

Summary
Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) have been shown to function downstream of several peptide hormones and cytokines that are required for postnatal development and secretory function of the mammary gland. As part of an extended network, these signal transducers can engage in crosstalk with other pathways to facilitate synergistic, and sometimes antagonistic, actions of different growth factors. Specifically, signaling through the JAK2/STAT5 cascade has been demonstrated to be indispensable for the specification, proliferation, differentiation, and survival of secretory mammary epithelial cells. Following a concise description of major cellular programs in mammary gland development and the role of growth factors that rely on JAK/STAT signaling to orchestrate these programs, this review highlights the significance of active STAT5 and its crosstalk with the PI3 kinase and AKT1 for mediating the proliferation of alveolar progenitors and survival of their functionally differentiated descendants in the mammary gland. Based on its ability to provide self-sufficiency in growth signals that are also capable of overriding intrinsic cell death programs, persistently active STAT5 can serve as a potent oncoprotein that contributes to the genesis of breast cancer. Recent experimental evidence demonstrated that, similar to normal developmental programs, oncogenic functions of STAT5 rely on molecular crosstalk with PI3K/AKT signaling for the initiation, and in some instances the progression, of breast cancer. The multitude by which STATs can interact with individual mediators of the PI3K/AKT signaling cascade may provide novel avenues for targeting signaling nodes within molecular networks that are crucial for the survival of cancer cells.
Keywords: Signal Transduction, Janus Kinase, STAT, Phosphatidylinositol 3-Kinases, Proto-Oncogene Proteins c-akt, Mammary Gland Development, Breast Cancer
The genesis of the mammary gland relies on a precisely controlled execution of distinct cellular programs
The development of the mammary gland is dependent on a number of precisely coordinated cellular programs that start with the determination of a specific cell fate and the proliferation of committed progenitors. As a skin appendage, the mammary gland starts to form as a placode on the surface of the ectoderm during embryogenesis. The committed mammary epithelial progenitors continue to proliferate, and the placode increases in size and invaginates the underlying mesenchyme (Robinson, 2007). Morphologically, the embryonic mammary gland becomes a bulb-shaped bud that subsequently elongates and bifuricates. This process culminates in the formation of a small rudimentary ductal system prior to birth. The mammary epithelium of perinatal or newborn females largely consists of quiescent epithelial progenitors that expresses both cytokeratin (CK) 14 and CK8 (Van Keymeulen et al., 2011; Sakamoto et al., 2012). These bipotent progenitors have been demonstrated to possess a high repopulation efficiency when transplanted into epithelial-free fat pads of adult recipient females (Spike et al., 2012).
A second major cellular program during mammary gland development is the initiation and implementation of a differentiation process. During the onset of puberty, the ducts elongate and branch at the leading proliferative zones called terminal end buds (TEBs). Two distinctly different epithelial cell types, i.e. cap cells and body cells, are present within TEBs, and these cells contribute to the mostly CK14-positive basal and CK8-positive luminal epithelial cell lineages. On the microscopic level, TEBs reveal a dramatic rate of cellular turnover illustrating the precise control of cellular homeostasis, which represents a third major cellular process that is essential to the development of the gland. While the leading edge of TEBs contains proliferating cells, many trailing body cells undergo programmed cell death (Humphreys et al., 1996). This controlled cellular turnover is imperative for the formation of the ductal lumen and the correct spatial arrangement of luminal and basal epithelial cells. The TEBs disappear after the ducts have completely penetrated the mammary fat pad. Nonetheless, the mammary glands of sexually mature females continue to undergo cycling changes with each estrus cycle. The terminal ends of tertiary side branches called lobuloalveolar units (rodents) or terminal ductal lobular units (humans) contain alveolar progenitors that proliferate, differentiate, and die in response to oscillating levels of ovarian hormones.
During pregnancy, alveolar progenitors exhibit an extraordinary burst of proliferation and the vast majority of them functionally differentiates into milk-producing, secretory epithelial cells (Hennighausen and Robinson, 2001; Grimm et al., 2002). Following the weaning of the offspring, the mammary gland undergoes a very rapid remodeling process, called involution, where most of the secretory epithelial cells die (Stein et al., 2007). Although it is generally accepted that the fully remodeled mammary gland of a parous female largely resembles that of a mature virgin, a full-term pregnancy and lactation cycle is known to cause sustained changes in the cellular composition of the epithelium as well as their gene expression profiles (D’Cruz et al., 2002; Wagner et al., 2002).
The postnatal development of the mammary gland is governed by hormones and locally produced cytokines
The execution of the various cellular programs at particular stages of mammary gland development are orchestrated by growth factor signaling networks within epithelial cells and the adjacent embryonic mesenchyme or the stroma of the postnatal gland. Signaling of Wnt (wingless-related MMTV integration site), fibroblast growth factor (FGF), neuregulin (NRG3), and parathyroid hormone-like hormone (PTHLH) have been shown to initiate the formation of the mammary placode, the proliferation of committed progenitors, and the genesis of the rudimentary mammary gland during embryogenesis (for a comprehensive review on this subject please refer to an article by Gertraud Robinson (Robinson, 2007)).
As females sexually mature, the increase in circulating levels of estrogen greatly propels the morphogenesis of the ductal tree. This is primarily the result of the direct action of estrogen on the mammary epithelium (Mallepell et al., 2006; Feng et al., 2007) and not through paracrine signaling of stromal cells that express the estrogen receptor as proposed earlier (Cunha et al., 2000). Essential functions of progesterone and prolactin (PRL) on the mammary epithelium are restricted to the formation of the lobuloalveolar units of the mature mammary gland (Brisken et al., 1998; Grimm et al., 2002). The various functions of these classical hormones during postnatal mammary gland development were highlighted in a previous review by Brisken and O’Malley (Brisken and O’Malley, 2010). In addition to the action of steroid and peptide hormones, locally synthesized cytokines have been shown to play key roles for the specification and differentiation as well as the postlactational remodeling of the mammary epithelium. Khaled and coworkers (Khaled et al., 2007) have shown recently that the commitment of epithelial cells to the luminal lineage is accompanied by a switch in the production of cytokines that are present in native T helper (Th) cells of the subtypes Th1 (IL-12, INFg) and Th2 (IL-4, IL-13, IL-5). In synergy with PRL, signaling of the Th2 cytokines IL-4 and IL-13 seems to support the development of alveolar cells during early to mid-pregnancy. It has been demonstrated in genetic models deficient in PRL or its receptor (PRLR) that this peptide hormone is essential for the proliferation, specification, and functional differentiation of alveolar cells (Horseman et al., 1997; Ormandy et al., 1997). PRL signaling also supports the survival of functionally differentiated alveolar cells throughout lactation.
Although circulating levels of PRL decline following the weaning of offspring, it is primarily the accumulation of milk within secretory alveoli that triggers the involution of the mammary gland (Li et al., 1997). This process requires a disengagement of PRL signaling from their downstream mediators within the epithelium as well as the production and signaling of interleukin-6 (IL-6)-class inflammatory cytokines, i.e., leukemia inhibitory factor (LIF) and oncostatin M (OSM). Following postlactational mammary gland remodeling, the developmental program of alveolar proliferation, differentiation, and cell death recurs with each subsequent full-term pregnancy and lactation cycle. In summary, the mammary gland develops in a female predominantly after the onset of puberty, and the growth and differentiation of the mammary epithelium is controlled by the synergistic action of steroid and peptide hormones as well as local growth factors.
Obligatory roles of JAK/STAT signaling cascades during mammary gland development
A commonality among peptide hormones and cytokines that orchestrate mammary gland development (i.e., PRL, IL-12, INFγ, IL-4, IL-13, IL-6, LIF, and OSM) is that they all rely on Janus kinases (JAKs) and signal transducers and activators of transcription (STATs) for intracellular signaling. Consequently, all major cellular programs during postnatal mammary gland development depend on one or more JAK/STAT signaling cascades. Of the seven STAT proteins that are present in mammalians, five (i.e., STAT1, 3, 5a, 5b, and 6) have been found to be expressed and activated at particular stages of mammary gland development (Watson and Neoh, 2008; Wagner and Schmidt, 2011). The activation of these STATs as defined by their tyrosine phosphorylation and nuclear translocation occurs in a sequential manner in response to the major cytokines that control a particular developmental program. The highest levels of STAT1 activation in the mammary epithelium were observed in nulliparous and nonpregnant parous females after postlactational remodeling as well as briefly during mid-pregnancy (Philp et al., 1996; Sakamoto et al., 2016b). STAT6 was reported to have a biologically significant role as a downstream effector of IL-4 and IL-13 in developing alveoli of pregnant females (Khaled et al., 2007). Despite the precise temporal control of the activation of STAT1 and STAT6, females that are deficient in these two STAT proteins are able to lactate, therefore STAT1 and STAT6 are not stringently required for ductal or alveolar morphogenesis (Khaled et al., 2007; Klover et al., 2010).
STAT5a and STAT5b exhibit a significant increase in their activation in response to elevated levels of circulating PRL during pregnancy and lactation (Liu et al., 1996), and studies in knockout mice have shown that the combined functions of both STAT5 isoforms are central for alveolar development and milk gene expression (Liu et al., 1998; Cui et al., 2004). The fact that the loss of STAT5a alone causes more severe phenotypic abnormalities compared to a STAT5b single knockout (Liu et al., 1997; Udy et al., 1997) is primarily a consequence of a disproportionally high expression of STAT5a in the mammary epithelium. The significance of STAT5a is best illustrated by its original name, ‘mammary gland factor’ or MGF, when this PRL-responsive signal transducer was identified in the early 1990s by the team of Bernd Groner (Wakao et al., 1992; Wakao et al., 1994). Irrespective of high levels of pituitary PRL in circulation, the maintenance of secretory capacity of the mammary gland and STAT5 activation requires a periodical discharge of milk. Milk stasis leads to a very rapid de-phosphorylation of STAT5 and a significant increase in the activation of STAT3 (Philp et al., 1996; Li et al., 1997; Watson and Neoh, 2008). This synchronous switch from STAT5 to STAT3 phosphorylation along with changes in the transcriptional expression of their respective downstream targets signifies the initiation of involution and postlactational remodeling. This mechanism explains on the molecular level what has been known since the domestication of cattle for milk production thousands of years ago: a cow has to be milked twice a day and evenly on all four quarters to ensure continuous and optimal lactation. Initially, the STAT5/3 switch is reversible when milk withdrawal is resumed. Studies in mice have shown that the local production of LIF increases during milk stasis and activates STAT3 during the reversible phase of mammary gland involution (Kritikou et al., 2003). Subsequently, active STAT3 enhances the expression of OSM and its receptor (Tiffen et al., 2008; Hughes et al., 2012; Sakamoto et al., 2016b). This autocrine loop ensures a sustained activation of STAT3, which then initiates the death of secretory alveolar cells and promotes the remodeling of the mammary gland, which, at this point, is nonreversible (Tiffen et al., 2008).
JAK1 and JAK2 have non-redundant functions in mammary gland development
Among the four upstream Janus tyrosine kinases (JAKs) that activate STAT proteins, JAK1 and JAK2 play pivotal roles in peptide hormone and cytokine signaling during mammary gland development. Due to their associations with multiple cytokine receptors, these two Janus kinases are suggested to have pleiotropic functions, and they are thought to be redundant for the activation of particular STATs in selected tissues (Kisseleva et al., 2002). However, a comparison of conditional knockout mice that lack either JAK1 or JAK2 in the mammary epithelium revealed that these tyrosine kinases have discrete roles in the activation of particular STAT proteins and non-redundant functions during mammary gland development (Wagner et al., 2004; Sakamoto et al., 2007; Sakamoto et al., 2016a; Sakamoto et al., 2016b). We and others have shown that the main role of JAK2 in the epithelium is to activate STAT5a and STAT5b in response to PRL signaling (Shillingford et al., 2002; Wagner et al., 2004). In support of this finding, a mammary-specific double knockout of STAT5a and STAT5b mirrors the phenotypic abnormalities that were observed in JAK2 deficient females (Cui et al., 2004) The collective results from these defined genetic models demonstrated that essential functions of JAK2/STAT5 signaling are restricted to the specification and proliferation of alveolar progenitors as well as the survival of their functionally differentiated descendants (Cui et al., 2004; Wagner et al., 2004). JAK2 has been generally perceived as the “JAK of all trades”, which is evident by the significantly higher number publications on this particular kinase (i.e., almost twice as many as all the other JAKs combined according to PubMed) as well as efforts to develop specific JAK2 inhibitors to treat a variety of malignancies.
JAK1 was thought to have a subordinate role for STAT activation, in particular STAT3, during mammary gland development and in breast cancer (Corcoran et al., 2011; Marotta et al., 2011; Abubaker et al., 2014). In contrast to this paradigm, a recent study using JAK1 conditional knockout mice has shown that this particular Janus kinase is required for the tyrosine phosphorylation of three of the five STAT proteins (STAT1, 3, and 6) that are active in the mammary epithelium (Sakamoto et al., 2016b). Despite the broader impact of JAK1 deficiency on the activity of several STATs, the biologically relevant functions of JAK1 are limited to postlactational mammary gland remodeling where a loss of this kinase uncouples IL-6-class cytokine signaling from its downstream effector STAT3. Therefore, a primary role of JAK1 is to mediate signals from inflammatory cytokines to the cell death machinery in the functionally differentiated mammary epithelium. It was somewhat surprising to note that the combined loss in the activation of STAT1, 3 and 6 in the mammary epithelium does not result in any additional, previously unrecognized phenotypic abnormalities. Possible reasons for this observation might be that a) the highest levels of activation of STAT1, 3, and 6 occur at different stages of development, and b) a strong nuclear accumulation of individual STATs appears to be confined to distinct epithelial subtypes in ducts versus alveoli. Deficiency in JAK1 closely phenocopies a mammary specific knockout of STAT3 (Chapman et al., 1999). However, despite the lack of STAT6 activation in the epithelium, the previously reported abnormalities in alveolar development associated with a complete loss of STAT6 (Khaled et al., 2007) were not apparent in JAK1 conditional knockout mice. Future experiments will have to clarify whether the observed phenotype in STAT6 deficient mice is primarily a consequence of the loss of this STAT protein in the epithelium or the stroma, which is known to exert paracrine effects on developing alveolar cells. It should also be considered that a deficiency in an upstream kinase and consequential lack of the tyrosine phosphorylation of its STAT target is functionally not the same as a complete loss of a STAT protein. Un-phosphorylated STATs have been suggested to play different roles in signaling (Yang and Stark, 2008), and, in the case of STAT1 and STAT3, these signal transducers can still be phosphorylated on the conserved serine 727 residue despite their lack of tyrosine phosphorylation in the JAK1 knockout (Sakamoto et al., 2016a; Sakamoto et al., 2016b). This clearly supports previous observations that the tyrosine phosphorylation of STAT1 may not be a prerequisite for other posttranslational modifications (Zhu et al., 1997; Milocco et al., 1999; Decker and Kovarik, 2000). In summary, the collective observations in JAK1 and JAK2 conditional knockout models have illustrated very clearly that JAKs have non-redundant functions for the tyrosine phosphorylation and activation of specific STATs, and therefore they control distinctly different developmental programs during mammogenesis.
Evidence for a crosstalk between active STAT5 and PI3K/AKT in response to PRL signaling in the mammary epithelium
As discussed earlier, female mice that are deficient in the PRL receptor, JAK2, or STAT5a/b are unable to nurse their offspring and are phenotypically quite similar as they exhibit a complete lack of alveolar proliferation and differentiation during pregnancy. This suggests that all biologically relevant functions of PRL signaling are mediated by the JAK2/STAT5 signaling pathway. Since PRL also activates the RAS/MAPK, AKT (protein kinase B), and phospholipase C-protein kinase C pathways (Erwin et al., 1995; Das and Vonderhaar, 1997; Grimley et al., 1999; Llovera et al., 2000), it was logical to assume that JAK2 or STAT5 control all other signaling events that converge on the PRLR. Surprisingly, the molecular analysis of JAK2 deficient mammary epithelial cells revealed that the SRC tyrosine kinase, the focal adhesion kinase (FAK), and MAP kinases could still be effectively activated in response to PRL stimulation (Sakamoto et al., 2007). This finding supports previous observations by Fresno Vara and colleagues (2000) that the PRLR-mediated activation of the c-SRC kinase occurs independently of JAK2 in human breast cancer cells. Since JAK2 deficient mammary epithelial cells lack active STAT5, it is evident that SRC does not activate STAT5 directly in a JAK2-independent manner. Although SRC and MAP kinases, i.e., signal transducers that are commonly associated with proliferative responses, were still active, a knockout of JAK2 caused a significant reduction in cell proliferation. As demonstrated by Brockman et al. (2005), STAT5 may play a direct role in the transcriptional activation of the Cyclin D1 (Ccnd1) gene. In JAK2 deficient mammary epithelial cells, however, the difference in the expression of Cyclin D1 was more pronounced on the level of the protein compared to the mRNA (Sakamoto et al., 2007). This was primarily a consequence of an accelerated, GSK-3β-mediated phosphorylation of Cyclin D1, which elicits the nuclear export and proteosomal degradation of this cell cycle regulator. The decline in Cyclin D1 levels in JAK2 deficient cells was likely caused by the reduced expression of AKT1, which is known to stabilize Cyclin D1 through inhibition of GSK-3β (Diehl et al., 1998). This initial finding that JAK2/STAT5 signaling engages in a crosstalk with the PI3K/AKT cascade was further substantiated by experimental evidence that showed that the proliferation of JAK2 deficient cells can be restored through expression of myristolated AKT1 or constitutively nuclear Cyclin D1 that cannot be phosphorylated by GSK-3β (Sakamoto et al., 2007; Creamer et al., 2010).
Despite evidence for signaling crosstalk, it was unclear how the PRL and its receptor might activate the PI3K/AKT pathway in a JAK2- or STAT5-dependent manner in the mammary epithelium. The fact that the PRL-mediated activation of SRC and MAP kinases was not blocked in the absence of JAK2 excluded them from being potential candidates that connect PRL signaling to PI3K and AKT as proposed previously for other signaling cascades (Rodriguez-Viciana et al., 1996; al Sakkaf et al., 1997). It has also been shown, that STAT proteins can form complexes with the p85 regulatory subunit of the PI3 kinase to modulate its activity (Pfeffer et al., 1997; Rosa Santos et al., 2000; Santos et al., 2001; Nyga et al., 2005). Indeed, active STAT5 can bind to the SRC-homology 2 (SH2) domain-containing p85α subunit of PI3K in a PRL-inducible and JAK2-dependent manner in mammary epithelial cells (Sakamoto et al., 2007). This association, however, was very weak and therefore not being considered to be the primary mechanism that vindicated the much lower expression and activation of AKT1 in JAK2-deficient cells.
PI3 kinase subunits and AKT isoforms are ubiquitously expressed, and the main mechanisms of action are generally being recognized to occur on the level of posttranslational modifications (i.e., phosphorylation), which is depicted in the pathway maps on posters that decorate the walls of many laboratories. It is less apparent, however, that the expression of the mRNAs of PI3K and AKT varies significantly between organs or even among different physiological states of the same tissue. In the mammary gland, the level of Akt1 mRNA appears to be inversely correlated to the transcriptional activation of Akt2 and Akt3 (Boxer et al., 2006). The Akt1 message and the total and active levels of the AKT1 kinase increase during pregnancy and lactation and decline sharply during involution (Schwertfeger et al., 2001; Boxer et al., 2006; Creamer et al., 2010). Similarly, the expression of certain PI3 kinase subunits fluctuates on the mRNA and protein level during mammary gland development, and Abell and colleagues (Abell et al., 2005) were first to demonstrate that active STAT3 directly controls the expression of the smaller PI3K regulatory subunits (p50α/p55α) during mammary gland involution. The expression of AKT1 as well as the regulatory and catalytic subunits of PI3K (p85α and p110α) mirror almost precisely the activation profile of the JAK2/STAT5 pathway in response to PRL signaling, and it has been demonstrated that active STAT5 binds to at least two consensus sequences within the mouse Akt1 locus in a growth factor-dependent manner (Creamer et al., 2010). During pregnancy and lactation, STAT5 greatly enhances the transcription of a unique Akt1 mRNA, called Akt1m, from a novel promoter located within the first intron. Like the mRNA message that originates from the upstream ubiquitously active promoter, the Akt1m transcript encodes the full-length AKT1 serine/threonine protein kinase. Notably, overexpression of a hyperactive mutant of STAT5a in cultured mammary epithelial cells and in the developing mammary gland of transgenic mice was sufficient to maintain an elevated expression of Akt1m and higher levels of the total and activated AKT1 kinase (Creamer et al., 2010). The sustained activation of AKT1 is mediated by the PI3 kinase through the transcriptional activation of the genes encoding p85α and p110α (Pik3r1 and Pik3ca) whose expression, like AKT1, is under control of active STAT5 (Schmidt et al., 2014a). These collective findings suggest that STAT transcription factors are able to enhance the expression of particular PI3 kinase subunits and AKT1 in the developing mammary gland (Figure 1). In particular, the sustained activation of the JAK2/STAT5 cascade greatly augments signaling through the PI3K/AKT1 pathway, which plays a significant biological role in the proliferation, survival, and possibly also the metabolism of alveolar cells in the mammary gland.
Figure 1. JAK/STAT signaling cascades control the survival or death of mammary epithelial cells by enhancing the expression and functionality of particular PI3 kinase subunits and AKT.
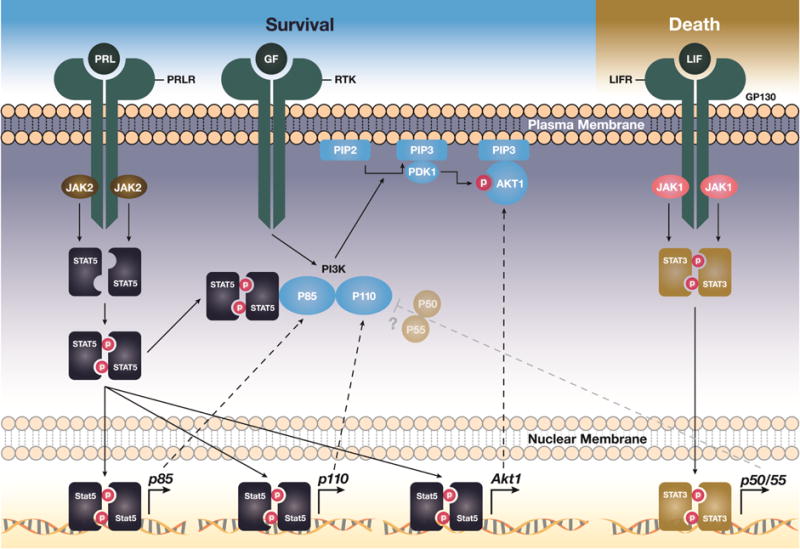
The activation of the JAK2/STAT5 pathway promotes cell proliferation and survival through transcriptional upregulation of p85α (Pik3r1), p110α (Pik3ca), and Akt1 as well as through direct interaction of tyrosine phosphorylated STAT5 with the p85α regulatory subunit of PI3K. The JAK1-mediated activation of STAT3 controls the expression of the shorter p50/55α (Pik3r1) regulatory subunits of PI3K during mammary gland involution. The persistent activation of STAT5, p110α, and AKT1 promotes a sustained survival of epithelial cells despite the tyrosine phosphorylation of STAT3 and transcriptional activation of its downstream targets. This may explain why these potent survival factors are protooncogenes. The precise mechanisms by which p50/55α may modulate PI3 kinase signaling and contribute mammary gland remodeling in the absence of active STAT5 needs further investigation.
JAK2/STAT5 signaling promotes the survival of mammary epithelial cells through PI3K and AKT1
It is well-known that diverse growth factor receptors can utilize common downstream signaling mediators, thereby engaging in signaling crosstalk. The extent of a specific crosstalk among downstream pathways might explain why an abnormal activation of diverse growth factor pathways can have very similar phenotypic responses. For example, an upregulation or constitutive activation of either PRLR or insulin-like growth factor 1 receptor (IGF-1R) accelerate the proliferation and differentiation of alveolar progenitors, and, more importantly, they greatly extend the survival of epithelial cells thereby delaying the postlactational remodeling of the gland (Sandgren et al., 1995; Neuenschwander et al., 1996; Weber et al., 1998; Gourdou et al., 2004).
Virtually identical to the deregulated expression of growth factor receptors, the overexpression of wildtype or hyperactive mutants of JAK2, STAT5, PI3K (p110), or AKT1 promotes a continuous survival of alveolar cells despite reduced circulating levels of PRL or the local induction of inflammatory cytokines (Hutchinson et al., 2001; Schwertfeger et al., 2001; Ackler et al., 2002; Iavnilovitch et al., 2002; Creamer et al., 2010; Vafaizadeh et al., 2010; Meyer et al., 2011; Caffarel et al., 2012; Yuan et al., 2013). The identified crosstalk between JAK2/STAT5 signaling and the PI3K/AKT1 cascade might explain why active STAT5 is a potent survival factor. The view that the PI3K/AKT1 pathway is a crucial downstream effector of JAK2/STAT5 signaling is supported by the observation that a conditional knockout of AKT1 averts the extended survival of mammary epithelial cells that express hyperactive STAT5 (Schmidt et al., 2014a). Also, overexpression of AKT1 causes a delay in involution without the sustained activation of STAT5 after circulating levels of PRL decline. In its role as a transcription factor that enhances the expression of p85α, p110α, and AKT1, STAT5 can greatly augment the activation of this prominent survival pathway in a direct manner as well as in an indirect manner through other receptor tyrosine kinases that activate PI3K (e.g., IGF-1R, ERBBs). In addition to its function on gene transcription, STAT5 binds to p85α via the Gab2 scaffolding adapter (Nyga et al., 2005), and thereby STAT5 may play another direct role within PI3K signaling complexes that activate AKT1 and promote cell survival.
The first step of a normal postlactational involution program is initiated by the swift dephosphorylation of STAT5 and the synchronous activation of STAT3 in response to milk stasis and increased levels of IL-6-class inflammatory cytokines. In the absence of active STAT5, a loss-of-function of STAT3 or its upstream regulator JAK1 significantly extends the functional competence of secretory epithelial cells and causes a delay in cell death and mammary gland remodeling (Chapman et al., 1999; Sakamoto et al., 2016b). The activation of STAT3 enhances the expression of p50α/p55α, and the analysis of an isoform specific knockout mouse model revealed that these smaller PI3 kinase regulatory subunits are involved in the programmed death of mammary epithelial cells (Figure 1), possibly through upregulation of cathepsin L (Pensa et al., 2014b) or though inhibition of autophagy (Pensa et al., 2014a). The model previously proposed by Abell et al. (2005) that p50α and p55α inhibit the p85α/p110α PI3K holoenzyme and AKT1 needs to be revisited since the loss of active STAT5 during the onset of involution causes a swift decline in p85α, p110α, and AKT1 expression. More importantly, a sustained activation of STAT5 during involution, which promotes an extended upregulation of p85α/p110α and AKT1, has been demonstrated to override active STAT3 and the downstream transcriptional upregulation of p50α/p55α (Creamer et al., 2010; Schmidt et al., 2014a). Therefore, the STAT3-mediated increase in p50α/p55α is likely not sufficient to inhibit a sustained activation of PI3K and AKT1 signaling. The collective findings from the analysis of genetic models clearly illustrates that JAK/STAT and PI3K/AKT signaling pathways are tightly interconnected (Figure 1). The normal involution program in the postlactational mammary gland requires a coordinated downregulation of the pro-survival JAK2/STAT5 cascade and PI3K (p85α/p110α)/AKT1 signaling as well as an upregulation of the cell death promoting JAK1/STAT3 pathway and its downstream effectors p50α/p55α. The interference of just one signaling mediator in these regulatory signaling networks can have severe consequences that manifest in developmental defects, mastitis, and even the initiation of breast cancer.
Aberrant STAT5 signaling and its crosstalk with the PI3K/AKT1 pathway contributes to breast cancer initiation
As a potent survival factor, active STAT5 contributes to two hallmarks for tumor initiation and malignant progression (Hanahan and Weinberg, 2000). First, STAT5 can largely override the STAT3-mediated cell death program in the normal mammary epithelium. In addition to controlling the mechanisms that lead to an evasion from canonical apoptosis or other forms of cell death, active STAT5 also provides self-sufficiency in growth signals. In support of its function as a protooncogene, tyrosine-phosphorylated STAT5 has been shown to be expressed in a significant subset of primary human breast cancers (Cotarla et al., 2004; Nevalainen et al., 2004), and the constitutive expression of hyperactive STAT5 in luminal epithelial cells as well as multipotent progenitors is sufficient to cause mammary cancer (Iavnilovitch et al., 2002; Vafaizadeh et al., 2010). Moreover, it was demonstrated that the activation of STAT5 through JAK2 is required for the initiation of mammary tumors in diverse genetic mouse models for breast cancer (Humphreys and Hennighausen, 1999; Ren et al., 2002; Sakamoto et al., 2009; Sakamoto et al., 2010; Haricharan et al., 2013). Collectively, these findings implicate oncogenic STAT5 with neoplastic transformation at the earliest stages of mammary carcinogenesis rather than cancer progression. In support of this notion, active STAT5 is more abundant in less invasive primary human breast cancers and largely absent in metastatic lesions (Nevalainen et al., 2004). Moreover, while constitutively active STAT5 causes mammary tumors in female mice, the neoplasms tend to occur after a long latency and rarely or never metastasize. To gain an understanding for this apparent discrepancy, it is important to recognize that STAT5 is not only essential for the survival but also the specification and differentiation of alveolar progenitors and their descendants. In its role as a survival factor, STAT5 neutralizes pro-apoptotic signaling pathways and is therefore able to counterbalance oncogenic stress responses and promote the manifestation and accumulation of subsequent mutations that drive neoplastic progression. As a differentiation factor, on the other hand, STAT5 has been shown to inhibit invasive characteristics of human breast cancer cells by stabilizing the expression of proteins mediating homotypic cell adhesion (Sultan et al., 2005). In the event that additional mutations take over STAT5’s critical role as a survival factor (e.g., Ras, p110, or AKT), more invasive cancer cells could emerge by acquiring yet another genetic or epigenetic alternation that prevents the activation of STAT5 or that promotes its dephosphorylation. This might explain why many metastatic breast cancer lesions lack active STAT5.
Like in many other human malignancies, the PI3K/AKT cascade including its negative regulator PTEN is one of the most frequently mutated signaling pathways in breast cancer. The recent observation that expression of hyperactive STAT5 can increase the incidence of PTEN-associated mammary tumorigenesis in a mouse model for human Cowden Syndrome underlines the significance of the crosstalk between JAK2/STAT5 signaling and the PI3K/AKT cascade in mammary carcinogenesis (Schmidt et al., 2014a). Additionally, deficiency in STAT5 completely prevented the onset of mammary cancer in mice that carry one copy of the mutant PtenG129E allele. These findings suggests that targeting the JAK2/STAT5 signaling might be a suitable alternative to prophylactic mastectomy in order to prevent the genesis of breast cancer in patients with Cowden Syndrome. Interestingly, the ligand-controlled downregulation of active STAT5 also resulted in a decelerated growth of established mammary tumors (Schmidt et al., 2014a). Neoplasms associated with mutant PTEN possess very high levels of active AKT1, and the outcome of that experiment might imply that, similar to cancer-initiating cells of the normal gland, the transcriptional activation of Akt1 (i.e., Akt1m) may still have been under the control of STAT5 in primary tumors. These findings imply that targeting this transcription factor or its upstream kinase JAK2 could be beneficial for therapy of a subset of primary mammary tumors that retain active STAT5, which controls the expression PI3K and AKT1. To some extent, this strategy may also apply for a subset of breast cancers that exhibit a rebound activation of AKT1 following mTOR inhibition (Fruman and Rommel, 2014). While a potential role of STAT5 as part of this feedback activation of AKT warrants further investigation, a recent study by Britschgi and coworkers (2012) showed that a compensatory increase in JAK2/STAT5 signaling can offset the inhibition of mTOR and PI3K in selected triple-negative breast cancer cell lines. Although deficiency in JAK2 alone does not seem to affect the growth of mammary tumors in genetic models, possibly due to the overexpression and activation of ERBB receptor tyrosine kinases (Sakamoto et al., 2009; Sakamoto et al., 2010), the findings by Britschgi et al. may highlight the importance to co-targeting JAK2/STAT5 signaling and the PI3K/AKT/mTOR cascade in more advanced, triple negative breast cancers.
In addition to therapeutic approaches that are aimed at disrupting the kinase functions or protein interactions within signaling cascades, it might be feasible to target particular signal transducers on the transcriptional level. The findings that STATs can control the expression of certain PI3 kinase subunits or AKT1 in mammary epithelial cells in a cytokine- or growth factor-dependent manner may open up alternative opportunities to modulate the expression of signal transducers on the transcriptional level. Studies are needed to determine whether the expression of AKT1 from the alternative Akt1m promoter in the mammary gland could be modulated by inhibiting STAT5 at various stages of neoplastic progression. The quantitative analysis of Akt1m mRNA expression in various murine cancer models showed that this specific Akt1 transcript was expressed in virtually all primary mammary tumors regardless of the cancer-initiating mutation or tumor subtype (Schmidt et al., 2014b). More interestingly, the human genome also contains a DNA sequence that is orthologues to the murine Akt1m. The putative promoter immediately upstream of the novel AKT1m exon is highly conserved and gives rise to four alternative splice variants that all encode the full-length AKT1 protein (Schmidt et al., 2014b). Although the majority of human breast cancer cell lines as well as a subset of primary human breast cancers exhibited an upregulated expression of the novel AKT1m RNAs, their biological significance has not yet been determined. Unfortunately, the expression of AKT1 does not seem to closely mirror the levels of active STAT5 in breast cancer cell lines. Nonetheless, the suggested significance of active STAT5 as part of a compensatory mechanism to counteract mTOR inhibition now provides a sound rationale to examine the transcriptional regulation of Akt1 as part of the rebound activation of this kinase.
Summary and Concluding Remarks
The development and physiology of the mammary gland relies on several cellular programs that are controlled in a sequential manner by hormones and locally synthesized cytokines. Upon binding to their corresponding receptors, these growth factors trigger the activation of a variety of common signal transduction cascades. Many peptide hormones and cytokines that are crucial for mammary gland development signal through receptor-associated Janus kinases and STAT proteins. In fact, there is not a single cellular program during postnatal mammary gland development that does not coincide with the activation of at least one JAK/STAT signaling cascade, and therefore the development of the gland is indeed a “STāT affair” as Lothar Hennighausen and colleagues remarked almost 20 years ago (Hennighausen et al., 1997). Among the five STAT proteins that are expressed and sequentially activated during mammary gland development, only the two STAT5 isoforms are indispensable for the genesis and secretory function of the gland. Following lactation, the synchronous deactivation of STAT5 and tyrosine phosphorylation of STAT3 are required for a coordinated remodeling of the mammary epithelium. The frequently stated view, however, that the activation of STAT3 alone is indicative for apoptosis or even sufficient to induce the cell death program is incorrect since active STAT5 can effectively sustain the survival of epithelial cells despite activation of STAT3 and its downstream effectors. This is a principal issue worth highlighting to gain a better comprehension why persistently active STAT5 is oncogenic. It is also evident that breast cancer cells express active STAT3 when STAT5 or other mechanisms promoting survival (e.g., increased RTK signaling, loss of PTEN, PI3K/AKT1 mutations) are engaged that override the intrinsic cell death program mediated by inflammatory cytokines.
STAT proteins are transcription factors that activate, and in some instances repress, numerous target genes. Among those targets, STAT5 and STAT3 enhance the expression of certain PI3 kinase subunits that seem to have discrete roles in the growth and physiology of the mammary gland. With its ability to enhance the expression of p85α, p110α and AKT1, STAT5 can augment signaling through the PI3K/AKT signal transduction cascade, which explains why a gain-of-function of STAT5 or AKT1 in the postlactational mammary gland causes very similar phenotypical abnormalities (i.e., sustained survival despite STAT3 activation). The binding of tyrosine phosphorylated STAT5 to the SH2 domain of the p85α regulatory subunit of PI3K in a PRL signaling-dependent manner suggests that STAT5 may also directly participate in the signaling of PI3K complexes. The collective observations in genetic models that overexpress or lack active STAT5 and AKT or that express mutant PTEN support the notion that central functions of STAT5 as a survival factor during normal mammary gland development and as an oncogene during mammary carcinogenesis are mediated by the PI3K/AKT pathway (Figure 1). Conditional knockout studies have revealed that STAT5 is solely activated by JAK2 in the mammary epithelium, whereas STAT1, 3 and 6 are controlled by receptor complexes that rely on the functionality of JAK1. This may provide a unique opportunity to target STATs and particular PI3K subunits via inhibition of their upstream Janus kinases in normal, preneoplastic, and perhaps malignant mammary epithelial cells.
Acknowledgments
Financial support: Public Health Service, grant numbers CA117930 (to K.U.W.) and CA009476 (to P.D.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: The authors have nothing to disclose.
References
- Abell K, Bilancio A, Clarkson RW, Tiffen PG, Altaparmakov AI, Burdon TG, Asano T, Vanhaesebroeck B, Watson CJ. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7:392–398. doi: 10.1038/ncb1242. [DOI] [PubMed] [Google Scholar]
- Abubaker K, Luwor RB, Zhu H, McNally O, Quinn MA, Burns CJ, Thompson EW, Findlay JK, Ahmed N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer. 2014;14:317. doi: 10.1186/1471-2407-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackler S, Ahmad S, Tobias C, Johnson MD, Glazer RI. Delayed mammary gland involution in MMTV-AKT1 transgenic mice. Oncogene. 2002;21:198–206. doi: 10.1038/sj.onc.1205052. [DOI] [PubMed] [Google Scholar]
- al Sakkaf KA, Dobson PR, Brown BL. Prolactin induced tyrosine phosphorylation of p59fyn may mediate phosphatidylinositol 3-kinase activation in Nb2 cells. J Mol Endocrinol. 1997;19:347–350. doi: 10.1677/jme.0.0190347. [DOI] [PubMed] [Google Scholar]
- Boxer RB, Stairs DB, Dugan KD, Notarfrancesco KL, Portocarrero CP, Keister BA, Belka GK, Cho H, Rathmell JC, Thompson CB, et al. Isoform-specific requirement for Akt1 in the developmental regulation of cellular metabolism during lactation. Cell Metab. 2006;4:475–490. doi: 10.1016/j.cmet.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Brisken C, O’Malley B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisken C, Park S, Vass T, Lydon JP, O’Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci USA. 1998;95:5076–5081. doi: 10.1073/pnas.95.9.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Brockman JL, Schuler LA. Prolactin signals via Stat5 and Oct-1 to the proximal cyclin D1 promoter. Mol Cell Endocrinol. 2005;239:45–53. doi: 10.1016/j.mce.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffarel MM, Zaragoza R, Pensa S, Li J, Green AR, Watson CJ. Constitutive activation of JAK2 in mammary epithelium elevates Stat5 signalling, promotes alveologenesis and resistance to cell death, and contributes to tumourigenesis. Cell Death Differ. 2012;19:511–522. doi: 10.1038/cdd.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int J Cancer. 2004;108:665–671. doi: 10.1002/ijc.11619. [DOI] [PubMed] [Google Scholar]
- Creamer BA, Sakamoto K, Schmidt JW, Triplett AA, Moriggl R, Wagner KU. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol Cell Biol. 2010;30:2957–2970. doi: 10.1128/MCB.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Wiesen JF, Werb Z, Young P, Hom YK, Cooke PS, Lubahn DB. Paracrine mechanisms of mouse mammary ductal growth. Adv Exp Med Biol. 2000;480:93–97. doi: 10.1007/0-306-46832-8_11. [DOI] [PubMed] [Google Scholar]
- D’Cruz CM, Moody SE, Master SR, Hartman JL, Keiper EA, Imielinski MB, Cox JD, Wang JY, Ha SI, Keister BA, Chodosh LA. Persistent parity-induced changes in growth factors, TGF-beta3, and differentiation in the rodent mammary gland. Mol Endocrinol. 2002;16:2034–2051. doi: 10.1210/me.2002-0073. [DOI] [PubMed] [Google Scholar]
- Das R, Vonderhaar BK. Prolactin as a mitogen in mammary cells. J Mammary Gland Biol Neoplasia. 1997;2:29–39. doi: 10.1023/a:1026369412612. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin RA, Kirken RA, Malabarba MG, Farrar WL, Rui H. Prolactin activates Ras via signaling proteins SHC, growth factor receptor bound 2, and son of sevenless. Endocrinology. 1995;136:3512–3518. doi: 10.1210/endo.136.8.7628388. [DOI] [PubMed] [Google Scholar]
- Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci USA. 2007;104:14718–14723. doi: 10.1073/pnas.0706933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno Vara JA, Carretero MV, Geronimo H, Ballmer-Hofer K, Martin-Perez J. Stimulation of c-Src by prolactin is independent of Jak2. Biochem J. 2000;345(Pt 1):17–24. doi: 10.1042/0264-6021:3450017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdou I, Paly J, Hue-Beauvais C, Pessemesse L, Clark J, Djiane J. Expression by transgenesis of a constitutively active mutant form of the prolactin receptor induces premature abnormal development of the mouse mammary gland and lactation failure. Biol Reprod. 2004;70:718–728. doi: 10.1095/biolreprod.103.019448. [DOI] [PubMed] [Google Scholar]
- Grimley PM, Dong F, Rui H. Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999;10:131–157. doi: 10.1016/s1359-6101(99)00011-8. [DOI] [PubMed] [Google Scholar]
- Grimm SL, Seagroves TN, Kabotyanski EB, Hovey RC, Vonderhaar BK, Lydon JP, Miyoshi K, Hennighausen L, Ormandy CJ, Lee AV, et al. Disruption of steroid and prolactin receptor patterning in the mammary gland correlates with a block in lobuloalveolar development. Mol Endocrinol. 2002;16:2675–2691. doi: 10.1210/me.2002-0239. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Haricharan S, Dong J, Hein S, Reddy JP, Du Z, Toneff M, Holloway K, Hilsenbeck SG, Huang S, Atkinson R, et al. Mechanism and preclinical prevention of increased breast cancer risk caused by pregnancy. Elife. 2013;2:e00996. doi: 10.7554/eLife.00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Signaling pathways in mammary gland development. Dev Cell. 2001;1:467–475. doi: 10.1016/s1534-5807(01)00064-8. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW, Wagner KU, Liu X. Developing a mammary gland is a stat affair. J Mammary Gland Biol Neoplasia. 1997;2:365–372. doi: 10.1023/a:1026347313096. [DOI] [PubMed] [Google Scholar]
- Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes K, Wickenden JA, Allen JE, Watson CJ. Conditional deletion of Stat3 in mammary epithelium impairs the acute phase response and modulates immune cell numbers during post-lactational regression. J Pathol. 2012;227:106–117. doi: 10.1002/path.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys RC, Hennighausen L. Signal transducer and activator of transcription 5a influences mammary epithelial cell survival and tumorigenesis. Cell Growth Differ. 1999;10:685–694. [PubMed] [Google Scholar]
- Humphreys RC, Krajewska M, Krnacik S, Jaeger R, Weiher H, Krajewski S, Reed JC, Rosen JM. Apoptosis in the terminal endbud of the murine mammary gland: a mechanism of ductal morphogenesis. Development. 1996;122:4013–4022. doi: 10.1242/dev.122.12.4013. [DOI] [PubMed] [Google Scholar]
- Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001;21:2203–2212. doi: 10.1128/MCB.21.6.2203-2212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavnilovitch E, Groner B, Barash I. Overexpression and forced activation of stat5 in mammary gland of transgenic mice promotes cellular proliferation, enhances differentiation, and delays postlactational apoptosis. Mol Cancer Res. 2002;1:32–47. [PubMed] [Google Scholar]
- Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ, Sprigg N, McKenzie AN, Watson CJ. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 2007;134:2739–2750. doi: 10.1242/dev.003194. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- Klover PJ, Muller WJ, Robinson GW, Pfeiffer RM, Yamaji D, Hennighausen L. Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia. 2010;12:899–905. doi: 10.1593/neo.10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS, Hennighausen L, Furth PA. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci USA. 1997;94:3425–3430. doi: 10.1073/pnas.94.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gallego MI, Smith GH, Robinson GW, Hennighausen L. Functional release of Stat5a-null mammary tissue through the activation of compensating signals including Stat5b. Cell Growth Differ. 1998;9:795–803. [PubMed] [Google Scholar]
- Liu X, Robinson GW, Hennighausen L. Activation of Stat5a and Stat5b by tyrosine phosphorylation is tightly linked to mammary gland differentiation. Mol Endocrinol. 1996;10:1496–1506. doi: 10.1210/mend.10.12.8961260. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11:179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- Llovera M, Touraine P, Kelly PA, Goffin V. Involvement of prolactin in breast cancer: redefining the molecular targets. Exp Gerontol. 2000;35:41–51. doi: 10.1016/s0531-5565(99)00078-9. [DOI] [PubMed] [Google Scholar]
- Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci USA. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 2011;71:4344–4351. doi: 10.1158/0008-5472.CAN-10-3827. [DOI] [PubMed] [Google Scholar]
- Milocco LH, Haslam JA, Rosen J, Seidel HM. Design of conditionally active STATs: insights into STAT activation and gene regulatory function. Mol Cell Biol. 1999;19:2913–2920. doi: 10.1128/mcb.19.4.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuenschwander S, Schwartz A, Wood TL, Roberts CT, Jr, Hennighausen L, LeRoith D. Involution of the lactating mammary gland is inhibited by the IGF system in a transgenic mouse model. J Clin Invest. 1996;97:2225–2232. doi: 10.1172/JCI118663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, Sauter G, Rui H. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–2060. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Nyga R, Pecquet C, Harir N, Gu H, Dhennin-Duthille I, Regnier A, Gouilleux-Gruart V, Lassoued K, Gouilleux F. Activated STAT5 proteins induce activation of the PI 3-kinase/Akt and Ras/MAPK pathways via the Gab2 scaffolding adapter. Biochem J. 2005;390:359–366. doi: 10.1042/BJ20041523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, Kelly PA. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- Pensa S, Lloyd-Lewis B, Sargeant TJ, Resemann HK, Kahn CR, Watson CJ. Signal transducer and activator of transcription 3 and the phosphatidylinositol 3-kinase regulatory subunits p55alpha and p50alpha regulate autophagy in vivo. Febs j. 2014a;281:4557–4567. doi: 10.1111/febs.13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensa S, Neoh K, Resemann HK, Kreuzaler PA, Abell K, Clarke NJ, Reinheckel T, Kahn CR, Watson CJ. The PI3K regulatory subunits p55alpha and p50alpha regulate cell death in vivo. Cell Death Differ. 2014b;21:1442–1450. doi: 10.1038/cdd.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer LM, Mullersman JE, Pfeffer SR, Murti A, Shi W, Yang CH. STAT3 as an adapter to couple phosphatidylinositol 3-kinase to the IFNAR1 chain of the type I interferon receptor. Science. 1997;276:1418–1420. doi: 10.1126/science.276.5317.1418. [DOI] [PubMed] [Google Scholar]
- Philp JA, Burdon TG, Watson CJ. Differential activation of STATs 3 and 5 during mammary gland development. FEBS Lett. 1996;396:77–80. doi: 10.1016/0014-5793(96)01069-1. [DOI] [PubMed] [Google Scholar]
- Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–4339. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- Robinson GW. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 2007;8:963–972. doi: 10.1038/nrg2227. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- Rosa Santos SC, Dumon S, Mayeux P, Gisselbrecht S, Gouilleux F. Cooperation between STAT5 and phosphatidylinositol 3-kinase in the IL-3-dependent survival of a bone marrow derived cell line. Oncogene. 2000;19:1164–1172. doi: 10.1038/sj.onc.1203418. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Creamer BA, Triplett AA, Wagner KU. The Janus kinase 2 is required for expression and nuclear accumulation of cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:1877–1892. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Lin WC, Triplett AA, Wagner KU. Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: Implications for cancer prevention and therapy. Cancer Res. 2009;69:6642–6650. doi: 10.1158/0008-5472.CAN-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Schmidt JW, Wagner KU. Generation of a novel MMTV-tTA transgenic mouse strain for the targeted expression of genes in the embryonic and postnatal mammary gland. PLoS One. 2012;7:e43778. doi: 10.1371/journal.pone.0043778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Triplett AA, Schuler LA, Wagner KU. Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene. 2010;29:5359–5369. doi: 10.1038/onc.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Wehde BL, Radler PD, Triplett AA, Wagner KU. Generation of Janus kinase 1 (JAK1) conditional knockout mice. Genesis. 2016a;54:582–588. doi: 10.1002/dvg.22982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Wehde BL, Yoo KH, Kim T, Rajbhandari N, Shin HY, Triplett AA, Radler PD, Schuler F, Villunger A, et al. Janus Kinase 1 Is Essential for Inflammatory Cytokine Signaling and Mammary Gland Remodeling. Mol Cell Biol. 2016b;36:1673–1690. doi: 10.1128/MCB.00999-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995;55:3915–3927. [PubMed] [Google Scholar]
- Santos SC, Lacronique V, Bouchaert I, Monni R, Bernard O, Gisselbrecht S, Gouilleux F. Constitutively active STAT5 variants induce growth and survival of hematopoietic cells through a PI 3-kinase/Akt dependent pathway. Oncogene. 2001;20:2080–2090. doi: 10.1038/sj.onc.1204308. [DOI] [PubMed] [Google Scholar]
- Schmidt JW, Wehde BL, Sakamoto K, Triplett AA, Anderson SM, Tsichlis PN, Leone G, Wagner KU. Stat5 regulates the phosphatidylinositol 3-kinase/Akt1 pathway during mammary gland development and tumorigenesis. Mol Cell Biol. 2014a;34:1363–1377. doi: 10.1128/MCB.01220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JW, Wehde BL, Sakamoto K, Triplett AA, West WW, Wagner KU. Novel transcripts from a distinct promoter that encode the full-length AKT1 in human breast cancer cells. BMC Cancer. 2014b;14:195. doi: 10.1186/1471-2407-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertfeger KL, Richert MM, Anderson SM. Mammary gland involution is delayed by activated Akt in transgenic mice. Mol Endocrinol. 2001;15:867–881. doi: 10.1210/mend.15.6.0663. [DOI] [PubMed] [Google Scholar]
- Shillingford JM, Miyoshi K, Robinson GW, Grimm SL, Rosen JM, Neubauer H, Pfeffer K, Hennighausen L. Jak2 is an essential tyrosine kinase involved in pregnancy-mediated development of mammary secretory epithelium. Mol Endocrinol. 2002;16:563–570. doi: 10.1210/mend.16.3.0805. [DOI] [PubMed] [Google Scholar]
- Spike BT, Engle DD, Lin JC, Cheung SK, La J, Wahl GM. A mammary stem cell population identified and characterized in late embryogenesis reveals similarities to human breast cancer. Cell Stem Cell. 2012;10:183–197. doi: 10.1016/j.stem.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T, Salomonis N, Gusterson BA. Mammary gland involution as a multi-step process. J Mammary Gland Biol Neoplasia. 2007;12:25–35. doi: 10.1007/s10911-007-9035-7. [DOI] [PubMed] [Google Scholar]
- Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760. doi: 10.1038/sj.onc.1208203. [DOI] [PubMed] [Google Scholar]
- Tiffen PG, Omidvar N, Marquez-Almuina N, Croston D, Watson CJ, Clarkson RW. A dual role for oncostatin M signaling in the differentiation and death of mammary epithelial cells in vivo. Mol Endocrinol. 2008;22:2677–2688. doi: 10.1210/me.2008-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafaizadeh V, Klemmt P, Brendel C, Weber K, Doebele C, Britt K, Grez M, Fehse B, Desrivieres S, Groner B. Mammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells. 2010;28:928–938. doi: 10.1002/stem.407. [DOI] [PubMed] [Google Scholar]
- Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, Sharma N, Dekoninck S, Blanpain C. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 2011;479:189–193. doi: 10.1038/nature10573. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129:1377–1386. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Krempler A, Triplett AA, Qi Y, George NM, Zhu J, Rui H. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol. 2004;24:5510–5520. doi: 10.1128/MCB.24.12.5510-5520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KU, Schmidt JW. The two faces of Janus kinases and their respective STATs in mammary gland development and cancer. J Carcinog. 2011;10:32. doi: 10.4103/1477-3163.90677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994;13:2182–2191. doi: 10.1002/j.1460-2075.1994.tb06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H, Schmitt-Ney M, Groner B. Mammary gland-specific nuclear factor is present in lactating rodent and bovine mammary tissue and composed of a single polypeptide of 89 kDa. J Biol Chem. 1992;267:16365–16370. [PubMed] [Google Scholar]
- Watson CJ, Neoh K. The Stat family of transcription factors have diverse roles in mammary gland development. Semin Cell Dev Biol. 2008;19:401–406. doi: 10.1016/j.semcdb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Weber MS, Boyle PL, Corl BA, Wong EA, Gwazdauskas FC, Akers RM. Expression of ovine insulin-like growth factor-1 (IGF-1) stimulates alveolar bud development in mammary glands of transgenic mice. Endocrine. 1998;8:251–259. doi: 10.1385/ENDO:8:3:251. [DOI] [PubMed] [Google Scholar]
- Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- Yuan W, Stawiski E, Janakiraman V, Chan E, Durinck S, Edgar KA, Kljavin NM, Rivers CS, Gnad F, Roose-Girma M, et al. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene. 2013;32:318–326. doi: 10.1038/onc.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wen Z, Xu LZ, Darnell JE., Jr Stat1 serine phosphorylation occurs independently of tyrosine phosphorylation and requires an activated Jak2 kinase. Mol Cell Biol. 1997;17:6618–6623. doi: 10.1128/mcb.17.11.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]