

Summary
Cassava (Manihot esculenta) feeds c. 800 million people world‐wide. Although this crop displays high productivity under drought and poor soil conditions, it is susceptible to disease, postharvest deterioration and the roots contain low nutritional content.
Here, we provide molecular identities for 11 cassava tissue/organ types through RNA‐sequencing and develop an open access, web‐based interface for further interrogation of the data.
Through this dataset, we consider the physiology of cassava. Specifically, we focus on identification of the transcriptional signatures that define the massive, underground storage roots used as a food source and the favored target tissue for transgene integration and genome editing, friable embryogenic callus (FEC). Further, we identify promoters able to drive strong expression in multiple tissue/organs.
The information gained from this study is of value for both conventional and biotechnological improvement programs.
Keywords: biotechnology, cassava (Manihot esculenta), food security, friable embryogenic callus, gene expression, organized embryogenic structures, RNA sequencing
Introduction
Cassava (Manihot esculenta) is the food security crop that feeds c. 800 million people worldwide (Liu et al., 2011; Howeler et al., 2013). Although this crop displays high productivity under drought and poor soil conditions, it is susceptible to disease, postharvest physiological deterioration and the roots contain low nutritional content (Gegios et al., 2010; Stephenson et al., 2010; Vanderschuren et al., 2014; Patil et al., 2015; Uarrota et al., 2016). Cassava improvement programs are focused on addressing these constraints but are hindered by the crop's high heterozygosity, difficulty in synchronizing flowering, low seed production and a poor understanding of the physiology of this plant (Ceballos et al., 2004). Among the major food crops, cassava is unique in its ability to develop massive, underground storage roots. Despite the importance of these structures, their basic physiology remains largely unknown, especially the molecular genetic basis of storage root development. Similarly, in cassava, the favored target tissue for transgene integration and genome editing is a friable embryogenic callus (FEC) (Taylor et al., 2001, 2012; Bull et al., 2009; Zainuddin et al., 2012; Nyaboga et al., 2013). Little is known concerning gene expression in this tissue, or its relatedness to the somatic organized embryogenic structures (OESs) from which it originates (Gresshoff & Doy, 1974; Taylor et al., 2012; Chauhan et al., 2015). Here, we provide molecular identities for 11 cassava tissue/organ types through RNA‐sequencing and develop an open access, web‐based interface for further interrogation of the data. Through this dataset, we report novel insight into the physiology of cassava and identify promoters able to drive specified expression profiles.
Materials and Methods
Plant material and tissues/organs sampled
Samples were taken from 3‐month‐old TME 204 cassava plants, grown in a glasshouse at the Donald Danforth Plant Science Center (St Louis, MO, USA). Plants were established from in vitro micropropagated plants (Taylor et al., 2012) and grown in a 12 h :12 h, light : dark photoperiod, 250–500 μmol s−1 m−2 irradiance. Day time temperatures ranged from 28 to 32°C with 70% relative humidity, and night time temperatures ranged from 25 to 27°C with 70% relative humidity. The following samples were harvested at 14:00 h: leaf blade, leaf midvein, petiole, stem, lateral buds, shoot apical meristem (SAM), storage roots, fibrous roots, and root apical meristem (RAM). For nonmeristem samples, c. 100 mg of material was collected in three separate biological replicates. For the SAM and RAM, six meristems were dissected and pooled for each of three biological replicates. For all sample types, razor blades and dissecting scopes were used to isolate the target material as much as possible though we note that clear organ boundaries do not exist for many of these sample types. All samples were frozen in liquid nitrogen after collection. Samples of TME 204 OES and FEC were generated as described previously (Chauhan et al., 2015). The OES induced on DKW/Juglans basal salts (PhytoTechnology Laboratories, Shawnee Mission, KS, USA) containing Murashige and Skoog (MS) vitamins and supplemented with 2% w/v sucrose, 50 μM picloram was sampled after 4 wk of culture. The OES was separated from the nonembryogenic materials and collected in 2 ml sampling tubes. FEC tissues were sampled after 3 wk of culture on Gresshoff and Doy basal medium supplemented with 2% w/v sucrose, 50 μM picloram, 500 μM tyrosine and 50 mg l−1 moxalactam. Approximately 200–250 mg of material was collected and the tubes containing the materials were immediately placed on dry ice.
Preparation of RNA‐seq libraries and Illumina sequencing
For nonmeristem samples, total RNA was isolated with the Spectrum Plant Total RNA Kit (Sigma). For SAM and RAM tissues, total RNA was isolated with the Arcturus PicoPure RNA Isolation Kit. RNA quality was assessed on an Agilent Bioanalyzer. For library preparation with samples other than SAM and RAM, 5 μg of RNA was used as input. For SAM and RAM, six samples were pooled to obtain a total of 500 to 600 ng each. The NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs, Ipswich, MA, USA) was used to isolate mRNA, which was then used for library prep using NEBNext mRNA Library Prep Master Mix Set for Illumina (New England BioLabs) with 13 cycles of PCR amplification. Standard library prep protocol was followed for all samples, except for the SAM and RAM in which 1 μl of fragmentation enzyme was used instead of 2 μl, and 0.5 μl of random primer instead of 1 μl. Library quality was assessed with the Agilent Bioanalyzer. In total, 32 RNA‐seq libraries were made from 11 different sample types with three biological replicates each, except for storage root which only had two biological replicates. All libraries were multiplexed into one lane of Illumina HiSeq2500.
Read mapping and gene expression analysis
Illumina RNA‐seq reads from each replicate were cleaned using Trimmomatic v.0.32 (Bolger et al., 2014). Using TopHat2 v.2.1.0 (Trapnell et al., 2009), these cleaned reads were then mapped to the version 6.1 draft assembly of Manihot esculenta AM560‐2 provided on Phytozome v.10.3 (http://phytozome.jgi.doe.gov/pz/portal.html). The read mapping output was linked to candidate gene models for each sample using Cufflinks v.2.2.1 (Trapnell et al., 2010). Cufflinks was run using default parameters and the –no‐faux‐reads flag. With this configuration, transcripts with less than 10 reads are counted as 0 and reads mapped to multiple genes are not included. The gene models from all samples of the experiment were merged into one gene model file using Cuffmerge v.2.2.1. Using the output from Cuffmerge and the read mapping files from each replicate, a differential expression analysis between sample types was performed using Cuffdiff v.2.2.1. Quality checks were performed on the Cuffdiff output using CummeRbund v.2.6.1 in R (R Core Team, 2015). The output of Cuffdiff was processed in Python with the pandas, numpy, and seaborn packages to visualize the expression data (McKinney, 2010).
Multivariate statistics
Analysis of transcript expression profiles began with those transcripts with (1) FPKM (fragments per kilobase of transcript per million reads mapped) values above a threshold of 1 FPKM and (2) those transcripts significantly differentially expressed in at least one pairwise comparison of all sample types against each other. For principal component analysis (PCA) of sample replicates, the prcomp() function in R was used with scaled FPKM values across transcripts as input. For the PCA of transcript profiles, the prcomp() function was again used with scaled mean FPKM values across samples as input. Self‐Organizing Maps (SOMs) were performed using the kohonen package in R. Scaled mean transcript expression values across samples were assigned to four nodes in a 2 × 2 hexagonal topology over 100 training iterations. To focus on those transcripts with expression profiles closest to over‐represented patterns of variance in the dataset, only those transcripts with distances from their respective nodes less than the median for the overall dataset were subsequently used and projected back onto the transcript PCA space. Data visualization for the above was carried out with the ggplot2 package in R, using geom_point() and geom_line() functions, among others. The color space for the above was determined using palettes from colorbrewer2.org.
Gene ontology (GO) enrichment analysis
GO enrichment analysis was completed using the Python goatools package (https://github.com/tanghaibao/goatools). goatools was run with the –fdr flag to calculate the False Discovery Rate (FDR) error corrected P‐value, and the –no_propagate_counts flag to prevent nodes at the root of the GO tree from being included in the analysis. GO terms for each gene were used from the annotation file provided on Phytozome. GO enrichment output was then filtered to include only enriched GO terms with a FDR error corrected P‐value < 0.001. For SOM node GO enrichment, each SOM node identified earlier was processed separately. The genes identified as part of the SOM node were used as the study group, and all genes expressed greater than 1 FPKM in at least one tissue/organ with significant differential expression in at least one pairwise comparison were used as the population or background group. For pairwise sample comparison GO enrichment, genes identified as significantly up‐regulated with a |log2(fold_change)| > 2 in one sample were treated as one study group to look at each sample separately. This resulted in two GO enrichment analyses for each pairwise comparison. Genes with at least 1 FPKM in either sample were used as the background dataset.
Identification of genes with strong, constitutive, and tissue/organ‐specific expression patterns
Custom Python code was used in a Jupyter notebook using the Pandas, NumPy, Seaborn, and SciPy packages to organize, process, and display the data (Supporting Information Notes S1). Genes with strong expression across all tissue/organ types were identified using expression values from the gene_exp.diff file produced by Cuffdiff. The genes were first checked for functional annotations, then shortened to a list of genes with a minimum expression of 300 FPKM in each material sampled. Specifically and constitutively expressed genes were identified using expression values from each replicate in the genes.read_group_tracking file produced by Cuffdiff. Genes used were annotated in the AM560‐2 v6.1 assembly on Phytozome v.10.3. For specifically expressed genes, this list was then subset by selecting genes with expression greater than 10 FPKM in the sample(s) specifically expressing the gene, and no more than 1 FPKM in all other samples. For a more relaxed analysis, genes were required to be expressed greater than 8 FPKM in the sample(s) specifically expressing the gene, and no more than 4 FPKM in all other samples. Constitutively expressed genes were identified using the replicate expression data. This list was filtered to include only genes with greater than 40 FPKM in all replicates, and then the coefficient of variation was calculated across all replicates for each gene.
Data availability
A graphical user interface was created using R shiny (v.0.13.2) to explore the tissue/organ‐specific data and discover trends therein. This application uses data from RNA‐seq differential expression analysis completed with the Tuxedo Suite pipeline (v.2.2.1), functional gene annotations from the Joint Genome Institute's Phytozome, and analysis from principle components (prcomp in R ‘stats’ package v.3.2.3) and self‐organizing maps (som in R ‘kohonen’ package v.2.0.19). The application has two main features: gene discovery based on gene expression patterns across samples; and creation of a tissue/organ‐specific heatmap of known or newly discovered genes for visualizing expression patterns. Detailed instructions are included in the application. The application can be found at: shiny.danforthcenter.org/cassava_atlas/. Additional R packages used in this application include: png (v.0.1‐7), grid (v.3.2.3), ggplot2 (v.2.1.0), shinyBS (v.0.61), shinydashboard (v.0.5.1), DT (v.0.1), stringr (v.1.0.0), mailR (v.0.4.1), and shinyjs (v.0.5.2).
In planta expression assays
Promoter fragments, listed in Notes S2, were cloned from cassava variety TME419 into a pCAMBIA vector upstream of GUS. Constructs were transformed into Agrobacterium tumefaciens strain LBA4404. Strains carrying the reporter constructs were re‐suspended in IM media (10 mM MES, pH5.6; 10 mM MgCl2; 150 μM Acetosyringeone), incubated at room temperature for 3 h and then infiltrated into Nicotiana benthemiana leaves at an OD600 = 0.1. Forty‐eight hours post‐inoculation, leaves were detached and placed in a petri dish. GUS staining solution (0.1 M NaPO4 pH7; 10 mM EDTA; 0.1% Triton X‐100; 1 mM K3Fe(CN)6; 2 mM X‐Gluc) was pipetted on to the detached leaf and a glass tube rolled across the leaf surface to lightly crush the leaf. Leaves were incubated overnight at 37°C. Before imaging, leaves were cleared of chlorophyll through several washes in 95% EtOH. To quantify GUS staining, multiple image processing steps were implemented using ImageJ to obtain the pixel statistics that are reported. The original RGB image was converted to HSL colorspace using the ‘Color Transform’ plugin and the lightness channel was extracted. The image look‐up table was changed to ‘thermal’ and a manually defined circular ROI was created whose size and shape remain constant when gathering the mean and standard deviation of the pixel intensities for each of the strains. Using the same ROI, the image was cropped for each of the strains to display the exact regions sampled.
Cassava transformation
Reporter constructs were introduced to cassava FEC cells by LBA4404 following our published methods (Chauhan et al., 2015).
Data are deposited in GEO repository: accession number GSE82279.
Results
To shed light on the development and physiology of cassava plants from a gene expression perspective, 11 tissue/organ types from cassava cultivar TME 204 were sampled for transcriptome profiling (Fig. 1). Gene expression patterns between storage and fibrous roots have previously been investigated with quantitative reverse transcription polymerase chain reaction (qRT‐PCR) (Yang et al., 2011). Expression profiles of 76 genes were considered for the previous dataset and that reported here and revealed a 96% consistency (i.e. higher expression in storage root or fibrous roots). Sample type relatedness was assessed based on Jensen–Shannon (JS) distances (Fig. 2) and PCA (Fig. 3). Biological replicates display a low squared coefficient of variation across expression values and cluster closely together in a PCA. These tests assess variation among the biological replicates and confirm the high quality of the dataset (Fig. S1A, Fig. 3a,b). Both analyses divided the 11 samples into three major groups: aerial (leaf, midvein, petiole, stem, lateral bud, and shoot apical meristem (SAM)), subterranean (storage root, fibrous root and root apical meristem (RAM)), and embryogenic (OES and FEC). Leaf and midvein, petiole and stem, lateral bud and SAM, and OES and FEC samples cluster together within the dendrogram (Fig. 2b), and occupy similar positions across the first four principal components (PCs), which collectively explain 67.3% of transcript expression level variance (Fig. 3a,b). These groupings are expected, representing leaf blade, vascular, shoot meristem, and callus‐associated tissues/organs. The root samples show more complicated relationships. Figure 2 indicates storage roots as distant from fibrous roots and RAM (Fig. 2b). Similarly, whereas the RAM, storage root and fibrous root samples cluster closely together when projected onto PC1 and PC2 (Fig. 3a), these samples occupy more disparate positions when evaluated by PC3 and PC4 (Fig. 3b). This indicates that while root samples share common gene expression patterns, sample specific signatures differentiate storage roots from the other subterranean samples.
Figure 1.
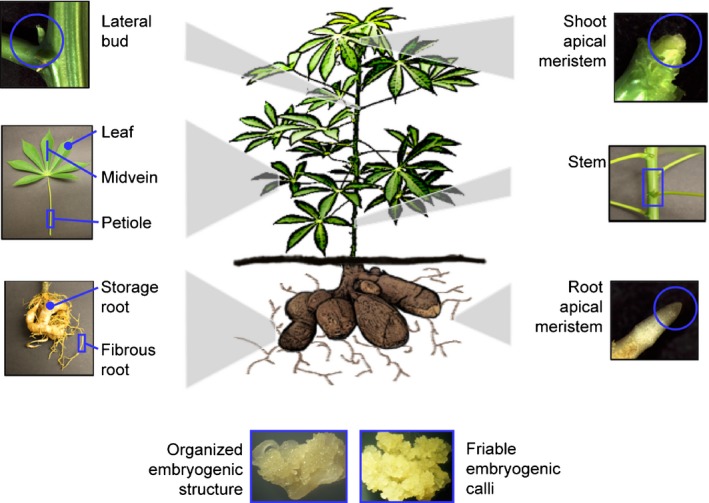
Cartoon and pictures of cassava (Manihot esculenta) tissues/organs sampled for gene expression atlas. Eleven sample types were dissected by hand and frozen in liquid nitrogen before processing for RNA sequencing library preparation.
Figure 2.
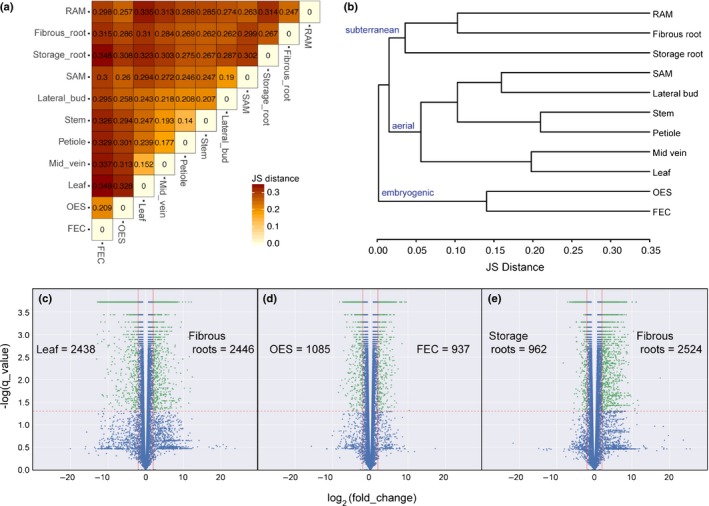
Comparison of global gene expression patterns across 11 cassava (Manihot esculenta) tissue/organ types. (a) Heatmap showing every pairwise comparison for the 11 tissue/organ types sampled, as produced by CummeRbund's csDistHeat() method. Lighter colors correspond to more closely related sample types. The numbers in each cell represent the Jensen–Shannon (JS) distance between those two samples using the mean expression values of the biological replicates. (b) Output of CummeRbund's csDendro() method. This dendrogram is created using the JS distances calculated between the consensus expression values of genes for each sample type. A low squared coefficient of variation for biological replicates was observed indicating the high quality of this dataset (Supporting Information Fig. S1A). (c–e) Volcano plots showing the differential expression of genes from leaf to fibrous roots (c), OES to FEC (d) and storage root to fibrous root (e) using FDR corrected P‐value as the y axis. Number of genes significantly up‐regulated in each sample type, for each comparison, are listed. Red vertical lines: ± log2(fold_change) = 2, red horizontal line: log score = 1.3. The green points indicate significantly differentially expressed genes based on these cutoffs. Shoot apical meristem (SAM), root apical meristem (RAM), organized embryogenic structure (OES) to friable embryogenic callus (FEC).
Figure 3.
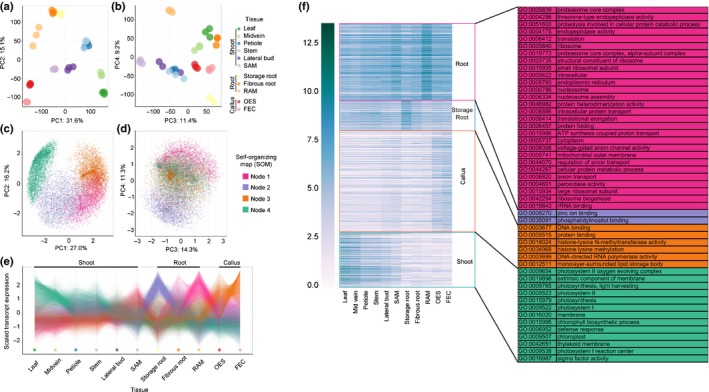
Transcript expression profiles across cassava (Manihot esculenta) samples. (a, b) Principal component analysis (PCA) performed on replicates of samples, using transcript expression levels. (c, d) PCA performed on transcript profiles, across samples. Colors correspond to self‐organizing map (SOM) nodes used to find transcripts with similar expression profiles, which cluster together in the PCA space. (e) Scaled transcript expression profiles of SOM nodes across tissue/organ types. (f) Heatmap of genes showing gene expression pattern corresponding to the nodes in (c, d). Gene ontology (GO) terms associated with these genes are listed on the right. Shoot apical meristem (SAM), root apical meristem (RAM), organized embryogenic structure (OES) to friable embryogenic callus (FEC).
Two tissue comparisons within the dataset: OES vs FEC and storage vs fibrous roots, are particularly intriguing, given how little is known about the features distinguishing their physiology. The results of a PCA on the expression profiles of individual transcripts was considered. A SOM was used to identify four main clusters of transcripts with similar expression profiles across samples, which was then projected back onto the PCA transcript space (Fig. 3c,d). To determine the identities of transcripts with shared expression profiles, we performed GO enrichment analysis for each SOM node (Fig. 3f). In addition, we directly examined the genes most highly differentially expressed between each comparison (Figs 2, 4).
Figure 4.
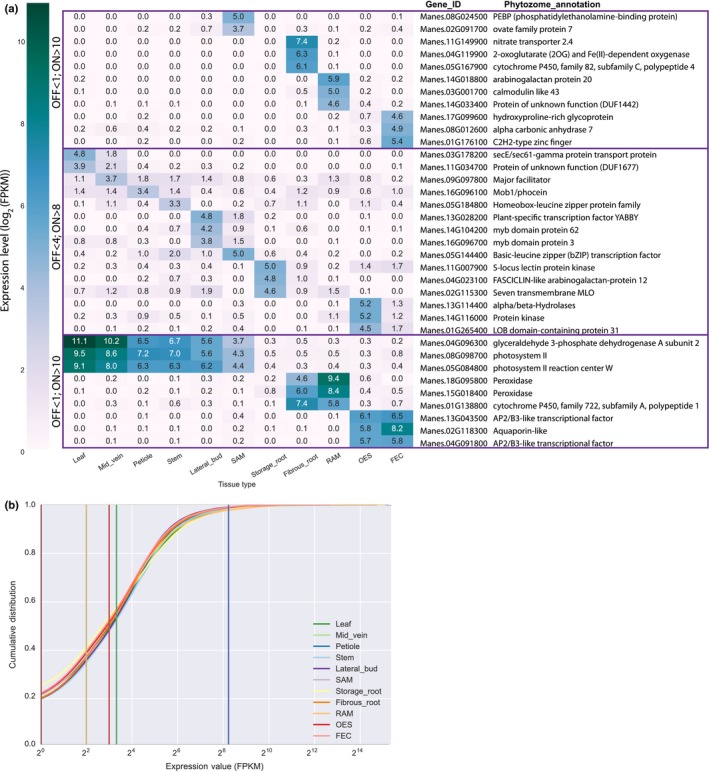
Identification of genes specifically expressed in a single or subset of cassava (Manihot esculenta) tissue/organ types. (a) Identification of genes with specified expression patterns. (top; middle) Heatmap of the most highly, uniquely expressed genes in each sample. Requirements for below and above the limits of detectable expression are listed on the left (OFF and ON, respectively). (bottom) Genes expressed highly in a subset of samples are reported. No genes specifically expressed across all subterranean samples (storage root, fibrous root and root apical meristem (RAM)) were identified so storage root was excluded from that group. (b) Cumulative distribution plot of FPKM (fragments per kilobase of transcript per million reads mapped) values of functionally annotated genes with expression in at least one sample type. The vertical lines represent three cutoffs used in the analysis: < 1 FPKM (maroon line) = below the limit of detection; < 4 FPKM (yellow line) = below the limit of detection (relaxed); > 8 FPKM (red line) = detected expression (relaxed); > 10 FPKM (green line) = detected expression; > 300 FPKM (blue line) = highly expressed genes. Shoot apical meristem (SAM), root apical meristem (RAM), organized embryogenic structure (OES) to friable embryogenic callus (FEC).
First, we used the earlier approach to examine gene expression patterns for well characterized samples and comparisons. Node 4 transcripts (teal) are highly expressed in the photosynthetic samples of the leaf and midvein (Fig. 3e). Similarly, comparison of leaf and fibrous roots revealed ~4900 genes differentially expressed greater than four‐fold (|log2(fold_change)| > 2) between samples (Fig. 2c; Notes S3). A similar number were up‐regulated in each tissue/organ and consistent with the GO term analysis presented in Fig. 3(f), the most highly up‐regulated genes in leaf samples pertained to photosynthesis activity while genes induced in fibrous root were related to lignin, ion binding, and transcription. We highlight that these analyses are complementary. The former takes an unbiased approach to identify variability within the dataset. The latter, directly looks at genes with maximum expression differences.
OES and FEC tissue are closely related with the latter generated from the former by a simple switch in the basal medium (Taylor et al., 2012). FEC tissues are highly disorganized and ultra‐juvenile in nature, consisting of proliferating, sub‐millimeter sized pre‐embryo units from which somatic embryos will regenerate on removal of auxin. Efficacy of FEC production from the OES is genotype dependent and can be challenging in some farmer‐preferred varieties, though this recalcitrance is poorly understood (Liu et al., 2011). Node 3 transcripts (burnt orange) are highly expressed in both callus samples, but especially the FEC (Fig. 3e). Node 3 transcripts are associated with GO terms related to epigenomic reprogramming (DNA methylation and histone modification). Over 2000 genes were identified as differentially expressed between OES and FEC samples (Fig. 2d; Notes S4). Genes up‐regulated in OES tissue are associated with GO tags for heme, iron and tetrapyrrole binding and oxidoreductase activity. By contrast, genes up‐regulated in FEC tissue are associated with sulfur and sulfate transport (Notes S5). Besides these key differences, our analyses emphasize the overall striking similarity between the two tissue types.
What distinguishes storage roots from other subterranean structures is ambiguous. A recent anatomical examination of these structures revealed that roots develop from the cut base of the stem cutting (basal) and from buried nodes (nodal), but that only the nodal roots will develop to form storage roots (Chaweewan & Taylor, 2015). Once initiated the storage roots develop by massive cell proliferation from the cambium to generate the central core that consists largely of xylem parenchyma in which starch is synthesized and stored. Node 1 transcripts (magenta) are highly expressed in the RAM and somewhat in the fibrous root while Node 2 transcripts (lavender) are highly expressed in the storage root suggesting that storage roots exhibit distinct gene expression patterns relative to RAM and fibrous roots. Node 1 transcripts, highly expressed in the RAM and the fibrous root, are enriched for GOs related to translation, proteolysis, and intracellular transport that might be expected for a tissue undergoing growth. Node 2 transcripts highly expressed in the storage root, are associated with zinc ion and phosphatidylinositol binding GO terms. In contrast to differentially expressed gene comparisons for leaf vs fibrous roots, and OES vs FEC, comparison of fibrous and storage roots revealed a significant shift towards gene induction in the former (Fig. 2e; Notes S6). Taken together, these data and analyses demonstrate that OES and FEC are highly similar tissue types and suggest that their difference may come mostly from the media on which they are cultured. By contrast, fibrous and storage roots appear as inherently distinct on a transcriptional level.
Promoters capable of driving gene expression in one or more defined tissue/organ types are essential for the successful application of biotechnology to improve crop plants. Currently, a limited set of promoters are available to achieve desired expression patterns for cassava in planta. For example, the root‐specific patatin promoter from Solanum tuberosum has been used to overexpress transgenes that enhance iron and zinc levels in cassava storage roots (Gaitan‐Solis et al., 2015; Narayanan et al., 2015). De Souza et al. (2006, 2009) has characterized the Pt2L4 gene (Manes.09G108300) and confirmed preferential expression in cassava storage roots but also in stems. This previously published expression pattern is consistent with the current dataset (Fig. S2). To identify cassava promoters capable of specific expression, we queried the dataset for genes expressed in a single sample type, henceforth referred to as uniquely expressed genes. To identify uniquely expressed genes, FPKM values of 1 and 10 were chosen to represent ‘below the limit of detection’ and ‘expressed’, respectively. These cutoffs were determined by investigating read mapping coverage for individual genes within our datasets. An FPKM value of less than one generally correlated with less than 1× coverage across a coding sequence. Genes expressed at greater than 10 FPKM had read mapping across the entire coding sequence. In addition, we choose an expression value of ≥ 300 FPKM as the cutoff for highly expressed genes which encompasses approximately the top 2% of expression values across our dataset. Below the limit of detection, expressed, and highly expressed cutoffs within the context of the entire dataset are displayed in Fig. 4(b). Uniquely expressed genes were identified as those expressed at greater than 10 FPKM in one sample, and less than 1 FPKM in all other samples (Fig. 4a). Applying the cutoff criteria, unique gene expression was observed for FEC, fibrous root, RAM and SAM, but not for the other seven samples. Using less stringent cutoff FPKM values (OFF < 4; ON > 8), we were able to identify uniquely expressed genes for all additional samples (Fig. 4a). In addition, we considered expression that would be constrained to the major groupings from the dendrogram in Fig. 2. Storage root was excluded from the subterranean group because of its distinct gene expression patterns (Figs 2b, 3).
In addition to identification of uniquely expressed genes, the data was queried to identify candidate promoters for driving strong gene expression within all surveyed sample types (constitutive). We identified genes that showed expression values of ≥ 300 FPKM across our entire dataset. This analysis resulted in a list of 31 genes (Fig. 5a). In order to test the in silica analysis, promoters from five of the 31 putative constitutively expressed genes were cloned and functionally validated by fusing to the uidA (GUS) reporter gene. These constructs were expressed transiently in Nicotiana benthamiana leaves and stably transformed into cassava FEC cells. Transgenic plantlets were regenerated and stained for GUS expression. At least two independent lines were stained for each construct and in all cases, similar results were observed. Four out of five tested promoters were confirmed to drive GUS expression in the six tested cassava tissue/organ types. One promoter fusion, Manes.11G159600, failed to drive expression in N. benthamiana, cassava leaves and midveins for unknown reasons (Fig. 5b, Fig. S3).
Figure 5.
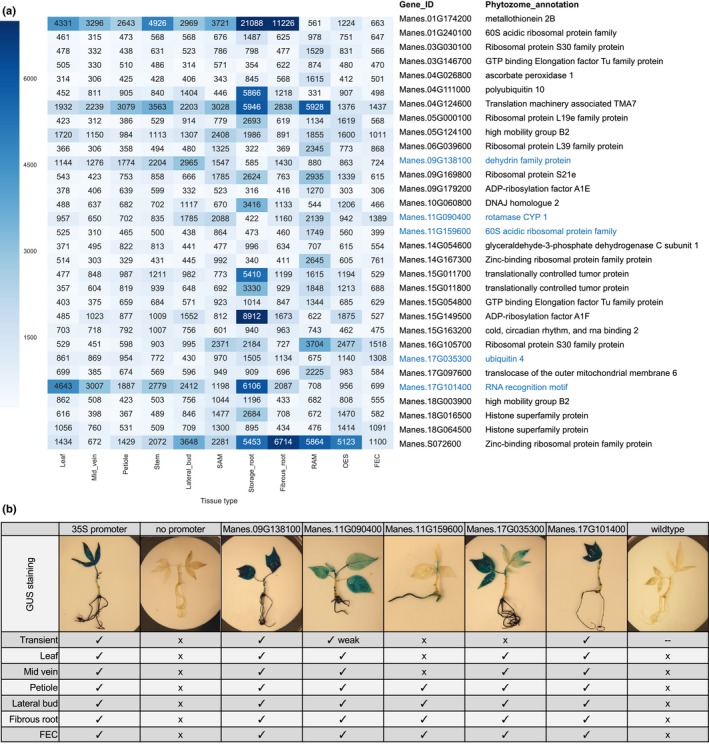
Identification of highly expressed genes in all tissue/organ types. (a) Heatmap displaying expression for 31 annotated genes expressed above 300 FPKM in all samples. Values greater than 7000 are condensed within the heatmap scale. (b) Promoter –GUS reporter gene constructs were stably transformed into cassava plants. Representative images from GUS stained, in vitro grown cassava plants are displayed. Expression (check) or no expression (x) is indicated below. Note that tissues that were difficult to stain (e.g. stems) or unavailable (e.g. storage roots, organized embryogenic structure (OES)) were not included. ‘Transient’ indicates the results of Agrobacterium mediated transient expression within Nicotiana benthamiana leaves. Raw data is presented in Supporting Information Fig. S2. Shoot apical meristem (SAM), root apical meristem (RAM), organized embryogenic structure (OES) to friable embryogenic callus (FEC).
A small collection of ‘housekeeping genes’ are routinely used for internal controls in qRT‐PCR experiments such as GTPb, PP2A, and UBQ10 (Moreno et al., 2011). While these genes are appropriate when comparing multiple samples collected from leaf material, data from the present study show that all three genes display significant variance between sample types (Fig. S4). The datasets described here were queried to identify candidate genes displaying medium level expression with low variance across the sample types. We identified genes with expression greater than 40 FPKM in all replicates with the lowest coefficient of variation in order to normalize for magnitude of expression. Figure S3 shows the top 10 candidates from our analysis in comparison to the three genes previously used. Additional research could adopt a similar approach to identify internal controls appropriate for various biotic and abiotic stress conditions.
To facilitate future analyses, a web application has been developed wherein users can specify a desired gene expression pattern across all sample types and receive a list of candidate genes. This application also allows users to visualize a heatmap of expression values for any gene of interest across each sample type. The queried gene is displayed in the PCA and overlaid SOM nodes. This application can be accessed at: shiny.danforthcenter.org/cassava_atlas/.
Discussion
To assist cassava improvement efforts, various genomic, transcriptomic and epigenomic resources have previously been described (Prochnik et al., 2012; Wang et al., 2014, 2015). Additional proteomic and metabolomics resources exist for cassava (Li et al., 2010; Uarrota et al., 2014; Vanderschuren et al., 2014; Uarrota & Maraschin, 2015). Our study provides a unique resource: we characterize the cassava transcriptome across a wide range of sample types. Comparison of gene expression patterns revealed a dramatic similarity between OES and FEC tissue. Storage roots were found to be significantly different from the other root samples, and closer examination of the data suggest that the majority of this difference comes from a lack of gene expression, consistent with the role of this organ as a sink. Our study provides new insight into cassava physiology, and the data will serve as a valuable resource for cassava researchers. In addition, we identify both genes that are constitutively expressed as well as those that are highly specific. While RNAseq has been established as a reliable method of determining expression patterns in most cases (Nagalakshmi et al., 2008), we encourage future researchers to perform functional validation on specific genes of interest. The promoters of these genes may be useful for diverse biotechnological applications, including those that seek to alter cassava metabolism and improve the value of cassava as a source of food for a large fraction of the world's population.
Author contributions
M.C.W. analyzed data and co‐wrote the manuscript. A.M.M. designed experiments and isolated samples for RNAseq analysis and co‐wrote the manuscript. A.W.H. designed experiments and made constructs. J.B. created the shiny application. R.D.C. created transgenic cassava plants. A.V. generated RNAseq libraries. N.J.T. and D.F.V. supervised the study and edited the manuscript. D.H.C. performed statistical analyses and co‐wrote the manuscript. R.S.B. designed experiments, supervised the study, and co‐wrote the paper.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Assessing variation among biological replicates.
Fig. S2 Gene expression profile for Manes.09G108300.
Fig. S3 Promoter:GUS fusion expression in Nicotiana benthamiana.
Fig. S4 Identification of constitutively expressed genes and assessment of expression variation across sample type.
Notes S1 Documentation of data analysis.
Notes S2 Promoter sequences used for GUS fusions.
Notes S3 Genes differentially expressed between leaf and fibrous roots samples.
Notes S4 Genes differentially expressed between OES and FEC samples.
Notes S5 Genes up‐regulated in FEC tissue.
Notes S6 Genes differentially expressed between storage roots and fibrous roots.
Acknowledgements
This research was supported by the Bill and Melinda Gates Foundation. Sequencing was performed at the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant #UL1 TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull SE, Owiti JA, Niklaus M, Beeching JR, Gruissem W, Vanderschuren H. 2009. Agrobacterium‐mediated transformation of friable embryogenic calli and regeneration of transgenic cassava. Nature Protocols 4: 1845–1854. [DOI] [PubMed] [Google Scholar]
- Ceballos H, Iglesias CA, Perez JC, Dixon AGO. 2004. Cassava breeding: opportunities and challenges. Plant Molecular Biology 56: 503–516. [DOI] [PubMed] [Google Scholar]
- Chauhan RD, Beyene G, Kalyaeva M, Fauquet CM, Taylor N. 2015. Improvements in Agrobacterium‐mediated transformation of cassava (Manihot esculenta Crantz) for large‐scale production of transgenic plants. Plant Cell, Tissue and Organ Culture (PCTOC) 121: 591–603. [Google Scholar]
- Chaweewan Y, Taylor N. 2015. Anatomical assessment of root formation and tuberization in cassava (Manihot esculenta Crantz). Tropical Plant Biology 8: 1–8. [Google Scholar]
- Gaitan‐Solis E, Taylor NJ, Siritunga D, Stevens W, Schachtman DP. 2015. Overexpression of the transporters AtZIP1 and AtMTP1 in cassava changes zinc accumulation and partitioning. Frontiers in Plant Science 6: 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegios A, Amthor R, Maziya‐Dixon B, Egesi C, Mallowa S, Nungo R, Gichuki S, Mbanaso A, Manary MJ. 2010. Children consuming cassava as a staple food are at risk for inadequate zinc, iron, and vitamin A intake. Plant Foods for Human Nutrition 65: 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresshoff PM, Doy CH. 1974. Derivation of a haploid cell line from Vitis vinifera and the importance of the stage of meiotic development of the anthers for haploid culture of this and other genera. Zeitschrift fur Pflanzenphysiologie 73: 132–141. [Google Scholar]
- Howeler R, Lutaladio N, Thomas G. 2013. Save and grow: cassava – a guide to sustainable production intensification. Rome, Italy: Food and Agriculture Organization of the United States of America. [Google Scholar]
- Li K, Zhu W, Zeng K, Zhang Z, Ye J, Ou W, Rehman S, Heuer B, Chen S. 2010. Proteome characterization of cassava (Manihot esculenta Crantz) somatic embryos, plantlets and tuberous roots. Proteome Science 8: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zheng Q, Ma Q, Gadidasu KK, Zhang P. 2011. Cassava genetic transformation and its application in breeding. Journal of Integrative Plant Biology 53: 552–569. [DOI] [PubMed] [Google Scholar]
- McKinney W. 2010. Data structures for statistical computing in python Proceedings of the 9th Python in Science Conference 2010: 51–56.
- Moreno I, Gruissem W, Vanderschuren H. 2011. Reference genes for reliable potyvirus quantitation in cassava and analysis of Cassava brown streak virus load in host varieties. Journal of Virological Methods 177: 49–54. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan N, Beyene G, Chauhan RD, Gaitan‐Solis E, Grusak MA, Taylor N, Anderson P. 2015. Overexpression of Arabidopsis VIT1 increases accumulation of iron in cassava roots and stems. Plant Science 240: 170–181. [DOI] [PubMed] [Google Scholar]
- Nyaboga E, Njiru J, Nguu E, Gruissem W, Vanderschuren H, Tripathi L. 2013. Unlocking the potential of tropical root crop biotechnology in east Africa by establishing a genetic transformation platform for local farmer‐preferred cassava cultivars. Front Plant Sci 4: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil BL, Legg JP, Kanju E, Fauquet CM. 2015. Cassava brown streak disease: a threat to food security in Africa. Journal of General Virology 96(Pt 5): 956–968. [DOI] [PubMed] [Google Scholar]
- Prochnik S, Marri PR, Desany B, Rabinowicz PD, Kodira C, Mohiuddin M, Rodriguez F, Fauquet C, Tohme J, Harkins T et al 2012. The Cassava genome: current progress, future directions. Tropical Plant Biology 5: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2015. R: a language and environment for statistical computing. Voemma, Austria: R Foundation for Statistical Computing; [WWW document] URL http://www.r-project.org/. [Google Scholar]
- de Souza CR, Aragao FJ, Moreira EC, Costa CN, Nascimento SB, Carvalho LJ. 2009. Isolation and characterization of the promoter sequence of a cassava gene coding for Pt2L4, a glutamic acid‐rich protein differentially expressed in storage roots. Genetics and Molecular Research 8: 334–344. [DOI] [PubMed] [Google Scholar]
- de Souza CR, Carvalho LJ, de Almeida ER, Gander ES. 2006. A cDNA sequence coding for a glutamic acid‐rich protein is differentially expressed in cassava storage roots. Protein and Peptide Letters 13: 653–657. [DOI] [PubMed] [Google Scholar]
- Stephenson K, Amthor R, Mallowa S, Nungo R, Maziya‐Dixon B, Gichuki S, Mbanaso A, Manary M. 2010. Consuming cassava as a staple food places children 2–5 years old at risk for inadequate protein intake, an observational study in Kenya and Nigeria. Nutrition Journal 9: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NJ, Gaitán‐Solís E, Moll T, Trauterman B, Jones T, Pranjal A, Trembley C, Abernathy V, Corbin D, Fauquet CM. 2012. A high‐throughput platform for the production and analysis of transgenic cassava (Manihot esculenta) plants. Tropical Plant Biology 5: 127–139. [Google Scholar]
- Taylor NJ, Masona MV, Carcamo R, Ho T, Schopke C, Fauquet CM. 2001. Production of embryogenic tissues and regeneration of transgenic plants in cassava (Manihot esculenta Crantz). Euphytica 120: 25–34. [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uarrota VG, Maraschin M. 2015. Metabolomic, enzymatic, and histochemical analyzes of cassava roots during postharvest physiological deterioration. BMC Research Notes 8: 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uarrota VG, Moresco R, Coelho B, Nunes Eda C, Peruch LA, Neubert Ede O, Rocha M, Maraschin M. 2014. Metabolomics combined with chemometric tools (PCA, HCA, PLS‐DA and SVM) for screening cassava (Manihot esculenta Crantz) roots during postharvest physiological deterioration. Food Chemistry 161: 67–78. [DOI] [PubMed] [Google Scholar]
- Uarrota VG, Nunes Eda C, Peruch LA, Neubert Ede O, Coelho B, Moresco R, Dominguez MG, Sanchez T, Melendez JL, Dufour D et al 2016. Toward better understanding of postharvest deterioration: biochemical changes in stored cassava (Manihot esculenta Crantz) roots. Food Sciences and Nutrition 4: 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren H, Nyaboga E, Poon JS, Baerenfaller K, Grossmann J, Hirsch‐Hoffmann M, Kirchgessner N, Nanni P, Gruissem W. 2014. Large‐scale proteomics of the cassava storage root and identification of a target gene to reduce postharvest deterioration. Plant Cell 26: 1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Beyene G, Zhai J, Feng S, Fahlgren N, Taylor NJ, Bart R, Carrington JC, Jacobsen SE, Ausin I. 2015. CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proceedings of the National Academy of Sciences, USA 112: 13729–13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Feng B, Xiao J, Xia Z, Zhou X, Li P, Zhang W, Wang Y, Møller BL, Zhang P et al 2014. Cassava genome from a wild ancestor to cultivated varieties. Nature Communications 5: 5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, An D, Zhang P. 2011. Expression profiling of cassava storage roots reveals an active process of glycolysis/gluconeogenesis. Journal of Integrative Plant Biology 53: 193–211. [DOI] [PubMed] [Google Scholar]
- Zainuddin IM, Schlegel K, Gruissem W, Vanderschuren H. 2012. Robust transformation procedure for the production of transgenic farmer‐preferred cassava landraces. Plant Methods 8: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Assessing variation among biological replicates.
Fig. S2 Gene expression profile for Manes.09G108300.
Fig. S3 Promoter:GUS fusion expression in Nicotiana benthamiana.
Fig. S4 Identification of constitutively expressed genes and assessment of expression variation across sample type.
Notes S1 Documentation of data analysis.
Notes S2 Promoter sequences used for GUS fusions.
Notes S3 Genes differentially expressed between leaf and fibrous roots samples.
Notes S4 Genes differentially expressed between OES and FEC samples.
Notes S5 Genes up‐regulated in FEC tissue.
Notes S6 Genes differentially expressed between storage roots and fibrous roots.
Data Availability Statement
A graphical user interface was created using R shiny (v.0.13.2) to explore the tissue/organ‐specific data and discover trends therein. This application uses data from RNA‐seq differential expression analysis completed with the Tuxedo Suite pipeline (v.2.2.1), functional gene annotations from the Joint Genome Institute's Phytozome, and analysis from principle components (prcomp in R ‘stats’ package v.3.2.3) and self‐organizing maps (som in R ‘kohonen’ package v.2.0.19). The application has two main features: gene discovery based on gene expression patterns across samples; and creation of a tissue/organ‐specific heatmap of known or newly discovered genes for visualizing expression patterns. Detailed instructions are included in the application. The application can be found at: shiny.danforthcenter.org/cassava_atlas/. Additional R packages used in this application include: png (v.0.1‐7), grid (v.3.2.3), ggplot2 (v.2.1.0), shinyBS (v.0.61), shinydashboard (v.0.5.1), DT (v.0.1), stringr (v.1.0.0), mailR (v.0.4.1), and shinyjs (v.0.5.2).