

Summary
Autoimmune diseases such as systemic lupus erythematosus (SLE) are associated with increased cardiovascular disease and reduced plasma high-density lipoproteins (HDL) levels. HDL mediates cholesterol efflux from immune cells via the ATP Binding Cassette Transporters A1 and G1 (ABCA1/G1). The significance of impaired cholesterol efflux pathways in autoimmunity is unknown. We observed that Abca1/g1 deficient mice develop enlarged lymph nodes (LNs) and glomerulonephritis suggestive of SLE. This lupus-like phenotype was recapitulated in mice with knockouts of Abca1/g1 in dendritic cells (DCs) but not in macrophages or T-cells. DC-Abca1/g1 deficiency increased LN and splenic CD11b+ DCs, which displayed cholesterol accumulation and inflammasome activation, increased cell surface levels of the GM-CSF receptor, and enhanced inflammatory cytokine secretion. Consequently, DC-Abca1/g1 deficiency enhanced T-cell activation, Th1 and Th17-cell polarization. NLRP3 inflammasome deficiency diminished the enlarged LNs and enhanced Th1-cell polarization. These findings identify an essential role of DC cholesterol efflux pathways in maintaining immune tolerance.
Keywords: autoimmunity, cholesterol efflux pathways, high-density-lipoprotein, lupus glomerulonephritis, inflammasome, dendritic cells
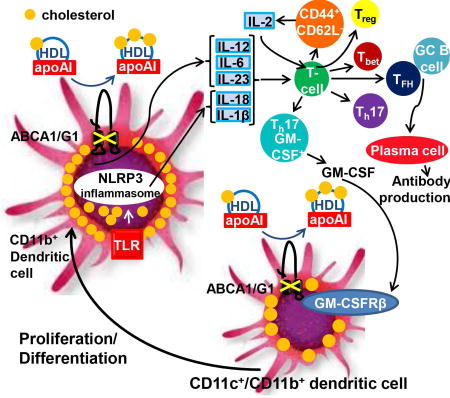
Autoimmune diseases such as systemic lupus erythematosus (SLE) are associated with reduced plasma high-density lipoproteins (HDL) levels. HDL mediates cholesterol efflux from immune cells via ATP Binding Cassette Transporters A1 and G1 (ABCA1/G1). We show that DC cholesterol accumulation in mice with DC Abca1/g1 deficiency promotes inflammasome activation and autoimmunity.
Introduction
Plasma HDL concentrations are reduced in patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) (Norata et al., 2012). These patients have an increased risk of cardiovascular disease (CVD) that cannot be explained by traditional risk factors (Asanuma et al., 2003; Roman et al., 2003; Skeoch and Bruce, 2015). Recent studies have suggested that the capacity of HDL to function as an acceptor for cholesterol efflux is a better predictor of incident CVD than plasma HDL cholesterol concentrations (Khera et al., 2011; Rohatgi et al., 2014). Interestingly, RA disease activity is inversely correlated with the capacity of HDL to function as an acceptor for cellular cholesterol efflux (Ronda et al., 2014). Apolipoprotein A1 (ApoA1) and HDL mediate the efflux of cholesterol from immune cells via the ATP Binding Cassette Transporters A1 and G1 (ABCA1 and ABCG1) (Yvan-Charvet et al., 2007). Whether low HDL and efflux pathways have a causal relationship to autoimmune diseases is unknown.
An almost complete absence of HDL due to deletion of the apoA1 gene, when combined with genetic and dietary hypercholesterolemia, leads to a partial autoimmune phenotype, characterized by cholesterol engorged, enlarged lymph nodes (LNs), increased plasma autoantibodies to dsDNA, and T-cell activation (Wilhelm et al., 2009), suggesting a link between cellular cholesterol accumulation and autoimmunity. Moreover, cholesterol efflux pathways mediated by ABCG1 in T cells have been implicated in T cell proliferative responses and acquired immunity (Armstrong et al., 2010; Bensinger et al., 2008). Autoimmune phenotypes including glomerulonephritis have also been observed in mice with macrophage deficiency of retinoid X receptor α (RXRα) or peroxisome proliferator activated receptor γ (PPARγ) or whole body deficiency of liver X receptor α and β (LXRα and LXRβ) (N-Gonzalez et al., 2009; Roszer et al., 2011). Conversely, treatment with an LXR activator inhibited the development of enlarged LNs and glomerulonephritis in a mouse model of SLE, thus ameliorating autoimmunity (N-Gonzalez et al., 2009). The lupus-like phenotypes were attributed to defective macrophage efferocytosis (the uptake of apoptotic bodies), reflecting reduced expression of MerTK, which is an LXR/RXR target mediating efferocytosis (N-Gonzalez et al., 2009; Roszer et al., 2011). The cholesterol efflux promoting ATP binding cassette transporters ABCA1 and ABCG1 are also key transcriptional targets of LXR/RXR (Costet et al., 2000; Kennedy et al., 2005; Wang et al., 2004), suggesting that defective cellular cholesterol efflux could be involved in the autoimmune phenotypes associated with LXR deficiency.
We have developed Abca1fl/flAbcg1fl/fl mice, which when crossed with appropriate Cre expressing strains lead to cholesterol accumulation in specific cell types (Westerterp et al., 2012). To investigate a potential role of immune cell cholesterol accumulation in autoimmunity, we first assessed autoimmune phenotypes in mice with Abca1/g1 deficiency. This revealed enlarged LNs and glomerulonephritis, suggestive of an autoimmune phenotype. However, autoimmune activation was not due to deficiency of transporters in macrophages or T-cells, but rather in DCs. DC-Abca1/g1 deficiency enlarged the CD11b+ DC population, reflecting increased cell surface expression of the common β subunit of the GM-CSF/IL-3 receptor and enhanced proliferation. Abca1/g1 deficient CD11b+ DCs displayed inflammasome activation, and enhanced secretion of inflammatory cytokines, leading to expansion of T and B-cell subsets in the spleen and LNs. Deficiency of the NOD-like receptor family pyrin domain containing 3 (Nlrp3) inflammasome partly reversed the enlarged LNs in DC-Abca1/g1 deficiency, as well as the increase in IL-1β secretion and Th1 expansion, indicating that this pathway contributes to aspects of the autoimmune phenotype in DC-Abca1/g1 deficiency.
Results
Abca1−/−Abcg1−/− mice and mice with DC Abca1/g1 deficiency develop an autoimmune phenotype
To assess a potential role of cellular cholesterol efflux pathways in acquired immune responses, we characterized mice with general deficiency of Abca1/g1 for autoimmune phenotypes. At 40 weeks of age, chow-fed Abca1−/−Abcg1−/− mice showed dramatic glomerulonephritis, with enlarged glomeruli, mesangial expansion, diffuse and global endocapillary proliferation, and obliterated capillary lumina. Higher power images showed infiltrating mononuclear and polymorphonuclear leukocytes in the glomeruli as well as fibrinoid necrosis (Figure 1A). Abca1−/−Abcg1−/− mice also showed enlarged lymph nodes (Figure 1B), consistent with autoimmunity.
Figure 1. Glomerulonephritis and enlarged lymph nodes in 40 weeks old Abca1−/−Abcg1−/− mice on chow diet.

Lymph nodes (LNs) and kidneys were collected. Kidneys were embedded in paraffin, sectioned and stained with H&E and PAS. (A) Glomerulonephritis in Abca1−/−Abcg1−/−and wild-type littermate mice. Fibrinoid necrosis is indicated. Representative pictures of n=5 mice. (B) Enlarged lymph nodes in Abca1−/−Abcg1−/− mice compared to wild-type controls. n=5; representative pictures of three independent experiments.
To determine which immune cell types might be responsible for these findings, we prepared mice with knockout of both transporters in macrophages, T-cells or DCs by crossing Abca1fl/flAbcg1fl/fl mice with LysmCre, LckCre or CD11cCre mice, respectively. These mice will be referred to as MAC-ABCDKO, T-ABCDKO or DC-ABCDKO mice, respectively, with the caveat that LysmCre and CD11cCre also partially delete in monocytes and neutrophils, and CD11cCre also in a subset of macrophages (Stranges et al., 2007; Westerterp et al., 2013). In each case, these mice were compared to floxed controls, all in the C57BL6/J background. Abca1 and Abcg1 mRNA expression were reduced by >90% in DCs from DC-ABCDKO mice, by >80% in CD4+ T-cells from T-ABCDKO mice (Figure S1A–C), and by >90% in macrophages from MAC-ABCDKO mice (Westerterp et al., 2013). Surprisingly, after 40 weeks of chow diet feeding, kidneys from DC-ABCDKO mice showed evidence of autoimmune glomerulonephritis (Figure 2A), while kidneys from MAC-ABCDKO or T-ABCDKO mice did not show significant pathology (Figure 2B and 2C). Ten out of ten kidneys from DC-ABCDKO mice showed evidence of autoimmune glomerulonephritis, indicating the consistency of this phenotype. Because of a gender-bias in autoimmune glomerulonephritis (Kim et al., 2011), we only included female mice in these studies. Further characterization of the glomeruli of DC-ABCDKO mice showed increased staining for various immunoglobulin classes (IgG, IgM, IgA) and complement components (C3 and C1) in the mesangium and peripheral glomerular capillary walls, compared to glomeruli from controls (Figure 2D). The “full house” positive staining for all these five immune reactants closely resembles that seen in active human lupus nephritis (Weening et al., 2004). The increased deposits of IgG, IgM, and IgA in glomeruli of DC-ABCDKO mice were associated with increased plasma auto-antibodies to IgG+IgM+IgA dsDNA, which was not observed in MAC-ABCDKO or T-ABCDKO mice (Figure 2E). In addition, DC-ABCDKO mice but not MAC-ABCDKO or T-ABCDKO mice exhibited enlarged lymph nodes (Figure 2F), and a temporal increase in plasma IgM and IgG (Figure S1D–E). At 20 weeks of age, DC-ABCDKO mice demonstrated only mild mesangial expansion, indicating that the phenotype develops with age, similar to other mouse models developing spontaneous lupus glomerulonephritis (Kim et al., 2011; N-Gonzalez et al., 2009), and paralleling the temporal increase in plasma IgM and IgG (Figure S1D–E). DC-ABCDKO mice also had compromised renal function, reflected by increased plasma levels of blood urea nitrogen (BUN), creatinine, and uric acid (Figure S1F–H). These characteristics all resemble an SLE phenotype. Increased plasma dsDNA antibodies combined with characteristic nephritis is a sufficient criterion for diagnosis of SLE in humans (Petri et al., 2012). Our findings suggest a major role for ABCA1 and ABCG1 expression in DCs in maintaining immune tolerance and preventing autoimmune glomerulnephritis.
Figure 2. Lupus glomerulonephritis and characteristics of an autoimmune phenotype in mice with DC but not macrophage or T-cell Abca1/g1 deficiency.

Mice were fed chow diet for 40 weeks and kidneys, plasma, and lymph nodes (LNs) were collected. Kidneys were embedded in paraffin, sectioned and stained with H&E and PAS or frozen in OCT, sectioned, and stained as indicated. (A) Glomerulonephritis in DC-ABCDKO mice. Wire loop deposits (middle panel), infiltrating leukocytes (upper arrow, lower panel) and mesangial hypercellularity (lower arrow, lower panel) are indicated. (B) and (C) No glomerulonephritis phenotype in MAC-ABCDKO (B) and T-ABCDKO (C) mice. (D) Immunoglobulin (IgG, IgM, IgA) and complement components 1 and 3 (C1 and C3) deposits are increased in glomeruli of DC-ABCDKO mice. (A) are representative pictures of n=10 mice; (B–D) are representative pictures of n=5 mice. (E) Increased plasma levels of auto-antibodies to dsDNA in DC-ABCDKO, but not MAC-ABCDKO or T-ABCDKO mice. n=6–10. ***P<0.001 by one-way ANOVA with Bonferroni post-test. (F) Increased lymph node mass in DC-ABCDKO, but not MAC-ABCDKO or T-ABCDKO mice. n=4–8. ***P<0.001 by one-way ANOVA with Bonferroni post-test. Data in E–F are presented as mean ± SEM. See also Figure S1.
Abca1/g1 deficiency enhances cholesterol accumulation in DCs
We evaluated lipid accumulation in Abca1/g1 deficient DCs. DC-Abca1/g1 deficiency increased BODIPY staining in splenic CD11b+ DCs (Figure S1I–J) and LNs from DC-ABCDKO mice showed marked enlargement and Oil Red O staining, suggesting neutral lipid accumulation in DCs i.e. DC “foam cells” (Figure S1K). DC-ABCDKO LN DCs isolated using CD11c+ beads and bone marrow (BM) derived DCs also showed evidence of unesterified cholesterol accumulation, as deduced from enzymatic assays or increased filipin staining (Figure S1L–N). Possibly as a result of these findings, plasma total cholesterol levels were decreased in DC-Abca1/g1 deficient mice (Figure S1O).
DC-Abca1/g1 deficiency increases T-cell activation
We next determined potential mechanisms responsible for development of autoimmunity in DC-ABCDKO mice. Blood CD4+ and CD8+ T-cells from DC-ABCDKO mice showed increased activation as reflected by a temporal increase in the percentage of CD44hiCD62Llo CD4+ and CD8+ T-cells (Figure 3A–D), accompanied by increased staining for CXCR3 (Figure 3E–F). DC-ABCDKO mice also showed increased CD4+ and CD8+ T-cell activation in lymph nodes (Figure 3G), and spleen (Figure 3H). The percentage of CD4+ and CD8+ T-cells in DC-ABCDKO mice was similar to controls in blood, and T-cell numbers were similar in spleen, but increased in LNs (Figure S2A–D). While T-cells from control mice did not show proliferation, we observed a low level of proliferation in LN T-cells from DC-ABCDKO mice, possibly accounting for the increase in LN size (Figure S2E–F). Likely, mainly the CD44hiCD62Llo T-cells show enhanced proliferation, as has been described (Tough et al., 1999). Splenic and LN regulatory T cells (Tregs) were also increased (Figure S2G–H), probably related to an increase in IL-2 mRNA levels (Figure S2I) in CD44hiCD62Llo CD4+ T-cells (Boyman and Sprent, 2012).
Figure 3. DC-ABCDKO mice show increased T-cell activation.

Blood, LNs, and spleen were collected; LNs and spleen digested using collagenase, and stained with the indicated antibodies and analyzed by flow cytometry. (A–B) Representative dot plot of activated CD44+CD62L− CD4+ (A) and CD8+ (B) T-cells in blood. (C–D) Time course of increased CD44+CD62L− CD4+ (C) and CD8+ (D) T-cell activation in blood of DC-ABCDKO mice. n=8. (E–F) Increased CXCR3 on CD4+ and CD8+ T-cells in blood. (E) Representative plot. (F) CXCR3+ CD4+ and CD8+ T-cells. n=8. Mice were sacrificed after 30 weeks of chow diet. (G) Increased CD44+CD62L− CD4+ and CD8+ T-cell activation in inguinal lymph nodes of DC-ABCDKO mice. n=5. (H) Increased CD44+CD62L− CD4+ and CD8+ T-cell activation in the spleen of DC-ABCDKO mice. n=7. ***P<0.001 and *P<0.05 by t-test. Data in C–D and F–H are presented as mean ± SEM. See also Figure S2–S3.
Expression profiling in DCs (Gautier et al., 2012) shows that all types of DCs have relatively high expression of Abcg1 with mostly lower expression of Abca1 (Figure S3A). However, ABCA1 and ABCG1 display overlapping functions and show mutual compensation (Yvan-Charvet et al., 2007), and we found that deficiency of both transporters was required for T-cell activation (Figure S3B–C), consistent with the previous finding that deletion of ABCA1 or ABCG1 alone did not lead to autoimmune glomerulonephritis (N-Gonzalez et al., 2009).
DC-Abca1/g1 deficiency does not affect antigen presentation
DCs present antigen to T-cells, which requires the interaction of the MHCII complex on DCs with the T cell receptor (TCR) on T-cells, as well as co-stimulatory signals such as those mediated by CD80/86 (Freeman et al., 1993). DC-Abca1/g1 deficiency increased CD11b+, but not CD8a+ DCs (Figure 4A–C), and did not affect DC surface levels of MHCII (Figure 4D–E), while moderately increasing DC surface levels of CD80/86, mainly on CD11b+ DCs (Figure S4A–B), and not affecting DC surface levels of PDL-1 on CD11b+ DCs (Figure S4C). We then investigated whether the CD11b+ DCs from DC-ABCDKO mice displayed enhanced antigen presentation. We first immunized DC-ABCDKO mice and their controls with ovalbumin (ova)-peptide and then performed an adoptive transplantation with CD90.1+CD4+ OT-II T-cells that specifically recognize ova-MHCII complexes. To monitor proliferation, CD90.1+OT-II CD4+ T-cells were labeled with carboxyfluorescein succinimidyl ester (CFSE). We observed similar dilution of the CFSE label in CD90.1+CD4+ T-cells in DC-ABCDKO mice and their controls, in both lymph nodes and spleen, indicating no difference in antigen presentation (Figure 4F–G). We found that the CD90.1+ marker was essential in distinguishing CFSE diluted CD4+OT-II cells from the enlarged resident T-cell population in DC-ABCDKO mice (results not shown), thus allowing for proper interpretation of the antigen presentation experiments. In vivo, several types of antigen presenting cells are present, and therefore we also investigated whether DC-Abca1/g1 deficiency affected antigen presentation in GM-CSF differentiated bone marrow derived CD11b+ DCs, with or without lipopolysaccharide treatment (mature and immature DCs, respectively). We incubated these DCs with ova-peptide in the presence of CFSE labeled CD4+OT-II T-cells and found a similar CFSE dilution of T-cells incubated with DC-ABCDKO compared to control DCs, both for immature and mature DCs (Figure S4D–E), indicating that DC-Abca1/g1 deficiency also does not affect antigen presentation in vitro. Hence, the increased T-cell activation in DC-ABCDKO mice cannot be explained by enhanced antigen presentation. These results are in line with previous observations showing that hypercholesterolemia in Ldlr−/− or Apoe−/− mice or cholesterol loading of DCs with modified LDL did not affect antigen presentation (Packard et al., 2008).
Figure 4. DC Abca1/g1 deficiency does not affect antigen presentation.

Control and DC-ABCDKO mice were 20 weeks old. Spleens were isolated, stained with the indicated antibodies, and analyzed by flow cytometry. (A–B) Gating strategy of splenic CD11c+MHCII+CD8a+ and CD11c+MHCII+CD11b+ DCs. (C) Total splenic CD8a+ and CD11b+ DCs. n=10. (D–E) Expression of MHCII on CD8a+ and CD11b+ DCs. n=6. (F–G) Antigen presentation. Mice were immunized with ova-peptide. At 24 h after injection, CD4+ OT-II cells from CD90.1 mice were isolated and labeled with CFSE. 2*106 cells were injected per mouse and 72 h after injection, CFSE dilution in CD90.1+CD4+ T-cells was assessed in spleen and inguinal lymph nodes. Representative CFSE dilutions are shown (F) and quantified (G). n=4. Data in C, E, and G are presented as mean ± SEM. **P<0.01, by t-test. See also Figure S4.
DC-Abca1/g1 deficiency activates the inflammasome in CD11b+ DCs
We next explored a potential role of inflammatory pathways in the lupus-like phenotype of DC Abca1/g1 deficiency. Abca1/g1 CD11b+ DCs show increased cytokine expression in response to Toll Like Receptor (TLR) 3/4 ligands (Westerterp et al., 2012). This may have contributed to T-cell activation and proliferation (Tough et al., 1999); however deficiency of Trif or Myd88 did not reverse T-cell activation (Figure S4F–G) or the enlarged LN size in DC-ABCDKO mice (results not shown), possibly because of mutual compensation. TLR activation together with cholesterol accumulation activates the NLRP3 inflammasome in immune cells (Sheedy et al., 2013), mediating the secretion of IL-1β and IL-18. The inflammasome is required for IL-18 production, and deficiency of its central enzyme, caspase-1, inhibits lupus glomerulonephritis in mice (Kahlenberg et al., 2014). We found that DC-Abca1/g1 deficiency enhanced intracellular pro-IL-1β levels in splenic CD11c+CD11b+MHCII+ cells (Figure 5A–B), as well as pro-IL-1β and Nlrp3 mRNA (Figure 5C–D), indicative of inflammasome priming and likely a consequence of TLR activation (Westerterp et al., 2012). To examine whether DC-Abca1/g1 deficiency activated the inflammasome, we used beads to isolate CD11b+ cells, >50% of which show expression of CD11c and MHCII (not shown), and measured caspase-1 cleavage, a hallmark of inflammasome activation. We observed that DC-Abca1/g1 deficiency enhanced caspase-1 cleavage in splenic CD11b+ DCs, and also in LN CD11b+ DCs (Figure 5E–H). MAC-Abca1/g1 deficiency did not affect caspase-1 cleavage in LN CD11b+ DCs (Figure 5G–H), indicating that the inflammasome activation in LNs was specific for DCs. DC-Abca1/g1 deficiency also increased IL-18 plasma levels (Figure 5I), further substantiating inflammasome activation in DC-ABCDKO mice. DC-Abca1/g1 deficiency also increased secretion of IL-1β and IL-18 from splenic CD11b+ cells (Figure 5J–K). This was reversed by treatment with reconstituted HDL (rHDL) (Figure 5J–K), which can promote cholesterol efflux via passive diffusion, confirming that inflammasome activation depends on cholesterol accumulation in Abca1/g1 deficient DCs. In addition, DC-Abca1/g1 deficiency increased splenic Tbet+CD4+ T-cells (Figure 5L, S5), suggesting skewing of T cells towards Th1-cells, consistent with increased DC IL-18 secretion (Ghayur et al., 1997). Thus, cholesterol accumulation in ABCDKO DCs leads to inflammasome activation, increased secretion of IL-1β and IL-18 and skewing of T-cells towards a Th1 phenotype. Interestingly, IL-18 levels are markedly elevated in human SLE, and inflammasome activation contributes to lupus glomerulonephritis in mice (Kahlenberg and Kaplan, 2014; Kahlenberg et al., 2014; Tucci et al., 2008), suggesting a role for inflammasome activation in the autoimmune phenotype of DC-ABCDKO mice.
Figure 5. DC Abca1/g1 deficiency activates the inflammasome and increases Th1-cells.

Mice used in all experiments were 20 weeks old unless indicated otherwise. (A–D). DC-Abca1/g1 deficiency enhances pro-IL-1β protein (A–B) and mRNA levels (C) and Nlrp3 mRNA levels (D) in splenic CD11c+MHCII+CD11b+ cells, indicative of inflammasome priming. FACS plot (A) and quantification (B) are shown. n=4. From mice of the indicated genotypes, CD11b+ cells were isolated from spleen (E–F) or LNs (G–H) using CD11b positive beads. Cells were lysed, and caspase-1 cleavage was assessed using Western blot. Representative images (E, G) are shown and caspase-1 cleavage was calculated as the ratio cleaved caspase-1 (p20) to pro-caspase-1 (p45) (F, H). n=4. (I) IL-18 plasma levels were assessed in 40 week old DC-ABCDKO and control mice using ELISA. Each datapoint indicates a single mouse. n=7–10. (J–K) CD11b+ cells were isolated from the spleens of control and DC-ABCDKO mice using CD11b positive beads, and incubated with or without rHDL (o/n). Media was collected and the secretion of IL-1β (J) and IL-18 (K) was assessed using ELISA, and corrected for cell protein. n=4. (L) Splenic cell homogenates were stained with an antibody to CD4, fixed and permeabilized, and stained for Tbet (L), or its isotype control. For quantification, values were corrected for their respective isotype controls. n=5. Data in B-D, F, and H, J–L are presented as mean ± SEM. B–D, F, I, and L, *P<0.05, **P<0.01, ***P<0.001, by t-test. H, J, K, *P<0.05, **P<0.01, ***P<0.001, by oneway ANOVA with Bonferroni post-test. See also Figure S5.
Deficiency of the NLRP3 inflammasome partly reverses the autoimmune phenotype
To assess a contribution of the inflammasome to autoimmune phenotypes, we generated DC-ABCDKO mice with Nlrp3 deficiency. This partly reversed the increase in LN size compared to DC-ABCDKONlrp3+/− littermate controls (Figure 6A–B), indicating an important contribution of the NLRP3 inflammasome to aspects of the autoimmune phenotype of DC-ABCDKO mice. Consistent with the role of the inflammasome in regulating IL-1β secretion, Nlrp3 deficiency decreased IL-1β secretion from DC-ABCDKO CD11b+ cells (Figure 6C). Nlrp3 deficiency also decreased splenic Tbet+CD4+ T-cells in DC-ABCDKO mice (Figure 6D–E), likely due to a decrease in local IL-18 secretion from splenic DC-ABCDKONlrp3−/− DCs. These results thus suggest that the Nlrp3 inflammasome contributes to aspects of the autoimmune phenotype in DC-ABCDKO mice.
Figure 6. Nlrp3 deficiency reverses part of the phenotype of DC-ABCDKO mice.

Abca1fl/flAbcg1fl/flNlrp3+/−, Abca1fl/flAbcg1fl/flNlrp3−/− (referred to as controls), DC-ABCDKONlrp3+/−, and DC-ABCDKONlrp3−/− mice were 10 weeks old. (A–B). LNs were isolated. Representative pictures are shown (A) and LNs were weighed (B). n=11–25. Splenic CD11b+ cells were isolated and IL-1β secretion (o/n) was assessed (C). n=5–10. Splenic cell homogenates were stained with an antibodies to CD4, fixed and permeabilized, and stained for Tbet, or its isotype control. CD4+Tbet+ T-cells were assessed and values were corrected for their respective isotype controls (D–E). n=5–8. Data in B, C, E are presented as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, by one-way ANOVA with Bonferroni post-test.
Increased GM-CSF receptor-β and cytokine secretion from CD11b+ ABCDKO DCs
Since Nlrp3 deficiency only partly reversed the autoimmune phenotype of DC-ABCDKO mice, we sought to define additional mechanisms regulating inflammation. We had observed that the CD11b+ DC population was expanded in DC-ABCDKO mice (Figure 4C). Abca1/g1 deficiency in hematopoietic stem and progenitor cells increases the cell surface expression of the common β subunit of the IL-3/GM-CSF receptor, increasing the proliferation of these cells (Yvan-Charvet et al., 2010). We found that DC-Abca1/g1 deficiency increased the cell surface but not mRNA levels of the common β subunit of the IL-3/GM-CSF receptor on splenic CD11b+ DCs (Figure 7A–B, S6A–C) together with CD11b+ DC proliferation (Figure 7C–D), likely accounting for the increased number of CD11b+ DCs (Figure 4C), and suggesting enhanced GM-CSF induced differentiation of CD11c+ into CD11b+ DCs.
Figure 7. DC Abca1/g1 deficiency increases surface expression of the common β subunit of the GM-CSF receptor, DC proliferation, secretion of interleukins with reversal by rHDL, and T- and B-cell subsets.

Mice used in all experiments were 20 weeks old unless indicated otherwise. Spleens were isolated (A–D), stained for DC markers in combination with the indicated antibodies, and analyzed by flow cytometry. (A–B) DC-Abca1/g1 deficiency increases common β subunit of the GM-CSF receptor on splenic CD11b+ DCs. n=6. (C–D) DC-Abca1/g1 deficiency increased the % of CD11b+ DCs in the G2M phase, calculated using the Watson model. n=4. (E–G) CD11b+ cells were isolated from the spleens of control and DC-ABCDKO mice using CD11b positive beads, and incubated with or without rHDL (o/n). Media was collected and the secretion of IL23p19 (E), IL12p70 (F), and IL-6 (G) was assessed using ELISA, and corrected for cell protein. n=4. (H–L) Splenic cell homogenates were stained with antibodies to CD4 (H–L), TCRβ and CD8 (K–-L), fixed and permeabilized, stained for IL17A (H–J) or GM-CSF (K–L), and Th17 cells (defined as CD4+IL17A+) (H–J) and GM-CSF+ T-cells (K-L) were measured using flow cytometry. n=5. Data in B, D–G, J, and L are presented as mean ± SEM. B, J, and L *P<0.05, **P<0.01, ***P<0.001, by t-test. D–G *P<0.05, **P<0.01, ***P<0.001, by one-way ANOVA with Bonferroni post-test. See also Figure S6–S7.
DC secretion of IL-23, IL-12, and IL-6 contributes to autoimmune phenotypes. Previous studies have suggested that GM-CSF induces the secretion of these cytokines by DCs (Codarri et al., 2011; El-Behi et al., 2011; Sonderegger et al., 2008), linking GM-CSF signaling with autoimmunity. We found that DC-Abca1/g1 deficiency in splenic CD11c+CD11b+MHCII+ cells markedly enhanced intracellular expression of IL-6 and p40, the subunit of both IL-23 and IL-12, as well as IL-23p19 mRNA expression (Figure S6D–H), likely due to enhanced TLR activation in these cells (Westerterp et al., 2012). The secretion of these cytokines from splenic CD11b+ cells was also increased, with reversal by rHDL treatment (Figure 7E–G). We also found that DC-Abca1/g1 deficiency enhanced GM-CSF induced secretion of IL-23, IL-12, and IL-6 in splenic CD11c+ DCs (Figure S6I–K). This may reflect GM-CSF induced differentiation of CD11c+ into inflammatory CD11b+ DCs (Campbell et al., 2011) in vivo, as DC-ABCDKO CD11b+ DCs were increased in numbers (Figure 4C) and showed increased interleukin secretion without a requirement for added GM-CSF ex vivo (Figure 7E–G). These studies thus link the increased expression of the common β subunit of the GM-CSF receptor on Abca1/g1 deficient DCs with proliferation of CD11b+ DCs that show inflammasome activation and enhanced secretion of a variety of inflammatory cytokines.
DC-Abca1/g1 deficiency increases T- and B-cell subsets
We then investigated whether T- and B-cell subsets that are known to be responsive to these DC interleukins were expanded. IL-1β, IL-6, and IL-23 induce Th17 differentiation (Acosta-Rodriguez et al., 2007). We observed that splenic CD4+IL17A+ T-cells were dramatically increased over time in DC-ABCDKO mice compared to controls, reflecting Th17 expansion (Figure 7H–J). Similar results were found in LNs (not shown). Even though MAC-ABCDKO mice also showed an increase in plasma IL-23 and IL-17 in previous studies, these mice did not develop lupus glomerulonephritis in the current study (Figure 2B), likely reflecting modest increases in plasma IL-23 and IL-17 compared to DC-ABCDKO mice (Westerterp et al., 2012). Consistent with Th17 cell expansion and T-cell activation (Campbell et al., 2011; Codarri et al., 2011; El-Behi et al., 2011), we also found that DC-Abca1/g1 deficiency enhanced splenic GM-CSF+CD4+ and GM-CSF+CD8+ T-cells (Figure 7K–L); 80% of GM-CSF+CD4+ T-cells were positive for IL17A, suggesting they are a subset of Th17 cells (Figure S7A–B). These T-cells by secreting GM-CSF could sustain the differentiation of CD11c+ into CD11b+ DCs that secrete T-cell activating cytokines, suggestive of a previously identified positive feedback loop in which GM-CSF sustains interleukin production by DCs (El-Behi et al., 2011). To test the hypothesis that these effects on T-cells were caused by Abca1/g1 deficiency in DCs, we co-incubated splenic DCs from DC-ABCDKO mice and controls with naïve T-cells. After 5 days, DC-Abca1/g1 deficiency increased differentiation of naïve T-cells into Th17 or GM-CSF+ T-cells (Figure S7C–F), indicating the effect on T-cells was secondary to the DC Abca1/g1 deficiency.
We next investigated whether DC-Abca1/g1 deficiency affected T follicular helper (TFH) and germinal center (GC) B cells, since a previous study suggested that IL-6 secretion by DCs led to expansion of these cells and caused SLE (Kim et al., 2011). Splenic TFH and GC B cells were increased in DC-Abca1/g1 deficient mice compared to their controls (Figure S7G–J), and we also observed an increase in short-lived plasma cells that are present in GCs (Figure 7K–L). Further supporting B-cell proliferation and differentiation, and contributing to TFH accumulation (Coquery et al., 2015), plasma levels of B-cell activating factor (BAFF) were increased in DC-Abca1/g1 deficient mice compared to their controls (Figure S7M). In sum, these findings show that deficiency of cholesterol efflux pathways in DCs promotes inflammasome activation and inflammatory cytokine secretion that leads to a dramatic expansion of Th17 cells, and increases in Th1 T-cells, TFH cells, GC B-cells, plasma cells, and in BAFF plasma levels, changes which have previously been linked to the development of SLE (Kim et al., 2011; Kyttaris et al., 2010; Pisitkun et al., 2012).
Discussion
Our findings indicate that cholesterol efflux pathways in DCs have a key role in preventing cholesterol accumulation and maintaining immune tolerance. Mice with DC Abca1/g1 deficiency in the non-perturbed homeostatic state developed a marked autoimmune phenotype. In contrast, deletion of these transporters in macrophages or T-cells did not lead to auto-immunity indicating a mechanism distinct from previous observations involving reduced MerTK in LXR, PPARγ or RXR deficient macrophages or ABCG1 deficiency in T cells (Bensinger et al., 2008; N-Gonzalez et al., 2009; Roszer et al., 2011). DC Abca1/g1 deficiency activated the inflammasome, increased cell surface expression of the common β subunit for the GM-CSF receptor, CD11b+ DC proliferation, and secretion of a variety of cytokines from CD11b+ DCs, leading to expansion of activated T-cells, Th17 cells, GM-CSF+ T-cells, Th1-cells, TFH cells, GC B-cells, and plasma cells, all changes that have been implicated in the pathogenesis of SLE. Thus, cholesterol accumulation in DCs promotes auto-immunity, predominantly through activation of inflammatory signaling pathways.
Deficiency of the Nlrp3 inflammasome diminished the increases in LN size, DC IL-1β secretion and Tbet+ T-cells in DC-ABCDKO mice, indicating an essential contribution of the inflammasome to key aspects of the autoimmune phenotype in DC-ABCDKO mice. The NLRP3 inflammasome is activated in several autoimmune diseases including RA and SLE. Factors that induce lupus glomerulonephritis, such as neutrophil extracellular traps, self dsDNA, and complement C3a activate the NLRP3 inflammasome in human monocytes and macrophages (Asgari et al., 2013; Kahlenberg et al., 2013; Mathews et al., 2014; Shin et al., 2013). Polymorphisms in NLRP3 or IL-18 (Kahlenberg and Kaplan, 2014) have been associated with SLE risk and plasma IL-18 levels with disease activity (Kahlenberg et al., 2014; Tucci et al., 2008). In mouse models, inhibition of the P2X7 receptor that is essential for ATP mediated NLRP3 inflammasome activation, caspase-1 deficiency, and IL-18 receptor deficiency protect against lupus glomerulonephritis (Kahlenberg et al., 2014; Kinoshita et al., 2004; Zhao et al., 2013). Our study provides direct evidence for a role for the NLRP3 inflammasome in regulating aspects of the autoimmune phenotype in DC-ABCDKO mice, and for the first time relates inflammasome activation in DCs to cholesterol accumulation. Inflammasome priming is likely increased by enhanced TLR signaling via cholesterol-enriched membrane lipid rafts as shown previously in Abca1/g1 deficient cells (Westerterp et al., 2012; Yvan-Charvet et al., 2008), similar to macrophages (Sheedy et al., 2013). However, we did not observe inflammasome activation in LN CD11b+ cells from MAC-ABCDKO mice, probably reflecting a paucity of inflammatory CD11b+ cells in the LNs.
In addition to the inflammasome mediated secretion of IL-1β and IL-18, cholesterol accumulation in splenic Abca1/g1 deficient CD11b+ DCs enhanced the secretion of IL-6, IL-23p19, and IL-12p70, which have been shown to promote autoimmunity (Sonderegger et al., 2008). Lupus patients also show an increase in Th17 cells and deficiency of the IL-23 receptor or IL-17 impairs the development of lupus glomerulonephritis in mice (Kyttaris et al., 2010; Pisitkun et al., 2012). It is thus likely that cholesterol-induced secretion of IL-1β, IL-6, and IL-23p19 by DCs, which induce Th17 expansion (Acosta-Rodriguez et al., 2007) contributes to lupus glomerulonephritis in DC-Abca1/g1 deficiency. Since inflammasome activation is involved in IL-1β secretion it may also link to the observed increased in GM-CSF+ Th17 cells. Multiple mechanisms of inflammatory cytokine production likely explain why the reversal of autoimmunity by Nlrp3 deficiency was only partial. Complexity and heterogeneity may also explain the failure of several interventions targeting specific cytokines in human SLE (Narain and Furie, 2016).
Our data suggest that cholesterol accumulation in DCs leads to both increased GM-CSF receptor levels and increased cytokine secretion and thus engages a positive feedback loop wherein GM-CSF-secreting Th17-cells promote differentiation of CD11c+ into CD11b+ DCs expressing high levels of the GM-CSF receptor, enhancing proliferation, and further cytokine production (Graphical Abstract), as suggested in earlier studies of autoimmune diseases (El-Behi et al., 2011; Lin et al., 2016). GM-CSF enhances cytokine secretion by CD11c+ DCs (Sonderegger et al., 2008), presumably because of its crucial role in differentiating CD11c+ into CD11b+ DCs (Campbell et al., 2011). Our findings suggest that in addition to previous autoimmune models such as EAE, this positive feedback loop plays a role in lupus.
Although our findings were made using a specific model in which cholesterol efflux pathways are deleted in DCs, similar principles may be involved in other settings in which hypercholesterolemia is associated with autoimmunity (Wilhelm et al., 2009). Apoe−/− mice show increased plasma autoantibodies to dsDNA and accelerated glomerulonephritis when crossed with gld mice, or simply in response to feeding a high-cholesterol diet. This was attributed to decreased apoptotic cell clearance (Aprahamian et al., 2004; Wang et al., 2014). Apoa1 and Apoe deficiency decrease plasma HDL levels in mice, potentially leading to DC inflammasome activation and enhanced cytokine secretion that activate T-cells, as suggested by our study. Apoe−/− mice also show increased splenic Th17 cells (Westerterp et al., 2012), which could have contributed to the lupus phenotype. A recent study showed that mice with whole body deficiency of Apoe and Lxrβ develop an autoimmune phenotype, associated with lipid accumulation in CD11c+ cells. Autoimmunity was attributed to enhanced antigen presentation as shown in LXRαβ−/− mice (Ito et al., 2016). Similar to our study, CD11c+MHCII+ cells were expanded in Apoe−/−CD11cCreLXRβfl/fl mice and BAFF levels were increased. In contrast, we did not observe any difference in antigen presentation with either in vivo or in vitro assays (Figure 4F–G, S4D–E). The reason for this apparent discrepancy is unclear. Previous studies have shown that hypercholesterolemia in Apoe−/− mice and cholesterol accumulation in DCs induced by loading with modified lipoproteins does not affect antigen presentation (Packard et al., 2008).
In general, inflammatory stimuli, such as IFNγ (Panousis and Zuckerman, 2000) or TLR3/4 activation (Castrillo et al., 2003) suppress cholesterol efflux pathways. Our studies suggest that in autoimmune disorders such as SLE or RA, inflammatory suppression of HDL levels and of cholesterol efflux pathways and cholesterol accumulation in DCs amplifies autoimmune responses. In this setting, treatments employing LXR activators to up-regulate ABCA1/G1, or cholesterol poor reconstituted HDL which mediate diffusional, non-transporter-dependent cholesterol efflux (Cuchel et al., 2010) and were shown to be anti-inflammatory in CD11b+ DCs in the present study, may have therapeutic benefit. The role of statins in ameliorating autoimmune phenotypes remains unclear (Khattri and Zandman-Goddard, 2013), but also warrants systematic investigation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Marit Westerterp (mw2537@columbia.edu or m.westerterp@umcg.nl).
EXPERIMENTAL MODEL DETAILS
Animals
Abca1−/−Abcg1−/− mice and wild-type littermate controls in the mixed DBA/C57Bl6/J background were generated as described previously (Yvan-Charvet et al., 2007) by crossbreeding Abcg1−/− mice (DeltaGen; subsequently backcrossed 10 times into the C57Bl/6J background) with Abca1−/− mice in the DBA background (Jackson Laboratories, stock number 003897). All other mice were in the C57Bl6/J background. We have deposited Abca1fl/flAbcg1fl/fl mice at Jackson Laboratories (stock number 021067). LysmCreAbca1fl/flAbcg1fl/fl, CD11cCreAbca1fl/flAbcg1fl/fl , and LckCreAbca1fl/flAbcg1fl/fl mice were generated by crossbreeding Abca1fl/flAbcg1fl/fl mice with LysmCre, CD11cCre, and LckCre mice (Jackson Laboratories, stock numbers 004781, 008068, and 003802, respectively). CD11cCreAbca1fl/flAbcg1fl/flMyd88fl/fl, Abca1fl/flAbcg1fl/flMyd88fl/fl, CD11cCreAbca1fl/flAbcg1fl/flTrif−/−, Abca1fl/flAbcg1fl/flTrif−/− , CD11cCreAbca1fl/flAbcg1fl/flNlrp3−/−, and Abca1fl/flAbcg1fl/flNlrp3−/− mice were generated by cross-breeding CD11cCreAbca1fl/flAbcg1fl/fl with Myd88fl/fl, Trif−/−, or Nlrp3−/− mice (Jackson Laboratories, stock numbers 008888, 005037, and 021302, respectively). OT-II mice were from Jackson Laboratories (stock number 004194) and CD90.1 OT-II mice were kindly provided by Dr. Hao Wei Li and Dr. Megan Sykes and were bred at Columbia University by intercrossing CD90.1 (also referred to as Thy1.1) mice (Jackson Laboratories, stock number 000406) with OT-II mice.
For all studies, littermates were used, and mice were housed under SPF conditions in cages (4–5 mice per cage) with micro-isolator tops with a 12 h light cycle (7 am −7 pm) and fed an irradiated chow diet (Purina Mills Diet 5053). Housing temperatures were kept within a range of 71–73 °F (21.7–22.8°C). Water and cages were autoclaved. Cages were changed once weekly, and the health status of the mice was monitored using a dirty bedding sentinel program. The mouse genotype did not cause changes in weight (mouse weight between 20–30 g, depending on gender and age), health or immune status. Female littermates (lupus glomerulonephritis studies) or littermates from both sexes were randomly assigned to experimental groups, unless stated otherwise. The age and number of the mice used for the experiments are indicated for each experiment in the figure legends. In general, n=4–10 mice were used per group and experiments were repeated at least once if n=4 mice were used per experiment to confirm the reproducibility of the results. No inclusion or exclusion criteria were used. All protocols were approved by the Institutional Animal Care and Use Committee of Columbia University.
METHOD DETAILS
Kidney and LN analysis, ELISAs
At 20 or 40 weeks of age, Abca1−/−Abcg1−/− or wild-type female mice were sacrificed. At 40 weeks of age DC-ABCDKO, MAC-ABCDKO, and T-ABCDKO, and their respective littermate control groups were sacrificed (all females). Kidneys were dissected, and one kidney per mouse was fixed in 10% phosphate buffered formalin. Kidneys were then embedded in paraffin, 2 µm sections were made, which were stained with hematoxylin & eosin (H&E) and periodic acid-Schiff (PAS). The other kidney was embedded in OCT, frozen, and 2 µm sections were made, and stained with FITC-conjugated rabbit antibodies to IgG, IgM, IgA, complement C3 or complement C1 (Dako, Carpinteria, CA). These procedures were performed in a blinded fashion, i.e. the analysis was done by an independent observer, unaware of the genotypes. n=5–10 mice were used per genotype.
Inguinal lymph nodes (LNs) were isolated concomitantly with the kidneys, and adipose tissue was removed from LNs. LNs were weighed and pictures were taken from Abca1−/−Abcg1−/− and wild-type LNs (and in later experiments also from LNs of DC-ABCDKONlrp3+/−, DC-ABCDKONlrp3−/− mice, and their littermate controls). LNs from DC-ABCDKO mice and their controls were fixed and embedded in OCT compound and immediately frozen on dry ice. Frozen cross-sections were made (5 µm), stained for Oil Red O, and counterstained with haematoxylin. Pictures were taken using an Olympus IX-70 microscope equipped with a mercury 100-W lamp (CHIU Technical Corp.), an Olympus LCPlanF1 ×100 objective, DP Manager Basic imaging software (version 3.1; Olympus), and an Olympus DP71 CCD camera. To assess cholesterol accumulation in LN DCs from DC-ABCDKO mice and their controls, LNs were meshed in PBS on a 40 µm filter, cells were collected and DCs were isolated using CD11c+ beads (Miltenyi Biotec, CA). Lipids were extracted using the Folch method, and cholesterol levels analyzed using an enzymatic assay (Wako Diagnostics, USA). n=4–8 mice per genotype were used for each experiment described above, and n=11–25 mice per genotype for experiments in DC-ABCDKONlrp3+/−, DC-ABCDKONlrp3−/− mice, and their littermate controls.
From the same abovementioned DC-ABCDKO, MAC-ABCDKO, T-ABCDKO, and their respective controls, also blood was drawn using heparin coated capillaries and kept on ice. Blood was then centrifuged at 10000 rpm for 10 min at 4°C and serum was collected, and immediately assayed for auto-antibodies to dsDNA (IgA+G+M; Alpha Diagnostic International). For serum from DC-ABCDKO mice, uric acid, BUN, and creatinine were measured using specific detection kits in conjunction with a Cobas C311 analyzer (Roche Diagnostics). In addition to these measurements, in serum from 10, 20, and 40 weeks old control and DC-ABCDKO mice (littermates from both sexes randomly assigned to control and DC-ABCDKO group), BAFF, IgM, and IgG levels were measured using ELISA (IgM and IgG, Alpha Diagnostic International; BAFF, R&D Systems). n=10 mice per genotype were used for these experiments.
Differentiation of dendritic cells in vitro
Bone marrow (BM) was isolated from DC-ABCDKO and control mice. Red blood cells (RBCs) were lysed (BD Pharm Lyse, BD Bioscience), and BM was incubated in DMEM containing 10% FBS and 1% pen strep supplemented with 10 ng/ml GM-CSF and 10 ng/ml IL-4 (Peprotech). Cells from one femur and one tibia per animal (either male or female) were incubated in 10 ml of medium in a non-tissue culture coated 100 mm dish. Fresh GM-CSF and IL-4 were added to the media on a daily basis. At day 3, another 10 ml of fresh full medium containing 10 ng/ml GM-CSF and 10 ng/ml IL-4 was added to the culture dish. At day 6 and 8, half of the culture supernatant was collected, centrifuged, and the cell pellet resuspended in 10 ml fresh full medium containing 10 ng/ml GM-CSF and 10 ng/ml IL-4, and added back to the original plate. At day 8, DCs were ready for use in experiments.
Filipin staining
BM derived DCs from DC-ABCDKO and control mice were fixed in 4% paraformaldehyde. Cells were washed, and incubated in 1.5 mg glycine/ml to quench the paraformaldehyde. Cells were stained using 100 µg/mL filipin (Sigma) for 1 h at room temperature. After another wash, cells were mounted using Mowiol (Calbiochem) and visualized with an Axioskop 2 FS MOT upright confocal microscope (Zeiss). Images were obtained by implementing z-scanning and analyzed using Image J software. n=5 fields.
Flow cytometry
Blood samples were collected by tail bleeding into EDTA coated tubes. For analysis of blood T-cells, tubes were kept at 4°C for the whole procedure unless stated otherwise. Red blood cells (RBCs) were lysed (BD Pharm Lyse, BD Bioscience) and white blood cells (WBCs) were centrifuged, washed, and resuspended in HBSS (0.1% BSA, 5 mM EDTA). Cells were stained with a cocktail of antibodies against CD45-APC-Cy7, Ly6-C/G-PerCP-Cy5.5 (BD Pharmingen), CD115-PE, CD44-PE-Cy7, CD62L-APC or CXCR3-APC, CD8-FITC (eBioscience), and TCRβ-PB (Biolegend). CD4+ T-cells were identified as CD45+Ly6-C/G−CD115−TCRβ+CD8− and CD8+ T-cells as CD45+Ly6-C/G−CD115−TCRβ+CD8+. Among these populations, activated T-cells were identified as CD44+CD62L− or CXCR3+. The same stainings were carried out on LN and splenic homogenates, after incubation of axillary and inguinal LNs in collagenase D (clostridium histolyticum, Roche) for 30 min at 37°C or RBC lysis of spleens. LN and splenic homogenates were stained with the Mouse Regulatory T Cell Staining Kit (eBioscience) for analysis of Tregs. n=7–8 mice per genotype were used for each experiment. For analysis of DCs, splenic homogenates were stained with a cocktail of antibodies against F480-PE-Cy7, MHCII-FITC (eBioscience), CD11c-PerCP-Cy5.5 (BD Pharmingen), CD11b-APC, CD8-APC-Cy7, CD86-PE, PD-L1-PE (eBioscience) and CD80-PB (Biolegend), or the common β subunit for the IL-3/GM-CSF receptor (BD Pharmingen). For intracellular cytokine staining, splenic homogenates were stained with CD11c-PE-Cy7, MHCII-FITC (BD Pharmingen), CD11b-APC or CD11b-PB (eBioscience), then fixed-permeabilized using the BD Cytofix/Cytoperm kit (BD Biosciences), and subsequently stained with pro-IL-1β-PE, p40-APC, or IL-6-PB, or their isotype controls (eBioscience). n=4–6 mice per genotype were used for each experiment.
To analyze neutral lipid accumulation in DCs, Bodipy-FITC (Thermo Fisher) was used. For analysis of spleen GM-CSF+ T-cells, T-cell levels of Tbet, or Th17-cells, spleen homogenates were first stained with TCRβ-PB (Biolegend), CD4-APC, CD8-FITC (eBioscience), or for CD4-PB (Th17 cells; eBioscience) then fix-permeabilized, and subsequently stained with GM-CSF-PE, Tbet-PE, or IL17A–FITC, or their isotype controls (eBioscience). For analysis of T follicular helper cells, spleen homogenates were stained with TCRβ-PB (Biolegend), CD4-APC, CXCR5-PerCP-Cy5.5, and PD1-FITC (eBioscience). T follicular helper cells were defined as TCRβ+CD4+CXCR5+PD1+. For analysis of GC B-cells, spleen homogenates were stained with B220-FITC (Biolegend) and GL7-APC (eBioscience). GC B-cells were defined as B220+GL7+. For analysis of plasma cells, spleen homogenates were stained with CD19-PE (Biolegend) and CD138-APC (BD Pharmingen). Plasma cells were defined as CD19−CD138+. n=5–8 mice per genotype were used for these experiments. All samples were analyzed on an LSRII (BD Biosciences), running FACSDiVa software.
To assess Abca1 and Abcg1 mRNA expression in T-cells from T-ABCDKO mice and their littermate controls, blood CD45+CD115−Ly6-C/G−TCRβ+ cells were isolated using flow cytometry and sorted directly into RNeasy lysis buffer (Qiagen) using the FACSAria, running FACSDiVa software. The same procedure was followed to assess Abca1 and Abcg1 mRNA expression in CD45+CD115−Ly6-C/G−CD11c+MHCII+ splenic DCs, or to assess mRNA levels of IL-23p19, GM-CSFRβ, pro-IL-1β, and Nlrp3 in CD45+CD115−Ly6-C/G−CD11c+CD11b+MHCII+ splenic DCs. The cells were lysed in RNeasy lysis buffer, and RNA was then extracted using an RNeasy Micro Kit (Qiagen) and cDNA synthesized using SuperScript VILO (Invitrogen). mRNA levels were assessed using qPCR on a Stratagene Mx3000P (Agilent Technologies), and initial differences in RNA quantity were corrected for using the housekeeping gene m36B4. n=4–6 mice were used per genotype.
Microarray
Microarray analysis was carried out as part of the Immunological Genome Project (http://www.immgen.org), GSE#15907, as described (Gautier et al., 2012).
Antigen presentation in vivo
DC-ABCDKO and control female mice were injected with or without 20 µg 332–339 ovalbumin peptide (OT-II peptide; Anaspec CA). At 24 h after injection, mice were injected with 2*106 cells 10 µM CFSE-PB labeled CD90.1+CD4+ OT-II T-cells. Briefly, CFSE-PB labeled CD4+ OT-II cells were prepared by isolating CD4+ T-cells from the spleen homogenates of CD90.1 OT-II female or male mice using CD4 coated magnetic beads (Miltenyi Biotec, CA) according to the manufacturer’s instructions. CD90.1+CD4+ OT-II T-cells were labeled with 10 µM CFSE-PB (Life Technologies) according to the manufacturer’s instructions. At 72 h after injection with the CD90.1+CD4+ OT-II T-cells, mice were sacrificed and LNs and spleens were isolated. Following homogenization over a 40 µm filter, LN and splenic T-cells were stained using CD4-APC, CD90.1-PE, and CD90.2-FITC (eBioscience), and CFSE-PB dilution was assessed using flow cytometry. All samples were analyzed on an LSRII (BD Biosciences), running FACSDiVa software. n=4 mice per genotype were used for this experiment.
Antigen presentation in vitro
BM derived DCs from DC-ABCDKO and control mice were incubated with or without lipopolysaccharide (LPS) for a period of 24 h (250 ng/ml). Subsequently, cells were incubated with CD4+OTII+ T-cells that had been isolated as described above for in vivo antigen presentation, and were labeled with 5 µM CFSE-PB (Life Technologies) according to the manufacturer’s instructions. At 72 h after incubation, T-cells were stained using CD4-FITC (eBioscience) and CFSE dilution was assessed using flow cytometry. All samples were analyzed on an LSRII (BD Biosciences), running FACSDiVa software. n=4 mice per genotype were used for this experiment.
Primary DC isolation and analysis, DC-T-cell co-incubation experiments
CD11c+ and CD11b+ cells were isolated from LN and spleen homogenates using CD11b and CD11c coated magnetic beads (Miltenyi Biotec, CA) according to the manufacturer’s instructions. Cells were cultured in 96 well plates in DMEM supplemented with 10% FBS and 1% pen-strep, and incubated with or without 20 ng/ml GM-CSF for 24 h (CD11c+) or with and without 50 µg/ml rHDL (CSL-111, a kind gift from Dr. Samuel Wright, CSL Australia) (CD11b+) for 24 h. Levels of IL-1β, IL-18, IL-23, IL-12p70, and IL-6 were measured in the media using ELISA (R&D systems), and corrected for cell protein. For Western blot to assess caspase-1 cleavage in CD11b+ cells, a primary rat anti-mouse caspase-1 antibody (eBioscience) and a rat secondary antibody (Cell-Signaling) were used. For co-incubation experiments, CD11c+ DCs were incubated with T-cells isolated from 8 week old wild-type mice using CD3ε+ beads (Miltenyi Biotec) in a ratio 1:5 for 5 days. Th17 and GM-CSF+ T-cells were assessed as described above in ‘flow cytometry’. For each experiment, one replicate of CD11c+ or CD11b+ DCs represented one mouse. n=4 mice per genotype were used for each experiment.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as means ± SEM. In each experiment, n defines the number of mice included, except for the filipin staining experiment, where n represents the number of fields. The statistical parameters (n, mean, SEM) can be found within the figure legends. The t-test was used to define differences between 2 datasets. To define differences between 3 or 4 datasets, One-way Analysis of Variance (ANOVA) was used with a Bonferroni multiple comparison post-test. The criterion for significance was set at P<0.05. No statistical method was used to determine whether the data met assumptions of the statistical approach. Statistical analyses were performed using GraphPad Prism version 5.01 (San Diego, CA).
Supplementary Material
Highlights.
Mice with deficiency of Abca1/g1 in DCs develop autoimmune glomerulonephritis.
Cholesterol-enriched DCs show inflammasome activation and enhanced cytokine secretion.
A GM-CSF driven crosstalk between Th17 cells and DCs promotes DC proliferation.
Acknowledgments
This work has been supported by NIH grants HL107653 (to A.R.T.), T32HL007343 (to M.M.M.), Netherlands Organization of Scientific Research VIDI-grant 917.15.350 (to M.W.), a UMCG Rosalind Franklin Fellowship (to M.W.), and American Heart Association SDG2160053 grant (to L.Y-C.). Flow cytometry experiments were performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH under awards S10RR027050. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. A. R. T. reports being a consultant to Amgen and CSL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
M.W., E.L.G., A.G., M.M.M., W.W., N.W., V.D.D., and L.Y-C. designed and performed experiments. G.J.R. was involved in the experimental design of the project. M.W. and A.R.T. wrote the manuscript. All authors reviewed the manuscript. A.R.T. supervised, designed, and reviewed the entire project.
The other authors report no conflicts of interest.
References
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J Immunol. 2010;184:173–183. doi: 10.4049/jimmunol.0902372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, Linton MF, Raggi P, Stein CM. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–2415. doi: 10.1056/NEJMoa035611. [DOI] [PubMed] [Google Scholar]
- Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, Cook HT, Kemper C. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122:3473–3481. doi: 10.1182/blood-2013-05-502229. [DOI] [PubMed] [Google Scholar]
- Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, Tontonoz P. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12:180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- Campbell IK, van Nieuwenhuijze A, Segura E, O’Donnell K, Coghill E, Hommel M, Gerondakis S, Villadangos JA, Wicks IP. Differentiation of inflammatory dendritic cells is mediated by NF-kappaB1-dependent GM-CSF production in CD4 T cells. J Immunol. 2011;186:5468–5477. doi: 10.4049/jimmunol.1002923. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Coquery CM, Loo WM, Wade NS, Bederman AG, Tung KS, Lewis JE, Hess H, Erickson LD. BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthr Rheumatol. 2015;67:773–784. doi: 10.1002/art.38950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- Cuchel M, Lund-Katz S, de la Llera-Moya M, Millar JS, Chang D, Fuki I, Rothblat GH, Phillips MC, Rader DJ. Pathways by which reconstituted high-density lipoprotein mobilizes free cholesterol from whole body and from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:526–532. doi: 10.1161/ATVBAHA.109.196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Borriello F, Hodes RJ, Reiser H, Hathcock KS, Laszlo G, McKnight AJ, Kim J, Du L, Lombard DB, et al. Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science. 1993;262:907–909. doi: 10.1126/science.7694362. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 2016;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Ito A, Hong C, Oka K, Salazar JV, Diehl C, Witztum JL, Diaz M, Castrillo A, Bensinger SJ, Chan L, Tontonoz P. Cholesterol Accumulation in CD11c+ Immune Cells Is a Causal and Targetable Factor in Autoimmune Disease. Immunity. 2016;45:1311–1326. doi: 10.1016/j.immuni.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217–1226. doi: 10.4049/jimmunol.1202388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. 2014;26:475–481. doi: 10.1097/BOR.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, Kaplan MJ. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthr Rheum. 2014;66:152–162. doi: 10.1002/art.38225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell metabolism. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Khattri S, Zandman-Goddard G. Statins and autoimmunity. Immunol Res. 2013;56:348–357. doi: 10.1007/s12026-013-8409-8. [DOI] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Zou YR, Goldstein J, Reizis B, Diamond B. Tolerogenic function of Blimp-1 in dendritic cells. J Exp Med. 2011;208:2193–2199. doi: 10.1084/jem.20110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K, Yamagata T, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M, Kanamaru A. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J Immunol. 2004;173:5312–5318. doi: 10.4049/jimmunol.173.8.5312. [DOI] [PubMed] [Google Scholar]
- Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Bradstreet TR, Schwarzkopf EA, Jarjour NN, Chou C, Archambault AS, Sim J, Zinselmeyer BH, Carrero JA, Wu GF, et al. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J Exp Med. 2016;213:251–271. doi: 10.1084/jem.20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Biologics in Rheumatoid Arthritis, G., Genomics Study, S. Eyre S, Churchman SM, Wilson AG, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2014;73:1202–1210. doi: 10.1136/annrheumdis-2013-203276. [DOI] [PubMed] [Google Scholar]
- N-Gonzalez A, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narain S, Furie R. Update on clinical trials in systemic lupus erythematosus. Curr Opin Rheumatol. 2016;28:477–487. doi: 10.1097/BOR.0000000000000311. [DOI] [PubMed] [Google Scholar]
- Norata GD, Pirillo A, Ammirati E, Catapano AL. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis. 2012;220:11–21. doi: 10.1016/j.atherosclerosis.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Packard RR, Maganto-Garcia E, Gotsman I, Tabas I, Libby P, Lichtman AH. CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103:965–973. doi: 10.1161/CIRCRESAHA.108.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panousis CG, Zuckerman SH. Interferon-gamma induces downregulation of Tangier disease gene (ATP-binding-cassette transporter 1) in macrophage-derived foam cells. Arterioscler Thromb Vasc Biol. 2000;20:1565–1571. doi: 10.1161/01.atv.20.6.1565. [DOI] [PubMed] [Google Scholar]
- Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthr Rheum. 2012;64:2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, Mayadas TN, Illei GG, Siebenlist U. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity. 2012;37:1104–1115. doi: 10.1016/j.immuni.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, Crow MK, Schwartz JE, Paget SA, Devereux RB, Salmon JE. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–2406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
- Ronda N, Favari E, Borghi MO, Ingegnoli F, Gerosa M, Chighizola C, Zimetti F, Adorni MP, Bernini F, Meroni PL. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 2014;73:609–615. doi: 10.1136/annrheumdis-2012-202914. [DOI] [PubMed] [Google Scholar]
- Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–631. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, Lazova R, Kang I. Self double-stranded (ds)DNA induces IL-1beta production from human monocytes by activating NLRP3 inflammasome in the presence of anti-dsDNA antibodies. J Immunol. 2013;190:1407–1415. doi: 10.4049/jimmunol.1201195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev Rheumatol. 2015;11:390–400. doi: 10.1038/nrrheum.2015.40. [DOI] [PubMed] [Google Scholar]
- Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205:2281–2294. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tough DF, Sun S, Zhang X, Sprent J. Stimulation of naive and memory T cells by cytokines. Immunol Revs. 1999;170:39–47. doi: 10.1111/j.1600-065x.1999.tb01327.x. [DOI] [PubMed] [Google Scholar]
- Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthr Rheum. 2008;58:251–262. doi: 10.1002/art.23186. [DOI] [PubMed] [Google Scholar]
- Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu H, Huang Z, Lin H, Lei Z, Chen X, Tang M, Gao F, Dong M, Li R, Lin L. Apolipoprotein E-knockout mice on high-fat diet show autoimmune injury on kidney and aorta. Biochem Biophys Res Commun. 2014;450:788–793. doi: 10.1016/j.bbrc.2014.06.060. [DOI] [PubMed] [Google Scholar]
- Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol. 2004;15:241–250. doi: 10.1097/01.asn.0000108969.21691.5d. [DOI] [PubMed] [Google Scholar]
- Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan-Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29:843–849. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via tolllike receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Wang H, Dai C, Wang H, Zhang H, Huang Y, Wang S, Gaskin F, Yang N, Fu SM. P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway. Arthr Rheum. 2013;65:3176–3185. doi: 10.1002/art.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.