

Abstract
Lymph node metastasis (LNM) of esophageal squamous cell carcinoma (ESCC) is well-known to be an early event associated with poor prognosis in patients with ESCC. Recently, tumor-specific aberrant DNA methylation of CpG islands around the promoter regions of tumor-related genes has been investigated as a possible biomarker for use in early diagnosis and prediction of prognosis. However, there are few DNA methylation markers able to predict the presence of LNM in ESCC. To identify DNA methylation markers associated with LNM of ESCC, we performed a genome-wide screening of DNA methylation status in a discovery cohort of 67 primary ESCC tissues and their paired normal esophageal tissues using the Illumina Infinium HumanMethylation450 BeadChip. In this screening, we focused on differentially methylated regions (DMRs) that were associated with LNM of ESCC, as prime candidates for DNA methylation markers. We extracted three genes, HOXB2, SLC15A3, and SEPT9, as candidates predicting LNM of ESCC, using pyrosequencing and several statistical analyses in the discovery cohort. We confirmed that HOXB2 and SEPT9 were highly methylated in LNM-positive tumors in 59 ESCC validation samples. These results suggested that HOXB2 and SEPT9 may be useful epigenetic biomarkers for the prediction of the presence of LNM in ESCC.
Keywords: esophageal squamous cell carcinoma, DNA methylation, predictive biomarker, lymph node metastasis, pyrosequencing analysis
INTRODUCTION
Esophageal squamous cell carcinoma (ESCC) is one of the most lethal cancers in spite of recent advancements in its diagnosis and therapeutics [1, 2]. One of the reasons for its poor prognosis is due to the development of lymph node metastasis (LNM) in the early phases [3]. Thus, early diagnosis and treatment are essential for ESCC patients. Predicting the presence of LNM before treatment is important for deciding appropriate therapy. Although endoscopic submucosal resection (ESD) is useful for minimally invasive treatment of ESCC, adaptation of the method is very limited. Indication of ESD requires early stage ESCC, in which the rate of LNM is low. If ESD is not indicated, esophagectomy with lymph node dissection is chosen as the next alternative for radical treatment. However, most patients are diagnosed at late-stage, and are often accompanied with extensive lymph node metastases. The degree of lymph node metastasis has been regarded as the most important prognostic factor in late stage disease [4, 5].
The presence and degree of lymph node metastasis have important clinical implications for the selection of proper treatment in patients with esophageal cancer. However, current imaging modalities, such as multi-detector-row computed tomography (MDCT), endoscopic ultrasonography (EUS) and positron emission tomography (PET), have limited diagnostic capabilities for precise detection of regional lymph node metastasis. The diagnostic sensitivity of MDCT for LNM, which is frequently used for ESCC staging, is said to be approximately 68%. If the presence of LNM can be predicted more precisely during the diagnostic phase prior to treatment, unnecessary invasive treatment can be avoided. However, despite numerous studies, no clinically useful LNM markers for ESCC have emerged. Thus, a powerful and less invasive LNM marker with high predictive power is required [6–8].
DNA methylation is one of the fundamental epigenetic processes. An epigenetic change is defined as a stable alteration in gene expression with no underlying modifications in the genetic sequence. Several epigenetic mechanisms regulate gene expression, including DNA methylation, histone modification, and non-coding RNA [9]. DNA methylation is one of the most widely and intensively studied processes in cancer epigenetics [10–12]. DNA hypermethylation involves the binding of methyl groups to CpG dinucleotides in the promotor region of a gene, controlling its expression. Aberrant DNA methylation of CpG islands in the promotor region of tumor suppressor genes has been widely reported in several types of cancer [13, 14]. Similarly, there have also been many studies on aberrant DNA methylation in ESCC [15]. Furthermore, some differentially methylated regions (DMR) associated with LNM in ESCC have previously been reported, such as UCHL1 [16, 17] and RARβ [18, 19].
These findings prompted us to investigate LNM-associated DMR, which may be useful for the selection of appropriate treatment in ESCC patients [20]. We performed genome-wide screening of DMR associated with LNM in ESCC patients, and extracted 10 candidate genes using methylation array data of 67 ESCC samples in a discovery cohort. Subsequently, Homeobox B2 (HOXB2), Solute carrier family 15 member 3 (SLC15A3), and Septin 9 (SEPT9) were narrowed down as final candidate genes for predicting LNM after several statistical analyses of the discovery cohort. The three candidate genes were validated as LNM predictive markers using 59 ESCC samples from another cohort. Finally, we identified HOXB2 and SEPT9 as LNM predictive markers by observing their methylation status. Thus, evaluation of HOXB2 and SEPT9 methylation status may facilitate earlier diagnosis of LNM in patients with ESCC.
RESULTS
Genome-wide screening of differentially methylated regions associated with lymph node metastasis in ESCC
To identify LNM-associated epigenetic biomarkers, we utilized the Illumina Infinium Human Methylation450 BeadChip array (Figure 1). Methylation information of the genome was obtained for a total of 485,577 CpG sites in 67 tumor and non-tumor paired ESCC frozen samples (Supplementary Table 1). The data were qualified by Genome studio software, and the output was saved as tab-separated files. The degree of cytosine methylation was scored by the beta value, which is the intensity ratio of methylated and unmethylated probes for each CpG site, ranging from 0 (unmethylated) to 1 (methylated). Sixty-seven samples were classified by N stage, and methylation profiles of representative genes are shown in Supplementary Figure 1A. Differences between beta values of tumor and normal tissue pairs, defined as the delta beta value and ranging from -1 to 1, were investigated to identify hyper- and hypo-methylation induced by carcinogenesis (Supplementary Figure 1B). Probes showing significant differences in delta beta values between N0 and N3 patients were identified as possible candidate predictors of LNM. Two approaches were utilized for extraction of candidate probes: identification of (i) single probes showing methylation status differences and (ii) probe clusters (groups of probes located within 1,000 bp of each other) showing methylation status differences. In the former approach, Student's t-test was performed and average delta beta values of all single probes were compared between the N0 and N3 groups, identifying 3,803 significantly methylated probes in the N3 group compared with the N0 group (p < 0.01). We have confirmed the normality of the data by Shapiro-Wilk test before Student's t-test. Based on p-value, six genes were selected as candidates: NEFL, SLC15A3, OBSL1, PLEC1, SEPT9, and HOXD9. The methylation profile heat map of each candidate gene is shown in Supplementary Figure 1A and 1B. For the latter approach assessing clustered methylation status, Student's t-test was performed in the same manner, and average delta beta values of all clusters were compared between the N0 and N3 groups. Three hundred and twelve probes with significant differences (p < 0.05) in methylation status were extracted and visualized as a heat map. Since we repeated hypothesis testing, some correction like Bonferroni's correction is necessary. However, we will create multivariate model for predicting LNM status and we want to minimize the false negatives. This is the reason why we used this relaxed significant level. Consequently, four genes, HOXB2, PAX6, MIR124-2, and KDM2B, were extracted (Supplementary Figure 1A and 1B). Thus, a total of 10 candidate genes were extracted as possible predictive markers of LNM by methylation array analysis (Supplementary Table 2).
Figure 1. Schematic strategy for the identification of useful epigenetic biomarkers for the prediction of the presence of LNM in primary ESCC cases.
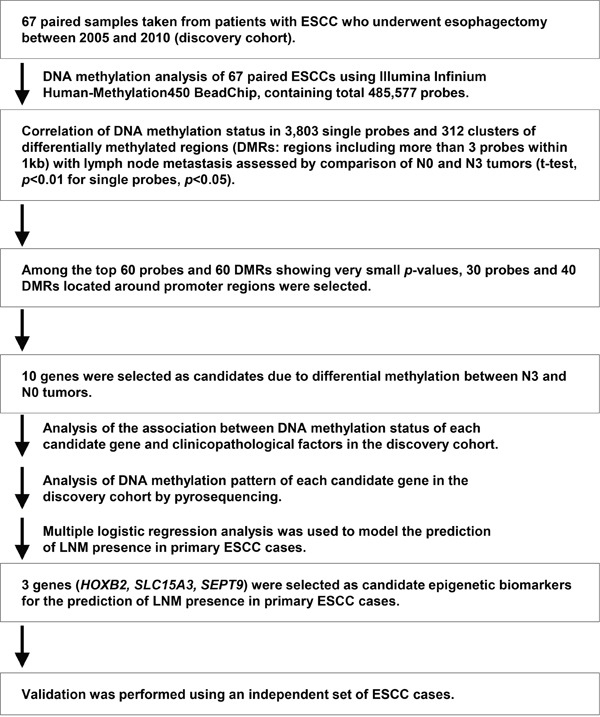
Extraction of candidate genes for prediction of LNM by methylome analysis of the discovery cohort
Pyrosequencing analysis was performed to validate the candidate genes extracted from the methylation array analysis. Pyrosequencing probes were designed using Pyromark Assay Design software to include all candidate probes. As an example, for HOXB2, three unique Illumina probes harboring target CpG sequences within the candidate gene were extracted by methylation array analysis (Supplementary Table 2), and forward and reverse primers were designed to cover all extracted array probes for PCR (Supplementary Table 3). Pyrosequencing allows assessment of CpG sequences in PCR products, and the methylation status of all CpG sites in the target regions of each candidate gene could be obtained. In the HOXB2 gene, seven CpG sites were identified within the designed sequence range (Figure 2A). The methylation statuses of all candidate genes were measured in both the tumor and non-tumor samples by pyrosequencing analysis, and compared in each tumor (all N stages) and non-tumor pair to evaluate the usefulness of these candidate genes as diagnostic biomarkers. Figure 2A shows representative results for HOXB2 in an N3 sample (upper), with hypermethylation, and an N0 sample (bottom). Subsequently, we evaluated the correlation among all probes in the extracted candidate genes, and observed that no probe showed correlation with any other probe (Figure 2B). Thus, the probes of each candidate gene may be useful as independent methylation markers.
Figure 2. DNA methylation analysis by pyrosequencing.
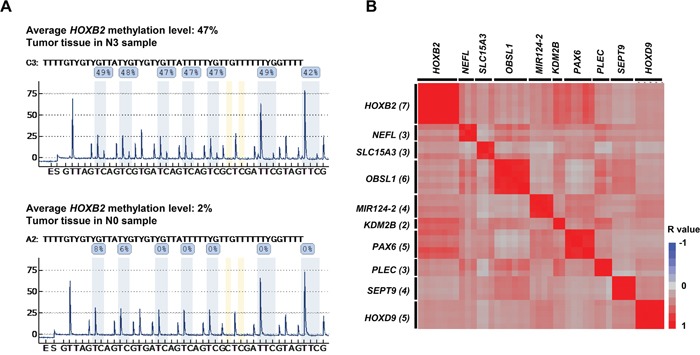
(A) Pyrosequencing was performed to measure the methylation level of candidate genes to validate the Illumina HumanMethylation450 assay results. Candidate CpGs in HOXB2 are shown. Average methylation was higher in N3 tumor tissue samples (upper: 47%) than in N0 tumor tissue samples (lower: 2%). (B) Correlation diagram of pyrosequencing data of each CpG site of the candidate genes. Matrix shows the correlation coefficient (r: -1 [blue] to 1 [red]) among all CpG sites within the sequencing areas of the pyrosequencing analysis. Each candidate gene contained multiple CpG sites. Rows and columns represent each CpG site of each candidate gene. The numbers in parentheses after gene name represent the number of CpG site which were within the sequence analyzed.
Next, differences in methylation status between non-tumor and tumor tissues were investigated in N0 and N3 samples, and data obtained by pyrosequencing were analyzed by paired t-test. 9 of the 10 candidate genes demonstrated significant differences in methylation status between tumor and non-tumor tissues in the N3 samples (Figure 3A), whereas 3 of the 10 genes showed significant differences in the N0 samples (Figure 3B). Thus, these results suggest those genes may be potentially useful as biomarkers of LNM in ESCC. Moreover, in all N stages, 9 of the 10 genes, except SLC15A3, showed significant differences in methylation status between tumor and non-tumor tissues (Supplementary Figure 2). These data suggested that nearly all of the extracted candidate genes may be used as possible predictors of not only LNM, but also the presence of cancer.
Figure 3. Analysis of DNA methylation of each candidate gene in the 67 ESCC patients of the discovery cohort.
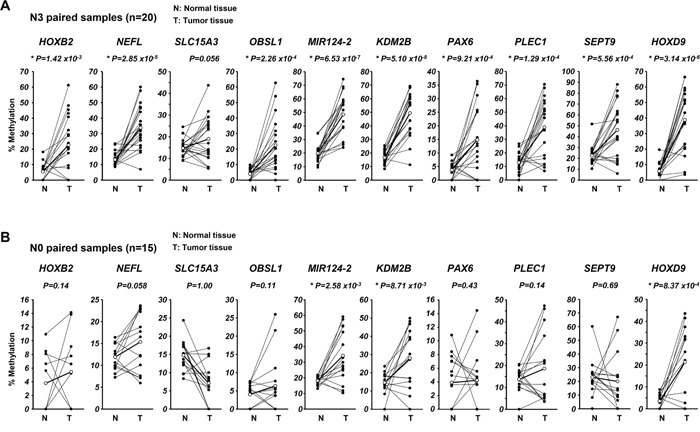
Differences in methylation status of each candidate gene between paired normal and tumor tissues in N3 samples (A) and N0 samples (B). Paired t-test was used for comparison of pyrosequencing results for each candidate.
The associations between clinicopathological characteristics and methylation status of the candidate genes were analyzed by Pearson's chi-square test. In 9 of the 10 genes, methylation status showed strong correlation with LNM status (Table 1). To evaluate whether methylation status of each candidate gene could predict the presence of LNM, univariate classification analysis was performed, and 8 of the 10 candidate genes showed to have certain level of predictive power. Moreover, ROC analysis and AUC showed moderate accuracy in 7 of the 10 candidate genes (Supplementary Table 4). To identify more powerful candidates for LNM prediction, multivariate analysis was performed using logistic regression and stepwise selection (Table 2). We employed AIC (Akaike's Information Criterion) on the selection in JMP9 software. Conclusively, three candidate genes, HOXB2, SLC15A3 and SEPT9, were extracted.
Table 1. Correlation between clinicopathological characteristics and methylation status of selected genes in primary ESCC cases.
| N | HOXB2 | NEFL | SLC15A3 | OBSL1 | miR-124-2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cutoff =16.3 | cutoff =21.3 | cutoff =13 | cutoff =12.6 | cutoff =39 | |||||||||||||
| Low | High | p-value | Low | High | p-value | Low | High | p-value | Low | High | p-value | Low | High | p-value | |||
| Total number | 67 | 42 | 24 | * total 66 | 43 | 24 | 32 | 35 | 33 | 34 | 25 | 42 | |||||
| Age | |||||||||||||||||
| Average 65.6 | <65 | 28 | 18 | 10 | 1.00 | 17 | 11 | 0.80 | 13 | 15 | 1.00 | 15 | 13 | 0.62 | 10 | 18 | 1.00 |
| (46-83) | ≧65 | 39 | 24 | 14 | 26 | 13 | 19 | 20 | 18 | 21 | 15 | 24 | |||||
| Gender | |||||||||||||||||
| Male | 62 | 38 | 23 | 0.65 | 39 | 23 | 0.65 | 28 | 34 | 0.18 | 31 | 31 | 1.00 | 23 | 39 | 1.00 | |
| Female | 5 | 4 | 1 | 4 | 1 | 4 | 1 | 2 | 3 | 2 | 3 | ||||||
| Histological grading | |||||||||||||||||
| GoodModerate | 53 | 32 | 20 | 0.56 | 35 | 18 | 0.55 | 25 | 28 | 1.00 | 26 | 27 | 1.00 | 18 | 35 | 0.35 | |
| Poor | 14 | 10 | 4 | 8 | 6 | 7 | 7 | 7 | 7 | 7 | 7 | ||||||
| TNM classification | |||||||||||||||||
| pT category | |||||||||||||||||
| T1+2 | 7 | 3 | 4 | 0.25 | 4 | 3 | 0.69 | 4 | 3 | 0.70 | 5 | 2 | 0.26 | 0 | 7 | 0.0401 | |
| T3+4 | 60 | 39 | 20 | 39 | 21 | 28 | 32 | 28 | 32 | 25 | 35 | ||||||
| pN category | |||||||||||||||||
| N- | 15 | 14 | 0 | 0.0011 | 14 | 1 | 0.0069 | 12 | 3 | 0.0073 | 12 | 3 | 0.0087 | 9 | 6 | 0.067 | |
| N+ | 52 | 28 | 24 | 29 | 23 | 20 | 32 | 21 | 31 | 16 | 36 | ||||||
| pM category | |||||||||||||||||
| M0 | 50 | 32 | 17 | 0.77 | 34 | 16 | 0.38 | 23 | 27 | 0.78 | 28 | 22 | 0.09 | 22 | 28 | 0.08 | |
| M1 | 17 | 10 | 7 | 9 | 8 | 9 | 8 | 5 | 12 | 3 | 14 | ||||||
| pStage | |||||||||||||||||
| I+II | 15 | 12 | 2 | 0.07 | 14 | 1 | 0.0069 | 11 | 4 | 0.0388 | 12 | 3 | 0.0087 | 7 | 8 | 0.55 | |
| III+IV | 52 | 30 | 22 | 29 | 23 | 21 | 31 | 21 | 31 | 18 | 34 | ||||||
Table 2. Logistic regression analysis and stepwise selection of candidate genes.
| Parameter | Intercept | df | Wald | Significance probability | |
|---|---|---|---|---|---|
| Intercept | -1.506 | 1.000 | 0.000 | 1.000 | |
| * | HOXB2 | 0.097 | 1.000 | 2.548 | 0.110 |
| NEFL | 0.000 | 1.000 | 1.754 | 0.185 | |
| * | SLC15A3 | 0.085 | 1.000 | 2.450 | 0.118 |
| OBSL1 | 0.000 | 1.000 | 0.189 | 0.664 | |
| MIR124-2 | 0.000 | 1.000 | 0.075 | 0.785 | |
| KDM2B | 0.000 | 1.000 | 0.107 | 0.744 | |
| PAX6 | 0.000 | 1.000 | 0.085 | 0.771 | |
| PLEC | 0.000 | 1.000 | 0.021 | 0.883 | |
| * | SEPT9 | 0.028 | 1.000 | 1.497 | 0.221 |
| HOXD9 | 0.000 | 1.000 | 0.765 | 0.382 |
*selected candidate gene by logistic regression analysis (stepwise method) df: degree of freedom.
| n | KDM2B | PAX6 | PLEC | SEPT9 | HOXD9 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cutoff =31.8 | cutoff =16.4 | cutoff =4.8 | cutoff =29.8 | cutoff =41.4 | |||||||||||||
| Low | High | p-value | Low | High | p-value | Low | High | p-value | Low | High | p-value | Low | High | p-value | |||
| Total number | 67 | 21 | 46 | 40 | 27 | 10 | 57 | 31 | 36 | 47 | 20 | ||||||
| Age | |||||||||||||||||
| Average 65.6 | <65 | 28 | 10 | 18 | 0.57 | 16 | 12 | 0.80 | 5 | 23 | 0.73 | 13 | 15 | 1.00 | 23 | 5 | 0.10 |
| (46-83) | ≧65 | 39 | 11 | 28 | 24 | 15 | 5 | 34 | 18 | 21 | 24 | 15 | |||||
| Gender | |||||||||||||||||
| Male | 62 | 19 | 43 | 0.65 | 38 | 24 | 0.39 | 9 | 53 | 0.57 | 29 | 33 | 1.00 | 43 | 19 | 1.00 | |
| Female | 5 | 2 | 3 | 2 | 3 | 1 | 4 | 2 | 3 | 4 | 1 | ||||||
| Histological grading | |||||||||||||||||
| GoodModerate | 53 | 17 | 36 | 1.00 | 32 | 21 | 1.00 | 9 | 44 | 0.67 | 24 | 29 | 0.77 | 37 | 16 | 1.00 | |
| Poor | 14 | 4 | 10 | 8 | 6 | 1 | 13 | 7 | 7 | 10 | 4 | ||||||
| TNM classification | |||||||||||||||||
| pT category | |||||||||||||||||
| T1+2 | 7 | 3 | 4 | 0.67 | 4 | 3 | 1.00 | 1 | 6 | 1.00 | 3 | 4 | 1.00 | 4 | 3 | 0.42 | |
| T3+4 | 60 | 18 | 42 | 36 | 24 | 9 | 51 | 28 | 32 | 43 | 17 | ||||||
| pN category | |||||||||||||||||
| N- | 15 | 9 | 6 | 0.0110 | 13 | 2 | 0.0181 | 6 | 9 | 0.0061 | 12 | 3 | 0.0037 | 15 | 0 | 0.0031 | |
| N+ | 52 | 12 | 40 | 27 | 25 | 4 | 48 | 19 | 33 | 32 | 20 | ||||||
| pM category | |||||||||||||||||
| M0 | 50 | 17 | 33 | 0.55 | 32 | 18 | 0.26 | 9 | 41 | 0.43 | 27 | 23 | 0.0475 | 36 | 14 | 0.56 | |
| M1 | 17 | 4 | 13 | 8 | 9 | 1 | 16 | 11 | 6 | 11 | 6 | ||||||
| pStage | |||||||||||||||||
| I+II | 15 | 8 | 7 | 0.06 | 12 | 3 | 0.08 | 7 | 8 | 0.0006 | 10 | 5 | 0.09 | 14 | 1 | 0.0281 | |
| III+IV | 52 | 13 | 39 | 28 | 24 | 3 | 49 | 21 | 31 | 33 | 19 | ||||||
p-values are from χ2 or Fisher's exact test as appropriate, and were statistically significant when < 0.05 (two-sided).
Methylation status of HOXB2 and SEPT9 may be useful as predictive biomarkers for the presence of LNM in ESCC
To determine whether HOXB2, SLC15A3, and SEPT9 can predict the presence of LNM in another cohort, pyrosequencing analysis of these genes was performed in an independent set of ESCC cases comprising 59 ESCC samples (hereafter called the validation cohort). The clinicopathological characteristics of the validation cohort are shown in Supplementary Table 5. The methylation statuses of HOXB2 and SEPT9 showed significant differences between LNM-negative and -positive samples (Figure 4), whereas SLC15A3 did not. Thus, HOXB2 and SEPT9 were identified as possible DNA methylation predictive biomarkers of LNM in ESCC.
Figure 4. Analysis of DNA methylation of HOXB2, SLC15A3, and SEPT9 in the 59 ESCC patients of the independent set of ESCC cases.
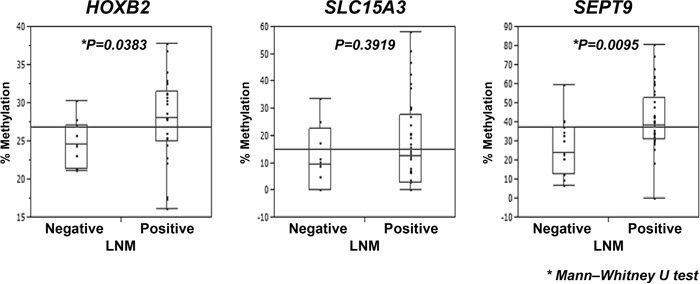
Validation of the methylation status of three candidate genes, HOXB2, SLC15A3, and SEPT9, by pyrosequencing in the independent set of ESCC cases. The methylation status in the LNM-negative and -positive groups were analyzed using the Mann–Whitney U-test. The horizontal lines represent the means of the whole samples for each genes.
DISCUSSION
We performed a genome-wide screening of DNA methylation status in a discovery cohort of 67 primary ESCC tissues and their paired normal esophageal tissues. We extracted three genes, HOXB2, SLC15A3, and SEPT9, as more powerful candidates predicting LNM in ESCC, using several statistical analyses and pyrosequencing analysis in a discovery cohort. Finally, we confirmed that HOXB2 and SEPT9 were highly methylated in LNM-positive tumors in 59 ESCC validation samples. Generally, endoscopic resection, which is a less invasive treatment, is indicated for treatment of early stage ESCC. This indication is based on evidence of reduced LNM possibility within the mucosa of tumor depth (T1a in TNM 7th), as well as the belief that cervical lymph node resection can be omitted in cases of lower esophagus ESCC without obvious LNM. Therefore, accurate predictive biomarkers for LNM are important for the indication of less invasive therapy for ESCC patients. Thus, the present results may be useful for the prediction of LNM in ESCC.
DNA methylation is one of the most heavily studied phenomena of epigenetics, and current research focuses on its usefulness as a clinical diagnostic or prognostic marker, as well as a possible therapeutic target [21]. Our methylation-array analysis identified 10 genes (HOXB2, NEFL, SLC15A3, OBSL1, MIR124-2, KDM2B, PAX6, PLEC, SEPT9, HOXD9) as candidates for predictive biomarkers of LNM in a discovery cohort of 67 ESCC cases. In previous studies, 8 of the 10 candidate genes, HOXB2 [22], NEFL [23], SLC15A3 [24], MIR124-2 [25], KDM2B [26], PAX6 [27], SEPT9 [28, 29] and HOXD9 [30], have been reported to be associated with DNA methylation. To extract candidate genes suitable for clinical use, we performed pyrosequencing analysis, a common method for examining methylation status, and performed univariate and multivariate analyses. Comparing LNM-positive and -negative groups, 9 of the 10 candidate genes showed significant hypermethylation in LNM-positive cases (Table 1). In addition, 3 of the 10 candidate genes, HOXB2, SLC15A3, and SEPT9, were identified as possible predictive biomarkers by multivariate logistic regression analysis with stepwise variable selection (Table 2, Supplementary Table 6). Subsequently, by evaluation of an independent set of ESCC cases (the validation cohort), we concluded that the methylation status of HOXB2 and SEPT9 may be able to predict the presence of LNM. Although, we have calculated cut-off value according to their clinical information in each candidate gene (Table 1), we consider them inappropriate to adapt to the analysis for the validation cohort. Because we obtained samples from frozen resected specimen in the discovery cohort and from paraffin embedded specimen in the validation cohort. Since the quality of extracted DNA was different between the discovery cohort and the validation cohort, the cut-off values lead from discovery cohort could not apply to the validation cohort. Finally, we have compared the methylation status of candidates in the validation cohort. We also consider it will be necessary to evaluate the prediction power of combination of these three genes using another independent set of ESCC cases.
We next analyzed methylation status of these candidate genes in the TCGA database. There were 93 ESCC samples without chemotherapy in the database and patients of those included multiracial populations. The result of the statistical analyses showed no significance among three candidates in the dataset (Supplementary Figure 3). We concluded thtat further analyses for the dataset with same race and admirable sample conditions are required.
HOXB2 encodes a nuclear protein with a homeobox DNA-binding domain [31]. The relationship between HOXB2 expression and cancer progression has been reported in several studies, and HOXB2 has been shown to have bifunctional roles, as an oncogene and as a tumor suppressor gene. Overexpression of HOXB2 was shown to be associated with cancer progression in cervical cancer, pancreatic cancer, and lung adenocarcinoma [32–34]. On the other hand, suppression of HOXB2 increased tumor growth in mice xenograft models of breast cancer cell lines, and overexpression of HOXB2 induced apoptotic cell death in vitro in acute myeloid leukemia (AML) [35]. In addition, it was reported that expression of HOXB cluster genes was repressed by DNA methylation in oral cancer cell lines [22]. At present, there have been no reports on the association between epigenetic regulation of HOXB2 and LNM in ESCC. In the present study, we examined only methylation status, and not gene expression. Our HM450 methylation array results (Supplementary Figure 1), demonstrated that the methylation status of some probes in tumor tissues were significantly hypermethylated compared with the paired normal tissue, and the intensity of methylation became higher with increasing LNM. Furthermore, pyrosequencing analysis of the validation cohort demonstrated that the methylation status of HOXB2 was significantly higher in ESCC samples with LNM than those without LNM. Thus, DNA methylation status of HOXB2 may be a predictive biomarker for the detection of ESCC.
SEPT9, a member of the septin family, regulates cytokinesis and the cell cycle, and has multiple splicing variants. It has been reported that SEPT9 is abnormally expressed in ESCC, ovarian, breast, prostate, and colorectal cancers (CRC) and acts as an oncogene or tumor suppressor gene [28, 29, 36–38]. SEPT9 is known to be frequently methylated in ESCC and CRC [28, 29]. Detection of methylated SEPT9 in blood plasma has been used as an aid in the diagnostic evaluation of CRC [29]. This method can detect presence of CRC at all stages, whereas almost adenomas were unable to be detected. Our data showed that methylation of SEPT9 significantly differed between normal and ESCC tumor tissues of all stages (Supplementary Figure 2), and the presence of LNM in ESCC could be predicted by SEPT9 methylation status with significance (Figure 3 and 4). Thus, in addition to HOXB2, methylation of SEPT9 may be a predictive biomarker in ESCC.
SLC15A3 encodes a protein involved in the transport of glucose and other sugars, bile salts, organic acids, metal ions, and amine compounds [39]. However, there are few studies reporting an association between SLC15A3 and cancer or methylation [24, 40]. In our discovery cohort, methylation status of SLC15A3 significantly differed between LNM-positive and -negative samples. However, in the validation cohort, although the methylation status of SLC15A3 in LNM-positive samples tended to be higher than that of the LNM-negative samples, this difference was not significant. This may be partially due to sample problems or a batch effect, because the DNA in the validation cohort was extracted from paraffin-embedded samples, whereas the DNA of the discovery cohort was extracted from frozen tissue samples.
In conclusion, the present results suggest that DNA methylation of HOXB2 and SEPT9 may be useful as predictive biomarkers of LNM in ESCC. The presence of LNM in ESCC is highly associated with poor prognosis, and prediction of LNM presence before treatment is important in clinical practice. Clinically, because the use of LNM predictive methylation biomarkers requires only a small amount of DNA from the tumor tissue of the ESCC patient, the diagnosis of the presence of LNM should be easy and may be applicable for use with liquid biopsies. Use of such biomarkers may also allow detection of micrometastases unable to be detected by CT. In the present study, we investigated the association between methylation status and presence of LNM in ESCC, and observed that the methylation status of HOXB2 and SEPT9 may be useful as diagnostic or prognostic biomarkers in ESCC.
MATERIALS AND METHODS
Primary tumor samples
A total of 67 ESCC frozen primary tumors and paired non-cancerous tissue samples were obtained from patients with ESCC had undergone esophagectomy with lymph node dissection at Tokyo Medical and Dental University between 2005 and 2010 (discovery cohort) (Supplementary Table 1). After approval by the local ethics committee of the Medical Research Institute and Faculty of Medicine, Tokyo Medical and Dental University, formal written consent was obtained from all patients. The average age of the discovery cohort was 65.6 years [range, 46-83 years], and comprised 62 males and 5 females. No patients had undergone prior chemotherapy or radiotherapy. Lymph node metastasis status was N0 in 15 patients, N1 in 15 patients, N2 in 17 patients, and N3 in 20 patients. Additionally, 59 paraffin-embedded ESCC tumor samples from patients who had undergone esophagectomy at Kyoto Prefectural University of Medicine between 2003 and 2013 were included as an independent set of ESCC cases for the validation. After approval by the local ethics committee of Kyoto Prefectural University of Medicine, formal written consent was obtained from all patients. In this cohort, T1 (TNM classification) cases were omitted due to the difficulty of extracting DNA from the paraffin-embedded samples. Specimens were classified by the TNM classification of the Union for International Cancer Control (UICC 7th) [41].
DNA extraction and bisulfite treatment
DNA from frozen samples were extracted by the phenol chloroform method and treated by bisulfite conversion with the EZ DNA Methylation kit (Zymo Research) according to the manufacturer's recommendations. DNA from paraffin-embedded samples were extracted using the DNeasy Blood & Tissue kit (QIAGEN, KJ Venlo, Netherlands) according to the manufacturer's recommendations.
Illumina infinium HumanMethylation450 BeadChip array
For the discovery round, the Human Methylation450 BeadChip (Illumina, San Diego, CA, USA), which covers 96% of the known CpG islands and 485,577 CpG sites, was employed. Approximately 500 ng of bisulfite-converted DNA from the discovery cohort was applied to the array and analyzed. The GenomeStudio software (Illumina) was used for the quality check of the raw data of each probe and for data normalization. We did not remove SNP-associated probes and those corresponding to the sex chromosomes. Extracted data were analyzed by JMP9 (SAS Institute Inc., Cary, NC, USA).
Pyrosequencing for quantitative measurement of DNA methylation level and statistical analysis
The DNA methylation statuses of the 10 candidate genes that were extracted from the discovery cohort previously analyzed by Illumina Human Methylation450 were validated by pyrosequencing analysis. Primers for pyrosequencing assays were designed by PyroMark Assay Design software (version 2.0.1.15 QIAGEN). PCR was performed under standard conditions with biotin-labeled primers using the PyroMark PCR kit and following the manufacturer's instructions. The biotin-labeled PCR products were assessed by electrophoresis and subjected to the PyroMark Vacuum Prep Tool (Biotage, Sweden) and PyroMark Q96 ID pyrosequencer (QIAGEN). The pyrosequencing data were analyzed by PyroMark Q96 software (version 2.5.8) and JMP9. Uni- and multivariate analyses and logistic regression analysis were performed using JMP9. Multiplicity corrections were performed by BH method.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Acknowledgments
The authors thank Ayako Takahashi and Rumi Mori for their technical assistance.
Dedication
This paper is dedicated to the memory of one of coauthors, Professor Ken-ichi Kozaki, who passed away on May 29, 2016.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
FUNDING
This study was supported in part by a Grant-in Aid for Scientific Research <KAKENHI> for Innovative Areas (22134002, 15H05908) (Integrative Systems Understanding of Cancer for Advanced Diagnosis, Therapy and Prevention and Conquering cancer through NEO-dimensional systems understandings), Scientific Research (A) (22240090, 25250019) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct), the Tailor-Made Medical Treatment with the BioBank Japan Project (BBJ) and the Practical Research for Innovative Cancer Control (15Ack0106017h0002) from the Japan Agency for Medical Research and Development (AMED). This study was supported by the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University (TMDU). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Baba Y, Watanabe M, Baba H. Review of the alterations in dna methylation in esophageal squamous cell carcinoma. Surg Today. 2013;43:1355–1364. doi: 10.1007/s00595-012-0451-y. [DOI] [PubMed] [Google Scholar]
- 2.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bollschweiler E, Baldus SE, Schröder W Prenzel K, Gutschow C, Schneider PM, Hölscher AH. High rate of lymph-node metastasis in submucosal esophageal squamous-cell carcinomas and adenocarcinomas. Endoscopy. 2006;38:149–156. doi: 10.1055/s-2006-924993. [DOI] [PubMed] [Google Scholar]
- 4.Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, Pinheiro DG, Torres C, de Carvalho AF, Cury PM, Nunes FD, Nishimoto IN, Soares FA, et al. Epigenetic silencing of crabp2 and mx1 in head and neck tumors. Neoplasia. 2009;11:1329–1339. doi: 10.1593/neo.91110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniel H, Kottra G. The proton oligopeptide cotransporter family slc15 in physiology and pharmacology. Pflugers Arch. 2004;447:610–618. doi: 10.1007/s00424-003-1101-4. [DOI] [PubMed] [Google Scholar]
- 6.Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH, Å Hedman, Sandling JK, Li LA, Irvin MR, Zhi D, Deloukas P, Liang L, et al. Epigenome-wide association study (ewas) of bmi, bmi change and waist circumference in african american adults identifies multiple replicated loci. Hum Mol Genet. 2015;24:4464–4479. doi: 10.1093/hmg/ddv161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 8.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 9.Gilad R, Meir K, Stein I, German L, Pikarsky E, Mabjeesh NJ. High sept9_i1 protein expression is associated with high-grade prostate cancers. PLoS One. 2015;10:e0124251. doi: 10.1371/journal.pone.0124251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gyobu K, Yamashita S, Matsuda Y, Igaki H, Niwa T, Oka D, Kushima R, Osugi H, Lee S, Suehiro S, Ushijima T. Identification and validation of dna methylation markers to predict lymph node metastasis of esophageal squamous cell carcinomas. Ann Surg Oncol. 2011;18:1185–1194. doi: 10.1245/s10434-010-1393-5. [DOI] [PubMed] [Google Scholar]
- 11.Ibragimova I, Ibáñez de Cáceres I, Hoffman AM, Potapova A, Dulaimi E, Al-Saleem T, Hudes GR, Ochs MF, Cairns P. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila) 2010;3:1084–1092. doi: 10.1158/1940-6207.CAPR-10-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inamura K, Togashi Y, Okui M, Ninomiya H, Hiramatsu M, Satoh Y, Okumura S, Nakagawa K, Shimoji T, Noda T, Ishikawa Y. Hoxb2 as a novel prognostic indicator for stage i lung adenocarcinomas. J Thorac Oncol. 2007;2:802–807. doi: 10.1097/JTO.0b013e3181461987. [DOI] [PubMed] [Google Scholar]
- 13.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 14.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 16.Kang S, Kim B, Park SB, Jeong G, Kang HS, Liu R, Kim SJ. Stage-specific methylome screen identifies that nefl is downregulated by promoter hypermethylation in breast cancer. Int J Oncol. 2013;43:1659–1665. doi: 10.3892/ijo.2013.2094. [DOI] [PubMed] [Google Scholar]
- 17.Kuo IY, Chang JM, Jiang SS, Chen CH, Chang IS, Sheu BS, Lu PJ, Chang WL, Lai WW, Wang YC. Prognostic cpg methylation biomarkers identified by methylation array in esophageal squamous cell carcinoma patients. Int J Med Sci. 2014;11:779–787. doi: 10.7150/ijms.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Mori M, Croce CM. Allele loss and promoter hypermethylation of vhl, rar-beta, rassf1a, and fhit tumor suppressor genes on chromosome 3p in esophageal squamous cell carcinoma. Cancer Res. 2003;63:3724–3728. [PubMed] [Google Scholar]
- 19.Ladabaum U, Allen J, Wandell M, Ramsey S. Colorectal cancer screening with blood-based biomarkers: Cost-effectiveness of methylated septin 9 dna versus current strategies. Cancer Epidemiol Biomarkers Prev. 2013;22:1567–1576. doi: 10.1158/1055-9965.EPI-13-0204. [DOI] [PubMed] [Google Scholar]
- 20.Lindblad O, Chougule RA, Moharram SA, Kabir NN, Sun J, Kazi JU, Rönnstrand L. The role of hoxb2 and hoxb3 in acute myeloid leukemia. Biochem Biophys Res Commun. 2015;467:742–747. doi: 10.1016/j.bbrc.2015.10.071. [DOI] [PubMed] [Google Scholar]
- 21.López R, Garrido E, Piña P, Hidalgo A, Lazos M, Ochoa R, Salcedo M. Hoxb homeobox gene expression in cervical carcinoma. Int J Gynecol Cancer. 2006;16:329–335. doi: 10.1111/j.1525-1438.2006.00350.x. [DOI] [PubMed] [Google Scholar]
- 22.Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, Kiyohara E, Wang J, Kawas NP, Donovan NC, Hata K, Wilmott JS, Murali R, et al. Epigenome-wide dna methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox d cluster associated with prognosis. Hum Mol Genet. 2014;23:226–238. doi: 10.1093/hmg/ddt420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montagna C, Lyu MS, Hunter K, Lukes L, Lowther W, Reppert T, Hissong B, Weaver Z, Ried T. The septin 9 (msf) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res. 2003;63:2179–2187. [PubMed] [Google Scholar]
- 24.Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, Ushijima T. The presence of aberrant dna methylation in noncancerous esophageal mucosae in association with smoking history: A target for risk diagnosis and prevention of esophageal cancers. Cancer. 2009;115:3412–3426. doi: 10.1002/cncr.24394. [DOI] [PubMed] [Google Scholar]
- 25.Puli SR, Reddy JB, Bechtold ML, Antillon D, Ibdah JA, Antillon MR. Staging accuracy of esophageal cancer by endoscopic ultrasound: A meta-analysis and systematic review. World J Gastroenterol. 2008;14:1479–1490. doi: 10.3748/wjg.14.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice TW, Blackstone EH, Rusch VW. 7th edition of the ajcc cancer staging manual: Esophagus and esophagogastric junction. Ann Surg Oncol. 2010;17:1721–1724. doi: 10.1245/s10434-010-1024-1. [DOI] [PubMed] [Google Scholar]
- 27.Rizk NP, Ishwaran H, Rice TW, Chen LQ, Schipper PH, Kesler KA, Law S, Lerut TE, Reed CE, Salo JA, Scott WJ, Hofstetter WL, Watson TJ, et al. Optimum lymphadenectomy for esophageal cancer. Ann Surg. 2010;251:46–50. doi: 10.1097/SLA.0b013e3181b2f6ee. [DOI] [PubMed] [Google Scholar]
- 28.Robertson KD. Dna methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 29.Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 30.Scott M, McCluggage WG, Hillan KJ, Hall PA, Russell SE. Altered patterns of transcription of the septin gene, sept9, in ovarian tumorigenesis. Int J Cancer. 2006;118:1325–1329. doi: 10.1002/ijc.21486. [DOI] [PubMed] [Google Scholar]
- 31.Segara D, Biankin AV, Kench JG, Langusch CC, Dawson AC, Skalicky DA, Gotley DC, Coleman MJ, Sutherland RL, Henshall SM. Expression of hoxb2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clin Cancer Res. 2005;11:3587–3596. doi: 10.1158/1078-0432.CCR-04-1813. [DOI] [PubMed] [Google Scholar]
- 32.Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin. 2010;60:376–392. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi H, Arimura Y, Masao H, Okahara S, Tanuma T, Kodaira J, Kagaya H, Shimizu Y, Hokari K, Tsukagoshi H, Shinomura Y, Fujita M. Endoscopic submucosal dissection is superior to conventional endoscopic resection as a curative treatment for early squamous cell carcinoma of the esophagus (with video) Gastrointest Endosc. 2010;72:255–264. 264.e251-–252. doi: 10.1016/j.gie.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 34.Toyota M, Suzuki H, Yamashita T, Hirata K, Imai K, Tokino T, Shinomura Y. Cancer epigenomics: Implications of dna methylation in personalized cancer therapy. Cancer Sci. 2009;100:787–791. doi: 10.1111/j.1349-7006.2009.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vieille-Grosjean I, Huber P. Transcription factor gata-1 regulates human hoxb2 gene expression in erythroid cells. J Biol Chem. 1995;270:4544–4550. doi: 10.1074/jbc.270.9.4544. [DOI] [PubMed] [Google Scholar]
- 36.Wang P, Chen L, Zhang J, Chen H, Fan J, Wang K, Luo J, Chen Z, Meng Z, Liu L. Methylation-mediated silencing of the mir-124 genes facilitates pancreatic cancer progression and metastasis by targeting rac1. Oncogene. 2014;33:514–524. doi: 10.1038/onc.2012.598. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Fang MZ, Liao J, Yang GY, Nie Y, Song Y, So C, Xu X, Wang LD, Yang CS. Hypermethylation-associated inactivation of retinoic acid receptor beta in human esophageal squamous cell carcinoma. Clin Cancer Res. 2003;9:5257–5263. [PubMed] [Google Scholar]
- 38.Xavier FC, Destro MF, Duarte CM, Nunes FD. Epigenetic repression of hoxb cluster in oral cancer cell lines. Arch Oral Biol. 2014;59:783–789. doi: 10.1016/j.archoralbio.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Yoon YC, Lee KS, Shim YM, Kim BT, Kim K, Kim TS. Metastasis to regional lymph nodes in patients with esophageal squamous cell carcinoma. Ct versus fdg pet for presurgical detection prospective study. Radiology. 2003;227:764–770. doi: 10.1148/radiol.2281020423. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Yang X, Wang J, Liang T, Gu Y, Yang D. Down-regulation of pax6 by promoter methylation is associated with poor prognosis in non small cell lung cancer. Int J Clin Exp Pathol. 2015;8:11452–11457. [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao SL, Zhu ST, Hao X, Li P, Zhang ST. Effects of dna methyltransferase 1 inhibition on esophageal squamous cell carcinoma. Dis Esophagus. 2011;24:601–610. doi: 10.1111/j.1442-2050.2011.01199.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.