

Abstract
Aims
Severe postprandial hypoglycemia with neuroglycopenia is an increasingly recognized, debilitating complication of Roux-en-Y gastric bypass (RYGB) surgery. Increased secretion of insulin and incretin hormones are implicated in its pathogenesis. Histopathologic examination of pancreas has demonstrated increased islet size and/or nuclear diameter in post-RYGB patients who underwent pancreatectomy for severe refractory hypoglycemia with neuroglycopenia (RYGB+NG). We aimed to determine whether β-cell proliferation or apoptosis are altered in RYGB+NG.
Methods
We performed an observational study to analyze markers of proliferation, apoptosis, and cell cycle, and transcription factor expression in pancreatic tissue from affected RYGB+NG patients (n=12), normoglycemic patients undergoing pancreatic surgery for benign lesions (controls, n=6, and individuals with hypoglycemia due to insulinoma (n=52).
Results
Proliferative cell nuclear antigen (PCNA) expression was increased in insulin-positive cells in RYGB+NG patients (4.5-fold increase, p<0.001 vs. controls) and correlated with β-cell mass. Ki-67 immunoreactivity was low in both RYGB+NG and controls, but did not differ between groups. Phospho-histone H3 levels did not differ between RYGB+NG and controls. PCNA and Ki-67 were both significantly lower in both controls and RYGB+NG than insulinomas. Markers of apoptosis and cell cycle (M30, p27, and p21) did not differ between groups. PDX1 and menin exhibited similar expression patterns, while FOXO1 appeared to be more cytosolic in RYGB+NG.
Conclusions
Markers of proliferation are heterogeneous in patients with severe post-RYGB hypoglycemia. Increased β-cell proliferation in some individuals may contribute to increased β-cell mass observed in severely affected patients.
Keywords: islet, hypoglycemia, human, gastro-entero pancreatic factors
Introduction
Bariatric surgery is increasingly performed for treatment of both obesity and type 2 diabetes (T2D). Randomized clinical trials comparing bariatric surgery with medical management have demonstrated that surgery achieves not only weight loss, but also improved glycemic control while reducing the need for medications [1–3]. One rare but increasingly recognized complication of Roux-en-Y gastric bypass (RYGB) surgery is severe hypoglycemia with neuroglycopenia [4, 5]. While severe hypoglycemia may occur in fewer than 1% of patients [6–8], mild, often unrecognized, hypoglycemia is more frequent [9, 10] and may contribute to increased appetite and weight regain after surgery [11]. More severely affected patients can develop profound neuroglycopenia, with falls, motor vehicle accidents, loss of consciousness, and seizures refractory to dietary management. Neuroglycopenic symptoms typically appear two or more years after RYGB surgery, well after weight loss has stabilized. Hypoglycemia usually occurs in the postprandial period and is accompanied by inappropriately high plasma insulin [12]. Higher levels of glucagon-like peptide-1 (GLP1) and other incretins may promote excessive insulin secretion [13, 14]. Increased insulin-independent glucose uptake may also contribute to hypoglycemia [15]. Some patients initially improve with medical nutritional therapy but may require progressive pharmacologic strategies, including acarbose to reduce carbohydrate absorption, and somatostatin analogs, diazoxide, and/or calcium channel blockade to reduce insulin secretion [16]. Surgical procedures which increase gastric restriction [17, 18] or tube feeding into the gastric remnant [19] have been successful for some patients. Reversal of gastric bypass may improve hypoglycemia, but is not uniformly successful [4, 20].
Partial pancreatectomy was previously performed in a small number of patients with severe neuroglycopenia refractory to medical management, in an attempt to reduce the frequency and severity of hypoglycemia [4, 5]. While some patients experience initial improvements following partial pancreatectomy, hypoglycemia often recurs [4, 21], suggesting progressive β-cell dysregulation and inappropriate insulin secretion. Pathologic examination of pancreatic tissue from severely affected patients who underwent pancreatectomy reveals large and small islets, often of irregular shape and clustered near ducts, with nuclear atypia, similar to findings seen with nesidioblastosis unrelated to gastric surgery [4, 5, 22]. Insulinoma has also been reported rarely in post-bypass patients [23]; whether this is linked to stimulation of proliferation and clonal expansion or to increased diagnostic attention in post-RYGB patients remains uncertain. β-cell mass is quantitatively increased in some [24] but not all series [25], potentially due to small cohort sizes or heterogeneity among case or control samples.
To determine whether cell proliferation or apoptosis are altered in pancreatic β-cells from patients with post-RYGB hypoglycemia with neuroglycopenia (RYGB+NG), we examined pancreatic samples obtained from patients who had undergone partial pancreatectomy for refractory hypoglycemia. Results were compared with those obtained in pancreatic samples from patients with normoglycemia undergoing pancreatic resection for benign mass lesions or other diseases, and from patients with hypoglycemia due to insulinoma.
Research Design and Methods
Patients
Consecutive patients presenting to either Joslin Diabetes Center (n=8) or University of Texas Health Science Center at San Antonio (n=4) for surgical management of post-RYGB hypoglycemia refractory to medical management were included in this cross-sectional observational study. Diagnosis of post-RYGB+NG was made on the basis of documented venous hypoglycemia with neuroglycopenia, inappropriately high insulin levels at the time of spontaneous hypoglycemia, and normal glucose and appropriately suppressed plasma insulin after an overnight fast or prolonged inpatient fast. Sample size was determined by the availability of specimens from patients with post-RYGB+NG who underwent palliative partial pancreatectomy in an attempt to control severe hypoglycemia; this number is limited, as pancreatic surgery is no longer performed in our institution for this condition due to incomplete efficacy. Control pancreatic tissues were obtained from patients without diabetes who underwent pancreatic surgery for benign pancreatic mass lesions (n=6) at Brigham and Women’s Hospital or Beth Israel Deaconess Medical Center. Patients requiring surgery for malignancy were excluded. Fifty-two sporadic insulinomas in patients with severe hypoglycemia treated at Haukeland University Hospital (n=7) or at Ospedale di Circolo (n=45) were also analyzed.
The study was approved by the Institutional Review Board or Regional Ethics Committee at each institution, and performed according to the Helsinki Declaration.
Surgical collection of samples
Partial pancreatectomy was performed for patients with severe post-RYGB hypoglycemia after extensive hormonal evaluation, noninvasive imaging, and arteriography with selective calcium-stimulated insulin secretion testing to rule out insulinoma and define the anatomic site of dominant insulin secretion. Routine diagnostic evaluation of pancreatic specimens was performed by pathologists at each institution, and detailed gross and microscopic analysis to rule out insulinoma or other neuroendocrine tumors was performed in all patients. Some cases included in our study were previously described in clinical case series on post-RYGB hypoglycemia [4], analysis of GLP1R expression [24], or prior studies of insulinoma [26, 27]. Patient gender and age are provided in Table 1; for one control and two post-RYGB patients, only de-identified information was available. For insulinomas, tumors and adjoining pancreatic parenchyma were resected at laparotomy. Large or infiltrating tumors were resected using standard surgical procedures (Whipple resection, middle or distal pancreatectomy), whereas small (≤ 2cm) well-defined, encapsulated and superficially localized insulinomas were enucleated. Insulinomas were classified according to the 2010 WHO classification of tumors of the digestive system [28].
Table 1.
Characteristics of patients from whom pancreatic samples were obtained at surgery. n/a, data not available.
| Gender | Age (years) | BMI at RYGB | BMI at Pancreatic Surgery | Pathology |
|---|---|---|---|---|
| CONTROLS (no RYGB) | ||||
| F | 28 | – | 18 | Microcystic serous cystadenoma |
| F | 38 | – | 25.1 | Leiomyoma, normal pancreas |
| n/a | n/a | – | n/a | Localized pancreatic tumor |
| M | 69 | – | n/a | Serous cystadenoma |
| M | 65 | – | n/a | Intraductal papillary mucinous adenoma |
| F | 43 | – | n/a | Ampullary adenoma, normal pancreas |
| POST-RYGB NEUROGLYCOPENIC HYPOGLYCEMIA | ||||
| M | 46 | 40.6 | 23.1 | Islet hyperplasia |
| F | 42 | n/a | 30 | Islet hyperplasia |
| F | 35 | 42.3 | 37.5 | Islet hyperplasia |
| F | 37 | n/a | n/a | Islet hyperplasia |
| F | 29 | 46.8 | 26.3 | Islet hyperplasia |
| F | 33 | 40 | 25.8 | Islet hyperplasia |
| F | 49 | 43.5 | 29.7 | Islet hyperplasia |
| F | 69 | n/a | n/a | Islet hyperplasia |
| n/a | n/a | n/a | n/a | Islet hyperplasia |
| F | 54 | n/a | n/a | Islet hyperplasia |
| F | 36 | n/a | n/a | Islet hyperplasia |
| n/a | n/a | n/a | n/a | Islet hyperplasia |
Immunostaining protocols
Pancreas tissues were fixed in 10% buffered formalin at 4°C overnight, and embedded in paraffin. Serial sections (5 μm thick) were used for all immunostainings. After being microwaved for 20 minutes in 10 mM sodium citrate, sections were blocked with donkey serum (5%) and incubated with primary antibodies against: a) PCNA (Dako, mouse monoclonal, clone PC10, 1:400), b) phosphohistone H3 (Millipore, 06570), c) Ki-67 (Dako, mouse monoclonal, clone B56 or MIB1, 1:100 dilution), d) P27 (BD, mouse monoclonal, clone 57, 1:25), e) P21 (Dako, mouse monoclonal, clone SX118, 1:20), anti-caspase cleavage product of cytokeratin 18 (M30 CytoDeath, Hoffmann-LaRoche mouse monoclonal, clone M30, 1:100), f) PDX1 (Cell Signaling, rabbit polyclonal, 1:400), g) FoxO1 (Cell Signaling, rabbit polyclonal, 1:50), h) Menin (Abcam, rabbit polyclonal, 1:1600), and i) insulin (Abcam, Guinea pig polyclonal, 1:200, or Cell Signaling, rabbit polyclonal, 1:400 or BioGenex, mouse monoclonal, clone AE9D6, 1:100). Specific signals were detected using fluorescence-conjugated secondary antibodies (Jackson Immunoresearch, Alexa 488 or Alexa 594, 1:400). P27, P21 and PCNA were detected by polymer-HRP/AP-conjugated secondary antibody (Dako), followed by color development with 0.03% 3,3′ diaminobenzidine tetrahydrochloride (DAB) or permanent red substrate. For quantification of Ki-67 or PCNA positivity, sections were co-stained with PCNA or Ki-67 and insulin, and then counterstained with DAPI.
Slides were examined under AXIO Imager A2 Fluorescence/Bright Field microscope (Zeiss) and images were captured with the same background setting by a single observer masked to group. Cell counting was conducted using Image J software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2009). For each sample, 1000–2000 β-cells (insulin positive cells) were counted; the number also positive for PCNA, pHH3, or Ki-67 was counted and the percentage of insulin-positive cells was calculated [28]. The number of patients analyzed for each antibody is indicated in figure legends. M30 immunostainings were performed as described previously [29].
β-cell area and nuclear area
Immunofluorescent staining for insulin was carried out by using guinea pig anti-insulin primary antibody (Abcam) and Alexa-conjugated secondary antibody. Images were captured using Olympus BX60 fluorescence microscope under both 4× and 20× magnification for the whole pancreas and all of the individual islets. Nuclear area was measured in 500 randomly selected β-cells from each section. Images were analyzed using Image J software. β-cell area was expressed as a fraction of the pancreas area (multiplied by 100).
Statistics
Student’s t-tests (2-tailed, unequal variance) or χ2 tests were used to assess between-group comparisons. Linear regression was used to assess the relationship between proliferative markers and islet mass. Results were considered significant if P<0.05.
Results
Islet morphology and proliferation were evaluated in 12 patients with RYGB+NG and 6 normoglycemic controls without malignancy (Table 1). For comparison, 52 patients with sporadic insulinoma were also analyzed (Supplementary Table 1).
Morphology
Fractional β-cell area was increased in pancreas samples from RYGB+NG patients versus controls (5.13±0.42 vs. 1.74±0.46%, p<0.001)(Figure 1A). Since nuclear area has been suggested to reflect β-cell activity [25], we measured nuclear area in 500 randomly selected β-cells from each section. There was no difference in nuclear diameter between RYGB+NG and controls (Figure 1B and Supplementary Figure 1).
Figure 1.
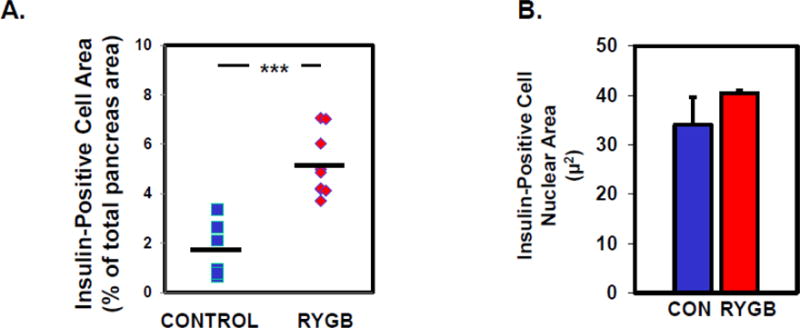
A. Area (% of total pancreas area) and B. nuclear area (μm) in insulin-positive cells in controls (n=6) and patients with post-RYGB hypoglycemia (RYGB+NG, n=9). Nuclear size was assessed in 500 cells per patient.
Proliferation of Insulin-Positive Cells
To determine whether increased β-cell mass in this population is accompanied by increased proliferation, pancreas sections were co-immunostained with insulin and either PCNA and Ki-67. The percentage of cells co-immunostaining for PCNA and insulin was 4.5 fold higher in pancreatic tissue from RYGB+NG patients with neuroglycopenia as compared with control pancreas (36.5±4.1% versus 8.1±1.8%, p<0.0001, Figure 2A). The percentage of PCNA-positive insulin-positive cells correlated with β-cell mass (r=0.60, p=0.02, Supplementary Figure 2). Similarly, the percentage of insulin-positive cells also positive for the mitosis marker phospho-histone H3 (Serine 10) was numerically higher in a subset of 6 patients with RYGB + NG (0.68 ± 0.18) as compared with 5 controls (0.29 ± 0.15, p=0.13, Supplementary Figure 3A/B).
Figure 2.
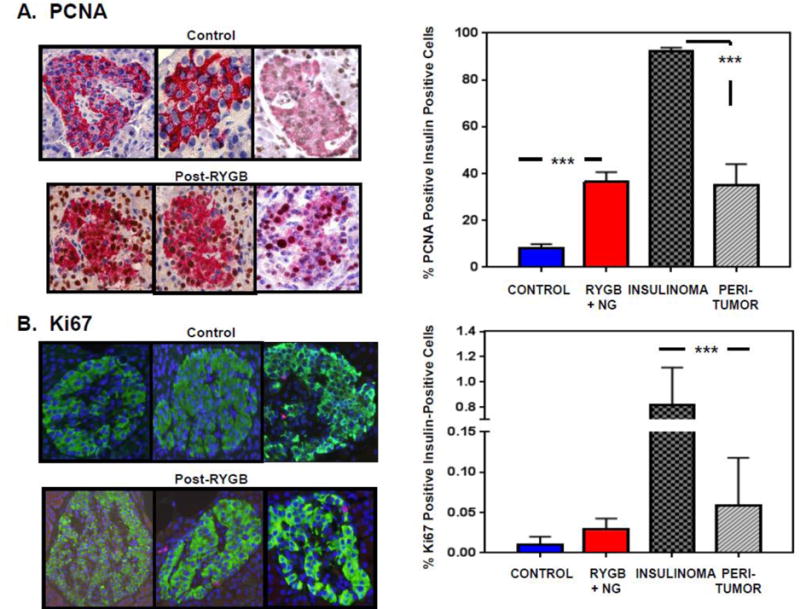
Representative images (left) and quantification (right) for (A) PCNA and (B) Ki-67 staining, expressed as percentage of insulin-positive cells in samples from controls (n=6), patients with RYGB+NG (n=11 for PCNA, 12 for Ki-67). These data are compared with neoplastic cells from patients with insulinoma (n=7), and with surrounding non-neoplastic islets. 1000–2000 β-cells were counted for each patient. *** indicates p<0.0001.
Ki-67-positive β-cells were less abundant (range 0–0.05%, Figure 2B) than PCNA-positive β-cells in all samples. Four of 12 samples from patients with RYGB+NG had detectable Ki-67 immunoreactivity, compared to only one of 6 control samples (p=0.44, Fisher’s exact test).
PCNA and Ki-67 data were compared to values in patients with insulinoma, since both insulinoma and RYGB+NG are characterized by hyperinsulinemia, and increased levels of insulin or signaling through the insulin/IGF-1 receptors could potentially contribute to proliferation in non-neoplastic pancreas [30]. As predicted, tumor samples had a higher percentage of positivity for the proliferation markers PCNA (92.2±1.4%) and Ki-67 (0.8±0.3%) as compared with non-neoplastic islets in the same individuals and with both RYGB+NG and controls (Figure 2A/2B and Supplementary Figure 4). Similarly higher proliferation (Ki-67) was observed in a larger independent series of insulinoma, with a mean Ki-67 index of 2.4±0.4% (n=45). In islets outside of, but adjacent to, the insulinoma (at least 25 μm away from the tumor), the percentage of PCNA-positive insulin-positive cells was 35.0±9.0%, and the percentage of Ki-67-positive insulin-positive cells was 0.06±0.06% (Figure 2A/2B, right column, and Supplementary Figure 4). These values were lower than for the adjacent tumor, as expected, but higher than control samples and quantitatively similar in magnitude to RYGB+NG islets. Percent positivity for PCNA correlated with percent positivity for Ki-67 across the cohort of both insulinoma and post-RYGB patients (r=0.34, p=0.03).
Apoptosis
Reduced apoptosis could also contribute to increased β-cell mass. To investigate this possibility, consecutive pancreatic sections were immunostained with the apoptosis marker M30 antibody. No significant M30 positivity was detectable in either RYGB+NG patients or controls (representative sections, Figure 3A). Similarly, there were no differences in overall immunoreactivity or subcellular distribution for the additional apoptosis markers p27 or p21 (Figure 3B/C and Supplementary Figure 5). In insulinoma samples, there were no detectable differences in p27 immunoreactivity in comparison with surrounding non-neoplastic islets (Supplementary Figure 5A), p21 was 7-fold higher in tumor (p<0.01, Supplementary Figure 5B/C).
Figure 3.
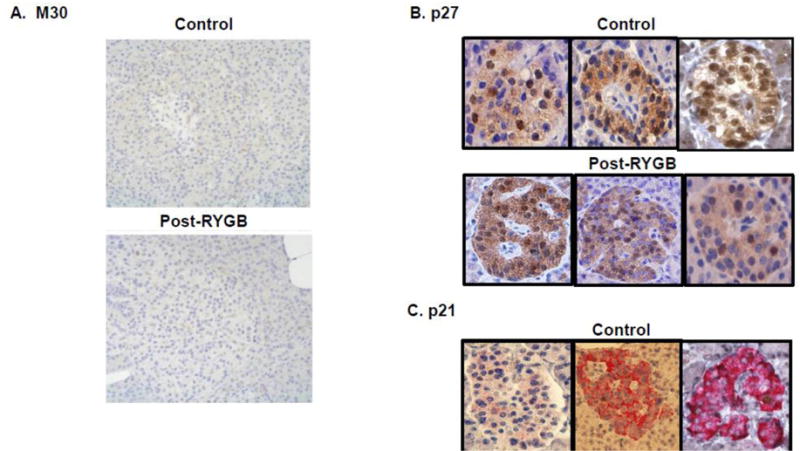
Representative images for the apoptosis markers and related proteins (A) M30, (B) p27 and (C) p21. 1000–2000 β-cells were counted.
Transcription Factors
Pancreatic sections were immunostained for key transcription factors that regulate β-cell development and differentiation, including pancreatic and duodenal homeobox 1 (PDX1), forkhead box O1 (FOXO1), and menin. PDX-1 expression in β-cells from RYGB+NG patients was both nuclear and cytoplasmic in distribution, whereas it was localized in the nucleus of control β-cells (Figure 4, top row). Likewise, FoxO1 was largely excluded from the nucleus in RYGB+NG subjects; by contrast, in control samples the majority of insulin-positive β-cells exhibited nuclear FoxO1 expression with minimal cytoplasmic immunoreactivity (Figure 4, middle row). Menin, implicated in β-cell replication and MEN1-linked tumorigenesis [31], was similarly expressed in the nucleus of all β-cells and non-islet cells in both RYGB+NG and control patients (Figure 4, lower row).
Figure 4.
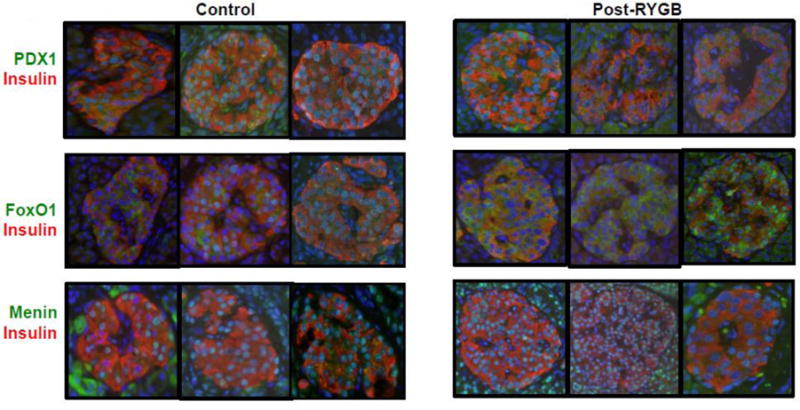
Representative images of immunostaining for the transcription factors PDX1, FOXO1, and menin (green), and insulin (red).
In pancreas sections from patients with insulinoma, the transcription factors PDX1, FOXO1, and menin were detected in both tumor and peri-tumor islets. Both nuclear and cytoplasmic expression of all factors was observed in tumor cells (Supplementary Figure 6A–C, upper panels), while expression was predominantly nuclear in peri-tumor islets (Supplementary Figure 6A–C, lower panels).
Discussion
Hypoglycemia following RYGB performed for severe obesity is a challenging and sometimes serious clinical syndrome. Increased postprandial insulin secretion, together with increased insulin-dependent and insulin-independent glucose disposal, may contribute to postprandial hypoglycemia [13, 14]. Unfortunately, severely affected patients do not fully respond to currently available therapies aimed at reducing insulin and incretin secretion, underscoring the need to better understand the pathophysiology of this syndrome. Interestingly, hypoglycemia can also be observed following sleeve gastrectomy but rarely after gastric banding [32].
Partial pancreatectomy has previously been utilized in severely affected patients with post-bariatric hypoglycemia in an attempt to reduce hypoglycemia and improve patient safety. This approach is no longer routinely employed due to high morbidity and poor long-term efficacy [21]. However, some severely affected patients have achieved reductions in hypoglycemia frequency and severity with partial pancreatectomy, raising the possibility that increased islet mass could contribute to the pathophysiology of this syndrome. Using surgical samples from these patients, we aimed to determine whether β-cell mass, proliferation, and apoptosis are altered, as compared with normal pancreas tissue from surgical resections for benign pancreatic lesions, and compared to pathology with hypoglycemia due to insulinoma.
Histologic analysis of pancreatic samples from this patient cohort revealed significant increases in insulin-positive cells, as compared with surgical controls. Increased islet mass has been detected in some [4, 5, 24] but not all studies [25]. Whether this reflects the heterogeneity of pancreatic pathology and β-cell mass in this syndrome, or the wide variation in β-cell mass even in healthy individuals across a spectrum of obesity [33], remains uncertain. We cannot exclude a role for prior severe obesity as driver of increased β-cell mass in individuals with post-RYGB hypoglycemia.
Increases in β-cell mass in severely affected post-RYGB patients could reflect alterations in proliferation or apoptosis. Markers of apoptosis, including M30, p27, and p21, were low in all patient groups, and did not differ between controls and post-RYGB patients with neuroglycopenia. By contrast, PCNA was 4.5 fold increased in insulin-positive cells from patients with RYGB+NG, as compared with normoglycemic patients with benign pancreatic lesions. Similarly, phospho-histone H3 and Ki-67 expression were numerically higher in insulin-positive cells in neuroglycopenic patients; these values were within a range similar to those reported by us previously and others [34, 35], suggesting variable extent of increased mitoses and proliferation in this population. Additionally, expression levels of Ki-67 were low in all noninsulinoma samples (range 0–0.095% for Ki-67). While reasons for differences between the magnitude of PCNA elevation and those for the mitotic markers PHH3 and Ki-67 remain uncertain, Ki-67 has recently been demonstrated to be particularly susceptible to loss of immunoreactivity during processing [36]. Alternatively, increases in PCNA may reflect not only S phase entry into the cell cycle, but also DNA repair synthesis after damage [37] or growth factor-stimulated protein expression [38]. Despite these mechanistic possibilities and differences in magnitude, PCNA and Ki-67 markers correlated across all samples, consistent with prior studies showing overall concordance of these markers in human pancreatic tissue [39]. Moreover, PCNA positivity correlated positively with β-cell area in patients with post-RYGB neuroglycopenia (Supplementary Figure 2).
Rapid changes in systemic metabolism, growth factors, improved insulin sensitivity, or paracrine factors could contribute to increased PCNA and/or proliferation of insulin-positive cells after RYGB. For example, activation of intracellular insulin signaling has been shown to increase β-cell PCNA [40], while infusion of transforming growth factor (TGF) or epidermal growth factor (EGF) in healthy rats increases PCNA expression in both pancreatic islets and ducts independent of 3H-thymidine incorporation [38]. Animal studies demonstrate rapid changes in β-cell mass after bariatric surgery, suggesting induction of proliferation. Longitudinal studies from a pig model of gastric bypass demonstrate 2-fold increases in β-cell mass as early as three weeks postoperatively, with increased islet number, extra-islet β-cells, and GLP1 receptor density, independent of weight loss [41]. Likewise, both duodenal-jejunal bypass and vertical sleeve gastrectomy in rodents result in increased β-cell mass 3 months postoperatively, assessed by positron emission tomography [42]. One potential contributor to both increased PCNA, proliferation, and insulin secretion in post-RYGB patients could be the incretin hormone GLP1. While GLP1 is associated with β-cell proliferation in rodents, it remains unknown whether the increased endogenous secretion of GLP1 observed after RYGB could also contribute to β-cell proliferative responses in humans [30]. Indeed, levels of GLP1 are markedly increased, up to 10-fold higher after meals in post-RYGB patients as compared with nonsurgical controls, and are even higher in patients with hypoglycemia [12]. While GLP1 receptors are expressed in human islets, expression is not increased in patients with post-bariatric hypoglycemia as compared with controls [24]. Nevertheless, it remains possible that exceedingly high levels of GLP1 in post-bypass patients could contribute to GLP1-dependent responses. We acknowledge that alterations in β-cell mass are not consistently observed in patients with post-RYGB hypoglycemia [25], indicating that increased proliferation and/or β-cell mass may not be absolutely essential for functional dysregulation of insulin secretion observed in this syndrome.
We find localization of FOXO1 is more cytosolic in those with post-RYGB neuroglycopenia compared to nonsurgical controls. Potential mediators of this pattern include increases in levels or action of growth factors, such as insulin or hepatocyte growth factor [43–45]. Since these localization patterns of FOXO1 differed between insulinoma and post-RYGB hypoglycemia patients, additional factors beyond high circulating insulin concentrations are likely to contribute in the post-RYGB setting. For example, increased expression of insulin-like growth factor-2 (IGF2) and insulin-like growth factor-1 receptor-α (IGF1Rα) have been reported in post-RYGB patients [22] and could also contribute to both altered FOXO1 regulation and islet function.
A single case report indicated increased PDX1 expression [46] in a post-RYGB patient, as seen in rodent models [47]; however, PDX1 expression patterns did not differ in our series of post-RYGB hypoglycemic patients. Menin was localized to the nucleus in both β-cells and ductal cells, implicating a potential functional role in both cell types [31]. However, immunostaining for menin did not differ in post-bypass hypoglycemia patients compared to control, indicating menin is not likely to be a major contributor to the post-RYGB hypoglycemia syndrome.
Parallel analysis of samples from patients with insulinoma, who have hypoglycemia due to excessive and autonomous insulin secretion by tumor cells, revealed the expected increases in both Ki-67 and PCNA, thus serving as a positive control. Levels of Ki-67 were not as high in adjacent normal islets, consistent with prior data [48]. However, PCNA was also increased in adjacent normal islets, with magnitude similar to that of RYGB+NG islets. These findings raise the possibility that endocrine or paracrine effects of excessive insulin secretion by either tumor or non-tumor mechanisms could contribute to PCNA positivity in β-cells.
Both insulinoma tumor and adjacent cells showed presence of nuclear PDX1, FOXO1, and menin. As noted, these patterns are distinct from those in post-RYGB neuroglycopenia, despite common increases in insulin levels. Cellular phenotypes exclusive to insulinoma could be linked to transformation or transdifferentiation; the effects of sustained increases in insulin/IGF-1 signaling via phosphoinositide 3-kinase (PI3K) or Akt-dependent pathways [4], the autocrine effects of local growth factors, alteration in regulation of cell cycle regulatory proteins [49] or additional mechanisms exclusive to tumor cells (e.g. increases in GLP1 receptor density) [24] will require additional investigation.
In summary, we find the proliferation marker PCNA is increased in parallel with increased β-cell area in patients with post-RYGB neuroglycopenia. Whether such alterations in pancreatic pathology are linked to dysregulation of postprandial insulin secretion, potentially via common upstream mechanisms, or are independent of each other, remains uncertain and cannot be addressed by examination of tissue pathology. We recognize our study is limited by sample size, heterogeneity between patients, and the inability to compare pre- and post-RYGB pancreatic samples from the same patient. Moreover, these data may not be generalizable to the entire population of less severely affected patients with post-RYGB hypoglycemia, or to those post-RYGB patients without clinically recognized hypoglycemia. Future studies will be required to analyze potential transcriptional contributors to increased insulin secretion.
More broadly, understanding processes that may lead to improved β-cell function is an area of significant interest given the need to find novel ways to counter progression of T2D. Multiple studies provide convincing evidence that rodent β-cells can be stimulated to proliferate, leading to increased functional β-cell mass. A series of emerging reports support this possibility in human β-cells [34, 35, 50–52]. Whether the ability to proliferate can be harnessed to modulate the overall functional β-cell mass for therapeutic purposes warrants further investigation and should continue to spur efforts to identify both systemic and paracrine mediators of islet mass and functional capacity.
Supplementary Material
Supplementary Figure 1. Representative images of β-cells stained with DAPI (blue) and for insulin (red).
Supplementary Figure 2. Correlation between % PCNA-positive insulin-positive cells and β-cell mass (% of area). Red symbols denote RYGB+NG, while black symbols indicate controls. Spearman r=0.68, p<0.01.
Supplementary Figure 3. A. Representative images of proliferation marker phospho-histone H3 in pancreas sections, expressed as percentage of insulin-positive cells in samples from controls (n=5) and patients with RYGB+NG (n=6). Arrows indicate cells selected for display in higher-magnification inset. B. Quantification of phosph-histone H3 positive, insulin-positive cells.
Supplementary Figure 4. Representative images of proliferation markers (A) PCNA and (B) Ki-67 in pancreas sections from 7 patients with insulinoma (INS1–7). Upper panels depict normal pancreas (adjacent to tumor) within the surgical specimen, while lower panels shows insulinoma tumor tissue. Arrows indicate cells selected for display in higher-magnification inset.
Supplementary Figure 5. Representative images of apoptosis markers (A) p27 and (B) p21 in pancreas sections from 7 patients with insulinoma (INS1–7). Upper panels depict normal pancreas (adjacent to tumor) within the surgical specimen, while lower panels shows insulinoma tumor tissue. Arrows indicate cells selected for display in higher-magnification inset. (C) Quantification of percentage of p21+ cells in insulinoma and adjacent non-neoplastic insulin-positive cells. *indicates p=0.004.
Supplementary Figure 6. Representative immunostaining images for the transcription factors (A) PDX1, (B) FOXO1, and (C) menin (green) and insulin (red) in normal and tumor tissue from patients with insulinoma. Arrows indicate cells selected for display in higher-magnification inset.
Supplementary Table 1. Characteristics of patients with insulinoma.
Acknowledgments
We gratefully acknowledge grant support from the Juvenile Diabetes Research Foundation (MEP), American Society of Metabolic and Bariatric Surgery (MEP), the Graetz Foundation (ABG), RC1 DK086918 (ABG), R56 DK095451 (ABG), Novo Nordisk Foundation (AM), KG Jebsen Foundation (AM), RCN FRIMEDBIO 240788 (AM), RO1 DK67536-10 (RNK), RO1 DK103215-01 (RNK), and P30 DK036836 (Diabetes Research Center, Joslin). Franco Folli is currently a Visiting Professor at FCM, OCRC, UNICAMP, supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
Abbreviations
- RYGB
roux-en-Y gastric bypass
- NG
neuroglycopenia
Footnotes
CONFLICT OF INTEREST: The authors declare that they have no conflict of interest.
STATEMENT OF HUMAN AND ANIMAL RIGHTS: All procedures performed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008. This article does not contain any studies with animal subject.
INFORMED CONSENT: Exemption from informed consent was obtained from the institutional review board as these studies were performed on discarded clinically obtained tissues.
AUTHOR CONTRIBUTIONS: MEP, ABG, and RNK wrote the manuscript. JH, DH, and SL performed immunohistochemical analysis. RNK, AM, and SL performed data analysis. JG and WHS contributed to sample procurement and discussion. AM, SL, and FF contributed to discussion and reviewed/edited manuscript.
References
- 1.Grover TR, Pallotto EK, Brozanski B, et al. Interdisciplinary teamwork and the power of a quality improvement collaborative in tertiary neonatal intensive care units. J Perinat Neonatal Nurs. 2015;29:179–186. doi: 10.1097/JPN.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 2.Schauer PR, Bhatt DL, Kirwan JP, et al. Bariatric Surgery versus Intensive Medical Therapy for Diabetes – 3-Year Outcomes. N Engl J Med. 2014 doi: 10.1056/NEJMoa1401329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halperin F, Ding SA, Simonson DC, et al. Roux-en-Y gastric bypass surgery or lifestyle with intensive medical management in patients with type 2 diabetes: feasibility and 1-year results of a randomized clinical trial. JAMA Surg. 2014;149:716–726. doi: 10.1001/jamasurg.2014.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patti ME, McMahon G, Mun EC, et al. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia. 2005;48:2236–2240. doi: 10.1007/s00125-005-1933-x. [DOI] [PubMed] [Google Scholar]
- 5.Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353:249–254. doi: 10.1056/NEJMoa043690. [DOI] [PubMed] [Google Scholar]
- 6.Marsk R, Jonas E, Rasmussen F, Naslund E. Nationwide cohort study of post-gastric bypass hypoglycaemia including 5,040 patients undergoing surgery for obesity in 1986–2006 in Sweden. Diabetologia. 2010 doi: 10.1007/s00125-010-1798-5. [DOI] [PubMed] [Google Scholar]
- 7.Kellogg TA, Bantle JP, Leslie DB, et al. Postgastric bypass hyperinsulinemic hypoglycemia syndrome: characterization and response to a modified diet. Surg Obes Relat Dis. 2008;4:492–499. doi: 10.1016/j.soard.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Sarwar H, Chapman WH, III, Pender JR, et al. Hypoglycemia after Roux-en-Y gastric bypass: the BOLD experience. Obes Surg. 2014;24:1120–1124. doi: 10.1007/s11695-014-1260-8. [DOI] [PubMed] [Google Scholar]
- 9.Kefurt R, Langer FB, Schindler K, Shakeri-Leidenmühler S, Ludvik B, Prager G. Hypoglycemia after Roux-En-Y gastric bypass: detection rates of continuous glucose monitoring (CGM) versus mixed meal test. Surg Obes Relat Dis. 2015;11:564–569. doi: 10.1016/j.soard.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Goldfine AB, Patti ME. How common is hypoglycemia after gastric bypass? Obesity (Silver Spring) 2016;24:1210–1211. doi: 10.1002/oby.21520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roslin M, Damani T, Oren J, Andrews R, Yatco E, Shah P. Abnormal glucose tolerance testing following gastric bypass demonstrates reactive hypoglycemia. Surg Endosc. 2011;25:1926–1932. doi: 10.1007/s00464-010-1489-9. [DOI] [PubMed] [Google Scholar]
- 12.Goldfine AB, Mun EC, Devine E, et al. Patients with neuroglycopenia after gastric bypass surgery have exaggerated incretin and insulin secretory responses to a mixed meal. J Clin Endocrinol Metab. 2007;92:4678–4685. doi: 10.1210/jc.2007-0918. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen NB, Dirksen C, Bojsen-Moller KN, et al. Exaggerated glucagon-like peptide 1 response is important for improved beta-cell function and glucose tolerance after Roux-en-Y gastric bypass in patients with type 2 diabetes. Diabetes. 2013;62:3044–3052. doi: 10.2337/db13-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salehi M, Gastaldelli A, D’Alessio DA. Blockade of glucagon-like peptide 1 receptor corrects postprandial hypoglycemia after gastric bypass. Gastroenterology. 2014;146:669–680. doi: 10.1053/j.gastro.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patti ME, Li P, Goldfine AB. Insulin response to oral stimuli and glucose effectiveness increased in neuroglycopenia following gastric bypass. Obesity (Silver Spring) 2015;23:798–807. doi: 10.1002/oby.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patti ME, Goldfine AB. Hypoglycemia after gastric bypass: the dark side of GLP-1. Gastroenterology. 2014;146:605–608. doi: 10.1053/j.gastro.2014.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Z’Graggen K, Guweidhi A, Steffen R, et al. Severe recurrent hypoglycemia after gastric bypass surgery. Obes Surg. 2008;18:981–988. doi: 10.1007/s11695-008-9480-4. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Esparrach G, Lautz DB, Thompson CC. Peroral endoscopic anastomotic reduction improves intractable dumping syndrome in Roux-en-Y gastric bypass patients. Surg Obes Relat Dis. 2010;6:36–40. doi: 10.1016/j.soard.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin T, Peck M, Holst J, Deacon C. Reversible hyperinsulinemic hypoglycemia after gastric bypass: a consequence of altered nutrient delivery. J Clin Endocrinol Metab. 2010;95:1851–1855. doi: 10.1210/jc.2009-1628. [DOI] [PubMed] [Google Scholar]
- 20.Lee CJ, Brown T, Magnuson TH, Egan JM, Carlson O, Elahi D. Hormonal response to a mixed-meal challenge after reversal of gastric bypass for hypoglycemia. J Clin Endocrinol Metab. 2013;98:E1208–E1212. doi: 10.1210/jc.2013-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanderveen KA, Grant CS, Thompson GB, et al. Outcomes and quality of life after partial pancreatectomy for noninsulinoma pancreatogenous hypoglycemia from diffuse islet cell disease. Surgery. 2010;148:1237–1245. doi: 10.1016/j.surg.2010.09.027. discussion 1245–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rumilla KM, Erickson LA, Service FJ, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis: histologic features and growth factor expression. Mod Pathol. 2009;22:239–245. doi: 10.1038/modpathol.2008.169. [DOI] [PubMed] [Google Scholar]
- 23.Mulla CM, Storino A, Yee EU, et al. Insulinoma After Bariatric Surgery: Diagnostic Dilemma and Therapeutic Approaches. Obes Surg. 2016;26:874–881. doi: 10.1007/s11695-016-2092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reubi JC, Perren A, Rehmann R, et al. Glucagon-like peptide-1 (GLP-1) receptors are not overexpressed in pancreatic islets from patients with severe hyperinsulinaemic hypoglycaemia following gastric bypass. Diabetologia. 2010;53:2641–2645. doi: 10.1007/s00125-010-1901-y. [DOI] [PubMed] [Google Scholar]
- 25.Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic Hypoglycemia After Gastric Bypass Surgery Is Not Accompanied by Islet Hyperplasia or Increased {beta}-Cell Turnover. Diabetes Care. 2006;29:1554–1559. doi: 10.2337/dc06-0392. [DOI] [PubMed] [Google Scholar]
- 26.Hoem D, Jensen D, Steine S, Thorsen TE, Viste A, Molven A. Clinicopathological characteristics and non-adhesive organ culture of insulinomas. Scand J Surg. 2008;97:42–49. doi: 10.1177/145749690809700106. [DOI] [PubMed] [Google Scholar]
- 27.La Rosa S, Klersy C, Uccella S, et al. Improved histologic and clinicopathologic criteria for prognostic evaluation of pancreatic endocrine tumors. Hum Pathol. 2009;40:30–40. doi: 10.1016/j.humpath.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Rindi GAR, Bosman FT, Capella C, Klimstra DS, Klöppel G, Komminoth P, Solcia E. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman TFCF, Hruban RH, Theise ND, editors. WHO Classification of Tumors of the Digestive System. International Agency for Research on Cancer (IARC); Lyon, France: 2010. p. 13. [Google Scholar]
- 29.Guardado Mendoza R, Perego C, Finzi G, et al. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas) Diabetologia. 2015;58:1814–1826. doi: 10.1007/s00125-015-3625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart AF, Hussain MA, Garcia-Ocana A, et al. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64:1872–1885. doi: 10.2337/db14-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scavini M, Pontiroli AE, Folli F. Asymptomatic hyperinsulinemic hypoglycemia after gastric banding. N Engl J Med. 2005;353:2822–2823. doi: 10.1056/NEJMc052356. [DOI] [PubMed] [Google Scholar]
- 33.Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia. 2011;54:1720–1725. doi: 10.1007/s00125-011-2118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang P, Alvarez-Perez JC, Felsenfeld DP, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. 2015;21:383–388. doi: 10.1038/nm.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dirice E, Walpita D, Vetere A, et al. Inhibition of DYRK1A Stimulates Human beta-Cell Proliferation. Diabetes. 2016;65:1660–1671. doi: 10.2337/db15-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan BA, Hollister-Lock J, Bonner-Weir S, Weir GC. Reduced Ki67 Staining in the Postmortem State Calls Into Question Past Conclusions About the Lack of Turnover of Adult Human beta-Cells. Diabetes. 2015;64:1698–1702. doi: 10.2337/db14-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyama T, Wilson DM., III DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardiner KR, Crockard AD, Halliday MI, Rowlands BJ. Class II major histocompatibility complex antigen expression on peripheral blood monocytes in patients with inflammatory bowel disease. Gut. 1994;35:511–516. doi: 10.1136/gut.35.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohler CU, Kreuter A, Rozynkowski MC, et al. Validation of different replication markers for the detection of beta-cell proliferation in human pancreatic tissue. Regul Pept. 2010;162:115–121. doi: 10.1016/j.regpep.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Stamateris RE, Sharma RB, Kong Y, et al. Glucose Induces Mouse beta-Cell Proliferation via IRS2, MTOR, and Cyclin D2 but Not the Insulin Receptor. Diabetes. 2016;65:981–995. doi: 10.2337/db15-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindqvist A, Spegel P, Ekelund M, et al. Gastric Bypass Improves beta-Cell Function and Increases beta-Cell Mass in a Porcine Model. Diabetes. 2014;63:1665–1671. doi: 10.2337/db13-0969. [DOI] [PubMed] [Google Scholar]
- 42.Inabnet WB, Milone L, Harris P, et al. The utility of [(11)C] dihydrotetrabenazine positron emission tomography scanning in assessing beta-cell performance after sleeve gastrectomy and duodenal-jejunal bypass. Surgery. 2010;147:303–309. doi: 10.1016/j.surg.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 44.Al-Masri M, Krishnamurthy M, Li J, et al. Effect of forkhead box O1 (FOXO1) on beta cell development in the human fetal pancreas. Diabetologia. 2010;53:699–711. doi: 10.1007/s00125-009-1632-0. [DOI] [PubMed] [Google Scholar]
- 45.Assmann A, Ueki K, Winnay JN, Kadowaki T, Kulkarni RN. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol. 2009;29:3219–3228. doi: 10.1128/MCB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabiee A, Magruder JT, Salas-Carrillo R, et al. Hyperinsulinemic hypoglycemia after Roux-en-Y gastric bypass: unraveling the role of gut hormonal and pancreatic endocrine dysfunction. J Surg Res. 2011;167:199–205. doi: 10.1016/j.jss.2010.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Zhang HY, Lv LX, et al. Roux-en-Y gastric bypass promotes expression of PDX-1 and regeneration of beta-cells in Goto-Kakizaki rats. World J Gastroenterol. 2010;16:2244–2251. doi: 10.3748/wjg.v16.i18.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montemurro C, Kohler CU, Uhl W, et al. Endogenous hyperinsulinaemia in insulinoma patients is not associated with changes in beta-cell area and turnover in the tumor-adjacent pancreas. Regul Pept. 2010;165:180–185. doi: 10.1016/j.regpep.2010.07.164. [DOI] [PubMed] [Google Scholar]
- 49.Ueberberg S, Tannapfel A, Schenker P, et al. Differential expression of cell-cycle regulators in human beta-cells derived from insulinoma tissue. Metabolism. 2016;65:736–746. doi: 10.1016/j.metabol.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Kondegowda NG, Fenutria R, Pollack IR, et al. Osteoprotegerin and Denosumab Stimulate Human Beta Cell Proliferation through Inhibition of the Receptor Activator of NF-kappaB Ligand Pathway. Cell Metab. 2015;22:77–85. doi: 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boucher J, Softic S, El Ouaamari A, et al. Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function. Diabetes. 2016 doi: 10.2337/db16-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhawan S, Dirice E, Kulkarni RN, Bhushan A. Inhibition of TGF-beta Signaling Promotes Human Pancreatic beta-Cell Replication. Diabetes. 2016;65:1208–1218. doi: 10.2337/db15-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Representative images of β-cells stained with DAPI (blue) and for insulin (red).
Supplementary Figure 2. Correlation between % PCNA-positive insulin-positive cells and β-cell mass (% of area). Red symbols denote RYGB+NG, while black symbols indicate controls. Spearman r=0.68, p<0.01.
Supplementary Figure 3. A. Representative images of proliferation marker phospho-histone H3 in pancreas sections, expressed as percentage of insulin-positive cells in samples from controls (n=5) and patients with RYGB+NG (n=6). Arrows indicate cells selected for display in higher-magnification inset. B. Quantification of phosph-histone H3 positive, insulin-positive cells.
Supplementary Figure 4. Representative images of proliferation markers (A) PCNA and (B) Ki-67 in pancreas sections from 7 patients with insulinoma (INS1–7). Upper panels depict normal pancreas (adjacent to tumor) within the surgical specimen, while lower panels shows insulinoma tumor tissue. Arrows indicate cells selected for display in higher-magnification inset.
Supplementary Figure 5. Representative images of apoptosis markers (A) p27 and (B) p21 in pancreas sections from 7 patients with insulinoma (INS1–7). Upper panels depict normal pancreas (adjacent to tumor) within the surgical specimen, while lower panels shows insulinoma tumor tissue. Arrows indicate cells selected for display in higher-magnification inset. (C) Quantification of percentage of p21+ cells in insulinoma and adjacent non-neoplastic insulin-positive cells. *indicates p=0.004.
Supplementary Figure 6. Representative immunostaining images for the transcription factors (A) PDX1, (B) FOXO1, and (C) menin (green) and insulin (red) in normal and tumor tissue from patients with insulinoma. Arrows indicate cells selected for display in higher-magnification inset.
Supplementary Table 1. Characteristics of patients with insulinoma.