2174/1567201813666160816105408. [DOI] [PubMed] [Google Scholar]
- 12.Nakase I., Tanaka G., Futaki S. Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells. Mol. Biosyst. 2013;9:855–861. doi: 10.1039/c2mb25467k. [DOI] [PubMed] [Google Scholar]
- 13.Wang F., Wang Y., Zhang X., Zhang W., Guo S., Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release. 2014;174:126–136. doi: 10.1016/j.jconrel.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Copolovici D.M., Langel K., Eriste E., Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8:1972–1994. doi: 10.1021/nn4057269. [DOI] [PubMed] [Google Scholar]
- 15.Margus H., Padari K., Pooga M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol. Ther. 2012;20:525–533. doi: 10.1038/mt.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen M., Kilk K., Langel U. Predicting cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008;60:572–579. doi: 10.1016/j.addr.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Brooks N., Esparon S., Pouniotis D., Pietersz G.A. Comparative immunogenicity of a cytotoxic T cell epitope delivered by penetratin and TAT cell penetrating peptides. Molecules. 2015;20:14033–14050. doi: 10.3390/molecules200814033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlagel L.J., Bors L., Mitchell G.W., King J.L., Cao L., Kirk M., Whitaker J.N. Immunological effects of an arginine side chain contaminating synthetically prepared peptides. Mol. Immunol. 1997;34:185–194. doi: 10.1016/s0161-5890(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 19.Riedl P., Reimann J., Schirmbeck R. Peptides containing antigenic and cationic domains have enhanced, multivalent immunogenicity when bound to DNA vaccines. J. Mol. Med. (Berl.) 2004;82:144–152. doi: 10.1007/s00109-003-0502-3. [DOI] [PubMed] [Google Scholar]
- 20.Won Y.-W., Yoon S.-M., Lee K.-M., Kim Y.-H. Poly(oligo-D-arginine) with internal disulfide linkages as a cytoplasm-sensitive carrier for siRNA delivery. Mol. Ther. 2011;19:372–380. doi: 10.1038/mt.2010.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.End C., Bikker F., Renner M., Bergmann G., Lyer S., Blaich S., Hudler M., Helmke B., Gassler N., Autschbach F. DMBT1 functions as pattern-recognition molecule for poly-sulfated and poly-phosphorylated ligands. Eur. J. Immunol. 2009;39:833–842. doi: 10.1002/eji.200838689. [DOI] [PubMed] [Google Scholar]
- 22.Casella C., Tuttolomondo M., Høilund-Carlsen P.F., Mollenhauer J. Natural pattern recognition mechanisms at epithelial barriers and potential use in nanomedicine. Eur. J. Nanomed. 2014;6:141–155. [Google Scholar]
- 23.Mollenhauer J., Wiemann S., Scheurlen W., Korn B., Hayashi Y., Wilgenbus K.K., von Deimling A., Poustka A. DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat. Genet. 1997;17:32–39. doi: 10.1038/ng0997-32. [DOI] [PubMed] [Google Scholar]
- 24.Holmskov U., Lawson P., Teisner B., Tornøe I., Willis A.C., Morgan C., Koch C., Reid K.B. Isolation and characterization of a new member of the scavenger receptor superfamily, glycoprotein-340 (gp-340), as a lung surfactant protein-D binding molecule. J. Biol. Chem. 1997;272:13743–13749. doi: 10.1074/jbc.272.21.13743. [DOI] [PubMed] [Google Scholar]
- 25.Mollenhauer J., Herbertz S., Holmskov U., Tolnay M., Krebs I., Merlo A., Schrøder H.D., Maier D., Breitling F., Wiemann S. DMBT1 encodes a protein involved in the immune defense and in epithelial differentiation and is highly unstable in cancer. Cancer Res. 2000;60:1704–1710. [PubMed] [Google Scholar]
- 26.Prakobphol A., Xu F., Hoang V.M., Larsson T., Bergstrom J., Johansson I., Frängsmyr L., Holmskov U., Leffler H., Nilsson C. Salivary agglutinin, which binds Streptococcus mutans and Helicobacter pylori, is the lung scavenger receptor cysteine-rich protein gp-340. J. Biol. Chem. 2000;275:39860–39866. doi: 10.1074/jbc.M006928200. [DOI] [PubMed] [Google Scholar]
- 27.Holmskov U., Mollenhauer J., Madsen J., Vitved L., Grønlund J., Tornøe I., Kliem A., Reid K.B., Poustka A., Skjodt K. Cloning of gp-340, a putative opsonin receptor for lung surfactant protein D. Proc. Natl. Acad. Sci. USA. 1999;96:10794–10799. doi: 10.1073/pnas.96.19.10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller H., Hu J., Popp R., Schmidt M.H.H., Müller-Decker K., Mollenhauer J., Fisslthaler B., Eble J.A., Fleming I. Deleted in malignant brain tumors 1 is present in the vascular extracellular matrix and promotes angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2012;32:442–448. doi: 10.1161/ATVBAHA.111.239830. [DOI] [PubMed] [Google Scholar]
- 29.Mollenhauer J., Holmskov U., Wiemann S., Krebs I., Herbertz S., Madsen J., Kioschis P., Coy J.F., Poustka A. The genomic structure of the DMBT1 gene: evidence for a region with susceptibility to genomic instability. Oncogene. 1999;18:6233–6240. doi: 10.1038/sj.onc.1203071. [DOI] [PubMed] [Google Scholar]
- 30.Mollenhauer J., Müller H., Kollender G., Lyer S., Diedrichs L., Helmke B., Holmskov U., Ligtenberg T., Herbertz S., Krebs I. The SRCR/SID region of DMBT1 defines a complex multi-allele system representing the major basis for its variability in cancer. Genes Chromosomes Cancer. 2002;35:242–255. doi: 10.1002/gcc.10115. [DOI] [PubMed] [Google Scholar]
- 31.Bikker F.J., Ligtenberg A.J.M., Nazmi K., Veerman E.C.I., van’t Hof W., Bolscher J.G.M., Poustka A., Nieuw Amerongen A.V., Mollenhauer J. Identification of the bacteria-binding peptide domain on salivary agglutinin (gp-340/DMBT1), a member of the scavenger receptor cysteine-rich superfamily. J. Biol. Chem. 2002;277:32109–32115. doi: 10.1074/jbc.M203788200. [DOI] [PubMed] [Google Scholar]
- 32.Bikker F.J., Ligtenberg A.J.M., End C., Renner M., Blaich S., Lyer S., Wittig R., van’t Hof W., Veerman E.C., Nazmi K. Bacteria binding by DMBT1/SAG/gp-340 is confined to the VEVLXXXXW motif in its scavenger receptor cysteine-rich domains. J. Biol. Chem. 2004;279:47699–47703. doi: 10.1074/jbc.M406095200. [DOI] [PubMed] [Google Scholar]
- 33.Hansen P.L., Blaich S., End C., Schmidt S., Moeller J.B., Holmskov U., Mollenhauer J. The pattern recognition molecule deleted in malignant brain tumors 1 (DMBT1) and synthetic mimics inhibit liposomal nucleic acid delivery. Chem. Commun. (Camb.) 2011;47:188–190. doi: 10.1039/c0cc02186e. [DOI] [PubMed] [Google Scholar]
- 34.van Asbeck A.H., Beyerle A., McNeill H., Bovee-Geurts P.H.M., Lindberg S., Verdurmen W.P.R., Hällbrink M., Langel U., Heidenreich O., Brock R. Molecular parameters of siRNA--cell penetrating peptide nanocomplexes for efficient cellular delivery. ACS Nano. 2013;7:3797–3807. doi: 10.1021/nn305754c. [DOI] [PubMed] [Google Scholar]
- 35.Law M., Jafari M., Chen P. Physicochemical characterization of siRNA-peptide complexes. Biotechnol. Prog. 2008;24:957–963. doi: 10.1002/btpr.13. [DOI] [PubMed] [Google Scholar]
- 36.Reddi K.K., Holland J.F. Elevated serum ribonuclease in patients with pancreatic cancer. Proc. Natl. Acad. Sci. USA. 1976;73:2308–2310. doi: 10.1073/pnas.73.7.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heffron S., Moe G.R., Sieber V., Mengaud J., Cossart P., Vitali J., Jurnak F. Sequence profile of the parallel beta helix in the pectate lyase superfamily. J. Struct. Biol. 1998;122:223–235. doi: 10.1006/jsbi.1998.3978. [DOI] [PubMed] [Google Scholar]
- 38.Khvorova A., Osborn M.F., Hassler M.R. Taking charge of siRNA delivery. Nat. Biotechnol. 2014;32:1197–1198. doi: 10.1038/nbt.3091. [DOI] [PubMed] [Google Scholar]
- 39.Colombo S., Zeng X., Ragelle H., Foged C. Complexity in the therapeutic delivery of RNAi medicines: an analytical challenge. Expert Opin. Drug Deliv. 2014;11:1481–1495. doi: 10.1517/17425247.2014.927439. [DOI] [PubMed] [Google Scholar]
- 40.Farkhani S.M., Valizadeh A., Karami H., Mohammadi S., Sohrabi N., Badrzadeh F. Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides. 2014;57:78–94. doi: 10.1016/j.peptides.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 41.Kurrikof K., Gestin M., Langel Ü. Recent in vivo advances in cell-penetrating peptide-assisted drug delivery. Expert Opin. Drug Deliv. 2015;13:373–387. doi: 10.1517/17425247.2016.1125879. [DOI] [PubMed] [Google Scholar]
- 42.Ligtenberg A.J.M., Karlsson N.G., Veerman E.C.I. Deleted in malignant brain tumors-1 protein (DMBT1): a pattern recognition receptor with multiple binding sites. Int. J. Mol. Sci. 2010;11:5212–5233. doi: 10.3390/ijms1112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zlotnick A., Porterfield J.Z., Wang J.C.Y. To build a virus on a nucleic acid substrate. Biophys. J. 2013;104:1595–1604. doi: 10.1016/j.bpj.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kivenson A., Hagan M.F. Mechanisms of capsid assembly around a polymer. Biophys. J. 2010;99:619–628. doi: 10.1016/j.bpj.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ku Z., Ye X., Huang X., Cai Y., Liu Q., Li Y., Su Z., Huang Z. Neutralizing antibodies induced by recombinant virus-like particles of enterovirus 71 genotype C4 inhibit infection at pre- and post-attachment steps. PLoS ONE. 2013;8:e57601. doi: 10.1371/journal.pone.0057601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nangia S., Sureshkumar R. Effects of nanoparticle charge and shape anisotropy on translocation through cell membranes. Langmuir. 2012;28:17666–17671. doi: 10.1021/la303449d. [DOI] [PubMed] [Google Scholar]
- 47.Hong Z., Merino E.G., Reis R.L., Mano J.F. Novel rice-shaped bioactive ceramic nanoparticles. Adv. Eng. Mater. 2009;11:B25–B29. [Google Scholar]
- 48.Karaman D.S., Desai D., Senthilkumar R., Johansson E.M., Råtts N., Odén M., Eriksson J.E., Sahlgren C., Toivola D.M., Rosenholm J.M. Shape engineering vs organic modification of inorganic nanoparticles as a tool for enhancing cellular internalization. Nanoscale Res. Lett. 2012;7:358. doi: 10.1186/1556-276X-7-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenfield N., Fasman G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 1969;8:4108–4116. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- 51.Liou Y.C., Tocilj A., Davies P.L., Jia Z. Mimicry of ice structure by surface hydroxyls and water of a beta-helix antifreeze protein. Nature. 2000;406:322–324. doi: 10.1038/35018604. [DOI] [PubMed] [Google Scholar]
- 52.Andrade M.A., Perez-Iratxeta C., Ponting C.P. Protein repeats: structures, functions, and evolution. J. Struct. Biol. 2001;134:117–131. doi: 10.1006/jsbi.2001.4392. [DOI] [PubMed] [Google Scholar]
- 53.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 54.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 55.Ebbesen M.F., Olesen M.T.J., Gjelstrup M.C., Pakula M.M., Larsen E.K.U., Hansen I.M., Hansen P.L., Mollenhauer J., Malle B.M., Howard K.A. Tunable CD44-specific cellular retargeting with hyaluronic acid nanoshells. Pharm. Res. 2015;32:1462–1474. doi: 10.1007/s11095-014-1552-7. [DOI] [PubMed] [Google Scholar]
- 56.Arthanari Y., Pluen A., Rajendran R., Aojula H., Demonacos C. Delivery of therapeutic shRNA and siRNA by Tat fusion peptide targeting BCR-ABL fusion gene in chronic myeloid leukemia cells. J. Control. Release. 2010;145:272–280. doi: 10.1016/j.jconrel.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 57.Crombez L., Aldrian-Herrada G., Konate K., Nguyen Q.N., McMaster G.K., Brasseur R., Heitz F., Divita G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.