

Abstract
Cardiac damage associated with iron overload is the most common cause of morbidity and mortality in patients with hereditary hemochromatosis, but the precise mechanisms leading to disease progression are largely unexplored. Here we investigated the effects of iron overload and age on cardiac hypertrophy using 1-, 5- and 12-month old Hfe-deficient mice, an animal model of hemochromatosis in humans. Cardiac iron levels increased progressively with age, which was exacerbated in Hfe-deficient mice. The heart/body weight ratios were greater in Hfe-deficient mice at 5- and 12-month old, compared with their age-matched wild-type controls. Cardiac hypertrophy in 12-month old Hfe-deficient mice was consistent with decreased alpha myosin and increased beta myosin heavy chains, suggesting an alpha-to-beta conversion with age. This was accompanied by cardiac fibrosis and up-regulation of NFAT-c2, reflecting increased calcineurin/NFAT signaling in myocyte hypertrophy. Moreover, there was an age-dependent increase in the cardiac isoprostane levels in Hfe-deficient mice, indicating elevated oxidative stress. Also, rats fed high-iron diet demonstrated increased heart-to-body weight ratios, alpha myosin heavy chain and cardiac isoprostane levels, suggesting that iron overload promotes oxidative stress and cardiac hypertrophy. Our findings provide a molecular basis for the progression of age-dependent cardiac stress exacerbated by iron overload hemochromatosis.
Introduction
Iron is an essential micronutrient in almost all living organisms for various metabolic processes, including oxygen transport, oxidative phosphorylation and neurotransmitter homeostasis1, 2. Abnormal iron metabolism due to either iron deficiency or overload results in multiple organ dysfunctions3. For example, iron deficiency causes microcytic anemia, cognitive impairment and growth retardation, while excess iron promotes the generation of reactive oxygen species (ROS) and increases oxidative stress that consequently damages parenchymal tissues3. Thus, iron levels must be maintained within physiological limits in order to avoid pathological manifestations.
Hereditary hemochromatosis (HH) is an iron overload disorder that occurs primarily due to a deficiency in hepcidin4, 5, the principal iron regulatory hormone that maintains systemic iron homeostasis by limiting intestinal iron absorption and iron efflux from the macrophages6. While loss of function in several iron modulators (e.g. hepcidin, hemojuvelin, transferrin receptor 2) is associated with decreased hepcidin expression and increased iron absorption1, mutations in the HFE (hyperferremia) gene are the most common cause of iron overload hemochromatosis7, which affects 0.5% of the North American populations8. In addition, iron overload occurs due to repeated blood transfusions in several anemias, including β thalassemia, sickle cell anemia and myelodysplastic syndrome9, making iron overload disorders a global health problem10.
Since there is no regulated pathway for iron excretion, the absorbed iron progressively accumulates in various organs, including liver, heart and pancreas, leading to liver cirrhosis, cardiomyopathy, diabetes and arthritis11. In particular, cardiomyopathy is the most common cause of morbidity and mortality in patients with iron overload disorders9, 12, 13. For instance, Engle et al. described the prevalence of iron overload associated cardiomyopathy and heart failure in patients with thalassemia14. Moreover, cardiovascular diseases contribute to the morbidity and mortality of patients with HH15, 16. In contrast, some studies indicate no association between cardiac dysfunction and hemochromatosis17, 18. Carpenter et al. showed that there is no evidence for hypertrophy in patients with β thalassemia and hereditary hemochromatosis, when compared with healthy subjects17. These findings suggest that iron overload alone may not promote cardiac damage and that other factor(s) could increase the susceptibility to the progression of cardiovascular disorders associated with iron loading.
In iron overload disorders, iron uptake by the heart is much slower than that by the liver, and thus cardiac iron accumulation occurs much later than hepatic iron overload19, 20. While iron is stored in the heart as ferritin, excessive or labile iron can promote the formation of ROS, resulting in tissue damage and subsequent organ failure19. Importantly, iron stores increase with age21, and it has been well-documented that aging contributes to increased oxidative stress22–24. For example, elevated ROS by the aged mitochondria along with concomitant decreases in the antioxidant defense mechanisms promote the damage of various tissues, including the heart25. Several studies have also shown a strong association between oxidative stress and the development of cardiac hypertrophy26, 27. These lines of evidence suggest that iron overload could increase age-associated oxidative stress and predispose to cardiac damage.
Despite a large body of evidence that patients with iron overload suffer from heart-related problems, the precise mechanisms of cardiotoxicity in iron overload hemochromatosis is not clearly understood. The present study was aimed at characterizing the underlying mechanisms involved in cardiac hypertrophy in iron overload hemochromatosis using Hfe-deficient mice, a mouse model of HH in humans, and to evaluate the influence of age on the disease progression. The role of iron loading in the development of cardiac hypertrophy was also verified by a rat model of dietary iron overload. Our results provide important evidence that individuals with iron overload HH could be more susceptible to age-associated cardiac stress, likely due to increased ROS mediated by elevated iron stores in the heart.
Materials and Methods
Animal care and procedures
Breeders of Hfe-deficient (Hfe −/−)28–30 and wild-type (Hfe +/+) control mice on the 129S6/SvEvTac background were kindly provided by Dr. Nancy Andrews (Duke University Medical Center, NC). The Hfe −/− mice display the same iron loading HH phenotype observed in humans28–35. Weanling mice were fed facility chow (250 mg iron/kg diet) and water ad libitum. To examine the effect of age on iron-related cardiac stress, age-matched male Hfe −/− and Hfe +/+ mice (1, 5 and 12 months of age) were used for the study. Mice were euthanized by isoflurane overdose, followed by exsanguination and the removal of heart and liver. To examine the effect of iron overload on cardiac hypertrophy without an influence of Hfe deficiency, an animal model of dietary iron overload was included: weanling Sprague-Dawley rats were fed iron overload diet (10,000 mg carbonyl iron/kg diet; Harlan Teklad) or basal diet (50 mg iron/kg diet) for 5 weeks, followed by euthanasia for tissue collection. All experiments were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Northeastern University Animal Care and Use Committee.
Non-heme iron analysis
Liver and heart tissues were incubated in a 15-fold volume of acid solution (10% trichloroacetic acid, 3 M HCl) in 65 °C water bath for 20 h. Samples (0.08 mL) were mixed with a reaction buffer (10% thioglycolic acid and 1% bathophenanthroline disulfonic acid in saturated sodium acetate) for colorimetric reaction36. Serum iron was determined as previously described36. The optical density was measured using UV/Vis spectrophotometer at 535 nm. Non-heme iron concentration was determined based on serially-diluted iron standard solutions. Data were presented in ppm (i.e. µg iron per gram of wet tissue weight).
Fluorescence microscopy
Heart tissues were cryopreserved using tissue freezing medium (OCT). Tissue sections (10 µm) were fixed with acetone and stained with bispecific anti-myosin antibody37, followed by incubation with a polymer which was conjugated to anti-dithiopropionic acid-rhodamine isothiocyanate. The samples were then imaged using fluorescence microscopy (Nikon Eclipse E400). Polymer controls were performed for all of the fluorescence microscopy experiments to confirm the specificity of detection. The relative degree of myosin expression was evaluated by four researchers who were blinded to the experiments, based on the rank order of fluorescence signal intensity of all individual microscope images. The combined scores of the signal intensities were used to quantify and compare the differences among the groups.
Western blot analysis
Snap-frozen heart tissues were homogenized in RIPA buffer (50 mM Tris, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, pH 7.5) containing protease inhibitors (Complete Mini, Roche) with 0.5 mM phenylmethanesulfonylfluoride. Tissue homogenates were centrifuged at 16,000 g for 6 min at 4 °C. Protein concentrations in heart homogenates were determined by the Bradford assay. The tissue extracts (20–80 µg protein) were electrophoresed on 10% gels and transferred to nitrocellulose membranes for 150 mA for 3 h. The membranes were incubated with blocking solution (0.05% Tween 20, 5% non-fat milk in TBS) for 1 h at room temperature, followed by incubation with primary antibodies in 2% non-fat milk at 4 °C for overnight. Antibodies used were mouse anti-alpha myosin heavy chain (AMHC, 1:500, Abcam) and mouse anti-beta myosin heavy chain (BMHC, 1:1,000, Sigma-Aldrich). Blots were probed with mouse anti-actin (MP Biomedicals) as a loading control. Secondary antibodies were sheep anti-mouse antibodies (GE Healthcare). Immunoreactivity was detected using ECL West Dura substrate (Thermo Scientific). Protein bands were visualized by ChemiDoc XRS (Bio-Rad) and intensities of protein bands were quantified using Image Lab (version 4.1, Bio-Rad).
Histopathological analysis
Heart tissues from 12-month old Hfe −/− mice and their age-matched wild-type mice were fixed in 4% formalin, paraffin-embedded and sectioned into thin slices (5 µm), which were mounted on microscope slides for H&E and Masson’s trichrome staining.
Real-time qPCR
RNA was isolated from snap-frozen tissues of Hfe −/− and Hfe +/+ mice using TRI reagent (Sigma-Aldrich) as per the manufacturer’s instructions. RNA (1 µg) was reversely transcribed into cDNA, which was used for real-time polymerase chain reaction assays. The iScriptTM reverse transcription supermix and iTaqTM universal SYBR® green supermix were obtained from Bio-Rad, USA. Primers for the nuclear factor of activated T-cells (NFAT) c1, c2, c3, and c438 were obtained from Eurofins, MWG Operon. The expression levels of the NFAT subtypes were normalized to those of β-actin to compare the relative expression of NFAT subtype mRNA levels between Hfe −/− and Hfe +/+ mice.
Isoprostane analysis
Free isoprostane levels in the heart tissues were measured using 8-isoprostane EIA assay kit (Cayman Chemical). Briefly, tissues (20–30 mg) were homogenized in 0.1 M Tris buffer (pH 7.4) with 1 mM EDTA and 0.005% butylated hydroxytoluene. The homogenates were centrifuged at 8,000 g for 10 min at 4 °C. The supernatant was further diluted with EIA buffer and loaded into the strips pre-coated with mouse anti-rabbit IgG. The tracer (acetylcholinesterase linked to 8-isoprostane) and antiserum to 8-isoprostane were added to the samples in the wells and incubated for 18 h at 4 °C. After wash, the color was developed by incubating the plate in Ellman’s reagent for 2 h and the absorbance was measured with a spectrophotometer at 410 nm. Concentrations of isoprostane were calculated based on the standard curve.
Statistical analysis
Values reported were expressed as means ± SEM. Two-way ANOVA was employed to assess the effects of age and the Hfe gene, as well as interaction effects (age x Hfe gene), followed by the Tukey’s post-hoc analysis for pairwise comparisons (Systat; version 13). Differences were considered significant at p < 0.05.
Results
Hfe−/− mice display cardiac hypertrophy with age
The body weight progressively increased in an age-dependent manner in both Hfe +/+ and Hfe −/− mice (Table 1). However, there were no significant differences in the body weight between Hfe −/− mice and their age-matched wild-type controls. Liver and heart weights also significantly increased as the age increased. Notably, Hfe −/− mice displayed significantly greater heart-to-body weight ratios at 5-month old (12% increase, p = 0.022) and 12-month-old (15% increase, p = 0.003) when compared with their age-matched wild-type counterparts (Table 1). These results indicate a significant association between Hfe deficiency and age in cardiac hypertrophy. The liver-to-body weight ratios were greater in 12-month-old Hfe −/− mice compared with age-matched Hfe +/+ controls. Combined, these results demonstrate that loss of Hfe function results in enlarged heart and liver with age.
Table 1.
Physiological characteristics of Hfe-deficient mice at 1, 5 and 12 months of age.
| N | 1-month | 5-month | 12-month | ||||
|---|---|---|---|---|---|---|---|
| Hfe +/+ | Hfe −/− | Hfe +/+ | Hfe −/− | Hfe +/+ | Hfe −/− | ||
| Body weight† (g) | 8–12 | 14.5 (0.4) | 15.6 (0.3) | 27.4 (0.5) | 26.6 (0.5) | 34.4 (1.3) | 33.4 (0.4) |
| Heart weight†,#,^ (g) | 8–12 | 0.081 (0.002) | 0.079 (0.001) | 0.124 (0.003) | 0.135 (0.003) | 0.162 (0.009) | 0.187* (0.005) |
| Liver weight†,# (g) | 8–12 | 0.62 (0.04) | 0.73 (0.02) | 1.03 (0.05) | 0.98 (0.03) | 1.10 (0.06) | 1.25* (0.02) |
| Heart/Body weight†,^ (%) | 8–12 | 0.56 (0.02) | 0.51* (0.01) | 0.45 (0.01) | 0.51* (0.01) | 0.49 (0.02) | 0.56* (0.01) |
| Liver/Body weight†,# (%) | 8–12 | 4.26 (0.19) | 4.68* (0.10) | 3.77 (0.15) | 3.67 (0.10) | 3.20 (0.15) | 3.76* (0.08) |
Data are presented as the means (SEM). Two-way ANOVA was employed to assess the main effects (age and Hfe gene) as well as interaction effects (age x Hfe gene), followed by the Tukey’s post-hoc analysis for pairwise comparisons (Systat; version 13).
†p < 0.05, age effect.
#p < 0.05, Hfe effect.
^p < 0.05, age x Hfe effect.
*p < 0.05, Hfe −/− vs. age-matched Hfe +/+ mice.
Hfe deficiency elevates iron in the heart with age
Levels of serum iron were significantly higher in Hfe −/− mice than in their age-matched wild-type controls at 5-months (p < 0.001) and 12-months old (p < 0.001), but not at 1-month old (Fig. 1A), suggesting a gradual iron loading in the circulation over time in the absence of Hfe. The liver non-heme iron content gradually increased with age in both Hfe +/+ and Hfe −/− mice, but was significantly increased in Hfe −/− mice at all ages studied compared with their age-matched wild-type controls (Fig. 1B). There was an age-dependent, progressive increase in the steady-state levels of iron in the heart, regardless of Hfe expression, suggesting that cardiac iron content increases with age (Fig. 1C). Moreover, cardiac non-heme iron levels were significantly higher in Hfe −/− mice at the age of 12 months (36% increase; p < 0.001), but not of 1 month or 5 months, when compared with age-matched wild-type mice (Fig. 1C). These results demonstrate that the heart becomes loaded with iron with age, which is exacerbated in Hfe deficiency.
Figure 1.
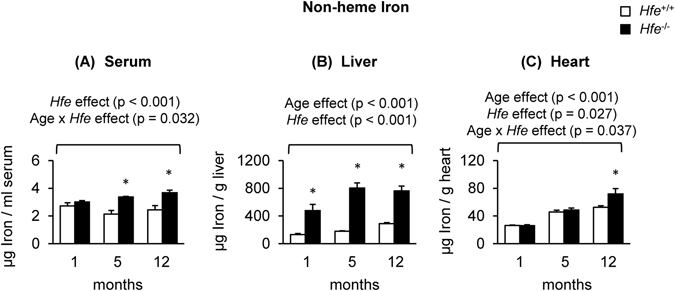
Non-heme iron levels in serum, liver and heart of Hfe +/+ and Hfe −/− mice. Non-heme iron content in serum (A), liver (B) and heart (C) was measured colorimetrically using bathophenanthroline (n = 5–9 per group). Data are presented as the means ± SEM. *p < 0.05 between Hfe −/− and Hfe +/+ mice assessed by the two-way ANOVA, followed by the Tukey’s post-hoc analysis.
Myosin levels are increased in the heart of Hfe−/− mice
Our observation of cardiac hypertrophy upon Hfe deficiency in older ages prompted us to determine the levels of α- and β-myosin heavy chains, indicative of cardiac hypertrophy, in the heart of these mice. We first characterized the expression levels of cardiac myosins by fluorescence microscopy using heart cryosections; myosin levels appeared to be increased in 5-month and 12-month old mice, but not in 1-month old mice, upon Hfe deficiency (Fig. 2A). This reflects an increased expression of myosins by loss of Hfe function with age. To obtain a more quantitative interpretation, we employed western blot analysis and found significantly lower levels of α-myosin heavy chains (42% decrease) and increased levels of β-myosin heavy chain expression in 12-month old Hfe −/− mice (653% increase) (Fig. 2B). Taken together, our results show that increased cardiac hypertrophy in Hfe deficiency is associated with elevated expression of myosin heavy chains in the heart and that there is a shift in cardiac myosin expression from α- to β-myosin heavy chains as the age increases.
Figure 2.
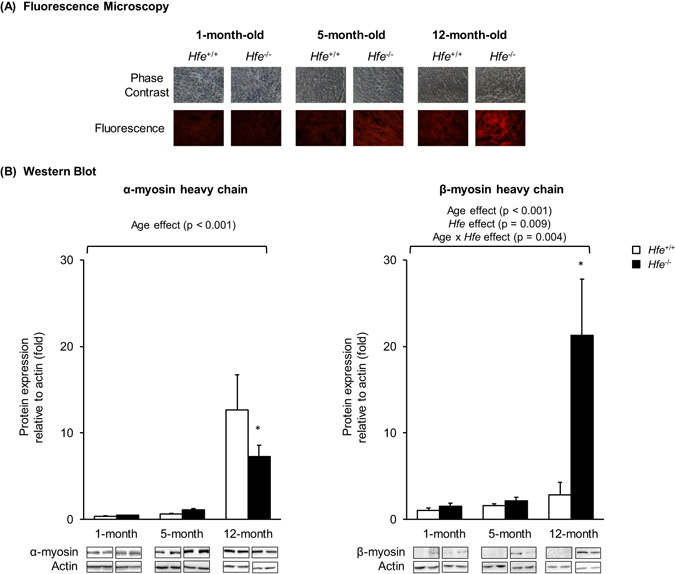
Effect of age on the expression of cardiac myosin heavy chains in Hfe +/+ and Hfe −/− mice. (A) Cardiac myosin expression in 1-month, 5-month and 12-month old Hfe +/+ and Hfe −/− mice (n = 3–5 per group) was evaluated by fluorescence microscopy. Frozen heart tissues were sectioned using cryostat (10 µm thickness) and stained with bispecific myosin antibody polymer, which was conjugated to anti-dithiopropionic acid-rhodamine isothiocyanate. Sections stained without bispecific polymer were used as background control. (B) Representative immunoblots are shown for α- and β-myosin heavy chains in heart tissues of 1-month, 5-month and 12-month old Hfe +/+ and Hfe −/− mice (n = 4–6 per group). The bar graph represents the relative expression in the protein level of α- and β-myosin heavy chains, normalized to that of actin. Data are presented as the means ± SEM. *p < 0.05 between Hfe −/− and Hfe +/+ mice assessed by the two-way ANOVA, followed by the Tukey’s post-hoc analysis.
Cardiac fibrosis is increased in Hfe−/− mice
Since 12-month old Hfe −/− mice showed signs of cardiac hypertrophy along with increased iron content, heart tissues from 12-month old animals were further examined for histopathological characteristics. H&E and Masson’s trichrome staining demonstrated increased cardiac fibrosis in Hfe −/− mice compared with age-matched Hfe +/+ mice (Fig. 3).
Figure 3.
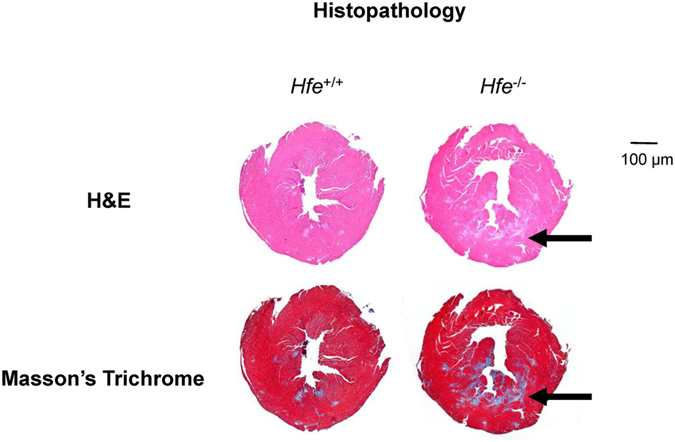
Cardiac fibrosis in Hfe +/+ and Hfe −/− mice. Heart tissues from 12-month old Hfe −/− mice and their age-matched wild-type mice were fixed in 4% formalin, embedded in paraffin and sectioned into 5 µm thin slices, which were mounted on microscope slides for H&E and Masson’s trichrome staining. Arrows indicate the areas of fibrosis.
NFAT-c2 expression is increased in Hfe−/− mice
To explore the role of cardiac iron and age in the progression of cardiac hypertrophy and up-regulation of cardiac myosin heavy chains, we compared mRNA levels of NFAT, a marker of cardiac hypertrophy, in the heart from Hfe −/− and Hfe +/+ mice at the age of 12-month old. While there were no significant changes in the expression of NFAT-c1, NFAT-c3 or NFAT-c4 between Hfe −/− and Hfe +/+ mice, the levels of NFAT-c2 were significantly up-regulated in Hfe −/− mice (Fig. 4).
Figure 4.
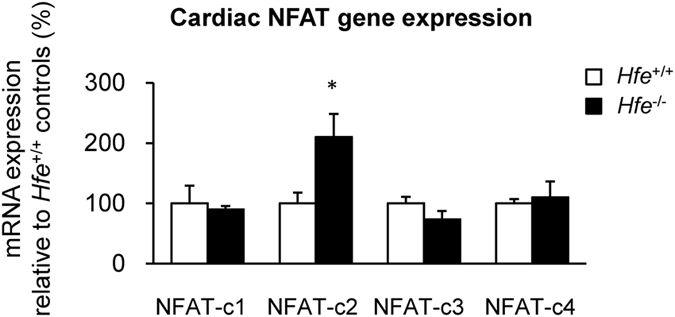
NFAT expression in Hfe +/+ and Hfe −/− mice. The expression levels of the NFAT genes in the heart from Hfe +/+ and Hfe −/− mice (n = 5 per group) at 12-months of age were quantified by real-time qPCR. Data are presented as the means ± SEM. *p < 0.05 between Hfe −/− and Hfe +/+ mice assessed by the two-sample t-test.
Cardiac isoprostane levels are increased in Hfe−/− mice
Levels of isoprostane, a marker of oxidative stress, were not different among all age groups in Hfe +/+ mice. However, Hfe −/− mice showed a progressive increase in cardiac isoprostane levels with age (Fig. 5). In addition, isoprostane levels were significantly greater in Hfe −/− mice at 12 months of age compared with age-matched wild-type mice, indicating elevated oxidative stress in the heart with age in iron overload hemochromatosis.
Figure 5.
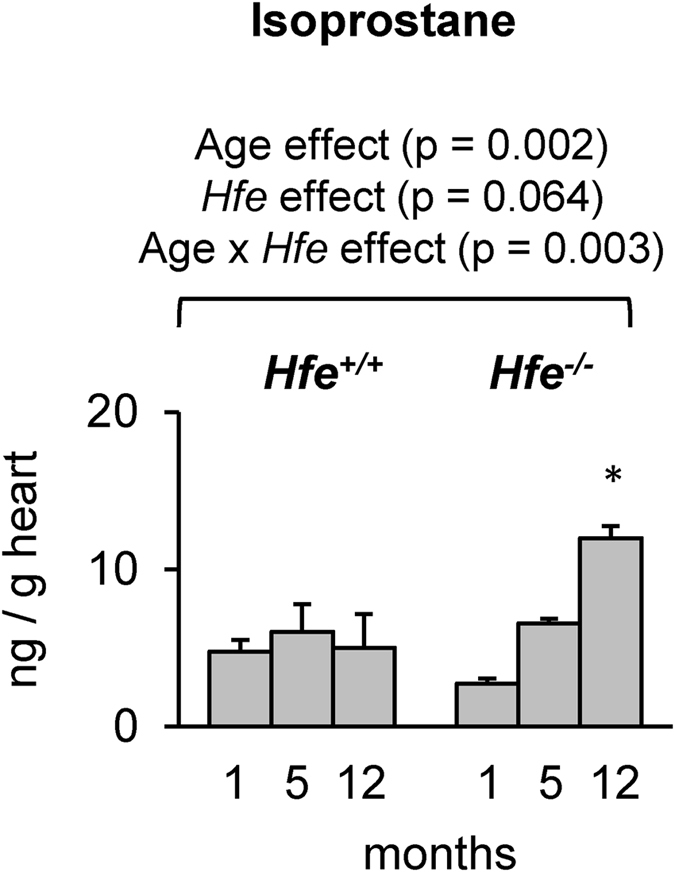
Levels of isoprostane in Hfe +/+ and Hfe −/− mice. Levels of isoprostane in the heart of Hfe +/+ and Hfe −/− mice (n = 5 per group) at 1, 5, and 12 months of age were determined by 8-isoprostane kit. Data are presented as the means ± SEM. *p < 0.05 between Hfe −/− and Hfe +/+ mice assessed by the two-way ANOVA, followed by the Tukey’s post-hoc analysis.
Dietary iron overload promotes cardiac hypertrophy
To examine if cardiac hypertrophy in Hfe −/− mice resulted from iron loading in the heart or from a potential direct effect of Hfe deficiency, we used rats fed iron overload diet. Consistent with the results from genetic iron overload animals (i.e. Hfe −/− mice), rats fed iron overload diet displayed increased heart/body weight ratios (Fig. 6A; p = 0.003), which was associated with an up-regulation of α-myosin heavy chains (Fig. 6B, p = 0.031). Again, isoprostane levels were increased in rats fed with iron overload diet (Fig. 6C, p = 0.032). Combined, these data support an idea that iron loading promotes cardiac hypertrophy and potentially increases vulnerability to heart injury in iron overload hemochromatosis.
Figure 6.
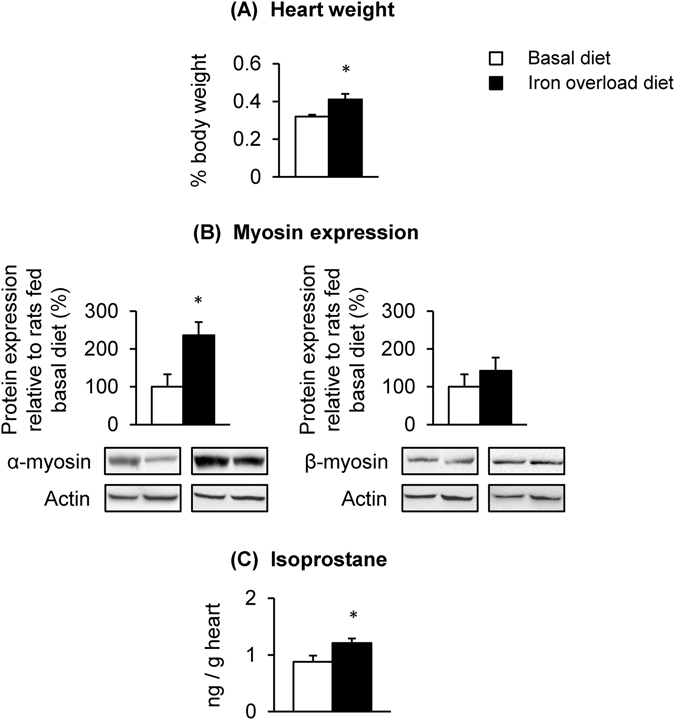
Effect of dietary iron overload on cardiac hypertrophy and oxidative stress in rats. Sprague-Dawley rats were fed basal diet (50 mg iron/kg diet) or iron overload diet (10,000 mg carbonyl iron/kg diet) for 5 weeks (n = 4 per group) to determine the heart-to-body weight ratios (A), levels of α- and β-myosin heavy chains (B) and isoprostane levels (C) in the heart. Data are presented as the means ± SEM. *p < 0.05 between rats fed basal diet and those fed iron overload diet assessed by the two-sample t-test.
Discussion
In the present study, we explored the effects of iron loading and age on cardiac hypertrophy. Since HFE-related hemochromatosis represents the most prevalent iron overload disorder7, we used Hfe −/− mice with three different age groups: 1, 5 and 12 months of age. We observed an age-dependent increase in the heart-to-body weight ratio in both Hfe +/+ and Hfe −/− mice. Moreover, the increase was greater in Hfe deficiency, which was associated with increased cardiac iron content and oxidative stress, as well as elevated levels of cardiac β-myosin heavy chains and fibrosis. To our knowledge, this is the first study to define the role of cardiac iron in age-related cardiac hypertrophy in a mouse model of hemochromatosis. Our findings suggest that elevated cardiac iron due to hemochromatosis could increase the age-associated risks of cardiovascular diseases in mice.
Consistent with previous studies29, 33, 39, our Hfe −/− mice showed increased levels of serum and liver iron, when compared with their age-matched wild-type mice. Notably, cardiac iron content was significantly elevated in Hfe −/− mice only at 12 months of age, but not at earlier time points (1 and 5 months old), suggesting that iron accumulation in the heart occurs more slowly than that in the liver19, 20. In contrast, Miranda et al.40 have shown elevated cardiac iron content in Hfe −/− mice at 8 weeks of age. This difference could result from several factors that influence iron transport and metabolism, including differences in sex and dietary iron content, as well as different methods of measurement of cardiac iron. For example, we used male mice fed standard facility chow containing 250 mg iron/kg diet, whereas Miranda et al. studied female mice with no information available on dietary iron content. In addition, we employed bathophenanthroline assay to measure “non-heme” iron content36, while Miranda et al. used atomic absorption spectrophotometry, which determines both heme and non-heme iron. It remains to be elucidated the effects of Hfe on heme iron metabolism in relation to cardiac function. While the mechanism by which cardiac iron stores are increased in hemochromatosis is not completely understood, several iron transporters, including the transferrin receptor, divalent metal transporter 1, ZRT/IRT-like protein 14 (Zip14), have been implicated in cardiac iron homeostasis41, 42. Also, a recent report demonstrated that an iron exporter ferroportin plays an important role in cardiac iron homeostasis43. In addition, studies have shown a potential role of L-type Ca2+ channels in iron transport in the heart44, 45. Further studies are warranted to investigate the molecular mechanism of cardiac iron transport in iron overload disorders and the effect of age on the expression of these transporters. Combined, our study suggests that Hfe deficiency increases iron levels in the circulation and hepatic iron stores, and later promotes iron accumulation into the heart.
The heart requires a large supply of energy, and thus, cardiomyocytes are rich in mitochondria and consume large amounts of oxygen46. However, cardiomyocytes have decreased levels of antioxidant enzymes compared with other organs47. Therefore, increased demands for oxygen along with inappropriately low levels of antioxidant enzymes make the heart highly susceptible to oxidative injury46. Importantly, iron in excess acts as a catalyst in the formation of toxic hydroxyl radicals from superoxide anion and hydrogen peroxide, resulting in the damage of macromolecules, such as DNA and proteins48, and organ dysfunction49, 50. Consistent with this idea, we found elevated levels of isoprostane, which are prostaglandin-like substances produced as a result of peroxidation of arachidonic acid by free radicals51, in 12-month old Hfe −/− mice. Notably, Hfe +/+ mice displayed increased cardiac iron with no change in isoprostane levels as the age progresses, suggesting the existence of protective mechanism(s) against age-associated oxidative stress in the presence of Hfe. Together, our study affirms that there is an age-dependent increase in cardiac oxidative stress in iron overload, which could contribute to the development of cardiac hypertrophy and thereby increase the risk of cardiovascular diseases in the elderly.
Since myocardial remodeling occurs during stress conditions52, we explored if loss of Hfe function could alter the expression of α and β myosin heavy chains, which are abundantly expressed in the heart and serve as the molecular motors. The α myosin heavy chain (AMHC) has a higher ATPase activity when compared with the β myosin heavy chain (BMHC) and thus the contractile velocity of the heart is proportional to the relative levels of AMHC and BMHC53. The AMHC accounts for >90% of total myosin heavy chains in the rodent heart54–56. In the present study, we found that Hfe −/− mice have decreased cardiac AMHC and increased BMHC at 12-months of age, suggesting an AMHC-to-BMHC transition in the production of myosin heavy chains. Studies have shown changes in the expression levels of AMHC and BMHC under stress53, 57, 58. Increased BMHC is the hallmark of cardiac hypertrophy59. Moderate induction of mean aortic pressure significantly increases the expression of BMHC in rats54. Previous reports suggest that an increase in BMHC could be an adaptive response for the optimum function of contraction/relaxation cycle60, but the underlying molecular mechanisms are poorly understood. Importantly, studies in humans and rats have also shown that the levels of BMHC increase as the age progresses53, 61. Therefore, age-associated up-regulation of BMHC, which was even greater in Hfe deficiency and correlated with cardiac iron, could predispose to cardiac hypertrophy and other types of cardiovascular diseases. Combined, our study provides important evidence that cardiac iron loading can accelerate the natural aging process of the heart, especially cardiac hypertrophy and fibrosis, and potentially heart failure, which occurs in several iron overload disorders (e.g. hemochromatosis, thalassemia and sickle cell anemia)9–11. Further, our study suggests an importance of therapeutic intervention to reduce cardiac iron in a timely manner.
To further understand the molecular mechanism of cardiac hypertrophy in Hfe −/− mice, we measured mRNA expression of cytoplasmic subunits of the NFAT transcription complex. Calcineurin/NFAT signaling has been shown to have a variety of functions in different tissues. A recent study showed that NFAT-c2 is implicated in calcineurin-mediated myocyte hypertrophy62. Our finding of elevated NFAT-c2 expression upon Hfe deficiency suggests that increased cardiac iron could activate the calcineurin-NFAT signaling pathway, which promotes cardiac hypertrophy63, 64. Further studies are necessary to better understand the mechanism by which iron regulates the calcineurin/NFAT signaling pathway and initiates hypertrophic responses. While NFAT-c1 is involved in regulating the expression levels of genes for heart valve development65, we did not observe any significant changes in the mRNA levels of the NFAT-c1 gene in Hfe −/− mice. Unlike NFAT-c1 and c2, c3 and c4 are involved in the transcriptional activation of many genes in lymphocytes and in the brain, respectively65. Together, our study suggests an important role of the NFAT-c2 gene in the development of cardiac hypertrophy, likely via the calcium-dependent signaling pathway. Alternatively, iron overload could enhance the expression of atrial natriuretic peptide/brain natriuretic peptide and/or influence hypertrophic events mediated by proinflammatory cytokines (e.g. TNFα and IL-6)66. Future studies are warranted to investigate the role of Hfe in the regulation of these events.
Since loss of Hfe function results in iron overload, we attempted to separate iron loading effect from a genetic influence of Hfe deficiency on cardiac hypertrophy by treating rats with high iron diet for 5 weeks. We used rats rather than mice since several reports have demonstrated that mice fed iron overload diet fail to show a significant increase in heart iron67 or cardiac hypertrophy68, whereas rats with dietary iron overload display increased cardiac iron and heart damage41, 69. These studies indicate that rats are a relevant rodent model to evaluate the role of acquired iron loading in the progression of cardiac hypertrophy. Our results demonstrated that dietary iron overload in rats increases heart-to-body weight ratios along with elevated isoprostane levels, supporting the idea that cardiac hypertrophy and oxidative stress in Hfe −/− mice are likely induced by iron loading, and not by a direct/intrinsic effect of Hfe deficiency. Moreover, iron overload induced by high iron diet for 5 weeks was correlated with increased AMHC content, but not BMHC, suggesting that prolonged exposure to high iron (e.g. nutrition, age, Hfe mutation, transfusion) could promote an AMHC-to-BMHC transition and heart damage. It remains to be explored how genetic alternations (Hfe deficiency) and external stimulus (iron loading) trigger the conversion of AMHC to BHMC.
The present study defines a significant role of age-associated cardiac iron loading in HFE-related hemochromatosis. Our results provide an improved understanding of the pathogenesis of cardiac hypertrophy, which will contribute to the development of novel therapeutic strategies for the treatment of iron overload-associated cardiomyopathy.
Acknowledgements
The authors are grateful to Junchi Huang, Qi Ye and Wen Ni for help during animal experiments, to Xiadong Wang for help with fluorescence microscopy, and to Dr. Yingfang Fan for help with histopathological analysis. This work was supported in part by the NIH K99/R00 ES017781 and Northeastern University TIER 1 Interdisciplinary Grant (J.K.).
Author Contributions
A.S., J.C., and J.K. designed the experiments; A.S., J.C., M.H., and S.M. performed the experiments; A.S., J.C., M.H., S.M., B.-A.K., and J.K. analyzed the data; A.S., J.C., and J.K. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Abitha Sukumaran and JuOae Chang contributed equally to this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Andrews NC, Schmidt PJ. Iron homeostasis. Annu. Rev. Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Wessling-Resnick M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem. 2014;25:1101–1107. doi: 10.1016/j.jnutbio.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews NC. Disorders of iron metabolism. N. Engl. J. Med. 1999;341:1986–1995. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 4.Fleming RE, Sly WS. Hepcidin: a putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA. 2001;98:8160–8162. doi: 10.1073/pnas.161296298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolas G, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 7.Feder JN, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 8.Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. Am. J. Epidemiol. 2001;154:193–206. doi: 10.1093/aje/154.3.193. [DOI] [PubMed] [Google Scholar]
- 9.Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J. Card. Fail. 2010;16:888–900. doi: 10.1016/j.cardfail.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y. Iron overload cardiomyopathy: better understanding of an increasing disorder. J. Am. Coll. Cardiol. 2010;56:1001–1012. doi: 10.1016/j.jacc.2010.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietrangelo, A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterol. 139, 393–408, 408 e391–392 (2010). [DOI] [PubMed]
- 12.Wood JC, et al. Physiology and pathophysiology of iron cardiomyopathy in thalassemia. Ann. N. Y. Acad. Sci. 2005;1054:386–395. doi: 10.1196/annals.1345.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zurlo MG, et al. Survival and causes of death in thalassaemia major. Lancet. 1989;2:27–30. doi: 10.1016/S0140-6736(89)90264-X. [DOI] [PubMed] [Google Scholar]
- 14.Engle MA, Erlandson M, Smith CH. Late Cardiac Complications of Chronic, Severe, Refractory Anemia with Hemochromatosis. Circulation. 1964;30:698–705. doi: 10.1161/01.CIR.30.5.698. [DOI] [PubMed] [Google Scholar]
- 15.Cecchetti G, et al. Cardiac alterations in 36 consecutive patients with idiopathic haemochromatosis: polygraphic and echocardiographic evaluation. Eur. Heart J. 1991;12:224–230. doi: 10.1093/oxfordjournals.eurheartj.a059873. [DOI] [PubMed] [Google Scholar]
- 16.Palka P, Macdonald G, Lange A, Burstow DJ. The role of Doppler left ventricular filling indexes and Doppler tissue echocardiography in the assessment of cardiac involvement in hereditary hemochromatosis. J. Am. Soc. Echocardiogr. 2002;15:884–890. doi: 10.1067/mje.2002.118032. [DOI] [PubMed] [Google Scholar]
- 17.Carpenter JP, et al. Right ventricular volumes and function in thalassemia major patients in the absence of myocardial iron overload. J. Cardiovasc. Magn. Reson. 2010;12:24. doi: 10.1186/1532-429X-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellervik C, Tybjaerg-Hansen A, Appleyard M, Ibsen H, Nordestgaard BG. Haemochromatosis genotype and iron overload: association with hypertension and left ventricular hypertrophy. J. Intern. Med. 2010;268:252–264. doi: 10.1111/j.1365-2796.2010.02217.x. [DOI] [PubMed] [Google Scholar]
- 19.Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011;124:2253–2263. doi: 10.1161/CIRCULATIONAHA.111.050773. [DOI] [PubMed] [Google Scholar]
- 20.Noetzli LJ, Carson SM, Nord AS, Coates TD, Wood JC. Longitudinal analysis of heart and liver iron in thalassemia major. Blood. 2008;112:2973–2978. doi: 10.1182/blood-2008-04-148767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairweather-Tait SJ, Wawer AA, Gillings R, Jennings A, Myint PK. Iron status in the elderly. Mech. Ageing Dev. 2014;136-137:22–28. doi: 10.1016/j.mad.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 23.Knight JA. The biochemistry of aging. Adv. Clin. Chem. 2000;35:1–62. doi: 10.1016/s0065-2423(01)35014-x. [DOI] [PubMed] [Google Scholar]
- 24.De la Fuente M. Effects of antioxidants on immune system ageing. Eur. J. Clin. Nutr. 2002;56(Suppl):3. doi: 10.1038/sj.ejcn.1601476. [DOI] [PubMed] [Google Scholar]
- 25.Andriollo-Sanchez M, et al. Age-related oxidative stress and antioxidant parameters in middle-aged and older European subjects: the ZENITH study. Eur. J. Clin. Nutr. 2005;59(Suppl):2. doi: 10.1038/sj.ejcn.1602300. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto M, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Invest. 2003;112:1395–1406. doi: 10.1172/JCI200317700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Date MO, et al. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J. Am. Coll. Cardiol. 2002;39:907–912. doi: 10.1016/S0735-1097(01)01826-5. [DOI] [PubMed] [Google Scholar]
- 28.Ajioka RS, Levy JE, Andrews NC, Kushner JP. Regulation of iron absorption in Hfe mutant mice. Blood. 2002;100:1465–1469. doi: 10.1182/blood-2001-11-0037. [DOI] [PubMed] [Google Scholar]
- 29.Levy, J. E., Montross, L. K. & Andrews, N. C. Genes that modify the hemochromatosis phenotype in mice. J. Clin. Invest. 105, 1209–1216 (2000). [DOI] [PMC free article] [PubMed]
- 30.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 31.Alsulimani HH, Ye Q, Kim J. Effect of Hfe Deficiency on Memory Capacity and Motor Coordination after Manganese Exposure by Drinking Water in Mice. Toxicol. Res. 2015;31:347–354. doi: 10.5487/TR.2015.31.4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claus Henn B, et al. Associations of iron metabolism genes with blood manganese levels: a population-based study with validation data from animal models. Environ. Health. 2011;10:97. doi: 10.1186/1476-069X-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim, J., Buckett, P. D. & Wessling-Resnick, M. Absorption of manganese and iron in a mouse model of hemochromatosis. PLoS One8 (2013). [DOI] [PMC free article] [PubMed]
- 34.Ye Q, Kim J. Loss of hfe function reverses impaired recognition memory caused by olfactory manganese exposure in mice. Toxicol. Res. 2015;31:17–23. doi: 10.5487/TR.2015.31.1.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye Q, Kim J. Effect of olfactory manganese exposure on anxiety-related behavior in a mouse model of iron overload hemochromatosis. Environ. Toxicol. Pharmacol. 2015;40:333–341. doi: 10.1016/j.etap.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang J, Kueon C, Kim J. Influence of lead on repetitive behavior and dopamine metabolism in a mouse model of iron overload. Toxicol. Res. 2014;30:267–276. doi: 10.5487/TR.2014.30.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patil V, et al. Imaging small human prostate cancer xenografts after pretargeting with bispecific bombesin-antibody complexes and targeting with high specific radioactivity labeled polymer-drug conjugates. Eur. J. Nucl. Med. Mol. Imaging. 2012;39:824–839. doi: 10.1007/s00259-011-2050-3. [DOI] [PubMed] [Google Scholar]
- 38.Wilkins BJ, et al. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol. Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhury C, et al. Accelerated transferrin degradation in HFE-deficient mice is associated with increased transferrin saturation. J. Nutr. 2006;136:2993–2998. doi: 10.1093/jn/136.12.2993. [DOI] [PubMed] [Google Scholar]
- 40.Miranda CJ, et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003;102:2574–2580. doi: 10.1182/blood-2003-03-0869. [DOI] [PubMed] [Google Scholar]
- 41.Nam H, et al. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica. 2013;98:1049–1057. doi: 10.3324/haematol.2012.072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W, et al. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell. Rep. 2015;13:533–545. doi: 10.1016/j.celrep.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lakhal-Littleton S, et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA. 2015;112:3164–3169. doi: 10.1073/pnas.1422373112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oudit GY, et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 2003;9:1187–1194. doi: 10.1038/nm920. [DOI] [PubMed] [Google Scholar]
- 45.Oudit GY, Trivieri MG, Khaper N, Liu PP, Backx PH. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. (Berl) 2006;84:349–364. doi: 10.1007/s00109-005-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gammella, E., Recalcati, S., Rybinska, I., Buratti, P. & Cairo, G. Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxid. Med. Cell. Longev. 2015, 230182 (2015). [DOI] [PMC free article] [PubMed]
- 47.Doroshow, J. H., Locker, G. Y. & Myers, C. E. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J. Clin. Invest. 65, 128–135 (1980). [DOI] [PMC free article] [PubMed]
- 48.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit. Rev. Clin. Lab Sci. 2008;45:1–23. doi: 10.1080/10408360701713104. [DOI] [PubMed] [Google Scholar]
- 50.Puntarulo S. Iron, oxidative stress and human health. Mol. Aspects Med. 2005;26:299–312. doi: 10.1016/j.mam.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Montuschi P, Barnes PJ, Roberts LJ., II Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 52.Grossman W, Paulus WJ. Myocardial stress and hypertrophy: a complex interface between biophysics and cardiac remodeling. J. Clin. Invest. 2013;123:3701–3703. doi: 10.1172/JCI69830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J. Clin. Invest. 1997;100:2362–2370. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izumo S, et al. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy. Interaction between hemodynamic and thyroid hormone-induced signals. J. Clin. Invest. 1987;79:970–977. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lompre AM, et al. Myosin isoenzyme redistribution in chronic heart overload. Nature. 1979;282:105–107. doi: 10.1038/282105a0. [DOI] [PubMed] [Google Scholar]
- 56.Mercadier JJ, et al. Myosin isoenzyme changes in several models of rat cardiac hypertrophy. Circ. Res. 1981;49:525–532. doi: 10.1161/01.RES.49.2.525. [DOI] [PubMed] [Google Scholar]
- 57.Lopez JE, et al. beta-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ. Res. 2011;109:629–638. doi: 10.1161/CIRCRESAHA.111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J. Clin. Invest. 1989;84:1693–1700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandya K, Smithies O. beta-MyHC and cardiac hypertrophy: size does matter. Circ. Res. 2011;109:609–610. doi: 10.1161/CIRCRESAHA.111.252619. [DOI] [PubMed] [Google Scholar]
- 60.Machackova J, Barta J, Dhalla NS. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can. J. Cardiol. 2006;22:953–968. doi: 10.1016/S0828-282X(06)70315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carnes CA, Geisbuhler TP, Reiser PJ. Age-dependent changes in contraction and regional myocardial myosin heavy chain isoform expression in rats. J. Appl. Physiol. (1985) 2004;97:446–453. doi: 10.1152/japplphysiol.00439.2003. [DOI] [PubMed] [Google Scholar]
- 62.Bourajjaj M, et al. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J. Biol. Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 63.Wilkins BJ, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 64.Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy: where have we been? Where are we going? J. Physiol. 2002;541:1–8. doi: 10.1113/jphysiol.2002.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crabtree, G. R. Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 276, 2313–2316 (2001). [DOI] [PubMed]
- 66.Cheng CF, Lian WS. Prooxidant Mechanisms in Iron Overload Cardiomyopathy. BioMed Res. Int. 2013;2013:740573. doi: 10.1155/2013/740573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Enculescu, M. et al. Modelling Systemic Iron Regulation during Dietary Iron Overload and Acute Inflammation: Role of Hepcidin-Independent Mechanisms. PLoS Comput. Biol. 13, e1005322 (2017). [DOI] [PMC free article] [PubMed]
- 68.Omara FO, Blakley BR, Wanjala LS. Hepatotoxicity associated with dietary iron overload in mice. Hum. Exp. Toxicol. 1993;12:463–467. doi: 10.1177/096032719301200603. [DOI] [PubMed] [Google Scholar]
- 69.Whittaker P, Hines FA, Robl MG, Dunkel VC. Histopathological evaluation of liver, pancreas, spleen, and heart from iron-overloaded Sprague-Dawley rats. Toxicol. Pathol. 1996;24:558–563. doi: 10.1177/019262339602400504. [DOI] [PubMed] [Google Scholar]